

Abstract
Introduction/Aims
ALS is a heterogeneous disease that may be complicated or in part driven by inflammation. NP001, a regulator of macrophage activation, was associated with slowing disease progression in those with higher levels of the plasma inflammatory marker C‐reactive protein (CRP) in phase 2A studies in ALS. Here, we evaluate the effects of NP001 in a phase 2B trial, and perform a post hoc analysis with combined data from the preceding phase 2A trial.
Methods
The phase 2B trial enrolled 138 participants within 3 y of symptom onset and with plasma hs‐CRP values >1.13 mg/L. They were randomized 1:1 to receive either placebo or NP001 for 6 mo. Change from baseline ALSFRS‐R scores was the primary efficacy endpoint. Secondary endpoints included vital capacity (VC) change from baseline and percentage of participants showing no decline of ALSFRS‐R score over 6 mo (non‐progressor).
Results
The phase 2B study did not show significant differences between placebo and active treatment with respect to change in ALSFRS‐R scores, or VC. The drug was safe and well tolerated. A post hoc analysis identified a 40‐ to 65‐y‐old subset in which NP001‐treated patients demonstrated slower declines in ALSFRS‐R score by 36% and VC loss by 51% compared with placebo. A greater number of non‐progressors were NP001‐treated compared with placebo (p = .004).
Discussion
Although the phase 2B trial failed to meet its primary endpoints, post hoc analyses identified a subgroup whose decline in ALSFRS‐R and VC scores were significantly slower than placebo. Further studies will be required to validate these findings.
Keywords: amyotrophic lateral sclerosis (ALS), C‐reactive protein (CRP), inflammation, NP001, respiratory function
Abbreviations
- AE
adverse event
- ALS
amyotrophic lateral sclerosis
- ALSFRS‐R
ALS Functional Rating Scale‐Revised
- ECG
electrocardiogram
- FVC
forced vital capacity
- HLA‐DR
human leukocyte antigen – DR isotype
- HO‐1
heme oxygenase‐1
- hs‐CRP
high sensitivity C‐Reactive Protein
- IL‐18
interleukin‐18
- LPS
lipopolysaccharide
- mITT
modified intent‐to‐treat
- NADPH
nicotinamide adenine dinucleotide phosphate
- NF‐κB
nuclear factor kappa B
- PROACT
Pooled Resource Open‐Access ALS Clinical Trials
- SVC
slow vital capacity
- TauCl
taurine chloramine
- TEAE
treatment‐emergent adverse events
- VC
vital capacity
- wr‐CRP
wide range C‐reactive protein
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is associated with inappropriate immune system dysfunction involving nuclear factor kappa B (NF‐kB) activation, proinflammatory factor production and progressive changes in motor neuron function. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 Factors elaborated by spinal cord microglia have been identified that both damage and inhibit repair of neurons injured by the accumulation of misfolded proteins as a general model. 9 The recent description of ALS patient monocyte subsets becoming more activated over time as reflected by human leukocyte antigen – DR isotype (HLA‐DR) cell surface levels 10 confirms our earlier studies implicating innate immune dysfunction in disease progression. 11 Therefore, drugs that interfere with or regulate this process could have a therapeutic impact on disease.
NP001 is a proprietary formulation of pH stabilized, purified chlorite. In the presence of heme‐associated iron, presumably from the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex on the surface of phagocytic cells, it is converted from a prodrug through a hypochlorite intermediate, to an intracellular form of taurine chloramine (TauCl). 12 , 13 , 14 TauCl is a long‐lived effector molecule within macrophages that down‐regulates NF‐kB expression and inhibits production of pro‐inflammatory cytokines in part through activation of heme oxygenase‐1 (HO‐1). 12 , 13 , 15 A phase 1 controlled trial of NP001 in patients with ALS demonstrated the safety, tolerability, and dose dependent down‐regulation of monocyte activation. 16
A prior phase 2A study assessed the safety, tolerability, and preliminary efficacy of NP001 for slowing progression of ALS. 17 A modest, non‐significant trend in slowing of progression was noted, greatest in those patients with elevated systemic inflammation at baseline. Elevated inflammation was defined as having plasma wide‐range C‐reactive protein (wr‐CRP) values >1.13 mg/L in whom the change from baseline on the ALSFRS‐R scale was −2.2 points over the 6‐mo treatment period compared with −5.1 points in the placebo group. Additionally, more than twice as many patients on high dose NP001 (25%) as on placebo (11%) did not progress during 6 mo of treatment. Most “non progressors” had an elevated baseline biomarker of inflammation. Elevated inflammation associated interleukin‐18 (IL‐18) and lipopolysaccharide (LPS) levels at baseline normalized following treatment with 2 mg/kg NP001. 17
The study objectives of phase 2B were to evaluate the safety, tolerability, and efficacy of NP001 in ALS patients with elevated plasma CRP. The study evaluated ALSFRS‐R score and slow vital capacity (SVC) change from baseline and quantitated the number of patients that did not lose any ALSFRS‐R score over 6 mo. The goal of the post hoc combined trial analysis was to identify characteristics of ALS patients responsive to NP001.
2. METHODS
2.1. Clinical trials overview
This report covers a previously unpublished phase 2B clinical trial for treatment of ALS patients with NP001 or placebo. In addition, data from this trial were merged with data from an earlier phase 2A clinical trial for the treatment of ALS. 17 These two 6‐mo phase 2 trials were conducted in ALS participants by Neuraltus Pharmaceuticals, Inc. (Palo Alto, CA), and were registered at ClinicalTrials.gov: NCT01281631 for phase 2A 17 and NCT02794857 for phase 2B. All of the chemical manufacturing, toxicology, and clinical records from these two trials were obtained from Neuraltus by Neuvivo Inc. (Palo Alto, CA) in 2019.
Both phase 2A and 2B studies were approved by the clinical site institutional ethics committees, and informed consent was obtained from all participants.
2.2. Methods for NP001 2B trial
2.2.1. Phase 2B participants
The phase 2B trial was conducted from August 2016 through December 2017 at 21 sites in the United States and one in Canada. The study was fully enrolled and did not terminate early. Participants were men and women who were diagnosed with probable or definite ALS according to the Revised El Escorial criteria. 18 Participants were required to have an onset of ALS‐related weakness within 3 y, SVC > 65% of predicted, and a clinically estimated life expectancy of > 6 mo. All participants were required to have high sensitivity CRP (hs‐CRP) levels at screening greater than 1.13 mg/L, the median value noted in the phase 2A trial. Participants receiving riluzole had to be on a stable dose for 30 days. Individuals on continuous positive airway pressure or bilevel positive airway pressure, those with active pulmonary disease, and those who had received recent immunotherapy were excluded.
2.2.2. Phase 2B design
The study was randomized, double‐blind, and placebo‐controlled, and study drug was administered over 6 cycles. Participants were allocated 1:1 to NP001 2 mg/kg/day, or placebo, and stratified by ALS site of onset (bulbar/limb). Study drug was infused over 30 min by an infusion pump. Participants received a total of 20 infusions over 6 monthly cycles as previously defined. 17 There was 1 mo between the start of each cycle. Cycle 1 consisted of five consecutive daily infusions. Cycles 2, 3, 4, 5, and 6 each consisted of 3 consecutive daily infusions. The randomized population who received at least one dose of NP001 or placebo and had at least one post‐baseline ALSFRS‐R total score assessment was pre‐defined as the modified intent‐to‐treat (mITT) population.
Investigators, site staff, and ALSFRS‐R raters remained blinded to treatment allocation throughout the study. An independent data monitoring committee periodically evaluated safety during the trial.
2.2.3. Safety evaluation
All randomized participants who received at least one dose of study drug are included in the analysis and summaries of safety data. Tolerability and safety were assessed via adverse event (AE) reports, vital signs, electrocardiograms (ECGs), laboratory parameters, physical examinations, and formal phlebitis scoring.
2.2.4. Phase 2B efficacy endpoints
Participants were seen for efficacy and safety assessments at monthly intervals. The primary efficacy measure in this study was the mean change from baseline in the ALSFRS‐R score following 6 mo of treatment. Secondary efficacy measures included the mean percentage change from baseline in predicted SVC following 6 mo of treatment, and percentage of participants who did not worsen as assessed by the change from baseline in the ALSFRS‐R score following 6 months of treatment (non‐progressors).
2.3. Methods for post hoc analysis
The primary endpoint in each of the phase 2 trials was change from baseline ALSFRS‐R score in participants receiving 2 mg/kg NP001 or placebo over the 6‐mo studies. The secondary endpoint in each trial was percent change in predicted vital capacity (VC) over the six‐months of study, which serves as a predictor of mortality in ALS patients. 19 The phase 2A trial assessed the percent predicted VC change in forced vital capacity (FVC), whereas the phase 2B assessed percent predicted VC change in SVC. Percent predicted VC change from baseline values between FVC and SVC are comparable 20 ; therefore, the evaluation of NP001 effects on VC combined changes from both trials normalized to “percent of predicted VC change from baseline [100 × (predicted VC at study end − predicted VC at baseline)/predicted VC at baseline]”. The plasma concentrations of wr‐CRP as exploratory biomarkers were measured for the phase 2A. For the phase 2B, the plasma hs‐CRP values were used as one of the entry criteria. To combine two sets of CRP data from the phase 2A and phase 2B for data analysis, the plasma wr‐CRP values from the phase 2A were converted to hs‐CRP using a simple calibration equation from Ziv‐Baran et al 21 to adjust the wr‐CRP values (Adjusted wrCRP = 0.3136 + 0.8803 × wrCRP). Analyses of baseline disease clinical factors and demographic factors were conducted by treatment group.
2.4. Statistical analyses
Statistical analysis was performed using SAS 9.4 and JMP Pro 16 (SAS Institute, Cary, North Carolina, USA). In general, data were summarized using counts and percentages for categorical data and using standard univariate descriptive statistics (number of participants, mean, SD, median) for continuous data. Fisher exact test was used to compare percentages between placebo and NP001 with respect to non‐progressors. Analysis of covariance models was used to compare placebo to each NP001 group for continuous data. For all analyses, a two‐sided p‐value < 0.05 was considered statistically significant.
3. RESULTS
3.1. Phase 2B results
3.1.1. Demographics analysis
There were no relevant differences in demographics or disease characteristics between the two treatment groups in the mITT population with the exception that the placebo group had a higher percentage of patients with familial ALS compared with the NP001 group (Table 1). Genetic testing information of familial ALS is shown in the Supporting Information Table S1, which is available online.
TABLE 1.
Baseline demographics of phase 2B mITT population
| Placebo (N = 68) | NP001 (N = 68) | Total (N = 136) | |
|---|---|---|---|
| Male/female, n (%male) | 46/22 (67.6%) | 45/23 (66.2%) | 91/45 (66.9%) |
| Age in years, mean (SD) a | 56.6 (10.75) | 57.8 (10.89) | 57.2 (10.80) |
| Months since ALS symptom onset (SD) b | 18.83 (8.13) | 19.07 (8.40) | 18.97(8.23) |
| Familial/sporadic ALS, n (%familial) c | 14/54 (20.6%) | 2/66 (2.9%) | 16/120 (11.8%) |
| Bulbar, n (%) | 10 (14.7%) | 7 (10.3%) | 17 (12.5%) |
| Limbs, n (%) | 58 (85.3%) | 61 (89.7%) | 119 (87.5%) |
| Concurrent riluzole use, n (%) | 45 (66.2%) | 45 (66.2%) | 90 (66.2%) |
| ALSFRS‐R score at baseline, mean (SD) | 36.7 (5.64) | 37.7 (5.07) | 37.2 (5.35) |
| Hs‐CRP at baseline, mg/L, mean (SD) | 3.99 (5.49) | 4.38 (4.35) | 4.18 (4.94) |
3.1.2. Safety and tolerability
A total of 300 subjects were screened to yield 68 randomized to placebo and 70 to NP001; 117 participants completed planned dosing. Figure 1 shows the flowchart of individual disposition in the phase 2B trial. A summary of the overall disposition of the randomized participants with the definition of different populations in the phase 2B trial also is shown in the Supporting Information Table S2. One placebo and three NP001 participants discontinued the trial to initiate treatment with edaravone, which had been approved for the treatment of ALS after initiation of the trial. The overall summary of common clinical treatment‐emergent adverse events (TEAEs) of NP001 compared with placebo in the phase 2B is in the Supporting Information Table S3. The only significant difference in TEAEs between the NP001 and placebo groups was the higher rate of infusion related side effects in the NP001 group. Infusion related sensations of burning resolved for most patients with a slowing of the infusion rate.
FIGURE 1.
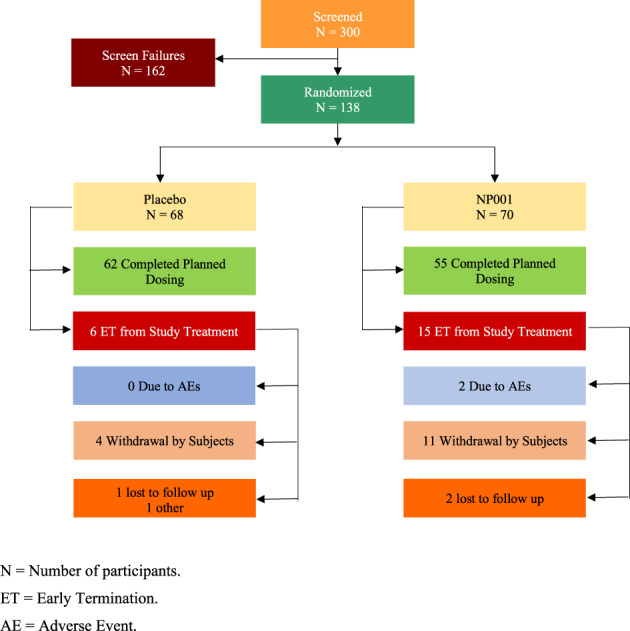
Participant disposition in phase 2B trial. N , number of participants; ET , early termination
3.1.3. Phase 2B mITT analysis of primary and secondary endpoints
The change from baseline in NP001 versus placebo in both the ALSFRS‐R score as well as the % change in predicted vital capacity showed similar rates of decline in the mITT population (Figure 2A,B). The analysis of % non‐progressors in those completing the study showed no difference between NP001 and placebo groups (Figure 2C).
FIGURE 2.
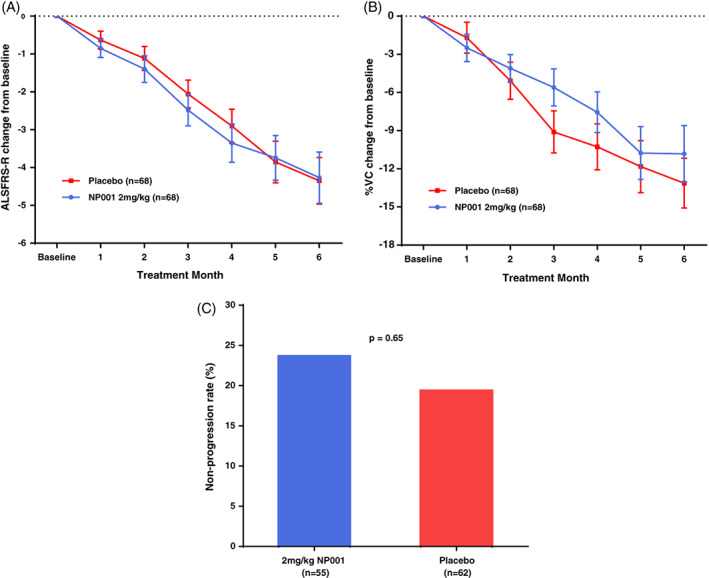
Efficacy of NP001 in the phase 2B mITT population. A, ALSFRS‐R score from baseline over the 6‐mo study in participants treated with NP001 compared with Placebo in the phase 2B mITT population. ALSFRS‐R score change from baseline for participants treated with NP001 depicted in blue (n = 68) and compared with placebo group depicted in red (n = 68). Bars represent mean of ALSFRS‐R score change from baseline ± SEM. No differences were seen between NP001 and placebo groups by the end of study (NP001 = −4.3 vs. Placebo = −4.3) (Wilcoxon test, p = .71). B, Change in % of baseline SVC over 6 mo in participants on NP001 compared with placebo in the phase 2B mITT population. Mean change from baseline in percent predicted SVC for participants treated with NP001(n = 68) is depicted in blue and compared with the placebo group (n = 68) depicted in red. Bars represent mean of % predicted SVC change from baseline ± SEM. There was no significant difference between NP001 treated patients and placebos by the end of study (NP001 = −10.8% vs. Placebo = −13.1%) (Wilcoxon test, p = .15). C, Bar chart of NP001 Non‐progression rate versus placebo group in those who had completed the phase 2B study. Non‐progression rate with non‐progressors defined as no decrease in ALSFRS‐R score at baseline to 6 mo. The proportion of non‐progressors (non‐progression rate) in 2 mg/kg NP001 treatment (13 out of 55) was similar to placebo group (12 out of 62) (Fisher exact test, p = .65)
3.2. Results from post hoc analyses
3.2.1. Age distribution of NP001 and placebo non‐progressors
Thirteen participants in the phase 2B NP001 and 12 in placebo did not progress out of a total of 117 participants who completed the study. A demographic analysis of these participants identified a non‐random age association with NP001 non‐progressors as compared with placebo non‐progressors (Fisher exact test, p = .002). 13/13 NP001 treated non‐progressors were within a 40‐ to 65‐y‐old age range. Placebo non‐progressors were arrayed across the range (32–76 y) of ages within the phase 2B study (Figure 3). This non‐random age association with NP001 treatment was confirmed with a follow up evaluation of NP001 non‐progressors in the phase 2A study; 6/6 were in the 40‐ to 65‐y age range.
FIGURE 3.
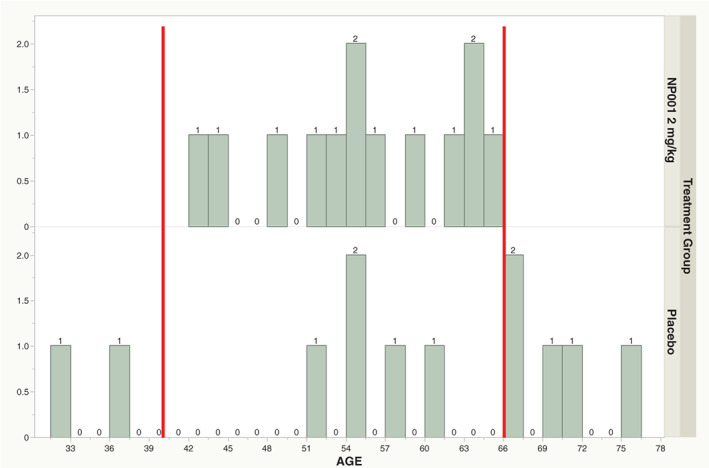
Age distribution of participants who did not progress during 6 mo in phase 2B trial. Age distribution plot with increasing age (years) of participant on the X‐axis and the number of participants in particular age categories on the Y axis. The top panel represents the age distribution of NP001 treated non‐progressors, 13 of NP001 treated non‐progressors in the phase 2B group (all of the phase 2B NP001 non‐progressors) fell into the 40‐ to 65‐y‐old age range (top panel, n = 13), however, only 5 out of 12 placebo non‐progressors were in the 40‐ to 65‐y age range (bottom panel, n = 12) (Fisher exact test, p = .002)
3.2.2. Demographics of merged phase 2A and 2B participants for post hoc analysis
154 participants with baseline plasma hs‐CRP of >1.13 mg/L had completed either the phase 2A or 2B trials. Table 2 shows the baseline demographics and characteristics of the 154 participants included in the post hoc evaluation by treatment group. Based on the initial observations of non‐progressor characteristics in the phase 2B, a subset of 40‐ to 65‐y‐old individuals from this combined 154‐participant cohort from both NP001 and placebo treated groups were chosen for efficacy evaluations. The selection process for this final group is shown in Figure 4. Approximately 76% (117) of the 154 participants who completed the trials fell into the 40‐ to 65‐y‐old age group all of whom had plasma CRP > 1.13 mg/L. The demographics of this smaller group were similar to the overall phase 2 combined demographics (Supporting Information Table S4).
TABLE 2.
Baseline demographics and characteristics of participants in the phase 2A and 2B trials included in post hoc evaluation by treatment group
| NP001 2 mg/kg | Placebo | Overall | |
|---|---|---|---|
| Characteristics | (N = 72) | (N = 82) | (N = 154) |
| Sex, N (%) | |||
| Female | 24 (33.3%) | 23 (28.0%) | 47 (30.5%) |
| Male | 48 (66.7%) | 59 (72.0%) | 107 (69.5%) |
| Age at enrollment in years, mean ± SD | 56.3 ± 10.6 | 56.0 ± 9.9 | 56.1 ± 10.2 |
| Type of ALS, N (%) | |||
| Familial | 1 (1.4%) | 15 (18.3%) | 16 (10.4%) |
| Sporadic | 71 (98.6%) | 67 (81.7%) | 138 (89.6%) |
| Site of ALS onset, N (%) | |||
| Bulbar | 9 (12.5%) | 14 (17.1%) | 23 (14.9%) |
| Limb | 63 (87.5%) | 68 (82.9%) | 131 (85.1%) |
| Revised El Escorial criteria for ALS, N (%) | |||
| Definite | 32 (44.4%) | 35 (42.7%) | 67 (43.5%) |
| Probable | 29 (40.3%) | 35 (42.7%) | 64 (41.6%) |
| Probable laboratory supported | 5 (6.9%) | 6 (7.3%) | 11 (7.1%) |
| Possible | 6 (8.3%) | 6 (7.3%) | 12 (7.8%) |
| ALSFRS‐R score at baseline, mean ± SD | 38.4 ± 4.6 | 37.5 ± 5.5 | 37.9 ± 5.1 |
| Vital capacity at baseline, mean ± SD | 93.3 ± 19.4 | 89.9 ± 18.4 | 91.5 ± 18.9 |
| Months since ALS symptom onset a , mean ± SD | 19.62 ± 8.45 | 18.14 ± 8.12 | 18.83 ± 8.28 |
| Hs‐CRP at baseline (mg/L), mean ± SD | 4.08 ± 3.46 | 3.97 ± 5.17 | 4.02 ± 4.44 |
Abbreviation: N, number of participants.
Months from ALS symptom onset to baseline.
FIGURE 4.
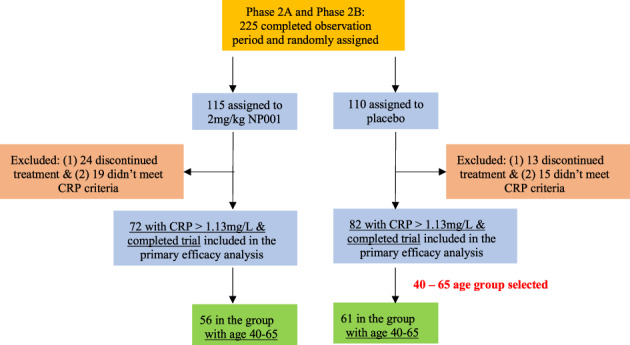
NP001 phase 2A and 2B trials combined participant assignments
3.2.3. Post hoc study efficacy evaluation
Among the 117 evaluable 40‐ to 65‐y‐old participants, the change in ALSFRS‐R score and % predicted VC score change from baseline to study end were calculated by treatment (Figure 5). As compared with the placebo group, the NP001 treatment group showed a slower decline in ALSFRS‐R score (36%, Figure 5A) (p = .01). In addition, NP001 treated patients had a 51% slowing of VC loss compared with placebo (Figure 5B) (p < .001).
FIGURE 5.
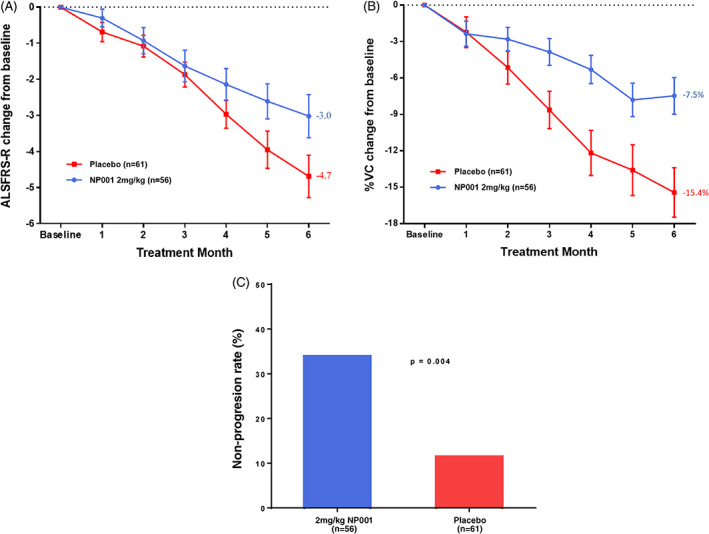
Post hoc analysis of NP001 efficacy in participants with plasma CRP > 1.13 mg/L and age between 40 and 65 y at baseline in phase 2A and 2B trials. A, Participants treated with NP001 experienced a slower decline in ALSFRS‐R score from baseline compared with placebo in those with plasma CRP > 1.13 mg/L and age between 40 and 65 y at baseline in phase 2A and 2B trials. ALSFRS‐R score change from baseline for participants treated with NP001 (n = 56) depicted in blue and compared with placebo group (n = 61) depicted in red. Bars represent mean of ALSFRS‐R score change from baseline ± SEM. The NP001 treatment group showed a 36% slower progression rate by the end of study (Wilcoxon test, p = .01). B, Percent change in vital capacity (VC) over 6 mo in participants on NP001 compared with placebo in those with plasma CRP > 1.13 mg/L and age between 40 and 65 y at baseline in phase 2A and 2B trials. Percent predicted VC change from baseline for participants treated with NP001 (n = 56) is depicted in blue and compared with the placebo group (n = 61) depicted in red. Bars represent mean of % predicted VC change from baseline ± SEM. Average %VC lost over the 6 mo of study: NP001: −7.5% (−1.3% per month); Placebo: −15.4% (−2.6% per month). The NP001 treatment arm lost 51% less respiratory function than the placebo arm by the end of study (Wilcoxon test, p < .001). C, NP001 treatment was associated with stabilized disease in participants over 6‐mo studies. Non‐progressors defined as having no decrease in ALSFRS‐R score from baseline to 6 mo, by treatment group, restricted to those with plasma CRP > 1.13 mg/L and age between 40 and 65 y at baseline in phase 2A and 2B trials. The proportion of non‐progressors (non‐progression rate) in 2 mg/kg NP001 treatment (19 out of 56) was significantly greater than that of the placebo group (7 out of 61) (Fisher exact test, p = .004). In participants with plasma CRP > 3 mg/L at baseline, the non‐progression rate for NP001 treated was 46% (13/28) versus 4.5% (1/22) in the placebo group (Fisher exact test, p = .001)
3.2.4. Statistical confirmation of selection: NP001 Recipients more likely to be non‐progressors than placebos
Participants showing no loss of ALSFRS‐R score over 6 mo (non‐progressors from both treated and placebo groups) were significantly more likely to have had NP001 treatment (Figure 5C) (p = .004). Compared with non‐progressors in placebo group, higher levels of baseline CRP were observed in NP001‐treated non‐progressors (Table S4). In participants with even higher CRP levels, > 3 mg/L at baseline, there was a 10‐fold selectivity for drug treated compared with placebo (p = .001).
4. DISCUSSION
Although the phase 2B clinical trial failed, the study did provide insight into various roles inflammation might play in ALS pathogenesis. The ALS literature has suggested that patients with elevated levels of CRP progress more rapidly. 4 , 22 CRP is generally made in the liver in response to infection or tissue damage and represents an indirect measurement of these types of disease activity. 23 , 24 CRP also augments the function of the innate immune system at clearing infected and apoptotic cells. 25 The NP001 phase 2B trial designers hypothesized that using elevated CRP levels at baseline as a selection criterion would select for a group of participants with more rapid disease progression, and that they would be most likely to respond to NP001. Instead, both the placebo and drug treated arms of the study progressed more slowly than many prior trials, leading to reduced statistical power to detect slowing of disease progression. Furthermore, it is becoming increasingly clear that not all ALS patients share the same type or contribution of inflammation to ALS disease progression. 26 The post hoc analyses presented in the second half of the manuscript were designed to explore in a more select group of participants disease characteristics responsive to NP001.
Given the slowly progressive nature of the patients with hs‐CRP > 1.13 mg/L described in these studies, another selection is needed to differentiate drug “responders” from exceedingly slow progressors, in order to document, if present, meaningful disease regulatory activity. In both NP001 trials a novel endpoint was defined, one in which patients who lost 0 units of ALSFRS‐R score in the 6‐mo study and were termed “non‐progressors”. All of the NP001“non‐progressors” were between the ages of 40–65. This age restriction identified the 117‐patient cohort evaluated in the post hoc efficacy study, representing 76% of all of those completing the trials. It was this subset that showed significant changes in ALSFRS‐R score and % change in VC decline.
The proportion of non‐progressors in both the phase 2B and the phase 2A were similar, representing 23%–25% of the overall treated patient population. These non‐progressor patients could be representative of the ALS population; the PROACT database reported similar proportions of non‐progressors. 27 Though the overall proportions of non‐progressors were balanced between NP001 and placebo arms in the phase 2A and 2B trials, a focus on participants with elevated CRP and restricted age range revealed a significantly higher proportion of non‐progressors in the NP001 arm.
Although a subgroup of ALS patients with elevated neuroinflammation appears to exist, identifying this group and selecting them for a clinical trial at screening is a challenge. The data presented in this manuscript may have helped further define a patient population with elevated inflammation who are more likely to be a slowly progressive inflammatory subset, responsive to immune regulatory approaches.
In ALS patients, loss of respiratory vital capacity translates directly to poor survival. 19 Similar to the variability seen in ALSFRS‐R score comparisons, respiratory function changes in ALS patients are reproducible over time, whether the measures are FVC or SVC. Natural history studies of VC over time in ALS patients confirm an average loss of 2.5%–3% of respiratory function per month. 19 , 20 In this post hoc analysis, NP001 slowed respiratory VC loss to 1.3% per month compared with the placebo group with a loss of 2.6%/mo.
The final assessment of NP001 activity showed that the percent of non‐progressors over the 6 mo of study was significantly higher in the NP001 treatment arm than in the placebo arm. In patients with clinically significant levels 24 of plasma CRP (> 3 mg/L) there was a 10:1 response advantage for those treated with NP001 compared with placebo controls. Furthermore, consistent with the anti‐inflammatory activities of NP001, the patients who benefitted from treatment were more likely to be those with higher level of inflammation as defined by blood CRP levels. Follow‐up studies of inflammatory biomarkers in cryopreserved plasma samples from both phase 2A and 2B trials may allow a better understanding how NP001 works in ALS pathogenesis.
Analyses by age showed that the population between 40 and 65 y of age was more likely to respond to NP001 treatment. It is plausible that the disease mechanism and trajectory for early age at onset (<40 y) differs from older patients and is more likely to have a genetic or heritable component as is often seen in other chronic conditions such as cancer and cardiovascular disease, although this has not yet been proven. With regard to the exclusion of the older age groups, it appears that CRP levels increase with age 23 and that the CRP sensitivity cutoffs applied to the NP001 phase 2 trials may have allowed an over representation of older ALS patients for whom the CRP values measured other disease activities, unrelated to ALS, thus obscuring the ALS specific activities of NP001.
5. LIMITATIONS OF THE STUDY
The role of inflammation in ALS pathogenesis may need to be studied in greater detail to understand why different inflammatory signals or pathways track with subsets of ALS patients, potentially targetable with appropriate therapeutics. Even though the results presented were largely post hoc, use of absolute changes in ALSFRS‐R scores as a measure of the magnitude of “response” may need to be revisited. The 1.7 units of ALSFRS‐R slowing over 6 mo in a slowly progressive cohort (36% slowing of decline) might be as clinically meaningful as absolute changes of 2.5 units observed in studies of other drugs conducted among a more rapidly progressive ALS patient subset. Further studies of this population and the relationship to other forms of ALS will be required to validate it as a unique target suitable for drug development.
AUTHOR CONTRIBUTIONS
All authors contributed to the manuscript for intellectual content, and approved the final manuscript for submission. Robert G. Miller was the principal investigator of the Neuraltus phase 2B as well as the phase 2A NP001 clinical trials. Dr. Jeremy Shefner was the co‐medical monitor and site monitor for the phase 2B trial. Drs. Richard Barohn, Richard Bedlack, Michael Benatar, James D. Berry, Merit Cudkowicz, Edward J. Kasarskis, Hiroshi Mitsumoto, Georgios Manousakis and David Walk, directly participated as clinical site investigators for the Neuraltus phase 2B clinical trial and all provided valuable comments on the contents of this manuscript. Rongzhen Zhang, Paige M. Bracci, Ari Azhir and Michael S. McGrath contributed to the writing of the manuscript, study design, data analysis and/or interpretation of the post hoc analysis.
CONFLICT OF INTEREST
R.G. Miller, the author reports no conflict of interest. R. Zhang, the author reports no conflict of interest. P.M. Bracci, the author reports no conflict of interest. A. Azhir, CEO of Neuvivo Inc. R. Barohn has received research grants from NIH and consulting support from NuFactor. R. Bedlack has received research support from ALSA, Healey Center, MediciNova. He has received consulting support from AB Science, ALSA, Alexion, Amylyx, Apellis, Biogen, Biohaven, Brainstorm, Clene, Corcept, Cytokinetics, GenieUS, Guidepoint, ITF Pharma, Mallinkrodt, Orphazyme, Shinkei, Woolsey Pharma. M. Benatar, the author reports no conflict of interest. J.D. Berry, the author reports no conflict of interest. M. Cudkowicz, the author reports no conflict of interest. E.J. Kasarskis, the author reports no conflict of interest. H. Mitsumoto has received research funding from NIH, ALS Association, SPF, MDA Wings, Tsumura & Co, Mitsubishi‐Tanabe Pharma. He has participated in advisory boards for Amylyx, PTC Bio (one time). G. Manousakis, the author reports no conflict of interest, D. Walk, the author reports no conflict of interest. B. Oskarsson the author, reports no conflict of interest. J. Shefner has received grant/research/clinical trial support from Amylyx, Biogen, Cytokinetics Incorporated, Mitsubishi Tanabe Pharma America, Sanofi, Novartis, Annexon, Jannsen. Alexion, Medicinova, Ionis, and Alector. He has received personal compensation from Amylyx, Apic Biosciences, Neurosense, Cytokinetics, Denal, GSK, Mitsubishi Tanabe Pharma America, RRD, Swanbio, Helixsmith, Novartis, Sanofi, and Sawei. M.S. McGrath, Founder and Chief scientific officer of Neuvivo Inc. Did not personally access the Neuvivo database for the primary analysis described in this manuscript.
ETHICS STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENTS
We thank all of the clinical sites involved in the phase 2B study, their principal investigators, study coordinators, support staff and data management center. The clinical site information and affiliated principal investigators (PIs) listed below are those identified in Neuraltus phase 2B clinical summary documents. The Participating Sites and Study Principal Investigators are listed here: Barrow Neurological Institute/PI: Shafeeq Ladha, MD. Massachusetts General Hospital/PI: James Berry, MD, MPH. Cleveland Clinic Foundation/PI: Erik Pioro, MD, PhD, FRCPC. McGill University/PI: Angela Genge, MD, FRCPC. Cedars‐Sinai Medical Center/PI: Richard Lewis, MD. Ohio State University/PI: Stephen Kolb, MD, PhD. Carolinas Medical Center/PI: Benjamin Brooks, MD. Providence ALS Center/PI: Kimberly Goslin, MD, PhD. Columbia University Medical Center/PI: Jinsy Andrews, MD, MSc. University of California Irvine Medical Center/PI: Namita Goyal, MD. California Pacific Medical Center/PI: Jonathan Katz, MD. University of Kansas Medical Center/PI: Richard Barohn, MD & Mazen Dimachkie, MD. Duke Medical Center/PI: Richard Bedlack, MD. University of Kentucky Hospital/PI: Edward Kasarskis, MD, PhD. Emory University School of Medicine/PI: Christina Fournier, MD, MSc. University of Miami Miller School ofMedicine/PI: Michael Benatar, MD. Houston Methodist Hospital/PI: Ericka Simpson, MD. University of Minnesota/PI: Georgios Manousakis, MD. Mayo Clinic Arizona/PI: Mark Ross, MD. Mayo Clinic Florida/PI: Bjorn Oskarsson.
Miller RG, Zhang R, Bracci PM, et al. Phase 2B randomized controlled trial of NP001 in amyotrophic lateral sclerosis: Pre‐specified and post hoc analyses. Muscle & Nerve. 2022;66(1):39-49. doi: 10.1002/mus.27511
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy restrictions.
REFERENCES
- 1. Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377:942‐955. [DOI] [PubMed] [Google Scholar]
- 2. Abe K, Itoyama Y, Sobue G, et al. Confirmatory double‐blind, parallel‐group, placebo‐controlled study of efficacy and safety of edaravone (MCI‐186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:610‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Butovsky O, Siddiqui S, Gabriely G, et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest. 2012;122:3063‐3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lunetta C, Lizio A, Maestri E, et al. Serum C‐reactive protein as a prognostic biomarker in amyotrophic lateral sclerosis. JAMA Neurol. 2017;74:660‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hooten KG, Beers DR, Zhao W, Appel SH. Protective and toxic Neuroinflammation in amyotrophic lateral sclerosis. Neurotherapeutics. 2015;12:364‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu CH, Davidson S, Harapas CR, et al. TDP‐43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell. 2020;183:636‐649.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kallstig E, McCabe BD, Schneider BL. The links between ALS and NF‐kappaB. Int J Mol Sci. 2021;22(8):3875. 10.3390/ijms22083875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Natale G, Biagioni F, Busceti CL, Gambardella S, Limanaqi F, Fornai F. TREM receptors connecting bowel inflammation to neurodegenerative disorders. Cells. 2019;8(10):1124. 10.3390/cells8101124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jin X, Liu MY, Zhang DF, et al. Natural products as a potential modulator of microglial polarization in neurodegenerative diseases. Pharmacol Res. 2019;145:104253. [DOI] [PubMed] [Google Scholar]
- 10. McGill RB, Steyn FJ, Ngo ST, et al. Monocyte CD14 and HLA‐DR expression increases with disease duration and severity in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2021;1‐8. 10.1080/21678421.2021.1964531 [DOI] [PubMed] [Google Scholar]
- 11. Zhang R, Gascon R, Miller RG, et al. Evidence for systemic immune system alterations in sporadic amyotrophic lateral sclerosis (sALS). J Neuroimmunol. 2005;159:215‐224. [DOI] [PubMed] [Google Scholar]
- 12. Giese T, McGrath MS, Stumm S, Schempp H, Elstner E, Meuer SC. Differential effects on innate versus adaptive immune responses by WF10. Cell Immunol. 2004;229:149‐158. [DOI] [PubMed] [Google Scholar]
- 13. McGrath MS, Kahn JO, Herndier BG. Development of WF10, a novel macrophage‐regulating agent. Curr Opin Investig Drugs. 2002;3:365‐373. [PubMed] [Google Scholar]
- 14. Schempp H, Reim M, Dornisch K, Elstner EF. Chlorite‐hemoprotein interaction as key role for the pharmacological activity of the chlorite‐based drug WF10. Arzneimittelforschung. 2001;51:554‐562. [DOI] [PubMed] [Google Scholar]
- 15. Joo K, Lee Y, Choi D, et al. An anti‐inflammatory mechanism of taurine conjugated 5‐aminosalicylic acid against experimental colitis: taurine chloramine potentiates inhibitory effect of 5‐aminosalicylic acid on IL‐1beta‐mediated NFkappaB activation. Eur J Pharmacol. 2009;618:91‐97. [DOI] [PubMed] [Google Scholar]
- 16. Miller RG, Zhang R, Block G, et al. NP001 regulation of macrophage activation markers in ALS: a phase I clinical and biomarker study. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:601‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miller RG, Block G, Katz JS, et al. Randomized phase 2 trial of NP001‐a novel immune regulator: safety and early efficacy in ALS. Neurol Neuroimmunol Neuroinflamm. 2015;2:e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brooks BR, Miller RG, Swash M, Munsat TL. World Federation of Neurology Research Group on motor neuron D. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293‐299. [DOI] [PubMed] [Google Scholar]
- 19. Andrews JA, Meng L, Kulke SF, et al. Association between decline in slow vital capacity and respiratory insufficiency, use of assisted ventilation, tracheostomy, or death in patients with amyotrophic lateral sclerosis. JAMA Neurol. 2018;75:58‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pinto S, de Carvalho M. SVC is a marker of respiratory decline function, similar to FVC, in patients with ALS. Front Neurol. 2019;10:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ziv‐Baran T, Shenhar‐Tsarfaty S, Etz‐Hadar I, et al. The ability of the wide range CRP assay to classify individuals with low grade inflammation into cardiovascular risk groups. Clin Chim Acta. 2017;471:185‐190. [DOI] [PubMed] [Google Scholar]
- 22. Beers DR, Zhao W, Neal DW, et al. Elevated acute phase proteins reflect peripheral inflammation and disease severity in patients with amyotrophic lateral sclerosis. Sci Rep. 2020;10:15295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cui C, Sun J, Pawitan Y, et al. Creatinine and C‐reactive protein in amyotrophic lateral sclerosis, multiple sclerosis and Parkinson's disease. Brain Commun. 2020;2:fcaa152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huysai K, Gecgel SK. Comparison of high sensitive C‐reactive protein levels with other biomarkers in cardiovascular disease risk assessment. Int J Clin Exp Med. 2019;12:4339‐4346. [Google Scholar]
- 25. Ngwa DN, Agrawal A. Structure‐function relationships of C‐reactive protein in bacterial infection. Front Immunol. 2019;10:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goyal NA, Berry JD, Windebank A, et al. Addressing heterogeneity in amyotrophic lateral sclerosis CLINICAL TRIALS. Muscle Nerve. 2020;62:156‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bedlack RS, Vaughan T, Wicks P, et al. How common are ALS plateaus and reversals? Neurology. 2016;86:808‐812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy restrictions.