

Abstract
Aims
To compare the airway potency, systemic activity and therapeutic index of three inhaled corticosteroids that differ in glucocorticoid receptor binding affinity, physicochemical and pharmacokinetic properties.
Methods
This escalating‐dose, placebo‐controlled, cross‐over study randomised adults with asthma to 1 or 2 treatment periods with ≥25 days washout in‐between. Each treatment period comprised five 7‐day dose escalations (μg/d): fluticasone furoate (FF; 25 → 100 → 200 → 400 → 800), fluticasone propionate (FP; 50 → 200 → 500 → 1000 → 2000), budesonide (BUD; 100 → 400 → 800 → 1600 → 3200) or placebo. Airway hyperresponsiveness to adenosine‐5'‐monophosphate (AMP PC20) was assessed on day 8. Plasma cortisol was assessed on day 1 (predose baseline) and from pre‐PM dose on day 6 to pre‐PM dose day 7 (24‐h weighted mean).
Results
Fifty‐four subjects were randomised. FF showed greater airway potency than FP and BUD (AMP PC20 dose at which 50% of the maximum effect is achieved [ED50] values: 48.52, 1081.27 and 1467.36 μg/d, respectively). Systemic activity (cortisol suppression) ED50 values were 899.99, 1986.05 and 1927.42 μg/d, respectively. The therapeutic index (ED50 cortisol suppression/ED50 AMP PC20) was wider for FF (18.55) than FP (1.84) and BUD (1.31). FF 100 μg/d and 200 μg/d were both comparable in terms of airway potency with high doses of FP (≥1000 μg twice daily [BID]) and BUD (≥1500 μg/BID). The systemic activity of FF 100 μg/d and 200 μg/d (cortisol suppression: 7.41% and 14.28%, respectively) was comparable with low doses of FP (100 μg/BID and 250 μg/BID) and BUD (100 μg/BID and 200 μg/BID).
Conclusion
This study provides evidence that FF can provide more protection against airway hyperresponsiveness, with less systemic activity, than FP or BUD. This suggests that all inhaled corticosteroids are not therapeutically similar and may differ in their therapeutic index. (203162; NCT02991859).
Keywords: AMP challenge, asthma, budesonide, fluticasone furoate, fluticasone propionate, therapeutic index
What is already known about this subject
Inhaled corticosteroid (ICS) molecules are known to vary in their pharmacodynamic, physicochemical and pharmacokinetic properties, but are widely regarded as therapeutically similar.
Potency differences between ICS are thought to be overcome by administering larger doses of a less potent drug to achieve therapeutic equivalence.
Is this assumption correct?
What this study adds
This study provides evidence that ICS may differ in their therapeutic index.
When compared at therapeutic doses, fluticasone furoate, with higher anti‐inflammatory activity and greater lung retention, has greater airway potency with lower systemic activity than fluticasone propionate and budesonide, which exhibit lower airway potency with more systemic activity.
1. INTRODUCTION
Inhaled corticosteroids (ICS) are the mainstay of asthma pharmacotherapy. 1 Currently available ICS molecules for clinical use have a wide range of pharmacodynamic, physicochemical and pharmacokinetic properties. 2 Despite these differences, all ICS molecules are widely regarded as therapeutically similar, where potency differences are thought to be overcome by administering larger doses of a less potent ICS to achieve therapeutic equivalence. This view is also reflected in dose equivalence tables within asthma treatment guidelines. 1 An article describing the theoretical basis for why this assumption is likely to be incorrect was previously published in this Journal. 2
We now present the results of a clinical trial that tests this earlier hypothesis, which proposed that the molecular structural features of ICS that increase glucocorticoid receptor (GR) binding affinity and selectivity also result in physicochemical and pharmacokinetic changes that together may enhance pulmonary targeting and reduce systemic exposure. Therefore, ICS molecules with greater anti‐inflammatory potency (greater GR binding affinity and selectivity) are also likely to have longer lung retention and enhanced tissue permeability, and should achieve efficacy with lower and less frequent dosing. 2 A lower dose regimen would also have the advantage of reduced systemic exposure. Together, these characteristics should confer a therapeutic advantage for newer ICS molecules with these features compared with older ICS molecules, the latter requiring higher and more frequent dosing due to their lower potency and shorter lung retention. 2
Fluticasone furoate (FF), fluticasone propionate (FP) and budesonide (BUD) are ICS approved for pharmacotherapy of asthma. 3 , 4 , 5 Of these, FF has the highest relative GR binding affinity (2989) compared to FP (1775), BUD (935) and dexamethasone (100). 2 Other important differences when assessing the relative airway and systemic potency of these molecules, are that FF and FP have negligible oral bioavailability and similar lung and systemic drug delivery when administered via their respective dry powder inhalers (ELLIPTA and DISKUS), whereas BUD has significant oral bioavailability and approximately 2‐fold greater lung and systemic drug delivery via the Turbuhaler dry powder inhaler. 2
This study was designed to assess the airway potency, systemic activity and therapeutic index (TI) of these three ICS molecules, which were selected on the basis of their different GR binding affinities and lung retention times. Airway hyperresponsiveness is a key feature of asthma, and adenosine‐5’‐monophosphate (AMP) challenge is an established method for evaluating the protective effects of ICS in reducing airway inflammation and thereby hyperresponsiveness in asthma clinical trials. 6 , 7 , 8 The most important systemic adverse effect of ICS is suppression of the hypothalamic–pituitary–adrenal (HPA) axis, and the use of cortisol as a marker for the unwanted systemic activity of ICS is well established. 9 , 10 In this study, we compared the dose–responses for topical efficacy via airway hyperresponsiveness to AMP challenge, and dose–responses for systemic activity via 24‐hour plasma cortisol suppression, and thereby the relative TI (systemic activity/airway potency ratio) for FF, FP and BUD in subjects with asthma.
2. METHODS
2.1. Study design
This randomised, escalating‐dose, placebo‐controlled, incomplete‐block, 2‐period, cross‐over study was conducted at 2 clinical research centres in the UK and 1 centre in Germany (GlaxoSmithKline plc. Study 203162; ClinicalTrials.gov NCT02991859).
Subjects were randomised to undertake 1 or 2 treatment periods separated by a 25–42 day washout period. Randomisation schedules were generated by the sponsor using validated internal software (RandAll NG, GlaxoSmithKline plc.). Each treatment period comprised 5 consecutive dose escalations each of 7 days' duration, for a total of 35 days: FF ELLIPTA (25 → 100 → 200 → 400 → 800 μg/d); FP DISKUS (50 → 200 → 500 → 1000 → 2000 μg/d); BUD Turbuhaler (100 → 400 → 800 → 1600 → 3200 μg/d); or placebo (either ELLIPTA or DISKUS) administered to match the active treatments. Salbutamol rescue medication use was permitted from screening and throughout the study as required.
This study was partially blinded. All treatments were supplied packaged and labelled in an open‐label fashion. An unblinded pharmacist at each clinic site blinded the supplies before dispensing them to the subjects. The subjects were provided with a dry powder inhaler for each dose‐escalation phase, either active treatment or placebo. DISKUS placebo and ELLIPTA placebo were used in this study; a Turbuhaler placebo was not used in the study, but subjects were unaware of this. In addition to the subjects, the investigators, nurses, technicians and other staff who interacted with the subjects were blinded to the treatment designations, as were the wider study team.
Doses on days 1, 6, 7 and 8 of each treatment period that were taken in the clinical unit were supervised. Doses on the other days of each treatment period were self‐administered by subjects at home and were recorded by subjects on a diary card; entries were checked and reconciled against the inhaler dose counter. It was prespecified that if subjects missed critical doses or made dosing errors, the data for that dose‐escalation period would be excluded from the data analysis.
All FF doses, FP 50 μg and BUD 100 μg were administered once daily in the evening; all other FP and BUD doses were administered twice daily (morning and evening, 12 h apart). This reflects the indicated posology for FF, FP and BUD. The selected doses included those both below and above the approved doses for asthma, to allow the full efficacy and systemic activity dose–response to be explored, but were within the ranges used in previous dose‐ranging studies. 11 , 12 , 13 , 14
The study was conducted in accordance with International Conference on Harmonisation Good Clinical Practice guidelines. 15 The study was approved by local ethics committees for the participating institutions (Ethikkommission des Landes Berlin, Berlin, Germany [reference number 17/0432‐EK 10], and North West – GM South, NHS Health Research Authority, Manchester, UK [reference number 16/NW/0781]). All subjects provided written informed consent.
2.2. Subjects
Subjects were aged 18–65 years, with body weight ≥50 kg, body mass index 18–35 kg/m2, and a history of bronchial asthma first diagnosed at least 6 months prior to screening. Subjects had to demonstrate a prebronchodilator forced expiratory volume in 1 second (FEV1) ≥65% predicted at screening (to be maintained at ≥65% predicted at the predose baseline assessment) and have documented sensitivity to AMP with a provocative concentration of AMP resulting in a decline of ≥20% in FEV1 (AMP PC20) of ≤80 mg/mL at screening. Subjects could have received short‐acting β2‐agonists for at least 12 weeks prior to screening. Those on prescribed low‐dose ICS were eligible after a 4‐week ICS washout period prior to a screening AMP challenge (provided asthma symptoms remained stable). Light smokers (≤20 cigarettes per week or equivalent) and those with smoking history <10 pack‐years were also eligible; these subjects had to satisfy all other screening and inclusion criteria and were required to refrain from smoking for ≥1 hour prior to lung function tests.
Key exclusion criteria were a history of life‐threatening asthma and other significant pulmonary diseases within 6 months of screening. Subjects who received long‐acting β2‐agonist, long‐acting muscarinic antagonist or leukotriene receptor antagonist therapy within 3 months, or biological therapies within 6 months, prior to study start were excluded.
All subjects received inhaler training prior to the first dose of each treatment period, with reinforcement of proper technique at subsequent visits prior to further dose administration.
2.3. Objectives
The study objectives were: to characterise the dose–response and relative potency of FF, FP and BUD in reducing airway hyperresponsiveness assessed via AMP challenge (AMP PC20); to characterise the dose–response and relative potency for FF, FP and BUD on 0–24 hour plasma cortisol suppression, expressed as plasma cortisol suppression relative to placebo; and to determine the TI for FF, FP and BUD, as measured by ED50 0–24 hour weighted mean plasma cortisol suppression (dose at which 50% of the maximum plasma cortisol suppression is achieved)/ED50 for AMP PC20 (dose at which 50% of the maximum protection against airway hyperresponsiveness [AMP PC20] is achieved).
Safety and tolerability were also assessed.
2.4. Assessments
2.4.1. AMP challenge
After each 7‐day dose‐escalation phase of each treatment period, an AMP challenge was performed on the morning of day 8, 12 ± 2 hours after cessation of dosing. Evening dosing was chosen for all FF doses, FP 50 μg and BUD 100 μg doses; other doses were administered morning and evening to allow the AMP challenge to be performed 12 hours postdose and at the same time of day for each treatment. This allowed estimation of true potency differences between treatments (which would have been confounded had AMP challenge been performed at different times post‐dose for FF, FP and BUD). The AMP challenge agent was produced according to Good Manufacturing Practice standard and provided in vials of different dilutions (0.04, 0.08, 0.16, 0.32, 0.63, 1.25, 2.5, 5.0, 10.0, 20.0, 40.0, 80.0, 160.0 and 320.0 mg/mL) by Stockport Pharmaceuticals (Stockport, UK).
An initial assessment established the baseline FEV1 (≥65% predicted required). Subjects then inhaled 5 breaths of 0.9% diluent, via a nebuliser with breath‐activated dosimeter of known output, by inspiring slowly from functional residual capacity to total lung capacity over 3 seconds, followed by breath holding for 6 seconds. FEV1 was repeated after 60 and 180 seconds post‐diluent nebulisation, with the higher value recorded as the post‐diluent FEV1. Following the same process, subjects were then administered doubling concentrations of AMP (from 0.04 to 320.0 mg/mL) until a ≥20% fall in FEV1 from the post‐diluent value was achieved, i.e. the AMP PC20 value. Where necessary this required extrapolation or interpolation between adjacent AMP concentrations on a log scale.
2.4.2. Plasma cortisol
Blood samples for determination of plasma cortisol concentration were taken on day 1 (predose; baseline) and over the entire 24‐hour dose interval between day 6 and day 7 of each dose escalation (day 6 pre‐evening dose, then 1, 2, 3, 5, 10, 12, 14, 16, 18 and 24 hours postdose). Samples were analysed by liquid chromatography–mass spectrometry at Covance Bioanalytical Services (Indianapolis, IN, USA).
2.4.3. Safety
Adverse events (AEs) were monitored throughout the study. During each treatment period and washout period, subjects were given a peak flow meter for home use to measure peak expiratory flow rate (PEFR) prior to each dose (morning and evening, as appropriate). Subjects recorded the highest PEFR of 3 measurements before each dose in a paper diary. Safety assessments also included ongoing assessment of asthma stability by the investigator to identify worsening asthma, including asthma exacerbations.
2.5. Statistical analysis
Following a protocol amendment (3 October 2017) to increase study recruitment, subjects were enrolled to complete either 1 or 2 treatment periods. The sample size was planned such that 48 subjects would complete the study; 24 subjects were to be enrolled to complete 2 treatment periods and a further 24 to complete 1 treatment period. Determination of sample size was based on estimates of variability from earlier studies and simulations using the Emax model, 16 , 17 which were then used to estimate the predicted width of the 95% confidence intervals (CIs) for the model parameter estimates in the study (described below). The AMP PC20 endpoint was chosen for sample size estimation since it was shown in earlier studies that 24‐hour weighted mean cortisol suppression is a less variable endpoint. 18 , 19 Precision estimates were calculated on the feasible sample size per study treatment, based on 12 subjects in a cross‐over design and 6 subjects in a parallel design, and the combined estimates for 18 subjects in total per treatment.
Baseline characteristics and safety were analysed in the total study population (all randomised subjects who received at least 1 dose of study medication). AMP challenge and plasma cortisol endpoints were analysed in the pharmacodynamics (PD) population (subjects in the total study population who had at least 1 postdose PD measurement).
Prior to the dose–response analysis, the AMP challenge and plasma cortisol suppression results for placebo across the 5 dose‐escalation phases were analysed using a repeated measures analysis of variance model. The weighted mean plasma cortisol values for the placebo treatment arms showed no observable changes across the weekly assessments. There was also no observable effect of repeated weekly AMP challenge on placebo responses and, as such, the dose–response modelling was conducted with the assumption that each of the placebo responses in the 5 dose‐escalation phases were the same. Therefore, the means of pooled placebo data for AMP challenge and for cortisol suppression, irrespective of the escalation phase, were used as E0 (response at zero dose) for the respective dose–response models.
For the AMP PC20 dose–response analysis, it was planned to fit a 3‐parameter maximum effect (Emax) model with subject term fitted as a random effect (equivalent to adding a random coefficient to the E0 parameter), and to include a common Emax if the estimated Emax values appeared similar, otherwise the model was to be fitted with different Emax for each ICS. However, after unblinding, visual inspection of the observed data vs fitted plot, showed the corresponding model was not best fit. The planned model was modified and fitted by adding random effects for Emax and ED50, assuming a common Emax across FF, FP and BUD, and with an unstructured variance–covariance matrix, and including log2‐transformed predose AMP PC20 value as an additional covariate. The final Emax model implemented took the form: log2 (PC20) = E0 + Emax/(1 + {((ED50)/dose) ^power)}) + log2 (predose PC20). Point estimates and corresponding 95% CIs were constructed for each of the 3 parameters from the Emax model (response at E0 and Emax, and the ED50 for each ICS).
The planned analysis for the plasma cortisol suppression data was to fit a 3‐parameter Emax model. However, after unblinding, the Emax model was deemed inappropriate, since visual inspection showed no evidence of a saturating minimal (Emax) effect with increasing dose of ICS. Therefore, an inhibitory exponential power‐law model was fitted for cortisol in lieu of Emax and 100% inhibition at infinite dose was assumed, which was implemented in the form: loge (cortisol 24‐h weighted mean) = E0 + slope*log e (dose)^power + predose.
The parameters of the dose–response models (ED50 weighted mean cortisol suppression/ED50 AMP PC20) were used to calculate the TI for FF, FP and BUD. As an additional parameter, the dose of FF, FP and BUD that resulted in 20% cortisol suppression was also estimated as the ED20 weighted mean cortisol suppression.
Analyses were conducted using SAS v.9.4 (SAS Institute Inc., Cary, NC, USA).
Additional methodological details, including full study eligibility criteria, study randomisation and masking, permitted and prohibited medications and non‐drug therapies, and withdrawal/stopping criteria are provided in the supplementary material.
2.6. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.
3. RESULTS
Between 9 February 2017 and 20 December 2018, 56 subjects were enrolled; of these, 54 were randomised to treatment (26 consented to 1 treatment period; 28 consented to 2 treatment periods) and received at least 1 dose of study medication (total study population). Fifty‐two subjects were included in the PD population. Forty‐five subjects (83%) completed the study as planned and 9 (17%) withdrew prematurely, mostly due to withdrawal of consent (n = 7, 13%; Figure 1). Mean age was 37.9 years, 41 subjects (76%) were male and 19 subjects (35%) were current or former smokers (Table 1). No subjects had a reported past medical condition, while 3 had a current medical condition (two [4%] hypertension, 1 [2%] hypercholesterolaemia). Most subjects were taking a concomitant medication during the study or within 28 days prior to screening, the most frequent of which were salbutamol (43 subjects [80%]), paracetamol (19 subjects [35%]), beclometasone dipropionate (9 subjects [17%]), ibuprofen (7 subjects [13%]) and loratadine (5 subjects [9%]; all other concomitant medications were used by ≤2 subjects).
FIGURE 1.
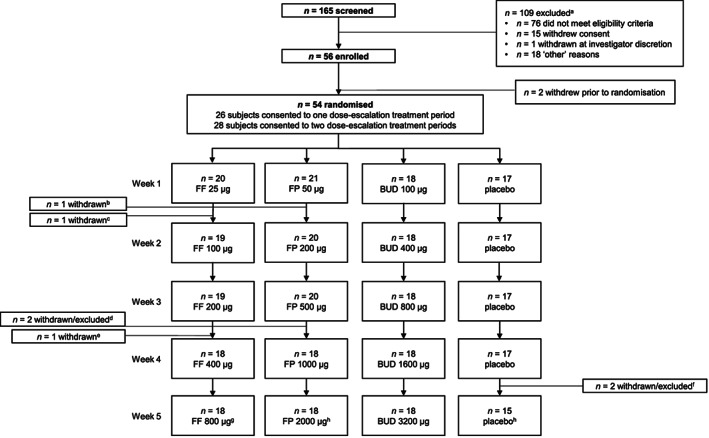
Subject flow through the study. Note: dose refers to total daily dose of the relevant treatment. aReasons for screening failure were not mutually exclusive; subjects could have more than 1 reason for exclusion at the screening stage. bOne subject withdrew during treatment period 1, FP 50 μg. cOne subject withdrew during treatment period 1, FF 25 μg. dOne subject withdrew during treatment period 1, FP 500 μg; a second subject completed treatment period 1, FP 500 μg, but withdrew from the study before entering FP 1000 μg. eOne subject withdrew during treatment period 1, FF 200 μg. fOne subject withdrew during treatment period 2, placebo 4; another subject did not enter treatment period 1, placebo 5, but was not withdrawn from the study and continued into treatment period 2. gOne subject completed treatment period 2, FF 800 μg, but withdrew from the study before completing final study assessments. hTwo subjects completed treatment period 1 (placebo 5 and FP 2000 μg, respectively), but withdrew during the washout period before entering treatment period 2. BUD = budesonide; FF = fluticasone furoate; FP = fluticasone propionate
TABLE 1.
Subject demographics and baseline characteristics (total study population)
| Characteristic | n = 54 |
|---|---|
| Mean age (SD), y | 37.9 (13.96) |
| Male, n (%) | 41 (76) |
| Mean BMI (SD), kg/m2 | 26.07 (3.65) |
| Race, n (%) | |
| White, white/Caucasian/European | 39 (72) |
| Black or African American | 7 (13) |
| Other | 8 (15) |
| Mean FEV1, L (SD) | 3.27 (0.81) |
| Mean FEV1% predicted, L (SD) | 85.53 (12.91) |
| Mean AMP PC20, mg/mL (SD) | 15.15 (16.30) |
| Smoking history, n (%) a | |
| Never | 35 (65) |
| Current smoker | 3 (6) |
| Former smoker | 16 (30) |
Sum of percentages for individual categories exceeds 100% due to rounding.
AMP PC20 = provocative concentration of adenosine‐5′‐monophosphate causing a ≥20% decline in FEV1; BMI = body mass index; FEV1 = forced expiratory volume in 1 second; SD = standard deviation.
FF showed greater airway potency than either FP or BUD (Table 2 and Figure 2), with AMP PC20 ED50 μg/d values of 48.52 (95% CI: 18.21–129.32), 1081.27 (95% CI: 448.00–2609.66) and 1467.36 (95% CI: 546.51–3939.84), respectively. AMP PC20 point estimates showed that the efficacy of FF 100 and 200 μg/d were both comparable with FP ≥2000 μg/d and BUD ≥3000 μg/d. AMP PC20 values for FF doses ≥200 μg/d exceeded the response for any FP or BUD doses, and at their lowest approved doses for asthma, FF provided 3.5–4 times more airway protection than FP or BUD.
TABLE 2.
AMP PC20 by total daily dose and treatment (pharmacodynamic population)
| Total daily dose | AMP PC20 values (mg/mL) | |
|---|---|---|
| Geometric mean a | 95% CI | |
| FF 25 μg (n = 19) | 33.45 | 19.10–58.60 |
| FF 100 μg (n = 19) | 81.45 | 44.65–148.58 |
| FF 200 μg (n = 18) | 115.69 | 66.82–200.31 |
| FF 400 μg (n = 18) | 145.97 | 85.02–250.59 |
| FF 800 μg (n = 17) | 167.26 | 95.36–293.37 |
| FP 50 μg (n = 20) | 15.19 | 10.80–21.36 |
| FP 200 μg (n = 20) | 20.47 | 13.94–30.07 |
| FP 500 μg (n = 19) | 31.39 | 18.88–52.19 |
| FP 1000 μg (n = 17) | 48.67 | 27.30–86.78 |
| FP 2000 μg (n = 17) | 76.35 | 43.21–134.91 |
| BUD 100 μg (n = 18) | 16.00 | 11.41–22.44 |
| BUD 400 μg (n = 18) | 23.91 | 15.08–37.90 |
| BUD 800 μg (n = 18) | 34.62 | 19.28–62.16 |
| BUD 1600 μg (n = 18) | 54.33 | 28.40–103.93 |
| BUD 3200 μg (n = 18) | 84.17 | 45.48–155.79 |
The analysis method was a 3‐parameter Emax model with log2‐transformed AMP PC20 as the outcome variable, assuming common Emax across FF, FP and BUD, and with an unstructured variance–covariance matrix. Random effects were included for E0, ED50 and Emax, and log2‐transformed predose AMP PC20 value was included as an additional covariate.
n refers to the number of subjects with available data for each dose‐escalation phase.
Estimate of geometric mean and 95% CIs for AMP PC20 were obtained on the log2 scale and back‐transformed.
AMP PC20 = provocative concentration of adenosine‐5′‐monophosphate causing a ≥20% decline in forced expiratory volume in 1 second; BUD = budesonide; CI = confidence interval; E0 = response at zero dose; ED50 = dose at which 50% of the maximum effect is achieved; Emax = maximum effect; FF = fluticasone furoate; FP = fluticasone propionate
FIGURE 2.
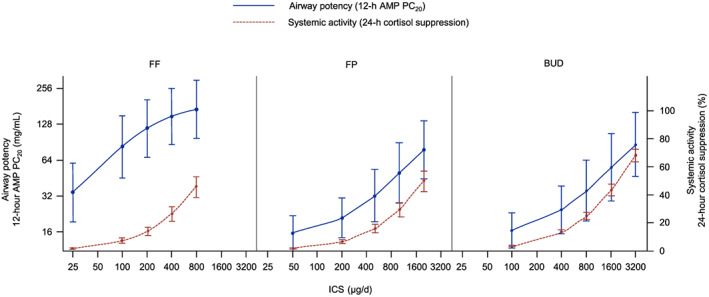
Airway potency (AMP PC20) and systemic activity (0–24 h weighted mean plasma cortisol suppression) by total daily dose and treatment (pharmacodynamic population). Note: Estimates and 95% CIs for AMP PC20 were obtained on the log2 scale and back‐transformed. Error bars represent the 95% CIs. AMP PC20 = provocative concentration of adenosine‐5′‐monophosphate causing a ≥20% decline in forced expiratory volume in 1 second; BUD = budesonide; CI = confidence interval; FF = fluticasone furoate; FP = fluticasone propionate; ICS = inhaled corticosteroid
All FF, FP and BUD doses produced some degree of systemic activity as measured by 0–24‐hour weighted mean plasma cortisol suppression (Table 3 and Figure 2), with ED50 μg/d values of 899.99 (95% CI: 698.36–1101.62), 1986.05 (95% CI: 1574.70–2397.39) and 1927.42 (95% CI: 1698.47–2156.37), respectively. Point estimates showed FF 100 and 200 μg/d to be comparable in systemic activity (cortisol suppression; 7.41 and 14.28%, respectively) with FP 200 and 500 μg/d (6.74 and 16.01% cortisol suppression, respectively). FF 100 and 200 μg/d were also similar to estimates for BUD 200 μg/d and 400 μg/d (≈7 and 13.4% cortisol suppression, respectively). FP 1000 μg/d had similar efficacy to BUD 1600 μg/d, but with less cortisol suppression (29.46 vs 43.75%; Figure S1).
TABLE 3.
Dose–response analysis for estimated 0–24 hour weighted mean plasma cortisol (nmol/L) and % cortisol suppression relative to placebo (pharmacodynamic population)
| Total daily dose | Estimated 0–24 hour weighted mean plasma cortisol, nmol/L | % cortisol suppression relative to placebo | ||
|---|---|---|---|---|
| Geometric mean a | 95% CI | Geometric mean a | 95% CI | |
| Placebo b (n = 17) | 176.09 | 162.87–190.38 | – | – |
| FF 25 μg (n = 19) | 172.73 | 159.88–186.62 | 1.91 | 1.48–2.33 |
| FF 100 μg (n = 19) | 163.03 | 151.01–176.02 | 7.41 | 5.80–9.00 |
| FF 200 μg (n = 18) | 150.95 | 139.48–163.37 | 14.28 | 11.27–17.18 |
| FF 400 μg (n = 18) | 129.40 | 117.82–142.12 | 26.51 | 21.26–31.41 |
| FF 800 μg (n = 17) | 95.09 | 82.26–109.93 | 46.00 | 38.00–52.96 |
| FP 50 μg (n = 20) | 173.04 | 160.15–186.97 | 1.73 | 1.37–2.08 |
| FP 200 μg (n = 20) | 164.22 | 152.11–177.29 | 6.74 | 5.38–8.08 |
| FP 500 μg (n = 19) | 147.89 | 136.59–160.13 | 16.01 | 12.92–18.99 |
| FP 1000 μg (n = 17) | 124.21 | 112.88–136.68 | 29.46 | 24.17–34.38 |
| FP 2000 μg (n = 17) | 87.62 | 75.37–101.85 | 50.24 | 42.50–56.94 |
| BUD 100 μg (n = 18) | 169.87 | 157.23–183.52 | 3.53 | 3.12–3.94 |
| BUD 400 μg (n = 18) | 152.50 | 141.26–164.63 | 13.40 | 11.91–14.87 |
| BUD 800 μg (n = 18) | 132.06 | 122.05–142.90 | 25.00 | 22.39–27.52 |
| BUD 1600 μg (n = 18) | 99.05 | 90.24–108.72 | 43.75 | 39.77–47.47 |
| BUD 3200 μg (n = 18) | 55.71 | 48.26–64.31 | 68.36 | 63.73–72.40 |
The analysis method was an inhibitory exponential power‐law model with loge‐transformed cortisol as the outcome variable, assuming 100% inhibition at infinite doses. A random effect was included for E0 and loge‐transformed predose cortisol value was included as an additional covariate. The power parameter was set to 1, to reduce correlation between the estimates.
Difference in cortisol value from placebo was estimated for each ICS dose on the loge scale as dose*slope. Percent cortisol suppression was then calculated for each dose as: (100 × [1 – exp {difference in cortisol value from placebo}]).
n refers to the number of subjects with available data for each dose‐escalation phase.
Estimate of geometric mean and 95% CIs were obtained on the loge scale and back‐transformed.
Pooled placebo analysis (ELLIPTA and DISKUS placebo inhalers).
BUD = budesonide; CI = confidence interval; E0 = response at zero dose; FF = fluticasone furoate; FP = fluticasone propionate; ICS = inhaled corticosteroid
FF had a higher airway potency to systemic activity ratio (Figure 3), and therefore a wider TI compared with FP and BUD. The TI value, based on ED50 for 0–24‐hour weighted mean plasma cortisol suppression/ED50 for AMP PC20, for FF was 18.55 vs 1.84 for FP and 1.31 for BUD (Table 4).
FIGURE 3.
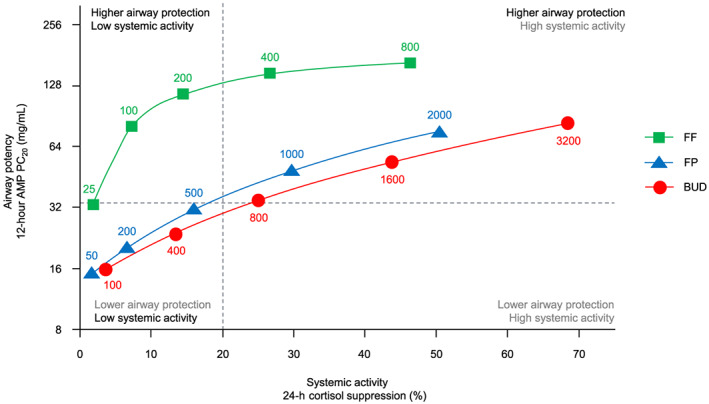
Comparison of airway potency and systemic activity by total daily dose of FF, FP and BUD (pharmacodynamic population). Note: The horizontal dashed reference line represents the cut‐off for high‐dose ICS, as defined by GINA (FP >500 μg/d and BUD >800 μg/d). The vertical dashed line is a reference point for a low level of plasma cortisol suppression (20% or ED20 for cortisol suppression, as estimated in this study). AMP PC20 = provocative concentration of adenosine‐5′‐monophosphate causing a ≥20% decline in forced expiratory volume in 1 second; BUD = budesonide; ED20 = dose at which 20% of the maximum effect is reached; FF = fluticasone furoate; FP = fluticasone propionate; GINA = Global Initiative for Asthma; ICS = inhaled corticosteroid
TABLE 4.
Summary of dose–response analysis for ED50 0–24 hour weighted mean plasma cortisol suppression/ED50 for AMP PC20 (pharmacodynamic population)
| Treatment | Parameter | Geometric mean a | 95% CI |
|---|---|---|---|
| FF | ED50 0–24 hour weighted mean plasma cortisol suppression | 899.99 μg/d | 698.36–1101.62 |
| ED50 for AMP PC20 | 48.52 μg/d | 18.21–129.32 | |
| TI (ED50 cortisol suppression/ED50 AMP PC20) | 18.55 b , e , f | 6.43–53.50 | |
| FP | ED50 0–24 hour weighted mean plasma cortisol suppression | 1986.05 μg/d | 1574.70–2397.39 |
| ED50 for AMP PC20 | 1081.27 μg/d | 448.00–2609.66 | |
| TI (ED50 cortisol suppression/ED50 AMP PC20) | 1.84 c , g | 0.706–4.78 | |
| BUD | ED50 0–24 hour weighted mean plasma cortisol suppression | 1927.42 μg/d | 1698.47–2156.37 |
| ED50 for AMP PC20 | 1467.36 μg/d | 546.51–3939.84 | |
| TI (ED50 cortisol suppression/ED50 AMP PC20) | 1.31 d | 0.487–3.54 |
Estimate of geometric mean and 95% CIs were obtained on the log scale and back‐transformed.
Difference vs unity P < 0.0001.
Difference vs unity P =0.1.
Difference vs unity P = 0.3.
Difference FF vs FP P = 0.00075.
Difference FF vs BUD P = 0.00018.
Difference FP vs BUD P = 0.683.
AMP PC20 = provocative concentration of adenosine‐5′‐monophosphate causing a ≥20% decline in forced expiratory volume in 1 second; BUD = budesonide; CI = confidence interval; ED50 = dose at which 50% of the maximum effect is achieved; FF = fluticasone furoate; FP = fluticasone propionate; TI = therapeutic index.
On‐treatment AE rates were 59% placebo (pooled across escalation phases), 30–42% FF, 20–48% FP and 17–44% BUD (Table S1). On‐treatment drug‐related AE rates were 12% placebo, 10–21% FF, 0–11% FP and 6–17% BUD. The most frequent drug‐related AE was headache (placebo 12%; FF 0–11%; FP 0%; BUD 0–11%). There was no apparent relationship between AE incidence and dose escalation for any study drug. There were no on‐treatment serious AEs, AEs leading to permanent treatment discontinuation, or AEs leading to dose reduction, interruption or delay during the study. Subjects self‐monitored PEFR throughout the clinical phase of the study; no recorded values were of clinical concern per investigator assessment.
4. DISCUSSION
Within their approved dose ranges in asthma, FF was more efficacious in reducing airway hyperresponsiveness with less systemic activity, and had the widest TI, compared with FP or BUD.
In the AMP challenge model, the potency of FF was greater than both FP and BUD. The numerical difference between the AMP PC20 ED50 values for FP and BUD was broadly in line with their relative GR‐binding affinities. 2 The airway efficacy (AMP PC20) of FP 1000 μg/d was also similar to BUD 1600 μg/d, which is in agreement with their established relative clinical efficacies. 1 For FF, the relative airway potency was greater than predicted from its relative GR‐binding affinity compared with FP and BUD. A similar finding was reported for FF in dose‐ranging studies that used trough FEV1 as the endpoint and FP as the comparator. 11 , 12 , 13 This is probably due to a combination of the physicochemical, pharmacokinetic and pharmacodynamic properties of FF. High lipophilicity and tissue permeability, low solubility and slow dissolution of inhaled drug particles, result in FF being absorbed approximately 3‐ and 6‐fold more slowly from the lung than FP and BUD, respectively. 2 In addition, FF has a high affinity for, and slow dissociation from, the GR. These factors may act multiplicatively to enhance the airway potency of FF and prolong its duration of action in reducing airway hyperresponsiveness, 20 making it suitable for once‐daily dosing with low doses.
Over the entire dose ranges studied, FF, FP and BUD demonstrated varying degrees of systemic activity. The ED20 values were 289.73, 639.36 and 620.49 μg for FF, FP and BUD, respectively. For FF, this dose is above the maximum approved dose for asthma (200 μg predispensed dose = 184 μg device emitted dose). For FP and BUD, the doses were well within their approved adult dose ranges for asthma (100–1000 μg twice daily for FP and 200–800 μg twice daily for BUD). The relative potencies for systemic activity were consistent with the relative GR binding affinities of FF, FP and BUD after allowing for differences in total drug delivery via the different inhalers and the oral bioavailability of the ICS molecules. The BUD Turbuhaler delivers approximately twice as much drug to the lung and systemically than the FP DISKUS inhaler and BUD has significant oral bioavailability; 21 , 22 whereas for FF and FP, the ELLIPTA and DISKUS inhalers have a similar drug delivery performance and both FF and FP have negligible oral bioavailability. 2 The ED50 values were 899.99, 1986.05 and 1927.42 μg for FF, FP and BUD, respectively (relative systemic potency ratio 2:1:1).
The considerably wider TI (ED50 cortisol suppression/ED50 AMP PC20) for FF (18.55) compared with FP (1.84) or BUD (1.31) was still evident even when the ED50 for efficacy was calculated using only last administered PM dose (rather than the total daily doses) for FP and BUD, which gave TI values for FP (3.67) and BUD (2.63), still much lower than FF (18.55). It was also notable that the higher efficacy (AMP PC20) of FF compared to FP and BUD was already apparent after the first week of dosing, during which all 3 ICS were administered once daily in the evening. This was despite FF being administered at 2‐fold and 4‐fold lower doses than FP and BUD, respectively. These findings are not explained by the higher GR binding affinity of FF alone, but more likely by a combination of higher GR affinity, a slower GR dissociation rate, higher tissue permeability and longer lung retention compared to FP and BUD. These characteristics permit lower and less frequent dosing of FF, resulting in less drug becoming available systemically. Although FF has the same potential for systemic activity as other ICS, this was not observed at the low doses sufficient for high efficacy in the lung. In this regard, these findings are in agreement with previous dose‐ranging studies 11 , 12 , 13 identifying FF 100 and 200 μg/d as the optimal doses for efficacy; lower doses were less efficacious, but higher doses offered little additional efficacy yet were associated with more systemic activity.
The study design employed had some key strengths and weaknesses. A perceived strength of the study was that it included a sufficiently wide dose range of each ICS to define both the efficacy that occurs with lower doses and the systemic activity that occurs with higher doses, and included 5 dose levels that allowed for a dose–response model to be used. This is a major improvement on previous studies that used only higher ICS doses and included at most 3 dose levels or employed pair‐wise comparisons rather than dose–response modelling. 23 , 24 Although all practical attempts were made to ensure that subjects adhered to their medication and administered it correctly throughout the study, no formal evaluation of adherence to study treatments was undertaken. Hence, adherence differences between treatment groups may have occurred. However, the individual plasma cortisol data provided evidence of high treatment adherence in this study.
A perceived weakness of the study is the short duration of the dose‐escalation phases in comparison to the durations used in conventional asthma efficacy trials. The 7‐day treatment period was based on the half‐lives of the 3 ICS all being <1 day 2 and steady‐state being achieved within 4 days (≥5 half‐lives). Many studies have measured the efficacy of ICS via reductions in airway hyperresponsiveness using a variety of challenge agents and where the duration of dosing has varied from a single dose to several weeks of treatment. 25 The timing of the challenge after cessation of dosing also varied from 1 hour to 2 weeks postdose. Even a single inhalation of ICS can reduce airway hyperresponsiveness, and after 72 hours of dosing 26 or 7 BID doses, 27 much of the achievable effect is seen. Notably, single‐dose FF 100 μg decreased methacholine airway hyperresponsiveness at 24 hours without significant further improvement with continued daily use over 7 days. 28 On cessation of dosing, the effect is lost quite rapidly following single and short‐term dosing, and even after 6 weeks of dosing most of the effect is lost within a week. 29 The specific question of whether a study design with short‐term dose‐escalations is sufficient to characterise ICS dose–response to airway hyperresponsiveness has previously been investigated using quadrupling dose‐escalation increments of 1 or 2 weeks duration. 6 The efficacy and dose–response relationships were similar to those seen after 3 weeks of continuous dosing at the highest dose. Based on these previous findings, we concluded that using ICS dose‐escalation phases of 7‐days' duration, together with a wide dose range, would be a suitable design for defining the relative dose–response whilst minimising any carry‐over from previous doses; that is, a 7‐day dose‐escalation phase would be a long enough time period to produce most of the efficacy achievable with longer dosing durations, but short enough to avoid the requirement for a longer washout period or the requirement for subjects to remain in the study for prolonged periods, which risks reduced adherence to study treatments, a higher rate of study withdrawals and increased variability due to temporal factors affecting disease variability. This appears to have been successful since there is no evidence from the study data or apparent from the Emax dose–response model fits that there were any confounding carry‐over effects due to the cumulative dose or cross‐over design. The dose‐escalation increments were the same for each ICS and over the entire 40‐fold dose range studied, the AMP PC20 increased from the lowest dose to the highest dose by a factor of 5 for all 3 ICS. Notably, the difference seen in AMP PC20 between the 3 ICS that was already apparent after the first week of each dose‐escalation phase was also maintained during the subsequent weeks of dose‐escalation. Any significant carry‐over effects, or a higher degree of carry‐over for 1 ICS in particular, would have been apparent in the shapes of the dose–response curves as the dose‐escalations progressed.
Another perceived strength of the study was the timing of the AMP challenge assessment following the cessation of dosing. In previous studies that have measured the efficacy of ICS via reductions in airway hyperresponsiveness, many were unable to demonstrate a clear dose–response relationship. 25 This is probably due to the doses being at the top of the dose–response curve, or because subjects were on ICS during run‐in and/or because the challenge assessment was conducted too soon following the last dose. Some published AMP challenge studies have not specified the exact time of the challenge relative to the last dose, but where it was specified, the endpoint was often measured between 1 and 26 hours postdose. 20 , 30 Longer times have been employed when looking at washout of effects. 29 It is apparent from these studies that when the AMP challenge is performed too soon after cessation of dosing (1 h) the dose–response is not defined. 23 This is probably due to the presence of a large amount of drug from the last dose inhibiting the release of inflammatory mediators, irrespective of the degree of underlying inflammation. Performing AMP challenge at later times minimises this effect, so the response is more likely to reflect the underlying inflammation and its reduction arising from the cumulative pharmacological effect from all the preceding doses. On cessation of dosing, this pharmacological effect appears to be maintained for 24–48 hours, but this is likely to be dose‐dependent and hence low doses may be difficult to distinguish from placebo much beyond 24 hours. 20 , 23 , 29 , 30 In our study, on cessation of dosing we selected a 12 ± 2 hour assessment window for the AMP challenge to maximise the chances of demonstrating a dose–response. Thus, we were able to demonstrate a clear dose–response for each ICS in reducing airway hyperresponsiveness due to the 40‐fold wide dose range and appropriate timing of the AMP challenge assessment relative to the cessation of dosing.
The cortisol suppression data and dose–response model fit also showed no evidence of any confounding carry‐over effects due to the cumulative dose and cross‐over design. The cortisol suppression values observed were entirely consistent with previously reported values including from studies without dose‐escalations or parallel‐group designs that had the same or longer treatment periods. 23 , 24 , 31 , 32 To assess ICS effects on the HPA axis, various cortisol endpoints have been used either during or after cessation of ICS dosing. Partial or complete 24‐hour urine sampling can be variable due to incomplete samples and impractical when required to be repeated on many occasions. Timed (8:00–10:00) AM blood (serum or plasma) sampling is also variable and more suited to diagnostic rather than dose–response analysis. HPA axis stimulation testing with adrenocorticotropic hormone is also more suited to diagnostic testing of adrenal insufficiency or suppression rather than dose–response analysis since short‐term dosing with therapeutic or low doses of ICS is unlikely to result in significant changes. A further consideration is that repeated adrenocorticotropic hormone testing in subjects with asthma is not advised as it risks hypersensitivity reactions. Therefore, we used 24‐hour serial blood sampling to assess plasma cortisol during days 6 and 7 whilst dosing was still ongoing. This is not a measure of adrenal insufficiency per se but cortisol suppression relative to placebo is a measure of systemic exposure to ICS and a clinically relevant endpoint suitable for dose–response modelling.
Historically, potency differences between corticosteroids are thought to be overcome by administering larger doses of a less potent drug to achieve therapeutic equivalence. 33 , 34 This principle may be valid for systemic corticosteroids, 35 but not necessarily for ICS, as we have demonstrated here. Our findings question the validity of the current approach to ICS dose equivalence embedded in asthma treatment guidelines, which do not consider efficacy and safety separately, and incorrectly assume that low‐ and high‐dose categories inevitably correspond with low and high risk of systemic and other side‐effects. For example, the Global Initiative for Asthma 1 low‐, medium‐ and high‐dose (μg/d) classifications for FF (low: 100, high: 200), FP (low: 100–250, medium: >250–500, high: >500) and BUD (low: 200–400, medium: >400–800, high: >800) imply that a high‐dose classification is associated with a higher risk of long‐term side‐effects compared to the low‐ and medium‐dose classifications. The high efficacy of FF 100 μg and 200 μg does not appear to be associated with a correspondingly high risk of systemic activity in this study or others. 11 , 12 , 13
In conclusion, this study provides evidence that all ICS molecules are not therapeutically similar. Across the approved doses for asthma, FF gave more protection against airway hyperresponsiveness with less systemic activity, and had a wider TI (systemic activity/airway potency ratio), than FP or BUD. These findings are relevant to asthma treatment guidelines that currently assume that there is no difference in TI between ICS molecules.
COMPETING INTERESTS
P.D.‐Y., N.Br., S.T., D.A., S.S. and N.Ba. disclose employment with, and stock/share ownership in, GlaxoSmithKline plc. T.H. reports receiving personal fees for advisory boards from GlaxoSmithKline plc., AstraZeneca, Synairgen and Vectura, as well as receiving fees for speaker meetings from AstraZeneca. D.S. reports receiving personal fees from AstraZeneca, Boehringer Ingelheim, Chiesi, Cipla, Genentech, GlaxoSmithKline plc., Glenmark, Menarini, Mundipharma, Novartis, Peptinnovate, Pfizer, Pulmatrix, Theravance and Verona.
CONTRIBUTORS
P.D.‐Y., N.Br., T.H., D.S. and N.Ba. were involved in study conception/design; S.S. was involved in data acquisition; P.D.‐Y., S.T., D.A. and N.Ba. were involved in data analysis and/or interpretation. All authors were involved in writing/critical review of draft versions of this manuscript and all approved the final version for submission for publication.
Supporting information
TABLE S1 On‐treatment adverse effects reported in ≥2 subjects in any study dose‐escalation phase (total study population)
FIGURE S1 Comparison of airway protection and systemic activity for approved therapeutic doses for asthma in adults of FF, FP and BUD (pharmacodynamic population). Note: the blue bars represent AMP PC20 values >80 mg/mL of the study inclusion definition for AMP hyperresponsiveness. The grey bars represent AMP PC20 values within the 80 mg/mL study inclusion definition for AMP hyperresponsiveness. The green bars represent cortisol suppression values below the reference point for a low level of plasma cortisol suppression (20% or ED20 for cortisol suppression as estimated in this study). The red bars represent values that approach or exceed 50% cortisol suppression. The amber bars represent an intermediate level of cortisol suppression. AMP: adenosine‐5′‐monophosphate; AMP PC20: provocative concentration of AMP causing a ≥ 20% decline in forced expiratory volume in 1 second; BUD: budesonide; ED20: dose at which 20% of the maximum effect is reached; FF: fluticasone furoate; FP: fluticasone propionate
ACKNOWLEDGEMENTS
This study was funded by GlaxoSmithKline plc. (study 203162; ClinicalTrials.gov identifier: NCT02991859). Editorial support (in the form of editorial suggestions to draft versions of this paper, assembling tables and figures, collating author comments, copyediting, referencing and graphic services) was provided by Emma Landers, PhD, of Gardiner‐Caldwell Communications (Macclesfield, UK) and was funded by GlaxoSmithKline plc. Trademarks are owned by or licensed to their respective owners (the GSK group of companies or AstraZeneca). Employees of the sponsor (GlaxoSmithKline plc.) were involved in the conception and/or design of the study, data acquisition, data analysis and/or interpretation, writing and critical review of the manuscript, approval of the final version and in the decision to submit the manuscript for publication. The corresponding author, P.D.‐Y., confirms that he had full access to all data in the study and had final responsibility for the decision to submit for publication. Authors did not receive any payment for writing this article. Dave Singh is supported by the National Institute for Health Research (NIHR) Manchester Biomedical Research Centre (BRC).
Daley‐Yates P, Brealey N, Thomas S, et al. Therapeutic index of inhaled corticosteroids in asthma: A dose–response comparison on airway hyperresponsiveness and adrenal axis suppression. Br J Clin Pharmacol. 2021;87:483–493. 10.1111/bcp.14406
Principal Investigator: The authors confirm that the Principal Investigator for this paper is Prof. Dave Singh and that he had direct clinical responsibility for subjects.
DATA AVAILABILITY STATEMENT
Anonymised individual participant data and study documents can be requested for further research from: www.clinicalstudydatarequest.com. Further information on GlaxoSmithKline's data sharing commitments can be found at: https://www.clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-GSK.aspx.
REFERENCES
- 1. Global Initiative for Asthma (GINA) . Global Strategy for Asthma Management and Prevention, 2019 update. https://ginasthma.org/wp-content/uploads/2019/06/GINA-2019-main-report-June-2019-wms.pdf. Accessed October 25, 2019.
- 2. Daley‐Yates PT. Inhaled corticosteroids: potency, dose equivalence and therapeutic index. Br J Clin Pharmacol. 2015;80(3):372‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. ARNUITY ELLIPTA (fluticasone furoate inhalation powder), for oral inhalation use . Highlights of prescribing information. Updated June 2019. https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Arnuity_Ellipta/pdf/ARNUITY%2010ELLIPTA%2010PI%2010PIL%2010IFU.PDF. Accessed November 27, 2019.
- 4. Flixotide 50, 125, 250 micrograms Evohaler . Fluticasone propionate. Summary of product characteristics. Updated 30 October 2019. https://www.medicines.org.uk/emc/product/3824/smpc. Accessed November 27, 2019.
- 5. Pulmicort Turbohaler 200 . Budesonide. Summary of product characteristics. Updated 15 June 2017. https://www.medicines.org.uk/emc/product/1385/smpc. Accessed November 27, 2019.
- 6. Phillips K, Oborne J, Harrison TW, Tattersfield AE. Use of sequential quadrupling dose regimens to study efficacy of inhaled corticosteroids in asthma. Thorax. 2004;59(1):21‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Joos GF, O'Connor B, Anderson SD, et al. Indirect airway challenges. Eur Respir J. 2003;21(6):1050‐1068. [DOI] [PubMed] [Google Scholar]
- 8. Singh D, Fairwood J, Murdoch R, et al. The reproducibility of adenosine monophosphate bronchial challenges in mild, steroid‐naive asthmatics. Br J Clin Pharmacol. 2008;66(2):261‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dahl R. Systemic side effects of inhaled corticosteroids in patients with asthma. Respir Med. 2006;100(8):1307‐1317. [DOI] [PubMed] [Google Scholar]
- 10. Pandya D, Puttanna A, Balagopal V. Systemic effects of inhaled corticosteroids: an overview. Open Respir Med J. 2014;8(1):59‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Busse WW, Bleecker ER, Bateman ED, et al. Fluticasone furoate demonstrates efficacy in patients with asthma symptomatic on medium doses of inhaled corticosteroid therapy: an 8‐week, randomised, placebo‐controlled trial. Thorax. 2012;67(1):35‐41. [DOI] [PubMed] [Google Scholar]
- 12. Bateman ED, Bleecker ER, Lötvall J, et al. Dose effect of once‐daily fluticasone furoate in persistent asthma: a randomized trial. Respir Med. 2012;106(5):642‐650. [DOI] [PubMed] [Google Scholar]
- 13. Bleecker ER, Bateman ED, Busse WW, et al. Once‐daily fluticasone furoate is efficacious in patients with symptomatic asthma on low‐dose inhaled corticosteroids. Ann Allergy Asthma Immunol. 2012;109(5):353‐358. [DOI] [PubMed] [Google Scholar]
- 14. Kaiser H, Aaronson D, Dockhorn R, Edsbäcker S, Korenblat P, Källén A. Dose‐proportional pharmacokinetics of budesonide inhaled via Turbuhaler. Br J Clin Pharmacol. 1999;48(3):309‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. International Conference on Harmonisation of technical requirements for registration of pharmaceuticals for human use. ICH harmonised guideline integrated addendum to ICH E6(R1): guideline for good clinical practice ICH E6(R2) ICH Consensus Guideline. https://ichgcp.net/. Accessed March 19, 2020.
- 16. GlaxoSmithKline . GSK Clinical Study Register. Study SIG102335 Scientific Results Summary. https://s3.amazonaws.com/ctr-gsk-7381/SIG102335/b8665aa1-690e-4514-967e-e2c2e0bd44cc/27c450e3-6733-452e-9ac2-0e5a0b91e78f/gsk-102335-synopsis-redact-v1.pdf Accessed March 30, 2020.
- 17. GlaxoSmithKline . GSK Clinical Study Register. Study SIG103337 Clinical Pharmacology Study Report. https://s3.amazonaws.com/ctr-gsk-7381/SIG103337/bd9b54da-4de9-4153-8a29-881304d0982a/f82610a1-5ed2-42c1-86d3-169fc32c2a86/gsk-sig103337-clinical-study-report-redact-v1.pdf Accessed March 30, 2020.
- 18. Nakahara N, Wakamatsu A, Kempsford R, et al. The safety, pharmacokinetics and pharmacodynamics of a combination of fluticasone furoate and vilanterol in healthy Japanese subjects. Int J Clin Pharmacol Ther. 2013;51(8):660‐671. [DOI] [PubMed] [Google Scholar]
- 19. Kempsford R, Allen A, Bareille P, Hamilton M, Cheesbrough A. The pharmacodynamics, pharmacokinetics, safety and tolerability of inhaled fluticasone furoate and vilanterol administered alone or simultaneously as fluticasone furoate/vilanterol. Clin Pharmacol Drug Dev. 2015;4(1):2‐11. [DOI] [PubMed] [Google Scholar]
- 20. van den Berge M, Luijk B, Bareille P, Dallow N, Postma DS, Lammers JWJ. Prolonged protection of the new inhaled corticosteroid fluticasone furoate against AMP hyperresponsiveness in patients with asthma. Allergy. 2010;65(12):1531‐1535. [DOI] [PubMed] [Google Scholar]
- 21. Agertoft L, Pedersen S. Lung deposition and systemic availability of fluticasone Diskus and budesonide Turbuhaler in children. Am J Respir Crit Care Med. 2003;168(7):779‐782. [DOI] [PubMed] [Google Scholar]
- 22. Cahn A, Allen A, Russell P, et al. Lower systemic exposure to corticosteroid for fluticasone propionate/salmeterol (500/50mcg bd) compared to budesonide/formoterol (400/12mcg bd) administered via combination dry powder inhalers in subjects with COPD [abstract]. Eur Respir J. 2006;28(Suppl 50):1229.16971403 [Google Scholar]
- 23. Derom E, Van De Velde V, Marissens S, Engelstätter R, Vincken W, Pauwels R. Effects of inhaled ciclesonide and fluticasone propionate on cortisol secretion and airway responsiveness to adenosine 5’monophosphate in asthmatic patients. Pulm Pharmacol Ther. 2005;18(5):328‐336. [DOI] [PubMed] [Google Scholar]
- 24. Nielsen LP, Dahl R. Therapeutic ratio of inhaled corticosteroids in adult asthma. A dose–range comparison between fluticasone propionate and budesonide, measuring their effect on bronchial hyperresponsiveness and adrenal cortex function. Am J Respir Crit Care Med. 2000;162(6):2053‐2205. [DOI] [PubMed] [Google Scholar]
- 25. van Grunsven PM, van Schayck CP, Molema J, Akkermans RP, van Weel C. Effect of inhaled corticosteroids on bronchial responsiveness in patients with “corticosteroid naive” mild asthma: a meta‐analysis. Thorax. 1999;54(4):316‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sovijärvi ARA, Haahtela T, Ekroos HJ, et al. Sustained reduction in bronchial hyperresponsiveness with inhaled fluticasone propionate within three days in mild asthma: time course after onset and cessation of treatment. Thorax. 2003;58(6):500‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ketchell RI, Jensen MW, Lumley P, Wright AM, Allenby MI, O'Connor BJ. Rapid effect of inhaled fluticasone propionate on airway responsiveness to adenosine 5′‐monophosphate in mild asthma. J Allergy Clin Immunol. 2002;110(4):603‐606. [DOI] [PubMed] [Google Scholar]
- 28. Okonkwo CS, Davis BE, Blais CM, Cockcroft DW. Short‐term effect of once‐daily fluticasone furoate on methacholine‐induced bronchoconstriction in mild asthmatics. Respir Med. 2019;156:53‐57. [DOI] [PubMed] [Google Scholar]
- 29. Vathenen AS, Knox AJ, Wisniewski A, Tattersfield AE. Time course of change in bronchial reactivity with an inhaled corticosteroid in asthma. Am Rev Respir Dis. 1991;143(6):1317‐1321. [DOI] [PubMed] [Google Scholar]
- 30. Luijk B, Kempsford RD, Wright AM, Zanen P, Lammers JWJ. Duration of effect of single‐dose inhaled fluticasone propionate on AMP‐induced bronchoconstriction. Eur Respir J. 2004;23(4):559‐564. [DOI] [PubMed] [Google Scholar]
- 31. Allen A. The relationship between fluticasone furoate systemic exposure and cortisol suppression. Clin Pharmacokinet. 2013;52(10):885‐896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Allen A, Schenkenberger I, Trivedi R, et al. Inhaled fluticasone furoate/vilanterol does not affect hypothalamic‐pituitary‐adrenal axis function in adolescent and adult asthma: randomised, double‐blind, placebo‐controlled study. Clin Respir J. 2013;7(4):397‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kamada AK, Szefler SJ, Martin RJ, et al. Issues in the use of inhaled glucocorticoids. The asthma clinical research network. Am J Respir Crit Care Med. 1996;153(6 Pt 1):1739‐1748. [DOI] [PubMed] [Google Scholar]
- 34. Kelly HW. Establishing a therapeutic index for the inhaled corticosteroids: part I: pharmacokinetic/pharmacodynamic comparison of the inhaled corticosteroids. J Allergy Clin Immunol. 1998;102(4 Pt 2):S36‐S51. [DOI] [PubMed] [Google Scholar]
- 35. Mager DE, Lin SX, Blum RA, Lates CD, Jusko WJ. Dose equivalency evaluation of major corticosteroids: pharmacokinetics and cell trafficking and cortisol dynamics. J Clin Pharmacol. 2003;43(11):1216‐1227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 On‐treatment adverse effects reported in ≥2 subjects in any study dose‐escalation phase (total study population)
FIGURE S1 Comparison of airway protection and systemic activity for approved therapeutic doses for asthma in adults of FF, FP and BUD (pharmacodynamic population). Note: the blue bars represent AMP PC20 values >80 mg/mL of the study inclusion definition for AMP hyperresponsiveness. The grey bars represent AMP PC20 values within the 80 mg/mL study inclusion definition for AMP hyperresponsiveness. The green bars represent cortisol suppression values below the reference point for a low level of plasma cortisol suppression (20% or ED20 for cortisol suppression as estimated in this study). The red bars represent values that approach or exceed 50% cortisol suppression. The amber bars represent an intermediate level of cortisol suppression. AMP: adenosine‐5′‐monophosphate; AMP PC20: provocative concentration of AMP causing a ≥ 20% decline in forced expiratory volume in 1 second; BUD: budesonide; ED20: dose at which 20% of the maximum effect is reached; FF: fluticasone furoate; FP: fluticasone propionate
Data Availability Statement
Anonymised individual participant data and study documents can be requested for further research from: www.clinicalstudydatarequest.com. Further information on GlaxoSmithKline's data sharing commitments can be found at: https://www.clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-GSK.aspx.