

ABSTRACT
Protein–protein interactions (PPIs) are crucial for various biological processes. Epstein-Barr virus (EBV) proteins typically form complexes, regulating the replication and persistence of the viral genome in human cells. However, the role of EBV protein complexes under physiological conditions remains unclear. In this study, we performed comprehensive analyses of EBV PPIs in living cells using the NanoBiT system. We identified 195 PPIs, many of which have not previously been reported. Computational analyses of these PPIs revealed that BLRF2, which is only found in gammaherpesviruses, is a central protein in the structural network of EBV tegument proteins. To characterize the role of BLRF2, we generated two BLRF2 knockout EBV clones using CRISPR/Cas9. BLRF2 knockout significantly decreased the production of infectious virus particles, which was partially restored by exogenous BLRF2 expression. In addition, self-association of BLRF2 protein was found, and mutation of the residues crucial for the self-association affected stability of the protein. Our data imply that BLRF2 is a tegument network hub that plays important roles in progeny virion maturation.
IMPORTANCE EBV remains a significant public health challenge, causing infectious mononucleosis and several cancer types. Therefore, the better understanding of the molecular mechanisms underlying EBV replication is of high clinical importance. As protein–protein interactions (PPIs) are major regulators of virus-associated pathogenesis, comprehensive analyses of PPIs are essential. Previous studies on PPIs in EBV or other herpesviruses have predominantly employed the yeast two-hybrid (Y2H) system, immunoprecipitation, and pulldown assays. Herein, using a novel luminescence-based method, we identified 195 PPIs, most of which have not previously been reported. Computational and functional analyses using knockout viruses revealed that BLRF2 plays a central role in the EBV life cycle, which makes it a valuable target for drug development.
KEYWORDS: BLRF2, EBV, PPI, lytic cycle, tegument
INTRODUCTION
Epstein-Barr Virus (EBV) is a common double-stranded DNA virus belonging to the family Herpesviridae (1, 2). EBV primarily infects B cells by binding to CD21 and is responsible for infectious mononucleosis. EBV infections have also been linked to various types of B-cell lymphoma, including Burkitt lymphoma and posttransplant lymphoproliferative disease (3). Less frequently, EBV infects T cells and NK cells, resulting in extranodal natural killer/T-cell lymphoma and chronic active EBV infection (4).
EBV infections can be broadly classified into latent or lytic infections. A switch from latent to lytic infection (reactivation) results in the production of virus particles by the infected cells (5). During reactivation, the expression of the basic leucine zipper (bZIP) transcription factor BZLF1 and the transcriptional activator BRLF1 is induced immediately. Those two immediate early gene products, BZLF1 and BRLF1, mediate the transcription of early genes, which are required for viral DNA replication. Immediate-early and early viral genes often function as complexes, which promote viral DNA synthesis. The viral DNA polymerase consists of BALF5, BMRF1, and BALF2, while the helicase/primase complex is composed of BBLF4, BSLF1, and BBLF2/3. These viral replication proteins, along with origin-binding protein BZLF1, accumulate in specific subnuclear regions called replication compartments, where the viral DNA is synthesized (6, 7). After the viral DNA has been replicated, the viral preinitiation complex (vPIC) promotes the expression of late genes. BcRF1, BDLF3.5, BDLF4, BGLF3, BFRF2, and BVLF1 accumulate at noncanonical TATA sequences in the promoters of late genes of newly synthesized viral DNA, promoting RNA pol II recruitment (8). Late genes include structural proteins, such as capsid, tegument, and envelope glycoproteins. Protein–protein interactions (PPIs) are essential for virus morphogenesis. The icosahedral EBV capsid is composed of BcLF1, BDLF1, BORF1, BFRF3, and BBRF1, while the viral procapsid also requires BVRF2 (protease/scaffold) and BdRF1 (scaffold) (9). Tegument proteins of herpesviruses form a complex network that mediates virion maturation and intracellular trafficking, as well as nucleocapsid budding (10, 11). In addition, some EBV glycoproteins, including gB, gH/gL, and gM/gN, also function as complexes (12). The nuclear egress complex (NEC) is composed of the late genes BFLF2 and BFRF1. NEC localizes at the nuclear rim, mediating the budding of nucleocapsids into the inner nuclear membrane (13), also known as primary envelopment. Subsequently, nucleocapsids are released into the cytoplasm, followed by re-envelopment, possibly at the cis- or trans-Golgi apparatus (secondary envelopment) (14). After fusion with the plasma membrane, EBV particles are released from the infected cells.
The yeast two-hybrid (Y2H) method has been widely used to analyze PPI networks in herpesviruses (15–19). Nevertheless, the screen may entail a risk for artifacts, because (i) the assay is not performed in mammalian cells, (ii) the bait and prey may not represent natural intracellular localization because the proteins are attached with exogenous nuclear localization signals, and (iii) the background is high in some cases due to auto-activation even in the absence of PPI. Although GST-pulldown and immunoprecipitation assays have also been used, these methods require cell lysis, further complicating the interpretation of the results. The NanoBiT system is a novel technology that enables measurement of PPIs in live cells. The luciferase gene encoded by O. gracilirostris is split into two subunits (LgBiT and SmBiT). The subunits do not emit light when separated, but when they are close, luciferase enzyme activity is reconstituted and the cells become luminous. By fusing two proteins of interest with LgBiT and SmBiT, respectively, interaction between the two proteins brings two BiT subunits in close proximity and leads to luminescence emission.
Herein, we used the NanoBiT system, analyzed EBV PPIs in living cells, and constructed a network based on these PPIs. In addition, we investigated the role of BLRF2 (also known as VCA-p23) in the interaction between tegument proteins, as well as in the life cycle of EBV.
RESULTS
Comprehensive analyses of EBV PPIs.
To find out candidates of PPIs between EBV proteins, we performed a large-scale screen using the NanoBiT system in living cells. To this end, we cloned 69 EBV ORFs into LargeBiT or SmallBiT vectors, allowing for the production of EBV proteins fused with LargeBiT or SmallBiT, respectively (Fig. 1A). The SmallBiT peptide contained 11 amino acids of Oplophorus gracilirostris luciferase (NanoLuc), while the LargeBiT peptide consisted of the remaining amino acids of NanoLuc. Hence, the interaction between two viral proteins brings LargeBiT and SmallBiT in close proximity, leading to luminescence emission.
FIG 1.
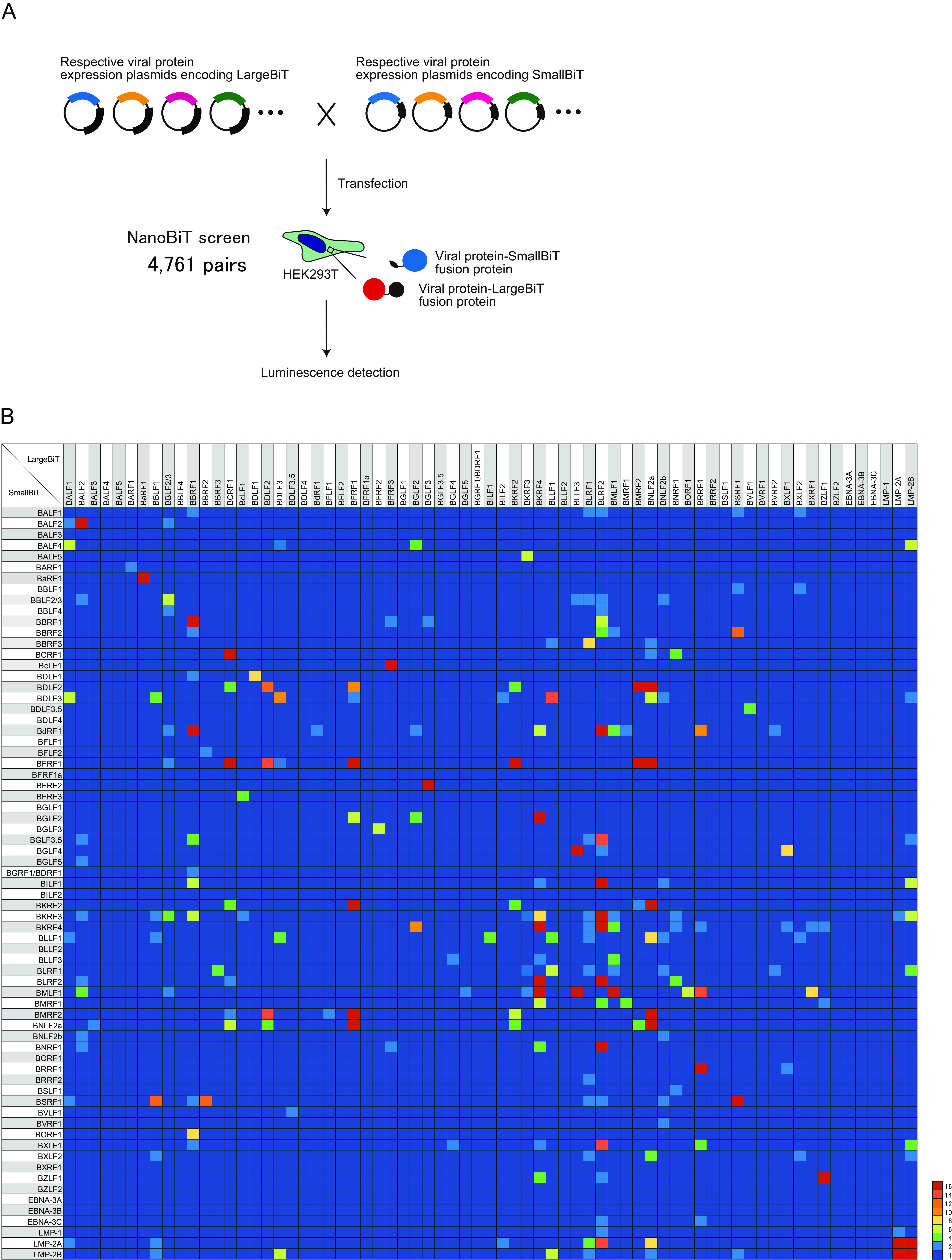
Comprehensive analyses of EBV protein interactions. (A) Schematic illustration of NanoBiT screening. All 69 EBV genes were cloned into LargeBiT and SmallBiT vectors. HEK293T cells were transfected with the NanoBiT plasmids in different pair combinations, as indicated. Luciferase activity was measured 24 h after transfection. (B) Heatmap showing the relative luminescence, which was calculated by dividing the luminescence of each combination by that of the negative control.
HEK293T cells were transfected with combinations of all LargeBiT and SmallBiT vectors, resulting in 4,761 combinations in total (Fig. 1A). PPI screening by luciferase assay revealed that 233 interactions led to significant luminescence emission (>2.0-fold compared with the control; Fig. 1B). These 233 PPIs included 38 protein pairs that resulted in luminescence emission after the fusion tags (LargeBiT and SmallBiT) were swapped (bidirectional). After eliminating such redundancies (bidirectional interactions), 195 pairs were identified.
To confirm the ability of the NanoBiT system to detect known protein–protein interactions, we compared our findings with known EBV PPIs from the literature. Of the 195 PPIs, 51 pairs (26.2%) were previously reported (15, 16). For example, the tegument proteins BSRF1 and BBRF2 were previously shown to interact (1), while the formation of homomeric BSRF1 complexes has not previously been reported to the best of our knowledge. Furthermore, some of these interactions have been reported for the homolog proteins of other herpesviruses, such as herpes simplex virus (HSV) and Kaposi’s sarcoma-associated herpesvirus (KSHV). In particular, the HSV portal protein UL6 has been shown to interact with the scaffold protein UL26.5 (2); nevertheless, the interaction of their EBV counterparts (BBRF1 and BdRF1) has not previously been shown. Among the 195 PPIs reported here, 65 (33.3%) were previously described in EBV and/or other herpesviruses, while the rest 130 (66.7%) were newly identified by our approach (Table 1).
TABLE 1.
The PPIs in this and other reports
| EBV |
KSHV |
HSV |
vZV |
mCMV |
Besides EBV |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| This report |
Caldwood et al., PNAS 2007. PMID 17446270 |
Fossum et al., PLOS pathog 2009. PMID 19730696 |
Other reports (PMID) |
Uetz et al., science 2006. PMID 16339411 |
Rozen et al., JV 2008. PMID 18321973 |
Lee et al., virology 2008. PMID 18602131 |
Fossum et al., PLOS pathog 2009. PMID 19730696 |
Uetz et al., science 2006. PMID 16339411 |
Fossum et al., PLOS pathog 2009. PMID 19730696 |
Other reports (PMID) |
||||||
| EBV PPIs | NanoBiT |
IP | Y2H | Y2H | IP | IP | Y2H | Y2H | IP | Y2H | Y2H | Y2H | Y2H | IP | ||
| L-S | S-L | |||||||||||||||
| BALF1 | BALF2 | 2.3 | ||||||||||||||
| BALF1 | BALF4 | 7.5 | ||||||||||||||
| BALF1 | BDLF3 | 6.5 | ||||||||||||||
| BALF1 | BLLF1 | 2.5 | ||||||||||||||
| BALF1 | BSRF1 | 2.545 | 2.8 | + | 30901892 | |||||||||||
| BALF1 | LMP-2A | 2.091 | ||||||||||||||
| BALF2 | BALF2 | 31 | ||||||||||||||
| BALF2 | BBLF2/3 | 2.5 | 2.636 | |||||||||||||
| BALF2 | BGLF3.5 | 2.17 | ||||||||||||||
| BALF2 | BGLF5 | 3.5 | 7747443 | |||||||||||||
| BALF2 | BKRF3 | 2.17 | ||||||||||||||
| BALF2 | BLRF2 | 2.17 | + | + | + | |||||||||||
| BALF2 | BMLF1 | 4.83 | 15582656 | |||||||||||||
| BALF2 | BNLF2b | 2 | ||||||||||||||
| BALF2 | BNRF1 | 3.5 | ||||||||||||||
| BALF3 | BNLF2a | 2 | ||||||||||||||
| BARF1 | BARF1 | 2 | 16647084 | |||||||||||||
| BaRF1 | BaRF1 | 37.333 | + | + | 17446270 | + | ||||||||||
| BBLF1 | BDLF3 | 4 | ||||||||||||||
| BBLF1 | BKRF3 | 3 | ||||||||||||||
| BBLF1 | BLLF1 | 2.375 | + | |||||||||||||
| BBLF1 | BSRF1 | 13 | 2 | + | ||||||||||||
| BBLF1 | BXLF2 | 2.875 | 2.4 | |||||||||||||
| BBLF1 | LMP-2A | 2.5 | ||||||||||||||
| BBLF1 | LMP-2B | 2.625 | ||||||||||||||
| BBLF2/3 | BBLF2/3 | 7.545 | ||||||||||||||
| BBLF2/3 | BBLF4 | 2.455 | 10580049 | |||||||||||||
| BBLF2/3 | BdRF1 | 3.727 | + | |||||||||||||
| BBLF2/3 | BFRF1 | 2.455 | + | |||||||||||||
| BBLF2/3 | BKRF3 | 4 | ||||||||||||||
| BBLF2/3 | BMLF1 | 2 | ||||||||||||||
| BBRF1 | BALF1 | 2.333 | ||||||||||||||
| BBRF1 | BBRF1 | 319.667 | 30710799,31467275 | |||||||||||||
| BBRF1 | BBRF2 | 2 | ||||||||||||||
| BBRF1 | BDLF1 | 2.167 | ||||||||||||||
| BBRF1 | BdRF1 | 29.5 | 12941896 | |||||||||||||
| BBRF1 | BDRF1/BGRF1 | 2 | 17392365 | |||||||||||||
| BBRF1 | BGLF3.5 | 4.833 | ||||||||||||||
| BBRF1 | BILF1 | 7.667 | ||||||||||||||
| BBRF1 | BKRF3 | 6.167 | ||||||||||||||
| BBRF1 | BSRF1 | 2.167 | + | |||||||||||||
| BBRF1 | BVRF2 | 8.5 | 12941896 | |||||||||||||
| BBRF1 | BXLF1 | 3.833 | ||||||||||||||
| BBRF2 | BFLF2 | 3 | + | |||||||||||||
| BBRF2 | BSRF1 | 12.4 | 12.2 | + | 30901892 | |||||||||||
| BBRF3 | BLRF1 | 5.778 | 8 | 9621013 | + | |||||||||||
| BcLF1 | BFRF3 | 4 | 30.6 | + | 19158247 | + | + | + | ||||||||
| BcLF1 | BKRF3 | 2 | ||||||||||||||
| BCRF1 | BCRF1 | 36.125 | 9159483 | |||||||||||||
| BCRF1 | BDLF2 | 5.25 | ||||||||||||||
| BCRF1 | BFRF1 | 37.25 | ||||||||||||||
| BCRF1 | BKRF2 | 5 | ||||||||||||||
| BCRF1 | BLRF2 | 2 | ||||||||||||||
| BCRF1 | BMRF2 | 3 | ||||||||||||||
| BCRF1 | BNLF2a | 6.5 | 2.5 | |||||||||||||
| BDLF1 | BDLF1 | 8.786 | + | + | ||||||||||||
| BDLF2 | BDLF2 | 12.444 | + | |||||||||||||
| BDLF2 | BFRF1 | 14 | 10.167 | + | ||||||||||||
| BDLF2 | BMRF2 | 15.111 | 91.75 | + | 18995876 | |||||||||||
| BDLF2 | BNLF2a | 4.111 | 35.5 | + | ||||||||||||
| BDLF3 | BALF4 | 3.625 | + | |||||||||||||
| BDLF3 | BDLF3 | 10.25 | ||||||||||||||
| BDLF3 | BFRF1 | 3.875 | 2.667 | |||||||||||||
| BDLF3 | BLLF1 | 5.375 | 14.2 | |||||||||||||
| BDLF3 | LMP-2B | 6.25 | 2.8 | |||||||||||||
| BDLF3.5 | BVLF1 | 2.5 | 4.267 | 30305361 | ||||||||||||
| BdRF1 | BdRF1 | 2.143 | 19158247 | 9188587 | ||||||||||||
| BFLF1 | BMRF2 | 3.167 | ||||||||||||||
| BFRF1 | BFRF1 | 359.667 | + | |||||||||||||
| BFRF1 | BGLF2 | 6.333 | ||||||||||||||
| BFRF1 | BKRF2 | 20.5 | 30.92 | + | ||||||||||||
| BFRF1 | BMRF2 | 21 | 29.375 | + | ||||||||||||
| BFRF1 | BNLF2a | 35.667 | 580.5 | + | ||||||||||||
| BFRF2 | BGLF3 | 7.143 | 16.6 | 31461506 | 31578296 | |||||||||||
| BFRF3 | BBRF1 | 2 | ||||||||||||||
| BFRF3 | BNRF1 | 2 | ||||||||||||||
| BGLF2 | BALF4 | 5.857 | + | + | + | |||||||||||
| BGLF2 | BdRF1 | 3.429 | + | |||||||||||||
| BGLF2 | BGLF2 | 4 | 26559845 | |||||||||||||
| BGLF2 | BKRF4 | 10.857 | 530.68 | 19158247 | + | + | ||||||||||
| BGLF3 | BBRF1 | 2 | ||||||||||||||
| BGLF4 | BLLF3 | 2.2 | 19.385 | + | + | |||||||||||
| BGLF4 | BXLF1 | 2 | 8.625 | + | ||||||||||||
| BGLF5 | BMLF1 | 2.233 | ||||||||||||||
| BILF1 | BLLF1 | 4.52 | ||||||||||||||
| BILF2 | BDLF3 | 2.2 | ||||||||||||||
| BILF2 | LMP-2A | 2.6 | ||||||||||||||
| BKRF2 | BDLF2 | 5.04 | + | + | + | |||||||||||
| BKRF2 | BKRF2 | 5.6 | ||||||||||||||
| BKRF2 | BMRF2 | 7.52 | 2.625 | |||||||||||||
| BKRF2 | BNLF2a | 4.56 | 101.625 | + | ||||||||||||
| BKRF3 | BALF5 | 7.2 | 24872582 | 20601642 | ||||||||||||
| BKRF3 | BKRF3 | 3.6 | ||||||||||||||
| BKRF3 | BLRF1 | 3.5 | 2.7 | |||||||||||||
| BKRF3 | BMLF1 | 2.7 | 2.25 | |||||||||||||
| BKRF4 | BdRF1 | 6.52 | ||||||||||||||
| BKRF4 | BILF1 | 2.08 | ||||||||||||||
| BKRF4 | BKRF3 | 8.6 | ||||||||||||||
| BKRF4 | BKRF4 | 22.6 | ||||||||||||||
| BKRF4 | BLLF3 | 2.12 | ||||||||||||||
| BKRF4 | BLRF2 | 16.68 | 57 | + | + | + | ||||||||||
| BKRF4 | BMLF1 | 30.08 | 4.5 | |||||||||||||
| BKRF4 | BMRF1 | 7.92 | ||||||||||||||
| BKRF4 | BNRF1 | 5.92 | 3.333 | + | ||||||||||||
| BKRF4 | BXLF1 | 3.52 | 2.25 | + | + | |||||||||||
| BKRF4 | BZLF1 | 5.96 | 3.286 | |||||||||||||
| BLLF1 | BBRF3 | 2.44 | ||||||||||||||
| BLLF1 | BLLF1 | 5.44 | + | |||||||||||||
| BLLF1 | BLRF1 | 7.2 | 3.7 | |||||||||||||
| BLLF1 | LMP-2B | 6.28 | ||||||||||||||
| BLLF3 | BBLF2/3 | 2.308 | ||||||||||||||
| BLLF3 | BMLF1 | 24.308 | 4 | + | ||||||||||||
| BLRF1 | BALF1 | 2.2 | ||||||||||||||
| BLRF1 | BBLF2/3 | 2.4 | ||||||||||||||
| BLRF1 | BGLF3.5 | 2.3 | ||||||||||||||
| BLRF1 | BKRF4 | 2.8 | + | + | ||||||||||||
| BLRF1 | BLRF1 | 2.3 | 9621013 | + | ||||||||||||
| BLRF1 | BRRF2 | 2.1 | ||||||||||||||
| BLRF1 | BSRF1 | 3.6 | ||||||||||||||
| BLRF1 | BXLF2 | 2 | ||||||||||||||
| BLRF1 | LMP-2A | 4 | ||||||||||||||
| BLRF1 | LMP-2B | 3.3 | 4.8 | |||||||||||||
| BLRF2 | BALF1 | 2.1 | ||||||||||||||
| BLRF2 | BBLF2/3 | 2.4 | ||||||||||||||
| BLRF2 | BBLF4 | 2.5 | ||||||||||||||
| BLRF2 | BBRF1 | 7 | ||||||||||||||
| BLRF2 | BBRF2 | 4.5 | 22090138 | |||||||||||||
| BLRF2 | BdRF1 | 85.4 | ||||||||||||||
| BLRF2 | BGLF3.5 | 14.8 | ||||||||||||||
| BLRF2 | BGLF4 | 2.1 | + | |||||||||||||
| BLRF2 | BILF1 | 22.8 | ||||||||||||||
| BLRF2 | BKRF3 | 36.8 | ||||||||||||||
| BLRF2 | BLRF2 | 260 | + | + | + | 22090138 | ||||||||||
| BLRF2 | BMRF1 | 5.4 | + | + | ||||||||||||
| BLRF2 | BNRF1 | 61.1 | 5.167 | + | + | + | + | + | ||||||||
| BLRF2 | BSRF1 | 2.9 | ||||||||||||||
| BLRF2 | BXLF1 | 15.6 | ||||||||||||||
| BLRF2 | BZLF1 | 2 | ||||||||||||||
| BLRF2 | EBNA-3C | 2.5 | ||||||||||||||
| BLRF2 | LMP-1 | 3.1 | + | |||||||||||||
| BLRF2 | LMP-2A | 15.7 | ||||||||||||||
| BMLF1 | BBRF2 | 2 | ||||||||||||||
| BMLF1 | BdRF1 | 4 | + | |||||||||||||
| BMLF1 | BLRF1 | 2.25 | ||||||||||||||
| BMLF1 | BMLF1 | 71 | + | + | 9765385 | + | + | |||||||||
| BMRF1 | BdRF1 | 2 | ||||||||||||||
| BMRF1 | BMRF1 | 4.857 | 19801550 | |||||||||||||
| BMRF2 | BNLF2a | 5.375 | 393.375 | + | ||||||||||||
| BNLF2a | BBRF3 | 3.75 | + | |||||||||||||
| BNLF2a | BDLF3 | 7.5 | ||||||||||||||
| BNLF2a | BLLF1 | 9.25 | ||||||||||||||
| BNLF2a | BNLF2a | 129.5 | + | + | ||||||||||||
| BNLF2a | BXLF2 | 5.875 | ||||||||||||||
| BNLF2a | LMP-2A | 8.125 | ||||||||||||||
| BNLF2a | LMP-2B | 3.125 | ||||||||||||||
| BNLF2b | BBLF2/3 | 2.8 | ||||||||||||||
| BNLF2b | BDLF3 | 2.6 | ||||||||||||||
| BNLF2b | BILF1 | 2.2 | ||||||||||||||
| BNLF2b | BLLF1 | 2.6 | ||||||||||||||
| BNLF2b | BLRF1 | 2.2 | ||||||||||||||
| BNLF2b | BNLF2b | 2.2 | ||||||||||||||
| BNLF2b | BSRF1 | 3.4 | ||||||||||||||
| BNLF2b | BVRF1 | 2.2 | ||||||||||||||
| BNRF1 | BCRF1 | 5.833 | ||||||||||||||
| BNRF1 | BKRF3 | 2 | ||||||||||||||
| BNRF1 | BSLF1 | 2 | ||||||||||||||
| BORF1 | BMLF1 | 7.1 | ||||||||||||||
| BRRF1 | BdRF1 | 11.2 | ||||||||||||||
| BRRF1 | BKRF4 | 3.7 | ||||||||||||||
| BRRF1 | BMLF1 | 14.4 | ||||||||||||||
| BRRF1 | BRRF1 | 53.2 | ||||||||||||||
| BRRF1 | BXLF1 | 4.4 | 2.25 | |||||||||||||
| BRRF1 | EBNA-3C | 2.5 | ||||||||||||||
| BSRF1 | BSRF1 | 25.9 | ||||||||||||||
| BSRF1 | LMP-2B | 2 | ||||||||||||||
| BVRF2 | BdRF1 | 3.286 | 19158247 | |||||||||||||
| BVRF2 | BLRF1 | 2.429 | ||||||||||||||
| BXLF2 | BALF1 | 2 | ||||||||||||||
| BXLF2 | BLLF1 | 2.4 | ||||||||||||||
| BXLF2 | BXLF2 | 2.6 | ||||||||||||||
| BXRF1 | BKRF4 | 3.8 | ||||||||||||||
| BXRF1 | BMLF1 | 8.6 | + | |||||||||||||
| BZLF1 | BMRF1 | 2.429 | 8764021 | |||||||||||||
| BZLF1 | BZLF1 | 79.143 | 2159531,2174563 | |||||||||||||
| LMP-2A | BKRF3 | 2.2 | ||||||||||||||
| LMP-2A | LMP-1 | 2.5 | ||||||||||||||
| LMP-2A | LMP-2A | 368.5 | 11602734 | |||||||||||||
| LMP-2A | LMP-2B | 81 | 295.6 | 17035319 | ||||||||||||
| LMP-2B | BALF4 | 7.4 | ||||||||||||||
| LMP-2B | BGLF3.5 | 3 | ||||||||||||||
| LMP-2B | BILF1 | 6.2 | ||||||||||||||
| LMP-2B | BKRF3 | 7.6 | ||||||||||||||
| LMP-2B | BXLF1 | 4.2 | ||||||||||||||
| LMP-2B | BXLF2 | 2.4 | ||||||||||||||
| LMP-2B | LMP-2B | 213 | 11602734 | |||||||||||||
Computational analyses of the EBV interactome network.
Based on the PPIs identified, we constructed the EBV interactome network using Cytoscape (20). The network was composed of 62 nodes and 195 edges (Fig. 2A; Table 2). The interaction networks were classified into four components, including two components representing BARF1 and BaRF1 homomeric complexes and one component representing the bi-directional interaction between BVLF1 and BDLF3.5.
FIG 2.
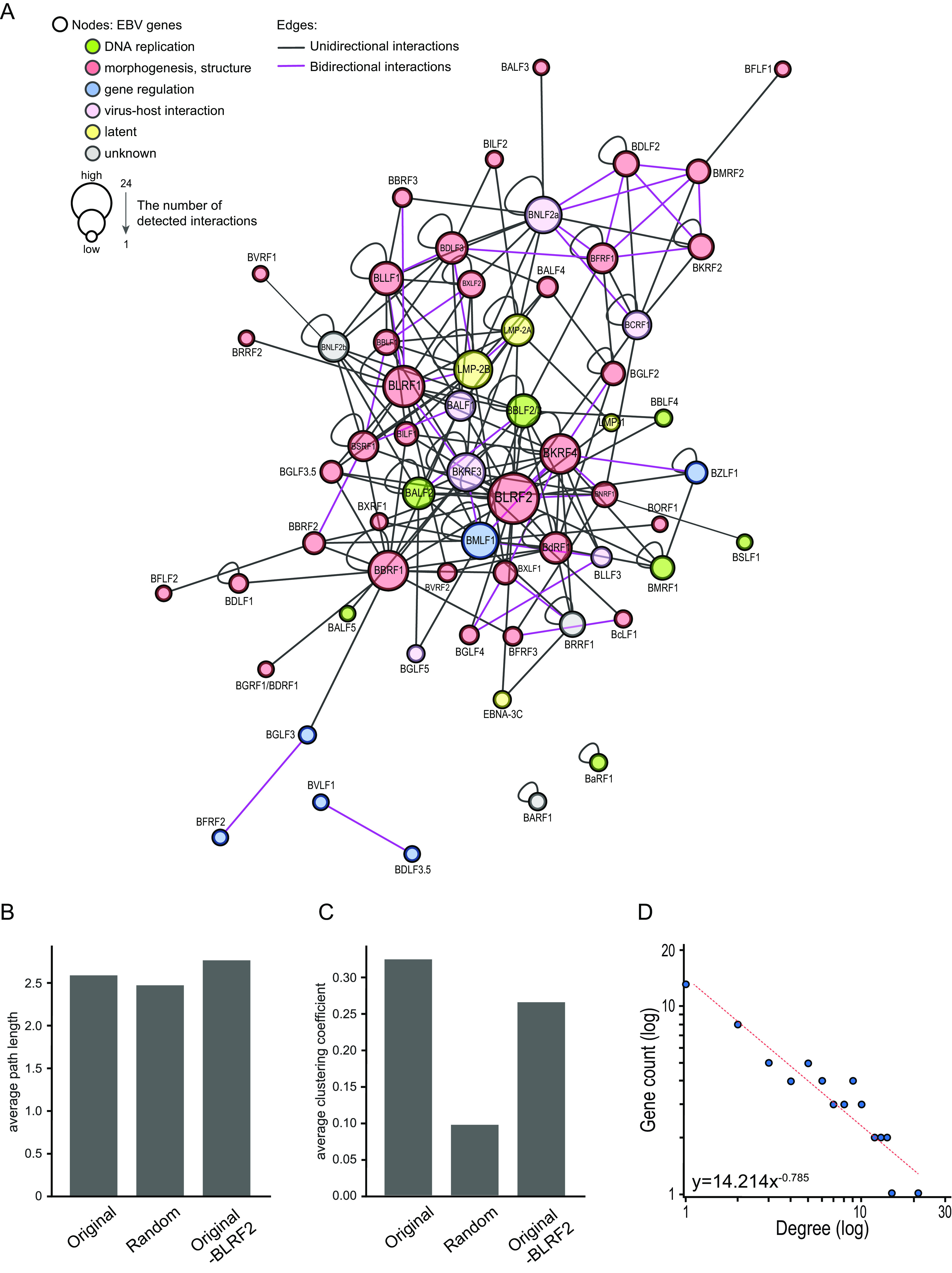
EBV interactome network. (A) Cytoscape EBV interactome network, in which EBV proteins (nodes) are connected by edges based on the PPIs identified by NanoBiT. Each node is labeled with the EBV protein name, and is color-coded according to the protein function: green, DNA replication-associated proteins; red, viral morphogenesis or structural proteins; blue, proteins regulating gene expression; pink, virus-host interaction proteins; yellow, latent proteins. The node size reflects the number of detected edges. (B, C) Clustering coefficient and shortest-path length of the main component of the original network, the corresponding random network, and the network excluding BLRF2 from the original component. The centrality values of 500 equivalent Erdős–Rényi random networks composed of the same number of nodes and edges as the main component of the original EBV protein network were calculated using the Python package NetworkX 2.3. (D) Degree distribution of EBV proteins.
TABLE 2.
The PPI network summary
| Network indices | Numerical data |
|---|---|
| No. of nodes | 62 |
| No. of edges | 195 |
| Bidirectional interactions | 38 |
| Unidirectional interactions | 157 |
| Avg degree | 5.39 |
| No. of self-loops | 28 |
| Network density | 0.088 |
| Isolated nodes | 2 |
| Connected components | 4 |
We examined whether the largest component consisting of 58 nodes and 166 edges exhibited the small-world property of a typical complex network. The small-world is a technical term of network science, where the average path length is comparable or smaller than that of the random model, which means that most nodes can be reached from other nodes within a small number of edges, and clustering coefficient is high; and for that matter, nodes tend to cluster at a high level. Put more simply, small world means that nodes are connected with edges more efficiently than just a random arrangement. To assess the topological characteristics of the network, we generated 500 equivalent Erdős–Rényi random networks, which were chosen uniformly at random from the set of all networks with the same number of nodes and edges in the original EBV protein network, except 26 self-loops. Then, we compared their average centrality values with the original network. Although the average shortest-path length of our original network was comparable with that of random networks (Fig. 2B), the clustering coefficient of the original network was approximately three times higher than that of random networks (Fig. 2C). Therefore, our EBV network was classified as a small-world network. Moreover, the small-world-ness index of our EBV network was 3.14, which is relatively high because a small-world-ness index > 1 is defined as “small-world” (21).
We then examined if our EBV PPIs network had the scale-free property. Scale-free means that edges are concentrated overwhelmingly to a limited number of nodes. This type of proteins (i.e., hubs) might have strong effects on the whole network. Many other nodes typically have a small number of edges. Here, most nodes in our EBV protein network exhibited a low-degree distribution, while a few nodes had more interacting partners; this power-law distribution is characteristic for scale-free networks (Table 3; Fig. 2D).
TABLE 3.
The PPI network summary of each gene
| Node | Degree | Bidirectional edge | SelfLoop | Clustering coefficient | Avg shortest path length | Betweenness centrality | Closeness centrality | Symbol |
|---|---|---|---|---|---|---|---|---|
| BALF1 | 11 | 1 | − | 0.26 | 2.1 | 0.049 | 0.47 | Virus-host interaction |
| BALF2 | 12 | 1 | + | 0.30 | 2.1 | 0.034 | 0.45 | DNA replication |
| BALF3 | 1 | 0 | − | 0 | 3.4 | 0 | 0.29 | morphogenesis, structure |
| BALF4 | 4 | 0 | − | 0.33 | 2.6 | 0.0038 | 0.38 | morphogenesis, structure |
| BALF5 | 1 | 0 | − | 0 | 2.9 | 0 | 0.34 | DNA replication |
| BaRF1 | 2 | 0 | + | 0 | 0 | 0 | 0 | DNA replication |
| BARF1 | 2 | 0 | + | 0 | 0 | 0 | 0 | unknown |
| BBLF1 | 9 | 2 | − | 0.42 | 2.4 | 0.0054 | 0.41 | morphogenesis, structure |
| BBLF2/3 | 13 | 1 | + | 0.28 | 2.0 | 0.060 | 0.49 | DNA replication |
| BBLF4 | 2 | 0 | − | 1 | 2.5 | 0 | 0.39 | DNA replication |
| BBRF1 | 16 | 0 | + | 0.12 | 2.0 | 0.17 | 0.48 | morphogenesis, structure |
| BBRF2 | 6 | 1 | − | 0.3 | 2.2 | 0.039 | 0.43 | morphogenesis, structure |
| BBRF3 | 4 | 1 | − | 0.66 | 2.6 | 0.0024 | 0.37 | morphogenesis, structure |
| BcLF1 | 3 | 1 | − | 0 | 2.8 | 0.0013 | 0.34 | morphogenesis, structure |
| BCRF1 | 10 | 1 | + | 0.52 | 2.3 | 0.071 | 0.43 | virus-host interaction |
| BDLF1 | 3 | 0 | + | 0 | 3.0 | 0 | 0.32 | morphogenesis, structure |
| BDLF2 | 10 | 3 | + | 1 | 2.8 | 0 | 0.34 | morphogenesis, structure |
| BDLF3 | 14 | 3 | + | 0.27 | 2.4 | 0.028 | 0.41 | morphogenesis, structure |
| BDLF3.5 | 2 | 1 | − | 0 | 1 | 0 | 1 | gene regulation |
| BdRF1 | 11 | 0 | + | 0.30 | 2.2 | 0.032 | 0.44 | morphogenesis, structure |
| BFLF1 | 1 | 0 | − | 0 | 3.8 | 0 | 0.26 | morphogenesis, structure |
| BFLF2 | 1 | 0 | − | 0 | 3.2 | 0 | 0.30 | morphogenesis, structure |
| BFRF1 | 15 | 5 | + | 0.39 | 2.4 | 0.042 | 0.41 | morphogenesis, structure |
| BFRF2 | 2 | 1 | − | 0 | 4 | 0 | 0.25 | gene regulation |
| BFRF3 | 4 | 1 | − | 0 | 2.7 | 0.0062 | 0.36 | morphogenesis, structure |
| BGLF2 | 7 | 1 | + | 0.16 | 2.5 | 0.012 | 0.39 | morphogenesis, structure |
| BGLF3 | 3 | 1 | − | 0 | 3.0 | 0.035 | 0.33 | gene regulation |
| BGLF3.5 | 5 | 0 | − | 0.3 | 2.2 | 0.0064 | 0.44 | morphogenesis, structure |
| BGLF4 | 5 | 2 | − | 0.33 | 2.6 | 0.0018 | 0.38 | morphogenesis, structure |
| BGLF5 | 2 | 0 | − | 1 | 2.9 | 0 | 0.34 | virus-host interaction |
| BGRF1/BDRF1 | 1 | 0 | − | 0 | 3.0 | 0 | 0.32 | morphogenesis, structure |
| BILF1 | 6 | 0 | − | 0.26 | 2.1 | 0.017 | 0.45 | morphogenesis, structure |
| BILF2 | 2 | 0 | − | 0 | 2.9 | 0.00022 | 0.33 | morphogenesis, structure |
| BKRF2 | 10 | 3 | + | 1 | 2.8 | 0 | 0.34 | morphogenesis, structure |
| BKRF3 | 17 | 2 | + | 0.25 | 1.9 | 0.11 | 0.51 | virus-host interaction |
| BKRF4 | 22 | 6 | + | 0.21 | 2.0 | 0.082 | 0.48 | morphogenesis, structure |
| BLLF1 | 14 | 2 | + | 0.42 | 2.3 | 0.019 | 0.43 | morphogenesis, structure |
| BLLF3 | 6 | 2 | − | 0.33 | 2.6 | 0.0035 | 0.37 | virus-host interaction |
| BLRF1 | 21 | 4 | + | 0.2 | 2.0 | 0.10 | 0.49 | morphogenesis, structure |
| BLRF2 | 25 | 2 | + | 0.15 | 1.7 | 0.23 | 0.57 | morphogenesis, structure |
| BMLF1 | 17 | 3 | + | 0.22 | 2.1 | 0.084 | 0.46 | gene regulation |
| BMRF1 | 6 | 0 | + | 0.83 | 2.5 | 0.00033 | 0.39 | DNA replication |
| BMRF2 | 10 | 4 | − | 0.66 | 2.8 | 0.035 | 0.34 | morphogenesis, structure |
| BNLF2a | 19 | 5 | + | 0.27 | 2.4 | 0.085 | 0.41 | virus-host interaction |
| BNLF2b | 10 | 0 | + | 0.21 | 2.3 | 0.043 | 0.42 | unknown |
| BNRF1 | 9 | 2 | − | 0.28 | 2.1 | 0.057 | 0.45 | morphogenesis, structure |
| BORF1 | 1 | 0 | − | 0 | 3.1 | 0 | 0.31 | morphogenesis, structure |
| BRRF1 | 8 | 1 | + | 0.4 | 2.6 | 0.0051 | 0.37 | unknown |
| BRRF2 | 1 | 0 | − | 0 | 3.0 | 0 | 0.33 | morphogenesis, structure |
| BSLF1 | 1 | 0 | − | 0 | 3.1 | 0 | 0.31 | DNA replication |
| BSRF1 | 13 | 3 | + | 0.32 | 2.1 | 0.025 | 0.45 | morphogenesis, structure |
| BVLF1 | 2 | 1 | − | 0 | 1 | 0 | 1 | gene regulation |
| BVRF1 | 1 | 0 | − | 0 | 3.3 | 0 | 0.30 | morphogenesis, structure |
| BVRF2 | 3 | 0 | − | 0.33 | 2.5 | 0.0025 | 0.38 | morphogenesis, structure |
| BXLF1 | 9 | 3 | − | 0.26 | 2.2 | 0.016 | 0.44 | morphogenesis, structure |
| BXLF2 | 9 | 1 | + | 0.6 | 2.4 | 0.0050 | 0.41 | morphogenesis, structure |
| BXRF1 | 2 | 0 | − | 1 | 2.9 | 0 | 0.34 | morphogenesis, structure |
| BZLF1 | 6 | 1 | + | 1 | 2.5 | 0 | 0.39 | gene regulation |
| EBNA-3C | 2 | 0 | − | 0 | 2.6 | 0.0011 | 0.37 | Latent |
| LMP-1 | 2 | 0 | − | 1 | 2.5 | 0 | 0.38 | Latent |
| LMP-2A | 12 | 1 | + | 0.27 | 2.1 | 0.051 | 0.47 | Latent |
| LMP-2B | 18 | 3 | + | 0.26 | 2.1 | 0.053 | 0.47 | Latent |
Among the 62 nodes, BLRF2 and BKRF4 had the highest degree centrality, suggesting that these two proteins have many interaction partners (Table 3; Fig. 3A). The clustering coefficient in the absence of BLRF2 was reduced by 18% compared with the original EBV protein network, suggesting that BLRF2 is essential for the small-world features of the network (Fig. 2C). Other centrality measures, including betweenness centrality and closeness centrality, also suggested that BLRF2 interacts with a number of proteins (Fig. 3B and C). Importantly, because BKRF4, the EBV protein with the second highest degree centrality (Fig. 3A), is crucial for virus production during the lytic cycle, interacting with many viral proteins (3, 22, 23), we hypothesized that BLRF2 also has an important role in virus particle production.
FIG 3.
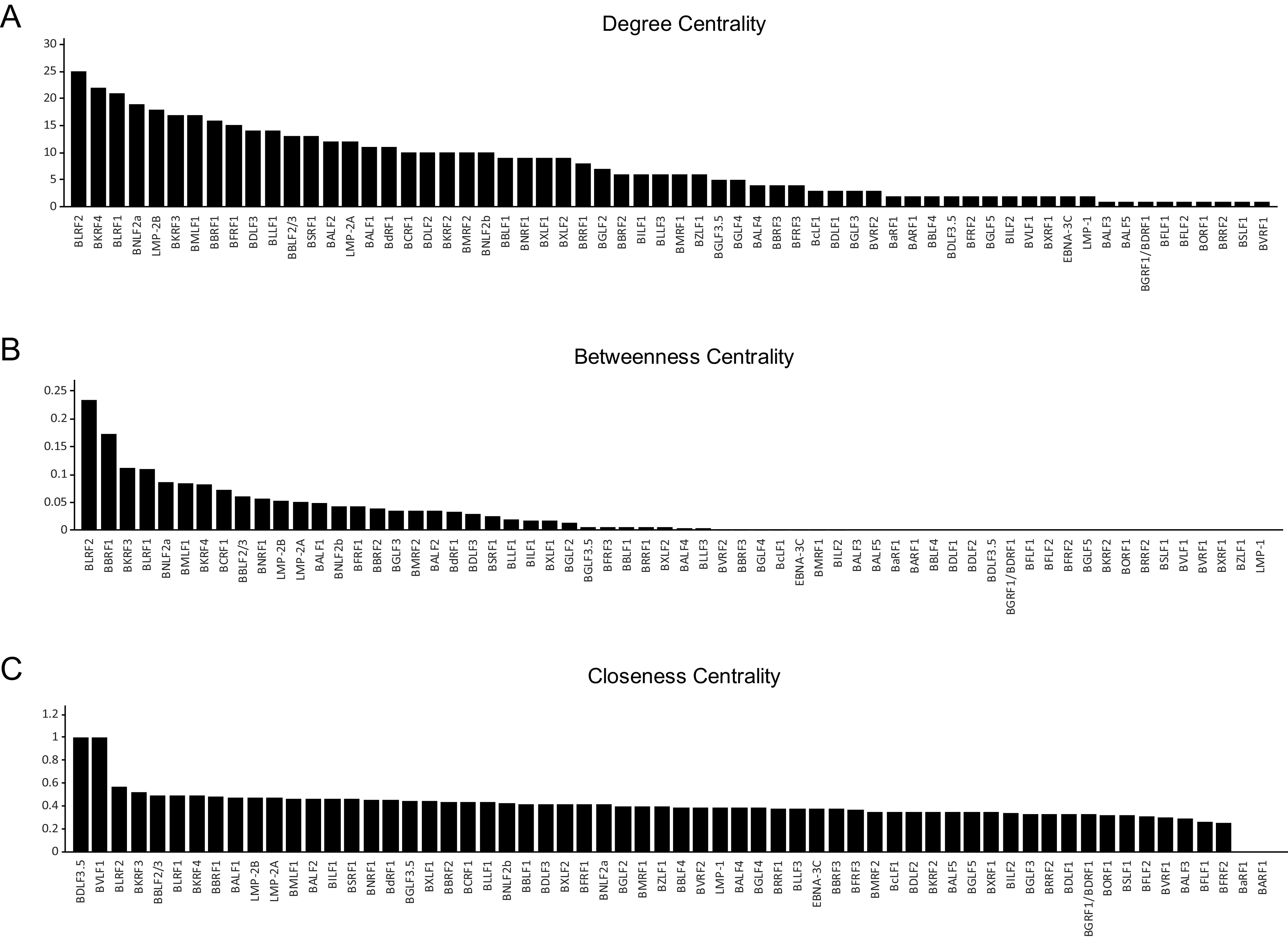
Centrality indices of nodes in the EBV PPIs network. (A to C) Bar charts showing the results of computational analyses of the three centrality measures, as determined by Cytoscape.
EBV protein interaction validation.
To further validate the PPIs identified by NanoBiT analyses, we investigated interactions between EBV proteins by immunoprecipitation. Because both BLRF2 and BKRF4 are tegument proteins, we focused on structural protein interaction networks (Fig. 4A). We further tested whether they interacted with factors associated with the DNA replication compartment (Fig. 4B). The local networks included previously reported interactions, such as BSRF1-BALF1, BSRF1-BBRF2, and BKRF4-BGLF2 (1, 23). Immunoprecipitation assays confirmed 14 additional interactions in the local network, namely, BDLF3-BALF4, BBLF1-BSRF1, BLRF2-BLRF2, BLRF2-BNRF1, BLRF2-BCRF1, BLRF2-BGLF3.5, BLRF2-BALF2, BLRF2-BMRF1, BALF4-BGLF2, BKRF4-BNRF1, BKRF4-BGLF2, BSRF1-BBRF1, and BSRF1-BBRF2 (Fig. 4C to P), further supporting the reliability of NanoBiT-based PPIs.
FIG 4.

Validation of EBV PPIs. (A) The local network centering BLRF2 and BKRF4 was isolated from the interactome network. Gene homologs in alpha- and beta-herpesviruses are shown. (B) The local network of BLRF2 with replication factors and latent proteins. (C to P) Validation of EBV PPIs by immunoprecipitation. HEK293T cells were transfected with the indicated expression vectors. Twenty-four hours after transfection, cell lysates were used for immunoprecipitation with an anti-Flag antibody. Immunoblotting was performed using the indicated antibodies. (Q) Validation of EBV PPIs by immunoprecipitation of endogenous proteins. HEK293EBV cells induced with BZLF1 expression vector were lysed and subjected for immunoprecipitation assay using anti-BLRF2 antibody.
We also tried to validate PPIs of endogenous EBV proteins in infected cells. Lysates were prepared from HEK293EBV cells transfected with BZLF1 expression vector to induce lytic cycle, and immunoprecipitation was performed using anti-BLRF2 antibody. Although we tested many viral proteins, we only could clearly demonstrate interaction of BLRF2 with BALF2 and LMP1 (Fig. 4Q), possibly because immunoprecipitation by using our anti-BLRF2 antibody was not very efficient.
Expression and localization of BLRF2 protein.
Next, we explored the relevance of BLRF2 in the viral life cycle. To detect endogenous BLRF2 expression, we developed a polyclonal antibody against BLRF2. BLRF2 protein levels were low during latent infection. Nevertheless, induction of the lytic cycle by anti-IgG or BZLF1 transfection in Akata and HEK293EBV cells, respectively, profoundly increased BLRF2 protein levels (Fig. 5A and B). Induction of the lytic cycle in B95–8 cells was performed using the chemical inducers TPA, A23187, and sodium butyrate (T/A/B). Although BLRF2 protein levels were low until 12 h, they were considerably higher from 24 h after induction (Fig. 5C). To examine the expression kinetics of BLRF2, we treated B95–8 cells with T/A/B in the presence or absence of PAA (Fig. 5D). The DNA synthesis inhibitor PAA suppressed the expression of the late gene gB, while it did not significantly affect the expression of the early genes BMRF1 and BALF2 (Fig. 5D, T/A/B+PAA). Furthermore, BLRF2 expression was markedly suppressed by PAA (Fig. 5D, T/A/B+PAA), suggesting that BLRF2 is expressed with late kinetics, corroborating previous findings (24).
FIG 5.

Expression of endogenous BLRF2 protein. (A) HEK293EBV cells were transfected with a BZLF1 expression vector to induce the lytic cycle followed by sample collection 48-h posttransfection. BLRF2 protein was detected by the immunoblotting using an anti-BLRF2 polyclonal antibody. (B) EBV-positive Akata cells were treated with an anti-human IgG antibody (1:500) for 48 h. (C) B95–8 cells were reacted with TPA (T, 20 ng/mL), A23187 (A, 1 μM), and sodium butyrate (B, 5 mM) for the indicated duration, followed by immunoblotting for BLRF2, BZLF1 (IE), BMRF1 (E), gB (L), and GAPDH proteins. (D) B95–8 cells were reacted with TPA (T, 20 ng/mL), A23187 (A, 1 μM), and sodium butyrate (B, 5 mM) in the presence or absence of PAA (400 μg/mL). Cells were harvested after 36 h and subjected to immunoblotting for BLRF2, BMRF1(E), BALF2 (E), gB (L), and GAPDH proteins.
To assess the intracellular localization of BLRF2, we overexpressed BLRF2 in the human cervical carcinoma cell line HeLa, followed by immunofluorescence. BLRF2 was predominantly detected in the cell nucleus and nuclear rim (Fig. 6A). A similar localization pattern was observed in the gastric carcinoma cell line AGS (Fig. 6B). These results coincide with previous reports that BLRF2 dominantly localized in the nucleus (25, 26).
FIG 6.

Subcellular localization of BLRF2. (A, B) HeLa and AGS cells were transiently transfected with BLRF2-HA-expressing vector and fixed 24-h posttransfection. Immunofluorescence staining was conducted using an anti-HA antibody (green). Cells were counterstained with DAPI (blue). (C) EBV-positive AGS cells were transfected with a BZLF1-expressing vector and incubated for 48 h. The cells were stained for BLRF2 (green) and BMRF1 (red). Then they were counterstained with DAPI (blue). (D) HEK293EBV cells transfected with BZLF1 were processed as in C, except that they were also labeled with actin (phalloidin-Alexa 647) to visualize the cytoplasm. (E, F) EBV-positive AGS and HEK293EBV cells were processed as in C and D, respectively, and fixed for immunostaining at 0, 24, 48, and 72 h after reactivation. (G) AGS EBV cells were transfected with BZLF1, fixed at 72 h after transfection, and then stained with Alexa555-labeled anti-BLRF2 antibody (red) and anti-BKRF4 antibody (green). (H) Confocal immunofluorescence imaging of BLRF2 (red), Giantin (green), and DAPI (blue) in AGS EBV cells in the virus production cycle. Scale bar = 10 μm.
In EBV-positive AGS and HEK293 cells, we detected endogenous BLRF2 in the cell nucleus and nuclear rim 48 h after reactivation (Fig. 6C and D). Moreover, we observed that 48 h after lytic induction, the replication compartments occupied a significant part of the cell nucleus. Consequently, the cellular genomic DNA was localized to the nuclear periphery. BLRF2 colocalized with BMRF1 in the core of the cell nucleus (Fig. 6C to F). It is possible that this colocalization is accounted by the BLRF2-BMRF1 interaction detected in our NanoBiT-based PPI and immunoprecipitation assays (Fig. 1B; Fig. 4K). We then carried out co-staining of BKRF4 and BLRF2 in EBV-positive AGS (Fig. 6G). BKRF4 predominantly localized at the nuclear periphery as reported previously (23) and BLRF2 was mainly found in the nucleus and nuclear rim, suggesting that these two tegument proteins interacted at the nuclear periphery (Fig. 6G).
A fraction of BLRF2 proteins were found in the cytoplasm after 48 h of reactivation (Fig. 6E and F). Because staining pattern of cytoplasmic BLRF2 was a reminiscent of the Golgi apparatus, BLRF2 was co-stained with a Golgi marker, Giantin (Fig. 6H). Although most of the BLRF2 protein existed outside the Giantin-positive organelle, at least a portion of BLRF2 colocalized with Giantin (Fig. 6H).
BLRF2 contributes to the production of infectious viral particles.
Information regarding the function of BLRF2 in the life cycle of EBV is based solely on evidence from exogenous overexpression of the protein in the ORF52-null mutant of murine gammaherpesvirus 68 (MHV68) (27). Therefore, to elucidate the relevance of EBV BLRF2, we generated a BLRF2-knockout virus by the CRISPR/Cas9 system using two different BLRF2-targeting sgRNAs (Fig. 7A). Akata cells were infected with two different BLRF2-knockout viral clones (Fig. 7B; BLRF2KO1 and BLRF2KO2) as per the previously described protocol (23). Although infectious viral particles were produced in Akata cells infected with BLRF2-knockout EBV, the virus titers were significantly lower (Fig. 7C). BLRF2 interacted with BALF2, a protein needed for viral DNA synthesis (Fig. 4J and Q), but BLRF2-knockout did not affect the DNA synthesis of the viral genome (Fig. 7D). In addition, the absence of BLRF2 from the viral genome did not affect the expression levels of the viral proteins BZLF1, BMRF1, and BALF4 (gB); BLRF2 was the only viral protein with impaired expression in cells infected by the knockout strains (Fig. 7E).
FIG 7.
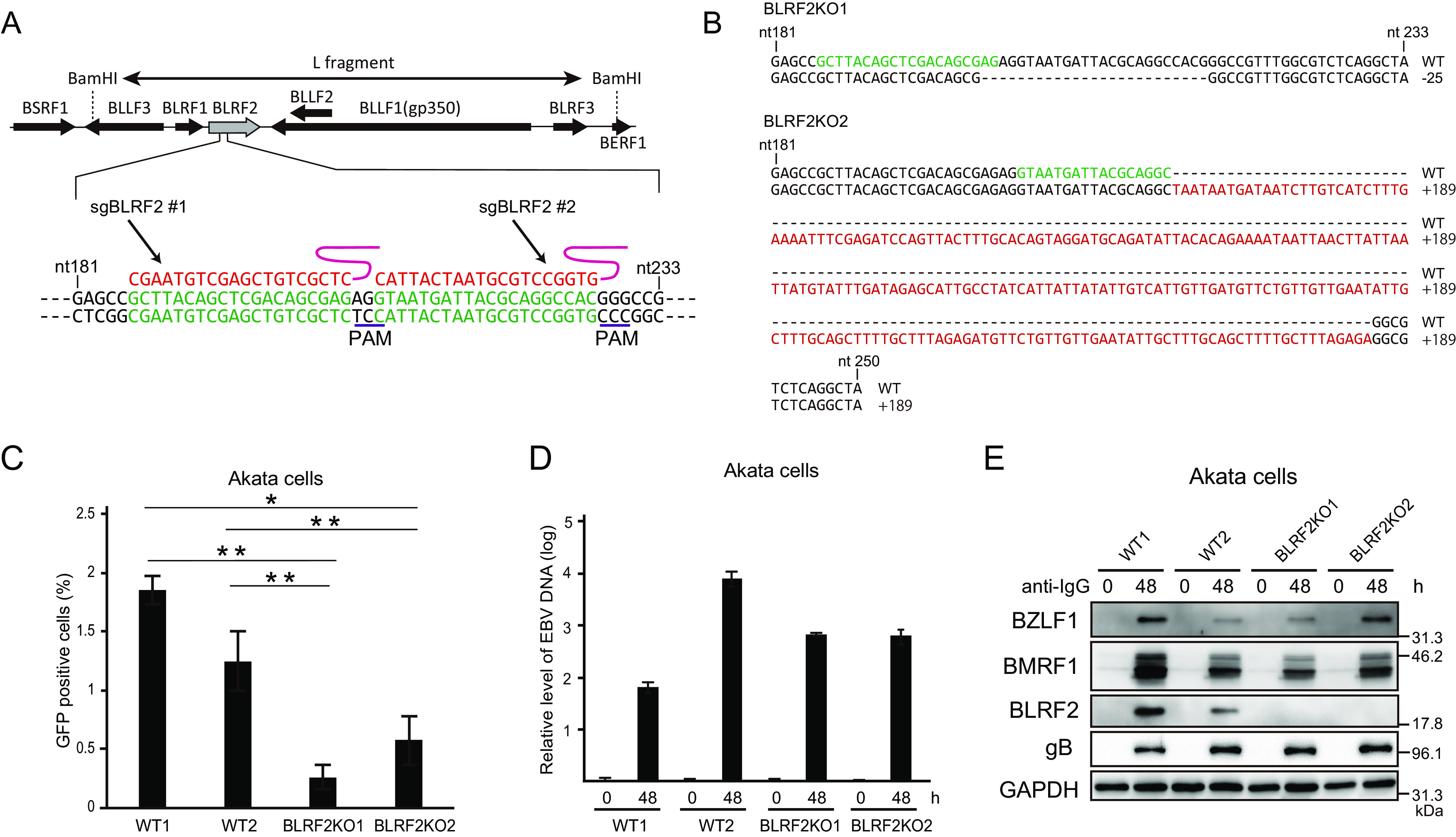
Functional relevance of BLRF2 in Akata cells. (A) The sequences of the two sgRNAs and PAM sites used are shown. AGS cells latently infected with EBV were transfected with the CRISPR/Cas9 vectors, followed by puromycin selection. Progeny virions were obtained from the cells, and infected to Akata(−) cells at low multiplicity of infection, followed by limited dilution and cell cloning in the presence of G418. We isolated two cell clones infected with different BLRF2 knockout EBVs. (B) Sequences of two BLRF2KO clones. The EBV in BLRF2KO1 cell line, obtained by using sgBLRF2#1, has 25 bp deletion. The EBV in BLRF2KO2 was made using sgBLRF2#2, having 189 bp insertion. The nucleotides in green color show corresponding sgRNA sequences. (C) Akata cells infected with BLRF2-knockout virus were stimulated with anti-human IgG, and the cells and culture media were harvested at 48 h after induction. The virus titers in the samples were determined. (D) Akata cells infected with BLRF2-knockout virus were treated as in (C) and cell-associated DNA was harvested at 0 and 48 h after induction for determination of viral DNA levels by real-time PCR. (E) Akata cells infected with BLRF2-knockout virus were treated as in (B) and cell-associated protein was harvested at 0 and 48 h after infection. Immunoblot analyses for BZLF1, BMRF1, BLRF2, gB, and GAPDH were performed.
Next, we infected AGS-CR2 cells with BLRF2-knockout EBV and observed endogenous BLRF2 (Fig. 8A) that was consistent with the BLRF2 localization shown in Fig. 6. The virus titers were significantly lower in AGS-CR2 cells infected with BLRF2-knockout EBV than those infected with wild-type (WT) EBV (Fig. 8B). No effects were observed on the viral DNA synthesis (Fig. 8C). The viral DNA level was higher in BLRF2KO1 (Fig. 8C), but we assume this is within a clonal variation.
FIG 8.

Functional relevance of BLRF2 in AGS cells. (A) EBV-negative AGS cells were infected with two BLRF2-knockout strains of EBV prepared from the two Akata cell lines in Fig. 7, followed by G418 selection. The resultant AGS cells latently infected with the knockout EBV were stimulated with BZLF1 transfection and harvested at 2 days after transfection. Scale bar = 10 μm. (B) AGS cells infected with BLRF2-knockout EBV were transduced with BZLF1, and the cells and culture supernatants were harvested at 72 h after transfection. The virus titer was determined. (C) AGS cells infected with BLRF2-knockout virus were transfected with BZLF1 and cell-associated DNA was harvested at 0 and 72 h after transfection for determination of viral DNA levels by real-time PCR. (D, E) Complementation assays. AGS cells infected with BLRF2-knockout virus were transfected with BLRF2-HA and/or BZLF1, and the cells and culture supernatants were harvested at 72 h after transfection. The virus titer was determined by flow cytometry, and protein levels were analyzed by Immunoblotting. (F) AGS cells infected with BLRF2-knockout EBV were transfected with BZLF1. After 72 h, cells and cell culture media were harvested altogether, and subjected to low-speed centrifugation. The supernatant was used as the source of “cell-free” virion. The cell pellet was added with the same volume of fresh media, and then the sample was frozen and thawed, followed by centrifugation. The supernatant from cell pellet was now used as the source of “cell-associated” virion. Progeny virus titers were examined by flow cytometry. (G) Quantification of EBV genome secreted into the culture media of AGS cells infected with BLRF2-knockout virus after lytic induction.
Then we examined whether exogenous expression of BLRF2 could rescue viral production in AGS-CR2 cells infected with BLRF2-knockout EBV. BLRF2 trans-complementation partly restored the viral titer (Fig. 8D and E). The reasons why exogenous expression of BLRF2 could not fully restore the decreased viral titer in the KO are not clear.
In Fig. 8B, we collected both cell-associated and cell-free virions, and found that infectious progeny titer was decreased by the knockout. To monitor the influence of BLRF2 on virus secretion, cells and media were collected separately, and analyzed the infectivity of cell-associated virions and cell-free virions (Fig. 8F). It was found that BLRF2 knockout significantly reduced the cell-free but not the cell-associated levels of infectious virus (Fig. 8F), suggesting that BLRF2 is likely involved in the extracellular release of infectious progeny virions. To be precise, this result indicates that BLRF2 knockout caused either (i) loss of infectivity of the extracellular virion particles without affecting the total number of extracellular virions, (ii) loss of extracellular secretion of total virion particles without affecting the ratio of infectious virion, or (iii) loss of both infectivity and total number of extracellular virions. To answer this question, we collected the culture media from cells infected with WT or the knockout virus after lytic induction, extracted the DNA in the media, and EBV genome DNA levels were quantified (Fig. 8G). Note that we treated the media with DNase before extraction of viral DNA, in order to eliminate the viral DNA that was not incorporated into virion particles. BLRF2-knockout did not affect the total number of virus particles in cell-free fraction (Fig. 8G), indicating that (i) is correct, where BLRF2 plays a role in the acquisition of viral infectivity. Overall, these results suggest that BLRF2 contributes to the production of infectious virus particles not only in lymphoid cells but also in epithelial cells.
Potential role of BLRF2 dimerization in infectious virus production.
Because our PPIs screen using NanoBiT system (Fig. 1), immunoprecipitation experiment (Fig. 4D), and previous GST pulldown assay (27) clearly demonstrated that BLRF2 interacts with itself, we lastly examined physiological significance of the self-association.
First, we confirmed whether endogenous BLRF2 could self-associate in infected cells (Fig. 9A). To this end, HEK293EBV cells were cotransfected with expression vectors for BZLF1 and Flag-tagged BLRF2, and immunoprecipitation was carried out by using anti-Flag antibody. Endogenous BLRF2 (about 20 kDa) protein was coprecipitated with overexpressed, Flag-tagged BLRF2 (Fig. 9A, endogenous).
FIG 9.

Self-association of BLRF2. (A) HEK293EBV cells infected with WT EBV were cotransfected with the BZLF1 expression vector together with Flag-tagged BLRF2 expression vector. Cells were lysed at 48 h after transfection and subjected to immunoprecipitation assays by using anti-Flag antibody. (B) Schematic diagram and amino acids sequence alignment of possible dimerization domain of gamma-herpesviruses. Conserved residues are indicated in black or gray. (C, D) HEK293T cells were cotransfected with LgBiT-tagged WT BLRF2 expression vector and SmBiT-tagged mutated BLRF2 vector. Cells were subjected to the NanoBiT assay. (E) HEK293T cells transfected with Flag-tagged WT BLRF2 and HA-tagged mutated BLRF2 vectors were lysed and subjected to immunoprecipitation by using anti-Flag antibody. (F) AGS cells infected with BLRF2-knockout EBV were transfected with BZLF1 vector, together with empty, WT, or I87AE88A BLRF2 vectors. Cell proteins were harvested for Immunoblotting. Progeny virions in the culture media were harvested at 72 h after transfection, and infected to Akata negative cells to determine the virus titer in the media by flow cytometry.
MHV68 ORF52, the homolog of EBV BLRF2, was reported to form a dimer through α2-helix domain in the central part of the protein (28, 29). Because this central region in ORF52 was conserved to some extent with EBV (Fig. 9B), we tested if EBV BLRF2 could dimerize through the same region (Fig. 9C). NanoBiT assays using LgBitT-WT BLRF2 and SmBiT-mutated BLRF2 showed that deletion of the region (del67-88) markedly decreased NanoBiT activity by more than 1 order of magnitude, compared with WT (Fig. 9C). To further analyze the motif needed for the self-association, a series of BLRF2 mutants, each lacking two amino acids in the domain, were prepared (Fig. 9C). Among the mutants, deletion of I87 and E88 (delI87E88) most efficiently decreased the luciferase activity (Fig. 9C). We then prepared alanine substitution mutants as indicated in Fig. 9D. Either of the single amino acid substitutions did not decrease the association notably, but substitution of both amino acids (I87AE88A) significantly blocked the activity (Fig. 9D).
We then carried out immunoprecipitation assays. HA-tagged WT BLRF2 was successfully coprecipitated with Flag-tagged WT BLRF2, but mutated HA-tagged BLRF2 proteins that exhibited very low NanoBiT activity (Fig. 9C and D) could not be copurified (Fig. 9E). However, Western blotting of input proteins revealed that expression of mutated BLRF2 proteins was reduced (Fig. 9E). Therefore, it is likely that inhibition of the dimer formation resulted in instability of the BLRF2 protein. Still, we cannot deny the possibility that BLRF2 self-association could not be detectable because the mutation of the protein caused lower expression. Either way, we tested if the mutant could restore the decreased progeny production. Exogenous expression of WT BLRF2 partially increased the progeny level, but the I87AE88A mutant did not (Fig. 9F).
Taken together, due to low-level expression of the mutant protein (Fig. 9F), our hypothesis that BLRF2 dimer formation promotes progeny production cannot be justified from these data. However, it may still be possible that self-association of BLRF2 might be important for stability of the protein and progeny production.
DISCUSSION
To date, there are only a few reports on EBV PPI networks, which are based primarily on Y2H screens (15, 16). Although the Y2H assay has been widely used for large-scale screenings, it relies on protein transport into the nucleus, which is not always the physiological subcellular localization of the proteins. In this study, we performed NanoBiT system-based PPI analyses, which allowed for the large-scale detection of protein interactions in living cells. Using this method, we identified 195 PPIs, most of which have not previously been reported (15, 16). Hence, NanoBiT is a promising method for identifying novel PPIs in living cells.
Interestingly, among the 69 EBV proteins, 28 (40.6%) formed homomeric complexes (Fig. 1B). Of these, 17 (63.0%) have previously been reported in EBV or other herpesviruses. The formation of homodimers or homo-oligomers is more common among virion structural components with repetitive structures, such as capsid and tegument proteins. The formation of homomeric complexes has also been reported for some membrane proteins and secreted proteins, such as BCRF1 (vIL-10) and BARF1 (vCSFR).
A major limitation of our NanoBiT assay is that among the 167 hetero-interactions, only 38 edges (22.8%) were identified as bidirectional (Table 2; Fig. 1B). This indicates that 129 from 167 hetero-PPIs (77.2%) were below the threshold value. This implies that LargeBiT and SmallBiT may not be able to interact due to structure constraints and that some interactions might not be detected by this method. In addition, we failed to clone some genes due to their large size, and the expression of some other genes was unsuccessful, possibly due to sequence complexity. Other genes such as BRLF1 (Rta) seemed to exhibit high auto activation, and thus were eliminated from the heatmap and network.
Our computational analyses identified BLRF2 as an EBV interactome hub (Fig. 2 and 3). Moreover, BLRF2 had the highest degree centrality and betweenness centrality values (Fig. 3A and B), and exhibited a high closeness centrality value (Fig. 3C). After BLRF2, BKRF4 was the node with the highest centrality values. We have previously shown that BKRF4 plays central roles in the tegument network and enhances progeny virion production functioning as a complex with other EBV proteins (22, 23, 30). Although their role in the tegument network is well documented in KSHV (11), to the best of our knowledge, this is the first study reporting EBV BLRF2 and BKRF4 as interactome network hubs. BLRF2 and BKRF4 are conserved among gammaherpesviruses but not in other subgroups of herpesviruses. We speculate that some other tegument proteins, such as UL48 and UL49, play central roles in the HSV tegument network (10), instead of BLRF2 and BKRF4.
BLRF2 has previously been reported to localize in the nucleus (27, 31); our immunofluorescence findings are consistent with those results (Fig. 6). We also found that BLRF2 was predominantly localized in the nuclear replication compartment. A recent study demonstrated that the replicated viral DNA was packaged into capsids in the replication compartments to form the mature nucleocapsids (32); thus, nuclear tegument proteins, including BLRF2, may bind to capsid proteins before primary envelopment of progeny nucleocapsids. Interestingly, we could also observe BLRF2 at the nuclear rim. Therefore, BLRF2 may also be involved in the primary envelopment and de-envelopment process at the nuclear membrane. The nuclear rim localization was reminiscent of another tegument network hub, BKRF4 (30), although it could not be detected in the nuclear replication compartment.
Disruption of the BLRF2 gene had little or no effect on viral gene expression and viral DNA synthesis but caused a significant decrease in the titer of infectious progeny (Fig. 7 and 8). Similar results were obtained for gene homologs in other gammaherpesviruses. Notably, ORF52, the BLRF2 homolog in KSHV and MHV68, is important for the acquisition of secondary envelope and efficient virus production (28, 33, 34). Because a portion of BLRF2 was localized in the Golgi apparatus and was involved in the production of extracellular virion (Fig. 6H and 8F), BLRF2 may also be involved in the acquisition of the secondary envelope. Intriguingly, the effects of viral BLRF2 and BKRF4 knockout on the phenotype of the target cells were similar (30). In addition, the rhesus monkey gammaherpesvirus ORF52 mediates the association of the BKRF4 homolog ORF45 to the nucleocapsids (35). Considering that BLRF2 and BKRF4 colocalized at the nuclear rim (Fig. 6G), these proteins likely act together to reinforce the transportation and virion morphogenesis of EBV at the nuclear rim. In addition to the function of BLRF2 in virion transportation and morphogenesis, KSHV ORF52 negatively regulates the innate immune response in infected cells by interacting with the DNA sensor molecule cGAS (36). BKRF4 has also been implicated in tumorigenesis by binding to histone proteins and suppressing DNA damage repair responses (37, 38). Hence, the tegument network hub proteins BLRF2 and BKRF4 or their homologs regulate the viral replication cycle and various target cell functions in multiple ways.
In conclusion, our findings provide novel insight into the complex intraviral PPI network in EBV infection. However, future comprehensive studies are required to identify novel PPIs in EBV and other gammaherpesviruses.
MATERIALS AND METHODS
Cell lines and reagents.
HEK293T and HeLa cells were cultured in Dulbecco minimal essential medium (DMEM; Sigma) supplemented with 10% fetal bovine serum (FBS). Akata, AGS, and B95–8 cells were maintained in RPMI 1640 (Sigma) supplemented with 10% FBS. TPA, A23187, and PAA were purchased from Sigma-Aldrich and dissolved in dimethyl sulfoxide (DMSO). Sodium butyrate was purchased from Wako and dissolved in DMSO. Goat anti-human IgG was purchased from Jackson Immuno Research Inc. and was used as an inducer of virus reactivation. Rabbit anti-BLRF2 antibodies were obtained by immunizing rabbits with the polypeptide NH2-C+RSRGREAKKVQISD-COOH conjugated with keyhole limpet hemocyanin (KLH); anti-BLRF2 antibodies were affinity-purified from the serum of immunized animals.
Immunoblotting.
To induce lytic infection, HEK293 cells and AGS cells latently infected with EBV were transfected with BZLF1 by electroporation or lipofection. Seventy-two hours after transfection, cells were harvested in lysis buffer (50 mM Tris-HCl, 2% SDS, 10% glycerol, 6% 2-mercaptoethanol, and 0.0025% bromophenol blue; pH 6.8). Equal amounts of proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis SDS-PAGE and then transferred onto PVDF membranes (Millipore). Antibodies against BZLF1, BMRF1, BALF2, BRRF2, and gB were prepared as previously described (20, 21). The anti-BLRF2 antibody was diluted 2,000 times in Can Get Signal solution (TOYOBO). Antibodies against GAPDH (14C10, Cell Signaling Technology) and α-tubulin (2144, Cell Signaling Technology) were used as endogenous controls. Horseradish peroxidase (HRP)-linked anti-rabbit and anti-mouse IgG (Cell Signaling Technology) were used as secondary antibodies. Forte (Merck) or Chemi-Lumi One Ultra (Nacalai Tesque, Inc.) was used for immunoblot signal development; EZ-Analyzer (ATTO) was used for chemiluminescent signal detection.
Expression vector.
EBV open reading frame (ORF) sequences were amplified using KOD One (TOYOBO). WT EBV bacterial artificial chromosome DNA or EBV expression vector library (22) was used as a template. The PCR products were subjected to agarose gel electrophoresis and then purified using the QIAquick Gel Extraction Kit (Qiagen). The NanoBiT expression vectors pBiT1.1-C(TK/LgBiT) and pBiT2.1-C(TK/SmBiT) were digested with EcoRI, and a NanoBiT library was constructed by homologous recombination using In-Fusion Cloning (TaKaRa) according to the manufacturer’s instructions. The BLRF2-HA expression vector has previously been described. The BLRF2-Flag expression vector was constructed by cloning the WT BLRF2 sequence into the EcoRI and XhoI sites of a pcDNA3-Flag vector (23). To generate a BLRF2 knockout virus, we generated a Cas9 expression vector pX459 (Addgene) carrying a BLRF2-targeting guide RNA (sgRNA). The two following oligonucleotide sequences were annealed and inserted into the px459 vector: sgBLRF2#1 forward (CACCGCTTACAGCTCGACAGCGAG), reverse (AAACCTCGCTGTCGAGCTGTAAGC); sgBLRF2#2 forward (CACCGTAATGATTACGCAGGCCAC), reverse (AAACGTGGCCTGCGTAATCATTAC).
NanoBiT assay.
The NanoBiT-based luciferase assay involves the use of the NanoLuc (19 kDa), which is derived from deep-sea shrimp (24). NanoBiT plasmids of 69 EBV genes were transfected into HEK293T cells using FuGENE (Promega) in combinations (4,761 pairs). Twenty-four hours after transfection, the NanoLuc substrate was added to the culture medium, and luminescence was detected using a microplate reader (PowerScan4; BioTek). The combination of the HaloTag-SmBiT vector and the LgBiT-PRKAR2A vector was used as a negative control. To calculate a relative luminance value, we divided the luminance signal detected for each combination by the signal of the negative control. Relative luminance values were visualized as a heatmap and imported into Cytoscape for network analyses.
Viral DNA quantification by real-time PCR.
The amount of viral DNA in the cells was quantified by real-time PCR as previously described (20). At the indicated time points after reactivation, EBV-positive AGS or Akata cells were solubilized in lysis buffer (10 mM Tris-HCL, 1 mM EDTA; pH 8) containing proteinase K. Real-time PCR was performed using FastStart Universal Probe Master (Rox) (Roche Applied Science), eukaryotic 18S rRNA primers (Applied Biosystems), custom-designed BALF2-specific primers, and BALF2-specific probes conjugated with FAM/TAMRA: BALF2 forward primer (5′-GCCCGTCCGGTTGTCA-3′), reverse primer (5′-AATATCTGGTTGTTGCCGTTGA-3′); BALF2 probe (5′-FAM-CTGCCAGTGACCATCAACAAGTACACGG-TAMRA-3′). Real-time PCRs were run on a 7500 Fast Dx system (Applied Biosystems).
Measurement of infectious virus titer by flow cytometry.
For recombinant EBV-positive Akata cells, 1 × 106 cells were treated with goat anti-human IgG (Jackson Immuno Research Inc.) for 48 h, and the medium was collected. For recombinant EBV-positive AGS cells, 5.0 × 105 cells were transfected with BZLF1 plasmid by using the Neon Transfection System (Thermo Fisher Scientific) and the cell culture medium was harvested at 72 h after transfection. After centrifugation, 1 mL harvested media, which contained progeny viruses, were inoculated with 1 × 106 EBV-negative Akata cells for 2.5 h with rotation, followed by a 2-day incubation in fresh cell culture medium. The cells were fixed with PBS containing 4% paraformaldehyde, and the percentage of GFP-positive cells was determined by flow cytometry (Gallios, Beckman Coulter). To obtain the cell-free virus, we centrifuged the cell culture medium at a low speed and collected the supernatant; the pellet was used to obtain the cell-associated virus. Freeze-thawed cell pellets were suspended in PBS, and suspended cells were centrifuged and filtered with a 0.45 μm membrane filter (Millipore).
Measurement of mature cell-free virion DNAs in lytic-induced cells.
To remove containing immature virion DNA, cell culture medium from lytic-induced AGS cells was reacted with Turbo DNase (Thermo Fisher Scientific) at 37°C for 1 h. The remaining mature virus particle was broken down by proteinase K treatment. The purified virus DNA was isolated using the DNeasy kit (Qiagen) according to the manufacturer’s recommendations. The purified DNA in mature virus particles was determined by amplification using EBV BALF2-specific primers on the 7500 Fast Dx system (Applied Biosystems).
Immunofluorescence staining.
HeLa, AGS, EBV-positive AGS, and EBV-positive HEK293 cells were seeded on coverslips coated with Poly d-Lysine (NEU), and plasmid transfection was performed 24 h later. Cells were fixed with 70% ethanol and permeabilized using 0.1% Triton X-100. After blocking with Protein Block (Agilent), cells were incubated with anti-HA (C29F4, Cell Signaling Technologies), anti-BMRF1 (clone R3, Sigma), anti-Giantin (9B6, Abcam), and anti-BLRF2 primary antibodies. In the case of co-staining for BKRF4 and BLRF2, anti-BLRF2 antibodies were labeled by Zenon Alexa555 rabbit IgG labeling kit (Invitrogen). After washing, cells were incubated with Alexa Fluor 488 anti-rabbit IgG and Alexa Fluor 546 anti-mouse IgG (Invitrogen) secondary antibodies. Alexa Fluor 647 Phalloidin (Invitrogen) was used to stain actin. After mounting Prolong Gold antifade reagent (Thermo Fisher Scientific), samples were observed under a fluorescence microscope (LSM880; Zeiss).
Immunoprecipitation.
HEK293T cells were cotransfected with HA-tag and Flag-tag expression vectors. Twenty-four hours posttransfection, cells were incubated in lysis buffer (20 mM Tris-HCl, 0.5% NP-40, 150 mM NaCl, 1 mM EDTA; pH 7.6) containing Complete Mini Protease Inhibitor Cocktail (Sigma). Cell lysates were incubated for 1 h with protein G agarose beads (Sigma) and then immunoprecipitated with anti-Flag antibody (F3165, Sigma) and protein G agarose beads for 3 to 24 h. For endogenous viral protein interactions, immunoprecipitation was performed with 2.4 μg of anti-BLRF2 antibody or normal rabbit IgG (Santa Cruz). Precipitated proteins were subjected to Western blotting.
Generation of BLRF2 knockout Akata cells.
Gastric carcinoma AGS cells latently infected with EBV were transduced with CRISPR/Cas9 vectors (px459) targeting the EBV BLRF2 gene. Successfully transduced cells were selected with puromycin (1 μg/mL) for 14 days and then transfected with BZLF1 to obtain infectious virus particles. Akata(−) cells were infected with the virus particles and selected by limiting dilution in the presence of G418 (Wako, 750 μg/mL). Successfully edited BLRF2 clones were confirmed by direct PCR using KOD One (TOYOBO) and sequencing, using a custom-designed primer: 5′-CAGCAATCTCGGCTGTCTG-3′ (forward) and 5′-CATATGATGACGCCGAGAC-3′ (reverse).
ACKNOWLEDGMENTS
We thank M. Miyata, S. Uchiayama, S. Kumagai, T. Kunogi, T. Kanda, H. Yoshiyama, Y. Narita, and T. Tsurumi for materials, technical assistance, and discussions. NanoBit screening, flow cytometry, and imaging analysis were performed at the Division for Medical Research Engineering, Nagoya University Graduate School of Medicine. This work was supported by grants-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (19K07580 to T.M., 21K15449 to T.W., 19K22560 and 20H03493 to H.K.), Japan Agency for Medical Research and Development (JP20wm0325012 to T.M.), the Takeda Science Foundation (to T.M.), and the Hori Sciences and Arts Foundation (to T.M., Y.S., and H.K.).
Contributor Information
Takahiro Watanabe, Email: t.nabe.watanabe@med.nagoya-u.ac.jp.
Hiroshi Kimura, Email: hkimura@med.nagoya-u.ac.jp.
Takayuki Murata, Email: tmurata@fujita-hu.ac.jp.
Lori Frappier, University of Toronto.
REFERENCES
- 1.Cohen JI. 2000. Epstein-Barr virus infection. N Engl J Med 343:481–492. 10.1056/NEJM200008173430707. [DOI] [PubMed] [Google Scholar]
- 2.Young LS, Yap LF, Murray PG. 2016. Epstein-Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer 16:789–802. 10.1038/nrc.2016.92. [DOI] [PubMed] [Google Scholar]
- 3.Murata T, Sato Y, Kimura H. 2014. Modes of infection and oncogenesis by the Epstein-Barr virus. Rev Med Virol 24:242–253. 10.1002/rmv.1786. [DOI] [PubMed] [Google Scholar]
- 4.Kimura H. 2018. EBV in T-/NK-Cell Tumorigenesis. Adv Exp Med Biol 1045:459–475. 10.1007/978-981-10-7230-7_21. [DOI] [PubMed] [Google Scholar]
- 5.Murata T, Sugimoto A, Inagaki T, Yanagi Y, Watanabe T, Sato Y, Kimura H. 2021. Molecular basis of Epstein-Barr virus latency establishment and lytic reactivation. Viruses 13:2344. 10.3390/v13122344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hammerschmidt W, Sugden B. 2013. Replication of Epstein-Barr viral DNA. Cold Spring Harb Perspect Biol 5:a013029. 10.1101/cshperspect.a013029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsurumi T, Fujita M, Kudoh A. 2005. Latent and lytic Epstein-Barr virus replication strategies. Rev Med Virol 15:3–15. 10.1002/rmv.441. [DOI] [PubMed] [Google Scholar]
- 8.Chakravorty A, Sugden B, Johannsen EC. 2019. An epigenetic journey: Epstein-Barr virus transcribes chromatinized and subsequently unchromatinized templates during its lytic cycle. J Virol 93. 10.1128/JVI.02247-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henson BW, Perkins EM, Cothran JE, Desai P. 2009. Self-assembly of Epstein-Barr virus capsids. J Virol 83:3877–3890. 10.1128/JVI.01733-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Owen DJ, Crump CM, Graham SC. 2015. Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses 7:5084–5114. 10.3390/v7092861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sathish N, Wang X, Yuan Y. 2012. Tegument proteins of Kaposi's sarcoma-associated herpesvirus and related gamma-herpesviruses. Front Microbiol 3:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai YC, Liao YT, Juan YT, Cheng YY, Su MT, Su YZ, Liu HC, Tsai CH, Lee CP, Chen MR. 2020. The novel nuclear targeting and BFRF1-interacting domains of BFLF2 are essential for efficient Epstein-Barr virus virion release. J Virol 94. 10.1128/JVI.01498-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nanbo A, Noda T, Ohba Y. 2018. Epstein-Barr virus acquires its final envelope on intracellular compartments with Golgi markers. Front Microbiol 9:454. 10.3389/fmicb.2018.00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calderwood MA, Venkatesan K, Xing L, Chase MR, Vazquez A, Holthaus AM, Ewence AE, Li N, Hirozane-Kishikawa T, Hill DE, Vidal M, Kieff E, Johannsen E. 2007. Epstein-Barr virus and virus human protein interaction maps. Proc Natl Acad Sci USA 104:7606–7611. 10.1073/pnas.0702332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fossum E, Friedel CC, Rajagopala SV, Titz B, Baiker A, Schmidt T, Kraus T, Stellberger T, Rutenberg C, Suthram S, Bandyopadhyay S, Rose D, von Brunn A, Uhlmann M, Zeretzke C, Dong YA, Boulet H, Koegl M, Bailer SM, Koszinowski U, Ideker T, Uetz P, Zimmer R, Haas J. 2009. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog 5:e1000570. 10.1371/journal.ppat.1000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347–354. 10.1016/j.virol.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 18.Rozen R, Sathish N, Li Y, Yuan Y. 2008. Virion-wide protein interactions of Kaposi's sarcoma-associated herpesvirus. J Virol 82:4742–4750. 10.1128/JVI.02745-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uetz P, Dong YA, Zeretzke C, Atzler C, Baiker A, Berger B, Rajagopala SV, Roupelieva M, Rose D, Fossum E, Haas J. 2006. Herpesviral protein networks and their interaction with the human proteome. Science 311:239–242. 10.1126/science.1116804. [DOI] [PubMed] [Google Scholar]
- 20.Narita Y, Murata T, Ryo A, Kawashima D, Sugimoto A, Kanda T, Kimura H, Tsurumi T. 2013. Pin1 interacts with the Epstein-Barr virus DNA polymerase catalytic subunit and regulates viral DNA replication. J Virol 87:2120–2127. 10.1128/JVI.02634-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe T, Narita Y, Yoshida M, Sato Y, Goshima F, Kimura H, Murata T. 2015. The Epstein-Barr virus BDLF4 gene is required for efficient expression of viral late lytic genes. J Virol 89:10120–10124. 10.1128/JVI.01604-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konishi N, Narita Y, Hijioka F, Masud H, Sato Y, Kimura H, Murata T. 2018. BGLF2 increases infectivity of Epstein-Barr virus by activating AP-1 upon de novo infection. mSphere 3. 10.1128/mSphere.00138-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masud H, Watanabe T, Sato Y, Goshima F, Kimura H, Murata T. 2019. The BOLF1 gene is necessary for effective Epstein-Barr viral infectivity. Virology 531:114–125. 10.1016/j.virol.2019.02.015. [DOI] [PubMed] [Google Scholar]
- 24.Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV. 2012. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol 7:1848–1857. 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yanagi Y, Masud H, Watanabe T, Sato Y, Goshima F, Kimura H, Murata T. 2019. Initial characterization of the Epstein–Barr virus BSRF1 gene product. Viruses 11:285. 10.3390/v11030285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newcomb WW, Thomsen DR, Homa FL, Brown JC. 2003. Assembly of the herpes simplex virus capsid: identification of soluble scaffold-portal complexes and their role in formation of portal-containing capsids. J Virol 77:9862–9871. 10.1128/jvi.77.18.9862-9871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duarte M, Wang L, Calderwood MA, Adelmant G, Ohashi M, Roecklein-Canfield J, Marto JA, Hill DE, Deng H, Johannsen E. 2013. An RS motif within the Epstein-Barr virus BLRF2 tegument protein is phosphorylated by SRPK2 and is important for viral replication. PLoS One 8:e53512. 10.1371/journal.pone.0053512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Guo H, Reyes N, Lee S, Bortz E, Guo F, Sun R, Tong L, Deng H. 2012. Distinct domains in ORF52 tegument protein mediate essential functions in murine gammaherpesvirus 68 virion tegumentation and secondary envelopment. J Virol 86:1348–1357. 10.1128/JVI.05497-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benach J, Wang L, Chen Y, Ho CK, Lee S, Seetharaman J, Xiao R, Acton TB, Montelione GT, Deng H, Sun R, Tong L. 2007. Structural and functional studies of the abundant tegument protein ORF52 from murine gammaherpesvirus 68. J Biol Chem 282:31534–31541. 10.1074/jbc.M705637200. [DOI] [PubMed] [Google Scholar]
- 30.Masud H, Watanabe T, Yoshida M, Sato Y, Goshima F, Kimura H, Murata T. 2017. Epstein-Barr virus BKRF4 gene product is required for efficient progeny production. J Virol 91. 10.1128/JVI.00975-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salsman J, Zimmerman N, Chen T, Domagala M, Frappier L. 2008. Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. PLoS Pathog 4:e1000100. 10.1371/journal.ppat.1000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugimoto A, Yamashita Y, Kanda T, Murata T, Tsurumi T. 2019. Epstein-Barr virus genome packaging factors accumulate in BMRF1-cores within viral replication compartments. PLoS One 14:e0222519. 10.1371/journal.pone.0222519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bortz E, Wang L, Jia Q, Wu TT, Whitelegge JP, Deng H, Zhou ZH, Sun R. 2007. Murine gammaherpesvirus 68 ORF52 encodes a tegument protein required for virion morphogenesis in the cytoplasm. J Virol 81:10137–10150. 10.1128/JVI.01233-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li W, Avey D, Fu B, Wu JJ, Ma S, Liu X, Zhu F. 2016. Kaposi's sarcoma-associated herpesvirus inhibitor of cGAS (KicGAS), encoded by ORF52, is an abundant tegument protein and is required for production of infectious progeny viruses. J Virol 90:5329–5342. 10.1128/JVI.02675-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson MS, Loftus MS, Kedes DH. 2014. Maturation and vesicle-mediated egress of primate gammaherpesvirus rhesus monkey rhadinovirus require inner tegument protein ORF52. J Virol 88:9111–9128. 10.1128/JVI.01502-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, Hand T, Ma S, Liu X, Miley W, Konrad A, Neipel F, Sturzl M, Whitby D, Li H, Zhu F. 2015. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18:333–344. 10.1016/j.chom.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ho TH, Sitz J, Shen Q, Leblanc-Lacroix A, Campos EI, Borozan I, Marcon E, Greenblatt J, Fradet-Turcotte A, Jin DY, Frappier L. 2018. A screen for Epstein-Barr virus proteins that inhibit the DNA damage response reveals a novel histone binding protein. J Virol 92. 10.1128/JVI.00262-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu G, Liu C, Zhou S, Li Q, Feng Y, Sun P, Feng H, Gao Y, Zhu J, Luo X, Zhan Q, Liu S, Zhu S, Deng H, Li D, Gao P. 2021. Viral tegument proteins restrict cGAS-DNA phase separation to mediate immune evasion. Mol Cell 81:2823–2837. 10.1016/j.molcel.2021.05.002. [DOI] [PubMed] [Google Scholar]