

Summary
During infection, animals exhibit adaptive changes in physiology and behavior aimed at increasing survival. Although many causes of infection exist, they trigger similar stereotyped symptoms such as fever, warmth seeking, loss of appetite and fatigue1,2. Yet exactly how the nervous system alters body temperature and triggers sickness behaviors to coordinate responses to infection remains unknown. Here we identify a previously uncharacterized population of neurons in the ventral medial preoptic area (VMPO) of the hypothalamus that are activated after lipopolysaccharide (LPS)- and Poly (I:C)- induced sickness and are critical for generating a fever response and other sickness symptoms such as warmth-seeking and loss of appetite. Single-nucleus RNA-sequencing and multiplexed error-robust fluorescent in situ hybridization (MERFISH) uncovered the identity and distribution of LPS-activated VMPO (VMPOLPS) neurons and non-neuronal cells. Gene expression and electrophysiological measurements suggest a paracrine mechanism in which the release of immune signals by non-neuronal cells during infection activates nearby VMPOLPS neurons. Finally, we show that VMPOLPS neurons exert a broad influence on the activity of brain areas associated with behavioral and homeostatic functions and are synaptically and functionally connected to circuit nodes controlling body temperature and appetite. Together these results uncover VMPOLPS neurons as a control hub that integrates immune signals to orchestrate multiple sickness symptoms in response to infection.
During infection, the brain orchestrates evolutionary conserved behavioral and physiological symptoms aimed at eliminating pathogens and increasing survival. Symptoms include fever, lethargy, appetite loss, warmth seeking, social withdrawal and increased pain sensitivity1. Fever, a key symptom of infection, is elicited by pathogen associated molecules that trigger the release of pro-inflammatory signals by immune cells2,3. These secreted signals interact with brain endothelial cells in circumventricular organs, where the blood brain barrier is permeable, enabling the further production and propagation of immune signals within the brain4. One such circumventricular organ, the vascular organ of lamina terminalis (OVLT), is located in the preoptic area of the hypothalamus, where major homeostatic functions such as body temperature, thirst, and sleep are regulated5. Specifically, thermoregulatory neurons in the preoptic area regulate body temperature by sensing ambient warmth and activating autonomic and behavioral circuits to decrease body temperature6–8. The preoptic area is also critical for fever generation9,10 and loss of pro-inflammatory prostaglandin E2 (PGE2) receptor 3 (EP3) in this region leads to a reduced febrile response11,12. However, which cell populations are responsible for generating fever, how they modulate circuits controlling body temperature, and whether they may affect other sickness symptoms is unknown. Here we seek to identify fever-generating neurons and determine their influence on sickness behaviors, leading to new insights into circuit mechanisms by which the brain responds to infection.
Brain areas activated during fever
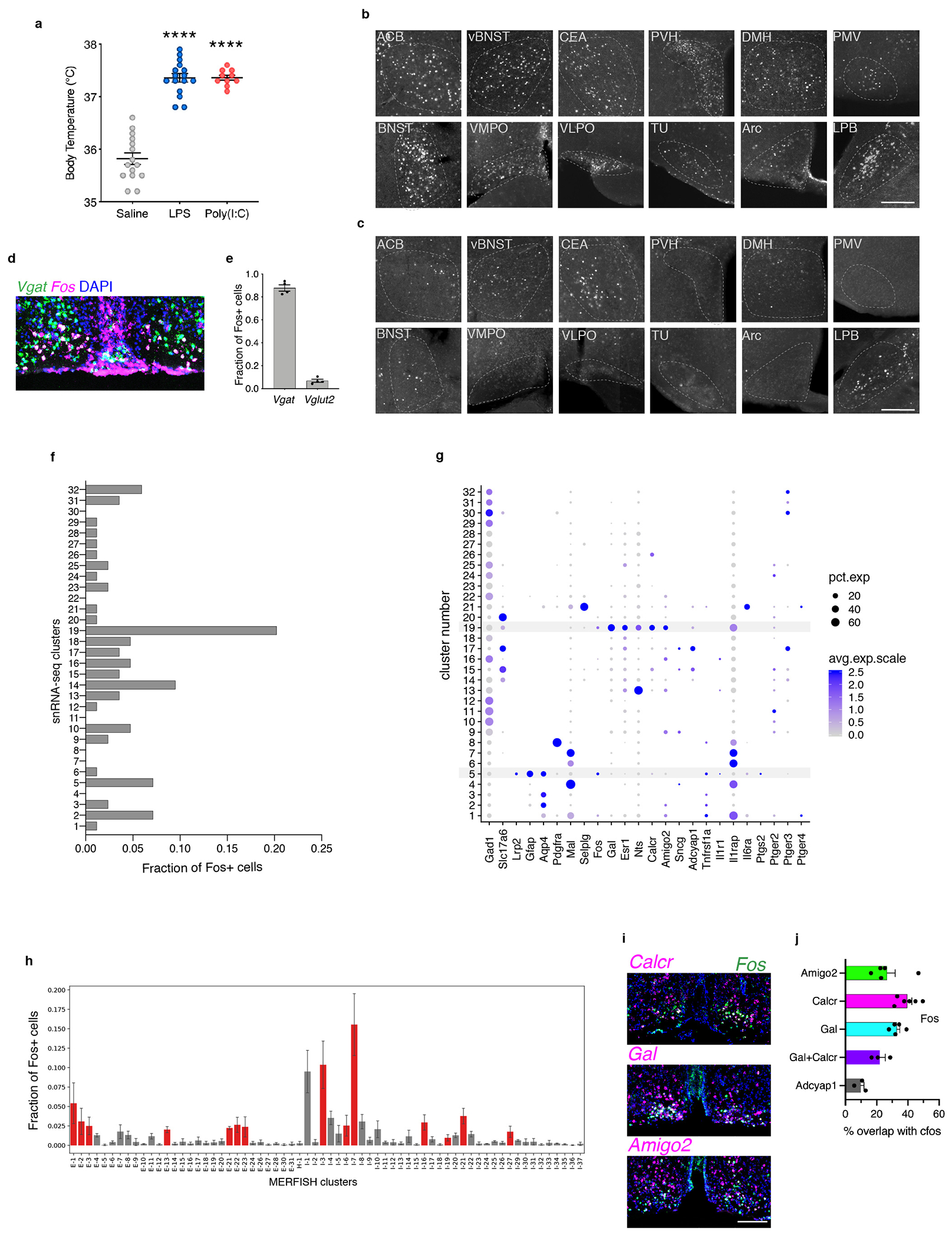
To generate fever we injected pro-inflammatory agents lipopolysaccharide (LPS) or polyinosinic:polycytidylic acid (Poly (I:C)), which mimic bacterial or viral infection, respectively. Body temperature peaks two hours following LPS and Poly (I:C) administration, with similar magnitude and timescale (Fig. 1a,b and Extended Data Fig. 1a). LPS was therefore used in all subsequent experiments. Twelve brain areas showed significant increase in Fos+ cells following LPS administration, including regions involved in appetite, metabolism, thermoregulation, sleep, stress and fear responses (Fig. 1a,c, Extended Data Fig.1b,c). Interestingly, cells within the ventral medial preoptic area (VMPO) (Fig. 1d–f arrowheads) and lining the adjacent ventricle (Fig. 1d–f arrow) were highly activated compared to controls. Activated VMPO neurons were inhibitory (~88% ± 1% vgat+, ~6.8% ± 1.4% vglut2+, Extended Data Fig.1d,e) and distinct from previously described warm-sensitive neurons located more antero-dorsal in the anteroventral periventricular nucleus (AVPe) and median preoptic nucleus (MnPO)6 (Fig. 1g). Based on their proximity to the OVLT circumventricular organ9, we hypothesized that LPS-activated VMPO neurons (VMPOLPS) may represent the long sought-after fever-generating neurons.
Figure 1. Activation of a specific VMPO neuronal population after LPS administration.
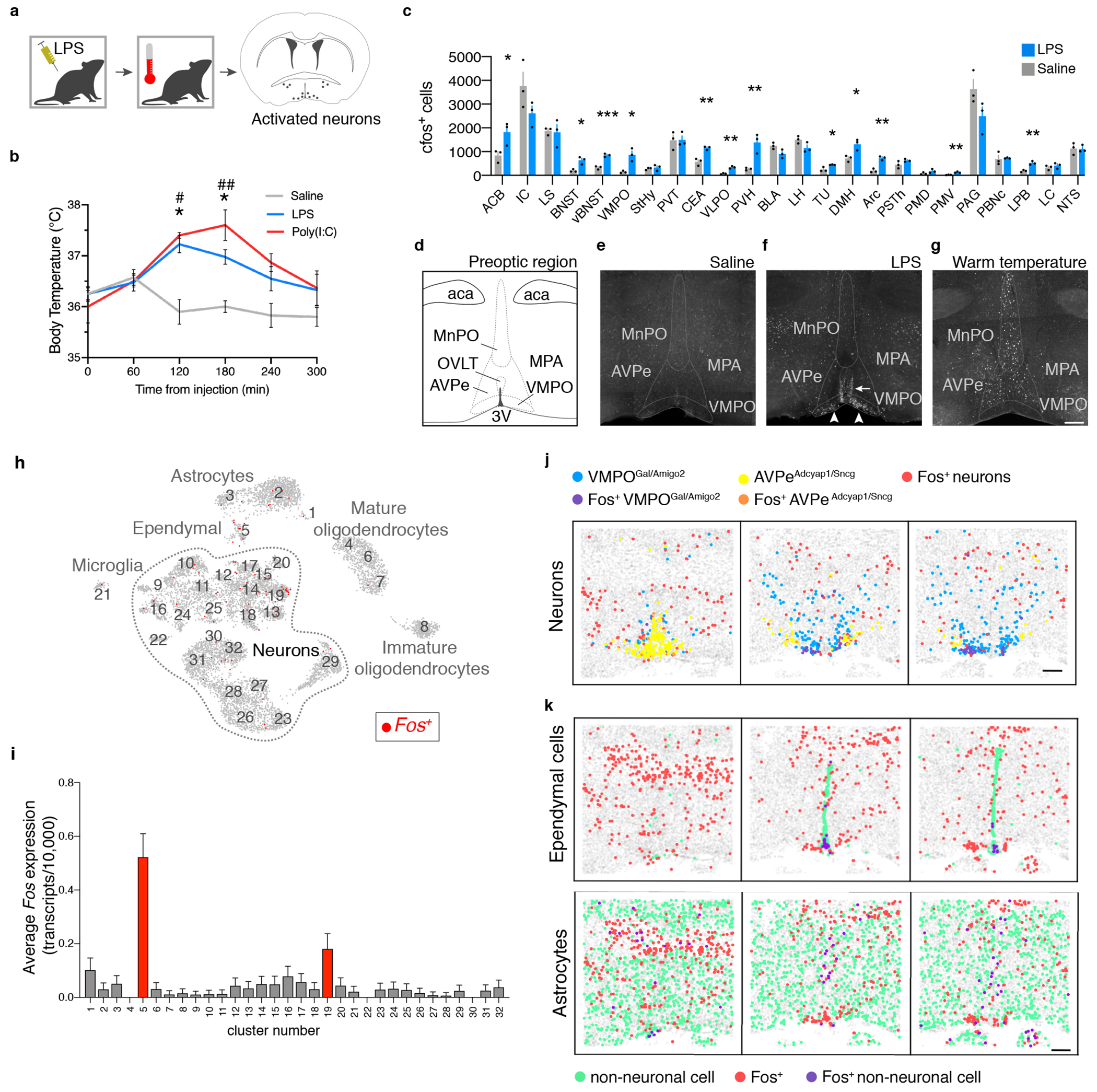
(a) Mice given an intraperitoneal injection of LPS or Poly(I:C) were monitored for changes in body temperature and brain Fos expression. (b) Mean body temperature following injection of saline, LPS or Poly(I:C), time zero is prior to injection, n=10/group. (c) Mean number of Fos+ cells per brain nucleus, n=3/group, Two-way ANOVA, for all graphs * =p<0.05, ** =p<0.01, *** =p<0.001. (d) Nuclei within the anterior preoptic region, see Extended Data Table 1 for brain area abreviations. (e-g) Fos expression after injection of saline (e), LPS (f) or warm temperature challenge (g), scale bar = 200μm, arrowheads: LPS-activated cells in the VMPO, arrow: LPS-activated cells lining the ventricle. (h) UMAP representation of 32 cell clusters identified by snRNA-sequencing. (i) Mean expression of Fos in snRNA-seq clusters, 16,430 total nuclei, 5 pooled mice, red bars indicate statistical significance, two-sided Mann-Whitney test, (p<0.05). (j) Representative spatial distribution of selected cell populations identified by MERFISH at approximately bregma +0.2mm, +0.0mm, −0.2mm. (k) Representative spatial distribution of ependymal cells (top) and astrocytes (bottom) identified by MERFISH in the preoptic region. Scale bars = 200μm. All error bars = SEM.
Characterization of VMPOLPS neurons
To characterize VMPOLPS cells we used a combination of single nuclear RNA-sequencing (snRNA-seq) and multiplexed error-robust fluorescent in situ hybridization (MERFISH)13,14. SnRNAseq data from 16,430 nuclei from VMPOs of saline- and LPS-injected mice integrating both conditions15 identified 32 neuronal and non-neuronal clusters (Fig. 1h). Fos expression was uniformly low in controls, enabling us to examine LPS-treated samples for average Fos expression levels and fraction of Fos+ cells within each cluster. Neuronal cluster #19 characterized by Galanin, Calcr, and Amigo2, and non-neuronal cluster #5, exhibited significant Fos enrichment (Fig.1h,i, Extended Data Fig.1f,g). Similarly, MERFISH on LPS-treated mice identified a VMPO neuronal cluster previously named I-713 (here referred to as VMPOgal/amigo2) characterized by the same genetic markers as snRNA-seq cluster #19 and displaying the highest fraction of Fos+ neurons13 (Fig.1j, Extended Data Fig.1h). By contrast, warm-sensitive neurons, identified as cluster E-3 or AVPeadcyap1/sncg were only weakly activated by LPS (Fig.1j, Extended Data Fig.1h). Accordingly, in situ expression of LPS-induced Fos displayed high overlap with Galanin, Calcr, and Amigo2, but not with Adcyap1 (Extended Data Fig.1I,j).
Endothelial and ependymal cells at circumventricular organs are involved in the signaling, production and translocation of pyrogenic cytokines and prostaglandins from the periphery to the brain4,16,17, with local astrocytes and microglia producing additional immune signals1,2. LPS-activated snRNAseq cluster #5 expressed markers for ependymal cells (Lrp2, Aqp4)13,18, and activated astrocytes (Gfap)19 (Fig.1i, Extended Data Fig.1g). Similarly, MERFISH identified astrocytes, endothelial cells, ependymal cells and mature oligodendrocytes as significantly enriched in Fos expression (Extended Data Fig. 2b). This activation appeared unique to LPS-induced responses, as there was no significant Fos enrichment in non-neuronal cells after aggression, mating or parenting episodes (Extended Data Fig. 2b).
How are VMPOLPS neurons activated? Strikingly, within the VMPOgal/amigo2 cluster ~90% of Fos+ neurons were localized within 200μm from the base of the brain, near the OVLT (Fig.1j, Extended Data Fig.2a). Additionally, strong Fos expression was seen in nearby non-neuronal cells raising the possibility of local activation of VMPOLPS neurons by secreted immune molecules from glial and epithelial cells 4,5. Indeed, MERFISH13,14 analysis indicated that ~70% of activated ependymal cells were also located within 200μm of the base of the brain, adjacent to VMPOgal/amigo2 neurons (Fig.1k, Extended Data Fig. 2c,d). Interestingly, astrocytes were more likely to be Fos+ near the midline both within the VMPO and more dorsally (Fig.1k, Extended Data Fig. 2c,d). Together, the close proximity between activated VMPOLPS neurons and secretory ependymal and astrocytes suggested that VMPOLPS neurons may be activated by signals secreted from local non-neuronal populations.
To identify the molecular mechanisms underlying VMPOLPS neuronal activation we examined expression of immune molecules and receptors previously implicated in fever and inflammation. Similar to previous studies, we found that prostaglandin-endoperoxide synthase 2 (Ptgs2, or COX-2), interleukin-1β (IL-1β) and chemokine ligand 2 (CCL2) were expressed near the VMPO 5,20–22 (Extended Data Fig.3a–c). Ptgs2 was identified in endothelial/pericyte cells of nearby vasculature 60 minutes, and in microglia 120 minutes, after LPS administration (Extended Data Fig.3j), while Il1β was found in ependymal cells and meningeal cells at the base of the brain 60 minutes and in microglia 120 minutes after LPS administration (Extended Data Fig.3k). Ccl2 was found in ependymal cells as well as in Gfap+ astrocytes near nearby capillaries (Extended Data Fig.3c,l). These data suggest that epithelial cells may stimulate an initial immune signal production later amplified by local microglia.
SnRNAseq data showed neurons from cluster #19 expressed interleukin-1 receptor accessory protein (Il1rap) (Extended Data Fig.1g), which is required for IL-1β signaling23–25 and we confirmed expression of Il1rap by VMPOLPS neurons in situ (Extended Data Fig. 3e). Although PGE2 receptor EP3 has been implicated in the generation of fever11, we did not detect EP3 in cluster #19 (Extended Data Fig.1g). However, snRNA-seq and in situ expression analysis identified expression of PGE2 receptor EP2 throughout the preoptic region, including by VMPOLPS neurons (Extended Data Fig.1g, 3d,g–i). Finally, chemokine ligand 2 receptor (Ccr2) was also expressed by VMPOLPS neurons (Extended Data Fig.3f). Thus, VMPOLPS neurons are likely sensitive to cytokines and prostaglandins released during infection and produced by nearby non-neuronal cells.
Immune activation of VMPOLPS neurons
We next sought to determine the effect of CCL2, PGE2 and IL-1β on VMPOLPS neuron activity using the TRAP2 transgenic line in which an inducible Cre-recombinase (2A-iCreERT2) is inserted into the Fos locus26,27. To confirm that we could “TRAP” (“induce Cre expression in”) LPS activated neurons, TRAP2;Ai9 mice were injected with LPS and 4-hydroxytamoxifen (4-OHT) inducing significant expression of tdTomato in VMPO of LPS-TRAP compared to saline-TRAP mice (Fig.2a,b). We quantified the overlap of tdTomato+ LPS-TRAP cells with Fos expression following a second LPS injection two weeks after the TRAP procedure. Similar to previous reports of TRAP2 labeling26, 82% of Fos+ cells in the VMPO were tdTomato+, and 52% of tdTomato+ cells expressed Fos (Fig. 2c,d), indicating efficient and specific labelling of LPS-activated cells in TRAP2 mice.
Figure 2. Effect of CCL1, PGE2 and IL-1β on intrinsic and synaptic properties of VMPOLPS neurons.
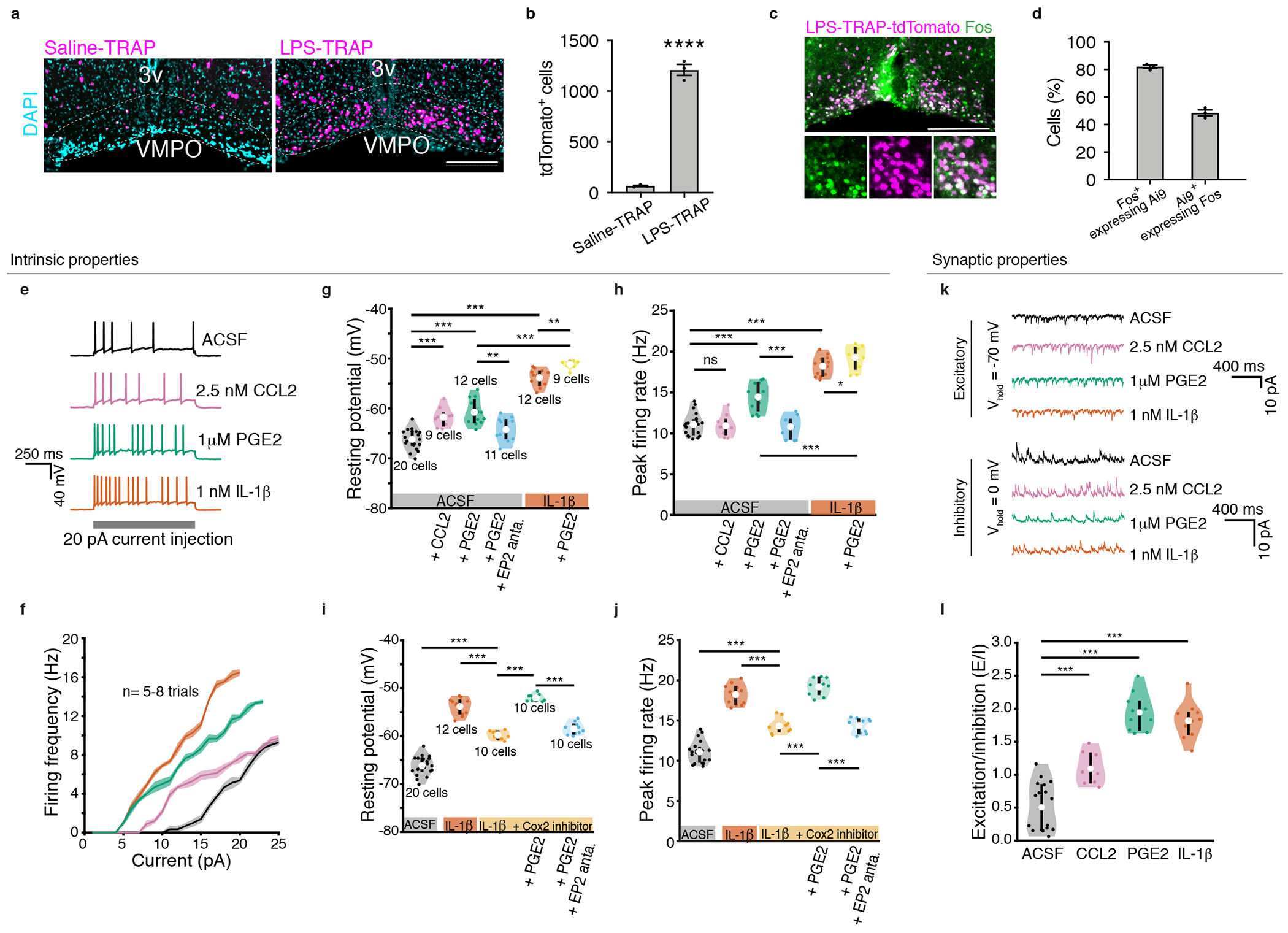
(a) Reporter expression in the VMPO following Saline-TRAP or LPS-TRAP. 3v= 3rd ventricle. (b) Average tdTomato+ cells in the VMPO, n=3/group, students t-test, p=<0.0001. (c) Overlap of LPS-TRAP neurons and subsequent LPS-induced Fos expression. (d) Mean percent of signal overlap in (c), n=3/group. All scale bars = 200μm (e) Examples of current clamp recordings of VMPOLPS neurons, control (black), 2.5nM CCL2 (pink), 1μM PGE2 (green) and 1nM IL-1β (orange). (f) Firing rate as a function of current injection; control n=8 trails, CCL2 n=5 trials, PGE2 n=5 trials, IL-1β n=7 trials. (g) Membrane resting potential and (h) peak firing rate of VMPOLPS neurons, control (black), CCL2 (pink), PGE2 (green), and addition of EP2 antagonist (blue) in presence of PGE2, IL-1β (orange) and IL-1β with additional PGE2 (yellow). (i) Resting membrane potential and (j) peak firing rate of VMPOLPS neurons after IL-1β (orange), addition of COX-2 inhibitor (light orange), addition of PGE2 (light green) and addition of EP2 antagonist (light blue). (k) Voltage clamp recordings, control (black), CCL2 (pink), PGE2 (green) and IL-1β (orange) showing mEPSCs (top) and mIPSCs (bottom). (l) Changes in excitatory to inhibitory charge transfer for control (black), CCL2 (pink, mean=1.10 ± 0.06, p = 2.20 * 10−04), PGE2 (green, mean=1.95 ± 0.09, p = 1.77 * 10−05) and IL-1β (orange, mean=1.82 ± 0.08, p = 1.83 * 10−05). See Extended Data Table 1 for all values and statistical tests. For violin plots: mean = central white circle, black line = interquartile range. All error bars = SEM. P values = two-sided Wilcoxon rank sum test with * = p<0.05, ** = p<0, *** = p<0.005 and ns = p>0.05. See Statistics and Reproducibility section for n’s.
Using tdTomato reporter expression in VMPOLPS neurons of LPS-TRAP mice we performed whole cell patch clamp in live brain slices using (Fig. 2e–j, Extended Data Fig.4a,b). Application of CCL2 resulted in significant depolarization of VMPOLPS neurons (Fig. 2g) and reduced rheobase current, indicating an increased propensity of VMPOLPS neurons to fire action potentials (Extended Data Fig.4b, Extended Data Table 1), but had no impact on the peak firing rate (Fig. 2h). Application of PGE2 or IL-1β also increased the excitability of VMPOLPS neuron by depolarizing membrane potential (Fig.2g) and reducing rheobase current (Extended Data Fig.4b). Moreover, PGE2 or IL-1β applications resulted in elevated peak firing rates (Fig.2h). PGE2 application in the presence of IL-1β additively increased the excitability of VMPOLPS neurons by further depolarizing the neurons (Fig.2g), reducing rheobase current (Extended Data Fig.4b), and increasing peak firing rate (Fig.2h), suggesting PGE2 and IL-1β cooperatively enhance the excitability of VMPOLPS neurons. The effects of PGE2 were blocked by the addition of an EP2 antagonist, confirming a role for this receptor (Fig.2g,h and Extended Data Fig.4b).
IL-1β can enhance biosynthesis of PGE2 by inducing COX-228. Using a selective blocker of COX-2, IL-1β mediated depolarization of VMPOLPS was partially blocked (Fig.2i,j, Extended Data Fig.4c). This partial block was alleviated by application of PGE2, an effect that was abolished by an EP2 antagonist (Fig.2i,j and Extended Data Fig.4c), suggesting that IL-1β regulates intrinsic properties of VMPOLPS neurons by direct increased excitability and indirect local PGE2 biosynthesis. Consistent with this hypothesis, IL-1β receptor 1 (il1r1) is expressed by endothelial cells, and COX-2 is induced in these cells after LPS administration (Extended data Fig.3j,m).
Next, we investigated the effect of CCL2, IL-1β and PGE2 on synaptic inputs to VMPOLPS neurons using voltage-clamp recordings to analyze miniature excitatory post-synaptic currents (mEPSCs) and miniature inhibitory post-synaptic currents (mIPSCs) (Fig. 2k, Extended Data Fig 4d–k). Application of CCL2 increased the amplitude and frequency of mEPSCs (Extended Data Fig.4d–e) and the frequency of mIPSCs (Extended Data Fig.4f–g). By analyzing the ratio of excitatory to inhibitory charge transfer (E/I ratio, Fig.2l and Extended Data Fig.4l–m, Extended Data Table 2) we found that CCL2 induces a net positive charge (Fig.2l). Similarly, PGE2 and IL-1β applications resulted in increased frequencies of mEPSCs (Extended Data Fig.4e), decreased frequencies of mIPSCs (Extended Data Fig.4i), and increased amplitudes of mIPSCs (Extended Data Fig.4h). The effect of PGE2 on mEPSCs was abolished by an EP2 antagonist (Extended Data Fig.4e). The E/I ratio showed that both PGE2 and IL-1β resulted in a positive synaptic charge transfer to VMPOLPS neurons (Fig.2l). However, COX-2 inhibitor abolished the effects of IL-1β (Extended Data Fig.4f–g,j–k), indicating PGE2 is required for IL-1β-mediated changes in synaptic activity (Extended Data Fig.4f–g,j–k). Together, PGE2, IL-1β and CCL2 both directly increase the excitability of VMPOLPS neurons and indirectly increase excitatory input to VMPOLPS neurons through changes in vesicle release and synaptic strengthening.
VMPOLPS neurons generate fever
To determine whether VMPOLPS neurons play a role in generating fever, we expressed the excitatory chemogenetic receptor hM3Dq in VMPOLPS neurons of TRAP2 mice. Specificity of viral injections was verified post-mortem (Extended Data Fig.5a) and we confirmed that clozapine-n-oxide (CNO) robustly activated M3Dq-expressing VMPOLPS neurons in slice preparations (Extended Data Fig.6a–c). In vivo chemogenetic activation of TRAPed VMPOLPS neurons significantly increased body temperature compared to controls (Fig.3a–c). Similarly, chemogenetic activation of the VMPO in Calcr-Cre and Gal-Cre mice increased body temperature (Extended Data Fig.5b). Thus, activation of VMPOLPS neurons in the absence of inflammatory signals is sufficient to induce fever.
Figure 3. VMPOLPS neurons drive LPS-induced generation of fever, warmth-seeking behavior and appetite suppression.
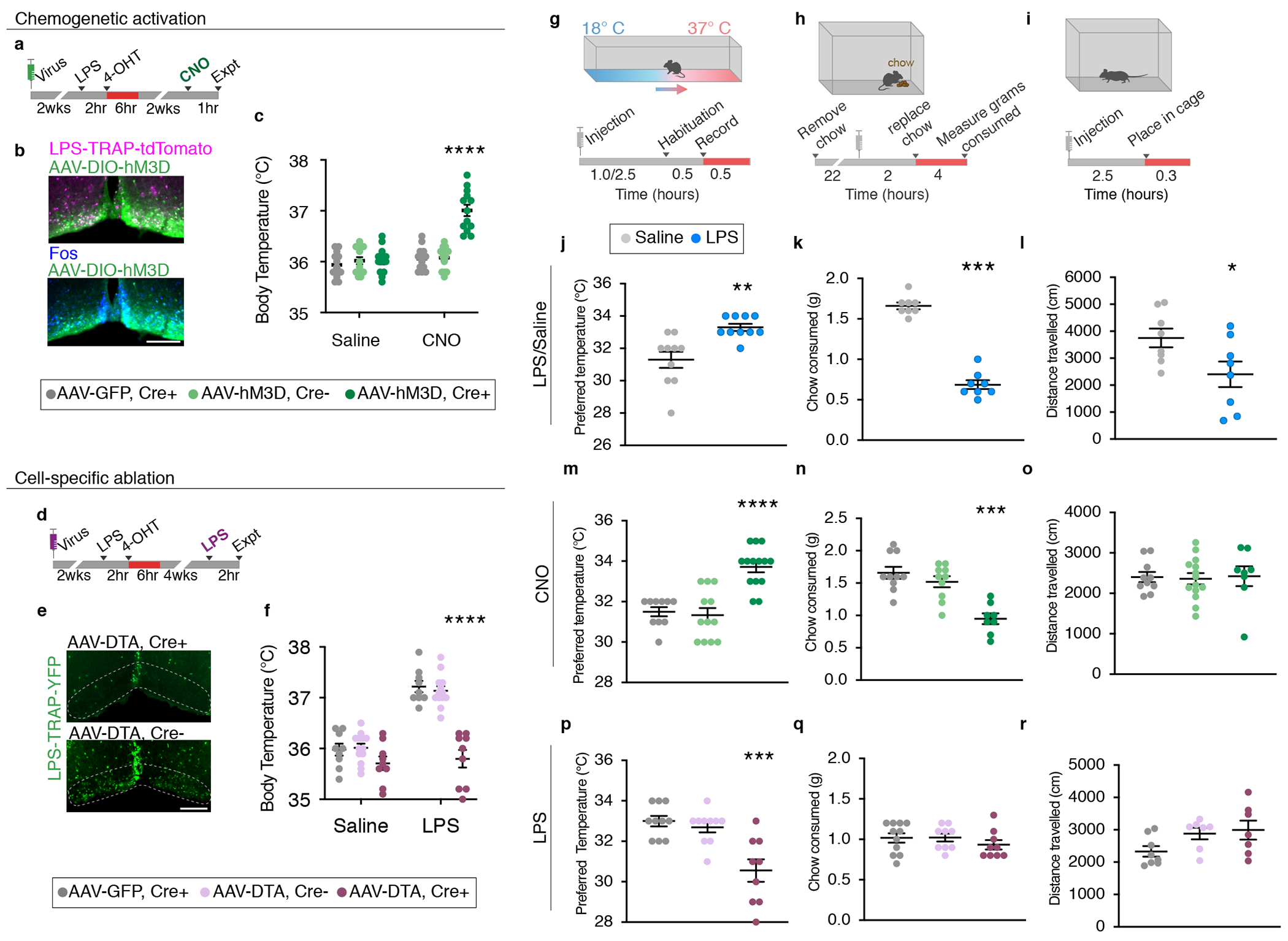
(a) Experimental procedure for VMPOLPS chemogenetic activation. (b) Expression of chemogenetic activating receptor hM3D (green) in LPS-TRAP cells in the VMPO (magenta) and CNO-induced Fos expression (blue). (c) Mean body temperature post- saline or CNO injection in Cre+ mice expressing hM3D (dark green), Cre− mice expressing hM3D (light green) or Cre+ mice expressing GFP (grey) controls, Two-way ANOVA, p=<0.0001. (d) Experimental procedure for VMPOLPS cell ablation by diphtheria toxin (DTA). (e) LPS-TRAP cells (green) in the VMPO after cell ablation (top panel) compared to control (bottom panel). (f) Mean body temperature following saline or LPS injection in DTA-ablated mice (dark purple) and Cre− mice injected with AAV-DTA (light purple) and Cre+ mice expressing GFP (grey) controls, Two-way ANOVA, p=<0.0001. (g-i) Procedures to test sickness behaviors: temperature preference (g), change in appetite (h), change in locomotion (i). (j-l) Quantification of behavioral changes in wildtype mice following saline (grey) or LPS (blue) injection; (j) Median preferred external temperature, p=0.0016, (k) mean appetite, p=<0.0001, (l) mean locomotion, p=0.0372, n=8/group, two-tailed t-test. (m-o) Quantification of behavioral changes following CNO injection in mice with activated VMPOLPS neurons and controls; (m) preferred median temperature, p=<0.0001, (n) mean appetite, p=0.0007, (o) mean locomotion, Kruskal-Wallis Test with Dunn’s multiple comparison. (p-r) Quantification of behavioral changes in mice with ablation of VMPOLPS neurons and controls; (p) preferred median temperature, p=0.0002, (q) mean appetite, (r) mean locomotion, Kruskal-Wallis Test with Dunn’s multiple comparison. All scale bars = 200μm. See Statistics and Reproducibility section for precise n values. in All error bars = SEM.
To test whether VMPOLPS neurons are required for fever generation we used selective ablation with viral-mediated expression of cre-dependent diphtheria toxin subunit A (DTA) 29 (Fig. 3d,e). Following VMPOLPS neuron ablation, mice exhibited no increase in body temperature in response to LPS (Fig.3f). Importantly, VMPOLPS ablation had no effect on maintenance of normal body temperature and VMPOLPS ablation following Saline-TRAP did not affect fever or temperature preference (Fig.3f, Extended Data Fig.5e–g). Thus, VMPOLPS neurons are critical for fever generation.
The role of VMPOLPS neurons in sickness
To determine whether VMPOLPS neurons control other sickness behaviors, we quantified LPS-induced changes in preferred temperature, appetite and locomotion30–32. We found that 2.5 hours post LPS-injection, mice preferred a ~2°C increase in external temperature suggesting increased warmth-seeking (Fig. 3g,j). In addition, LPS-injected mice consumed ~60% less chow (Fig.3h,k) and travelled ~40% less compared to controls, indicating appetite suppression and increased lethargy (Fig.3i,l).
To determine whether VMPOLPS neurons affect warmth-seeking, appetite or lethargy behaviors we chemogenetically activated VMPOLPS neurons in TRAP2, Gal-Cre and CalcR-Cre mouse lines and then tested animals in each assay. VMPOLPS neuron activation in TRAP2 mice resulted in increased preferred temperature and reduced appetite compared to controls but no change in locomotion (Fig.3m–o). Activating Gal+ neurons significantly increased preferred temperature but did not affect appetite while activating Calcr+ neurons decreased appetite but did not affect preferred temperature (Extended Data Fig.5c,d), suggesting cell heterogeneity within the VMPOLPS population. Indeed, while most LPS-induced Fos+ neurons expressed either Gal or Calcr, a smaller subset was Gal+Calcr+ (Extended Data Fig.1j), pointing to the advantage of the TRAP2 system to target all cell types activated by a given stimulus.
We next tested the requirement of VMPOLPS neurons to induce sickness behaviors using DTA-mediated ablation. VMPOLPS ablated mice no longer exhibited increased temperature preference after LPS administration but there was no change in appetite or locomotion (Fig.3p–r). The apparent lack of effect on appetite may be explained by a higher threshold of activated cells required to control appetite compared to warmth-seeking behavior or may result from additional circuit mechanisms controlling loss of appetite. Altogether, VMPOLPS neurons are critical for warmth-seeking behavior during sickness and sufficient to induce loss of appetite.
LPS-induced changes in thermoregulation
To determine the mechanisms by which VMPOLPS neurons modulate homeostatic functions we identified their downstream targets using a conditional virus expressing synaptophysin-mRuby that delineates synapses from fibers of passage (Fig.4a–c). Projections to 12 brain areas were identified (Fig.4d) that included areas related to stress and aversion as well as thermoregulation, appetite, and sleep, thus representing possible nodes at which VMPOLPS neurons may exert their function during sickness.
Figure 4. VMPOLPS neurons regulate body temperature and appetite through direct and indirect synaptic connections.
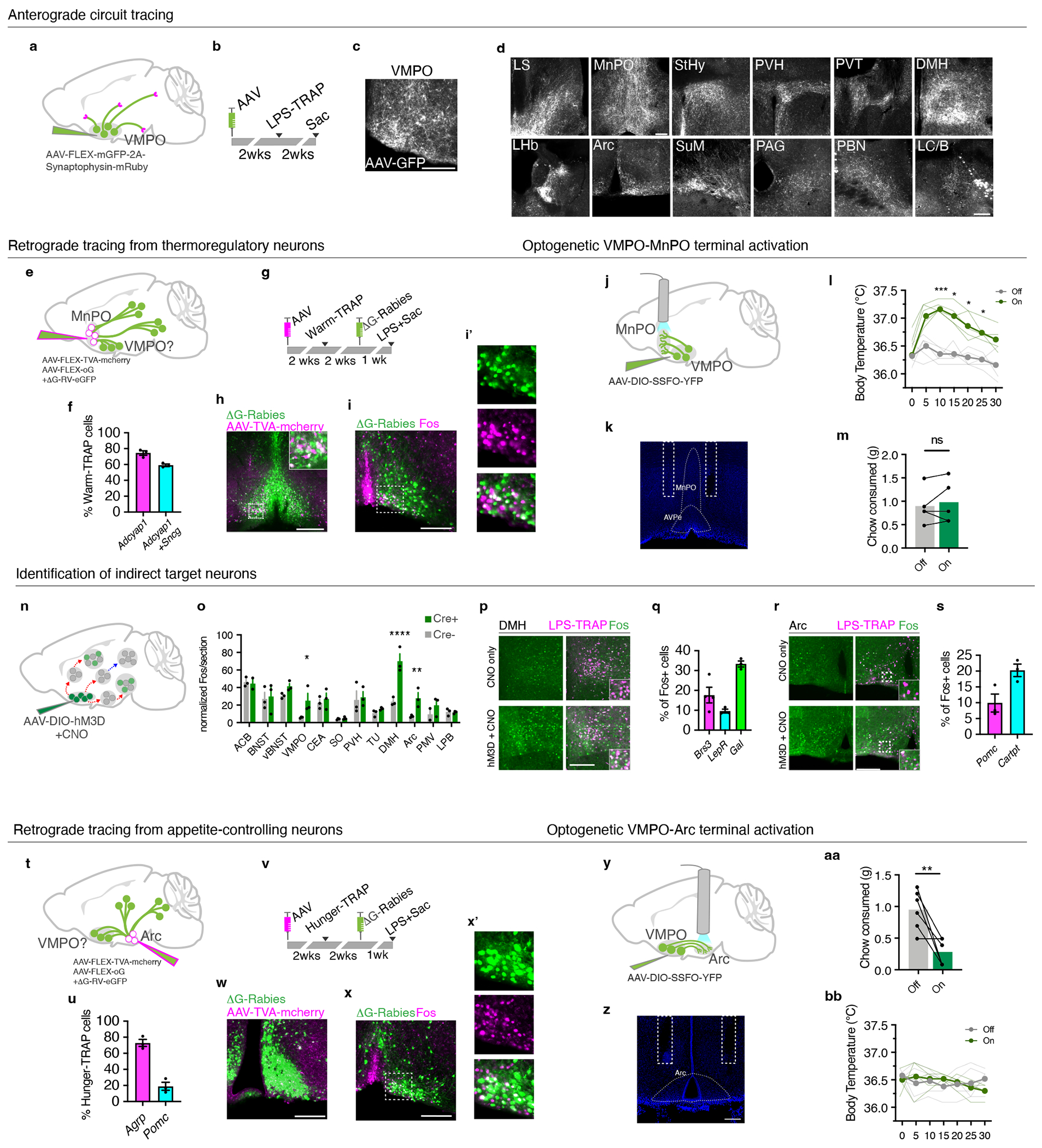
(a,b) Anterograde tracing from LPS-TRAP neurons in the VMPO(c). (d) Expression of GFP-labeled VMPOLPS axonal fibers, n=6 mice. (e) Retrograde tracing from warm-sensitive neurons in AVPe/MnPO (warm-TRAP). (f) Percent overlap of warm-TRAP neurons with adcyap1 and sncg, n=3. (g) Experimental timeline for retrograde tracing. (h) Starter cells in the AVPe/MnPO, green=g-deleted rabies, magenta=AAV-TVA-mcherry. (i) Overlap of rabies-infected input cells (green) and LPS-induced Fos (magenta). (i’) magnification of boxed region in (i). (j) Optogenetic activation of projections from VMPOLPS neurons to AVPe/MnPO. (k) Fiber placement. (l) Body temperature, stimulation (green) or no stimulation (grey) of VMPOLPS-AVPe/MnPO projections, light lines = individual mice, dark lines = mean, n=5, two-way ANOVA, * = p<0.05, ** = p<0.01, *** =p<0.001 (m) Chow consumed, n=5, ns=not significant, two-way Mann-Whitney test (n) Identification of downstream targets. (o) Mean Fos expression following CNO, n=3/group, two-way ANOVA, p=<0.0001 (p) Fos expression in LPS-TRAP neurons, control (top), VMPO activated (bottom). (q) Mean marker expression in DMH LPS-TRAP neurons, n=3 mice. (r) Fos expression following CNO. (s) Mean marker expression in LPS-TRAP neurons in the Arc, n=3 mice. (t) Retrograde tracing from appetite-controlling neurons in Arc. (u) Overlap of agrp and pomc in hunger-TRAP neurons, n=3/group. (v) Timeline of retrograde tracing. (w) Starter cells in the Arc. (x) Overlap of rabies-infected input neurons (green) and LPS-induced Fos (magenta). (x’) high magnification of (x). (y) Activation of VMPOLPS projections to Arc. (z) Fiber placement. (aa) Mean chow consumed following stimulation (on) or no stimulation (off) of VMPOLPS-Arc projections. dot-lines=individuals, n=6, p=0.0065, two-way Mann-Whitney test. (bb) Body temperature after VMPOLPS-Arc stimulation, (n=6). All scale bars = 200μm. All error bars = SEM.
How do VMPOLPS neurons alter body temperature? Warm-sensitive neurons in the AVPe/MnPO decrease body temperature in response to warm external temperatures via connections with the DMH, leading to inhibition of brown adipose tissue thermogenesis and behavioral thermoegulation6. To test whether VMPOLPS neurons form direct connections with warm-sensitive neurons we used Cre-dependent trans-synaptic retrograde rabies tracing from TRAPed warm-sensitive neurons (warm-TRAP) and looked for retrograde rabies in VMPOLPS neurons (Fig.4e). Warm-TRAP neurons expressed markers for warm-sensitive neurons: adcyap1 and sncg (Fig.4f, Extended Data Fig.5h)6,13. We expressed TVA receptor, optimized-G protein and G-deleted rabies virus in AVPe/MnPO warm-TRAP cells (Fig.4g,h) and found 38% (± 2.6%) overlap between LPS-mediated Fos and retrograde rabies signal in the VMPO (Fig.4i,i’). This direct connectivity makes VMPOLPS neurons ideally positioned to inhibit the activity of warm-sensitive neurons and thereby increase body temperature.
To confirm functional connectivity between VMPOLPS and warm-sensitive neurons, we optogenetically activated VMPOLPS→AVPe/MnPO projections (Fig. 4j) using a stabilized step function opsin (SSFO) too reduce heat generated by extended light pulses6,33. Current clamp recordings showed that SSFO activation triggered long-lasting increases in firing frequency in VMPOLPS neurons (Extended Data Fig.6d–f). To test the function of VMPOLPS→AVPe/MnPO projections in vivo, we implanted optic fiber cannulas above the AVPe/MnPO area (Fig.4k). Following a 2 second laser pulse, mice exhibited a significant increase in body temperature, which peaked at around 10 minutes6, but did not alter chow consumption (Fig. 4l,m). Altogether, VMPOLPS neurons alter body temperature via inhibitory connections with warm-sensitive thermoregulatory neurons.
In addition to the VMPO, multiple brain areas were activated by LPS (Fig.1c). To determine which of these brain areas are downstream of the VMPO circuit we chemogenetically activated VMPOLPS neurons and quantified the number of Fos+ cells in LPS-activated brain regions (Fig.4n,o). The thermoregulatory DMH6 and appetite controlling Arc34 appeared significantly activated with a ~100% overlap between CNO-induced Fos and LPS-TRAP reporter expression (Fig.4o,p,r). Activation of bombesin receptor 3 (Brs3) or leptin receptor (Lepr) expressing neurons in DMH has been shown to increase body temperature, while silencing of DMHBrs3 neurons inhibits LPS-mediated fever7,35. Indeed, a substantial fraction of LPS-activated cells in the DMH express Brs3 (18% ± 3.3%) and LepR (10% ± 0.95%), while a large population (34% ± 2.6%) of LPS-sensitive DMH neurons expressed Gal, a population of unknown function (Fig.4q). These data suggest stimulation of VMPOLPS neurons leads to an activation of DMH neurons involved in thermoregulation and metabolism.
LPS-induced changes in appetite
Next, we sought to determine whether VMPOLPS neurons were directly connected to hunger-generating neurons (Fig.4t). Neurons were TRAPed following food deprivation (hunger-TRAP) with the majority of hunger-TRAP Arcuate neurons expressing AGRP, while few expressed POMC (Fig.4u, Extended Data Fig.5i)34. Following rabies tracing from hunger-TRAP neurons, 22.5% (± 6%) Fos+ VMPOLPS neurons were labeled by rabies virus, indicating that VMPOLPS neurons form direct inhibitory connections with ArcAGRP neurons and thus may cause loss of appetite during sickness (Fig.5v–x’). We optogenetically activated those projections and found a dramatic decrease in chow consumption following stimulation, while their body temperature remained unchanged (Fig.4y–bb). Together, VMPOLPS neurons decrease appetite via inhibitory projections to ArcAGRP neurons.
Figure 5. Model of the control of body temperature and appetite by VMPOLPS neurons.
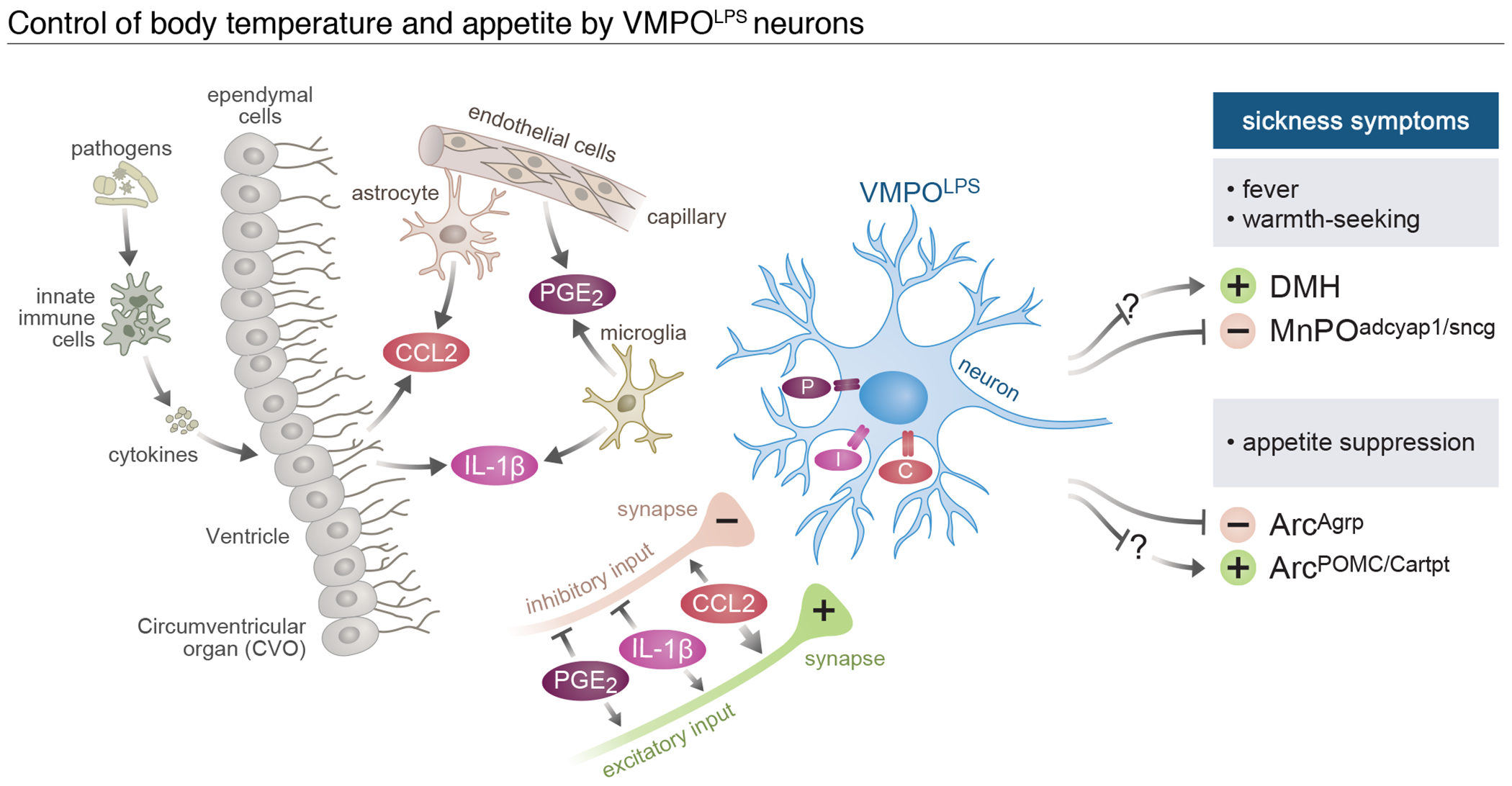
Activation of the immune system in periphery leads to circulating immune signals. These signals activate ependymal and endothelial cells lining the blood brain barrier, which react by secreting additional signals including IL-1b, PGE2 and CCL2, which are further amplified by local glial cells. VMPOLPS neurons are activated by these signals through their expression of the corresponding receptors as well as through activation of local synaptic inputs. In response, VMPOLPS neurons induce fever, warmth seeking and loss of appetite through direct and indirect connections to homeostatic brain circuits.
Interestingly, number of Fos+ cells in the Arc following VMPO activation appeared higher than in controls (Fig.4o,r). Because the Arc contains neurons that both induce and suppress appetite34 we hypothesized that sickness may increase activity in appetite-suppressing neurons, characterized by POMC and Cartpt expression36,37. Thus, we quantified the overlap between LPS-induced Fos expression and these markers in the Arc nucleus and found Fos+ cells do express POMC and Cartpt (Fig.4s), indicating appetite-suppressing neurons in the Arc are likely indirectly activated following stimulation of VMPOLPS neurons.
Summary
Altogether our data show that VMPOLPS neurons are necessary and sufficient to generate fever, directly sense immune signals, and coordinate sickness-induced increase in body temperature, warm seeking behavior and appetite suppression. Due to the location of activated non-neuronal cell populations and VMPOLPS neurons near the OVLT, we propose that activity in VMPOLPS neurons is triggered by a paracrine mechanism generated by locally secreted immune signals (Fig.5). Indeed, PGE2, IL-1β and CCL2 are locally produced by ependymal and endothelial cells as well as microglia and astrocytes and these molecules increase the excitability of VMPOLPS neurons through both direct changes of intrinsic properties and net increases in excitatory input. Circuit connectivity analysis and functional manipulations demonstrate how activity of VMPOLPS neurons leads to an increase in body temperature by directly inhibiting warm-sensitive neurons and through indirect multi-synaptic activation of DMHBrs3/LepR neurons, which in turn may activate brown adipose tissue thermogenesis and other thermoregulatory behaviors (Fig.5). Further, VMPOLPS neurons form direct inhibitory connections with appetite-increasing neurons to suppress feeding behavior and show indirect excitatory influence on appetite-suppressing neurons, suggesting a circuit mechanism by which VMPOLPS neurons may reduce appetite (Fig.5). In addition, VMPOLPS neurons project to 12 brain areas, some of which known to control thirst, pain sensation or social interactions, suggesting other sickness behaviors may also be affected by VMPOLPS neuron activity. Based on the data presented here we propose the function of VMPOLPS neurons is to translate the peripheral inflammatory state into changes in brain activity in order to elicit sickness symptoms. Our results indicate that immune signals have an immediate effect on the excitability of VMPOLPS neurons as well as plasticity in both VMPOLPS neurons and their upstream synaptic partners in a locally coordinated fashion. While there are likely other neuronal populations controlling sickness symptoms, our study reveals that at least some of these behaviors are altered in a co-dependent manner, reflecting the need for coordinated changes in homeostatic systems to increase survival during infection.
Methods
Animals
Mice were maintained on a 12h:12h dark light cycle with access to food and water ad libitum. The temperature was maintained at 22 degrees Celsius and the humidity was controlled between 30-70%. All experiments were performed in accordance with NIH guidelines and approved by the Harvard University Institutional Animal Care and Use Committee (IACUC). Due to the high degree of neural activity elicited in the hypothalamus during social interactions, all mice were single housed for 1 week prior to the start of experiments. As female mice show large natural fluctuations in body temperature linked to estrus, suggesting additional mechanism for body temperature regulation, only male mice were used for experiments. C57BL/6J mice were used for fever induction, Fos expression analysis, and cell-type identification experiments. TRAP2 transgenic mice (also called Fos2A-iCreER)25,26 are available from the Jackson Laboratory (Stock no. 030323). Gal-Cre BAC transgenic line was imported from the Mutant Mouse Regional Resource Center (MMUCD 031060-UCD). Calcr-IRES-Cre mice were generated by the Gene Targeting and Transgenics Center at the Janelia Research Campus using accession number NM_001042725.1. Transgenic reporter lines were obtained from Jackson Laboratory: Ai9(RCL-tdT), stock no. 007909 and R26R-EYFP, stock no. 006148. For all experiments only mice heterozygous for TRAP2 and reporter constructs were used. Sample sizes were chosen based on similar experiments from relevant studies.
Fever induction and body temperature monitoring
For intermittent temperature recordings we used implantable temperature transponders (IPTT-300, Bio Medic Data Systems) were inserted under the skin below the neckline in the upper abdominal region to the right of the midline. Mice were anesthetized using 2-4% isoflurane. Hair was shaved and skin was disinfected prior to transponder insertion. Mice recovered for 1 week prior to experiments. Fever was induced by intraperitoneal (IP) injection of either LPS (50ug/kg) or Poly(I:C) (10mg/kg). Mice were habituated in the experimental room in their home cage for 1 hour prior to injection. Body temperature was measured hourly or just prior to behavior experiments using a handheld transponder reader (DAS-7007, Bio Medic Data Systems). Statistical significance was measured using Kruskal-Wallis test to compare saline, LPS and Poly(I:C). For continuous temperature monitoring we implanted telemetry probes (E-Mitter G2, Starr Life Sciences Corp.) into the I.P. space of anesthetized mice. Body temperature was measured every 5 minutes for up to 24 hours. To compare body temperature of saline versus LPS groups in chemogenetic and ablation experiments, two-way ANOVA was used
Warm Challenge
Single housed mice in their home cages are placed into a veterinary incubator set at 30° C for 2 hours with access to food and hydrogel, then sacrificed for Fos expression analysis.
Single nuclear sequencing
The anterior ventral preoptic area was dissected from mice 2 hours after administration of either saline or LPS. Dissected tissues from 5 mice were pooled for each sample, dounce homogenized on ice and centrifuged at 500g for 6 minutes at 4 ° C. Supernatant was removed and passed through a 70μm filter then through a 20μm filter (MACS Smartstrainer) to remove large debris. DAPI was added to the suspension and nuclei were separated using fluorescence activated cell sorting (FACS). The resulting suspension was loaded into a 10x Genomics Chromium single-cell chip at a concentration of ~500 nuclei/μl with the aim of sequencing 7000-8000 nuclei per sample. Downstream preparation of sequencing libraries was carried out using the 10x Genomics Single Cell Kit V2. The libraries were sequenced on an Illumina NextSeq500 instrument using instructions provided by 10x Genomics. Paired-end sequencing with read lengths of 150nt was performed for all samples. Illumina sequencing reads were aligned to the mouse genome using the 10x Genomics CellRanger pipeline with the default parameters. Identification of cell type clusters was performed as previously described12. Briefly, we first filtered out non-nuclei such as mitochondria and red blood cells as well as obvious doublets. We selected variable genes using the Seurat command “FindVariableGenes” using y cutoff =1, x low cutoff = 0.0125 and x high cutoff = 3. 1500 variable genes were collected from each sample and the intersection of variable genes used to perform Canonical Correlation Analysis (CCA). Immediate early genes were removed from variable gene selection. CCA was performed using 50 components with the aim to cluster similar cell types across the saline and LPS conditions. To generate cell clusters, we utilized the Jaccard-Louvain community detection approach in the Seurat command “FindClusters” with k.param=15, and resolution set to 2. We used the following markers to identify major cell type classes: microglia (Selplg+), Astrocytes (Aqp4+), immature oligodendrocytes (Pdgfra+), mature oligodendrocytes (Mal+), and ependymal cells (Lrp2+, Aqp4+)12,16.
MERFISH
The MERFISH preoptic area probe libraries were designed and generated as previously described12. Mice 8-12 weeks old were individually housed for 1 week. After 1 hour of habituation, mice were injected with LPS (50ug/kg) and 2 hours later mice were euthanized and brains quickly dissected and frozen in Tissue-Tek O.C.T (VWR) and stored at −80° C before sectioning. The preoptic area was sectioned at −18° C on a cryostat (MICROM, HM550), generating 10um-thick coronal sections. Coverslips were treated and prepared as previously described12. Briefly, beginning at approximately +0.5 bregma, slices staggered approximately 50 μm apart along the anterior-posterior axis were placed onto silanized 40 mm #1.5 coverslips pre-treated with orange fiducial beads until slices spanning the extent of the VMPO had been collected. Sections were fixed in 4% PFA, washed in PBS and dehydrated in 70% ethanol. Samples were stained with a hybridization mixture containing encoding probes for MERFISH measurements and for the non-combinatorial, sequential FISH measurements, and a polyA-anchor probe, after which samples were embedded in a polyacrylamide gel matrix and cleared using an SDS-proteinase K treatment, as previously described12. Samples were washed extensively with 2X SSC following digestion and stored at 4 C in 2X SSC with 1:100 murine RNAse inhibitory for up to 1 week prior to imaging.
Samples were imaged as previously described, imaging the preoptic region from slices that contain the VMPO in 3 LPS-treated animals. Briefly, samples were stained with a readout hybridization mixture containing the readout probes for the first round of MERFISH measurements, along with a readout probe targeting the polyA anchor probe and DAPI to stain the cytoplasm and nucleus, respectively. Imaging was performed using a PlanApo 60X/1.3 NA silicon oil objective, taking a 9-by-9 grid of fields-of-view (FOVs) that cover a 1.8 mm by 1.8 mm region centered on the preoptic region present in each tissue slice. Such a grid of FOVs was taken for each slice present on a given coverslip. The first round of imaging was performed in the 750 nm, 650 nm, 560 nm, 488 nm, and 405 nm channels to image the Alexa750 readout probe, Cy5 readout probe, fiducial beads, polyA-probe, and DAPI, respectively. Subsequent rounds of imaging were performed in the 750 nm, 650 nm, and 560 nm channels only. After the first round of imaging, the sample was treated with TCEP to cleave the fluorescent dyes off the readout probes, hybridized with the readout probes for the second round of imaging, and then imaging was performed covering the same set of positions as previously. This process was repeated until rounds of imaging for both MERFISH and non-combinatorial, sequential FISH were completed. For all images of the fiducial beads a single image at the surface of the coverslip was obtained. For the MERFISH measurements of the 135 genes, and polyA probe, and DAPI imaging, seven 1.5 μm thick z-stacks were collected. For the non-combinatorial sequential FISH measurements of the 20 genes, a single image at 4.5 μm above the coverslip surface was taken.
MERFISH image analysis was performed using algorithms and approaches similar to those previously described. The revised pipeline used to perform analyses is freely available at github.com/ZhuangLab/MERlin. The output of this pipeline is a cell by gene matrix of expression, wherein each gene targeted by MERFISH is represented as absolute RNA counts, and each gene targeted by non-combinatorial, sequential FISH is represented as the total fluorescence intensity of all pixels within the cell boundary. Counts were volume-normalized and then data from each experiment was rescaled to reflect a typical counts density to minimize batch effects from different samples. Fluorescence intensities were normalized to the number of pixels contributing to each measurement, and then the background of each cell in the 650 nm and 750 nm channels were estimated as the median of all the non-combinatorial, sequential FISH measurements in that channel for a given cell (as no cell is expected to express more than a few of the 20 different genes analyzed in this manner) and subtracted from the signal in each round of the corresponding color. All data were then transformed as log10(x + 1). Cells without a segmentation boundary in the 4.5 μm z plane were discarded due to the absence of valid quantification for the non-combinatorial, sequential FISH genes. Additionally, cells were filtered on volume (discarding those with volumes less than 100 μm3 or greater than 3000 μm3, MERFISH mRNA counts (discarding those with 20 or fewer total MERFISH counts), and high non-combinatorial, sequential FISH signals (discarding those yielding high signals in more than 5 genes manual inspection suggested that these signals often arose from erroneous sources of signal).
Expression measurements from LPS-treated animals were normalized by calculating the z-score for each gene across all cells, and the same was done for the expression measurements of naïve animals collected previously12. A single layer, 100 node neural net classifier was trained on the z-scored expression measurements of the naïve animals using the 135 MERFISH genes measured by MERFISH and 20 genes measured by non-combinatorial, sequential multicolor FISH (Fos is not contained within these measurements) to predict the cluster label of each cell. The classifier was used to predict the cluster label for each cell in the LPS-treated data. Cells assigned to the “ambiguous” class were discarded, as well as those with prediction probability < 0.9. This left 92,990 of 204,319 cells with cluster assignments. A second round of classification was performed to recover unassigned LPS cells. In the second round, the assigned LPS cells were used to train a new single layer, 100 node neural net classifier to predict their assigned cluster labels. The classifier was then used to predict the unassigned cells in the LPS-treated data, again discarding predictions with probabilities < 0.9. A further 85,138 cells were assigned a cluster label, and the remaining 26191 were labeled as ambiguous.
To identify neurons and neuronal clusters activated by LPS injection, the Fos signal of all cells assigned an inhibitory or excitatory cluster label was normalized by calculating the z-score for cells from each replicate independently. All neurons with a Fos signal that is 2 or more standard deviations greater than the mean Fos signal of all the cells in the same animal (i.e. z-score >= 2) were considered Fos positive, and all other neurons were considered Fos−. In cases where non-neuronal cells were also analyzed, the same procedure was used except that z-score was calculated using all non-ambiguous cells. Clusters were determined to have significant enrichment of Fos+ cells/neurons based on a binomial test and a false discovery rate of <5%. Measurements of distances of selected cell populations to the midline or base of the brain were calculated for the 4 slices with the most VMPOgal/amigo2 cells in a given replicate and data from all replicates were combined in the cumulative distribution function, P-values were determined using a Mann-Whitney U test.
TRAP induction
4-hydroxytamoxifen (4-OHT) was dissolved at 20mg/mL in ethanol by shaking at 37° C for 15 minutes then mixed with corn oil (Sigma) at a concentration of 10mg/ml by vortexing. Ethanol was evaporated in a 37° C oven for 1 hour. The final 10mg/ml 4-OHT solution was injected intraperitoneally immediately after preparation at a dose of 50mg/kg.
LPS-TRAP/saline-TRAP:
Mice were habituated to the procedure room in their home cages for 1 hour. Saline or LPS (50ug/kg) was administered intraperitoneally then 2 hours later prepared 4-OHT was administered. Mice were left in the same environment for an additional 4-6 hours while 4-OHT is metabolized and they recover from the fever. Although subsets of non-neuronal cells expressed Fos following LPS administration, we did not see any viral infection of non-neuronal cells following the TRAP procedure, presumably due to the use of neuronal-specific viral serotypes and promotors.
Warm-TRAP:
mice were placed in 30° C veterinary incubator for 2 hours inside their home cages then 4-OHT was administered. Mice remained in the incubator for an additional 4 hours before returning to room temperature.
Hunger-TRAP:
Mice were food deprived for 22 hours prior to 4-OHT administration. Regular chow was returned 6 hours following the 4-OHT injection.
Electrophysiology
Solutions:
Modified ACSF (artificial cerebro-spinal fluid) contained (in mM): 105 choline chloride, 20 glucose, 24 NaHCO3, 2.5 KCl, 0.5 CaCl2, 8 MgSO4, 5 sodium ascorbate, 3 sodium pyruvate, 1.25 NaH2PO4 (osmolarity 290, pH 7.35) was used as cutting solution. All recordings were made in an oxygenated ACSF with composition (in mM): 115 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 1 MgSO4, 20 glucose, 2.0 CaCl2 (osmolarity 290, pH 7.35). See list of drugs table for more details.
Current-clamp internal solution contained (in mM): 120 potassium gluconate, 2.0 sodium gluconate, 10 HEPES, 4.0 Mg-ATP, 2.0 Na2-ATP, 0.3 Na3-GTP, and 4.0 NaCl (osmolarity 292, pH 7.34). Voltage clamp internal solution contained (in mM): 130 d-gluconic acid, 130 cesium hydroxide, 5.0 NaCl, 10 HEPES, 12 di-tris-phosphocreatine, 1 EGTA, 3.0 Mg-ATP, 0.2 Na3-GTP and 5 mM QX-314(osmolarity 291, pH 7.35). To visualize the neurons during recording, internal solutions contained 10 μM Alexa 488 or Alexa 594 as indicated. All chemicals were purchased from Sigma-Aldrich unless stated otherwise.
Acute brain slices:
In vitro slice physiology was performed in adult TRAP2:Ai9 (8-24 weeks old) mice 2-10 weeks after TRAP induction. Slices were prepared using methods described previously38. Mice were lightly anesthetized with isoflurane exposure using a vaporizer (Datex-Ohmeda) connected to a clear acrylic chamber for two min, and then deeply anesthetized with an i.p. injection of a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg). Mice were transcardially perfused with ice-cold modified ACSF cutting solution, and brains were dissected into the same solution. Coronal slices (300 μm thick) were obtained using a vibratome (VT1000S; Leica, Germany) and collected in ice-cold cutting solution. VMPO was identified based on its proximity to anatomical landmarks including ventricle, and ventral surface. VMPOLPS neurons were identified by the expression of tdTomato reporter. After cutting, slices were incubated in oxygenated ACSF solution at 35 °C for 45 min and then at room temperature for the duration of the experiment.
In vitro recordings:
Whole-cell current-clamp and voltage-clamp recordings were made using borosilicate glass patch pipettes (6-10 Mohm) filled with current-clamp internal and voltage-clamp internal solutions, respectively, and slices maintained at 35 °C in oxygenated ACSF. Slices were visualized under custom-built infrared optics on a BX51WI microscope (Olympus Optical, Tokyo, Japan). Recordings were obtained with a Multiclamp 700B amplifier (Molecular Devices, Palo Alto, CA), and physiological data were collected via software written in LabView (National Instruments) and pClamp 10.3 (Molecular Devices). Current and voltage clamp recordings were low-pass filtered at 1–2 kHz and digitized at 10-20 kHz. Membrane resting potential was recorded 5 minutes after breaking into the cells. For rheobase current calculations, neurons were injected with increasing steps 1 pA (2 s duration) till a single action potential was evoked in the recorded neuron. Same protocol with 1pA steps (2s duration) was used to measure the peak firing rates of the neuron. For our measurements of synaptic currents, voltage-clamp recordings were perform using a holding potential (−70 mV) that was near the reversal potential for chloride to record the excitatory currents. Similarly, to record the inhibitory currents, voltage-clamp recordings were perform using a holding potential (0 mV) that was near the reversal potential for AMPA/NMDA. Synaptic current measurements were performed in the presence of 1μM TTX to block sodium currents.
Pharmacology:
To synaptically isolate the neurons during current clamp recordings, synaptic blockers (20 μM AP-5, 50 μM CNQX and 20 μM Gabazine) were added to the bath solution. Similarly, to isolate miniature EPSCs and IPSCs, we used 1μM TTX in our bath solution during voltage clamp recordings.
Effects of Prostaglandin E2 and CCL2:
during our current clamp and voltage clamp recordings, bath application of 1 μM PGE2 and 2.5 nM CCL2 was used to determine the role of above-mentioned chemicals on the intrinsic properties and synaptic activity of VMPOLPS neurons. To analyze the contribution of EP2 receptors, we used a selective EP2 receptor antagonist (PF 04418948). Measurements were made 5-10 minutes after the start of perfusion of the drug.
Effects of IL-1β and NS-398:
to analyze the role of IL-1 qualitatively and quantitatively, we incubated the slices for 1 hour in regular ACSF at 35 °C in the presence of 1 nM IL-1β. To selectively block the activity of COX-2, we incubated the slices in the presence of 1 nM IL-1β and a selective COX-2 inhibitor (20 μM NS-398) for 1 hour. During our experiments involving whole cell patch, same concentrations of IL-1β and NS-398 were continuously perfused through the recording chamber.
Optogenetic validation:
TRAP2 mice 8-16 weeks were injected with 80-100 nL of AAV5-EF1a-DIO-hChR2(C128S/D156A)-EYFP (UNC Vector Core). Two to 8 weeks after TRAP induction, mice were sacrificed, and brain slices (300 μm thick) were made as described previously. We used a custom-built set up to deliver a single 20 ms pulse of blue light (460 ±10 nm, CBT-90-B-L11, Luminus) to activate hChR2(C128S/D156A) and used a single 50 ms green (525±9 nm, CBT-90-B-L11, Luminus) light pulse to inactivate the same.
Chemogenetic validation:
TRAP2 mice 8-16 weeks old were injected with100 nL of AAV8-hSyn-DIO-hM3D(Gq)-mCherry virus (Addgene). Two to 8 weeks after TRAP induction, mice were sacrificed, and brain slices (300 μm thick) were made as described previously. 1-10 μM CNO was bath applied during whole-cell current clamp recordings.
Analysis:
we used a custom written software (MATLAB R2017a) to measure the timing of action potentials and the firing rates of neurons. Template match algorithm in Axograph (1.7.6) was used to measure the amplitude, timing and inter event intervals of synaptic events. We used variable amplitude templates with τrise =0.5 ms and τdecay =2 ms for the excitatory events and τrise =2 ms and τdecay =5 ms for the inhibitory events.
Statistical test:
we used two-sided Wilcoxon rank sum test in MATLAB (R2017a) to compare the distribution.
Behavior assays
Mice were individually housed for at least 1 week prior to testing, unless otherwise noted. To avoid activity-related fluctuations in body temperature experiments were conducted during the light phase when mice are less active, exceptions stated below. For all behaviors, mice were tested with saline and LPS/CNO in a randomized order. No difference was found based on the order of the trials. Animals were tested for a single behavior per session with at least 1 week between trials. To avoid adapted responses to LPS, mice were not subjected to more than 3 LPS trials. All mice were habituated for 1 hour prior to trials to minimize stress-induced temperature fluctuations. Statistical differences between groups were analyzed using 2-way ANOVA for body temperature tests or with non-parametric Kruskal-Wallis Test with Dunn’s multiple comparison for behavioral tests. N=8-13/group depending on assay, see main text, figure legends and graphs with individual dots representing each mouse. For chemogenetic experiments, mice were used for multiple behavior experiments, allowing 4-7 days between CNO injections to allow full metabolism of CNO. Repeated CNO injections resulted in similar body temperatures for an individual mouse for each trial. For ablation experiments, mice were used for only one behavioral trial to circumvent adaptation to LPS administration.
Temperature preference
Mice were placed in a thermal gradient test box (Bio-TGT2, Bioseb) with one side set to 4° C and the other side set to 55° C either 1hr or 2.5hrs after injection (CNO and LPS, respectively), depending on the experiment. Mice were given 30 minutes to habituate then recorded for 30 minutes. Time in temperature zones between 18-37° C were recorded and analyzed using the Thermal Gradient Test Software (Bioseb). The median temperature zone was used as the preferred temperature.
Feeding behavior
Mice were food-deprived for 22 hours prior to the experiment. After 1 hour habituation, mice were injected with saline, LPS or CNO then a pre-weighed amount of regular chow was returned to the cage at the time of fever induction. The weight of remaining chow was measured 4 hours later.
Locomotion
After 1 hour habituation and fever induction mice were placed in a new cage and recorded for 10 minutes. Tests were recorded by Geovision surveillance system and the distance traveled was measured using Ethovision XT 8.0 (Noldus Information Technology).
Inter-male aggression
One hour after the onset of the dark cycle, under dim red lighting, castrated C57Bl6/J males (aged ~6-8 weeks) swabbed with intact adult male urine (100 ul) were introduced in to the home cage of an adult virgin male mouse (age ~8 weeks). Mice were sacrificed for tissue harvesting 30 minutes following aggressive attacking of intruder, characterized by rapid bouts of biting leading to defensive reactions by the intruder (rearing and escaping). Intruder males were removed from the cage 10 minutes after the commencement of the first attack.
Male Mating
Ovariectomized female mice (aged 6-8 weeks) were hormonally primed to be in estrous as previously described12. On test day, females were introduced into the home cage of an adult virgin male mouse (age ~8 weeks) one hour after the onset of the dark cycle, under dim red lighting. Male mice were sacrificed for tissue harvesting 30 minutes following intromission of the female. Female mice were removed from the cage 10 minutes after the first intromission.
Male Parenting
Virgin male mice were paired with females (age ~7 weeks) and co-housed until the birth of pups (~21 days). Fathers were allowed to remain with pups for 3 days before removal. Two days later, parental behavior was tested. After 1 hour of habituation, 1 hour after the onset of the dark cycle, under dim red lighting, a single mouse pup (age 2-3 days) was introduced into one corner of the home cage away from nesting material. A parental response was defined as retrieval of the pup to the nest combined with nesting, crouching, grooming and licking behaviors. Fathers were sacrificed for tissue harvest 30 minutes following the retrieval of the pup to the nest. Only mice displaying all of the above behavioral subroutines were selected for further processing.
Chemogenetics
TRAP2 mice 8-16 weeks old were used for these experiments. We injected 100 nL of AAV8-hSyn-DIO-HA-hM3D(Gq)-IRES-mCitrine virus (Duke Viral Vector Core) bilaterally into the VMPO (AP +0.1, ±ML 0.15, DV −5.2) using a Drummond Nanoject III. Mice recovered for two weeks prior to TRAP (CreER) induction or mock induction. Two weeks after Cre induction mice were administered 0.5mg/kg clozapine-n-oxide (CNO) dissolved in saline intraperitoneally. Temperature was recorded or behavior was measured 1 hour following CNO injection. 2-way ANOVA was used to compare experimental and control groups. See graphs with individual dots representing each mouse. Mice were used for multiple behavior experiments, allowing 4-7 days between CNO injections to allow full metabolism of CNO. Repeated CNO injections resulted in similar body temperatures for an individual mouse for each trial, regardless of experimental condition.
Optogenetics
TRAP2 mice 8-16 weeks were injected with 80-100 nL of AAV5-EF1a-DIO-hChR2(C128S/D156A)-EYFP (stabilized step function opsin, SSFO, UNC Vector Core) bilaterally into the VMPO (AP +0.1, ±ML 0.15, DV −5.2) using a Drummond Nanoject III. Optic fibers (Doric Lenses) were then implanted above the AVPe/MnPO (AP +0.5, ±ML 0.3, DV −4.6) or the Arc nucleus (AP −1.5, ±ML 0.4, DV −5.5). Fibers were cemented to the skull. Mice recovered for two weeks prior to LPS-TRAP (CreER) induction. Two weeks after Cre-induction mice underwent a habituation and testing protocol specific to the behavioral test (see below). Optogenetic stimulation was a single 2 second constant blue LED stimulation. Light intensity at the tip of the dual fiber was measured prior to each testing day and adjusted to ~10mW. Body temperature test: mice were habituated to the optogenetic setup with fiber connected for 1 hour on three consecutive days. On the 4th day, mice habituated for 30 minutes then half of the mice underwent the “light off” behavioral testing and half underwent “light on” testing. Body temperature was measured prior to stimulation and then every 5 minutes. On the 5th day mice underwent the opposite (“on” vs “off”) trials (30 min habituation and 30 min body temperature measurements). No difference was found when comparing the order of the “on” or “off” trials, n=5. Appetite test: mice were habituated for 1 hour for 3 consecutive days. Chow was removed from cages at 5:00pm on the 3rd day. On the 4th day mice were habituated for 30 minutes then half of mice underwent “on” and half underwent “off” trials. Weight of chow was measured before and after the 30 minute trial. Mice were allowed one day to recover and the following day (day 6) food was removed at 5:00pm. Day 7 mice underwent the opposite (“on” vs “off”) trial. No difference was found when comparing the order of “on” or “off” trials, n=6. Placement of fibers was verified post-mortem and those with incorrect fiber placements were removed from the study.
Cell ablation
AAV8-EF1a-mCherry-FLEX-dtA (UNC vector core) was stereotactically injected bilaterally into the VMPO of TRAP2 mice (100ul) (AP +0.1, ±ML 0.15, DV −5.2) using a Drummond Nanoject III. Mice recovered for 2 weeks and then underwent LPS-TRAP induction. Body temperature response to LPS/saline or behavior analyses were conducted 4-5 weeks later. The accuracy of virus injection as complete or near complete loss of reporter+ cells in the VMPO was used to determine the successful of ablation and subsequent inclusion for analysis. 2-way ANOVA was used to compare experimental and control groups. See graphs with individual dots representing each mouse. Mice were used for only one behavioral trial to circumvent adaptation to LPS administration.
Anterograde tracing
AAVDJ-hSyn-FLEX-mGFP-2A-Synaptophysin-mRuby (Stanford Vector Core) were stereotaxically injected bilaterally into the VMPO (AP +0.1, ±ML 0.15, DV −5.2) using a Drummond Nanoject III (100 nL) into adult TRAP2 mice. After 2 weeks, mice underwent TRAP/Cre induction and after an additional two weeks for viral/fluorophore expression, mice were transcardially perfused with 4% paraformaldehyde. Tissue will be cryoprotected (30% sucrose) and serial sections will be prepared on a freezing microtome (50 μm). Sections were mounted to superfrost plus slides, cover-slipped with Vectashield plus DAPI and imaged at 10X magnification using the AxioScan and Zen Blue 3.4 software (Zeiss).
Monosynaptic input tracing
A 1:1 ratio (100 nL) of AAV1-EF1a-FLEX-TVA-mCherry (avian TVA receptor, UNC Viral Vector Core) and AAV1-CAG-FLEX-oG-WPRE-SV40-PA (optimized rabies glycoprotein, Salk Institute) was stereotaxically injected into either the AVPe/MnPO (Bregma coordinates AP +0.5, ±ML 0.2, DV −5.0) or Arc (AP −1.5, ±ML 0.2, DV −5.8) of adult TRAP2 mice. Two weeks later TRAP/Cre is induced and then one week later, g-deleted rabies virus (deltaG-RV-eGFP; Janelia Viral Tools Facility) (200 nL) was injected into the same coordinates. Seven days later, mice were transcardially perfused with 4% paraformaldehyde. Tissue was post-fixed in PFA overnight and cryoprotected (30% sucrose) then serial sections were prepared on a freezing microtome (50 μm). Sections were imaged at 10X using the AxioScan (Zeiss).
In situ hybridization
Double and triple label fluorescent in situ hybridization was performed using the RNAscope assay V1 kit (Advanced Cell Diagnostics, ACD) according to the manufacturer’s instructions with a few exceptions. Fresh frozen brains were sectioned on a cryostat at 16μm and stored at −80° C. Slides were thawed and fixed in 4% PFA for 15 minutes followed by dehydration in 50%, then 75% then 100% ethanol. Cells were permeabilized for 30 minutes at room temperature using Protease III provided by ACD. Sections were then processed as suggested by the ACD protocol. Slides were imaged at 20x on an Axioimager Z2 using Axiovision Release 4.8 (Zeiss). All probes were made by ACD. Quantification of marker expression was done using an adapted CellProfiler v2.2 (Broad Institute) pipeline that identified cells using DAPI signal and identified cells with fluorescent signal corresponding to Fos and specific marker genes.
Immunohistochemistry
To visualize Fos protein, perfused tissue was sliced on a freezing microtome at 50μm, sections were incubated in blocking solution containing 5% goat serum, 3% fetal bovine serum and 0.3% TritonX-100 diluted in PBS for 1 hour at room temperature. Primary antibodies rabbit anti-Fos (9F6, Cell Signaling) and chicken anti-GFP (ab13970, Abcam were diluted 1:1000 in blocking solution and sections were incubated overnight at 4° C. After rinsing with PBS, secondary antibodies were applied at 1:1000 dilution in PBS for 2 hours at room temperature. Secondary antibodies used include Alexa-488 anti-rabbit (A21206, Invitrogen), Alexa-568 anti-rabbit (A11011, Invitrogen), Alexa-647 anti-rabbit (A21245), and Alexa-488 anti-chicken (A32921, Invitrogen). Sections were rinsed in PBS, mounted to superfrost plus slides, cover slipped with Vectashield containing DAPI, and imaged on a Axioscan at 10x (Zeiss) at 0.65um/pixel. Fos+ cell nuclei were identified algorithmically in serial section images by spatial filtering, thresholding and finding connected components. Spots of connected pixels ranging from approximately 30 to 315μm2 (6.5-20μm diameter) were counted as Fos+ nuclei. Contours of brain areas were manually drawn aided by local architecture/landmarks and an atlas of the mouse brain. Brain areas were measured in each section for 3 mice/group. Statistical significance between LPS and saline groups was determined using 2-way ANOVA with Fisher’s Test for multiple comparisons.
Statistics and Reproducibility
Statistical differences between groups were analyzed using 2-way ANOVA for body temperature tests or with non-parametric Kruskal-Wallis Test with Dunn’s multiple comparison for behavioral tests. Use of other statistical tests noted in the figure legends. All statistical tests are two-sided. See Extended Data Tables 5 and 6 for precise animal and cell numbers for Figures 2, 3 and Extended Data Figure 4.
Antibody staining with cFos in VMPO (Fig. 1e–g, Extended Data Fig.1b,c) was repeated independently 3 times with similar results. Visualization of reporter expression in the VMPO of Saline- and LPS-TRAP mice for the purpose of quantification was repeated independently 2 times (1-2 mice each) (Fig.2a). Visualization of projection patterns from VMPO neurons was repeated independently 4 times with similar results (Fig. 4c,d). Retrograde rabies tracing from warm-sensitive neurons and appetite-controlling neurons was repeated independently 2 times with similar results (Fig 4h,i,w,x). Identification of indirect target neurons was repeated 2 times independently with similar results (Fig. 4o–s). All RNAscope experiments were repeated 3 separate times independently with similar results (Extended data Fig.3 and 4h,i).
Data Availability
Raw and processed data have been deposited in the Gene Expression Omnibus and are available under accession number GSE197547.
Code Availability
The pipeline used to perform MERFISH analyses is freely available at github.com/ZhuangLab/MERlin. Other custom code available upon request.
Extended Data
Extended Data Figure 1. Specificity and identity of VMPOLPS neurons during inflammation.
(a) Mean body temperature 2 hours following injection of saline (n=13), LPS (n=16) or Poly(I:C) (n=10). (b-c) Representative images of Fos expression in brain areas displaying significant increases in number of Fos+ cells following LPS administration (b) or saline (c), scale bar for all panels = 200μm. (d) mRNA expression of inhibitory neuronal marker Vgat (green) and Fos (magenta) in the VMPO after LPS injection. (e) Fraction of Fos+ cells that express Vgat or Vglut2 (n=4). (f) Quantification of the fraction of Fos+ cells within individual snRNA-seq clusters in LPS-injected sample. (g) Dotplot of average expression of marker genes and genes with immunological significance in each cluster: dot size indicates percent of cells in cluster with measurable expression and color indicates average expression levels; ependymal cluster cluster #5 and neuronal cluster #19, found significantly activated after LPS injection are highlighted in red. (h) Quantification of the fraction of Fos+ cells in individual MERFISH neuronal clusters, with statically significant enrichment for Fos+ cells indicated in red, n=3. (i) mRNA expression of markers for VMPOLPS neurons (calcR, gal and amigo2) in LPS-injected mice. (j) Mean of overlap of markers for VMPOLPS neurons and for warm-sensitive neurons (adcyap1) with LPS-mediated Fos expression in the VMPO, n=3 mice/experiment. All scale bars = 200μm. All error bars = SEM.
Extended Data Figure 2. Spatial distribution of activated neuronal and non-neuronal cell type classes in the preoptic area during inflammation.
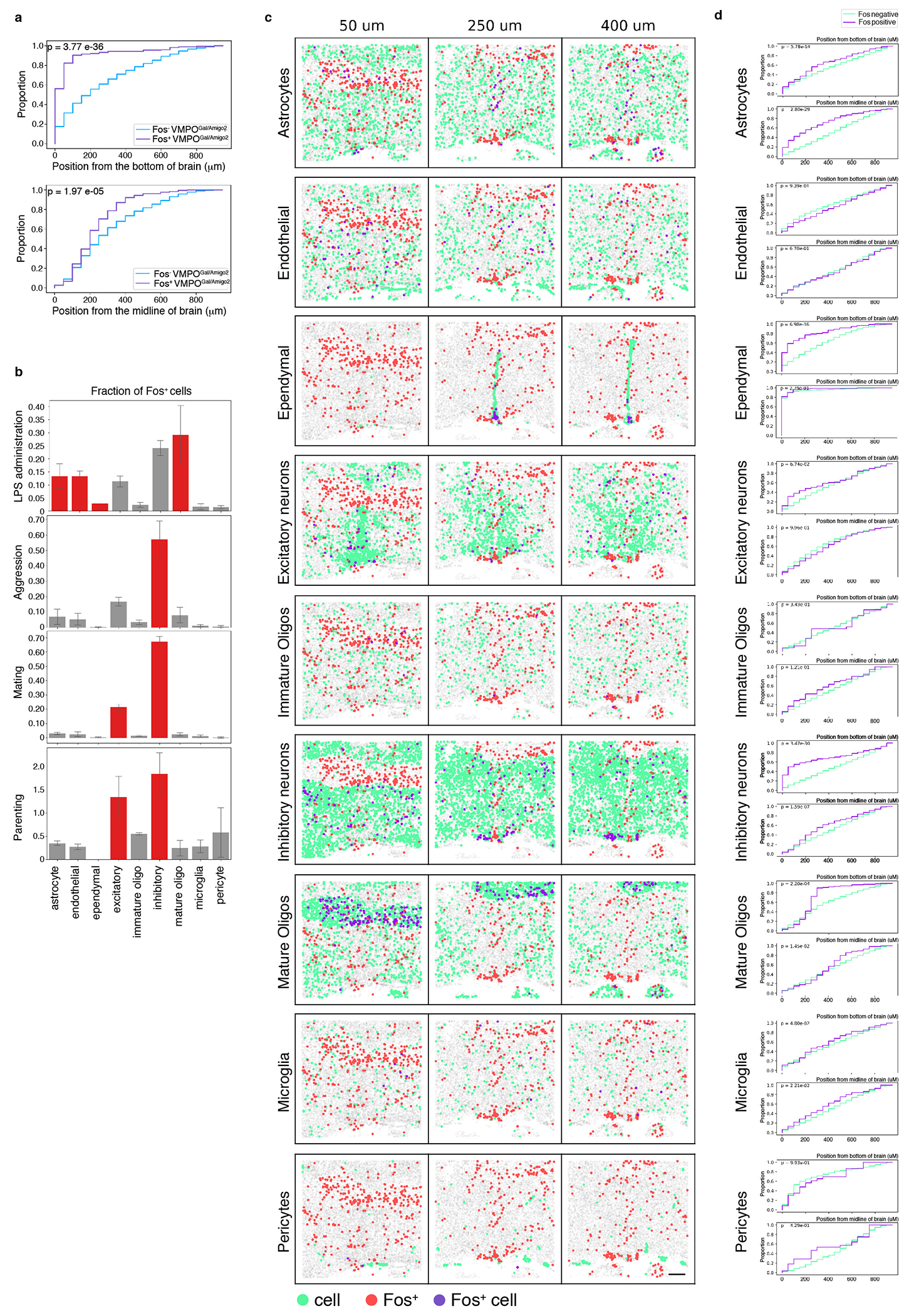
(a) Cumulative distribution of Fos+ (purple) and Fos− (blue) VMPOGal/Amigo2 neurons as a function of the distance to the bottom of the section (top) or the midline (bottom), n=3 replicates. (b) MERFISH analysis indicating the fraction of Fos+ cells in major cell type classes in samples from mice injected with LPS versus mice displaying other behaviors: aggression, mating or parenting. Significantly activated populations are indicated in red. (c) Spatial distribution of major cell type classes in MERFISH analysis, cell type (green), Fos+ cell (red), Fos+ cells in the indicated cell type (purple). (d) Cumulative distribution of Fos+ (purple) and Fos− (green) cells in major cell type classes as a function of the distance to the bottom of the section (top) or the midline (bottom), n=3 mice. All scale bars = 200μm. All error bars = SEM.
Extended Data Figure 3. Expression of key immune molecules and their receptors in the VMPO.
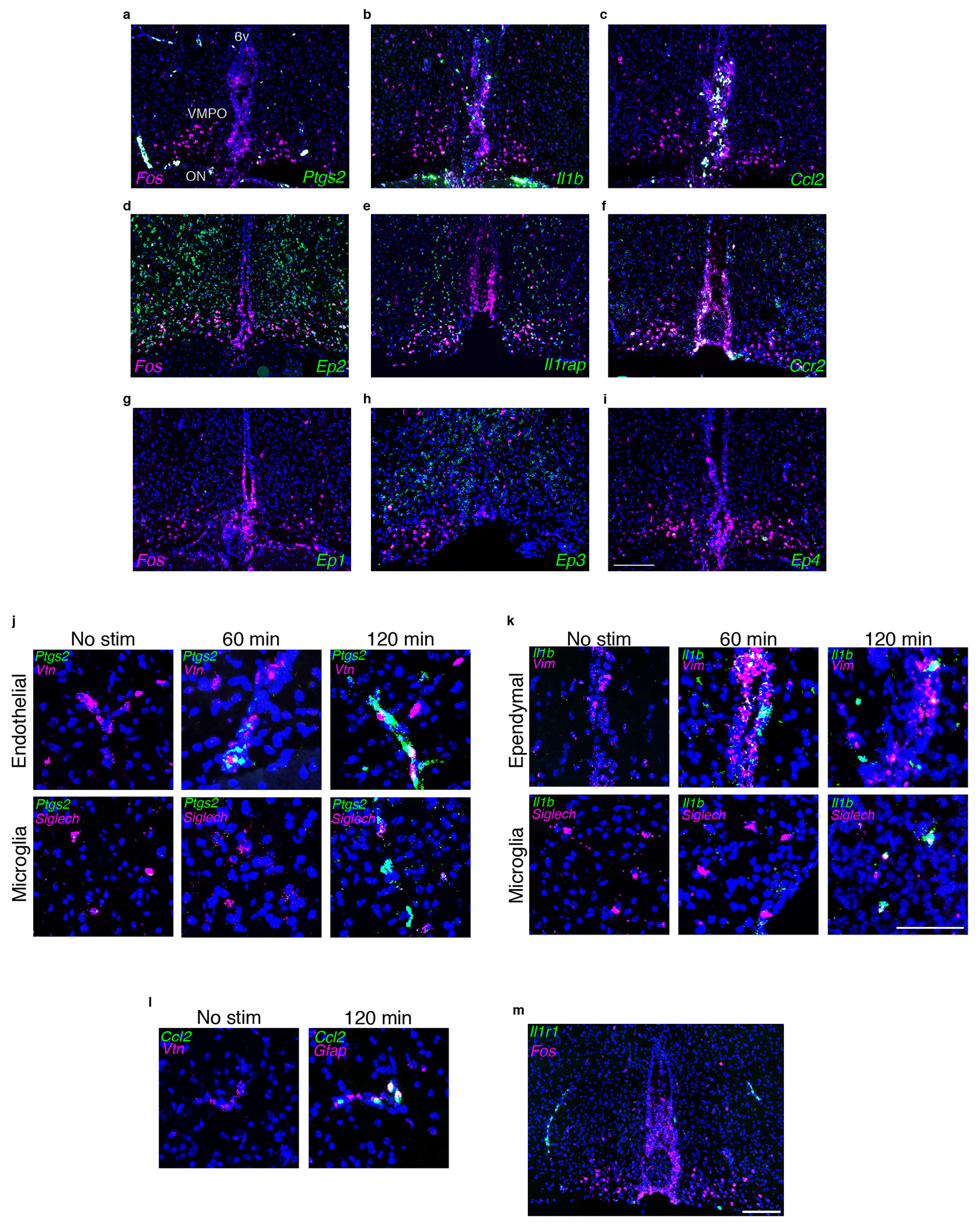
(a-i) mRNA expression in the VMPO at 2hrs post LPS administration, genes of interest in green, LPS-induced Fos is in magenta. Scale bar for a-i = 200μm (a) Prostaglandin E synthase 2 (ptgs2) and its receptors ep2 (d), ep1 (g), ep3 (h), ep4 (i). (b) interleukin-1β (il1β) and its receptor il1rap (e). (c) chemokine ligand 2 (ccl2) and it’s receptor chemkine receptor 2 (ccr2) (f). (j) Expression of ptgs2 in absence of LPS stimulation and at 60min and 120min post LPS. No or weak expression is found without LPS stimulation, and expression after LPS challenge shows overlap with markers for endothelial cells and microglia. (k) Expression of il1β with no stimulation and at 60min and 120min post LPS, overlap with markers for ependymal cells and microglia. Scale bar for j-l = 50μm (l) Expression of ccl2 with no stimulation and 120 min post LPS inejction, overlap with gfap+ astrocytes. (m) Expression of il1r1 in the VMPO after LPS injection, Scale bar = 200μm.
Extended data figure 4: Effects of PGE-2 and cytokines on the intrinsic properties and synaptic activity of VMPOLPS neurons.
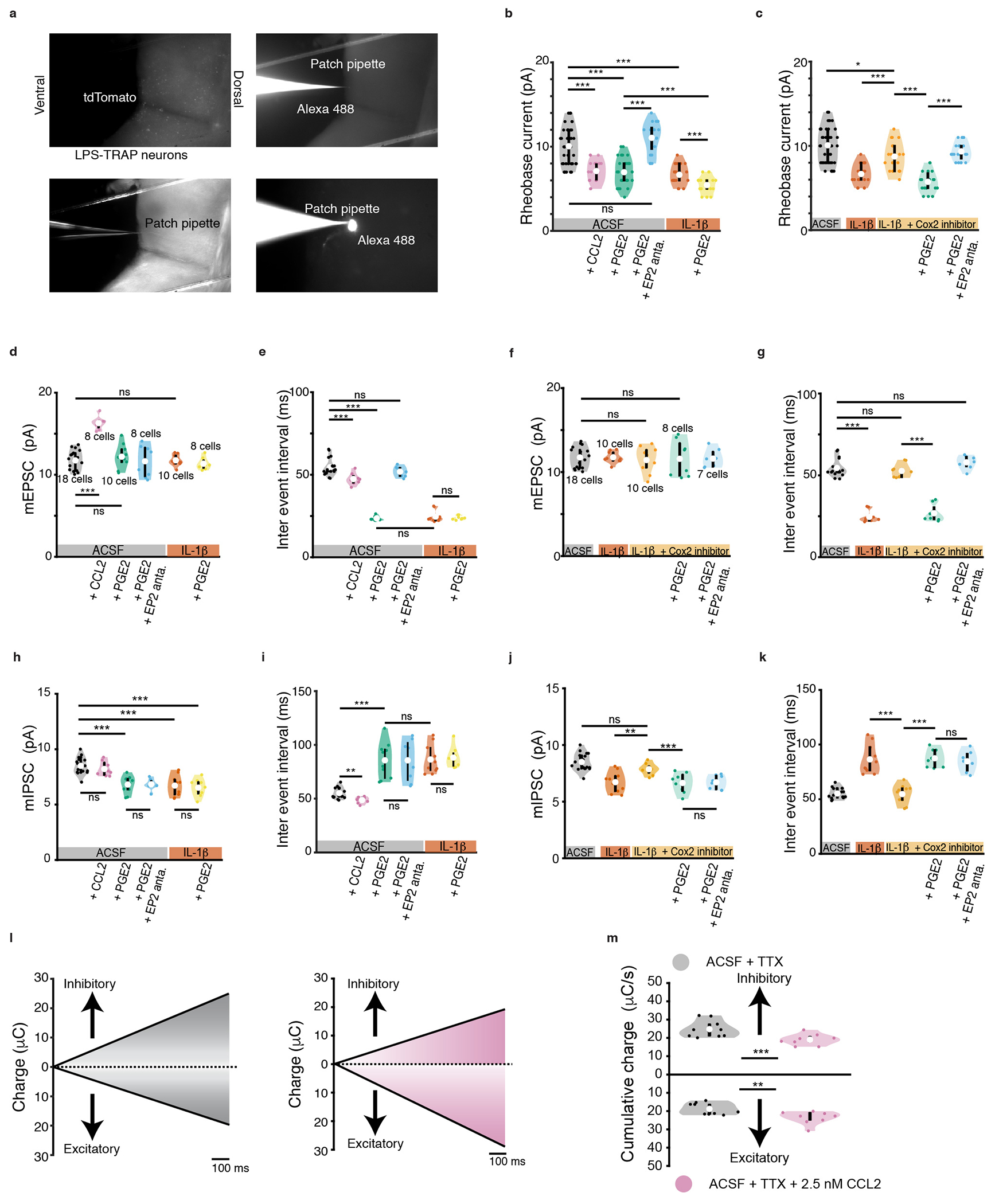
(a) Wide field microscope images depicting tdTomato expression in the VMPO of TRAP2;Ai9 during whole cell patch clamp recording with pipette containing Alexa 488. (b) Changes in rheobase current for VMPOLPS neurons in presence of ACSF (black), CCL2 (pink), PGE2 (green), addition of EP2 antagonist in presence of PGE2 (blue), IL-1β (orange) and further addition of PGE2 (yellow), and (c) IL-1β (orange), addition of COX-2 inhibitor in the presence of IL-1β (light orange), further addition of PGE2 (light green) and further addition of EP2 antagonist (light blue). During whole cell voltage clamp recordings, changes in (d) amplitude of miniature EPSCs, (e) inter event interval for miniature EPSCs, (f) amplitude of miniature IPSCs and (g) inter event interval for miniature IPSCs for VMPOLPS neurons in presence of ACSF (black), CCL2 (pink), PGE2 (green), addition of EP2 antagonist in presence of PGE2 (blue), IL-1β (orange) and further addition of PGE2 (yellow). Changes in (h) amplitude of miniature EPSCs, (i) inter event interval for miniature EPSCs, (j) amplitude of miniature IPSCs and (k) inter event interval for miniature IPSCs for VMPOLPS neurons in the presence of IL-1β and COX-2 inhibitor (light orange), further addition of PGE2 (light green) and further addition of EP2 antagonist (light blue). (l) Cumulative plots showing total charge transferred over 1s for inhibitory events and excitatory events for control (black) and CCL2 (pink). (m, top) Cumulative charge transferred by IPSCs for control (mean value of 24.81 ± 0.14 μC/s) and CCL2 (mean value of 19.06 ± 0.09 μC/s p = 2.0 * 10−03 for). (m, bottom) Cumulative charge transferred by miniature EPSCs for control (mean value of 18.81 ± 0.09 μC/s) and CCL2 (mean value of 23.37 ± 0.11 μC/s p = 9.0 * 10−03 ). All other values and statistical tests are catalogued in extended data table 2. For violin plots, central white circle depicts the mean value and thick black line depicts the interquartile range. All p values shown are for two-sided Wilcoxon rank sum test with * = p<0.05, ** = p<0, *** = p<0.005 and ns = p>0.05. All error bars = SEM. See Statistics and Reproducibility section for exact n values.
Extended Data Figure 5. Specificity of viral injections and cell type identity.
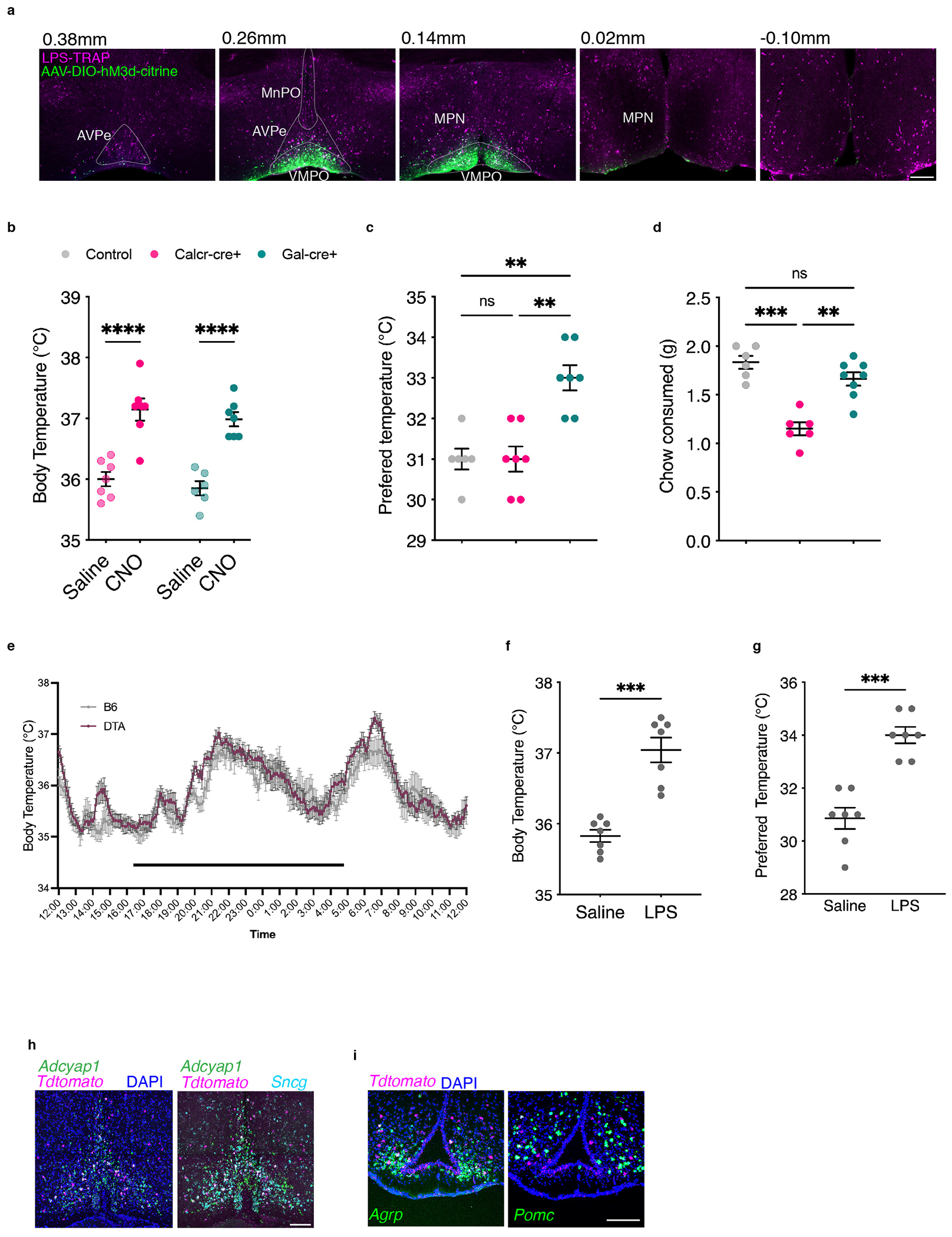
(a) Example of viral injection specificity that qualified for inclusion in the study. Note viral expression in the VMPO and absence in surrounding brain areas. Injections found to be more broadly dispersed were not included in analysis. (b-d) Chemogenetic activation of Calcr+ and Gal+ neurons in the VMPO. (b) CNO injection elicited strong increase in body temperature in both genetic backgrounds (n=6 mice/group). (c) Activation of Gal-Cre but not Calcr-Cre increased preferred temperature (n=7mice/group). (d) Activation of Calcr-Cre (n=6 mice) but not Gal-Cre (n=8) decreased chow consumed (saline n=6 mice). (e) DTA-mediated Ablation of VMPOLPS neurons had no effect on circadian temperatures, dark bar indicates dark phase, WT mice were used as controls. (f-g) Effect of DTA-mediated ablation of VMPO neurons in Saline-TRAP mice had no effect on body temperature or preferred temperature following LPS injection (n=7 mice/group). (h) Overlap of warm-TRAP reporter expression with markers of warm-sensitive neurons adcyap1 and sncg. (i) Overlap of hunger-TRAP-mediated reporter expression (magenta) with markers of appetite-increasing neurons, agrp, or appetite-decreasing neurons, pomc, (green). Scale bars = 200μm. All error bars = SEM. For all graphs * =p<0.05, ** =p<0.01, *** =p<0.001
Extended data figure 6: Chemogenetic and optogenetic activation of VMPOLPS neurons.
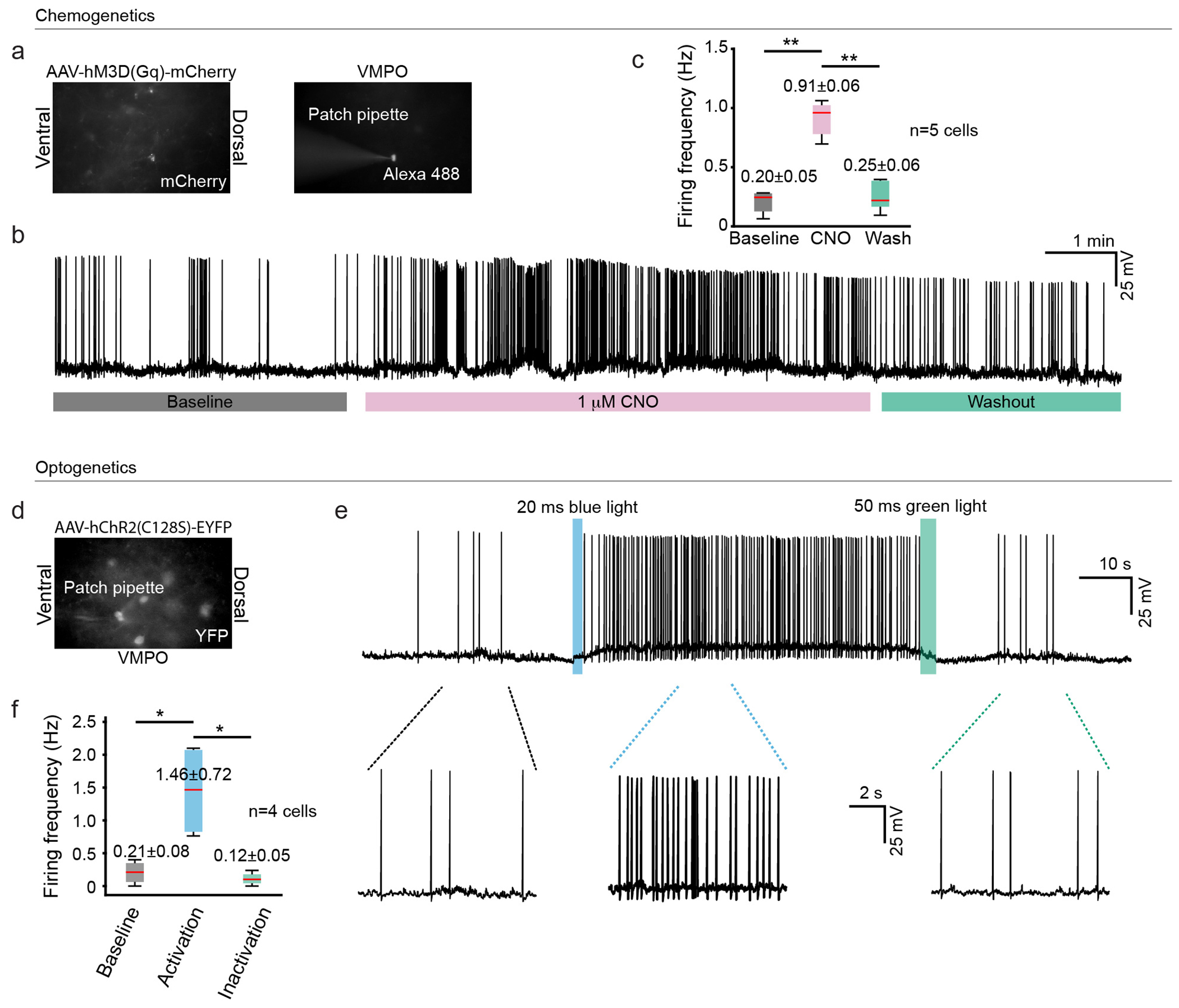
(a) Widefield microscope images of VMPOLPS neurons in the VMPO of TRAP2 injected with AAV8-hSyn-DIO-hM3D(Gq)-mCherry. (b) Whole cell current clamp recording of a VMPOLPS neuron showing the effects of bath application of 1mM CNO (baseline: black, CNO: pink and washout: green). (c) Effects of CNO application on the firing rate of VMPOLPS neurons; black (n= 5 cells with mean values of 0.20± 0.05 Hz for baseline), pink (0.91 ± 0.06 Hz for CNO application and p = 7.0 * 10−03 compared to baseline), green (0.25 ± 0.06 Hz for washout and p = 6.9 * 10−03 compared to CNO application), boxes represent the 25th and 75th percentile, whiskers extend to the minimum and maximum data points (d) Widefield microscope images of VMPOLPS neurons in the VMPO of TRAP2 injected with AAV5-EF1a-DIO-hChR2(C128S/D156A)-EYFP. (e) Whole cell current clamp recording of a VMPOLPS neuron showing activation of hChR2(C128S/D156A) by 20 ms blue light (460 ±10 nm) light pulse and inactivation by 50 ms green light (525±9 nm) pulse. (f) Firing rate of VMPOLPS neurons (n=4 cells) in response to activation of hChR2(C128S/D156A), baseline (black, mean firing frequency of 0.21±0.08 Hz), following blue light activation (blue, mean firing frequency of 1.46±0.72 Hz and p = 2.1 * 10−02 ) and following green light mediated inactivation of hChR2(C128S/D156A) (green, mean firing frequency of 0.12±0.05 Hz and p = 2.0 * 10−02 ), boxes represent the 25th and 75th percentile, whiskers extend to the minimum and maximum data points. All p values shown are for two-sided Wilcoxon rank sum test with * = p<0.05, ** = p<0, *** = p<0.005 and ns = p>0.05.
Extended data table 1:
Effects of prostaglandin E2 and cytokines (Il-1β and CCL2) on the intrinsic properties of VMPOLPS neurons.
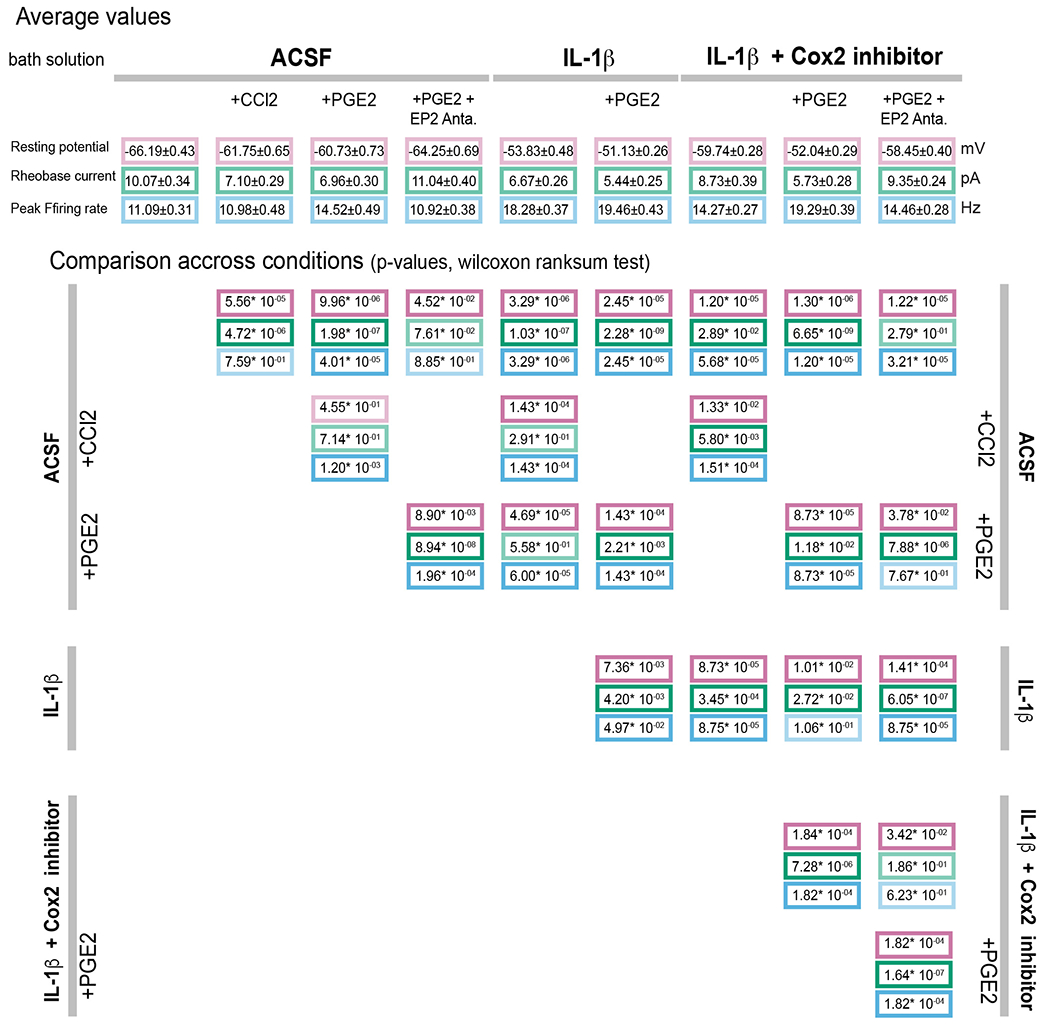
|
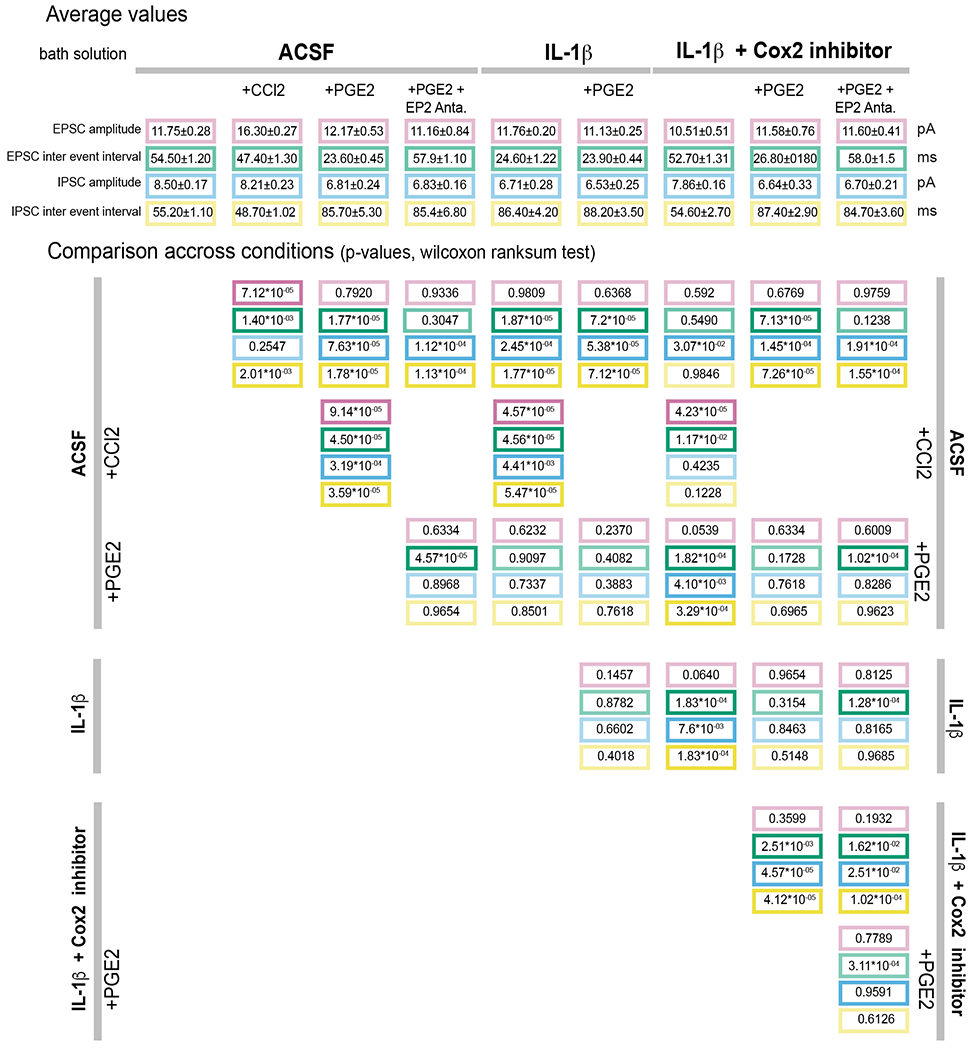
Extended data table 2:
Effects of prostaglandin E2 and cytokines (Il-1β and CCL2) on the synaptic inputs to VMPOLPS neurons.
|
Extended data table 3:
Abbreviations of brain areas
| ACB | nucleus accumbens |
| Arc | arcuate nucleus |
| BLA | basolateral amygdala |
| BNST | bed nucleus of the stria terminalis |
| CEA | central amygdala |
| DMH | dorsal medial hypothalamus |
| IC | insular cortex |
| LC | locus coeruleus |
| LH | lateral hypothalamus |
| LPB | lateral parabrachial nucleus |
| LS | lateral septum |
| NTS | nucleus tractus solitarius |
| PAG | periaqueductal gray |
| PBNc | parabrachial nucleus complex |
| PMD | dorsal premammillary nucleus |
| PMV | ventral premammillary nucleus |
| PSTh | parasubthalamic nucleus |
| PVH | paraventricular nucleus of the hypothalamus |
| PVT | paraventricular nucleus of the thalamus |
| StHy | striohypothalamic nucleus |
| TU | tuberal nucleus |
| vBNST | ventral part of the BNST |
| VLPO | ventral lateral preoptic area |
| VMPO | ventral medial preoptic area |
Extended Data Table 4:
List of drugs
| Drug name | Company name | Solvent | Stock conc. | Working Conc. |
|---|---|---|---|---|
| CNQX disodium salt | Hello Bio | Water | 20 mM | 20 μM |
| D-AP5 | Hello Bio | Water | 50 mM | 50 μM |
| Gabazine | Hello Bio | Water | 20 mM | 20 μM |
| TTX citrate | Hello Bio | Water | 1 mM | 1 μM |
| QX-314 bromide | Tocris | Recording solution | 50 mM | 5 mM |
| Prostaglandin E2 | Tocris | Ethanol | 20 mM | 1 μM |
| Mouse CCL2 | R&D systems | ACSF | 250 nM | 2.5 nM |
| IL-1β | R&D systems | ACSF | 50 nM | 1 nM |
| NS-398 (COX-2 inhibitor) | Cayman chemicals | DMSO | 80 mM | 20 μM |
| PF 04418948 (EP2 antagonist) | Cayman chemicals | DMSO | 50 mM | 10 μM |
| Clozapine N-oxide | Sigma Aldrich | DMSO | 10 mM | 1-10 μM |
Extended Data Table 5:
Precise animal numbers for Figure 3
| Figure 3 Panel: | C | F | M | N | O | P | Q | R |
|---|---|---|---|---|---|---|---|---|
| AAV-GFP, Cre+ | 13 | 9 | 10 | 10 | 10 | 10 | 11 | 8 |
| AAV-hM3D/DTA, Cre− | 12 | 13 | 12 | 10 | 14 | 10 | 9 | 7 |
| AAV-hM3D/DTA, Cre+ | 12 | 9 | 14 | 8 | 8 | 9 | 9 | 7 |
Extended Data Table 6:
Precise cell numbers for Figure 2 and Extended Data Figure 4
| Figure panels: | Fig. 2g–h, Ext. Data Fig. 4b | Ext. Data Fig. 4d–e, Ext Data Fig. 4h–i | Fig. 2i–j, Ext. Data Fig. 4c | Ext. Data Fig. 4f–g Ext. Data Fig. 4j–k | Fig. 2L | Ext. Data Fig. 4m |
| Control | 20 | 18 | 20 | 18 | 18 | 10 |
| CCL2 | 9 | 8 | 8 | 8 | ||
| PGE2 | 12 | 10 | 10 | |||
| PGE2 + EP2 antagonist | 11 | 8 | ||||
| IL-1β | 12 | 10 | 12 | 10 | 10 | |
| IL-1β + PGE2 | 9 | 8 | ||||
| IL-1β + COX2 inhibitor | 10 | 10 | ||||
| IL-1β + COX2 inhibitor +PGE2 | 10 | 8 | ||||
| IL-1β + COX2 inhibitor +PGE2 +EP2 antagonist | 10 | 7 |
Acknowledgements
We thank K. Prichett-Corning and other veterinary staff at the Harvard Office of Animal Resources for advice on working with sick mice, and R. Hellmiss and MCB Graphics for help with illustrations. We thank Ashley Emery and Josie Emery for assistance with histology and RNAscope experiments. We are grateful to members of the Dulac laboratory for helpful advice on experiments, analysis and manuscript and Drs. F. Engert, and R. Losick for comments on the manuscript.
This work was supported by NIH awards K99NS114107 to J.A.O. and F31MH120911 to E.V., NIH grant R01NS050835 to L.L, and NIH grant R01NS112399 and Simons Foundation Award 572189 to C.D. C.D., X.Z. and L.L. are investigators at the Howard Hughes Medical Institute.
Footnotes
Competing financial interests
X.Z. is an inventor on patents applied for by Harvard University related to MERFISH.
References
- 1.Konsman JP, Parnet P & Dantzer R Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci 25, 154–159, (2002). [DOI] [PubMed] [Google Scholar]
- 2.McCusker RH & Kelley KW Immune-neural connections: how the immune system’s response to infectious agents influences behavior. J Exp Biol 216, 84–98, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans SS, Repasky EA & Fisher DT Fever and the thermal regulation of immunity: the immune system feels the heat. Nat Rev Immunol 15, 335–349, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quan N & Banks WA Brain-immune communication pathways. Brain Behav Immun 21, 727–735, (2007). [DOI] [PubMed] [Google Scholar]
- 5.Nakamori T et al. Organum vasculosum laminae terminalis (OVLT) is a brain site to produce interleukin-1 beta during fever. Brain Res 618, 155–159, (1993). [DOI] [PubMed] [Google Scholar]
- 6.Tan CL et al. Warm-Sensitive Neurons that Control Body Temperature. Cell 167, 47–59 e15, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y et al. Leptin-receptor-expressing neurons in the dorsomedial hypothalamus and median preoptic area regulate sympathetic brown adipose tissue circuits. J Neurosci 31, 1873–1884, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao ZD et al. A hypothalamic circuit that controls body temperature. Proc Natl Acad Sci U S A 114, 2042–2047, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elmquist JK, Scammell TE, Jacobson CD & Saper CB Distribution of Fos-like immunoreactivity in the rat brain following intravenous lipopolysaccharide administration. J Comp Neurol 371, 85–103, (1996). [DOI] [PubMed] [Google Scholar]
- 10.Oka T et al. Relationship of EP(1-4) prostaglandin receptors with rat hypothalamic cell groups involved in lipopolysaccharide fever responses. J Comp Neurol 428, 20–32, (2000). [DOI] [PubMed] [Google Scholar]
- 11.Lazarus M et al. EP3 prostaglandin receptors in the median preoptic nucleus are critical for fever responses. Nat Neurosci 10, 1131–1133, (2007). [DOI] [PubMed] [Google Scholar]
- 12.Machado NLS, Bandaru SS, Abbott SBG & Saper CB EP3R-Expressing Glutamatergic Preoptic Neurons Mediate Inflammatory Fever. J Neurosci 40, 2573–2588, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moffitt JR et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konsman JP, Tridon V & Dantzer R Diffusion and action of intracerebroventricularly injected interleukin-1 in the CNS. Neuroscience 101, 957–967, (2000). [DOI] [PubMed] [Google Scholar]
- 17.Kis B et al. Effects of LPS stimulation on the expression of prostaglandin carriers in the cells of the blood-brain and blood-cerebrospinal fluid barriers. J Appl Physiol (1985) 100, 1392–1399, (2006). [DOI] [PubMed] [Google Scholar]
- 18.Zywitza V, Misios A, Bunatyan L, Willnow TE & Rajewsky N Single-Cell Transcriptomics Characterizes Cell Types in the Subventricular Zone and Uncovers Molecular Defects Impairing Adult Neurogenesis. Cell Rep 25, 2457–2469 e2458, (2018). [DOI] [PubMed] [Google Scholar]
- 19.Roessmann U, Velasco ME, Sindely SD & Gambetti P Glial fibrillary acidic protein (GFAP) in ependymal cells during development. An immunocytochemical study. Brain Res 200, 13–21, (1980). [DOI] [PubMed] [Google Scholar]
- 20.Duan L et al. PDGFRbeta Cells Rapidly Relay Inflammatory Signal from the Circulatory System to Neurons via Chemokine CCL2. Neuron 100, 183–200 e188, (2018). [DOI] [PubMed] [Google Scholar]
- 21.Quan N, Stern EL, Whiteside MB & Herkenham M Induction of pro-inflammatory cytokine mRNAs in the brain after peripheral injection of subseptic doses of lipopolysaccharide in the rat. J Neuroimmunol 93, 72–80, (1999). [DOI] [PubMed] [Google Scholar]
- 22.Wilhelms DB et al. Deletion of prostaglandin E2 synthesizing enzymes in brain endothelial cells attenuates inflammatory fever. J Neurosci 34, 11684–11690, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hojen JF et al. IL-1R3 blockade broadly attenuates the functions of six members of the IL-1 family, revealing their contribution to models of disease. Nat Immunol 20, 1138–1149, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis CJ et al. The neuron-specific interleukin-1 receptor accessory protein is required for homeostatic sleep and sleep responses to influenza viral challenge in mice. Brain Behav Immun 47, 35–43, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liege S, Laye S, Li KS, Moze E & Neveu PJ Interleukin 1 receptor accessory protein (IL-1RAcP) is necessary for centrally mediated neuroendocrine and immune responses to IL-1beta. J Neuroimmunol 110, 134–139, (2000). [DOI] [PubMed] [Google Scholar]
- 26.Allen WE et al. Thirst-associated preoptic neurons encode an aversive motivational drive. Science 357, 1149–1155, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeNardo LA et al. Temporal evolution of cortical ensembles promoting remote memory retrieval. Nat Neurosci 22, 460–469, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molina-Holgado E, Ortiz S, Molina-Holgado F & Guaza C Induction of COX-2 and PGE(2) biosynthesis by IL-1beta is mediated by PKC and mitogen-activated protein kinases in murine astrocytes. Br J Pharmacol 131, 152–159, (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu Z, Autry AE, Bergan JF, Watabe-Uchida M & Dulac CG Galanin neurons in the medial preoptic area govern parental behaviour. Nature 509, 325–330, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kozak W, Conn CA & Kluger MJ Lipopolysaccharide induces fever and depresses locomotor activity in unrestrained mice. Am J Physiol 266, R125–135, (1994). [DOI] [PubMed] [Google Scholar]
- 31.Akins C, Thiessen D & Cocke R Lipopolysaccharide increases ambient temperature preference in C57BL/6J adult mice. Physiol Behav 50, 461–463, (1991). [DOI] [PubMed] [Google Scholar]
- 32.Liu Y et al. Lipopolysacharide Rapidly and Completely Suppresses AgRP Neuron-Mediated Food Intake in Male Mice. Endocrinology 157, 2380–2392, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yizhar O et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andermann ML & Lowell BB Toward a Wiring Diagram Understanding of Appetite Control. Neuron 95, 757–778, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pinol RA et al. Brs3 neurons in the mouse dorsomedial hypothalamus regulate body temperature, energy expenditure, and heart rate, but not food intake. Nat Neurosci 21, 1530–1540, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farzi A et al. Arcuate nucleus and lateral hypothalamic CART neurons in the mouse brain exert opposing effects on energy expenditure. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Millington GW The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr Metab (Lond) 4, 18, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kapoor V, Provost AC, Agarwal P & Murthy VN Activation of raphe nuclei triggers rapid and distinct effects on parallel olfactory bulb output channels. Nat Neurosci 19, 271–282, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw and processed data have been deposited in the Gene Expression Omnibus and are available under accession number GSE197547.