

SUMMARY
Background
For patients with UC, flexible maintenance dosing therapy may confer advantages for safety, efficacy, costs and patient preference. Tofacitinib is an oral, small molecule JAK inhibitor for the treatment of UC.
Aim
To assess the efficacy and safety of tofacitinib dose de‐escalation and escalation in patients with UC.
Methods
We evaluated data (November 2017 data cut‐off) from OCTAVE Open, an ongoing, open‐label, long‐term extension study. The dose de‐escalation group comprised 66 tofacitinib induction responders in remission following 52 weeks' tofacitinib 10 mg b.d. maintenance therapy, subsequently de‐escalated to 5 mg b.d. in OCTAVE Open. The dose escalation group comprised 57 tofacitinib induction responders who experienced treatment failure while receiving 5 mg b.d. maintenance therapy, subsequently escalated to 10 mg b.d. in OCTAVE Open.
Results
After tofacitinib de‐escalation, 92.4% (61/66) and 84.1% (53/63) of patients maintained clinical response and 80.3% (53/66) and 74.6% (47/63) maintained remission, at months 2 and 12, respectively. After dose escalation, 57.9% (33/57) and 64.9% (37/57) of patients recaptured clinical response and 35.1% (20/57) and 49.1% (28/57) were in remission, at months 2 and 12, respectively. The incidence rate of herpes zoster with dose escalation (7.6 patients with events/100 patient‐years) was numerically higher than in the overall tofacitinib UC programme.
Conclusions
Following tofacitinib de‐escalation in patients already in remission on 10 mg b.d., most maintained remission, although 25.4% lost remission, at month 12. For induction responders who dose‐escalated following treatment failure on 5 mg b.d. maintenance therapy, 49.1% achieved remission by month 12. (ClinicalTrials.gov number: NCT01470612).
Short abstract
LINKED CONTENT
This article is linked to Amiot et al paper. To view this article, visit https://doi.org/10.1111/apt.15638.
1. INTRODUCTION
For patients with ulcerative colitis (UC), the goals of treatment are to induce and maintain disease remission, to improve patients’ quality of life, and to prevent both disease‐ and medication‐related complications.1, 2 It is not uncommon for therapy to be de‐escalated: (a) to reduce the risk of dose‐dependent side effects associated with long‐term immunosuppression; (b) for the management of intercurrent illnesses (such as bacterial or certain viral infections), for which concomitant, full‐dose immunosuppressant therapies might impede or delay recovery; (c) to reduce the burden of therapy for patients in sustained remission; and (d) to decrease the cost of treatment.3, 4 However, for patients who experience a disease flare or lose response, the ability to recapture the initial response by escalating the dose is an important consideration.
Tofacitinib is an oral, small molecule Janus kinase (JAK) inhibitor for the treatment of UC. Three global, phase 3, randomised, double‐blind, placebo‐controlled trials (OCTAVE Induction 1 and 2, NCT01465763 and NCT01458951; and OCTAVE Sustain, NCT01458574) reported the efficacy and safety of tofacitinib as induction and maintenance therapy for patients with moderately to severely active UC.5 OCTAVE Open (NCT01470612) is an ongoing, open‐label, long‐term extension study that enrolled patients who completed or lost response to treatment in OCTAVE Sustain, and also included non‐responders from OCTAVE Induction 1 and 2.6
The safety profile of tofacitinib in the UC clinical programme appeared similar to that reported in patients with rheumatoid arthritis and—with the exception of higher rates of herpes zoster infection—similar to biologic therapies for the treatment of UC.7 Dose‐related increases in the rate of herpes zoster infections observed during OCTAVE Sustain5, 7, 8 and an increased incidence of venous thromboembolic events manifested as pulmonary embolism events observed in patients treated with tofacitinib 10 mg twice daily (b.d.) compared to tofacitinib 5 mg b.d. or tumour necrosis factor inhibitors in one large ongoing randomised post‐authorisation safety surveillance study in patients with rheumatoid arthritis who were 50 years or older with at least one cardiovascular risk factor, provide a rationale for treating patients with the lowest effective dose for maintenance therapy. For patients who lose response to tofacitinib 5 mg b.d. as maintenance therapy, escalating the dose to 10 mg b.d. may be a suitable treatment strategy. However, tofacitinib 10 mg b.d. should be used with caution in patients for whom 10 mg b.d. is the recommended dose and in whom venous thromboembolic risk factors are identified.
We analysed the efficacy and safety of two adjustments of tofacitinib maintenance dosing for patients in OCTAVE Open: (a) dose de‐escalation to tofacitinib 5 mg b.d. for patients in remission following 52 weeks of maintenance therapy with tofacitinib 10 mg b.d.; and (b) dose escalation to tofacitinib 10 mg b.d. for patients who lost response while receiving tofacitinib 5 mg b.d. as maintenance therapy (Figure 1).
Figure 1.
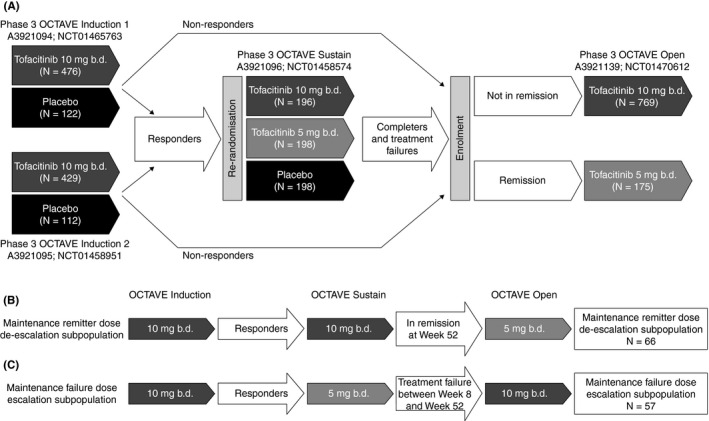
(A) Overview of the phase 3 OCTAVE programme, and treatment sequences for patients in (B) the tofacitinib maintenance remitter dose de‐escalation subpopulation and (C) the tofacitinib maintenance failure dose escalation subpopulation. OCTAVE Open is ongoing; data as of November 2017 data cut‐off. b.d., twice daily; N, number of patients included in each treatment group or subpopulation
2. PATIENTS AND METHODS
2.1. Patients and study design
OCTAVE Open is an ongoing, phase 3, multicentre, open‐label, long‐term extension study that enrolled patients who completed or demonstrated treatment failure in OCTAVE Sustain, as well as patients who completed OCTAVE Induction 1 and 2 without clinical response.6
Efficacy and safety data up to the November 2017 data cut‐off are presented. Analyses are presented for the following patients in OCTAVE Open (Figure 1): (a) the tofacitinib maintenance remitter dose de‐escalation subpopulation, defined as: clinical responders to tofacitinib 10 mg b.d. at week 8 of OCTAVE Induction 1 and 2; in remission following 52 weeks of maintenance therapy with tofacitinib 10 mg b.d. in OCTAVE Sustain; dose de‐escalation to tofacitinib 5 mg b.d. in OCTAVE Open; and (b) the tofacitinib maintenance failure dose escalation subpopulation, defined as: clinical responders to tofacitinib 10 mg b.d. at week 8 in OCTAVE Induction 1 and 2; treatment failure with tofacitinib 5 mg b.d. (between weeks 8 and 52) in OCTAVE Sustain; dose escalation to tofacitinib 10 mg b.d. in OCTAVE Open.
Clinical response was defined as a decrease from induction study baseline total Mayo score of ≥3 points and ≥30%, plus a decrease in rectal bleeding subscore of ≥1 point or an absolute rectal bleeding subscore of 0 or 1. Treatment failure was defined as increase from OCTAVE Sustain baseline total Mayo score of ≥3 points, plus an increase in rectal bleeding subscore and endoscopic subscore of ≥1 point and an absolute endoscopic subscore ≥2 after at least 8 weeks of maintenance therapy.
Patients who entered OCTAVE Open in remission (defined as a total Mayo score of ≤2 with no individual subscore >1, and a rectal bleeding subscore of 0) received tofacitinib 5 mg b.d. per protocol. Patients not in remission received tofacitinib 10 mg b.d. Accordingly, patients in the dose de‐escalation subpopulation qualified to receive tofacitinib 5 mg b.d. in OCTAVE Open, and patients in the dose escalation subpopulation qualified to receive tofacitinib 10 mg b.d. in OCTAVE Open due to treatment failure in OCTAVE Sustain while receiving tofacitinib 5 mg b.d. Patients who experienced a UC flare in OCTAVE Open could have their dose increased from 5 mg b.d. to 10 mg b.d. Flare in OCTAVE Open was defined by an increase in total Mayo score of ≥3 points from OCTAVE Sustain baseline, plus an increase in rectal bleeding subscore of ≥1 point and an increase in endoscopic subscore of ≥1 point (unless the endoscopic subscore was “3” at baseline and remained “3”) after a minimum of 8 weeks of treatment in OCTAVE Open.
Prohibited concomitant therapies in OCTAVE Open included azathioprine, mercaptopurine, methotrexate, tumour necrosis factor inhibitors and anti‐adhesion therapies. Concomitant therapy with oral 5‐aminosalicylates and sulfasalazine was permitted in OCTAVE Open. Oral corticosteroids were permitted for patients entering OCTAVE Open who were taking corticosteroids at the time of entry into the study (maximum dose of 25 mg/day of oral prednisone or equivalent).
2.2. Endpoints and analyses
2.2.1. Efficacy
Efficacy endpoints assessed at baseline, month 2, month 12 and month 24 of OCTAVE Open included: clinical response, mucosal healing (a Mayo endoscopic subscore of 0 or 1) and remission. Efficacy endpoints at baseline of OCTAVE Open were derived from the Mayo score, based on centrally read endoscopic subscore; efficacy endpoints at months 2, 12 and 24 were derived based on locally read endoscopic subscore.
The full analysis set comprised all patients who received at least one dose of study drug in OCTAVE Open. Efficacy was analysed in both subpopulations of the full analysis set. Non‐responder imputation was used for imputation of missing data. Patients were treated as non‐responders after the time of discontinuation up to the visit they would have reached if they had stayed in the study up to the time of the data cut‐off (ie up to November 2017). No imputation for missing data was applied for ongoing patients.
In order to identify factors that may have been associated with efficacy, we performed descriptive analyses of demographics and clinical characteristics at baseline of OCTAVE Induction and OCTAVE Open in the dose de‐escalation and dose escalation subpopulations, stratified by treatment success. Formal statistical comparison was not performed due to the small sample size. Treatment success for dose de‐escalation was defined as remaining in remission at month 12 of OCTAVE Open. Treatment success for dose escalation was defined as having clinical response at month 2 of OCTAVE Open. Neither set of treatment success criteria specifically required patients to be steroid‐free; however, per‐protocol tapering of corticosteroids was mandatory at the beginning of both OCTAVE Sustain and OCTAVE Open.
2.2.2. Safety
Safety endpoints evaluated included adverse events, serious adverse events and safety events of special interest (serious infections, herpes zoster, opportunistic infections, malignancies [excluding non‐melanoma skin cancer], non‐melanoma skin cancer, gastrointestinal perforations [excluding any fistulae and perianal abscesses] and major adverse cardiovascular events). Opportunistic infections, major adverse cardiovascular events, malignancy and gastrointestinal perforation events were adjudicated by external specialist review committees blinded to study treatment.
Safety was assessed in all patients who received at least one dose of study drug in OCTAVE Open, with no imputation for missing data. Safety data are presented for the dose de‐escalation and dose escalation subpopulations in OCTAVE Open.
Incidence rates (the number of unique patients with events per 100 patient‐years of exposure) and 95% confidence intervals for safety events of special interest were calculated using the Exact Poisson method.
2.3. Study ethics and patient consent
All studies were conducted in compliance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice Guidelines and were approved by the Institutional Review Boards and/or Independent Ethics Committees at each of the investigational centres participating in the studies, or a central Institutional Review Board. All patients provided written informed consent. All authors had access to the study data and reviewed and approved the final manuscript.
3. RESULTS
3.1. Patients
Baseline demographics and disease characteristics for the tofacitinib maintenance remitter de‐escalation subpopulation (n = 66) and the tofacitinib maintenance failure dose escalation subpopulation (n = 57) are summarised in Table 1. Fourteen patients (21.2%) in the de‐escalation subpopulation discontinued from OCTAVE Open. Thirty‐two patients (56.1%) in the dose escalation subpopulation discontinued from the study. Insufficient clinical response (including adverse events of worsening UC) was the most frequent reason for discontinuation in the de‐escalation subpopulation (6 patients; 9.1% of the total subpopulation) and in the dose escalation subpopulation (25 patients; 43.9%).
Table 1.
Baseline demographics and disease characteristics of patients in the tofacitinib dose de‐escalation and dose escalation subpopulations of OCTAVE Open
| Tofacitinib maintenance remitter dose de‐escalation subpopulation (N = 66) | Tofacitinib maintenance failure dose escalation subpopulation (N = 57) | |
|---|---|---|
| Age (y), mean (SD) | 45.6 (15.1) | 39.8 (12.5) |
| Male, n (%) | 32 (48.5) | 24 (42.1) |
| Mean body mass index, kg/m2 (SD) | 25.7 (4.4) | 25.0 (5.4) |
| Total Mayo score at baseline of OCTAVE Open, mean (SD) | 1.1 (0.8) | 9.1 (1.6) |
| Disease duration, n (%)a | ||
| <6 y | 32 (48.5) | 28 (49.1) |
| ≥6 y | 34 (51.5) | 29 (50.9) |
| Corticosteroid use at baseline of OCTAVE Open, n (%) | 1 (1.5) | 4 (7.0) |
| Prior corticosteroid failure, n (%)a | 52 (78.8) | 44 (77.2) |
| Prior immunosuppressant failure, n (%)a | 43 (65.2) | 51 (89.5) |
| Prior tumour necrosis factor inhibitor failure, n (%)a | 27 (40.9) | 28 (49.1) |
| Extent of disease, n (%)a | ||
| Proctosigmoiditis | 16 (24.6) | 7 (12.3) |
| Left‐sided colitis | 22 (33.8) | 20 (35.1) |
| Extensive colitis/pancolitis | 27 (41.5) | 30 (52.6) |
Abbreviations: N, number of evaluable patients; n, number of patients; SD, standard deviation; y, years.
Data at baseline of induction studies.
3.2. Efficacy
3.2.1. Maintenance remitter dose de‐escalation subpopulation
In the dose de‐escalation subpopulation, 100.0% (66/66) of patients had clinical response, mucosal healing and remission at baseline of OCTAVE Open (based on central read of endoscopy), reflecting that patients in this subpopulation had to be in remission at entry to OCTAVE Open. In the dose de‐escalation subpopulation, clinical response was maintained in 92.4% (61/66), 84.1% (53/63) and 77.8% (35/45) of patients at months 2, 12 and 24, respectively; mucosal healing was seen in 89.4% (59/66), 81.5% (53/65) and 71.7% (33/46) of patients, respectively; and remission in 80.3% (53/66), 74.6% (47/63) and 60.0% (27/45) of patients, respectively (Figure 2A).
Figure 2.
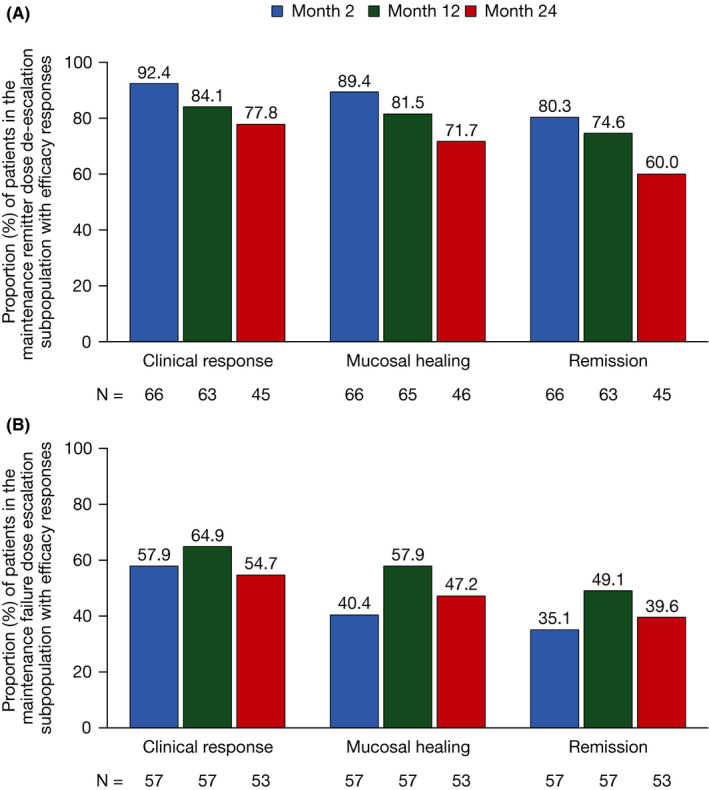
Proportion of patients in (A) the tofacitinib maintenance remitter dose de‐escalation and (B) the tofacitinib maintenance failure dose escalation subpopulations with clinical response, mucosal healing and remission over time in OCTAVE Open (non‐responder imputation). Data were based on local read of endoscopy. Missing data were imputed using non‐responder imputation; patients were treated as nonresponders after the time of discontinuation up to the visit they would have reached if they had stayed in the study. No imputation for missing data was applied for ongoing patients. N, number of evaluable patients
In the dose de‐escalation subpopulation, 19.7% (13/66) of patients had their dose increased from 5 mg b.d. back to 10 mg b.d. during OCTAVE Open. Based on the Kaplan‐Meier method, the 25th percentile of time to dose increase was 30 months after entry into OCTAVE Open. The median and inter‐quartile range for the time to dose escalation could not be estimated due to too few patients with dose increase back to 10 mg b.d.
3.2.2. Maintenance failure dose escalation subpopulation
In the dose escalation subpopulation, rates of clinical response (3.5%; 2/57), mucosal healing (0.0%; 0/0) and remission (0.0%; 0/57), based on central read of endoscopy, at baseline of OCTAVE Open reflected that these patients had failed treatment in OCTAVE Sustain. In the dose escalation subpopulation, clinical response was seen in 57.9% (33/57), 64.9% (37/57) and 54.7% (29/53) of patients at months 2, 12 and 24, respectively; mucosal healing in 40.4% (23/57), 57.9% (33/57) and 47.2% (25/53) of patients, respectively; and remission in 35.1% (20/57), 49.1% (28/57) and 39.6% (21/53) of patients, respectively (Figure 2B).
3.3. Baseline characteristics in the maintenance remitter dose de‐escalation subpopulation by remission status at month 12 of OCTAVE Open
To evaluate potential differences in baseline characteristics of patients who were able to maintain remission after 12 months following dose de‐escalation vs those who were not, baseline demographics and disease characteristics for the dose de‐escalation subpopulation by remission status at month 12 of OCTAVE Open are presented in Table 2 (descriptive statistics only; no formal statistical comparison was performed). A smaller proportion of patients in remission at month 12 (44.7%) were male vs patients not in remission at month 12 (57.9%). The proportions of patients with prior corticosteroid failure (78.7% vs 78.9%), prior immunosuppressant failure (63.8% vs 68.4%) and prior tumour necrosis factor inhibitor failure (42.6% vs 36.8%) were generally similar for those in remission at month 12 and those not in remission. A greater proportion of remitters had extensive colitis/pancolitis (50.0%) compared with patients not in remission (21.1%).
Table 2.
Baseline demographics and disease characteristics in the maintenance remitter dose de‐escalation subpopulation, stratified by whether patients were in remission at month 12 of OCTAVE Open
| Maintenance remitter dose de‐escalation subpopulation | ||
|---|---|---|
| In remission at month 12 of OCTAVE Open (N = 47) | Not in remission at month 12 of OCTAVE Open (N = 19) | |
| Age (y), mean (SD) | 45.9 (14.6) | 45.0 (16.6) |
| Male, n (%) | 21 (44.7) | 11 (57.9) |
| Mean body mass index, kg/m2 (SD) | 25.1 (4.1) | 27.2 (4.8) |
| Total Mayo score at baseline of OCTAVE Open, mean (SD) | 1.1 (0.8) | 1.1 (0.7) |
| Disease duration, n (%)a | ||
| <6 y | 23 (48.9) | 9 (47.4) |
| ≥6 y | 24 (51.1) | 10 (52.6) |
| Corticosteroid use at baseline of OCTAVE Open, n (%) | 0 (0.0) | 1 (5.3) |
| Prior corticosteroid failure, n (%)a | 37 (78.7) | 15 (78.9) |
| Prior immunosuppressant failure, n (%)a | 30 (63.8) | 13 (68.4) |
| Prior tumour necrosis factor inhibitor failure, n (%)a | 20 (42.6) | 7 (36.8) |
| Extent of disease, n/N1 (%)a | ||
| Proctosigmoiditis | 9/46 (19.6) | 7/19 (36.8) |
| Left‐sided colitis | 14/46 (30.4) | 8/19 (42.1) |
| Extensive colitis/pancolitis | 23/46 (50.0) | 4/19 (21.1) |
Remission status at month 12 of OCTAVE Open was defined based on local read of endoscopy, using nonresponder imputation.
Abbreviations: N, number of evaluable patients; N1, number of patients with non‐missing data; n, number of patients; SD, standard deviation; y, years.
Data at baseline of induction studies.
3.4. Baseline characteristics in the maintenance failure dose escalation subpopulation by clinical response status at month 2 of OCTAVE Open
To evaluate potential differences in baseline characteristics of patients who were able to recapture clinical response after 2 months following dose escalation vs those who were not, baseline demographics and disease characteristics for the dose escalation subpopulation by clinical response status at month 2 of OCTAVE Open are presented in Table 3 (descriptive statistics only; no formal statistical comparison was performed). A smaller proportion of patients with month 2 clinical response in OCTAVE Open were male (36.4%) vs patients without month 2 clinical response in OCTAVE Open (50.0%). A greater proportion of month 2 responders had disease duration ≥6 years (54.5%) compared with non‐responders at month 2 (45.8%). The proportion of patients with prior corticosteroid failure (75.8% vs 79.2%), prior immunosuppressant failure (90.9% vs 87.5%) and prior tumour necrosis factor inhibitor failure (48.5% vs 50.0%) was generally similar for patients with and without month 2 clinical response. A smaller proportion of patients with clinical response at month 2 had extensive colitis/pancolitis (48.5%) vs non‐responders (58.3%).
Table 3.
Baseline demographics and disease characteristics in the maintenance failure dose escalation subpopulation, stratified by whether patients had clinical response at month 2 of OCTAVE Open
| Maintenance failure dose escalation subpopulation | ||
|---|---|---|
| Clinical response at month 2 of OCTAVE Open (N = 33) | No clinical response at month 2 of OCTAVE Open (N = 24) | |
| Age (y), mean (SD) | 41.0 (12.3) | 38.3 (12.8) |
| Male, n (%) | 12 (36.4) | 12 (50.0) |
| Mean body mass index, kg/m2 (SD) | 24.5 (5.8) | 25.8 (4.8) |
| Total Mayo score at baseline of OCTAVE Open, mean (SD) | 8.9 (1.6) | 9.3 (1.8) |
| Disease duration, n (%)a | ||
| <6 y | 15 (45.5) | 13 (54.2) |
| ≥6 y | 18 (54.5) | 11 (45.8) |
| Corticosteroid use at baseline of OCTAVE Open, n (%) | 1 (3.0) | 3 (12.5) |
| Prior corticosteroid failure, n (%)a | 25 (75.8) | 19 (79.2) |
| Prior immunosuppressant failure, n (%)a | 30 (90.9) | 21 (87.5) |
| Prior tumour necrosis factor inhibitor failure, n (%)a | 16 (48.5) | 12 (50.0) |
| Extent of disease, n/N1 (%)a | ||
| Proctosigmoiditis | 4/33 (12.1) | 3/24 (12.5) |
| Left‐sided colitis | 13/33 (39.4) | 7/24 (29.2) |
| Extensive colitis/pancolitis | 16/33 (48.5) | 14/24 (58.3) |
Clinical response status at month 2 of OCTAVE Open was defined based on local read of endoscopy, using non‐responder imputation.
Abbreviations: N, number of evaluable patients; N1, number of patients with non‐missing data; n, number of patients; SD, standard deviation; y, years.
Data at baseline of induction studies.
3.5. Safety
The proportion of patients with adverse events, serious adverse events and discontinuation, and incidence rates for safety events of special interest are shown in Table 4 for the tofacitinib dose de‐escalation subpopulation and in Table 5 for the tofacitinib dose escalation subpopulation.
Table 4.
Summary of safety and incidence rates and 95% confidence intervals for safety events of special interest in the tofacitinib dose de‐escalation subpopulation of OCTAVE Open
| Tofacitinib maintenance remitter dose de‐escalation subpopulation (N = 66) (Overall exposure: 131.1 patient‐years)a | |
|---|---|
| Patients with adverse events, n (%) | 48 (72.7) |
| Patients with serious adverse events, n (%) | 8 (12.1) |
| Discontinuations, n (%) | 14 (21.2) |
| Due to adverse event (excluding adverse events of worsening UC) | 3 (4.5) |
| Due to insufficient clinical response (including adverse events of UC) | 6 (9.1) |
| n (%) | Incidence rate (95% confidence interval) | |
|---|---|---|
| Serious adverse events | 5 (7.6) | 4.0 (1.3, 9.4) |
| Serious infections | 1 (1.5) | 0.8 (0.0, 4.3) |
| Herpes zoster | 3 (4.5) | 2.3 (0.5, 6.8) |
| Herpes zoster serious adverse events | 0 (0.0) | 0.0 (0.0, 2.8) |
| Opportunistic infectionsb | 0 (0.0) | 0.0 (0.0, 2.8) |
| Malignancies (excluding non‐melanoma skin cancer)b | 1 (1.5) | 0.8 (0.0, 4.3) |
| Non‐melanoma skin cancerb | 2 (3.0) | 1.6 (0.2, 5.6) |
| Major adverse cardiovascular eventsb | 0 (0.0) | 0.0 (0.0, 2.8) |
| Gastrointestinal perforationsb | 0 (0.0) | 0.0 (0.0, 2.8) |
| Deaths | 0 (0.0) | 0.0 (0.0, 2.8) |
Abbreviations: N, number of evaluable patients; n, number of unique patients with one or more events; UC, ulcerative colitis.
Patient‐years of exposure for incidence rates was calculated up to the earliest of: day of the first event, time to data cut‐off or progression to the next study, or time to last dose plus 28 days. Exposure time per event could be different from overall exposure.
Per adjudication.
Table 5.
Summary of safety and incidence rates and 95% confidence intervals for safety events of special interest in the tofacitinib dose escalation subpopulation of OCTAVE Open
| Tofacitinib maintenance failure dose escalation subpopulation (N = 57) (Overall exposure: 102.1 patient‐years)a | |
|---|---|
| Patients with adverse events, n (%) | 43 (75.4) |
| Patients with serious adverse events, n (%) | 6 (10.5) |
| Discontinuations, n (%) | 32 (56.1) |
| Due to adverse event (excluding adverse events of worsening UC) | 1 (1.8) |
| Due to insufficient clinical response (including adverse events of UC) | 25 (43.9) |
| n (%) | Incidence rate (95% confidence interval) | |
|---|---|---|
| Serious adverse events | 5 (8.8) | 4.9 (1.6, 11.5) |
| Serious infections | 1 (1.8) | 1.0 (0.0, 5.5) |
| Herpes zoster | 7 (12.3) | 7.6 (3.0, 15.6) |
| Herpes zoster serious adverse events | 0 (0.0) | 0.0 (0.0, 3.6) |
| Opportunistic infectionsb | 0 (0.0) | 0.0 (0.0, 3.6) |
| Malignancies (excluding non‐melanoma skin cancer)b | 1 (1.8) | 1.0 (0.0, 5.5) |
| Non‐melanoma skin cancerb | 1 (1.8) | 1.0 (0.0, 5.5) |
| Major adverse cardiovascular eventsb | 0 (0.0) | 0.0 (0.0, 3.6) |
| Gastrointestinal perforationsb | 0 (0.0) | 0.0 (0.0, 3.6) |
| Deaths | 0 (0.0) | 0.0 (0.0, 3.6) |
Abbreviations: N, number of evaluable patients; n, number of unique patients with one or more events; UC, ulcerative colitis.
Patient‐years of exposure for incidence rates was calculated up to the earliest of: day of the first event, time to data cut‐off or progression to the next study, or time to last dose plus 28 days. Exposure time per event could be different from overall exposure.
Per adjudication.
In the tofacitinib dose de‐escalation subpopulation, one patient had a serious infection (viral gastroenteritis classified as gastroenteritis norovirus). One patient in the dose escalation subpopulation had a serious infection (lower respiratory tract infection). In the dose de‐escalation subpopulation, there were three patients with herpes zoster events, all of which had resolved at the time of reporting. In the dose escalation subpopulation, there were seven patients with herpes zoster events, six of which had resolved at the time of reporting. All cases of herpes zoster in both subpopulations were limited to cutaneous involvement over one or two adjacent dermatomes. One patient in the dose de‐escalation subpopulation and three patients in the dose escalation subpopulation temporarily stopped tofacitinib due to herpes zoster events. There were no herpes zoster serious adverse events and no cases of post‐herpetic neuralgia in either subpopulation, and no cases that required permanent tofacitinib discontinuation. No opportunistic infections were reported in either subpopulation.
There was one patient with malignancy (excluding non‐melanoma skin cancer) in the dose de‐escalation subpopulation. The patient—a 75 year‐old male, ex‐smoker, with prior use of immunosuppressant therapy—had lung cancer on day 269 of OCTAVE Open. Two patients (both with prior history of non‐melanoma skin cancer and prior thiopurine use) in the dose de‐escalation subpopulation had non‐melanoma skin cancer: one patient with two events of basal cell carcinoma and one event of squamous cell carcinoma (all with the same day of onset, day 175 of OCTAVE Open), and one patient with one event of basal cell carcinoma (day 81 of OCTAVE Open) and one event of squamous cell carcinoma (day 263 of OCTAVE Open). In the dose escalation subpopulation, there was one patient with malignancy (excluding non‐melanoma skin cancer): cervical cancer on day 489 of OCTAVE Open. One patient (with prior thiopurine use) in the dose escalation subpopulation had non‐melanoma skin cancer: squamous cell carcinoma on day 428 of OCTAVE Open.
There were no deaths, adjudicated major adverse cardiovascular events or gastrointestinal perforations in either the dose de‐escalation or the dose escalation subpopulations.
4. DISCUSSION
This analysis assessed the clinical effect of two tofacitinib dose‐adjustment paradigms for patients with moderate to severe UC. Patients in the tofacitinib dose de‐escalation subpopulation maintained clinically relevant rates of clinical response, mucosal healing and remission after 12 months of open‐label therapy—with a large proportion remaining in remission after 24 months in the open‐label study. Approximately one‐quarter of patients in the dose de‐escalation subpopulation had lost remission 12 months after initiating tofacitinib de‐escalation. For patients who lost initial clinical response to tofacitinib 10 mg b.d. induction therapy while on tofacitinib 5 mg b.d. maintenance therapy, dose escalation back to 10 mg b.d. recaptured clinical response, mucosal healing and remission in a substantial proportion of patients at 2, 12 and 24 months. However, approximately one‐third of patients in the dose escalation subpopulation had not recaptured response by month 12. Acknowledging the limitations of samples size and differences in patient characteristics when comparing patients in these two subpopulations with the overall open‐label study population, safety in the dose de‐escalation and dose escalation subpopulations was generally similar to that observed in the overall study population.6 The incidence rate of herpes zoster in the dose escalation subpopulation was numerically higher than that in the overall tofacitinib UC programme.
Demographics and baseline clinical characteristics of patients who had treatment success (as defined by remission at month 12 of OCTAVE Open for dose de‐escalation, and by clinical response at month 2 of OCTAVE Open for dose escalation) vs those without treatment success were generally similar, with only small differences observed. Analyses of treatment success were based on descriptive statistics, and no formal statistical comparisons were performed due to the small sample size. Accordingly, the clinical significance of these findings is not clear.
As a small molecule, tofacitinib is not expected to elicit the neutralising anti‐drug antibodies seen with biologic therapies, which can occur with or without dose de‐escalation, but are more likely to occur with lower drug exposure as a complication of dose de‐escalation.9 Secondary loss of response with biologic therapies may occur in patients treated with tumour necrosis factor inhibitor therapies, with loss of response often related to the formation of such neutralising anti‐drug antibodies.10 Dose escalation or switching to an alternative tumour necrosis factor inhibitor may recapture response in patients with UC, but the likelihood of lack of response with successive tumour necrosis factor inhibitor agents is increased.11, 12 Despite this, dose‐dependency of adverse events with tumour necrosis factor inhibitor agents has not been demonstrated in patients with UC,13, 14, 15, 16 which may be advantageous when considering dose escalation or intensification with these therapies. However, it should be noted that some patients require therapy with immunomodulators in combination with tumour necrosis factor inhibitor agents,17 and immunomodulators are associated with additional safety issues that require monitoring (eg non‐melanoma skin cancers and lymphomas).18, 19
In these analyses, rates of recapture of efficacy responses among patients who had their tofacitinib dose escalated were high, and suggest both that the initial loss of response during maintenance therapy can be recaptured without switching therapies or mechanisms of action or adding concomitant agents, and that the original loss of response may have been due to insufficient drug exposure in patients requiring higher doses to suppress the inflammatory burden.
In this analysis, the maintenance failure dose escalation subpopulation comprised clinical responders to tofacitinib 10 mg b.d. in OCTAVE Induction 1 and 2 who failed treatment with tofacitinib 5 mg b.d. between weeks 8 and 52 in OCTAVE Sustain. Accordingly, treatment failure may have occurred during the corticosteroid taper in conjunction with tofacitinib dose reduction (ie the combination of tofacitinib dose reduction and corticosteroid tapering may have made it more likely that these patients would lose response). It is also noteworthy that patients were able to recapture response while undergoing the mandatory corticosteroid tapering protocol in OCTAVE Open. The efficacy of tofacitinib in the dose de‐escalation subpopulation suggests the utility of dose de‐escalation as a treatment strategy for reducing drug exposure—and thus the risk of dose‐dependent side effects including herpes zoster or potential risk of thrombosis—in tofacitinib‐treated patients who were in remission following 52 weeks of maintenance therapy with tofacitinib 10 mg b.d.
The incidence rates for adverse events of special interest including serious infections, herpes zoster and malignancies observed in the dose de‐escalation and dose escalation subpopulations were generally consistent with those observed in the overall tofacitinib UC programme. However, due to the small sample size and differences in the disease characteristics of patients in these two subpopulations, comparison with the overall study population should be interpreted cautiously. The incidence rate of herpes zoster was numerically higher in the dose escalation subpopulation (who received tofacitinib 10 mg b.d. in OCTAVE Open) than in the overall tofacitinib UC programme.7 The majority of herpes zoster cases that occurred had resolved without complications, and patients were able to continue tofacitinib treatment or resume treatment following temporary discontinuation, consistent with the management of herpes zoster across other tofacitinib disease populations.20
When interpreting the efficacy results of this analysis, it should be noted that Mayo endoscopic subscores (at months 2, 12 and 24 of OCTAVE Open) were calculated based on local read of endoscopy, and not central read, as was the case in the primary analyses of the OCTAVE randomised controlled trials.5 Efficacy analyses reported in this manuscript were based on the use of non‐responder imputation, which gives a more conservative estimate of efficacy than observed case data. Efficacy and safety analyses in the dose de‐escalation and dose escalation subpopulations should be interpreted with caution because of the small sample sizes. In addition, in this analysis of open‐label, long‐term extension study data, there was no placebo treatment arm with which to compare safety data.
These analyses suggest that for patients with moderate to severe UC who had initial clinical response to induction therapy with tofacitinib 10 mg b.d. and who were in remission following 52 weeks of tofacitinib 10 mg b.d. maintenance therapy, dose de‐escalation to 5 mg b.d. was often a suitable option for long‐term maintenance of clinical response, mucosal healing and remission, with a manageable safety profile. They also demonstrate the utility of dose escalation to tofacitinib 10 mg b.d. for patients who lose response after reducing the dose of tofacitinib from 10 mg b.d. to 5 mg b.d. When compared with the rate of herpes zoster in the overall tofacitinib UC programme, a numerically higher rate of herpes zoster was observed in the dose escalation group although sample size and tofacitinib exposure in this subpopulation were limited. Overall, these data suggest that flexible dosing with tofacitinib 5 mg b.d. and tofacitinib 10 mg b.d. can be incorporated into long‐term disease management strategies for patients with ulcerative colitis. Patient‐level benefit‐risk assessment of tofacitinib dose‐changes should be made by the treating physician.
AUTHORSHIP
Guarantor of the article: Amy Marren.
Author contributions: Bruce E Sands, Alessandro Armuzzi, John K Marshall, William J Sandborn, Silvio Danese, Julián Panés, Brian Bressler and Jean‐Frédéric Colombel were investigators on the studies included in this analysis and made substantial contributions to the interpretation of the data. James O Lindsay made substantial contributions to the interpretation of the data. Nervin Lawendy, Eric Maller, Haiying Zhang, Gary Chan, Leonardo Salese, Konstantinos Tsilkos, Amy Marren and Chinyu Su contributed to the design of the analysis and interpreted the data. All authors contributed to the drafting of the manuscript, and critically reviewed/revised the manuscript for important intellectual content.
All authors approved the final version of the manuscript before submission.
ACKNOWLEDGEMENTS
The authors thank the patients, investigators and study teams who were involved in the OCTAVE clinical trial programme.
Declaration of personal interests: Bruce E Sands has received grant support, personal fees and non‐financial support from Pfizer Inc during the conduct of this study; personal fees from AbbVie, Akros Pharma, Allergan, Amgen, Arena Pharmaceuticals, AstraZeneca, Biopharma R&D, Boehringer‐Ingelheim, Bristol‐Myers Squibb, EnGene, Forward Pharma, Immune Pharmaceuticals, Ironwood Pharmaceuticals, Lycera, Lyndra, Receptos, Salix, Shire, Synergy Pharmaceuticals, Target PharmaSolutions, Theravance TiGenix and TopiVert Pharma; grants, personal fees and non‐financial support from Celgene, Janssen and Takeda; personal fees and non‐financial support from 4D Pharma, Capella Bioscience, Ferring, Gilead, Hoffman‐La Roche, Lilly, MedImmune, Oppilan Pharmaceuticals, Otsuka, Palatin Technologies, Prometheus Laboratories, Protagonist Therapeutics, Rheos Medicines, Seres Therapeutics, UCB and Vivelix Pharmaceuticals, outside the submitted work. Alessandro Armuzzi has received consulting and/or advisory board fees from AbbVie, Allergan, Amgen, Biogen, Bristol‐Myers Squibb, Celgene, Celltrion, Ferring, Hospira, Janssen, Lilly, MSD, Mundipharma, Mylan, Pfizer Inc, Roche, Samsung Bioepis, Sandoz, Sofar and Takeda; lecture and/or speaker bureau fees from AbbVie, Amgen, AstraZeneca, Chiesi, Ferring, Hospira, Janssen, Medtronic, MSD, Mitsubishi Tanabe, Mundipharma, Nikkiso, Otsuka, Pfizer Inc, Samsung Bioepis, Takeda, Tigenix and Zambon; and research funding from MSD, Pfizer Inc and Takeda. John K Marshall has received consulting fees from AbbVie, Allergan, Celgene, Celltrion, Ferring, Hospira, Janssen, Lilly, Merck, Pfizer Inc, Pharmascience, Shire and Takeda; and speaker fees from AbbVie, Allergan, Ferring, Janssen, Lupin, Pfizer Inc, Shire and Takeda. James O Lindsay has received consulting fees from AbbVie, Celgene, Ferring, GSK, Janssen, Merck, Pfizer Inc, Robarts Clinical Trials, Shire and Takeda; research grants from Hospira (Pfizer Inc), MSD, Shire and Takeda; lecture and/or speaker bureau fees from AbbVie, Allergan, Ferring, Janssen, MSD, Shire and Takeda; and advisory board fees from AbbVie, Atlantic Healthcare, Ferring, Hospira (Pfizer Inc), Janssen, MSD, NAP, Shire, Pfizer Inc, Takeda and Vifor. William J Sandborn has received grants, personal fees and non‐financial support from Pfizer Inc during the conduct of the study; and, outside of the submitted work, grants and personal fees from AbbVie, Amgen, Atlantic Pharmaceuticals, Boehringer Ingelheim, Celgene (Receptos), Genentech, Gilead Sciences, Janssen, Lilly, Nutrition Science Partners, Pfizer Inc, Prometheus Laboratories, Robarts Clinical Trials (owned by Health Academic Research Trust or HART) and Takeda; personal fees from Akros Pharma, Allergan, Ambrx Inc, Ardelyx, Arena Pharmaceuticals, Avxia, Biogen Idec, Bristol‐Myers Squibb, Conatus, Cosmo Technologies, Ferring Pharmaceuticals, Ferring Research Institute, Forward Pharma, Galapagos, Immune Pharmaceuticals, Index Pharmaceuticals, Kyowa Hakko Kirin Pharma, MedImmune, Mesoblast, Miraca Life Sciences, Nivalis Therapeutics, Novartis, Otsuka, Palatin, Paul Hastings, Salix Pharmaceuticals, Seattle Genetics, Seres Therapeutics, Shire, Sigmoid Biotechnologies, Theradiag, Theravance, Tigenix, Tillotts Pharma, UCB Pharma, Vascular Biogenics and Vivelix; and personal fees and other from Escalier Biosciences, Oppilan Pharma, Precision IBD, Progenity, Qu Biologics, Regeneron and Ritter Pharmaceuticals. Silvio Danese has been a speaker, consultant and advisory board member for AstraZeneca, Boehringer Ingelheim, Cosmo Pharmaceuticals, Ferring, Genentech, Grünenthal, Johnson and Johnson, Merck & Co, Millennium, Novo Nordisk, Pfizer Inc, Pharmacosmos, Takeda, TiGenix and Vifor. Julián Panés has received consulting fees from AbbVie, Arena Pharmaceuticals, Boehringer Ingelheim, Celgene, Ferring, Genentech/Roche, GoodGut, GSK, Janssen, MSD, Nestlé, Oppilan, Pfizer Inc, Progenity, Takeda, Theravance, TiGenix and Topivert. Brian Bressler has been an advisor and/or speaker for AbbVie, Celgene, Ferring, Genentech, Janssen, Merck, Pfizer Inc, Shire and Takeda; an advisor for Allergan, Amgen, Celgene, Microbiome Insights, Pendopharm and Protagonist; has received research support from AbbVie, Alvine, Amgen, Atlantic Pharmaceuticals, Boehringer Ingelheim, Bristol‐Myers Squibb, Celgene, Genentech, GSK, Janssen, Merck, Qu Biologic, RedHill Biopharm and Takeda; and holds stock options with Qu Biologic. Jean‐Frédéric Colombel has received research grants from AbbVie, Janssen Pharmaceuticals and Takeda; payment for lectures from AbbVie, Amgen, Ferring Pharmaceuticals, Shire and Takeda; consulting fees from AbbVie, Amgen, Boehringer Ingelheim, Celgene Corporation, Celltrion, Enterome, Ferring Pharmaceuticals, Genentech, Janssen Pharmaceuticals, Lilly, Medimmune, Merck, Novartis, Pfizer Inc, Protagonist Therapeutics, Sandoz, Second Genome, Seres Therapeutics, Shire, Takeda, Theradiag and Theravance Biopharma; and holds stock options in Genfit and Intestinal Biotech Development. Nervin Lawendy, Eric Maller, Haiying Zhang, Gary Chan, Leonardo Salese, Amy Marren and Chinyu Su are employees and stockholders of Pfizer Inc. Konstantinos Tsilkos was an employee of Pfizer Inc at the time the research was conducted.
APPENDIX 1.
AUTHORS’ COMPLETE AFFILIATIONS
Bruce E. Sands: Dr. Henry D. Janowitz Division of Gastroenterology, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Alessandro Armuzzi: IBD Unit, Presidio Columbus, Fondazione Policlinico Universitario A. Gemelli IRCCS, Università Cattolica del Sacro Cuore, Rome, Italy; John K. Marshall: Division of Gastroenterology, Department of Medicine, Farncombe Family Digestive Health Research Institute, McMaster University, Hamilton, ON, Canada; James O. Lindsay: Department of Gastroenterology, The Royal London Hospital, Barts Health NHS Trust, London, UK; William J. Sandborn: Division of Gastroenterology, University of California, San Diego, La Jolla, CA, USA; Silvio Danese: IBD Center, Department of Gastroenterology, Humanitas Research Hospital, Rozzano, Milan, Italy; Julián Panés: Hospital Clínic de Barcelona, IDIBAPS, CIBERehd, Barcelona, Spain; Brian Bressler: Division of Gastroenterology, Department of Medicine, University of British Columbia, Vancouver, BC, Canada; Jean‐Frédéric Colombel: Dr. Henry D. Janowitz Division of Gastroenterology, Icahn School of Medicine at Mount Sinai, New York, New York, USA; Nervin Lawendy, Eric Maller, Haiying Zhang, Gary Chan, Leonardo Salese, Konstantinos Tsilkos, Amy Marren, Chinyu Su: Pfizer Inc, Collegeville, PA, USA.
Sands BE, Armuzzi A, Marshall JK, et al. Efficacy and safety of tofacitinib dose de‐escalation and dose escalation for patients with ulcerative colitis: results from OCTAVE Open. Aliment Pharmacol Ther. 2020;51:271–280. 10.1111/apt.15555
The Handling Editor for this article was Dr Nicholas Kennedy, and it was accepted for publication after full peer‐review.
Authors’ complete affiliations are listed in Appendix section.
Funding information
These studies were sponsored by Pfizer Inc. Medical writing support, under the guidance of the authors, was provided by Daniel Binks, PhD, at CMC Connect, a division of McCann Health Medical Communications Ltd, Macclesfield, UK and was funded by Pfizer Inc, New York, NY, USA in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med. 2015; 163:461‐464).
LINKED CONTENT
This article is linked to Amiot et al paper. To view this article, visit https://doi.org/10.1111/apt.15638.
DATA AVAILABILITY STATEMENT
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programmes that have been terminated (ie development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
REFERENCES
- 1. Harbord M, Eliakim R, Bettenworth D, et al. Third European evidence‐based consensus on diagnosis and management of ulcerative colitis. part 2: current management. J Crohns Colitis. 2017;11:769‐784. [DOI] [PubMed] [Google Scholar]
- 2. Kornbluth A, Sachar DB; Practice Parameters Committee of the American College of Gastroenterology . Ulcerative colitis practice guidelines in adults: American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol. 2010;105:501‐523. [DOI] [PubMed] [Google Scholar]
- 3. Pariente B, Laharie D. Review article: why, when and how to de‐escalate therapy in inflammatory bowel diseases. Aliment Pharmacol Ther. 2014;40:338‐353. [DOI] [PubMed] [Google Scholar]
- 4. Torres J, Boyapati RK, Kennedy NA, Louis E, Colombel JF, Satsangi J. Systematic review of effects of withdrawal of immunomodulators or biologic agents from patients with inflammatory bowel disease. Gastroenterology. 2015;149:1716‐1730. [DOI] [PubMed] [Google Scholar]
- 5. Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376:1723‐1736. [DOI] [PubMed] [Google Scholar]
- 6. Lichtenstein GR, Loftus EV Jr, Bloom S, et al. Tofacitinib, an oral Janus kinase inhibitor, in the treatment of ulcerative colitis: open‐label, long‐term extension study [abstract]. Am J Gastroenterol. 2017;112(Suppl 1):S395. Abstract 714. [Google Scholar]
- 7. Sandborn WJ, Panés J, D’Haens GR, et al. Safety of tofacitinib for treatment of ulcerative colitis, based on 4.4 years of data from global clinical trials. Clin Gastroenterol Hepatol. 2019;17:1541‐1550. [DOI] [PubMed] [Google Scholar]
- 8. Winthrop KL, Melmed GY, Vermeire S, et al. Herpes zoster infection in patients with ulcerative colitis receiving tofacitinib. Inflamm Bowel Dis. 2018;24:2258‐2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mukherjee A, Hazra A, Smith MK, et al. Exposure‐response characterization of tofacitinib efficacy in moderate to severe ulcerative colitis: results from a dose‐ranging phase 2 trial. Br J Clin Pharmacol. 2018;84:1136‐1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yanai H, Hanauer SB. Assessing response and loss of response to biological therapies in IBD. Am J Gastroenterol. 2011;106:685‐698. [DOI] [PubMed] [Google Scholar]
- 11. Taxonera C, Rodríguez C, Bertoletti F, et al. Clinical outcomes of golimumab as first, second or third anti‐TNF agent in patients with moderate‐to‐severe ulcerative colitis. Inflamm Bowel Dis. 2017;23:1394‐1402. [DOI] [PubMed] [Google Scholar]
- 12. Olivera P, Danese S, Pouillon L, Bonovas S, Peyrin‐Biroulet L. Effectiveness of golimumab in ulcerative colitis: a review of the real world evidence. Dig Liver Dis. 2019;51:327‐334. [DOI] [PubMed] [Google Scholar]
- 13. Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353:2462‐2476. [DOI] [PubMed] [Google Scholar]
- 14. Reinisch W, Sandborn WJ, Hommes DW, et al. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: results of a randomised controlled trial. Gut. 2011;60:780‐787. [DOI] [PubMed] [Google Scholar]
- 15. Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab induces clinical response and remission in patients with moderate‐to‐severe ulcerative colitis. Gastroenterology. 2014;146:85‐95. [DOI] [PubMed] [Google Scholar]
- 16. Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab maintains clinical response in patients with moderate‐to‐severe ulcerative colitis. Gastroenterology. 2014;146:96‐109. [DOI] [PubMed] [Google Scholar]
- 17. Rubin DT, Ananthakrishnan AN, Siegel CA, Sauer BG, Long MD. ACG clinical guideline: ulcerative colitis in adults. Am J Gastroenterol. 2019;114:384‐413. [DOI] [PubMed] [Google Scholar]
- 18. Kotlyar DS, Lewis JD, Beaugerie L, et al. Risk of lymphoma in patients with inflammatory bowel disease treated with azathioprine and 6‐mercaptopurine: a meta‐analysis. Clin Gastroenterol Hepatol. 2015;13:847‐858.e4. [DOI] [PubMed] [Google Scholar]
- 19. Ariyaratnam J, Subramanian V. Association between thiopurine use and nonmelanoma skin cancers in patients with inflammatory bowel disease: a meta‐analysis. Am J Gastroenterol. 2014;109:163‐169. [DOI] [PubMed] [Google Scholar]
- 20. Colombel JF. Herpes zoster in patients receiving JAK inhibitors for ulcerative colitis: mechanism, epidemiology, management, and prevention. Inflamm Bowel Dis. 2018;24:2172‐2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programmes that have been terminated (ie development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.