

Summary
Background
Peroxisome proliferator‐activated receptors (PPARs) are ligand‐activated transcription factors known to regulate glucose and fatty acid metabolism, inflammation, endothelial function and fibrosis. PPAR isoforms have been extensively studied in metabolic diseases, including type 2 diabetes and cardiovascular diseases. Recent data extend the key role of PPARs to liver diseases coursing with vascular dysfunction, including nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH).
Aim
This review summarises and discusses the pathobiological role of PPARs in cardiovascular diseases with a special focus on their impact and therapeutic potential in NAFLD and NASH.
Results and Conclusions
PPARs may be attractive for the treatment of NASH due to their liver‐specific effects but also because of their efficacy in improving cardiovascular outcomes, which may later impact liver disease. Assessment of cardiovascular disease in the context of NASH trials is, therefore, of the utmost importance, both from a safety and efficacy perspective.
PPARs are nuclear receptors that are involved in many vascular and metabolical processes, which may be common in the development and progression of NASH. In this article, we review the possible roles that PPARs may have on NASH in order to propose new therapeutic targets for this disease.

1. BIOLOGY OF PEROXISOME PROLIFERATOR‐ACTIVATED RECEPTORS
Peroxisome proliferator‐activated receptors (PPARs) are a subfamily of nuclear receptors involved in metabolism and inflammation. There are three isotypes of PPARs (PPARα, PPARβ/δ and PPARγ) which are expressed in different cell types and tissues and have different functions (Figure 1).
FIGURE 1.
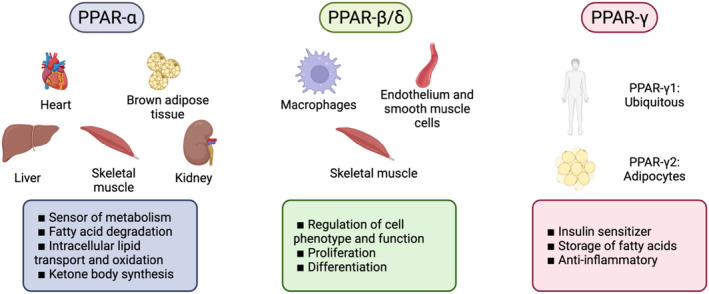
PPARs expression by tissue and their main functions
PPARα is mainly expressed in tissues with high metabolic rates (including the liver, skeletal muscle, heart, kidney or neural and brown adipose tissue). Natural ligands of PPARα are mostly products of lipid catabolism, lipogenesis or oxidation, as well as eicosanoid derivates. Therefore, PPARα can act as a sensor of metabolism‐regulating processes such as fatty acid degradation, intracellular lipid transport and oxidation or ketone body synthesis. 1 In rodents, it also regulates glucose metabolism, although these effects are less obvious in human. Global PPARα knockout in mice lead to overweight, whilst specific deletion in hepatocytes induced hepatic steatosis in ageing animals and had additional systemic effects on lipid homeostasis. 2
PPARβ/δ is probably the least studied of the PPARs. It is mainly expressed in the skeletal muscle, stimulating fatty acid oxidation, although it may also participate in cell proliferation and differentiation. This isotype of PPAR is also expressed in endothelial cells, smooth muscle cells and macrophages and may be involved in the regulation of their phenotype and function. 3
Finally, PPARγ is found ubiquitously, although one of its splicing isoforms (γ2) is exclusively found in adipocytes. In contrast to the other PPAR family members, which promote the catabolism of fatty acids, PPARγ induces the storage of fatty acids by acting as a strong insulin sensitizer. In addition, PPARγ plays an important anti‐inflammatory role. 4 In adipocytes, PPARγ regulates glucose metabolism, lipogenesis and adipocyte differentiation, whilst promoting the production of adiponectin. 5 Indeed, rare monogenic mutations in PPARγ in humans may lead to severe insulin resistance, partial lipodystrophy, type 2 diabetes mellitus (T2DM) and hypertension. 6
The expression of the different PPAR isoforms across tissues may vary in certain pathologic conditions. PPARs have been extensively studied in metabolic diseases, including T2DM, cardiovascular diseases and non‐alcoholic steatohepatitis (NASH). In the last years, NASH has become one of the major etiologies for chronic liver disease worldwide and it is an important risk factor for hepatocellular carcinoma. NASH is defined by hepatic steatosis associated with lobular inflammation and hepatocyte ballooning with or without liver fibrosis. 7 Severity of NASH in humans inversely correlates with hepatic PPARα and PPARγ expression, 8 suggesting that advanced patients may be less responsive to endogenous PPAR ligands. Therefore, pharmacologic modulation of PPAR expression in different pathologic conditions could be a rational approach to regulate their target pathways. However, as transcription factors, the activity of PPARs may be regulated at other multiple levels.
As members of the nuclear receptor superfamily, all isotypes of PPARs share a similar structure and function. 1 In order to regulate gene expression, PPARs usually require heterodimerization with the retinoid X receptor (RXR) in order to bind to the peroxisome proliferator response element (PPRE) of the DNA. In the unligated state, PPARs are bound to co‐repressors (such as nuclear receptor co‐repressor 1, NCoR1), which possess the histone‐deacetylase activity and, therefore, prevent transcription. 9 , 10 However, in the presence of ligands, co‐activators can modulate PPAR activity in various ways. On the one hand, they may have intrinsic histone acetylase activity (such as steroid receptor co‐activators, SRC or CBP/p300), directly affecting transcription. On the other hand, other co‐activators (such as the PPARγ co‐activator‐1a, PGC‐1a) may promote the recruitment of additional proteins with such transcriptional activity. 11 Additionally, PPAR activity may be further regulated through post‐translational modifications, such as phosphorylation at different sites (mediated by different protein kinases including MAPK, PKA or PKC) or SUMOylation, which can occur as a result of negative feedback induced by products of their target metabolic pathways. 10 , 12
Alternatively, PPARs may act as transrepressors of alternative molecular pathways. Indeed, PPARα may dimerize with other transcription factors such as nuclear factor κB (NF‐κB), activator protein‐1 (AP‐1) and signal transducers and activators of transcription (STATs), preventing them from interacting with the DNA and repressing their transcriptional activity. 13 This mechanism of transrepression has been associated with reduced expression of interleukin 6 (IL‐6) in vascular endothelial cells. 14 PPARα has also been reported to dimerize with Sirtuin 1 (Sirt1), thus competing with oestrogen‐related receptors and inhibiting their target genes. 15 In this regard, some studies have reported the existence of a truncated form of PPARα in various tissues that is unable to interact with the DNA, reinforcing its regulatory role independent of transcription. 16
2. PPARS AS RHEOSTATS OF LIVER VASCULAR CHANGES OCCURRING IN NASH
PPARs regulate main molecular pathways including metabolism, inflammation, fibrosis, and cell proliferation and differentiation, all of which play important roles in liver diseases' pathophysiology. For this reason, PPARs represent a promising therapeutic target for NASH. Considering that hepatic microvascular dysfunction is key in the development and aggravation of NASH, 17 and that its clinical consequences (including portal hypertension) have high morbidity and mortality, the development of new therapeutics targeting vascular dysfunction in NASH represents an unmet clinical need. Below we summarise the current knowledge regarding PPARs and vascular functionality, which indeed may be relevant for the treatment of the hepatic microvascular dysfunction occurring in NASH (Figure 2).
FIGURE 2.
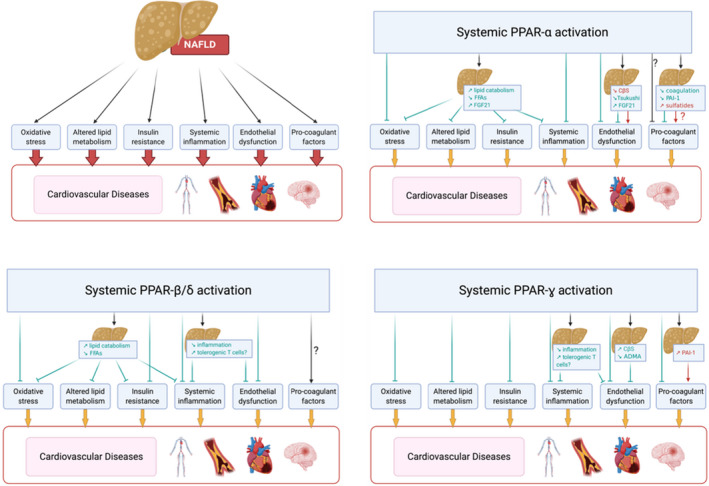
Schematic representation of potential mechanisms mediating increased CVD in NAFLD and how systemic activation of different PPARs could influence CVD progression.
2.1. PPARs and vascular relaxation
The hepatic microvasculature plays a crucial role in health and chronic liver disease. In the normal liver, liver sinusoidal endothelial cells (LSECs) are in tight communication with hepatic stellate cells (HSCs) in order to regulate intrahepatic blood flow. In this healthy scenario, LSECs synthesise nitric oxide (NO) and other vasodilators that are detected by HSCs, which subsequently induces vasodilation. However, during chronic liver disease, LSECs lose their specialised phenotype and their ability to produce NO decreases, which altogether with the reduced sensitivity of HSCs to vasodilators leads to microvascular dysfunction. 18 Activated HSC in diseased liver responds by cell contraction and proliferation, which further affects intrahepatic blood flow. These vascular abnormalities are known as the dynamic component of the increased intrahepatic vascular resistance in liver disease, being the primary factor in the development of portal hypertension. 19
All three subtypes of PPARs participate in the synthesis of NO by endothelial cells and prevent vascular dysfunction in diabetic animals 20 (Figure 3). Vascular dysfunction is indeed commonly associated with a pro‐oxidant phenotype of endothelial cells. 21 , 22 Studies using PPARβ/δ agonists describe the increased expression of the anti‐oxidant enzymes heme‐oxygenase 1 (HO‐1), superoxide dismutase (SOD), catalase and thioredoxin, leading to a reduction in reactive oxygen species (ROS). 3 , 23 PPARα activation showed similar effects, specifically in the liver microvasculature of cirrhotic rats. 24
FIGURE 3.
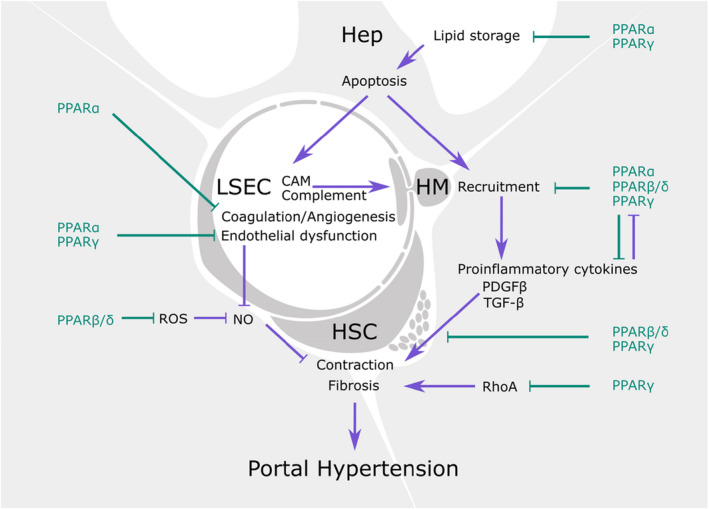
Potential effects and underlying mechanisms of PPAR agonists on liver vascular dysfunction. CAM, cell adhesion molecules; hep, hepatocyte; HM, hepatic macrophages; HSC, hepatic stellate cell; LSEC, liver sinusoidal endothelial cell.
On the contrary, specific PPARγ deletion in vascular smooth muscle cells led to exacerbated response to angiotensin‐II and vascular dysfunction. 25 These effects of PPARγ interference in smooth muscle cells may be due to impaired degradation of the small GTPase RhoA (involved in cytoskeleton contraction), leading to RhoA accumulation and an over‐contractile phenotype. 26 Furthermore, PPARα and PPARγ would prevent smooth muscle cell proliferation and vascular remodelling by blocking the PDGF and TGF‐β pathways, suggesting that these could be additional PPAR‐related mechanisms that regulate vascular tone. 27
Specifically, in the hepatic milieu, the pan‐PPAR agonist lanifibranor displayed beneficial effects on hepatocyte phenotype in an in vitro model of steatosis only when cocultured with non‐parenchymal cells, suggesting that PPAR signalling in the liver microvasculature could have paracrine effects on hepatocyte function in the context of NASH. 28 In fact, in a recent study, pan‐PPAR activation led to improvement in vascular dysfunction and portal hypertension in two pre‐clinical models of chronic liver disease, which was accompanied by ameliorated hepatic function and reduced inflammation. 29 Observations in pre‐clinical rodent models were further validated in primary liver cells isolated from human cirrhotic livers, altogether reinforcing the vasoprotective benefits of PPAR activation in chronic liver disease (Figure 3).
In agreement with these last observations, studies performed in pre‐clinical models of vascular diseases such as pulmonary arterial hypertension and cardiac fibrosis, which share common pathways with chronic liver disease (endothelial dysfunction, imbalance in vasoconstrictors/dilators, activation and proliferation of smooth muscle cells, inflammation, and vascular remodelling), demonstrated that PPARγ activation reduces arterial hypertension and improves NO‐dependent endothelial vasodilation and fibrosis. 30 This is in accordance with the observation that humans with dominant‐negative PPARγ mutations are typically hypertensive. 31
Although this review focuses on the intrahepatic vasculature as the main target for the treatment of NASH, it is worth noting that PPAR modulation could also have beneficial extra‐hepatic effects that indirectly regulate chronic liver disease and its complications. Indeed, in the partial portal vein ligation model (a model of non‐cirrhotic portal hypertension), treatment with pioglitazone (PPARγ agonist) improved portosystemic shunting due to amelioration of splanchnic inflammation and angiogenesis. 32 In pre‐clinical models of chronic liver disease, PPAR agonists also showed benefits, as demonstrated by improvement in splanchnic vasoactivity and neoangiogenesis. 33
2.2. PPARs and inflammation
The progression from non‐alcoholic fatty liver (NAFL) to NASH is characterised by hepatic inflammation, usually as a consequence of hepatocellular damage. In this scenario, LSECs express cell adhesion molecules, and immune cells are recruited to the liver, acquiring a proinflammatory phenotype and triggering fibrosis. 34
PPARα deficiency is known to promote NASH features in high‐fat diet (HFD)‐fed animals, such as increased triglyceride accumulation, hepatocyte ballooning, hepatic inflammation and elevated transaminases, whilst its activation protects these animals from NASH due to anti‐inflammatory effects in addition to increased lipid catabolism. 35 In this regard, both the pan‐PPAR agonist lanifibranor and the PPARα agonist fenofibrate reduced the recruitment of circulating macrophages in animal models of NAFLD‐NASH whilst having no effects on Kupffer cells (KC, the resident liver macrophages). 28 Furthermore, PPAR agonists seem to regulate the inflammatory profile of leukocytes, whilst PPARβ/δ regulates the pro‐inflammatory profile of macrophages, 28 suggesting profound changes in gene expression. In this regard, transcriptomic studies in mice and primates revealed that PPARα activation leads to a potent downregulation of the complement cascade 36 and upregulation of anti‐inflammatory mediators such as IL‐1ra and IκB. 37
In addition to direct activation of transcription, and as indicated above, PPARs may regulate the transcriptional activity of other transcription factors, such as NFκB, through direct protein–protein interactions. 38 On the contrary, IL‐1b treatment was shown to reduce the expression of PPARα, 39 which suggests that inflammation per se could induce negative feedback on PPAR anti‐inflammatory pathways. This is in accordance with the observations of reduced PPARα and PPARγ expression in the liver of NASH patients 40 and should be considered when using PPAR agonists.
2.3. PPARs and coagulation
Coagulation disorders constitute a key factor in the pathophysiology of liver diseases. Indeed, the hypercoagulable state of the cirrhotic liver actively contributes to disease progression, and its amelioration using anti‐coagulants improves chronic liver disease complications and patients' prognosis. 41 , 42 In NASH, in particular, there is a prothrombotic state derived both from the hepatic alterations of NASH and obesity and the metabolic syndrome, with some data supporting that these prothrombotic alterations may be relevant for the progression of the disease. 43 It has been shown, in humans, that treatment with fibrates (PPARα agonists) diminishes plasminogen concentration. 44 Transcriptomic studies performed in mouse and primate livers showed that the anti‐coagulation effects of fibrates are also observed at the mRNA level. Indeed, the coagulation pathway was one of the most downregulated ones in these studies, including the expression of fibrinogen, plasma kallikrein B and several coagulation factors. 36 , 45 Additionally, fibrates are known to potentiate the effect of the anticoagulant drug warfarin and increase bleeding risk when taken together. Therefore, these observations encourage further studies on the role of PPARs and their anticoagulation effects in NASH and the contribution of these effects to the modulation of disease progression.
2.4. PPARs in HSCs activation and fibrogenesis
Upon chronic liver damage, HSCs get activated and become the main source of hepatic extracellular matrix components. This activation may occur due to both increased inflammation or altered communication with LSECs. 46 As seen above, PPAR agonists may prevent the recruitment and activation of immune cells and confer a vasoprotective phenotype to endothelial cells. Therefore, most studies assessing PPAR agonists in NASH in vivo showed improved microvascular function or inflammation, accompanied by a reduction/prevention of fibrosis and liver stiffness. 29 , 35 , 47 , 48 , 49 , 50 , 51 , 52 This is also observed in non‐hepatic vascular diseases such as cardiac ischemia and reperfusion. 27
PPARs could also have a direct effect on HSCs. Indeed, PPARγ is normally expressed in HSCs, but its expression is reduced during their activation. Treatment of isolated primary HSCs with pan‐PPAR or PPARγ agonists prevents spontaneous or TGFβ‐induced in vitro activation, 53 and even promotes the de‐activation of HSCs isolated from human cirrhotic livers. 29 The mechanisms by which PPAR agonists achieve this include inhibition of proliferation, expression of extracellular matrix proteins, inhibition of senescence and even induction of hepatic autophagy, which has recently been described to play a complex role in liver fibrosis and NAFLD. 3 , 35
2.5. PPARs in NAFLD clinical trials
Given the aforementioned complex and key role of PPARs in mechanisms that are highly relevant for NAFLD pathophysiology and based on pre‐clinical evidence as mentioned, several PPAR agonistic drugs have been explored for their clinical utility. Fibrates did not result in histological improvement. The dual PPARα/δ agonist elafibranor showed an impact on steatohepatitis in patients with higher degrees of disease activity in phase 2, 54 but did not reach the endpoint of NASH resolution in Phase 3 (Harrison et al, oral communication). Pioglitazone clearly induces resolution of NASH after 18 months of treatment or longer, with a trend of improving also fibrosis. 55 Lanifibranor is to date the only compound that achieved both the endpoints of NASH resolution and fibrosis improvement after 24 weeks of treatment. 54 A detailed discussion of the reasons behind these results is beyond the scope of this review. Briefly, steatohepatitis is considered the driver of fibrogenesis and is itself driven by upstream metabolic derangements, including adipose tissue dysfunction. Despite their important role, tackling just the intrahepatic mechanisms, leaving extrahepatic drivers of disease untouched, will presumably not be powerful enough to achieve the high barrier endpoints of NASH resolution and/or fibrosis regression that are required by the regulatory authorities. Conversely, tackling the upstream drivers together with the intrahepatic mechanisms of inflammation and fibrogenesis has conceptually a higher likelihood of achieving positive results, seen to some extent with pioglitazone and even more convincingly with the panPPAR agonism of lanifibranor.
3. ROLE OF PPARS FOR THE INCREASE IN CARDIOVASCULAR DISEASE IN NAFLD
Cardiovascular diseases (CVD) are the first cause of death in patients with NAFLD, accounting for approximately 40% of deaths in total. There is a strong association between NAFLD and common cardiovascular risk factors and, interestingly, recent epidemiological data suggest that NAFLD is an independent CVD risk factor. 56 , 57 Nevertheless, this question is also subject to some controversy since some reports suggest no association between NAFLD and increased risk of stroke nor myocardial infarction, 58 or increased death from cardiovascular events. 59 However, this lack of association could be due to study limitations, showing NAFLD in only 0.5% of the population instead of the expected 23%. This question has been approached in detailed recent reviews. 60
With the data available today, which is mostly of epidemiological nature, it is difficult to entangle the effects due to extra‐hepatic CVD risk factors associated with NAFLD and the effects due to NAFLD in itself. There are several proposed mechanisms by which NAFLD progression could influence cardiovascular risk (Figure 4), as reviewed elsewhere. 61
FIGURE 4.
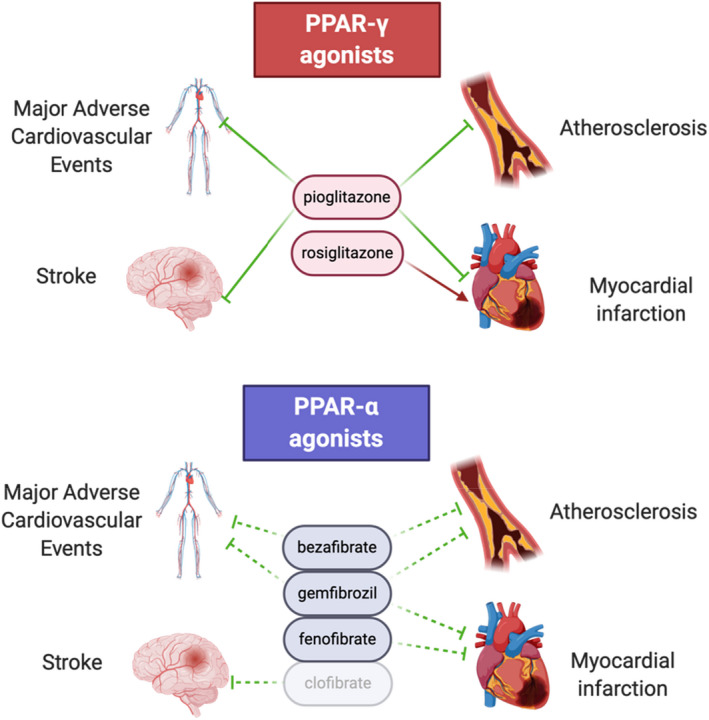
Effects of PPAR agonists on major adverse cardiovascular events, atherosclerosis, myocardial infarction and stroke. Dotted lines represent effects suggested in the literature but that warrant further studies. Rosiglitazone and clofibrate have been withdrawn from the market because of unwanted side effects.
Apart from their role in the liver, PPARs have pleiotropic actions in the body, amongst others in the heart, vessels and macrophages, where their activation exerts mostly protective effects.
4. MECHANISMS OF ACTION OF PPARS IN CVD: POSSIBLE ROLE OF THE LIVER
Assessing the effects of PPAR agonism on a specific diseased organ, independently of other organs, is challenging given the cross‐talk between organs. The effects of PPAR agonism observed in patients result from additive or synergic effects of these receptors on different organs including the liver but also adipose tissue, muscle, vessel and heart. As an additional level of complexity, distinct cell types in organs may respond via specific PPAR isotypes, whilst the specificity of PPAR agonists on effective PPAR activation vs off‐target effects is not usually assessed. The liver is a prominent example of this notion, since hepatocyte metabolism, endothelial dysfunction, macrophage activation and stellate cell transdifferentiation are triggered or hampered by different PPAR isotypes preferentially. Therefore, understanding the mechanisms and the exact contribution of each PPAR in each organ and cell type is crucial in the perspective of modulating the delicate balance of PPARs to improve patients' outcome. The underlying mechanisms mediating the effects of systemic PPARα and PPARγ activation on reduced risk for CVD are not clear. Because of their pleiotropic actions directly on the vasculature but also on the liver, action on PPAR proteins is appealing to modulate CVD risk in NAFLD. In the next section, we will detail mechanisms implicated in CVD progression and discuss whether they are susceptible to be liver‐mediated and implicate PPAR proteins (Figure 4).
4.1. Type II diabetes and systemic insulin resistance
T2DM is a risk factor for the development of CVD. PPARγ agonists thiazolidinediones have been used for diabetes mellitus management for years and are very potent insulin‐sensitising agents, as they have effects both on adipose tissue and beta‐cells. 62 Also, the dual agonist PPARα/γ saroglitazar improved insulin resistance more than pioglitazone alone in mice, suggesting a potential additive or synergistic effect of dual PPARα/γ agonism. 63 Moreover, a meta‐analysis of the use of saroglitazar in patients with diabetic dyslipidemia non‐invasively diagnosed with NAFLD demonstrated that it induces a statistically significant decrease in ALT levels (and liver stiffness in some patients) and improved cardiometabolic profile. 64 As there is no PPARβ/δ agonist available on the market for humans, most of the evidence is preclinical but indicates that PPARβ/δ favorises insulin sensitivity in adipocytes, skeletal muscle and hepatocytes. 65
Systemic insulin resistance has several hepatic‐related drivers, such as liver insulin resistance, closely linked to the pathophysiology of NAFLD. In insulin‐sensitive conditions, insulin signalling in the liver drives nutrient storage, in the forms of glycogen synthesis and de novo lipogenesis. In insulin‐resistant conditions, gluconeogenesis is diminished, excess glucose and circulating free fatty acids (FFAs) in the liver increase triglyceride (TG) synthesis, which in turn worsens insulin resistance by several mechanisms. One of these mechanisms is the activation by diacylglycerols (DAG) of protein kinase C epsilon (PKCε), which promotes inhibitory phosphorylation of insulin receptor substrate (IRS) by the insulin receptor, hindering insulin signalling. 66 Additionally, excess of FFAs in the liver causes lipotoxic inflammation, with increased levels of pro‐inflammatory cytokines such as tumour necrosis factor‐alpha (TNF‐α) and IL‐6, which are both promoted by different pathways inhibitory phosphorylation of IRS, contributing to shutting down insulin signalling. 67 PPARα has pleiotropic effects in the liver, particularly in fatty acid metabolism. PPARα activation enhances fatty acid catabolism, allowing the liver to adapt to excess FFAs. 2 Notably, PPARα activation decreases the quantity of DAG in the liver of mice fed a high fructose diet, 68 thereby contributing to the restoration of insulin signalling, whilst treatment with the dual PPARα/δ agonist elafibranor or the pan‐PPAR agonist lanifibranor have positive effects on hepatic and muscle insulin sensitivity 69 , 70 and improved glycemic control in NASH patients. 54 , 70 PPARβ/δ liver‐restricted overexpression also improved insulin resistance in mice fed a high fat, high carbohydrate diet 71 and similar effects were observed using the selective PPARβ/δ agonist seladelpar, which improved insulin sensitivity and steatohepatitis in mouse models of NAFLD. 72 In humans, the effects of seladelpar are mainly on atherogenic dyslipidaemia (e.g. a reduction of apolipoprotein B‐100 by 20%–38% and LDL cholesterol by 18%–43%) and are rather modest on insulin sensitivity or steatosis compared with other PPARβ/δ agonists. 73 Finally, thiazolidinediones, PPARγ agonists, have long been known to be highly effective insulin‐sensitising molecules, 74 although treatment of liver‐specific PPARγ‐defective mice fed a high‐fat diet with rosiglitazone remains effective, indicating that its main insulin‐sensitising action does not take place in the liver, but is probably due to effects in adipose tissue and beta‐cells. 75
4.2. Altered lipid metabolism
Lipid profile has an influence on CVD development: low high‐density lipoprotein (HDL) cholesterol and high triglycerides are recognised risk factors. 76 The liver occupies a central place in global lipid metabolism. Dyslipidemia and NAFLD are closely linked, both associated with metabolic syndrome. NAFLD is associated with higher fasting serum triglycerides and lower serum HDL‐C. 77 Whilst it is likely that this dyslipidaemia globally contributes to the CVD burden in NAFLD patients, the association between NAFLD and increased cardiovascular events remains significant even after adjustment for dyslipidaemia. 78 Agonism of all PPARs seems to improve atherogenic dyslipidemia. Mechanistically, PPARα has an important action activating lipids catabolism in the liver, but the implicated pathways are not as clear for the other PPARs and warrant further investigation.
The metabolic syndrome is usually associated with changes in the composition of the gut microbiota. Microbial byproducts contain small‐chain fatty acids, which are known PPAR activators, such as butyrate, 79 suggesting that changes in microbial composition would lead to PPAR deregulation. Indeed, faecal microbiota transplantation and microbiota byproducts have been shown to regulate PPARs and prevent hepatic fat accumulation. 79
4.3. Systemic inflammation
Metabolic syndrome and insulin resistance are clearly associated with systemic low‐grade inflammation with activation of inflammatory pathways in many organs. Importantly, and albeit CRP is produced by the liver, circulating markers of inflammation such as CRP poorly correlate with the severity of NAFLD and hence do not reflect liver inflammation but rather systemic inflammation. 80 One of the drivers of NAFLD development and progression is the excessive presence of pro‐inflammatory toxic lipids in the liver, such as cholesterol, FFAs, DAG or ceramides. They provoke hepatic insulin resistance and hepatocyte injury‐mediated liver inflammation. 81 Independent association between NAFLD and several circulating factors exhibiting systemic inflammation has also been consistently reported, 82 suggesting that liver inflammation can lead to systemic inflammation and vice versa.
The PPARα agonist fenofibrate decreased circulating levels of CRP and inflammatory cytokine IL‐6 in metabolic syndrome patients in whom the liver disease was not assessed, 83 and CRP in patients with impaired glucose tolerance, also decreasing pro‐inflammatory cytokines secretion by monocytes isolated from these patients after LPS stimulation. 84 Additionally, the PPARγ agonist rosiglitazone also decreased systemic inflammation in non‐diabetic patients with metabolic syndrome 85 and the dual PPARα/δ agonist elafibranor as well as the panPPAR agonist lanifibranor also achieved systemic anti‐inflammatory effects in patients with NASH vs the placebo group. 54 , 70 However, the underlying mechanisms are not clear, but it is possible that some of it are mediated by the anti‐inflammatory effects of PPARs in the liver, which have been described above.
In summary, activation of all PPARs plays anti‐inflammatory roles in the liver that possibly contribute to decrease systemic inflammation associated with NAFLD.
4.4. Endothelial dysfunction and activation
Endothelial dysfunction and control of vascular tone are one of the earliest detectable changes in the process of atherosclerotic lesion formation and in the pathogenesis of systemic hypertension. The implication of PPARγ in the pathogenesis of hypertension is demonstrated by the fact that patients with mutations in the DNA or ligand‐binding domains of PPARγ and endothelial PPARγ‐deficient mice develop severe early‐onset hypertension, 86 , 87 showing an effect at least partly directly mediated in the endothelium. However, PPARγ agonist pioglitazone treatment induced no change in systolic blood pressure and only a modest decrease in diastolic blood pressure in NAFLD patients in a meta‐analysis. 88
The main mediator of vascular dilatation is NO, but the endothelial function is also sensitive to certain circulating factors, some of which are altered in the case of liver disease. 18 As detailed above, PPAR proteins are protective against endothelial dysfunction, in particular by promoting NO production. NO production is especially sensitive to oxidative stress, which is defined as an imbalance between the production of reactive oxygen species (ROS) and the antioxidant capacities of the cell. In the presence of oxidative stress, NO is inactivated and transformed into peroxynitrite, and endothelial nitric oxide synthase (eNOS) becomes uncoupled and shifts from NO production to superoxide anion production, a very powerful ROS, 89 perpetuating a vicious circle. Therefore, endothelial dysfunction is closely linked to oxidative stress.
ROS are highly unstable molecules with half‐lives below one millisecond and exert their action at a cellular level. It is, therefore, highly unlikely that liver‐generated ROS can have a direct impact on heart and vasculature, nor that PPAR action on CVD is mediated through modulation of liver ROS production. More stable circulating mediators can induce oxidative stress at a distance, which explains the role played remotely by the liver modulating systemic endothelial oxidative stress. For instance, excess of circulating FFAs in NAFLD causes oxidative stress in the endothelium, 90 as well as pro‐inflammatory cytokines. 91 Both dyslipidemia and chronic inflammation associated with NAFLD are factors that can be modified by PPAR agonist treatments, and, therefore, may indeed modulate systemic ROS.
Another interesting example of a circulating mediator influencing endothelial dysfunction is homocysteine, a by‐product of methionine metabolism, which mainly takes place in the liver, making it a major source of circulating homocysteine. High homocysteinemia is a risk factor for CVD. 92 Several studies have demonstrated that homocysteine promotes endothelial dysfunction and atherosclerosis by different pathways such as NO synthesis impairment, deregulation of hydrogen sulfide signalling pathway or increased oxidative stress. 93 There is an association between high blood homocysteine levels and biopsy‐proven NAFLD, as demonstrated in a meta‐analysis. 94 Because of this association, increased circulating homocysteine could contribute to explain the increased incidence of CVD in NAFLD patients.
There is only scarce data available on the relationship of PPARs with homocysteine metabolism. It has been demonstrated that PPARα activation increases homocysteinemia in patients with dyslipidemia 95 and healthy mice and rats treated with fenofibrate, 96 , 97 although not in diabetic rats. 97 This could potentially be explained by PPARα‐mediated repression of the irreversible conversion of homocysteine into cystathionine, catalysed by the cystathionine‐β‐synthase (CβS) enzyme. 98 Indeed, it was demonstrated in the mouse that partial or total CβS deletion causes hyperhomocysteinemia. 99 Interestingly, treatment with rosiglitazone or troglitazone, PPARγ agonists, decrease hyperhomocysteinemia, respectively, in rats under methionine diet 100 and in hyperphagic rats. 101 It has been reported that either pioglitazone or rosiglitazone combined with antidiabetic medication glimepiride decrease blood homocysteine after 1 year of treatment in patients with diabetes mellitus and metabolic syndrome. 102 Interestingly, there is a report that rosiglitazone‐mediated PPARγ activation increases CβS activity in the rat, which would be an indirect mechanism of diminution of homocysteinemia. 103 Thus, PPARα and PPARγ seem to have opposite effects on the homocysteine blood level, although this subject warrants further research, as does the role of PPARβ/δ.
Hepatic methionine metabolism also influences hydrogen sulfide (H2S) levels. H2S, like NO, is a gaseous mediator implicated in endothelial vasorelaxation. Its half‐life is in the range of minutes in the blood, which makes an effect of liver production of H2S on plasma levels conceptually possible. It is synthesised by several alternative pathways, mainly CβS and gamma cystathionase. 104 It could be hypothesized that PPAR‐mediated modulation of CβS in the liver also influences H2S production and plasma concentration. There is no data on potential regulation of gamma cystathionase activity by PPAR proteins. As for homocysteine regulation, it warrants further investigations.
Asymmetric dimethylarginine (ADMA) is a competitive inhibitor of eNOS and has been associated in a recent meta‐analysis with an increased risk of a major adverse cardiovascular event and increased all‐cause mortality in patients with pre‐existing CVD. 105 ADMA is formed by methylation of arginine residues in proteins and released after proteolysis. ADMA can be degraded in the liver by dimethylarginine dimethylaminohydrolase 1 (DDAH1). ADMA plasma increase in NAFLD patients is not clearly established, but fenofibrate decreases ADMA plasma level in cholesterol‐fed rats 106 and rosiglitazone decreases plasma ADMA level in insulin‐resistant patients, 107 although their mode of action remains to be determined. Along this line, symmetric dimethylarginine (SDMA) can impact vascular tension via NO pathways, and its plasma levels correlate with the mortality risk in patients with liver cirrhosis. 108
In summary, endothelial PPARs are protective against endothelial dysfunction. The liver‐mediated action of PPARγ seems also mostly protective but this is less clear for PPARα, with a demonstrated increase in homocysteinemia in patients treated with fibrates. Globally, the effect of PPAR activation in the liver on endothelial function warrants further investigation.
4.5. Cardiovascular effect of hepatokines
The liver synthesises and secretes several proteins collectively called hepatokines that have a plethora of biological effects in multiple tissues across the body. Some of them may play a role in NAFLD co‐morbidities. 109
Fetuin A (TLR4 agonist) is a hepatokine that promotes endothelial dysfunction and activation, correlating with subclinical atherosclerosis in patients with NAFLD. 109 Interestingly, fetuin A is decreased by treatment with pioglitazone, 110 and its plasma levels in humans are associated with PPARα and PPARγ mutations. 111 Therefore, the protective effect of pioglitazone against CVD could be partly mediated by lowering the blood level of Fetuin A.
Fibroblast Growth Factor 21 (FGF21; FGFR1c agonist) is a well‐characterised hepatokine inducing systemic insulin sensitization. Treatment of cultured hepatocytes, mice, and diabetic patients with PPARα agonists increased FGF21 expression and secretion. 112 FGF21 also exerted a protective cardiac effect post‐myocardial infarction in mice, in a manner dependent on the liver—but not heart—IL‐22 signalling pathway. 113 This latter study is relevant because it conceptually demonstrates the importance of liver signalling on heart homeostasis in the mouse and supports the hypothesis of a direct effect of the liver on cardiovascular health.
Leukocyte cell‐derived chemotaxin‐2 (LECT2; CD209 agonist) is a hepatokine that is increased in NAFLD patients. 114 In vitro, LECT2 causes endothelial activation by increased pro‐inflammatory and adhesion proteins in endothelial cells and increased adhesion of monocytes, suggesting it can contribute to the progression of atherosclerosis. 115 Whilst its mRNA expression is not modified in PPARα‐deficient mouse hepatocytes, 116 whether it can be regulated by PPARγ or PPARβ/δ, remains to be investigated.
Tsukushi (TSK; Frizzled4 ligand) is a recently discovered hepatokine synthesised by the liver in response to NAFLD and obesity. 117 Its hepatic overexpression decreases plasma HDL cholesterol in the mouse and inhibits cholesterol efflux from J774 cells, used as a model of macrophages, 117 suggesting a detrimental effect in the development of atherosclerosis. Interestingly, TSK mRNA is higher in PPARα‐deficient mouse hepatocytes compared with normal hepatocytes, 116 suggesting that activation of PPARα could be protective against increased liver TSK expression. Whether TSK can be regulated by PPARγ or PPARβ/δ in the liver remains to be elucidated.
4.6. Coagulation
Whether there is a coagulation imbalance in NAFLD patients is a subject to some controversy, with studies describing comparable hemostatic profiles in NAFLD and control patients, 118 and others reporting pro‐coagulant imbalance. 119 However, independently of overall coagulation state, higher activity of liver‐produced fibrinogen, and factors VIII, IX, XI and XII has been observed in patients with NAFLD, 120 as well as higher plasma plasminogen activator inhibitor‐1 (PAI‐1), 82 , 121 most of which are associated with CVD. 122 In addition, platelets in the liver may serve as “sentinels” for immune activation in NASH progression. 123
PPARα is extensively involved in the regulation of coagulation by various mechanisms. It inhibits the increase of tissue factor and carboxypeptidase B2 liver production and plasma level 124 and, transcriptomics studies have shown downregulation of coagulation‐associated genes in the liver after PPARα agonists treatment in the mouse and cynomolgus monkey. 36 , 125 Fibrates also decrease plasma fibrinogen levels in humans. 126
PPARα can also indirectly modulate the coagulation state by increasing the level of sulfatides in the mouse liver and plasma by modulating synthesis and export proteins. 124 , 127 Sulfatides are a class of sphingolipids with a role in haemostasis debated for decades. 128 High plasma sulfatides are associated with intima‐media thickness in hypertensive patients 129 and patients with familial hypercholesterolemia, 130 and with major adverse cardiovascular events and in‐hospital deaths in patients with ST‐segment elevated myocardial infarction. 131
Overall, the regulation of coagulation by PPARα is complex and seems to require a systemic component which remains to be investigated, as a transcriptomics meta‐analysis showed that PPARα activation regulated coagulation pathways only in vivo (mouse) and not ex vivo (liver slices) nor in vitro (mouse primary hepatocytes). 132
PPARγ systemic activation exerts a general anti‐thrombotic action in rats. 133 Paradoxically, its activation results in increased liver production of PAI‐1 and blood secretion in several in vitro and in vivo models, opposite to PPARα action. 134 Remarkably, in the study by Verrijken and colleagues, only PAI‐1 and not fibrinogen, factor VII, factor VIII, factor XI nor von Willebrand factor was still associated with liver histological lesions after correction for metabolic factors, suggesting that its production could be linked to liver disease in itself rather than underlying metabolic causes. 135 Indeed, PPARs activation in patients with complex diseases like CVD and NAFLD will have a positive impact both on the metabolic (systemic) and hepatic underlying pathological mechanisms of the disease, which will occur simultaneously and might even be synergistic. Further studies are required to disentangle these organ / vascular bed specific contributions.
4.7. Extracellular vesicles
Extracellular vesicles (EVs) include apoptotic bodies, microvesicles and exosomes. EVs are vesicles released by all cells, under both physiological and pathological conditions and can act as vectors of information that regulate the function of target cells, which can be situated in other organs. 136 , 137
There is scarce data on the relationship between PPAR proteins and EVs. It has been demonstrated that fenofibrate treatment decreases the EV content of the atherosclerotic lesion but not of the liver of mice fed a western diet, 138 suggesting that the apparent action of PPARα on EVs in NAFLD is not mediated by the liver. PPARα can also inhibit tumour‐derived exosomal lipid‐induced dendritic cell dysfunction. 139 PPARγ was found in human circulating exosomes suggesting a potential for paracrine transfer of nuclear receptors. 140
5. CVD RISK AND NASH IN CLINICAL TRIAL DESIGN
As mentioned before, given the fact that patients with non‐cirrhotic NASH mainly die from cardiovascular disease and data indicate that NASH contributes to the development of cardiovascular disease, PPARs may be attractive for the treatment of NASH beyond their efficacy on liver‐centred clinical trial endpoints. Improving cardiovascular outcomes might be a differentiator for the choice of the drug once several compounds will be available on the market specifically for the treatment of NASH. 141 Assessment of CVD in the context of NASH trials is therefore of the utmost importance, both from a safety and efficacy perspective. To date, this has mainly been restricted to studying the impact on classical CV risk factors (mainly lipid profile and glycaemic control, as discussed previously) and the registration of CV events.
Weight gain is often observed with drugs with a PPARγ activity (with variable data, but usually reported to be 2%–4% of initial body weight after 6 months of treatment or longer). Usually, this is considered a “side‐effect”, as patients are encouraged to lose weight and weight loss is known to associate with histological improvement. For pioglitazone, it has been shown that this corresponds to a shift from visceral to more metabolically healthy subcutaneous fat and an improvement in the metabolic‐inflammatory (e.g. an improvement in insulin sensitivity) environment, despite the net weight gain. 142 Saroglitazar treatment also induces weight gain 143 which goes along with increases in adiponectin levels, a sign of improvement in adipose tissue function, and improved insulin sensitivity. Lanifibranor, even so, improves liver histology despite a modest increase in body weight, and also with an increase in adiponectin and improved insulin sensitivity. 70 It hence appears that the observed weight gain is related to an improvement in the capacity of the adipose tissue to store energy. This is also reflected in the fact that, for all the aforementioned drugs, insulin sensitivity To further assess this, a more detailed anthropometric assessment of the patients, both at baseline and in follow‐up, should be incorporated into the clinical trial design.
Fluid retention and heart failure have also been reported in association with PPAR drugs, in particular in those with PPARγ activity. As mentioned, pioglitazone reduces the risk of CV events, but significant confusion remains about its effects on cardiac function. Cardiac failure was reported in more (~2%) patients on pioglitazone than on placebo in the PROACTIVE trial. 144 This was, however, not observed in other placebo‐controlled studies. 145 , 146 A recent large RCT in 3851 patients did even so not find a difference in the 5‐year heart failure risk (4.1% pioglitazone, 4.2% placebo). 147 It has been shown that pioglitazone improves myocardial insulin sensitivity, left ventricular diastolic and systolic function in healthy patients with T2DM. 148 Nevertheless, undiagnosed “diastolic dysfunction” (i.e. heart failure with preserved left ventricular function) may occur in ≥10% of patients with longstanding obesity, T2DM and/or NASH, 149 if fluid retention occurs during pioglitazone therapy in such patients, it may unmask this subclinical heart disease. Very few cases of oedema were reported with lanifibranor 70 and no CV events with saroglitazar, 143 but this needs of course confirmation in larger trials.
Anyhow, this advocates for the incorporation of some assessment of cardiovascular function in clinical trials for NASH. Electrocardiograms and markers like N‐terminal‐prohormone B‐type natriuretic peptide can be useful. However, a more detailed analysis at baseline and follow‐up should be considered. Although within‐patient variability as well as intra‐ and interobserver variability of functional tests like flow‐mediated dilation hamper the interpretation of the data, especially in large multicenter trials, some tests like imaging test (ultrasound, coronary artery score on computed tomography, or cardiac MRI) allow for central reading that can help mitigating these methodological issues. This should, of course, be balanced against the logistical and financial implications, as well as the increased burden of examinations for the patients and workload for the clinical trial team. Given, however, the importance of this cardiovascular aspect, including a more in‐depth assessment of cardiovascular function in NASH clinical trial design should be mandatory, from both a safety and efficacy point of view. Assessment of cardiovascular benefit based on clinical event rate is not realistic with the current clinical trial design, given the sample size and study duration needed to obtain these results. In the future, when non‐invasive parameters will allow assessing treatment efficacy (which is currently based on liver biopsy in phase 3), larger trials with an appropriate sample size to assess CV benefit on clinical events will hopefully definitely answer the question of the complex link between NASH and CVD and the potential of PPAR drugs to beneficially impact hereon.
6. CONCLUSION
PPARs agonism may represent a promising treatment for NASH, having liver‐specific effects but also improving cardiovascular outcomes, which may later impact liver disease. Therefore, assessment of cardiovascular disease within NASH trials is of the utmost importance, both from a safety and efficacy perspective.
AUTHOR CONTRIBUTIONS
Sergi Guixé‐Muntet: Methodology (equal); writing – original draft (equal). Louise Biquard: Methodology (equal); writing – original draft (equal). Gyongyi Szabo: Validation (equal); writing – review and editing (equal). Jean‐Francois Dufour: Validation (equal); writing – review and editing (equal). F. Tacke: Validation (equal); writing – review and editing (equal). Sven Francque: Validation (equal); writing – review and editing (equal). Pierre‐emmanuel Rautou: Conceptualization (equal); supervision (equal); writing – review and editing (equal). Jordi Gracia‐Sancho: Conceptualization (equal); supervision (equal); writing – review and editing (equal).
FUNDING INFORMATION
G.S. is supported by NIH grants R01AA015576, R01AA017729 and R01AA0207440. S.F. holds a senior clinical research fellowship from the Research Foundation Flanders (1802154 N). P.E.R. is supported by the Institut National de la Santé et de la Recherche Médicale (ATIP AVENIR), the Agence Nationale pour la Recherche (ANR‐18‐CE14‐0006‐01, RHU QUID‐NASH, ANR‐18‐IDEX‐0001) and Émergence, Ville de Paris, and Fondation ARC. J.G.‐S. is supported by the Instituto de Salud Carlos III (FIS PI20/00220), the CIBEREHD, the Swiss National Science Funds (320030_189252), the Novartis Foundation for Medical‐Biological Research and the Foundation Suisse Contre le Cancer du Foie.
AUTHORSHIP
Guarantor of the article: Jordi Gracia‐Sancho.
ACKNOWLEDGEMENTS
Declaration of personal interests: G.S. is a scientific consultant for Durect Co, Evive Bio, Glympse, Novartis, Pandion, Surrozen, Terra Firma, Pfizer, Merk, Satellie bio, Zomagen, Cyta Therapeutics, and Pandion. She received royalty from Springer and UpToDate. Dr. Szabo has equity holding in Glympse bio and Ventyx. She is the Editor of Hepatology Communications. J.‐F.D. Advisory committees: Alentis, Astra‐Zeneca, Bayer, Bristol‐Myers Squibb, Enyo, Esai, Falk, Genfit, Gilead Sciences, Intercept, Inventiva, Ipsen, Lilly, Madrigal, Merck, Novartis, Novo‐Nordisk, Roche. Speaking and teaching: Bayer, Bristol‐Myers Squibb, Intercept, Gilead Sciences, Novartis, Roche. F.T. has received research funding from Gilead, Inventiva, BMS and Allergan. S.F. has acted as advisor and/or lecturer for Astra‐Zeneca, Roche, Gilead, Abbvie, Bayer, BMS, MSD, Janssen, Actelion, Astellas, Genfit, Inventiva, Intercept, Genentech, Galmed, Promethera, Coherus, Julius Clinical, CLS Behring, Galapagos, Madrigal and NGM Bio. P.E.R. has received research funding from Terrafirma and acted as a consultant for Mursla and Abbelight, provided training sessions for Cook and received speaker fees from Tillots pharma. J.G.‐S. has received research funding from Gilead, Inventiva, Conatus, Surrozen, GAT Tx and BrudyLab, and acted as a consultant for Ambys, Novo Seeds, Kintsugi Tx, BrudyLab and Terrafirma. Open access funding provided by Inselspital Universitatsspital Bern.
Guixé‐Muntet S, Biquard L, Szabo G, et al. Review article: vascular effects of PPARs in the context of NASH . Aliment Pharmacol Ther. 2022;56:209–223. 10.1111/apt.17046
Sergi Guixé‐Muntet and Louise Biquard share first authorship.
Pierre‐Emmanuel Rautou and Jordi Gracia‐Sancho are co‐senior authors.
The Handling Editor for this article was Dr Rohit Loomba, and this uncommissioned review was accepted for publication after full peer‐review.
Contributor Information
Pierre‐Emmanuel Rautou, Email: pierre-emmanuel.rautou@inserm.fr.
Jordi Gracia‐Sancho, Email: jordi.gracia@idibaps.org.
References
- 1. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non‐alcoholic fatty liver disease. J Hepatol. 62:720–33. [DOI] [PubMed] [Google Scholar]
- 2. Montagner A, Polizzi A, Fouché E, Ducheix S, Lippi Y, Lasserre F, et al. Liver PPARα is crucial for whole‐body fatty acid homeostasis and is protective against NAFLD. Gut. 2016;65:1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ding Y, Yang KD, Yang Q. The role of PPARδ signaling in the cardiovascular system. Progress in Molecular Biology and Translational Science. Volume 121. Elsevier B.V.; 2014. p. 451–73. [DOI] [PubMed] [Google Scholar]
- 4. Abdelrahman M, Sivarajah A, Thiemermann C. Beneficial effects of PPAR‐gamma ligands in ischemia‐reperfusion injury, inflammation and shock. Cardiovasc Res. 2005;65:772–81. [DOI] [PubMed] [Google Scholar]
- 5. Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, et al. PPARγ ligands increase expression and plasma concentrations of adiponectin, an adipose‐derived protein. Diabetes. 2001;50:2094–9. [DOI] [PubMed] [Google Scholar]
- 6. Meirhaeghe A, Amouyel P. Impact of genetic variation of PPARgamma in humans. Mol Genet Metab. 2004;83:93–102. [DOI] [PubMed] [Google Scholar]
- 7. Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2019;69:2672–82. [DOI] [PubMed] [Google Scholar]
- 8. De Gottardi A, Pazienza V, Pugnale P, Bruttin F, Rubbia‐Brandt L, Juge‐Aubry CE, et al. Peroxisome proliferator‐activated receptor‐α and ‐γ mRNA levels are reduced in chronic hepatitis C with steatosis and genotype 3 infection. Aliment Pharmacol Ther. 2006;23:107–14. [DOI] [PubMed] [Google Scholar]
- 9. Blanquart C, Mansouri R, Paumelle R, Fruchart JC, Staels B, Glineur C. The protein kinase C signaling pathway regulates a molecular switch between transactivation and transrepression activity of the peroxisome proliferator‐activated receptor α. Mol Endocrinol. 2004;18:1906–18. [DOI] [PubMed] [Google Scholar]
- 10. Pourcet B, Pineda‐Torra I, Derudas B, Staels B, Glineur C. SUMOylation of human peroxisome proliferator‐activated receptor α inhibits its trans‐activity through the recruitment of the nuclear corepressor NCoR. J Biol Chem. 2010;285:5983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Surapureddi S, Yu S, Bu H, Hashimoto T, Yeldandi A V., Kashireddy P, et al. Identification of a transcriptionally active peroxisome proliferator‐activated receptor α‐interacting cofactor complex in rat liver and characterization of PRIC285 as a coactivator. Proc Natl Acad Sci USA. 2002;99:11836–11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta Mol Cell Biol Lipids. 2007;1771:952–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grimaldi PA. Regulatory functions of PPARβ in metabolism: implications for the treatment of metabolic syndrome. Biochim Biophys Acta Mol Cell Biol Lipids. 2007;1771:983–90. [DOI] [PubMed] [Google Scholar]
- 14. Xu X, Otsuki M, Saito H, Sumitani S, Yamamoto H, Asanuma N, et al. PPARα and GR differentially down‐regulate the expression of nuclear factor‐κB‐responsive genes in vascular endothelial cells. Endocrinology. 2001;142:3332–9. [DOI] [PubMed] [Google Scholar]
- 15. Oka SI, Zhai P, Alcendor R, Park JY, Tian B, Sadoshima J. Suppression of ERR targets by a PPARα/Sirt1 complex in the failing heart. Cell Cycle. 2012;11:856–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beaumont J, Arias T, Ravassa S, Díez J. Overexpression of human truncated peroxisome proliferator‐activated receptor α induces apoptosis in HL‐1 cardiomyocytes. Cardiovasc Res. 2008;79:458–63. [DOI] [PubMed] [Google Scholar]
- 17. Francque S, Wamutu S, Chatterjee S, Van Marck E, Herman A, Ramon A, et al. Non‐alcoholic steatohepatitis induces non‐fibrosis‐related portal hypertension associated with splanchnic vasodilation and signs of a hyperdynamic circulation in vitro and in vivo in a rat model. Liver Int. 2010;30:365–75. [DOI] [PubMed] [Google Scholar]
- 18. Gibert‐Ramos A, Sanfeliu‐Redondo D, Aristu‐Zabalza P, Martínez‐Alcocer A, Gracia‐Sancho J, Guixé‐Muntet S, et al. The hepatic sinusoid in chronic liver disease: the optimal milieu for cancer. Cancers (Basel). 2021;13:5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gracia‐Sancho J, Marrone G, Fernández‐Iglesias A. Hepatic microcirculation and mechanisms of portal hypertension. Nat Rev Gastroenterol Hepatol. 2019;16:221–34. [DOI] [PubMed] [Google Scholar]
- 20. Cho DH, Choi YJ, Jo SA, Jo I. Nitric oxide production and regulation of endothelial nitric‐oxide synthase phosphorylation by prolonged treatment with troglitazone: evidence for involvement of peroxisome proliferator‐activated receptor (PPAR) γ‐dependent and PPARγ‐independent signaling. J Biol Chem. 2004;279:2499–506. [DOI] [PubMed] [Google Scholar]
- 21. Guixé‐Muntet S, Zhu CP, Xie WF, Gracia‐Sancho J. Novel therapeutics for portal hypertension and fibrosis in chronic liver disease. Pharmacol Therap. 2020;215. [DOI] [PubMed] [Google Scholar]
- 22. Gracia‐Sancho J, Laviña B, Rodríguez‐Vilarrupla A, García‐Calderó H, Fernández M, Bosch J, et al. Increased oxidative stress in cirrhotic rat livers: a potential mechanism contributing to reduced nitric oxide bioavailability. Hepatology. 2008;47:1248–56. [DOI] [PubMed] [Google Scholar]
- 23. Krönke G, Kadl A, Ikonomu E, Blüml S, Fürnkranz A, Sarembock IJ, et al. Expression of heme oxygenase‐1 in human vascular cells is regulated by peroxisome proliferator‐activated receptors. Arterioscler Thromb Vasc Biol. 2007;27:1276–82. [DOI] [PubMed] [Google Scholar]
- 24. Rodríguez‐Vilarrupla A, Laviña B, García‐Calderó H, Russo L, Rosado E, Roglans N, et al. PPARα activation improves endothelial dysfunction and reduces fibrosis and portal pressure in cirrhotic rats. J Hepatol. 2012;56:1033–9. [DOI] [PubMed] [Google Scholar]
- 25. Marchesi C, Rehman A, Rautureau Y, Kasal DA, Briet M, Leibowitz A, et al. Protective role of vascular smooth muscle cell PPARγ in angiotensin II‐induced vascular disease. Cardiovasc Res. 2013;97:562–70. [DOI] [PubMed] [Google Scholar]
- 26. Pelham CJ, Ketsawatsomkron P, Groh S, Grobe JL, De Lange WJ, Ibeawuchi SRC, et al. Cullin‐3 regulates vascular smooth muscle function and arterial blood pressure via PPARγ and RhoA/Rho‐kinase. Cell Metab. 2012;16:462–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van Bilsen M, Van Nieuwenhoven FA. PPARs as therapeutic targets in cardiovascular disease. Expert Opin Ther Targets. 2010;14:1029–45. [DOI] [PubMed] [Google Scholar]
- 28. Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A, et al. Differential effects of selective‐ and pan‐PPAR agonists on experimental steatohepatitis and hepatic macrophages. J Hepatol. 2020;73:757–70. [DOI] [PubMed] [Google Scholar]
- 29. Boyer‐Diaz Z, Aristu‐Zabalza P, Andres‐Rozas M, Robert C, Ortega‐Ribera M, Fernández‐Iglesias A, et al. Pan‐PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J Hepatol. 2020;74:1188–99. [DOI] [PubMed] [Google Scholar]
- 30. Afdal P, AbdelMassih AF. Is pulmonary vascular disease reversible with PPAR ɣ agonists? Microcirculation. 2018;25. [DOI] [PubMed] [Google Scholar]
- 31. Barroso I, Gurnell M, Crowley VEF, Agostini M, Schwabe JW, Soos MA, et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402:880–3. [DOI] [PubMed] [Google Scholar]
- 32. Schwabl P, Payer BA, Grahovac J, Klein S, Horvatits T, Mitterhauser M, et al. Pioglitazone decreases portosystemic shunting by modulating inflammation and angiogenesis in cirrhotic and non‐cirrhotic portal hypertensive rats. J Hepatol. 2014;60:1135–42. [DOI] [PubMed] [Google Scholar]
- 33. Tsai HC, Li TH, Huang CC, Huang SF, Liu RS, Yang YY, et al. Beneficial effects of the peroxisome proliferator‐activated receptor α/γ agonist aleglitazar on progressive hepatic and splanchnic abnormalities in cirrhotic rats with portal hypertension. Am J Pathol. 2018;188:1608–24. [DOI] [PubMed] [Google Scholar]
- 34. Li H, Zhou Y, Wang H, Zhang M, Qiu P, Zhang M, et al. Crosstalk between liver macrophages and surrounding cells in nonalcoholic steatohepatitis. Front Immunol. 2020;11:1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARα‐dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology. 2003;38:123–32. [DOI] [PubMed] [Google Scholar]
- 36. Cariello NF, Romach EH, Colton HM, Ni H, Yoon L, Falls JG, et al. Gene expression profiling of the PPAR‐alpha agonist ciprofibrate in the cynomolgus monkey liver. Toxicol Sci. 2005;88:250–64. [DOI] [PubMed] [Google Scholar]
- 37. Kleemann R, Gervois PP, Verschuren L, Staels B, Princen HMG, Kooistra T. Fibrates down‐regulate IL‐1‐stimulated C‐reactive protein gene expression in hepatocytes by reducing nuclear p50‐NFκB‐C/EBP‐β complex formation. Blood. 2003;101:545–51. [DOI] [PubMed] [Google Scholar]
- 38. Pawlak M, Baugé E, Bourguet W, De Bosscher K, Lalloyer F, Tailleux A, et al. The transrepressive activity of peroxisome proliferator‐activated receptor alpha is necessary and sufficient to prevent liver fibrosis in mice. Hepatology. 2014;60:1593–606. [DOI] [PubMed] [Google Scholar]
- 39. Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JEM, Van Rooijen N, et al. Kupffer cells promote hepatic steatosis via interleukin‐1β‐dependent suppression of peroxisome proliferator‐activated receptor α activity. Hepatology. 2010;51:511–22. [DOI] [PubMed] [Google Scholar]
- 40. Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, et al. PPARα gene expression correlates with severity and histological treatment response in patients with non‐alcoholic steatohepatitis. J Hepatol. 2015;63:164–73. [DOI] [PubMed] [Google Scholar]
- 41. Villa E, Cammà C, Marietta M, Luongo M, Critelli R, Colopi S, et al. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology. 2012;143:1253–1260.e4. [DOI] [PubMed] [Google Scholar]
- 42. Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, Kotsiliti E, et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med. 2019;25:641–55. [DOI] [PubMed] [Google Scholar]
- 43. Jiang ZG, Feldbrügge L, Tapper EB, Popov Y, Ghaziani T, Afdhal N, et al. Aspirin use is associated with lower indices of liver fibrosis among adults in the United States. Aliment Pharmacol Ther. 2016;43:734–43. [DOI] [PubMed] [Google Scholar]
- 44. Watts GF, Dimmitt SB. Fibrates, dyslipoproteinaemia and cardiovascular disease. Curr Opin Lipidol. 1999;10:561–74. [DOI] [PubMed] [Google Scholar]
- 45. Kramer JA, Blomme EAG, Bunch RT, Davila JC, Jackson CJ, Jones PF, et al. Transcription profiling distinguishes dose‐dependent effects in the livers of rats treated with clofibrate. Toxicol Pathol. 2003;31:417–31. [DOI] [PubMed] [Google Scholar]
- 46. Gracia‐Sancho J, Caparrós E, Fernández‐Iglesias A, Francés R. Role of liver sinusoidal endothelial cells in liver diseases. Nat Rev Gastroenterol Hepatol. 2021;18:411–31. [DOI] [PubMed] [Google Scholar]
- 47. Staels B, Rubenstrunk A, Noel B, Rigou G, Delataille P, Millatt LJ, et al. Hepatoprotective effects of the dual peroxisome proliferator‐activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. 2013;58:1941–52. [DOI] [PubMed] [Google Scholar]
- 48. Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARα agonist, Wy‐14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39:1286–96. [DOI] [PubMed] [Google Scholar]
- 49. Abdelmegeed MA, Yoo SH, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song BJ. PPARα expression protects male mice from high fat‐induced nonalcoholic fatty liver. J Nutr. 2011;141:603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jain MR, Giri SR, Bhoi B, Trivedi C, Rath A, Rathod R, et al. Dual PPARα/γ agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. 2018;38:1084–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Daniels SJ, Leeming DJ, Detlefsen S, Bruun MF, Hjuler ST, Henriksen K, et al. Biochemical and histological characterisation of an experimental rodent model of non‐alcoholic steatohepatitis – effects of a peroxisome proliferator‐activated receptor gamma (PPAR‐γ) agonist and a glucagon‐like peptide‐1 analogue. Biomed Pharmacother. 2019;111:926–33. [DOI] [PubMed] [Google Scholar]
- 52. Nakajima A, Eguchi Y, Yoneda M, Imajo K, Tamaki N, Suganami H, et al. Randomised clinical trial: pemafibrate, a novel selective peroxisome proliferator‐activated receptor α modulator (SPPARMα), versus placebo in patients with non‐alcoholic fatty liver disease. Aliment Pharmacol Ther. 2021;54:1263–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati‐Baroni G, et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology. 2002;122:1924–40. [DOI] [PubMed] [Google Scholar]
- 54. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator‐activated receptor‐α and ‐δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150:1147–1159.e5. [DOI] [PubMed] [Google Scholar]
- 55. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz‐Lopez C, et al. Long‐term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med. 2016;165:305–15. [DOI] [PubMed] [Google Scholar]
- 56. Baratta F, Pastori D, Angelico F, Balla A, Paganini AM, Cocomello N, et al. Nonalcoholic fatty liver disease and fibrosis associated with increased risk of cardiovascular events in a prospective study. Clin Gastroenterol Hepatol. 2020;18:2324–2331.e4. [DOI] [PubMed] [Google Scholar]
- 57. Henson JB, Simon TG, Kaplan A, Osganian S, Masia R, Corey KE. Advanced fibrosis is associated with incident cardiovascular disease in patients with non‐alcoholic fatty liver disease. Aliment Pharmacol Ther. 2020;51:728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Alexander M, Loomis AK, Van Der Lei J, Duarte‐Salles T, Prieto‐Alhambra D, Ansell D, et al. Non‐alcoholic fatty liver disease and risk of incident acute myocardial infarction and stroke: findings from matched cohort study of 18 million European adults. BMJ. 2019;367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Targher G, Byrne CD, Lonardo A, Zoppini G, Barbui C. Non‐alcoholic fatty liver disease and risk of incident cardiovascular disease: a meta‐analysis. J Hepatol. 2016;65:589–600. [DOI] [PubMed] [Google Scholar]
- 60. Targher G, Byrne CD, Tilg H. NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut. 2020;69:1691–705. [DOI] [PubMed] [Google Scholar]
- 61. Francque SM, van der Graaff D, Kwanten WJ. Non‐alcoholic fatty liver disease and cardiovascular risk: pathophysiological mechanisms and implications. J Hepatol. 2016;65:425–43. [DOI] [PubMed] [Google Scholar]
- 62. Reynaert H, Geerts A, Henrion J. Review article: the treatment of non‐alcoholic steatohepatitis with thiazolidinediones. Aliment Pharmacol Ther. 2005;22:897–905. [DOI] [PubMed] [Google Scholar]
- 63. Kumar DP, Caffrey R, Marioneaux J, Santhekadur PK, Bhat M, Alonso C, et al. The PPAR α/γ agonist saroglitazar improves insulin resistance and steatohepatitis in a diet induced animal model of nonalcoholic fatty liver disease. Sci Rep. 2020;10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kaul U, Parmar D, Manjunath K, Shah M, Parmar K, Patil KP, et al. New dual peroxisome proliferator activated receptor agonist – saroglitazar in diabetic dyslipidemia and non‐alcoholic fatty liver disease: integrated analysis of the real world evidence. Cardiovascular Diabetol. 2019;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wettstein G, Luccarini J‐M, Poekes L, Faye P, Kupkowski F, Adarbes V, et al. The new‐generation pan‐peroxisome proliferator‐activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol Commun. 2017;1:524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Investig. 2016;126:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Patel BM, Goyal RK. Liver and insulin resistance: new wine in old bottle!!! Eur J Pharmacol. 2019;862. [DOI] [PubMed] [Google Scholar]
- 68. Chan SMH, Sun RQ, Zeng XY, Choong ZH, Wang H, Watt MJ, et al. Activation of PPARα ameliorates hepatic insulin resistance and steatosis in high fructose‐fed mice despite increased endoplasmic reticulum stress. Diabetes. 2013;62:2095–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cariou B, Hanf R, Lambert‐Porcheron S, Zaïr Y, Sauvinet V, Noël B, et al. Dual peroxisome proliferator‐activated receptor α/δ agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care. 2013;36:2923–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A randomized controlled trial of the pan‐PPAR agonist lanifibranor in NASH. N Engl J Med. 2021;385:1547–58. [DOI] [PubMed] [Google Scholar]
- 71. Liu S, Hatano B, Zhao M, Yen CC, Kang K, Reilly SM, et al. Role of peroxisome proliferator‐activated receptor δ/β in hepatic metabolic regulation. J Biol Chem. 2011;286:1237–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Haczeyni F, Wang H, Barn V, Mridha AR, Yeh MM, Haigh WG, et al. The selective peroxisome proliferator‐activated receptor‐delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol Commun. 2017;1:663–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bays HE, Schwartz S, Littlejohn T, Kerzner B, Krauss RM, Karpf DB, et al. MBX‐8025, a novel peroxisome proliferator receptor‐δagonist: lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J Clin Endocrinol Metab. 2011;96:2889–97. [DOI] [PubMed] [Google Scholar]
- 74. Alam F, Islam MA, Mohamed M, Ahmad I, Kamal MA, Donnelly R, et al. Efficacy and safety of pioglitazone monotherapy in type 2 diabetes mellitus: a systematic review and meta‐analysis of randomised controlled trials. Sci Rep. 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, et al. Liver peroxisome proliferator‐activated receptor γ contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278:34268–76. [DOI] [PubMed] [Google Scholar]
- 76. Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140:e596–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. DeFilippis AP, Blaha MJ, Martin SS, Reed RM, Jones SR, Nasir K, et al. Nonalcoholic fatty liver disease and serum lipoproteins: the multi‐ethnic study of atherosclerosis. Atherosclerosis. 2013;227:429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mantovani A, Csermely A, Petracca G, Beatrice G, Corey KE, Simon TG, et al. Non‐alcoholic fatty liver disease and risk of fatal and non‐fatal cardiovascular events: an updated systematic review and meta‐analysis. Lancet Gastroenterol Hepatol. 2021;6:903–913. [DOI] [PubMed] [Google Scholar]
- 79. Penny Oh HY, Visvalingam V, Wahli W. The PPAR‐microbiota‐metabolic organ trilogy to fine‐tune physiology. FASEB J. 2019;33:9706–30. [DOI] [PubMed] [Google Scholar]
- 80. Zimmermann E, Anty R, Tordjman J, Verrijken A, Gual P, Tran A, et al. C‐reactive protein levels in relation to various features of non‐alcoholic fatty liver disease among obese patients. J Hepatol. 2011;55:660–5. [DOI] [PubMed] [Google Scholar]
- 81. Heymann F, Tacke F. Immunology in the liver‐from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13:88–110. [DOI] [PubMed] [Google Scholar]
- 82. Sookoian S, Castaño GO, Burgueño AL, Rosselli MS, Gianotti TF, Mallardi P, et al. Circulating levels and hepatic expression of molecular mediators of atherosclerosis in nonalcoholic fatty liver disease. Atherosclerosis. 2010;209:585–91. [DOI] [PubMed] [Google Scholar]
- 83. Belfort R, Berria R, Cornell J, Cusi K. Fenofibrate reduces systemic inflammation markers independent of its effects on lipid and glucose metabolism in patients with the metabolic syndrome. J Clin Endocrinol Metab. 2010;95:829–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Okopień B, Krysiak R, Herman ZS. Effects of short‐term fenofibrate treatment on circulating markers of inflammation and hemostasis in patients with impaired glucose tolerance. J Clin Endocrinol Metab. 2006;91:1770–8. [DOI] [PubMed] [Google Scholar]
- 85. Samaha FF, Szapary PO, Iqbal N, Williams MM, Bloedon LT, Kochar A, et al. Effects of rosiglitazone on lipids, adipokines, and inflammatory markers in nondiabetic patients with low high‐density lipoprotein cholesterol and metabolic syndrome. Arterioscler Thromb Vasc Biol. 2006;26:624–30. [DOI] [PubMed] [Google Scholar]
- 86. Auclair M, Vigouroux C, Boccara F, Capel E, Vigeral C, Guerci B, et al. Peroxisome proliferator‐activated receptor‐γ mutations responsible for lipodystrophy with severe hypertension activate the cellular renin‐angiotensin system. Arterioscler Thromb Vasc Biol. 2013;33:829–38. [DOI] [PubMed] [Google Scholar]
- 87. Guignabert C, Alvira CM, Alastalo TP, Sawada H, Hansmann G, Zhao M, et al. Tie2‐mediated loss of peroxisome proliferator‐activated receptor‐γ in mice causes PDGF receptor‐β‐dependent pulmonary arterial muscularization. Am J Physiol ‐ Lung Cell Mol Physiol. 2009;297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Musso G, Cassader M, Rosina F, Gambino R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non‐alcoholic fatty liver disease (NAFLD): a systematic review and meta‐analysis of randomised trials. Diabetologia. 2012;55:885–904. [DOI] [PubMed] [Google Scholar]
- 89. Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. 2017;120:713–35. [DOI] [PubMed] [Google Scholar]
- 90. Zhang H, Dellsperger KC, Zhang C. The link between metabolic abnormalities and endothelial dysfunction in type 2 diabetes: an update. Basic Res Cardiol. 2012;107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc Pharmacol. 2018;100:1–19. [DOI] [PubMed] [Google Scholar]
- 92. Chrysant SG, Chrysant GS. The current status of homocysteine as a risk factor for cardiovascular disease: a mini review. Expert Rev Cardiovasc Ther. 2018;16:559–65. [DOI] [PubMed] [Google Scholar]
- 93. Esse R, Barroso M, Almeida IT, De Castro R. The contribution of homocysteine metabolism disruption to endothelial dysfunction: state‐of‐the‐art. Int J Mol Sci. 2019;20:867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dai Y, Zhu J, Meng D, Yu C, Li Y. Association of homocysteine level with biopsy‐proven non‐alcoholic fatty liver disease: a meta‐analysis. J Clin Biochem Nutr. 2016;58:76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Melenovsky V, Stulc T, Kozich V, Grauova B, Krijt J, Wichterle D, et al. Effect of folic acid on fenofibrate‐induced elevation of homocysteine and cysteine. Am Heart J. 2003;146:110. [DOI] [PubMed] [Google Scholar]
- 96. Legendre C, Caussé E, Chaput E, Salvayre R, Pineau T, Edgar AD. Fenofibrate induces a selective increase of protein‐bound homocysteine in rodents: a PPARα‐mediated effect. Biochem Biophys Res Commun. 2002;295:1052–6. [DOI] [PubMed] [Google Scholar]
- 97. Olukman M, Sezer ED, Ülker S, Szmen EY, Çnar GM. Fenofibrate treatment enhances antioxidant status and attenuates endothelial dysfunction in streptozotocin‐induced diabetic rats. Exp Diabetes Res. 2010;2010:828531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Dahlhoff C, Desmarchelier C, Sailer M, Fürst RW, Haag A, Ulbrich SE, et al. Hepatic methionine homeostasis is conserved in c57bl/6n mice on high‐fat diet despite major changes in hepatic one‐carbon metabolism. PLoS One. 2013;8:e57387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tyagi N, Qipshidze N, Sen U, Rodriguez W, Ovechkin A, Tyagi SC. Cystathionine beta synthase gene dose dependent vascular remodeling in murine model of hyperhomocysteinemia. Int J Physiol Pathophysiol Pharmacol. 2011;3:210–22. [PMC free article] [PubMed] [Google Scholar]
- 100. Murthy S, Fonseca V, McNamara D. Hyperhomocysteinemia exacerbates the development of intimal hyperplasia in Sprague‐Dawley rats: alleviation by rosiglitazone. Exp Clin Cardiol. 2005;10:154–9. [PMC free article] [PubMed] [Google Scholar]
- 101. Fonseca V, Keebler M, Dicker‐Brown A, DeSouza C, Poirier LA, Murthy SN, et al. The effect of troglitazone on plasma homocysteine, hepatic and red blood cell S‐adenosyl methionine, and S‐adenosyl homocysteine and enzymes in homocysteine metabolism in Zucker rats. Metabolism. 2002;51:783–6. [DOI] [PubMed] [Google Scholar]
- 102. Derosa G, Cicero AFG, D’Angelo A, Gaddi A, Ciccarelli L, Piccinni MN, et al. Effects of 1 year of treatment with pioglitazone or rosiglitazone added to glimepiride on lipoprotein (a) and homocysteine concentrations in patients with type 2 diabetes mellitus and metabolic syndrome: a multicenter, randomized, double‐blind. Controlled Clin Ther. 2006;28:679–88. [DOI] [PubMed] [Google Scholar]
- 103. Murthy SN, Obregon DF, Chattergoon NN, Fonseca NA, Mondal D, Dunne JB, et al. Rosiglitazone reduces serum homocysteine levels, smooth muscle proliferation, and intimal hyperplasia in Sprague‐Dawley rats fed a high methionine diet. Metabolism. 2005;54:645–52. [DOI] [PubMed] [Google Scholar]
- 104. Cao X, Ding L, Xie ZZ, Yang Y, Whiteman M, Moore PK, et al. A review of hydrogen sulfide synthesis, metabolism, and measurement: is modulation of hydrogen sulfide a novel therapeutic for cancer? Antioxid Redox Signal. 2019;31:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhou S, Zhu Q, Li X, Chen C, Liu J, Ye Y, et al. Asymmetric dimethylarginine and all‐cause mortality: a systematic review and meta‐analysis. Sci Rep. 2017;7:44692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Sun B, Xie Y, Jiang J, Wang Y, Xu X, Zhao C, et al. Pleiotropic effects of fenofibrate therapy on rats with hypertriglycemia. Lipids Health Dis. 2015;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Stühlinger MC, Abbasi F, Chu JW, Lamendola C, McLaughlin TL, Cooke JP, et al. Relationship between insulin resistance and an endogenous nitric oxide synthase inhibitor. J Am Med Assoc. 2002;287:1420–6. [DOI] [PubMed] [Google Scholar]
- 108. Pilz S, Putz‐Bankuti C, Meinitzer A, März W, Kienreich K, Stojakovic T, et al. Association of homoarginine and methylarginines with liver dysfunction and mortality in chronic liver disease. Amino Acids. 2015;47:1817–26. [DOI] [PubMed] [Google Scholar]
- 109. Gehrke N, Schattenberg JM. Metabolic inflammation—a role for hepatic inflammatory pathways as drivers of comorbidities in nonalcoholic fatty liver disease? Gastroenterology. 2020;158:1929–1947.e6. [DOI] [PubMed] [Google Scholar]
- 110. Mori K, Emoto M, Araki T, Yokoyama H, Lee E, Teramura M, et al. Effects of pioglitazone on serum fetuin‐A levels in patients with type 2 diabetes mellitus. Metabolism. 2008;57:1248–52. [DOI] [PubMed] [Google Scholar]
- 111. Márkus B, Vörös K, Supák D, Melczer Z, Cseh K, Kalabay L. Association of PPAR alpha intron 7 G/C, PPAR gamma 2 Pro12Ala, and C161T polymorphisms with serum fetuin‐a concentrations. PPAR Res. 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos‐Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–37. [DOI] [PubMed] [Google Scholar]
- 113. Tang TT, Li YY, Li JJ, Wang K, Han Y, Dong WY, et al. Liver‐heart crosstalk controls IL‐22 activity in cardiac protection after myocardial infarction. Theranostics. 2018;8:4552–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Yoo HJ, Hwang SY, Choi JH, Lee HJ, Chung HS, Seo JA, et al. Association of leukocyte cell‐derived chemotaxin 2 (LECT2) with NAFLD, metabolic syndrome, and atherosclerosis. PLoS ONE. 2017;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hwang HJ, Jung TW, Hong HC, Seo JA, Kim SG, Kim NH, et al. LECT2 induces atherosclerotic inflammatory reaction via CD209 receptor‐mediated JNK phosphorylation in human endothelial cells. Metabolism. 2015;64:1175–82. [DOI] [PubMed] [Google Scholar]
- 116. Smati S, Régnier M, Fougeray T, Polizzi A, Fougerat A, Lasserre F, et al. Regulation of hepatokine gene expression in response to fasting and feeding: influence of PPAR‐α and insulin‐dependent signalling in hepatocytes. Diabetes Metab. 2020;46:129–36. [DOI] [PubMed] [Google Scholar]
- 117. Mouchiroud M, Camiré É, Aldow M, Caron A, Jubinville É, Turcotte L, et al. The hepatokine Tsukushi is released in response to NAFLD and impacts cholesterol homeostasis. JCI Insight. 2019;4:e129492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Potze W, Siddiqui MS, Boyett SL, Adelmeijer J, Daita K, Sanyal AJ, et al. Preserved hemostatic status in patients with non‐alcoholic fatty liver disease. J Hepatol. 2016;65:980–7. [DOI] [PubMed] [Google Scholar]
- 119. Tripodi A, Fracanzani AL, Primignani M, Chantarangkul V, Clerici M, Mannucci PM, et al. Procoagulant imbalance in patients with non‐alcoholic fatty liver disease. J Hepatol. 2014;61:148–54. [DOI] [PubMed] [Google Scholar]
- 120. Kotronen A, Joutsi‐Korhonen L, Sevastianova K, Bergholm R, Hakkarainen A, Pietiläinen KH, et al. Increased coagulation factor VIII, IX XI and XII activities in non‐alcoholic fatty liver disease. Liver Int. 2011;31:176–83. [DOI] [PubMed] [Google Scholar]
- 121. Targher G, Bertolini L, Scala L, Zenari L, Lippi G, Franchini M, et al. Plasma PAI‐1 levels are increased in patients with nonalcoholic steatohepatitis. Diabetes Care. 2007;30. [DOI] [PubMed] [Google Scholar]
- 122. Lowe G, Rumley A. The relevance of coagulation in cardiovascular disease: what do the biomarkers tell us? Thromb Haemost. 2014;112:860–7. [DOI] [PubMed] [Google Scholar]
- 123. Ramadori P, Klag T, Malek NP, Heikenwalder M. Platelets in chronic liver disease, from bench to bedside. JHEP Reports. 2019;1:448–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Lu Y, Harada M, Kamijo Y, Nakajima T, Tanaka N, Sugiyama E, et al. Peroxisome proliferator‐activated receptor α attenuates high‐cholesterol diet‐induced toxicity and pro‐thrombotic effects in mice. Arch Toxicol. 2019;93:149–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Lu Y, Boekschoten MV, Wopereis S, Müller M, Kersten S. Comparative transcriptomic and metabolomic analysis of fenofibrate and fish oil treatments in mice. Physiol Genomics. 2011;43:1307–18. [DOI] [PubMed] [Google Scholar]
- 126. Bairaktari ET, Tzallas CS, Tsimihodimos VK, Liberopoulos EN, Miltiadous GA, Elisaf MS. Comparison of the efficacy of atorvastatin and micronized fenofibrate in the treatment of mixed hyperlipidemia. J Cardiovasc Risk. 1999;6:113–6. [DOI] [PubMed] [Google Scholar]
- 127. Kimura T, Nakajima T, Kamijo Y, Tanaka N, Wang L, Hara A, et al. Hepatic cerebroside sulfotransferase is induced by PPARα activation in mice. PPAR Res. 2012;2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kyogashima M. The role of sulfatide in thrombogenesis and haemostasis. Arch Biochem Biophys. 2004;426:157–62. [DOI] [PubMed] [Google Scholar]
- 129. Li G, Hu R, Gu J, Wu HZ. Relationship between carotid artery atherosclerosis and sulfatide in hypertensive patients. Genet Mol Res. 2015;14:4840–6. [DOI] [PubMed] [Google Scholar]
- 130. Li G, Hu R. Association between serum sulfatide and carotid intima media thickness in patients with familial hypercholesterolemia. Glycoconj J. 2014;31:587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Li G, Hu R, Guo Y, He L, Zuo Q, Wang Y. Circulating sulfatide, a novel biomarker for st‐segment elevation myocardial infarction. J Atheroscler Thromb. 2019;26:84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Szalowska E, Tesfay HA, van Hijum SAFT, Kersten S. Transcriptomic signatures of peroxisome proliferator‐activated receptor α (PPARα) in different mouse liver models identify novel aspects of its biology. BMC Genomics. 2014;15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Li D, Chen K, Sinha N, Zhang X, Wang Y, Sinha AK, et al. The effects of PPAR‐γ ligand pioglitazone on platelet aggregation and arterial thrombus formation. Cardiovasc Res. 2005;65:907–12. [DOI] [PubMed] [Google Scholar]
- 134. Oishi K, Uchida D, Ohkura N, Horie S. PPARα deficiency augments a ketogenic diet‐induced circadian PAI‐1 expression possibly through PPARγ activation in the liver. Biochem Biophys Res Commun. 2010;401:313–8. [DOI] [PubMed] [Google Scholar]
- 135. Verrijken A, Francque S, Mertens I, Prawitt J, Caron S, Hubens G, et al. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2014;59:121–9. [DOI] [PubMed] [Google Scholar]
- 136. Kostallari E, Valainathan S, Biquard L, Shah VH, Rautou PE. Role of extracellular vesicles in liver diseases and their therapeutic potential. Adv Drug Deliv Rev. 2021;175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Szabo G, Momen‐Heravi F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nat Rev Gastroenterol Hepatol. 2017;14:455–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Baron M, Leroyer AS, Majd Z, Lalloyer F, Vallez E, Bantubungi K, et al. PPARα activation differently affects microparticle content in atherosclerotic lesions and liver of a mouse model of atherosclerosis and NASH. Atherosclerosis. 2011;218:69–76. [DOI] [PubMed] [Google Scholar]
- 139. Yin X, Zeng W, Wu B, Wang L, Wang Z, Tian H, et al. PPARα inhibition overcomes tumor‐derived exosomal lipid‐induced dendritic cell dysfunction. Cell Rep. 2020;33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Looze C, Yui D, Leung L, Ingham M, Kaler M, Yao X, et al. Proteomic profiling of human plasma exosomes identifies PPARγ as an exosome‐associated protein. Biochem Biophys Res Commun. 2009;378:433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Francque S, Vonghia L. Pharmacological treatment for non‐alcoholic fatty liver disease. Advan Therapy. 2019;36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Gastaldelli A, Harrison S, Belfort‐Aguiar R, Hardies J, Balas B, Schenker S, et al. Pioglitazone in the treatment of NASH: the role of adiponectin. Aliment Pharmacol Ther. 2010;32:769–75. [DOI] [PubMed] [Google Scholar]
- 143. Gawrieh S, Noureddin M, Loo N, Mohseni R, Awasty V, Cusi K, et al. Saroglitazar, a PPAR‐α/γ agonist, for treatment of nafld: a randomized controlled double‐blind phase 2 trial. Hepatology. 2021. 10.1002/hep.31843 [DOI] [PubMed] [Google Scholar]
- 144. Dormandy JA, Charbonnel B, Eckland DJA, Erdmann E, Massi‐Benedetti M, Moules IK, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial in macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–89. [DOI] [PubMed] [Google Scholar]
- 145. Mazzone T, Meyer PM, Feinstein SB, Davidson MH, Kondos GT, D’Agostino RB, et al. Effect of pioglitazone compared with glimepiride on carotid intima‐media thickness in type 2 diabetes: a randomized trial. J Am Med Assoc. 2006;296:2572–81. [DOI] [PubMed] [Google Scholar]
- 146. Nissen SE, Nicholls SJ, Wolski K, Nesto R, Kupfer S, Perez A, et al. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial. J Am Med Assoc. 2008;299:1561–73. [DOI] [PubMed] [Google Scholar]
- 147. Young LH, Viscoli CM, Schwartz GG, Inzucchi SE, Curtis JP, Gorman MJ, et al. Heart failure after ischemic stroke or transient ischemic attack in insulin‐resistant patients without diabetes mellitus treated with pioglitazone. Circulation. 2018;138:1210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Clarke GD, Solis‐Herrera C, Molina‐Wilkins M, Martinez S, Merovci A, Cersosimo E, et al. Pioglitazone improves left ventricular diastolic function in subjects with diabetes. Diabetes Care. 2017;40:1530–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lehrke M, Marx N. Diabetes Mellitus and Heart Failure. Am J Med. 2017;130:S40–50. [DOI] [PubMed] [Google Scholar]