

Summary
Background
Combination nivolumab plus ipilimumab was efficacious in patients with asymptomatic melanoma brain metastases (MBM) in CheckMate 204, but showed low efficacy in patients with symptomatic MBM. Here, we provide final 3-year follow-up data from the trial.
Methods
This open-label, multicentre, phase 2 study (CheckMate 204) included adults (aged ≥18 years) with measurable MBM (0·5–3·0 cm in diameter). Asymptomatic patients (cohort A) had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 and no neurological symptoms or baseline corticosteroid use; symptomatic patients (cohort B) had an ECOG performance status of 0–2 with stable neurological symptoms and could be receiving low-dose dexamethasone. Nivolumab 1 mg/kg plus ipilimumab 3 mg/kg was given intravenously every 3 weeks for four doses, followed by nivolumab 3 mg/kg every 2 weeks for up to 2 years, until disease progression or unacceptable toxicity. The primary endpoint was intracranial clinical benefit rate (complete responses, partial responses, or stable disease lasting ≥6 months) assessed in all treated patients. Intracranial progression-free survival and overall survival were key secondary endpoints. This study is registered with ClinicalTrials.gov, NCT02320058.
Findings
Between Feb 19, 2015, and Nov 1, 2017, 119 (72%) of 165 screened patients were enrolled and treated: 101 patients were asymptomatic (cohort A; median follow-up 34·3 months [IQR 14·7–36·4]) and 18 were symptomatic (cohort B; median follow-up 7·5 months [1·2–35·2]). Investigator-assessed intracranial clinical benefit was observed in 58 (57·4% [95% CI 47·2–67·2]) of 101 patients in cohort A and three (16·7% [3·6–41·4]) of 18 patients in cohort B; investigator-assessed objective response was observed in 54 (53·5% [43·3–63·5]) patients in cohort A and three (16·7% [3·6–41·4]) patients in cohort B. 33 (33%) patients in cohort A and three (17%) patients in cohort B had an investigator-assessed intracranial complete response. For patients in cohort A, 36-month intracranial progression-free survival was 54·1% (95% CI 42·7–64·1) and overall survival was 71·9% (61·8–79·8). For patients in cohort B, 36-month intracranial progression-free survival was 18·9% (95% CI 4·6–40·5) and overall survival was 36·6% (14·0–59·8). The most common grade 3–4 treatment-related adverse events (TRAEs) were increased alanine aminotransferase and aspartate aminotransferase (15 [15%] of 101 patients each) in cohort A; no grade 3 TRAEs occurred in more than one patient each in cohort B, and no grade 4 events occurred. The most common serious TRAEs were colitis, diarrhoea, hypophysitis, and increased alanine aminotransferase (five [5%] of each among the 101 patients in cohort A); no serious TRAE occurred in more than one patient each in cohort B. There was one treatment-related death (myocarditis in cohort A).
Interpretation
The durable 3-year response, overall survival, and progression-free survival rates for asymptomatic patients support first-line use of nivolumab plus ipilimumab. Symptomatic disease in patients with MBM remains difficult to treat, but some patients achieve a long-term response with the combination.
Funding
Bristol Myers Squibb.
Introduction
Melanoma brain metastases (MBM) frequently exist at diagnosis or develop during the course of the disease.1–3 Historically, median overall survival following diagnosis of brain metastases was 3–13 months.4 Despite recent improvements in survival outcomes associated with the introduction of immune checkpoint inhibitors and targeted therapy, MBM remain a major cause of morbidity and mortality.5,6 Only a few clinical trials with these therapies have been done in patients with untreated MBM, and those have mainly included patients with asymptomatic MBM and precluded corticosteroid use or previous systemic therapy. Immune checkpoint inhibitors have shown durable intracranial activity with single-agent ipilimumab and single-agent anti-PD-1 treatments, albeit with modest response rates.7–10 Targeted therapy with dabrafenib plus trametinib in patients with BRAF-mutant MBM showed increased, but less durable, intracranial activity.11 Indeed, a recent systematic literature review and meta-analysis on therapy in patients with MBM concluded that combination immunotherapy increases long-term progression-free and overall survival compared with single-agent immunotherapy and combination targeted therapies.12
CheckMate 204 investigated the use of combination nivolumab plus ipilimumab in patients with MBM in both asymptomatic patients and patients with neurological symptoms with or without corticosteroids at baseline.13,14 Results from asymptomatic patients in CheckMate 204 demonstrated intracranial activity of nivolumab plus ipilimumab that was equivalent to extracranial activity, with a greater than 50% objective response rate (up to a median follow-up of 20·6 months). In addition, more than 85% of responses are durable as evidenced by both median progression-free survival and overall survival not being reached at a median follow-up of 20·6 months (minimum follow-up 11 months). The Anti-PD-1 Brain Collaboration study independently conducted in Australia demonstrated similar results with nivolumab plus ipilimumab,9,10 and a 27-patient cohort in the phase 3 NIBIT-M2 trial also showed the efficacy of this combination, with a median overall survival of 29·2 months (95% CI 0–65·1).15 On the basis of these trials, nivolumab plus ipilimumab is widely recognised as a standard of care for most patients with asymptomatic MBM who are candidates for immunotherapy. Patients with neurological symptoms, including those treated with corticosteroids at baseline, showed only a modest response in CheckMate 204, reinforcing the fact that these patients’ disease remains difficult to treat.14
The US Food and Drug Administration (FDA) Guidance for Industry supports an independent review of tumour endpoints and provides recommendations for blinded imaging evaluations,16,17 in part, because local investigators have access to clinically relevant observations that have the potential to cause bias in the reading. This bias is especially important with intracranial disease because of the complexities involved in these typically MRI-based evaluations. To date, there has been little consistency in how to measure intracranial response beyond the consensus that MRI imaging should be used to improve the accuracy of the evaluation.18 Published in July, 2021, Guidance for Industry from the US FDA for evaluating cancer drugs in patients with central nervous system metastases has recommended standard response criteria (including modified Response Evaluation Criteria in Solid Tumors [RECIST] version 1.1) and blinded independent central review (BICR) assessment.19 Here, we report 3-year data from CheckMate 204 along with the first results of BICR imaging data for both response and progression-free survival, as well as concordance results for the investigator-assessed and BICR-assessed data.
Methods
Study design and participants
CheckMate 204 was an open-label, multicentre, phase 2 study done at 28 sites in the USA (appendix pp 23–24). Patients aged at least 18 years with histologically confirmed metastatic melanoma with at least one non-irradiated brain metastasis measuring 0·5–3·0 cm in diameter (as assessed by MRI) were included. Previous stereotactic radiotherapy or excision of up to three brain metastases was permitted if treatment was completed at least 3 weeks before the start of treatment and at least one lesion remained unirradiated for the assessment of intracranial response. Previous approved adjuvant systemic therapies were allowed, including ipilimumab, if the last dose was administered at least 6 months before the first dose of study drug. Previous use of BRAF and MEK inhibitors in the advanced setting was allowed after a 4-week washout period. Patients were excluded if they had known leptomeningeal involvement or autoimmune disease.
Enrolment started with asymptomatic patients (cohort A) who had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, no neurological symptoms, and no systemic corticosteroid therapy for at least 10 days before treatment initiation. The protocol was amended on Aug 15, 2016, to include a separate symptomatic group of patients (cohort B) who had neurological signs and symptoms, with or without baseline corticosteroid use of up to 4 mg dexamethasone or equivalent per day, and had an ECOG performance status of 0–2. Safety laboratory tests to determine eligibility included serum chemistry (creatinine, bilirubin, aspartate aminotransferase, and alanine aminotransferase; although amylase and lipase levels were to be collected, the results did not require review prior to dosing) and complete blood count with differential.
The protocol and amendments for this trial (appendix) were reviewed and approved by the review board for each institution. The trial was conducted in accordance with Good Clinical Practice guidelines, as specified by the International Conference on Harmonisation. Prior to enrolment, all patients provided written informed consent.
Procedures
Patients received nivolumab 1 mg/kg combined with ipilimumab 3 mg/kg both intravenously once every 3 weeks for 12 weeks (for a total of four doses; induction phase), followed by nivolumab 3 mg/kg intravenously every 2 weeks for a total of 24 months or until progression or unacceptable toxicity (maintenance phase). Dose reductions or escalations were not permitted, but dosing could be delayed for treatment-related adverse events or stereotactic treatment. Laboratory monitoring (complete blood count with differential, liver function tests, blood urine nitrogen or serum urea concentration, creatinine, Ca, Mg, Na, Cl, lactate dehydrogenase, glucose, amylase, lipase, and thyroid-stimulating hormone) was required at week 1 and at week 4. Patients who had grade 3 or 4 adverse events during the induction phase could be treated with nivolumab monotherapy during the maintenance phase.
Response was determined by radiographic assessment every 6 weeks for the first year and then every 12 weeks thereafter until documented disease progression. Intracranial lesions were assessed by gadolinium-enhanced MRI using RECIST version 1.1, modified to allow the measurement of up to five intracranial target lesions 5–30 mm in longest diameter.9 Extracranial lesions were assessed by CT using RECIST version 1.1 with up to five baseline target lesions of at least 10 mm in diameter.20 Global responses were assessed using a combination of both types of lesions and included up to five target intracranial and up to five target extracranial lesions. Complete and partial response, as well as progression, was confirmed at least 4 weeks after the initial assessment, and stable disease was defined as lasting at least 6 months following treatment initiation. Both investigator-assessed and BICR-assessed responses were collected and analysed. Investigator assessment consisted of the institutional radiologist’s clinical reading as interpreted by the investigator. BICR analysis was performed at Bioclinica (Princeton, NJ, USA) by two independent neuroradiologists for each patient, who were masked to patient demographics, site assessment of response, site choice of target and non-target lesions, as well as the identification of new lesions, clinical history, and read number of results. All on-study images required by the protocol that were submitted to Bioclinica were reviewed and assessed according to the same response criteria as the investigator review. Patients’ information was not sent for BICR evaluation if the patient had died, had disease progression, or withdrew consent prior to having the requisite scans available. In the case of non-agreement between the two independent radiologists, adjudication was performed by a radiologist at Bioclinica who was not involved in the primary review and who was blinded to the identity of the two primary readers. The resulting BICR response data (intracranial, extracranial, and global) were transferred to the sponsor for analysis. Per protocol, rates of BICR-assessed responses were calculated based on all treated patients, and patients for whom data were not available for BICR were categorised as non-responders.
Safety was evaluated in all treated patients using Common Terminology Criteria for Adverse Events version 4.0. In addition to treatment-related adverse events collected up to 30 days after the last dose, immune-mediated adverse events, reported between the first dose and 100 days after the last dose of study therapy, were collected. These included events for which immune-modulating medication was initiated (non-endocrine events) and endocrine events, which were included regardless of treatment and without the requirement of specific laboratory criteria.
Outcomes
The primary endpoint was intracranial clinical benefit rate, defined as the percentage of patients with complete responses, partial responses, or stable disease lasting at least 6 months (per modified RECIST version 1.1), along with a sensitivity analysis to determine intracranial clinical benefit rate per independent review analysis. Secondary endpoints were extracranial and global clinical benefit rate; intracranial, extracranial, and global objective response rate (defined as the percentage of patients with complete responses or partial responses); intracranial, extracranial, and global progression-free survival (defined as the time between the date of first study drug dose and the first date of documented progression, as determined by the investigator, or death, whichever occurred first); overall survival (defined as the time between the date of first study drug dose and the date of death); and safety. Sensitivity analyses were included for response-derived secondary endpoints per independent review assessment. Exploratory endpoints included median time to objective response (intracranial) and duration of response (intracranial, extracranial, and global) for both investigator-assessed and BICR-assessed responses, and to evaluate associations between BRAF mutation status and response or survival (in asymptomatic patients) and between dexamethasone and treatment effect in patients treated with corticosteroids (symptomatic patients). Safety analysis in patients with previous or on-study stereotactic radiotherapy (a protocol-specified secondary endpoint) has not been presented because too few patients met this criterion.
Statistical analysis
The analyses in this study are based on a Dec 18, 2020, data cutoff and represent a minimum follow-up of 34 months. Primary analyses for the study have previously been published.13,14 The planned sample size of 110 patients ensured that the maximum width of the exact 90% CI for any given estimate of the clinical benefit rate did not exceed 18% and that of the 95% CI did not exceed 20%. All enrolled patients were included in both the efficacy and safety analyses. Data for asymptomatic and symptomatic patients were analysed separately (per protocol) because of differences in patient characteristics. Clinical benefit rates were calculated to yield clinically meaningful results with respect to the lower bounds of the Clopper-Pearson exact two-sided 95% CI. Time-to-event analyses were estimated using the Kaplan-Meier method, with medians presented along with 95% CIs based on the Brookmeyer and Crowley method. Post-hoc analyses included 6-month and 12-week landmark analyses to assess overall survival in patients with or without an investigator-assessed response as well as an analysis of response rates across subgroups.
Concordance between investigator-assessed and BICR-assessed responses were assessed numerically, both on an individual response level and by grouping responder (complete response or partial response) results and non-responder results. In addition, concordance was determined per Cohen’s kappa coefficient analysis to normalise the random chance of the dataset.21
Analyses were performed using SAS software (version 9.2). This study is registered with ClinicalTrials. gov, NCT02320058.
Role of the funding source
The study was originally developed by the Cytokine Working Group and was then expanded under Bristol Myers Squibb as the study sponsor. The study was designed by academic authors who were members of the study steering committee, along with sponsor physicians and staff. The steering committee was established in lieu of a data and safety monitoring committee. Data were collected by the sponsor and analysed and interpreted in collaboration with the authors. The study sponsor paid for medical writing and editorial support.
Results
165 patients were enrolled between Feb 19, 2015, and Nov 1, 2017, of whom 119 (72%) were treated (101 asymptomatic patients in cohort A and 18 symptomatic patients in cohort B; table 1, appendix p 15). 16 (89%) of the 18 symptomatic patients had neurological symptoms or signs at baseline; two symptomatic patients were not reported to have neurological symptoms at baseline but were on 2 mg and 4 mg of dexamethasone.
Table 1:
Baseline characteristics
| Asymptomatic patients (n=101) | Symptomatic patients* (n=18) | |
|---|---|---|
| Age, years | 59·0 (51·0–66·0) | 59·5 (50·0–70·0) |
| Sex | ||
| Female | 33 (33%) | 5 (28%) |
| Male | 68 (67%) | 13 (72%) |
| Lactate dehydrogenase | ||
| ≤ULN | 60 (59%) | 9 (50%) |
| >ULN | 41(41%) | 8 (44%) |
| ≤2xULN | 90 (89%) | 15 (83%) |
| >2xULN | 11 (11%) | 2 (11%) |
| Not reported | 0 | 1 (6% ) |
| PD-L1 expression† | ||
| ≥1% | 46/91 (51%) | 6/16 (38%) |
| <1% | 37/91 (41%) | 8/16 (50%) |
| Indeterminant or not evaluable | 8/91 (9%) | 2/16 (13%) |
| BRAF mutation status | ||
| Mutant | 66 (65%) | 8 (44%) |
| Wild-type | 33 (33%) | 8 (44%) |
| Not reported | 2 (2%) | 2 (11%) |
| NRAS mutation status | ||
| Mutant | 7 (7%) | 1 (6%) |
| Wild-type | 19 (19%) | 1 (6%) |
| Not reported | 75 (74%) | 16 (89%) |
| Previous systemic therapy | ||
| Adjuvant‡ | 11 (11%) | 2 (11%) |
| Metastatic§ | 6 (6%) | 2 (11%) |
| Previous SRT | ||
| 0 | 92 (91%) | 15 (83%) |
| 1 | 5 (5%) | 3 (17%) |
| 2 | 3 (3%) | 0 |
| ≥3 | 1 (1%) | 0 |
| Sum of intracranial target lesion diameters, mm | 15·0 (8·0–27·6) | 26·0 (13·6–34·0) |
| Intracranial target lesions‖ | ||
| No lesions | 1 (1%) | 0 |
| 1–2 lesions | 78 (77%) | 11 (61%) |
| ≥3 lesions | 22 (22%) | 7 (39%) |
Data are median (IQR) or n (%). SRT=stereotactic radiotherapy. ULN=upper limit of normal.
16 (89%) of 18 patients had neurological symptoms or signs at baseline and two (11%) had symptoms of night sweats and anorexia recorded (ie, not definitively neurological); one of these two patients had neurological symptoms or signs recorded within 1 month of screening.
Expression assessed with a validated automated immunohistochemical assay (PD-L1 IHC 28–8 pharmDx; Dako, an Agilent Technologies company, Santa Clara, CA, USA).
Including four patients with targeted therapy (one monotherapy and three combination) in asymptomatic patients.
Including five patients with targeted therapy combination (asymptomatic patients); both patients in the symptomatic cohort received targeted therapy combination.
Per investigator assessment; inclusion of one patient in the asymptomatic cohort with no lesion was a protocol deviation.
At a minimum follow-up for the total population (ie, the time from the last patient’s first dose to the clinical cutoff date) of 34·2 months, median follow-up (the median time between the first dose date and the date of death or last known date alive) was 34·3 months (IQR 14·7–36·4) in 101 asymptomatic patients in cohort A and 7·5 months (IQR 1·2–35·2) in 18 symptomatic patients in cohort B. 59 (58%) patients in cohort A and five (28%) patients in cohort B remained in follow-up at database cutoff (appendix p 15). Patients in cohort A had an overall median duration of therapy of 3·4 months (IQR 1·4–20·7) and 58 (57%) patients entered the maintenance phase; median duration of therapy in cohort B was 0·7 months (0·03–1·4) with four (22%) patients entering the maintenance phase and the other 14 (78%) receiving between one and two total doses. Subsequent systemic therapy was received by 19 (19%) patients in cohort A and four (22%) patients in cohort B (appendix p 2).
Consistent with previous results, intracranial clinical benefit per investigator assessment was observed in 58 (57·4% [95% CI 47·2–67·2]) of 101 asymptomatic patients, and objective responses were achieved in 54 patients (53·5% [43·3–63·5]; table 2). Per BICR evaluation, 54 patients (53·5% [43·3–63·5]) achieved intracranial clinical benefit and 50 patients (49·5% [39·4–59·6]) had intracranial objective responses based on the entire cohort population of 101 patients (appendix p 3). Response data for extracranial and global disease were similar to those for intracranial disease (table 2, appendix p 3).
Table 2:
Response to treatment (investigator assessment)
| Asymptomatic patients (n=101) |
Symptomatic patients (n=18) |
|||||
|---|---|---|---|---|---|---|
| Intracranial | Extracranial | Global | Intracranial | Extracranial | Global | |
| Best overall response* | ||||||
| Complete response | 33 (33%) | 16 (16%) | 17 (17%) | 3 (17%) | 1 (6%) | 1 (6%) |
| Partial response | 21 (21%) | 33 (33%) | 35 (35%) | 0 | 3 (17%) | 3 (17%) |
| Stable disease ≥6 months | 4 (4%) | 5 (5%) | 4 (4%) | 0 | 0 | 0 |
| Progressive disease | 30 (30%) | 17 (17%) | 26 (26%) | 11 (61%) | 7 (39%) | 10 (56%) |
| Not evaluable for clinical benefit rate | 13 (13%) | 30 (30%) | 19 (19%) | 4 (22%) | 7 (39%) | 4 (22%) |
| Death prior to first on-study assessment | 2 (2%) | 3 (3%) | 3 (3%) | 2 (11%) | 1 (6%) | 1 (6%) |
| Early discontinuation due to study toxicity | 1 (1%) | 1 (1%) | 1 (1%) | 0 | 0 | 0 |
| Stable disease <6 months | 6 (6%) | 14 (14%) | 10 (10%) | 2 (11%) | 3 (17%) | 1 (6%) |
| No extracranial disease at baseline | NA | 7 (7%) | 0 | NA | 1 (6%) | 0 |
| Other† | 4 (4%) | 5 (5%) | 5 (5%) | 0 | 2 (11%) | 2 (11%) |
| Objective response rate‡ | 54/101 (53·5, 43·3–63·5) | 49/101 (48·5, 38·4–58·7) | 52/101 (51·5, 41·3–61·6) | 3/18 (16·7, 3·6–41·4) | 4/18 (22·2, 6·4–47·6) | 4/18 (22·2, 6·4–47·6) |
| Clinical benefit rate§ | 58/101 (57·4, 47·2–67·2) | 54/101 (53·5, 43·3–63·5) | 56/101 (55·4, 45·2–65·3) | 3/18 (16·7, 3·6–41·4) | 4/18 (22·2, 6·4–47·6) | 4/18 (22·2, 6·4–47·6) |
| Duration of response | ||||||
| Ongoing responders/patients with objective response (%) | 46/54 (85%) | 38/49 (78%) | 40/52 (77%) | 3/3 (100%)‖ | 4/4 (100%) | 4/4 (100%) |
| Median (95% CI), months | NR (NR-NR) | NR (32·8-NR) | NR (32·8-NR) | NR (NR-NR) | NR (NR-NR) | NR (NR-NR) |
Data are n (%) or n/N (%, 95% CI), unless otherwise stated. NA=not applicable. NR=not reached.
Best overall response was assessed by the investigators in accordance with Response Evaluation Criteria in Solid Tumors version 1.1 (modified criteria were used for intracranial response).
Asymptomatic: total of five patients for all three response categories (intracranial, extracranial, and global): one patient withdrew consent (all three categories), one patient stopped study (extracranial and global categories), one patient for gamma knife therapy (all three categories), one patient for extracranial lesion procedure not done (extracranial and global); one patient did not receive any on-study scans (all three categories); symptomatic: insufficient radiographical scan data (two patients; extracranial and global).
Data include patients with a complete response or partial response; 95% CI based on Clopper-Pearson method.
Data include patients with a complete response, partial response, or stable disease for 6 months or longer; 95% CI based on Clopper-Pearson method.
Previously reported as 3/4 because one patient had disease progression followed by a response;14 currently reported per the analysis for concordance with blinded independent central review.
Per BICR assessment, patients in cohort A had a median reduction in tumour volume of −69·0% (appendix p 16), and 42 (84%) of the 50 patients with an intracranial response had an ongoing response (appendix p 17). Median duration of response had not been reached (appendix p 3), with the majority of responses (29 [58%] of 50 patients) lasting more than 2 years (appendix p 17) and most first responses occurring early in treatment (median 2·8 months [IQR 1·3–4·1] per BICR and 1·4 months [1·2–2·8] per investigator). Per investigator assessment in a post-hoc analysis, response rates were consistently above 42% across subgroup categories (appendix pp 4–5).
In cohort A, median intracranial progression-free survival was not reached by investigator assessment (40 events in 101 patients in cohort A) and was 39·3 months (95% CI 7·5–45·8) by BICR (45 events in 101 patients). Progression-free survival at 36 months was similar in both intracranial investigator-based assessments (54·1% [95% CI 42·7–64·1]) and BICR-based assessments (52·5% [41·4–62·4]), and across intracranial, extracranial, and global disease (figure 1, appendix p 6). With 29 events in 101 patients, 36-month overall survival was 71·9% (61·8–79·8; figure 1, appendix p 6). 36-month overall survival was similar among patients with BRAF-mutant tumours (73·0%, 60·2–82·3) or wild-type tumours (68·8%, 49·7–81·8; appendix p 18). Overall survival as a landmark post-hoc analysis 6 months from enrolment showed a 24-month overall survival of 91·8% (79·6–96·8) for responders and 62·3% (43·3–76·6) for non-responders, and results were similar in a 12-week landmark post-hoc analysis (appendix p 19). Across the asymptomatic patient group, three (6%) of 54 responders and 16 (34%) of 47 non-responders received subsequent systemic therapy.
Figure 1: Kaplan-Meier estimates of investigator-assessed progression-free survival (A), BICR-assessed progression-free survival (B), and overall survival (C) in patients with asymptomatic melanoma brain metastases.
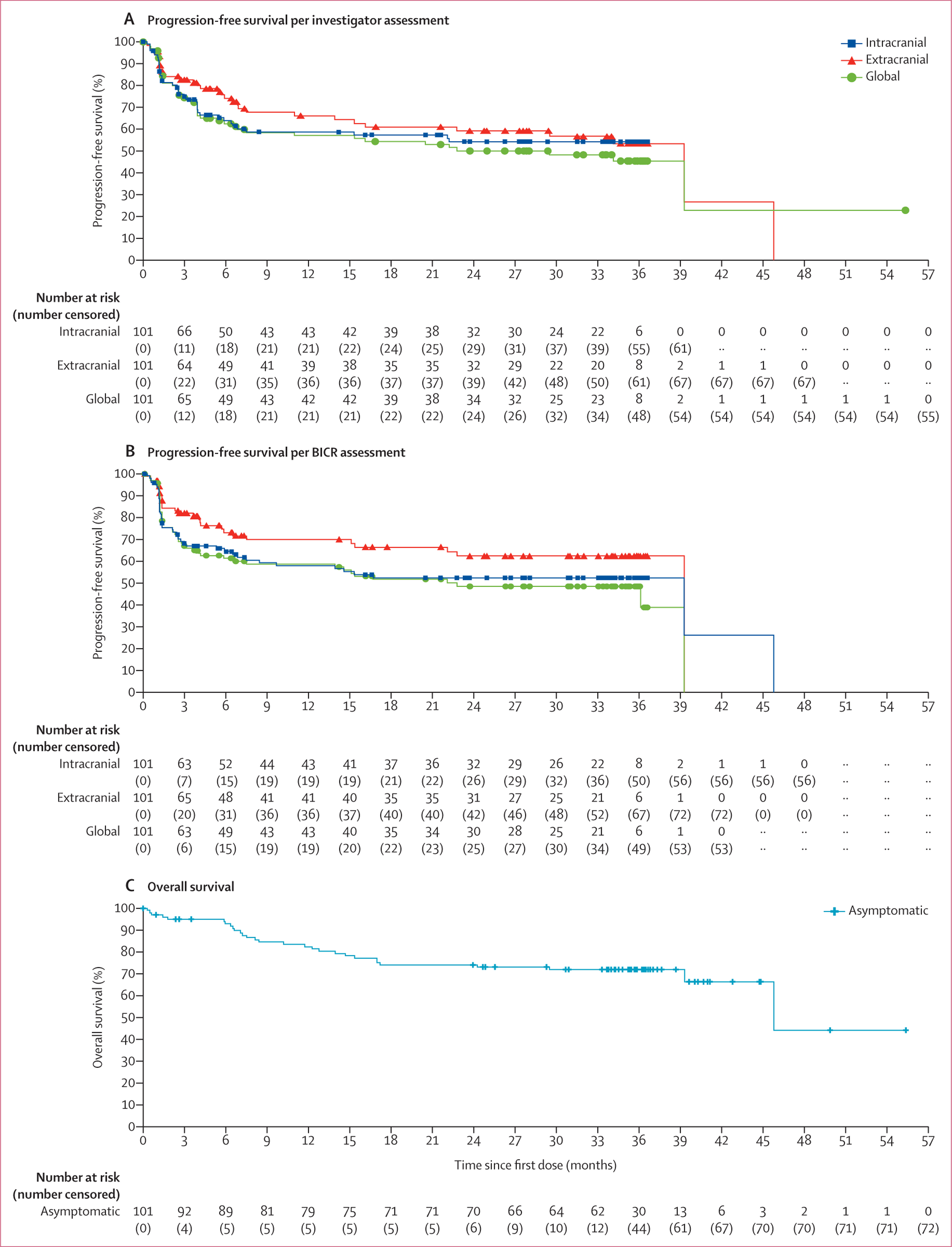
BICR=blinded independent central review.
Of the 18 patients with symptomatic disease in cohort B, three (16·7% [95% CI 3·6–41·4]) had an intracranial response and four (22·2% [6·4–47·6]) had both extracranial and global responses per investigator assessment (table 2). BICR-assessed responses were similar to investigator-assessed responses, with intracranial, extracranial, and global objective response in four (22·2% [6·4–47·6]) of 18 patients (appendix p 7) based on the whole population. All responses were ongoing at the time of the database lock, so the median duration of response had not been reached (table 2, appendix p 7). Per BICR assessment, patients in cohort B had an overall median increase in tumour volume of 23·0% (appendix p 20). Responses in this cohort lasted more than 2·5 years, and median time to response was 2·0 months (IQR 1·2–2·7) per BICR and 5·5 months (1·0–7·0) per investigators (appendix p 21). Response rates across subgroup categories were not estimable due to the low patient numbers in each subgroup and therefore the data are not presented.
For symptomatic patients, median intracranial progression-free survival was 1·2 months (95% CI 0·7–1·2) by investigator assessment (13 events in 18 patients) and 1·2 months (95% CI 0·7–not reached) by BICR (12 events in 18 patients; figure 2). Progression-free survival at 36 months for intracranial disease was 18·9% (95% CI 4·6–40·5) by investigator assessment and 28·2% (9·6–50·5) by BICR assessment; 28·2% (8·9–51·5) and 36·3% (12·3–61·2) for extracranial disease; and 23·9% (7·5–45·5) and 25·9% (8·1–48·3) for global disease (appendix p 8). With ten events in 18 patients, 36-month overall survival was 36·6% (14·0–59·8; figure 2, appendix p 8). We have previously shown that two of 12 patients who received baseline corticosteroids achieved a response versus two of six who did not.14 Here, we show that 24-month overall survival was 32·4% (95% CI 8·0–60·5) in the 12 patients with baseline dexamethasone use versus 66·7% (19·5–90·4) in the six patients without such use (appendix p 22).
Figure 2: Kaplan-Meier estimates of investigator-assessed progression-free survival (A), BICR-assessed progression-free survival (B), and overall survival (C) in patients with symptomatic melanoma brain metastases.
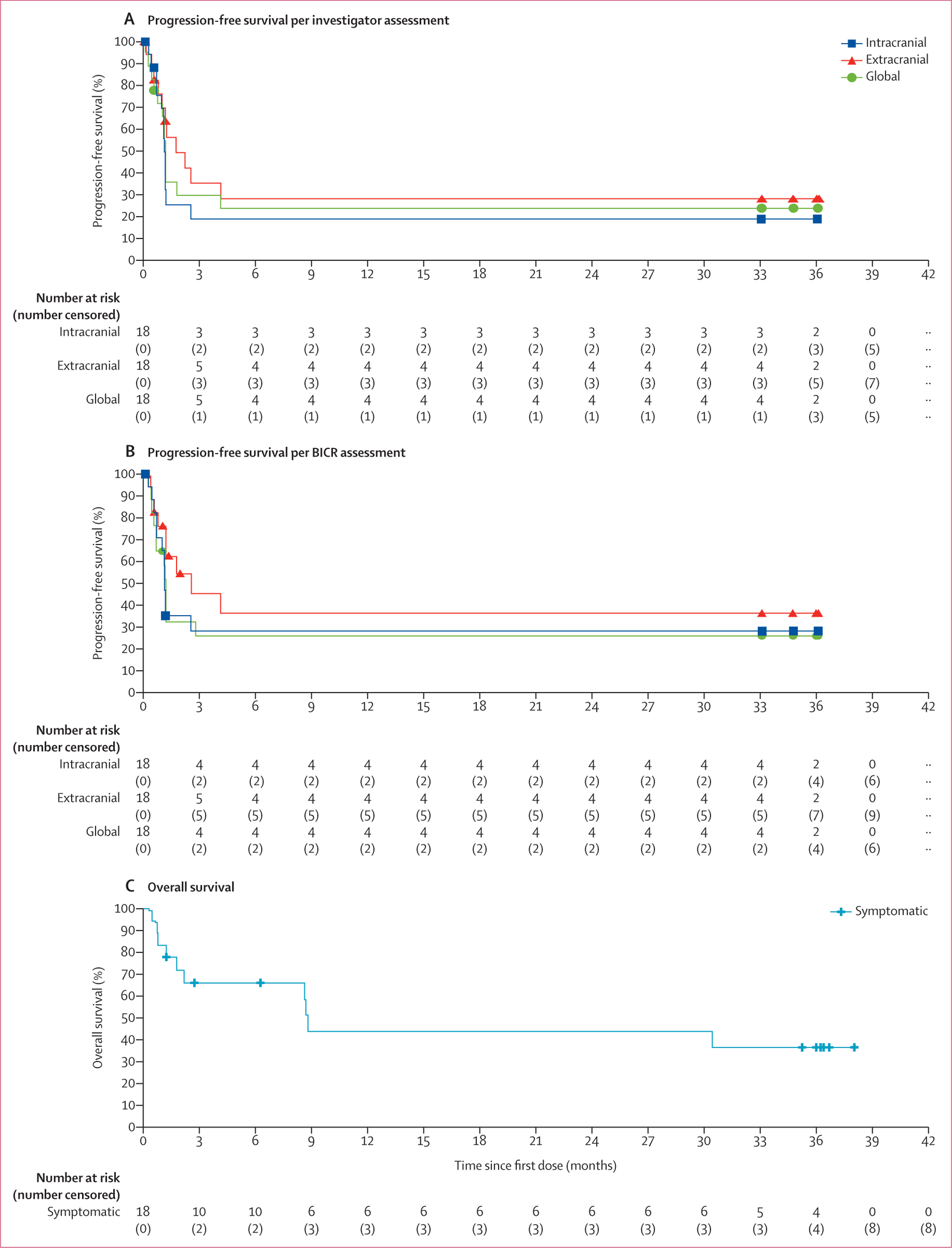
BICR=blinded independent central review.
For asymptomatic patients, concordance between the investigator and BICR intracranial assessments based on responder and non-responder categories was 81 (85%) of 95 patients (table 3), with a Cohen’s kappa value of 0·70. Concordance based on matching individual response categories ranged from 67 to 72 (71–76%) of 95 patients across the three disease groups (appendix p 9), with a Cohen’s kappa value of 0·60 for intracranial disease. For symptomatic patients, the concordance for intracranial disease was 16 (94%) of 17 patients by responder and non-responder categories (Cohen’s kappa 0·82; table 3) and 14 (82%) of 17 patients by individual response categories (0·68; appendix p 10). Of the overall combined population of 119 patients, 33 (28%) and 28 (24%) patients in the BICR analysis required adjudication between the two neuroradiologists for intracranial and extracranial assessments, respectively (data not shown).
Table 3:
Concordance between investigator-assessed and BICR-assessed response
| Investigator-assessed |
Concordance rate of responders, n/N (%)* | |||
|---|---|---|---|---|
| Responders (CR or PR) | Non-responders | Not evaluable | ||
| BICR-assessed | ||||
| Asymptomatic intracranial | 81/95 (85%) | |||
| Responders (CR or PR) | 45 (47%) | 5 (5%) | 0 | |
| Non-responders | 8 (8%) | 25 (26%) | 1 (1%) | |
| Not evaluable | 1 (1%) | 4 (4%) | 6 (6%) | |
| Asymptomatic extracranial | 84/95 (88%) | |||
| Responders (CR or PR) | 44 (46%) | 3 (3%) | 3 (3%) | |
| Non-responders | 1 (1%) | 14 (15%) | 4 (4%) | |
| Not evaluable | 4 (4%) | 5 (5%) | 17 (18%) | |
| Asymptomatic global | 88/95 (93%) | |||
| Responders (CR or PR) | 47 (49%) | 2 (2%) | 0 | |
| Non-responders | 4 (4%) | 25 (26%) | 5 (5%) | |
| Not evaluable | 1 (1%) | 3 (3%) | 8 (8%) | |
| Symptomatic intracranial | 16/17 (94%) | |||
| Responders (CR or PR) | 3 (18%) | 1 (6%) | 0 | |
| Non-responders | 0 | 9 (53%) | 1 (6%) | |
| Not evaluable | 0 | 1 (6%) | 2 (12%) | |
| Symptomatic extracranial | 17/17 (100%) | |||
| Responders (CR or PR) | 4 (24%) | 0 | 0 | |
| Non-responders | 0 | 4 (24%) | 2 (12%) | |
| Not evaluable | 0 | 3 (18%) | 4 (24%) | |
| Symptomatic global | 17/17 (100%) | |||
| Responders (CR or PR) | 4 (24%) | 0 | 0 | 17/17 (100%) |
| Non-responders | 0 | 7 (41%) | 3 (18%) | |
| Not evaluable | 0 | 3 (18%) | 0 | |
BICR=blinded independent central review. CR=complete response. PR=partial response.
Quantifies the frequency with which investigator-assessed and BICR-assessed response agreed on classification of a patient as responder versus non-responder or not-evaluable (ie, matches included responder to responder, non-responder to non-responder, not evaluable to not evaluable, and not evaluable to non-responder) as a proportion of the total number of patients with assessments by both the investigator and BICR (n=95 for asymptomatic and n=17 for symptomatic).
With more than 90% of patients already having stopped treatment at the last database lock in 2018, there were few changes in the treatment-related adverse events reported from those previously reported.14 Grade 3 or grade 4 treatment-related adverse events occurred in 56 (55%) of 101 patients in cohort A and 12 (67%) of 18 patients in cohort B, with the most common being increased alanine or aspartate aminotransferase (15 [15%] each) in cohort A; in cohort B, no grade 3 adverse events occurred in more than one patient each, and no grade 4 events occurred (table 4). 29 (29%) of 101 patients discontinued treatment due to any-grade treatment-related adverse events in cohort A, most commonly due to increased alanine or aspartate aminotransferase (both in eight [8%] patients) and diarrhoea in six (6%); there were three such patients in cohort B (one each due to pustular rash, nephritis, or pneumonitis). Grade 3 or 4 neurological treatment-related adverse events occurred in seven (7%) of 101 patients in cohort A and three (17%) of 18 patients in cohort B (appendix p 11). The profile of immune-mediated adverse events was similar in cohort A and cohort B patients, with the most common categories being hepatitis, rash, and hypothyroidism (appendix p 12). The most common serious treatment-related adverse events were colitis, diarrhoea, hypophysitis, and increased alanine aminotransferase (five [5%] of each among the 101 patients in cohort A; no serious treatment-related adverse events occurred in more than one patient each in cohort B (appendix p 13). In addition, late emergent treatment-related adverse events voluntarily reported more than 100 days after end of therapy were rare and are listed in the appendix (p 14). There was one treatment-related death in the asymptomatic cohort (grade 5 myocarditis; previously reported).13,22 Overall, there were 29 deaths in the asymptomatic cohort (21 due to disease, one to study drug toxicity, seven to other or unknown causes) and ten deaths in the symptomatic cohort (eight due to disease and two to other or unknown causes).
Table 4:
Treatment-related adverse events
| Asymptomatic patients (n=101) |
Symptomatic patients (n=18) |
|||||
|---|---|---|---|---|---|---|
| Grade 1–2 | Grade 3 | Grade 4 | Grade 1–2 | Grade 3 | Grade 4 | |
| Treatment-related adverse events* | 40 (40%) | 42 (42%) | 14 (14%) | 4 (22%) | 12 (67%) | 0 |
| Fatigue | 42 (42%) | 4 (4%) | 0 | 2 (11%) | 1 (6%) | 0 |
| Pruritus | 39 (39%) | 0 | 0 | 4 (22%) | 0 | 0 |
| Diarrhoea | 31 (31%) | 6 (6%) | 0 | 4 (22%) | 1 (6%) | 0 |
| Maculopapular rash | 30 (30%) | 8 (8%) | 0 | 2 (11%) | 1 (6%) | 0 |
| Nausea | 26 (26%) | 2 (2%) | 0 | 3 (17%) | 0 | 0 |
| Increased alanine aminotransferase | 23 (23%) | 13 (13%) | 2 (2%) | 2 (11%) | 0 | 0 |
| Arthralgia | 22 (22%) | 0 | 0 | 0 | 0 | 0 |
| Hypothyroidism | 22 (22%) | 1 (1%) | 0 | 1 (6%) | 0 | 0 |
| Increased aspartate aminotransferase | 20 (20%) | 13 (13%) | 2 (2%) | 2 (11%) | 0 | 0 |
| Headache | 17 (17%) | 3 (3%) | 0 | 0 | 1 (6%) | 0 |
| Pyrexia | 17 (17%) | 0 | 0 | 2 (11%) | 1 (6%) | 0 |
| Decreased appetite | 16 (16%) | 1 (1%) | 0 | 2 (11%) | 0 | 0 |
| Vomiting | 11 (11%) | 2 (2%) | 0 | 1 (6%) | 0 | 0 |
| Hyperthyroidism | 10 (10%) | 2 (2%) | 1 (1%) | 0 | 0 | 0 |
| Cough | 9 (9%) | 0 | 0 | 2 (11%) | 0 | 0 |
| Rash | 9 (9%) | 2 (2%) | 0 | 3 (17%) | 0 | 0 |
| Increased lipase | 8 (8%) | 5 (5%) | 5 (5%) | 0 | 1 (6%) | 0 |
| Pneumonitis | 8 (8%) | 2 (2%) | 0 | 1 (6%) | 1 (6%) | 0 |
| Abdominal pain | 7 (7%) | 1 (1%) | 0 | 0 | 0 | 0 |
| Anaemia | 7 (7%) | 1 (1%) | 0 | 0 | 0 | 0 |
| Adrenal insufficiency | 6 (6%) | 2 (2%) | 0 | 0 | 0 | 0 |
| Hypophysitis | 6 (6%) | 5 (5%) | 0 | 1 (6%) | 0 | 0 |
| Increased amylase | 6 (6%) | 7 (7%) | 0 | 0 | 0 | 0 |
| Increased blood bilirubin | 6 (6%) | 1 (1%) | 0 | 0 | 0 | 0 |
| Pruritic rash | 5 (5%) | 0 | 0 | 1 (6%) | 1 (6%) | 0 |
| Decreased lymphocyte count | 4 (4%) | 0 | 1 (1%) | 1 (6%) | 0 | 0 |
| Hyponatraemia | 4 (4%) | 1 (1%) | 1 (1%) | 0 | 0 | 0 |
| Increased blood creatinine | 3 (3%) | 0 | 0 | 2 (11%) | 0 | 0 |
| Influenza-like illness | 3 (3%) | 1 (1%) | 0 | 1 (6%) | 0 | 0 |
| Dehydration | 2 (2%) | 1 (1%) | 0 | 0 | 0 | 0 |
| Dermatitis acneiform | 2 (2%) | 0 | 0 | 2 (11%) | 0 | 0 |
| Hyperglycaemia | 2 (2%) | 1 (1%) | 0 | 0 | 0 | 0 |
| Macular rash | 2 (2%) | 1 (1%) | 0 | 1 (6%) | 0 | 0 |
| Stomatitis | 2 (2%) | 0 | 0 | 0 | 1 (6%) | 0 |
| Haemorrhage intracranial | 1 (1%) | 0 | 1 (1%) | 0 | 0 | 0 |
| Hypotension | 1 (1%) | 2 (2%) | 0 | 0 | 0 | 0 |
| Myositis | 1 (1%) | 1 (1%) | 0 | 0 | 0 | 0 |
| Nephritis | 1 (1%) | 0 | 0 | 0 | 1 (6%) | 0 |
| Pancreatitis | 1 (1%) | 1 (1%) | 0 | 0 | 0 | 0 |
| Acute kidney injury | 0 | 2 (2%) | 0 | 0 | 0 | 0 |
| Amnesia | 0 | 0 | 0 | 0 | 1 (6%) | 0 |
| Brain oedema | 0 | 0 | 2 (2%) | 0 | 0 | 0 |
| Colitis | 0 | 7 (7%) | 0 | 0 | 1 (6%) | 0 |
| Confusional state | 0 | 0 | 0 | 0 | 1 (6%) | 0 |
| Decreased blood phosphorus | 0 | 1 (1%) | 0 | 0 | 0 | 0 |
| Duodenitis | 0 | 0 | 1 (1%) | 0 | 0 | 0 |
| Dysarthria | 0 | 0 | 0 | 0 | 1 (6%) | 0 |
| Gastritis | 0 | 0 | 1 (1%) | 0 | 0 | 0 |
| Gastroenteritis | 0 | 0 | 0 | 0 | 1 (6%) | 0 |
| Hepatitis acute | 0 | 1 (1%) | 0 | 0 | 0 | 0 |
| Hypersensitivity | 0 | 0 | 0 | 0 | 1 (6%) | 0 |
| Immune-mediated hepatitis | 0 | 2 (2%) | 0 | 0 | 0 | 0 |
| Immune-mediated pancreatitis | 0 | 1 (1%) | 0 | 0 | 0 | 0 |
| Increased transaminases | 0 | 1 (1%) | 0 | 0 | 0 | 0 |
| Lymphocytic hypophysitis | 0 | 1 (1%) | 0 | 0 | 0 | 0 |
| Mucosal inflammation | 0 | 0 | 0 | 0 | 1 (6%) | 0 |
| Oral disorder | 0 | 0 | 0 | 0 | 1 (6%) | 0 |
| Partial seizures | 0 | 0 | 0 | 0 | 1 (6%) | 0 |
| Peripheral motor neuropathy | 0 | 1 (1%) | 0 | 0 | 0 | 0 |
| Rhabdomyolysis | 0 | 1 (1%) | 0 | 0 | 0 | 0 |
| Rash pustular | 1 (1%) | 0 | 0 | 0 | 1 (6%) | 0 |
| Syncope | 0 | 1 (1%) | 0 | 0 | 1 (6%) | 0 |
| Tumour pseudoprogression | 0 | 0 | 0 | 0 | 1 (6%) | 0 |
| Type 1 diabetes | 0 | 0 | 1 (1%) | 0 | 0 | 0 |
| Upper respiratory tract infection | 0 | 0 | 0 | 0 | 1 (6%) | 0 |
| Uveitis | 0 | 1 (1%) | 0 | 0 | 0 | 0 |
| Warm-type haemolytic anaemia | 0 | 0 | 1 (1%) | 0 | 0 | 0 |
Data are n (%).
Shown are treatment-related adverse events of any grade that occurred in at least 5% of patients or any treatment-related adverse events of grade 3 or 4. One patient in the asymptomatic cohort died from grade 5 myocarditis.
Discussion
At about 3 years of follow-up, the intracranial activity of the combination of nivolumab plus ipilimumab continued to show a high rate of durable responses in patients with MBM, with 85% of intracranial responses ongoing at the time of data cutoff in asymptomatic patients. Given that durable responses occurred in more than 50% of asymptomatic patients with unirradiated MBM, we confirmed that median progression-free survival and overall survival continued to not have been reached, with 3-year intracranial progression-free survival of 54·1% and overall survival of 71·9%. In patients with stable symptomatic MBM, the objective response rate was modest (16·7%); however, patients who did achieve a response maintained durable disease control, suggesting that interventions designed to relieve these patients of their symptoms or wean steroid therapy use might improve their responsiveness. In both cohorts, a high concordance rate was observed between investigator-assessed and BICR-assessed responses, suggesting the validity of the investigators’ assessments. The safety profile of nivolumab plus ipilimumab for both asymptomatic and symptomatic patients was similar to that of patients without MBM, with no new safety signals compared with the primary analysis.
Taking into consideration the caveats of cross-trial comparisons, the results reported here in patients with asymptomatic MBM are in agreement with those reported in the phase 2 Australian ABC trial10 in a cohort of 35 patients with asymptomatic MBM treated with nivolumab plus ipilimumab, who had an intracranial objective response rate of 51%, 3-year overall survival of 57%, and 5-year overall survival of 51%. In addition, a phase 3 study cohort of 27 patients responded to the combination with an intracranial objective response rate of 44·4% and a 4-year overall survival rate of 41%.15 Although patient selection is a general caveat of non-randomised, single-arm studies such as CheckMate 204, the characteristics of our patient population were similar to the two studies mentioned.10,15 In the CheckMate 204 study, 58 (57%) of 101 patients were able to enter the maintenance phase. The intracranial objective response rate of 53·5% and the 3-year overall survival rate of 71·9% in asymptomatic patients were similar to the results of CheckMate 067, in which patients with metastatic melanoma without brain metastases treated with nivolumab plus ipilimumab had an objective response rate of 58% and 3-year overall survival of 58%.23 Moreover, overall survival at 3 years in patients from CheckMate 204 with BRAF-mutated tumours and MBM (73%) was similar to that observed in patients from CheckMate 067 with BRAF-mutated tumours without MBM (68%), which established the combination as a standard for overall survival in these patients.23 Although the percentage of asymptomatic patients with BRAF mutations in CheckMate 204 was higher than that of CheckMate 067 (65% vs 32%),20 it is similar to other studies of patients with MBM10,15 and it is important clinically to highlight that responders in CheckMate 204 maintained durable responses similar to those of patients without MBM. In addition, as shown in a landmark analysis of patients who had survived to 6 months, some non-responders also had favourable overall survival, with 2-year rates of 62·3% compared with 91·8% in patients with a response, which could be a result of subsequent therapy use. Overall, non-responders received more subsequent systemic therapy than did responders (34% vs 6%). Alternatively, patients might be deriving clinical benefit that is not identifiable with the current imaging techniques and response criteria, highlighting the need to develop a better understanding of the patterns of radiographic response to immunotherapy and to evolve our response criteria to better identify patients benefiting from treatment.
Unsurprisingly, patients with symptomatic MBM, with or without corticosteroid use, had worse therapeutic outcomes compared with those who were asymptomatic and steroid-free. Many patients progressed rapidly and, in CheckMate 204, most patients received only one or two doses of the combination. Nevertheless, the few durable responses in our small cohort of patients suggests that the combination might be active in some patients with neurological symptoms, although steroid dependence at the start of immunotherapy appeared to be a determinant of unfavourable outcomes, with patients not on corticosteroids seeming to have improved progression-free and overall survival.14 Strategies that could allow these patients to discontinue corticosteroid use are needed to overcome the potent immune suppressive effects of corticosteroids when utilised at doses that can control intracranial oedema. Possible strategies to allow patients to discontinue corticosteroids prior to starting immunotherapy include treatment with agents that impact cerebral and peritumoural oedema (such as anti-VEGF antibodies prior to checkpoint inhibitor treatment in the trials NCT02681549 [bevacizumab] and NCT04955743 [lenvatinib]), deploying initial stereotactic radiotherapy (as in the ongoing ABC-X study [NCT03340129]), or surgical resection (as indicated in a retrospective study in patients with MBM).24 Other potential strategies to improve outcomes in these patients include combination with newer checkpoint inhibitors, such as LAG3 antibodies, which seem to be less toxic than CTLA-4 antibodies,25 or combinations or sequential dosing of BRAF-targeted therapy in BRAF-mutated tumours with checkpoint inhibitors. In addition, many of these same methods could be of benefit in patients with advanced brain metastases that cause mass effect even in the absence of significant oedema by rapidly reducing tumour volume.
BICR assessment is a US FDA recommendation when the primary endpoint is based on tumour assessment, and patient-level concordance rates for progression status of 71–76% between BICR and investigators are common in drug approvals.26 The current trend is moving towards sample-based BICR, although a full BICR is recommended in some situations,27 as was the case for CheckMate 204. The complications involved in analysing intracranial lesions, in addition to the possibility of checkpoint inhibitor-induced cerebral oedema, made a BICR analysis particularly important in this study. Moreover, the high rates of discordance in the BREAK-MB study9 of patients with MBM treated with dabrafenib plus trametinib, in which investigator-assessed and review committee-assessed results were discordant for almost half (42%) of the patients, increased the need for a BICR assessment here. In our study, the concordance rate for responders in both patients with asymptomatic and with symptomatic disease were high, and the Cohen’s kappa values were both in the range characterised as good (0·70 and 0·82),21 confirming the results obtained in the study. Overall, the results presented here in both asymptomatic and symptomatic patients suggest that if a patient with MBM has a response to nivolumab plus ipilimumab, the response is durable.
The small population in our symptomatic cohort limited the ability to draw firm conclusions from the data. In addition, the exclusion of patients with unstable neurological symptoms, recent seizures, or a higher corticosteroid dose, prevented gathering data on this higher-risk patient population, possibly reducing the generalisability of the data to patients with symptomatic disease in real-world clinical practice. Symptomatic patients with MBM have generally been excluded from clinical trials, limiting the relevant data available in the literature and making it more difficult to identify optimal treatment approaches for these patients. A recent systematic literature review and meta-analysis highlighted the continued unmet need in patients with MBM.28
These final 3-year results support the continued use of nivolumab 1 mg/kg plus ipilimumab 3 mg/kg as first-line standard of care for asymptomatic patients with MBM who are candidates for immunotherapy. In symptomatic patients whose overall outcomes are poorer than the asymptomatic patient group and the general melanoma population, there is a continued unmet need, as few patients had responses to nivolumab plus ipilimumab, although the responses were durable. Novel agents might be considered in triplet regimens or sequentially with nivolumab plus ipilimumab to further improve upon outcomes in this setting, as well as in combination with standard treatments such as stereotactic radiotherapy. Overall, the final results of the phase 2 CheckMate 204 study demonstrated that patients with MBM who achieve responses to nivolumab plus ipilimumab have durable responses leading to improved overall survival and that intracranial response interpretations were highly concordant between investigators and BICR.
Supplementary Material
Research in context.
Evidence before this study
We searched PubMed and congress abstracts focusing on the annual meetings of the American Society of Clinical Oncology, the European Society for Medical Oncology, and the Society for Melanoma Research for clinical study articles published in English up to June 1, 2021. We searched for studies evaluating checkpoint inhibitors and targeted therapy for patients with metastatic melanoma and brain metastases, using the search terms “melanoma brain metastases”, “checkpoint inhibitors”, “targeted therapy”, “nivolumab”, “pembrolizumab”, “ipilimumab”, “dabrafenib”, “trametinib”, “vemurafenib”, and “cobimetinib”. Early evidence of intracranial response from novel systemic agents had indicated that targeted therapy (single-agent dabrafenib, followed by combination dabrafenib plus trametinib or combination vemurafenib plus cobimetinib) induced objective response rates of 39–58% in patients with a BRAF mutation, including in patients with symptomatic brain metastases or those on corticosteroids. The responses were shorter in duration than extracranial responses, with reduced intracranial progression-free survival. Single-agent checkpoint inhibitor therapy also induced durable intracranial responses, with lower rates than in patients without melanoma brain metastases (MBM; ~20% for patients with intracranial disease vs 40% for patients without MBM), and responses of about 5% in patients on steroids. Combination therapy with nivolumab plus ipilimumab increases the objective response rate to 44–54%, with overall survival of more than 30 months at a minimum follow-up of 43 months. A publication and a congress report from the NIBIT-M2 and ABC studies reported long-term follow-up and confirmed durable responses to combination checkpoint inhibitors (ipilimumab plus nivolumab) beyond 3 years, with small sample sizes (27 and 35 patients, respectively) and without symptomatic patients or those on steroids treated with the combination (the ABC study did include a cohort of ten patients with neurological symptoms treated with nivolumab monotherapy). Finally, only two of the reported studies had a blinded independent central review (BICR; BREAK-MB and COMBI-MB); BREAK-MB presented patient-level data, had discordance in the measurement of intracranial response in 42% of cases, and required an adjudication committee.
Added value of this study
Our study (CheckMate 204) has, to our knowledge, the largest sample size of patients with MBM treated with combination nivolumab plus ipilimumab and continues to show durable responses in patients with asymptomatic brain metastases and high overall survival rates at 3 years. The study also suggested for the first time that combination immunotherapy has modest response rates in symptomatic patients, but that those with responses could derive long-term benefit.
Moreover, responses were largely concordant between investigator and blinded independent review analyses, even with the inherent complications involved in analysing brain metastases, possibly exacerbated here with checkpoint inhibitor-induced oedema.
Implications of all the available evidence
Analysis of intracranial disease response using modified Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 is reproducible between investigator assessment and BICR assessment, validating the results obtained in CheckMate 204. The results indicate that nivolumab plus ipilimumab combination can be considered for the first-line treatment of most patients with asymptomatic MBM who are candidates for immunotherapy, with long-term survival benefits. Some patients with symptomatic MBM can also derive long-term benefit, but there is a need for new approaches for these patients. Possible approaches include local therapies, such as stereotactic radiosurgery or added systemic treatment in combination with checkpoint inhibitors, with the intent of resolving symptoms and dependence on steroids before checkpoint inhibitor treatment.
Acknowledgments
This study was supported by Bristol Myers Squibb. We thank the patients and investigators who participated in the CheckMate 204 trial. This research was supported in part through the US National Institutes of Health/National Cancer Institute Cancer Center Support Grant P30 CA008748 (to MAP) and P30 CA016056 (to IP). We acknowledge Ono Pharmaceutical Company (Osaka, Japan) for contributions to nivolumab development and Dako, an Agilent Technologies company (Santa Clara, CA, USA) for collaborative development of the PD-L1 immunohistochemistry 28–8 pharmDx assay. We acknowledge David Leung from Bristol Myers Squibb for contributions to the design of the study and blinded independent central review (BICR), and for support of the BICR data acquisition and analyses. Professional medical writing and editorial assistance were provided by Melissa Kirk and Michele Salernitano at Ashfield MedComms, an Ashfield Health Company, funded by Bristol Myers Squibb.
Declaration of interests
HAT worked in a consulting/advisory role for Array BioPharma, Bristol Myers Squibb (BMS), Genentech/Roche, Merck, and Novartis; participated in a scientific advisory board for Kayopharm; received research/grant support from BMS, Celgene, Genentech/Roche, GlaxoSmithKline (GSK), and Merck; and received honoraria from Eisai. PAF worked in a consulting role for AbbVie, Boehringer Ingelheim, NCI Neuro-Oncology Branch Peer Review, Novellus, Physical Sciences Oncology Network, Tocagen, and Ziopharm; received honoraria from BTG, NCRI, Tocagen, and Ziopharm; worked in an advisory role for Bayer, Inovio, Novocure, and BTG; and received research/grant support from CDMRP, NIH/NCI, Department of Defense, Pfizer, State of Florida Bankhead Coley, and Moffitt Center of Excellence Celgene Project. FSH worked in a consulting role for BMS, Corner Therapeutics, Eisai, EMD Serono, Genentech/Roche, Gossamer, Idera, Kairos, Merck, Novartis, Psioxus Therapeutics, Pieris Pharmaceutical, Takeda, and Sanofi; worked in an advisory role for 7 Hills Pharma, Aduro, Apricity, Bicara, Checkpoint Therapeutics, Pionyr, and Torque; received research/grant support from BMS and Novartis; received royalties from BMS and Novartis; and holds equity in Apricity. APA worked in an advisory role for Array BioPharma, OncoSec Medical, and Regeneron; received research/grant support from Acerta, Amgen, AstraZeneca, BMS, Dynavax, Genentech, Idera, Incyte, ISA, LOXO, Merck, Novartis, OncoSec Medical, Regeneron, Sensei, and Tessa; received travel/accommodations/expenses support from OncoSec Medical; and holds stock in OncoSec Medical and Valitor Biosciences. OH worked in a consulting role for and received research/grant support from Aduro, Akeso, Amgen, Beigene, Bioatla, BMS, Genentech, GSK, Immunocore, Idera, Incyte, Janssen, Merck, NextCure, Novartis, Pfizer, Sanofi Regeneron, Seagen, Tempus, and Zelluna; received research/grant support from BMS; and served as a speaker for BMS, Novartis, Pfizer, and Sanofi Regeneron. CDL received research/grant support from Novartis and Merck; worked in an advisory role for BMS and Immunocore; and received travel/accommodations/expenses support from BMS and Immunocore. SJM received research/grant support from Amgen, Merck, and Syndax Pharmaceuticals; worked in a consulting role for iTeos Therapeutics and EMD Serono; and participated in a Data Safety Monitoring board for IQVIA. MBA worked in a consulting role for Adagene, Agenus, AstraZeneca, Calithera, Exelixis, Idera, Immunocore, Iovance, Neoleukin, Sanofi, SeaGen, and Takeda; and worked in an advisory role for Apexigen, Aveo, BMS, Eisai, Elpis, Genentech/Roche, Leads Biopharma, Merck, Novartis, Pfizer, PACT, Pyxis Oncology, and Werewolf Therapeutics. KL received research/grant support from BMS, Merck, Roche/Genentech, Pfizer, and Regeneron; and worked in a consulting role for Merck, Roche/Genentech, and Pfizer. MAP worked in a consulting/advisory role for Aduro, BMS, Eisai, Incyte, Merck, NewLink Genetics, Novartis, and Pfizer; received research/grant support from Array BioPharma, AstraZeneca, BMS, Infinity, Merck, Novartis, and RGenix; and received honoraria from BMS and Merck. SJ worked in a consulting/advisory role for Array Biopharma, BMS, EMD Sorono, Genentech, Novartis, Sanofi, Sun Biopharma, and Pfizer. NIK worked in an advisory role for Array BioPharma, BMS, EMD Serono, Genentech, HUYA Bioscience International, Immunocore, Merck, Regeneron, and Jounce; received research/grant support from Amgen, BMS, Celgene, GSK, HUYA Bioscience International, Merck, Novartis, Replimmune, and Regeneron; received honoraria from BMS and Sanofi; holds stock in Amarin Corporation, Bellicum Pharmaceuticals, Mazor Robotics, and Asensus Surgical (formerly TransEnterix); participated in a Data Safety Monitoring board for AstraZeneca and Incyte; and participated in the Scientific Review Committee for NCCN (from Pfizer) and study steering committees for BMS, Regeneron, and Nektar. ACP worked in a consulting/advisory role for BMS, Merck, Regeneron, and Sanofi/Regeneron; received research/grant support from BMS, Merck, Regeneron, and Replimune; and worked on a speaker’s bureau for BMS. MSE participated in a Data Safety Monitoring board for BMS; and holds stock or stock options in BMS. DAR worked in a consulting/advisory role for AbbVie, Advantagene, Agenus, Amgen, Bayer, BMS, Boston Biomedical, Celldex, DelMar, EMD Serono, Genentech/Roche, Inovio, Medicenna, Merck, Merck KGaA, Monteris, Oncorus, Oxigene, Novocure, Regeneron, Stemline, and Taiho; and received research/grant support from Tragara, Acerta Pharmaceuticals, Agenus, Celldex, EMD Serono, Incyte, Inovio, Midatech, and Omniox. RK worked in a consulting/advisory role for Array BioPharma, BMS, Immunocore, Merck, Novartis, Pfizer, and Regeneron; received research/grant support from BMS, Merck, and Regeneron; received honoraria from Array BioPharma and BMS; and received travel/accommodations/expenses support from BMS. AT worked in a consulting/advisory role for Array Biopharma, BioNTech, BMS, Clinigen, EMD Serono, Genentech/Roche, Immunocore, Merck, NewLink Genetics, Novartis, Partner Therapeutics, Pfizer, and Sanofi-Genzyme/Regeneron; and received research/grant support from BMS, Genentech/Roche, OncoSec Medical, Merck, Sanjofi-Genzyme, Regeneron, Clinigen, and CheckMate. CC received research/grant support from Siemens Healthineers and Raysearch Laboratories; and received honorarium from Elekta. CR and PD are employees of BMS and hold BMS stock or stock options. MA is an employee of BMS. IP worked in a consulting role for Amgen, Merck, and Nouscom. KAM worked in a consulting/advisory role for ImaginAb, Oncosec, Werewolf, Xilio, and Tentarix; participated in a Data Safety Monitoring board for CheckMate Pharmaceuticals; and received research/grant support from ImaginAb. RPT and JG report no competing interests.
Footnotes
Data sharing
BMS policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html. Data are generally available 2 years after completion of the study and will be made available to qualified researchers who submit an in-scope proposal approved by the Independent Review Committee, with available information dependent upon the individual request. The deidentified and anonymised datasets may be accessed within a secured portal if the proposal is approved and upon execution of the agreement.
Contributor Information
Prof Hussein A Tawbi, University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Prof Peter A Forsyth, Department of Neuro-Oncology, Moffitt Cancer Center & Research Institute, Tampa, FL, USA.
Prof F Stephen Hodi, Dana-Farber Cancer Institute, Boston, MA, USA.
Alain P Algazi, Melanoma Center, University of California—San Francisco, San Francisco, CA, USA.
Omid Hamid, Melanoma Center, The Angeles Clinic and Research Institute, Los Angeles, CA, USA.
Prof Christopher D Lao, Department of Dermatology, University of Michigan, Ann Arbor, MI, USA.
Stergios J Moschos, Division of Hematology & Oncology, The University of North Carolina Lineberger Comprehensive Cancer Center, Chapel Hill, NC, USA.
Prof Michael B Atkins, Department of Medical Oncology, Georgetown-Lombardi Comprehensive Cancer Center, Washington DC, USA.
Karl Lewis, Department of Medical Oncology, University of Colorado Comprehensive Cancer Center, Aurora, CO, USA.
Michael A Postow, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY, USA.
Reena P Thomas, Department of Neurology, Stanford University Cancer Center, Stanford, CA, USA.
Prof John Glaspy, Jonsson Comprehensive Cancer Center, University of California, Los Angeles, CA, USA.
Sekwon Jang, Inova Schar Cancer Institute, Fairfax, VA, USA.
Nikhil I Khushalani, Department of Cutaneous Oncology, H Lee Moffitt Cancer Center, Tampa, FL USA.
Anna C Pavlick, Department of Medical Oncology, Weill Cornell Medicine, New York, NY, USA.
Prof Marc S Ernstoff, Department of Immuno-Oncology, Division of Cancer Treatment and Diagnosis, National Cancer Institute at the National Institutes of Health, Rockville, MD, USA.
David A Reardon, Center for Neuro-Oncology, Dana-Farber Cancer Institute, Boston, MA, USA.
Ragini Kudchadkar, Department of Hematology and Medical Oncology, Winship Cancer Institute, Emory University School of Medicine, Atlanta, GA, USA.
Prof Ahmad Tarhini, Departments of Cutaneous Oncology and Immunology, Moffitt Cancer Center & Research Institute, Tampa, FL, USA.
Caroline Chung, University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Corey Ritchings, Bristol Myers Squibb, Princeton, NJ, USA.
Piyush Durani, Bristol Myers Squibb, Princeton, NJ, USA.
Margarita Askelson, Bristol Myers Squibb, Princeton, NJ, USA.
Prof Igor Puzanov, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA.
Prof Kim A Margolin, Department of Medical Oncology, City of Hope, Duarte, CA, USA.
References
- 1.Davies MA, Liu P, McIntyre S, et al. Prognostic factors for survival in melanoma patients with brain metastases. Cancer 2011; 117: 1687–96. [DOI] [PubMed] [Google Scholar]
- 2.Cagney DN, Martin AM, Catalano PJ, et al. Incidence and prognosis of patients with brain metastases at diagnosis of systemic malignancy: a population-based study. Neuro-oncol 2017; 19: 1511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fidler IJ, Schackert G, Zhang RD, Radinsky R, Fujimaki T. The biology of melanoma brain metastasis. Cancer Metastasis Rev 1999; 18: 387–400. [DOI] [PubMed] [Google Scholar]
- 4.Sperduto PW, Kased N, Roberge D, et al. Summary report on the graded prognostic assessment: an accurate and facile diagnosis-specific tool to estimate survival for patients with brain metastases. J Clin Oncol 2012; 30: 419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berghoff AS, Preusser M. Target therapies for melanoma brain metastases. Curr Treat Options Neurol 2017; 19: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutzmer R, Vordermark D, Hassel JC, et al. Melanoma brain metastases - interdisciplinary management recommendations 2020. Cancer Treat Rev 2020; 89: 102083. [DOI] [PubMed] [Google Scholar]
- 7.Margolin K, Ernstoff MS, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol 2012; 13: 459–65. [DOI] [PubMed] [Google Scholar]
- 8.Kluger HM, Chiang V, Mahajan A, et al. Long-term survival of patients with melanoma with active brain metastases treated with pembrolizumab on a phase II trial. J Clin Oncol 2019; 37: 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long GV, Atkinson V, Lo S, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicentre randomised phase 2 study. Lancet Oncol 2018; 19: 672–81. [DOI] [PubMed] [Google Scholar]
- 10.Long GV, Atkinson V, Lo S, et al. Five-year overall survival from Anti-PD1 Brain Collaboration (ABC study): randomised phase 2 study of nivolumab or nivolumab+ipilimumab in patients with melanoma brain metastases. American Society of Clinical Oncology (ASCO) Congress; June 8–12, 2021. (abstr 9508).
- 11.Davies MA, Saiag P, Robert C, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol 2017; 18: 863–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rulli E, Legramandi L, Salvati L, Mandala M. The impact of targeted therapies and immunotherapy in melanoma brain metastases: a systematic review and meta-analysis. Cancer 2019; 125: 3776–89. [DOI] [PubMed] [Google Scholar]
- 13.Tawbi HA, Forsyth PA, Algazi A, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med 2018; 379: 722–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tawbi HA, Forsyth PA, Hodi FS, et al. Safety and efficacy of the combination of nivolumab plus ipilimumab in patients with melanoma and asymptomatic or symptomatic brain metastases (CheckMate 204). Neuro-oncol 2021; noab094. [DOI] [PMC free article] [PubMed]
- 15.Di Giacomo AM, Chiarion-Sileni V, Del Vecchio M, et al. Primary analysis and 4-year follow-up of the phase III NIBIT-M2 trial in melanoma patients with brain metastases. Clin Cancer Res 2021; 27: 4737–45. [DOI] [PubMed] [Google Scholar]
- 16.US Food and Drug Administration. Clinical trial endpoints for the approval of cancer drugs and biologics: guidance for industry. December, 2018. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-trial-endpoints-approval-cancer-drugs-and-biologics (accessed July 8, 2021).
- 17.Food US and Administration Drug. Guidance document: developing medical imaging drug and biological products, part 3: design, analysis, and interpretation of clinical studies. June, 2004. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/developing-medical-imaging-drug-and-biological-products-part-3-design-analysis-and-interpretation (accessed July 8, 2021).
- 18.Lin NU, Lee EQ, Aoyama H, et al. Response assessment criteria for brain metastases: proposal from the RANO group. Lancet Oncol 2015; 16: e270–78. [DOI] [PubMed] [Google Scholar]
- 19.Food US and Administration Drug. Evaluating cancer drugs in patients with central nervous system metastases. Guidance for industry. July, 2021. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/evaluating-cancer-drugs-patients-central-nervous-system-metastases (accessed July 8, 2021).
- 20.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015; 373: 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altman DG. Practical statistics for medical research. London: Chapman and Hall, 1991. [Google Scholar]
- 22.Johnson DB, Balko JM, Compton ML, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med 2016; 375: 1749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017; 377: 1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alvarez-Breckenridge C, Giobbie-Hurder A, Gill CM, et al. Upfront surgical resection of melanoma brain metastases provides a bridge toward immunotherapy-mediated systemic control. Oncologist 2019; 24: 671–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lipson EJ, Tawbi HA, Schadendorf D, et al. Relatlimb plus nivolumab (NIVO) versus NIVO in first-line advanced melanoma: primary phase III results from RELATIVITY-047 (CA224–047). American Society of Clinical Oncology (ASCO) Congress; June 8–12, 2021 (abstr 9503).
- 26.Ford R, Schwartz L, Dancey J, et al. Lessons learned from independent central review. Eur J Cancer 2009; 45: 268–74. [DOI] [PubMed] [Google Scholar]
- 27.Amit O, Mannino F, Stone AM, et al. Blinded independent central review of progression in cancer clinical trials: results from a meta-analysis. Eur J Cancer 2011; 47: 1772–78. [DOI] [PubMed] [Google Scholar]
- 28.Tawbi H, Long GV, Meyer N, et al. Treatment outcomes in patients with melanoma brain metastases undergoing systemic therapy: a systematic literature review and meta-analysis. American Society of Clinical Oncology (ASCO) Congress; June 8–12, 2021. (abstr 9561).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.