

Abstract
Aims
SAR247799 is a selective G‐protein‐biased sphingosine‐1 phosphate receptor‐1 (S1P1) agonist with potential to restore endothelial function in vascular pathologies. SAR247799, a first‐in‐class molecule differentiated from previous S1P1‐desensitizing molecules developed for multiple sclerosis, can activate S1P1 without desensitization and consequent lymphopenia. The aim was to characterize SAR247799 for its safety, tolerability, pharmacokinetics and pharmacodynamics (activation and desensitization).
Methods
SAR247799 was administered orally to healthy subjects in a double‐blind, randomized, placebo‐controlled study with single (2.5–37.5 mg) or 2‐week once‐daily (0.5–15 mg) doses. An open‐label single dose pilot food‐interaction arm with 10 mg SAR247799 in cross‐over design was also performed.
Results
SAR247799 was well tolerated and, at the higher end of the dose ranges, caused the expected dose‐dependent pharmacodynamics associated with S1P1 activation (heart rate reduction) and S1P1 desensitization (lymphocyte count reduction). SAR247799 demonstrated dose‐proportional increases in exposure and was eliminated with an apparent terminal half‐life of 31.2–33.1 hours. Food had a small effect on the pharmacokinetics of SAR247799. SAR247799 had a low volume of distribution (7–23 L), indicating a potential to achieve dose separation for endothelial vs cardiac S1P1 activation pharmacology. A supratherapeutic dose (10 mg) of SAR247799 produced sustained heart rate reduction over 14 days, demonstrating cardiac S1P1 activation without tachyphylaxis. Sub‐lymphocyte‐reducing doses (≤5 mg) of SAR247799, which, based on preclinical data, are projected to activate S1P1 and exhibit endothelial‐protective properties, had minimal‐to‐no heart rate reduction and displayed no marked safety findings.
Conclusion
SAR247799 is suitable for exploring the biological role of endothelial S1P1 activation without causing receptor desensitization.
Keywords: endothelium, pharmacodynamics, pharmacokinetics, SAR247799, sphingosine 1‐phosphate
What is already known about this subject
The multiple sclerosis drugs fingolimod and siponimod cause sphinogosine‐1 phosphate receptor‐1 (S1P1) desensitization and consequent lymphopenia.
Cardiac S1P1 activation causes heart rate reduction; an effect that can be rapidly desensitized.
Preclinically, SAR247799 activates S1P1 without desensitization and its endothelial‐protective profile suggests potential as a new treatment for vascular diseases.
What this study adds
Fourteen‐day treatment with 10 mg SAR247799 activated cardiac S1P1 without tachyphylaxis.
SAR247799 had a low volume of distribution suitable for separating endothelial vs cardiac S1P1‐activating doses.
Sub‐lymphocyte‐reducing doses of SAR247799 had minimal‐to‐no heart‐rate‐reducing effect and, in accordance with animal studies, are suitable to explore the biological role in endothelial function.
1. INTRODUCTION
Endothelial dysfunction is a hallmark of many vascular diseases. 1 Activation of sphingosine‐1 phosphate receptor‐1 (S1P1) has been implicated in preserving endothelial barrier structure and function. 2 SAR247799 is an oral, selective, S1P1 agonist with a mechanism of action making it a potential drug candidate for diseases associated with endothelial dysfunction including coronary and peripheral artery disease, diabetic complications (e.g. retinopathy, nephropathy, foot ulcers), heart failure, microvascular angina, myocardial infarction, stroke, sickle cell disease, vascular dementia, chronic rheumatic disorders (e.g. Raynaud's, lupus), acute lung injury, acute kidney injury, and vascular leak/septic shock associated with severe viral infectious diseases.
S1P1 is a G‐protein‐coupled receptor that can be activated by classical G‐protein coupling and by β‐arrestin activation; the former associated with endothelial‐protective effects and the latter contributing to receptor desensitization. S1P1 desensitization prevents lymphocyte egress from lymphoid organs causing peripheral blood lymphocyte reduction, 3 , 4 and this has been capitalized in the approval of 3 S1P1‐desensitizing drugs for the treatment of multiple sclerosis; fingolimod (a nonselective S1P receptor modulator) 5 and more recently, siponimod and ozanimod (selective S1P1/5 modulators). 6 , 7 Several other S1P1‐desensitizing molecules are in clinical development, the most advanced of which is ponesimod. 8 All of these molecules were designed to desensitize S1P1 and cause peripheral blood lymphocyte reduction for therapeutic benefit in multiple sclerosis and other autoimmune diseases, including inflammatory bowel disease, psoriasis and lupus where clinical effects have been shown. 9 , 10 , 11 At therapeutic and supra‐therapeutic doses, S1P1‐desensitizing agents have shown side‐effects in patients consistent with endothelial damage including macular oedema, lung dysfunction, renal dysfunction and impaired flow‐mediated dilation. 5 , 6 , 7 , 12 , 13 , 14 , 15 , 16 , 17 These clinical findings, together with a breadth of preclinical data, implicate S1P1 as a key regulator of endothelial homeostasis. 18 , 19 , 20 In contrast, SAR247799 is a G‐protein‐biased S1P1 agonist, capable of activating G‐protein pathways (e.g. inhibition of cyclic adenosine mono‐phosphate) more potently than β‐arrestin recruitment and receptor internalization, 21 and to our knowledge is the first molecule of its class to be evaluated in humans. SAR247799 is >100‐fold selective on S1P1 vs S1P2–5. 21 The preclinical profile of SAR247799 demonstrates bi‐phasic pharmacology; it activates endothelial S1P1 (increasing cellular impedance and phosphorylation of extracellular‐regulated kinase‐1/2, protein kinase‐B and endothelial nitric oxide synthase) at low concentrations, and S1P1 desensitization at higher concentrations. 21 Consequently, SAR247799 provides endothelial‐protective properties in rat and pig models of ischaemia/reperfusion‐induced renal and coronary injury, respectively, at doses that are devoid of lymphocyte‐reducing properties. 21 At supra‐endothelial‐protective doses, SAR247799 causes dose‐dependent lymphocyte reduction in preclinical models similar to fingolimod and siponimod. 21 The pharmacodynamic (PD) characterization of SAR247799 on lymphocytes is therefore useful to identify suitable (sub‐lymphocyte‐reducing) doses for evaluation in patients with endothelial dysfunction.
S1P1 on atrial myocytes activates an inwardly rectifying Gαi‐protein‐regulated potassium channel, 22 causing hyperpolarization similar to vagal stimulation, resulting in heart rate reduction and PR interval prolongation. This translates into higher rates of symptomatic bradycardia and atrioventricular block in susceptible patients treated with fingolimod and other S1P1 modulators. 8 The heart rate effects of previous S1P1 modulators are transient, and rapidly diminished once receptor desensitization is established. 23 , 24 , 25 Although both heart rate reduction and endothelial protection are associated with S1P1 activation, SAR247799 did not reduce heart rate in animals over the dose‐range that was endothelial protective. SAR247799 is an acidic molecule displaying a low volume of distribution in rats and pigs (0.2–0.5 L/kg), 21 suggesting that endothelial‐protective effects could be produced in humans at doses that have minimal‐to‐no effect on heart rate.
The safety and tolerability of various S1P1 modulators has been evaluated in humans. However, since all of these molecules were designed to desensitize S1P1 and cause lymphopenia, the safety and tolerability associated with specific S1P1 activation in the absence of marked S1P1 desensitization is currently unknown.
The current study was conducted to evaluate the effects of single and 2‐week repeated administration of SAR247799 over a broad dose range vs placebo on lymphocyte count and heart rate in healthy subjects. The objective was to define the doses of SAR247799 that provide minimal‐to‐no lymphocyte reduction, as these are expected to be endothelial‐protective based on preclinical projections, and to characterize the safety, tolerability and pharmacokinetics (PK) over this dose range.
2. METHODS
2.1. Study design
This randomized first‐in‐human single centre study was approved by an independent ethics committee (State Office of Health and Social Affairs Ethics Committee of Berlin, Germany) and was conducted by Parexel International GmbH (Berlin, Germany) in accordance with the clinical trial protocol and with ethical principles that have their origin in the Declaration of Helsinki. All subjects provided written informed consent before participating. The study dosing scheme is summarized in Figure 1.
FIGURE 1.
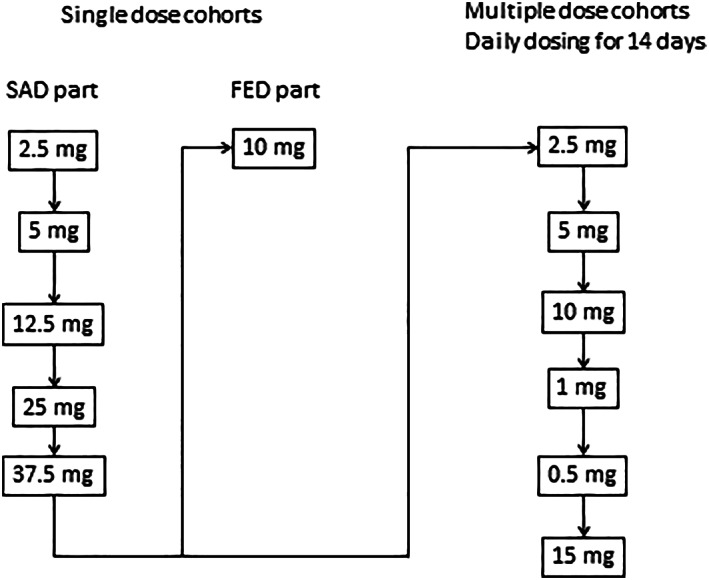
Study dosing scheme. Each cohort in the single ascending dose (SAD) part was composed of 6 subjects on SAR247799 and 2 subjects on placebo. The fed part had 6 subjects in a cross‐over design for the fed and fasted arms. Each cohort in the multiple‐dosing part had 9 subjects on SAR247799 (except n = 10 for the 2.5 mg dose) and 3 subjects on placebo. Each cohort had a 7 day follow‐up period
The first part of the study was designed to assess the safety, tolerability, PK and PD following oral single ascending doses (SAD) in fasted conditions, and to investigate the pilot food interaction at a single dose. Five dose groups of SAR247799 (2.5, 5, 12.5, 25 and 37.5 mg) were assessed in sequential ascending order (8 subjects per cohort; 6 on SAR247799 and 2 on placebo) in a randomized, double‐blind, placebo‐controlled design. The food interaction part was performed with 10 mg SAR247799 and with 6 subjects as an open‐label, randomized, 2‐treatment, 2 period (fasted and fed), cross‐over design (with at least 7 days between periods). Subjects in the single‐dose studies were evaluated for 5 days (day −1 to day 3, including an ambulatory visit on day 4), except for the 2 highest doses for which an additional PK sample was collected at the end of study (EOS) visit on day 7.
Following review of preliminary blinded safety and PK data up to day 7 from the SAD part, the multiple ascending dose (MAD) part of the study was initiated as a randomized, double‐blind, placebo‐controlled design, ensuring that daily doses and exposure in the MAD part did not exceed those of the SAD part. Subjects received oral doses of SAR247799 (in the following order 2.5, 5, 10, 1, 0.5 and 15 mg) or matching placebo in fasted conditions in the morning for 14 days (12 subjects per cohort; 9 on SAR247799 and 3 on placebo). EOS visit was on day 21.
Randomization occurred in the morning before study drug or matching placebo capsules were administered. After the screening period, subjects checked into the clinic on day −2 for baseline assessments prior to dosing (at approximately 09.00) on day 1 (SAD part) or days 1 to 14 (MAD part). In each cohort of the SAD and MAD, 2 sentinel subjects were dosed before the rest of the cohort, which according to the randomization block would result in at least 1 receiving active drug. There was at least 1 day separation between the 2 sentinel subjects, and between the second sentinel subject and the other subjects of the cohort.
2.2. Subjects
Healthy male subjects (SAD part) and healthy male or female subjects of non‐child‐bearing potential (MAD part) were eligible. Further eligibility criteria included age of 18–60 years, body weight 50–100 kg for males and 40–90 kg for females, body mass index 18–29.9 kg/m2, and vital signs after 10 minutes rest in supine position in the following ranges: systolic blood pressure 100–140 mmHg, diastolic blood pressure 50–90 mmHg, heart rate 50–100 beats/min. In addition, clinical laboratory evaluations, electrocardiography (ECG) recordings, pulmonary function tests (spirometry) and physical and ophthalmological examinations were to be normal or without any clinically relevant finding. Key exclusion criteria included current infection or relevant disease as judged by the investigator, use of any medication within 2 weeks of inclusion and significant abnormalities during 24‐hour Holter screening.
2.3. Assessments
Absolute lymphocyte count was measured using a standard white blood cell (WBC) counter with differential count capability. Baseline was defined as the day −1 (predose) value in SAD and MAD parts, and as the day −1 value for each period of the food‐effect part. In the SAD part absolute lymphocyte counts were determined at baseline and at 1, 2, 4, 6.5, 12, 24, 36, 48 and 72 hours, and EOS. In the MAD part, absolute lymphocyte count was determined at baseline, from day 2 to day 13, and on days 1 and 14 (1, 2, 4, 5, 6.5, 8, 12 and 24 h).
ECG parameters (heart rate, PR, QRS and QTc intervals) were measured using Cardiosoft (GE Medical Systems, Freiburg, Germany). Parameters were assessed at baseline (at day −1 or predose) and then at day 1 (SAD and MAD), from day 2 to day 13 (MAD), and day 14 (MAD) at the following time‐points: 0.5, 1, 2, 3, 4, 5, 6.5, 12 and 24 h. Additional time‐points in the SAD included 36, 48 and 72 hours and EOS.
Other safety assessments were performed throughout the study period: adverse events (AE) recording, physical examinations, vital signs, safety laboratory tests (haematology, biochemistry, urinalysis), continuous ECG telemetry, ECG Holters, pulmonary function tests, and ophthalmological examinations. AEs of special interest (AESIs) included grade‐2 lymphopenia, AV conduction disorder, bradycardia, dyspnoea and any persistent visual abnormality, and were based on the safety profile of fingolimod and more selective S1P1 modulators in development and/or the preclinical profile of SAR247799.
2.4. PK
SAR247799 concentrations were measured from plasma and urine from predose to 72 hours post‐dose (SAD) and from predose to 120 hours postdose at Day 14 (MAD). SAR247799 in plasma samples were determined using a validated liquid chromatography tandem mass spectrometry method with a lower limit of quantification of 5 ng/mL. SAR247799 concentrations in urine were determined using a nonvalidated liquid chromatography with tandem mass spectrometry, derived from the plasma method, with an lower limit of quantification of 5 ng/mL. The analytical method had an accuracy of −4.5 to 4.0% for within‐run and −4.0 to 2.8% for between‐run, and the precision was 0.6 to 6.0% for within‐run and 1.8 to 3.1% for between‐run. The following PK parameters were calculated from plasma and urine concentrations using noncompartmental methods. For SAD and MAD: maximum plasma concentration observed (Cmax), time to reach Cmax (tmax), area under the plasma concentration vs time curve calculated using the trapezoidal method over 24 hours (AUC0–24), terminal half‐life associated with the terminal slope (t1/2Z), apparent total body clearance (CL/F) and apparent volume of distribution (VZ/F). For MAD: time corresponding to the last concentration above the limit of quantification (tlast), apparent volume of distribution at the steady state (Vss/F), accumulation ratio (Rac[Cmax, AUC0–24]), trough concentration on day 14 (C24h), peak‐to‐trough ratio on day 14 and time to steady state (defined as time in days at which 90% of the estimated subject‐specific steady‐state trough concentration was reached).
2.5. Sample size considerations
As the main objective was to assess the overall safety of SAR247799, no formal sample size was provided, but it was based on empirical considerations.
2.6. Statistics
The safety analyses were conducted on the safety population and were based on the review of descriptive statistics (summary tables) and individual data for AEs, clinical laboratory, white blood cell count including lymphocyte count, vital signs, ECG parameters, pulmonary function tests including spirometry, and ophthalmological evaluations (including visual acuity Snellen score). AEs were coded using Medical Dictionary for Regulatory Activities (MedRA), and treatment‐emergent AEs (TEAEs) were summarized (counts and percentage) by primary system‐organ class (SOC) and preferred term (PT) for each treatment group. Subjects presenting TEAEs were also listed by treatment group, primary SOC and PT, and AE diagnosis. Potential clinically significant abnormalities (version dated 24 May 2014) for clinical safety laboratory parameters, vital sign, and ECG data and out‐of‐normal range values for clinical safety laboratory data were flagged, listed, and summarized in frequency tables by treatment group. For ECGs, vital signs, safety laboratory parameters, pulmonary function parameters, and ophthalmological parameters raw data and changes from baseline were summarized as descriptive statistics, and summary plots were provided for relevant parameters.
2.7. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.
3. RESULTS
3.1. Study population
Overall, 91 healthy subjects were exposed to SAR247799 and 28 to placebo, representing the safety population.
In the SAD part, 30 subjects were exposed to single SAR247799 doses of 2.5, 5, 12.5, 25 or 37.5 mg (6 subjects per dose). 6 subjects were administered 10 mg SAR247799 in the cross‐over food interaction arm.
In the MAD part, 55 subjects were exposed to daily doses of 0.5 (n = 9), 1 (n = 9), 2.5 (n = 10), 5 (n = 9), 10 (n = 9) and 15 mg (n = 9). Subjects randomized into the first 5 cohorts of the MAD part received all of their assigned multiple doses of SAR247799 or placebo for 14 days, with the exception of 2 subjects at 2.5 mg SAR247799 who withdrew treatment prematurely, 1 for personal reasons (subject was replaced) and 1 due to occurrence of a TEAE (second degree AV block Mobitz 1). In cohort 6 (15 mg SAR247799), 10 out of 12 subjects did not complete the study: 2 subjects, both receiving SAR247799, were withdrawn due to a TEAE (1 for a serious AE [SAE]) of obstructive pancreatitis for which the blind was broken, and 1 for nonserious asymptomatic sinus bradycardia) and 8 subjects (6 receiving SAR247799 and 2 placebo) after it was decided to stop cohort 6 due to the SAE reported on active treatment.
3.2. PK
Table 1 shows the PK parameters of SAR247799 on day 1 and day 14 (see also supplementary Figure S1 & S2 for graphical representation). Median tmax on day 1 was between 2.5 and 5 hours in the single and repeated‐dosing parts of the study. The apparent volume of distribution (Vz/F) ranged from 17.2 to 22.9 L following single dosing and the apparent volume of distribution at steady state (Vss/F) ranged from 7.56 to 10.5 L following repeated dosing. Mean estimates of terminal half‐life were 31.2 and 33.1 hours in the SAD and MAD parts, respectively. The median estimate for time to steady state was 7.05 days (90% confidence interval [CI] 6.09–7.68) following repeated dosing. The accumulation ratio (day 14/day 1) was 2.12 for Cmax and 2.37 for AUC. The peak‐to‐trough concentration ratio on day 14 was <2. All of the aforementioned parameters showed no apparent dose relationship.
TABLE 1.
Pharmacokinetic parameters of SAR247799 after single and multiple oral dosing
| Single doses | |||||||
|---|---|---|---|---|---|---|---|
| Fasted | Fasted | Fed | |||||
| Parameter (unit) | 2.5 mg [n = 6] | 5 mg [n = 6] | 12.5 mg [n = 6] | 25 mg [n = 6] | 37.5 mg [n = 6] | 10 mg [n = 6] | 10 mg [n = 6] |
| Cmax (ng/mL) | 133 ± 28.9 | 253 ± 24.0 | 874 ± 242 | 1770 ± 181 | 2620 ± 468 | 620 ± 182 | 402 ± 103 |
| Tmax (h) | 4.00 (2.00–4.03) | 3.01 (3.00–4.00) | 4.00 (3.00–4.00) | 4.00 (2.00–4.00) | 3.52 (2.00–4.02) | 3.00 (2.00–4.00) | 5.00 (4.00–10.00) |
| T1/2z | 27.5 ± 3.72 | 31.6 ± 4.61 | 35.3 ± 5.20 | 34.7 ± 6.56 | 29.6 ± 6.02 | 32.0 ± 13.8 | 30.5 ± 9.77 |
| AUC0–24 (ng h/mL) | 4410 ± 829 | 9410 ± 1030 | 35 300 ± 10 000 | 64 700 ± 14 600 | 85 600 ± 9230 | 19 300 ± 7020 a | 13 600 ± 3400 b |
| CL/F (L/h) | 0.581 ± 0.090 | 0.537 ± 0.061 | 0.378 ± 0.108 | 0.400 ± 0.073 | 0.443 ± 0.048 | ||
| Vz/F (L) | 22.7 ± 1.64 | 24.2 ± 2.48 | 18.7 ± 3.02 | 19.5 ± 2.28 | 18.8 ± 4.05 | ||
| Multiple doses | ||||||
| Day 1 | ||||||
| Parameter (unit) | 0.5 mg [n = 9] | 1 mg [n = 9] | 2.5 mg [n = 10] | 5 mg [n = 9] | 10 mg [n = 9] | 15 mg [n = 9] |
| Cmax (ng/mL) | 26.0 ± 4.96 | 53.7 ± 7.99 | 162 ± 48.0 | 339 ± 67.1 | 711 ± 113 | 961 ± 217 |
| Tmax (h) | 3.00 (2.00–5.02) | 3.00 (2.00–5.02) | 2.53 (1.52–4.02) | 3.02 (2.00–4.02) | 3.00 (1.50–4.03) | 3.00 (1.52–4.02) |
| AUC0–24h (ng h/mL) | 414 ± 83.4 | 864 ± 148 | 2400 ± 566 | 5210 ± 830 | 11 100 ± 1960 | 14 600 ± 2860 |
| Day 14 | ||||||
| Parameter (unit) | 0.5 mg [n = 9] | 1 mg [n = 9] | 2.5 mg [n = 8] | 5 mg [n = 9] | 10 mg [n = 9] | 15 mg [n = 1] |
| Cmax (ng/mL) | 51.4 ± 11.4 | 127 ± 41.1 | 343 ± 128 | 698 ± 165 | 1600 ± 447 | 1600 ± NC |
| Tmax (h) | 5.00 (2.00–5.05) | 4.97 (1.52–5.00) | 3.55 (2.03–5.03) | 4.03 (2.00–5.00) | 5.00 (3.00–5.07) | 2.02 (2.02–2.02) |
| Tlast (h) | 95.6 (48.1–96.4) | 120 (95.1–121) | 120 (118–121) | 120 (119–121) | 120 (119–122) | 120 (120–120) |
| T1/2z | 41.0 ± 30.6 | 35.2 ± 8.66 | 37.2 ± 9.12 | 34.4 ± 10.4 | 36.9 ± 6.60 | 25.4 ± NC |
| AUC0–24h (ng h/mL) | 875 ± 194 | 2210 ± 764 | 5690 ± 1970 | 11 700 ± 3440 | 27 100 ± 7600 | 26 600 ± NC |
| Vss/F (L) | 10.5 ± 1.67c | 9.23 ± 2.38 | 9.24 ± 2.18 | 8.64 ± 2.13 | 7.56 ± 1.67 | 9.66 ± NC |
| C24h (ng/mL) | 27.0 ± 6.88 | 73.9 ± 29.6 | 185 ± 74.4 | 385 ± 141 | 898 ± 299 | 860 ± NC |
| Peak/trough ratio | 1.93 ± 0.284 | 1.76 ± 0.191 | 1.89 ± 0.209 | 1.87 ± 0.198 | 1.83 ± 0.303 | 1.86 ± NC |
Cmax = maximum plasma concentration, Tmax = time to maximum plasma concentration, Tlast = time of last measurable plasma concentration, T1/2z = elimination half‐life, AUC0‐24 = area under curve of plasma concentration versus time from 0 to 24 hour, Cl/F = apparent total clearance from plasma, Vz/F = apparent volume of distribution during terminal phase, Vss/F = apparent volume of distribution at steady state, C24h = plasma concentration 24 hours after last administration. Data are geometric mean ± standard deviation, except for Tmax and Tlast, which are median (range)
Peak/trough ratio = Cmax/C24h.
NC, not calculated
a n = 5; b n = 4; c n = 6
Following single dose administration of SAR247799, Cmax and AUC increased with dose, consistent with dose‐proportionality. Similarly, at steady state on day 14, SAR247799 exposure parameters (Cmax, AUC and C24h) also increased in a dose‐dependent manner.
After administration of 10 mg SAR247799 under fed conditions, Cmax was decreased by 36% (estimate 0.64; 90%CI 0.53–0.77) compared to fasted conditions, and was associated with a 2‐hour delay in median tmax (5 vs 3 h). AUC was decreased by 18% (estimate 0.82; 90%CI 0.57–1.18).
No metabolites of SAR247799 were detected in plasma following 14‐day repeated administration of 1, 5 and 10 mg. As biliary excretion of unchanged drug was the main route of elimination (from preclinical studies), metabolites were not measured in urine, and about 1% of unchanged compound was found in urine.
3.3. Lymphocyte count
In single‐dose studies (Figure 2A), there was an increase in peripheral blood lymphocytes compared to placebo at the 2.5 mg dose (29% increase between 4 and 12 h). There was no clear change vs placebo at 5 or 12.5 mg. At the higher doses of 25 and 37.5 mg, lymphocytes decreased in a dose‐dependent manner with mean reductions of 24 and 44% vs placebo at 6.5 hours, respectively. Maximum changes in lymphocyte counts occurred at about 6.5 hours and returned to baseline values by 24 hours at all doses (Figures 2A, 4A). Baseline lymphocyte counts were comparable across groups, but slightly lower baseline values noted in the 2.5 mg group (Table S1) may have contributed to the increases seen at this dose. Based on the increase in lymphocytes observed at 2.5 mg, lower doses were selected for multiple dose studies.
FIGURE 2.
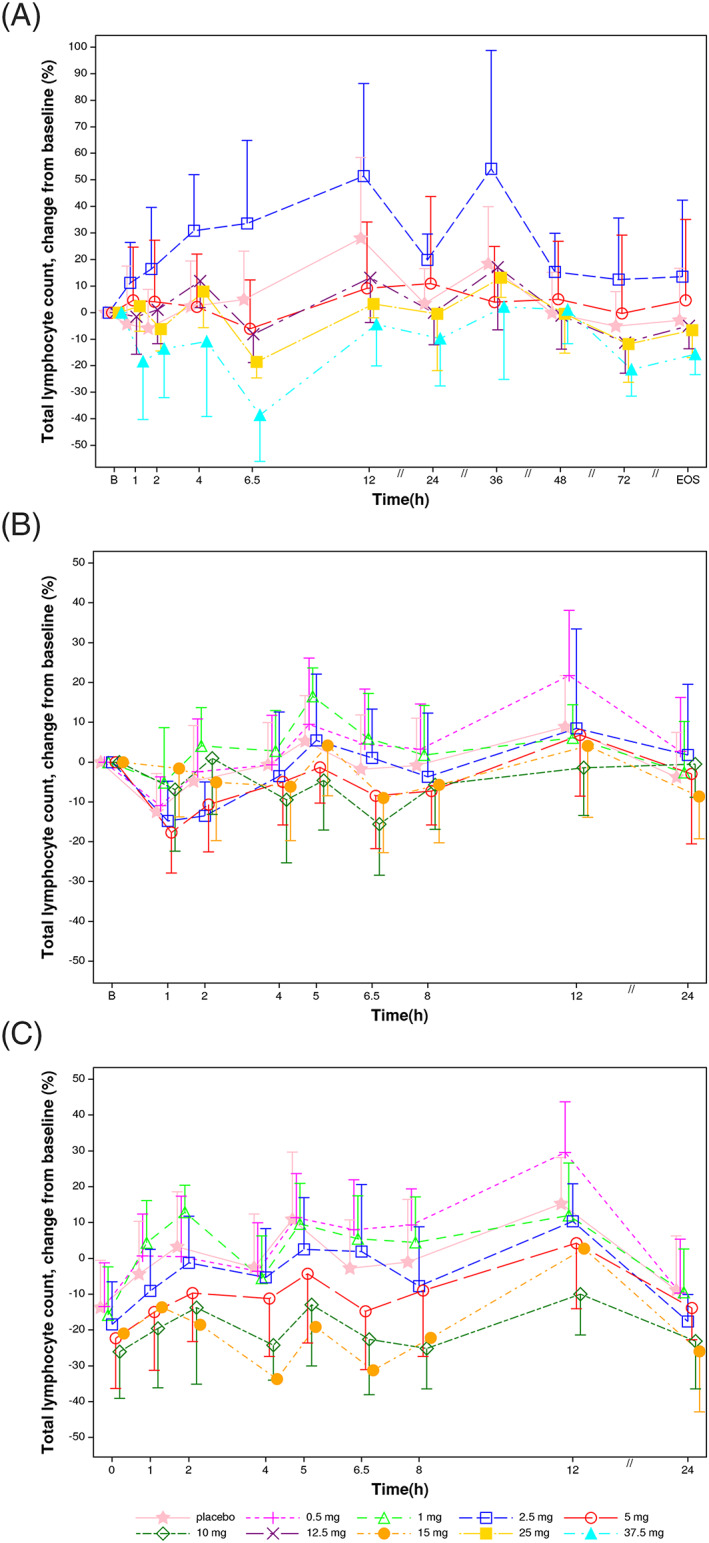
Lymphocyte changes vs baseline following single and 14‐day oral administration of SAR247799 to healthy subjects. A, Day 1 of single ascending dose (n = 6 per dose, 10 placebo). B, Day 1 of multiple‐dosing study (n = 9 at 0.5 mg and 1 mg, n = 10 at 2.5 mg, n = 9 at 5 mg, 10 mg and 15 mg, and n = 18 on placebo). C, Day 14 of multiple‐dosing study (n = 9 at 0.5 mg and 1 mg, n = 10 at 2.5 mg, n = 9 at 5 mg and 10 mg, n = 1 at 15 mg, and n = 16 on placebo). Arithmetic mean and standard deviation
FIGURE 4.
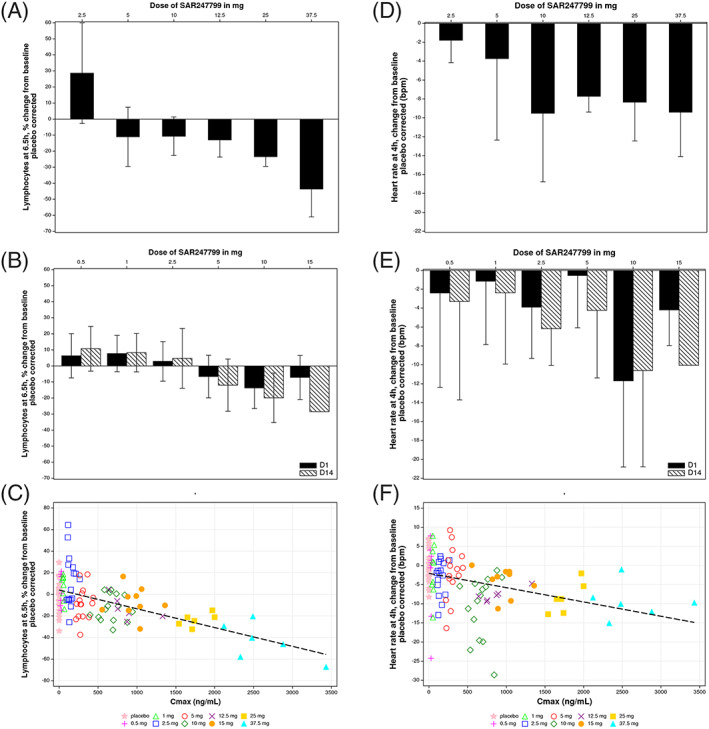
Changes from baseline in lymphocytes at 6.5 hours and heart rate at 4 hours, corrected for mean of time‐matched placebo. Lymphocyte and heart rate changes in single‐dose A, D, or 14‐day repeated‐dose studies B, E, with mean and standard deviation. Scatter plots for change of lymphocytes C, and heart rate F, for all subjects on day 1 of single ascending dose and multiple ascending dose parts, with linear regression lines as shown. Single doses had 6 subjects (10‐mg dose included from fasted arm of food interaction part). Multiple doses had 9 subjects at each time‐point, except for 2.5 mg (n = 10 at days 1 and 14) and 15 mg (n = 1 at day 14). Placebo correction was vs the mean heart rate change at 4 hours, or lymphocyte change at 6.5 hours, in placebo subjects; n = 10 (single doses), n = 18 (day 1 of multiple‐dose study) or n = 16 (day 14 of multiple‐dose study)
In multiple‐dose studies (Figures 2B,C, 4B), compared to placebo there were slight increases (up to 14%) at 0.5 and 1 mg, no change at 2.5 mg and dose‐related decrease from 5 to 15 mg. The decreases on day 14 at 5 and 10 mg reached 10 and 25%, respectively (mean difference vs placebo between 2 and 12 h; Figures 2C, 4B). (Note that only 1 subject completed day 14 at the 15 mg dose.)
Lymphocyte changes at 6.5‐hour post‐treatment showed a dose‐ and concentration (Cmax)‐effect relationship (Figure 4A–C).
3.4. Heart rate
Heart rate changes from baseline following single and repeated dose administration of SAR247799 or placebo are shown in Figure 3. An expected circadian effect on heart rate is illustrated in the various placebo‐treated groups (Figure 3A–C).
FIGURE 3.
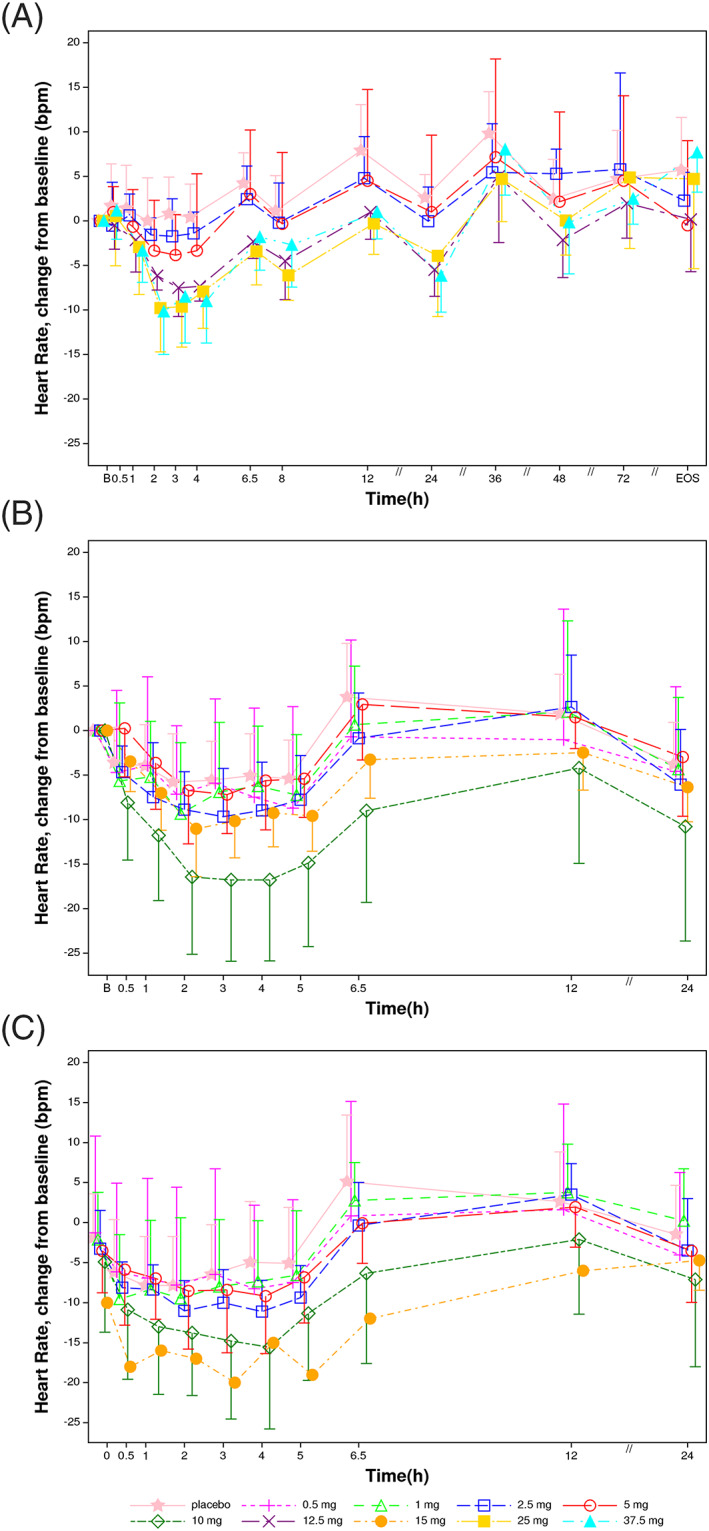
Heart rate changes vs baseline following single and 14‐day oral administration of SAR247799 to healthy subjects. A, Day 1 of single‐ascending dose (n = 6 per dose, 10 placebo. B, Day 1 of multiple‐dosing study (n = 9 at 0.5 mg and 1 mg, n = 10 at 2.5 mg, n = 9 at 5, 10 and 15 mg, and n = 18 on placebo). C, Day 14 of multiple‐dosing study (n = 9 at 0.5 and 1 mg, n = 10 at 2.5 mg, n = 9 at 5 and 10 mg, n = 1 at 15 mg, and n = 16 on placebo). Arithmetic mean and standard deviation. Baseline was defined as the average of replicate values at day 1 predose assessments
On day 1 of the SAD and MAD parts (Figure 3A, 3B), there was a dose‐dependent decrease in heart rate compared to placebo. The greatest reduction in heart rate at each dose occurred at approximately tmax (2–4 h postdose). Consequently, heart rate changes at 4 hours were re‐plotted by correcting for the circadian effect using the respective placebo time point (Figure 4D–F). At single doses of 5 mg or less, mean heart rate reductions were <5 beats/min, compared to placebo (Figure 4D,E). At higher single doses, mean reductions at 4 hours on day 1 were 8–11 beats/min compared to placebo (10, 12.5, 25 and 37.5 mg in SAD and 10 mg in MAD), with an apparent plateau of 11 beats/min mean reduction (Figure 3A,B; Figure 4D–F).
At doses up to 5 mg, day‐14 heart rate reductions compared to placebo were similarly <5 beats/min (Figure 3C, Figure 4E) and alongside the ~2‐fold increase in compound exposure over 14 days, mean heart rate reductions on day 14 were higher than the corresponding reductions on day 1 (Figure 4E). At 10 mg SAR247799, the mean decrease at 4 hour compared to placebo was 11 beats/min on day 14; an effect similar to that observed on day 1 (also −11 beats/min; Figure 4E). Cmax on day 14 with 10 mg (1600 + 447 ng/mL) approached Cmax on day 1 with 25 mg (1770 + 181 ng/mL; Table 1), and both of these doses/time‐points produced heart rate reductions close to the plateau seen in the study (−11 beats/min; Figure 3A–C). Consequently, doses of SAR247799 up to 10 mg can sustain the heart rate reduction over 14 days without evidence for tachyphylaxis.
Heart rate changes at 4 hours post‐treatment showed a dose‐ and concentration (Cmax)‐effect relationship (Figure 4E‐F).
No symptomatic bradycardia was reported in any subject.
3.5. PR interval
There were no clinically significant PR interval abnormalities on ECG. An increase of PR interval of 41 ms from baseline was reported in 1 subject at the 37.5 mg dose (SAD part), 3 hours post dose. However, all PR values in this subject remained within the normal range (and were between 125–172 ms).
No evidence of QRS interval modification or clinically‐relevant change from baseline in QTcF interval was observed. In particular, no subject reported QTcF >450 ms, or an increase from baseline >60 ms.
3.6. Other safety and tolerability assessments
SAR247799 at single doses (2.5–37.5 mg) and once daily doses for 14 days (0.5–15 mg) was well tolerated. There were no deaths or dose‐limiting toxicities in the study. The number of subjects reporting AEs and the total number of AEs were similar across all treatment groups including placebo (Tables 2 and 3).
TABLE 2.
Number of subjects with TEAE(s) by primary SOC and PT in single‐dose studies
| Primary SOC PT [n] | Single‐ascending dose part | Fed part | ||||||
|---|---|---|---|---|---|---|---|---|
| Placebo | SAR247799 | |||||||
| (N = 10) | 2.5 mg (N = 6) | 5 mg (N = 6) | 12.5 mg (N = 6) | 25 mg (N = 6) | 37.5 mg (N = 6) | Fasted 10 mg (N = 6) | Fed 10 mg (N = 6) | |
| Any class | 3 | 2 | 1 | 0 | 2 | 0 | 2 | 0 |
| Infections and infestations | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nasopharyngitis | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Rhinitis | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nervous system disorders | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Headache | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Cardiac disorders | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Ventricular extrasystoles | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| General disorders and administration site conditions | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Medical device site dermatitis | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Respiratory, thoracic and mediastinal disorders | 2 | 0 | 1 | 0 | 1 | 0 | 0 | 0 |
| Cough | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Hiccups | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nasal congestion | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Oropharyngeal pain | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Gastrointestinal disorders | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Abdominal pain upper | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Diarrhoea | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Dry mouth | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Injury, poisoning and procedural complications | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Fall | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hand fracture | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
TEAE: treatment‐emergent adverse event, SOC: system organ class, PT: preferred term MedDRA 19.1
N = number of subjects treated within each group; n = number of subjects with at least 1 TEAE in each category
Table sorted by SOC internationally agreed order and decreasing frequency of PT in SAR247799 37.5 mg group.
An adverse event is considered as treatment emergent if it occurred from the time of the first investigational medicinal product administration up to the end of study visit (included).
TABLE 3.
Number of subjects with TEAE(s) by primary SOC and PT in14‐day studies
| Primary SOC PT [n] | Placebo | SAR247799 | |||||
|---|---|---|---|---|---|---|---|
| (N = 18) | 0.5 mg (N = 9) | 1 mg (N = 9) | 2.5 mg (N = 10) | 5 mg (N = 9) | 10 mg (N = 9) | 15 mg (N = 9) | |
| Any class | 11 | 4 | 6 | 5 | 9 | 6 | 7 |
| Infections and infestations | 3 | 0 | 1 | 1 | 1 | 0 | 0 |
| Nasopharyngitis | 3 | 0 | 1 | 1 | 1 | 0 | 0 |
| Psychiatric disorders | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Restlessness | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Nervous system disorders | 3 | 2 | 0 | 2 | 2 | 1 | 2 |
| Head discomfort | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| Headache | 1 | 1 | 0 | 1 | 2 | 1 | 1 |
| Dizziness postural | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Paraesthesia | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Eye disorders | 1 | 0 | 0 | 0 | 1 | 0 | 0 |
| Accommodation disorder | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Eye pruritus | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cardiac disorders | 1 | 0 | 0 | 1 | 0 | 1 | 1 |
| Sinus bradycardia | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Atrioventricular block second degree | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Palpitations | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ventricular extrasystoles | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| General disorders and administration site conditions | 8 | 4 | 5 | 1 | 8 | 6 | 5 |
| Medical device site dermatitis | 8 | 4 | 5 | 1 | 8 | 6 | 4 |
| Catheter site haematoma | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Asthenia | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Vascular disorders | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Vein disorder | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Respiratory, thoracic and mediastinal disorders | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| Cough | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Oropharyngeal pain | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Gastrointestinal disorders | 0 | 0 | 0 | 0 | 1 | 0 | 2 |
| Abdominal discomfort | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Obstructive pancreatitis | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Diarrhoea | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Hepatobiliary disorders | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Cholelithiasis | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Skin and subcutaneous tissue disorders | 0 | 0 | 0 | 0 | 1 | 1 | 0 |
| Erythema | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Rash popular | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Musculoskeletal and connective tissue disorders | 3 | 1 | 0 | 0 | 1 | 0 | 0 |
| Arthralgia | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Back pain | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Musculoskeletal chest pain | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Neck pain | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Reproductive system and breast disorders | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Penile vascular disorder | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Investigations | 0 | 0 | 0 | 1 | 1 | 1 | 1 |
| Lymphocyte count decreased | 0 | 0 | 0 | 1 | 1 | 1 | 1 |
| Injury, poisoning and procedural complications | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Dental restoration failure | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
TEAE: treatment‐emergent adverse event; SOC: system organ class; PT: preferred term MedDRA 20.1
N = number of subjects treated within each group; n = number of subjects with at least 1 TEAE in each category
Table sorted by SOC internationally agreed order and decreasing frequency of PT in SAR247799 15 mg group.
An adverse event is considered as treatment emergent if it occurred from the time of the first investigational medicinal product administration up to the end of study visit (included).
In the SAD part there were no SAEs. All AEs were of mild to moderate intensity, with no evidence of any dose‐dependent safety signals.
In the MAD part, 1 SAE of obstructive pancreatitis was reported on day 7 in a subject in the 15‐mg dose, probably due to spontaneous passage of a gallstone and not treatment related. The subject recovered without any sequelae on day 32. Six nonserious AESIs were reported, of which 2 led to permanent treatment discontinuation: 1 event of 2second degree AV block Mobitz 1 after 2.5 mg SAR247799 (considered not clinically‐significant) and 1 event of nocturnal sinus bradycardia after 15 mg SAR2477999 (considered related to study treatment). Four AESIs of lymphopenia CTCAE Grade 2 (between 500 and 800/mm3) were reported, 1 in each of the 2.5, 5, 10 and 15 mg groups. All other AEs were of mild to moderate intensity, with no evidence of any dose‐dependent safety signals. There were no clinically relevant or significant findings compared to baseline in systolic or diastolic blood pressure, or spirometry in any subject after single or multiple dosing. Ophthalmological examinations did not show any relevant findings on fundoscopy, visual examination or optical coherence tomography.
4. DISCUSSION
SAR247799 is a G‐protein‐biased S1P1 agonist being developed for vascular diseases in which endothelial dysfunction is prevalent. As illustrated in Figure 5, S1P1 activation is associated with endothelial‐protective effects (which were not evaluated in this study) and heart rate reduction, whereas S1P1 desensitization is associated with lymphocyte reduction, heart rate desensitization and endothelial damage.
FIGURE 5.
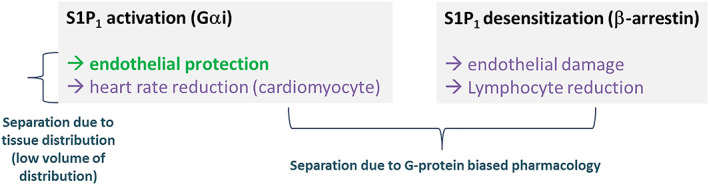
Model for achieving separated pharmacological profiles at S1P1 suitable for endothelial protection
SAR247799 was well tolerated up to the maximum doses tested; 37.5 mg at single dose and 15 mg once daily for 14 days. No severe or dose‐limiting AEs were observed except for the occurrence of obstructive pancreatitis in 1 subject administered with the 15 mg dose, considered to be unrelated to the treatment. Most TEAEs were not considered related to the investigational drug. Potential dose‐dependent safety findings in this study included asymptomatic bradycardia and lymphocyte reduction (at higher doses), which were as expected from the known safety profile of fingolimod, siponimod and other S1P1 modulators.
SAR247799 is the first molecule in its class that is being developed to activate endothelial S1P1, which is in contrast to previous molecules that were designed to desensitize S1P1 and cause resultant lymphopenia. While this first‐in‐human study did not set out to measure endothelial effects (which requires patients with endothelial damage), doses of SAR247799 <10 mg may be associated with endothelial efficacy based on the limited lymphocyte reduction observed at 10 mg. Lymphocyte reductions with 10 mg SAR247799 (up to 25% reduction at steady state on day 14) were considerably less than the approved doses of fingolimod and siponimod (~70% reduction at steady state).11,12 As preclinical studies in rats and pigs demonstrated that endothelial‐protective doses of SAR247799 had minimal‐to‐no effect at reducing lymphocytes (0–10% reduction), 21 10 mg SAR247799 is predicted to be a supratherapeutic dose for endothelial effects as it caused moderate lymphocyte reduction (25%). It is interesting to note that the lower ends of the dose range of SAR247799 (0.5 and 1 mg in the MAD, and 2.5 mg in the SAD) showed a tendency for lymphocyte increase, even if not consistent in all subjects. Since peripheral blood lymphocyte reduction is clearly a marker of lymphocyte S1P1 inactivation, is it tempting to speculate that lymphocyte increase may conversely be a marker for lymphocyte S1P1 activation. However, in healthy individuals, the plasma S1P levels are high, which might mask the ability to reliably measure further pharmacological S1P1‐activating effects. In contrast, plasma S1P levels are reduced in patients with endothelial dysfunction 26 , 27 , 28 , 29 , 30 : the resultant environment may allow the pharmacological restoration of S1P1 activation to be more readily measured.
Endothelial damage has been attributed to S1P1 desensitization. 20 Indeed the incidence of macular oedema in fingolimod‐treated patients dose‐dependently increased from the approved 0.5 mg dose. 15 Similarly, siponimod also increased the incidence of macular oedema at doses characterized by marked S1P1 desensitization. 13 Likewise, dose‐dependent pulmonary toxicity has been associated with fingolimod and ponesimod, mainly at supratherapeutic doses, manifested as reductions of up to 30% in forced expiratory volume over 1 second, accompanied by dose‐limiting chest discomfort and dyspnoea. 5 , 12 , 14 As expected by the moderate lymphocyte reduction achieved at the highest tested doses of SAR247799, there was no evidence for the aforementioned findings in this limited‐size study.
S1P1 activation on atrial myocytes has been associated with heart rate reduction. The ideal profile of an endothelial‐protective agent is to activate endothelial S1P1 while sparing or limiting cardiomyocyte S1P1 activation. SAR247799 dose‐dependently reduced heart rate (−11 beats/min at 10 mg and higher), consistent with other S1P1 modulators that have shown maximum reductions of up to 15–20 beats/min. 22 , 25 Other S1P1 modulators, designed to be desensitizing agents to reduce lymphocytes, have shown rapid desensitization of the heart rate effects in humans. In contrast, we have shown in this study, based on the PD for heart rate reduction, that 10 mg SAR247799 (a supratherapeutic dose) can activate cardiac S1P1 in a sustained manner over 14 days, raising the possibility that similar sustained activation of endothelial S1P1 could occur without desensitization at lower doses. In this regard, the moderate half‐life of SAR247799 (31.3–32.1 h) is ideal for maintaining stable concentrations without significant peak‐to‐trough variations. Consequently, SAR247799 displayed <2‐fold concentration variations over daily dosing intervals, allowing sufficient compound levels to achieve S1P1 activation, without excess concentrations to be reached that would promote S1P1 desensitization. Furthermore, food only had a small effect on the exposure of SAR247799 and, therefore, is unlikely to be a significant cause of PK variation.
The PK analysis revealed that SAR247799 had a lower volume of distribution (7–23 L) compared to previous clinical molecules; fingolimod (1738 L), 31 siponimod (124 L), 6 ponesimod (160 L) 32 and ozanimod (73–102 L/kg or 5000–7000 L). 33 Based on this preferred tissue distribution profile, SAR247799 is expected to activate S1P1 in the intravascular compartment (i.e. on endothelium) at lower doses than S1P1 activation in the heart (Figure 5).
The present study had several limitations. (i) The safety and PD of the 15 mg dose in the 2‐week study was not fully characterized, as only 1 subject completed day 14 of treatment due to the premature discontinuation of cohort 6. However, given that lymphocyte reduction and heart rate reduction have been extensively characterized with previous S1P1 modulators and are known markers of S1P1 desensitization and S1P1 activation, respectively, it is likely that supratherapeutic doses for endothelial effects have already been reached at 10–15 mg of SAR247799. (ii) The study did not have a clear PD marker to monitor endothelial S1P1 activation and hence understand the doses tested in the context of the desired endothelial pharmacology. Although the lymphocyte increases observed in some subjects may provide a surrogate indication of S1P1 activation at doses of 2.5 mg or less, we are not aware of a reliable endothelial marker suitable for assessment in healthy subjects. As such, the safety profile established in this study at the lower end of the dose range reflects primarily preclinical projections about endothelial‐protective doses. Whether this dose context is appropriate requires testing of SAR247799 in patients with endothelial damage. To this end, a study in type‐2 diabetes patients to measure endothelial function improvements with SAR247799 has been performed (clinicaltrials.gov NCT03462017).
Overall, SAR247799 was well tolerated at all doses evaluated. Sub‐lymphocyte‐reducing doses (≤5 mg), which are expected to activate endothelial S1P1, had minimal‐to‐no effect on heart rate and did not display any other marked safety findings. Such doses of SAR247799 are therefore suitable for exploring the role of S1P1 activation in patients with endothelial dysfunction.
COMPETING INTERESTS
Authors were employees of Sanofi or Paraxel at the time of conduct of the studies. Authors affiliated with Sanofi may have equity interest in Sanofi. A.A.P. is inventor of US patent number 9,782,411.
CONTRIBUTORS
Conceptualization: L.B., S.K., P.D., A.A.P. Methodology and design: L.B., S.A., G.G., O.V., F.H., M.S., F.P., D.R., C.G., A.J.M., S.K., P.D., A.A.P. Investigation: S.A., G.G.. Data curation, analysis and visualization: L.B., O.V., F.H., M.S., F.P., D.R., L.H., A.A.P. Project administration: A.T. Resources: L.B., S.A., G.G., A.T. Supervision: L.B., S.A., G.G., C.G., A.J.M., S.K., P.D., A.A.P. Funding acquisition: A.J.M., P.D., A.A.P. Writing—original draft: L.B., A.A.P. Writing—review and editing: S.A., G.G., A.T., O.V., F.H., M.S., F.P., D.R., C.G., L.H., A.J.M., S.K., P.D. Accountable for accuracy, integrity and approval of final version: all authors.
Supporting information
FIGURE S1 Plasma concentration–time profiles of SAR247799 in healthy subjects following administration of single doses (A) and following multiple doses on day 1 (B) or day 14 (C).
FIGURE S2 Trough concentrations of SAR247799 after repeated oral once daily administrations (0.5–15 mg) in fasted conditions
TABLE S1 Baseline lymphocyte count in each dose group of single ascending dose (SAD) and multiple ascending dose (MAD) study parts.
ACKNOWLEDGEMENTS
We thank Anaïs Audry, Rania Boiron, Charlène Buffat, Anne Charlier, Clementine Chopin, Carole Damon, Mélodie Hermier, Mitsuru Ishibai, Helene Joyeux, Christine Maestrini, Delphine Marin, Brigitte Molinier and Tadashi Sugihara for their active participation in this research.
This work was funded and sponsored by Sanofi.
Bergougnan L, Armani S, Golor G, et al. First‐in‐human study of the safety, tolerability, pharmacokinetics and pharmacodynamics of single and multiple oral doses of SAR247799, a selective G‐protein‐biased sphingosine‐1 phosphate receptor‐1 agonist for endothelial protection. Br J Clin Pharmacol. 2021;87:598–611. 10.1111/bcp.14422
The authors confirm that the Principal Investigators for this paper were Sara Armani and Georg Golor and that they had direct clinical responsibility for patients
The clinical study (phase 1 in healthy subjects) was performed in Germany and registered as per EU regulations with EudraCT N° 2015–003046‐96. The study was not registered on clinicaltrials.gov, as it was not under US disclosure regulation, nor applicable under Sanofi's Commitment to disclosure.
DATA AVAILABILITY STATEMENT
All relevant data are provided within the paper. Qualified researchers may request access to patient level data and related study documents including the clinical study report, blank case report form, statistical analysis plan and dataset specifications. Patient level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi's data sharing criteria, eligible studies and process for requesting access can be found at www.clinicalstudydatarequest.com.
REFERENCES
- 1. Rajendran P, Rengarajan T, Thangavel J, et al. The vascular endothelium and human diseases. Int J Biol Sci. 2013;9(10):1057‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sanchez T. Sphingosine‐1‐phosphate signaling in endothelial disorders. Curr Atheroscler Rep. 2015;18(6):31. 10.1007/s11883-016-0586-1 [DOI] [PubMed] [Google Scholar]
- 3. Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Pharmacol Ther. 2007;115:84‐105. [DOI] [PubMed] [Google Scholar]
- 4. Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355‐360. [DOI] [PubMed] [Google Scholar]
- 5. Gilenya (fingolimod) package insert . Basel, Switzerland. Novartis pharmaceuticals 2019.
- 6. Mayzent (siponimod) package insert. Basel, Switzerland. Novartis pharmaceuticals 2019.
- 7. Zeposia (ozanimod) package insert . New York, Bristol‐Myers Squibb pharmaceuticals 2020.
- 8. Comi G, Hartung HP, Bakshi R, Williams IM, Wiendl H. Benefit‐risk profile of Sphingosine‐1‐phosphate receptor modulators in relapsing and secondary progressive multiple sclerosis. Drugs. 2017;77(16):1755‐1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sandborn WJ, Feagan BG, Wolf DC, et al. Ozanimod induction and maintenance treatment for ulcerative colitis. N Engl J Med. 2016;374(18):1754‐1762. [DOI] [PubMed] [Google Scholar]
- 10. Hermann V, Batalov A, Smakotina S, Juif PE, Cornelisse P. First use of cenerimod, a selective S1P1 receptor modulator, for the treatment of SLE: a double‐blind, randomised, placebo‐controlled, proof‐of‐concept study. Lupus Sci Med. 2019;6(1):e000354. 10.1136/lupus-2019-000354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vaclavkova A, Chimenti S, Arenberger P, et al. Oral ponesimod in patients with chronic plaque psoriasis: a randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet. 2014;384(9959):2036‐2045. [DOI] [PubMed] [Google Scholar]
- 12. Hoch M, D'Ambrosio D, Wilbraham D, Brossard P, Dingemanse J. Clinical pharmacology of ponesimod, a selective S1P1 receptor modulator, after uptitration to supratherapeutic doses in healthy subjects. Eur J Pharm Sci. 2014;63:147‐153. [DOI] [PubMed] [Google Scholar]
- 13. Kappos L, Bar‐Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double‐blind, randomised, phase 3 study. Lancet. 2018;391(10127):1263‐1273. [DOI] [PubMed] [Google Scholar]
- 14. Kappos L, Radue EW, O'Connor P, et al. Aplacebo‐controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387‐401. [DOI] [PubMed] [Google Scholar]
- 15. Jain N, Bhatti T. Fingolimod‐associated macular edema: incidence, detection and management. Neurology. 2012;78(9):672‐680. [DOI] [PubMed] [Google Scholar]
- 16. Salvadori M, Budde K, Charpentier B, et al. FTY720 versus MMF with cyclosporine in de novo renal transplantation: a 1‐year, randomized controlled trial in Europe and Australasia. Am J Transplant. 2006;6(12):2912‐2921. [DOI] [PubMed] [Google Scholar]
- 17. Westhoff TH, Schmidt S, Glander P, et al. The impact of FTY720 (fingolimod) on vasodilatory function and arterial elasticity in renal transplant patients. Nephrol Dial Transplant. 2007;22(8):2354‐2358. [DOI] [PubMed] [Google Scholar]
- 18. Allende ML, Yamashita T, Proia RL. G‐protein‐coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood. 2003;102(10):3665‐3667. [DOI] [PubMed] [Google Scholar]
- 19. Awad AS, Ye H, Huang L, et al. Selective sphingosine 1‐phosphate 1 receptor activation reduces ischemia‐reperfusion injury in mouse kidney. Am J Physiol Renal Physiol. 2006;290(6):F1516‐F1524. [DOI] [PubMed] [Google Scholar]
- 20. Shea BS, Brooks SF, Fontaine BA, Chun J, Luster AD, Tager AM. Prolonged exposure to sphingosine 1‐phosphate receptor‐1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. Am J Respir Cell Mol Biol. 2010;43(6):662‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poirier B, Briand V, Kadereit D, et al. In pressA G‐protein‐biased S1P1 agonist, SAR247799, protects endothelial cells without affecting lymphocyte numbers. Sci Signal. 2020;13(634):eaax8050. 10.1126/scisignal.aax8050 [DOI] [PubMed] [Google Scholar]
- 22. Gergely P, Nuesslein‐Hildesheim B, Guerini D, et al. The selective sphingosine 1‐phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species‐specific effects on heart rate. Br J Pharmacol. 2012;167(5):1035‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Legangneux E, Gardin A, Johns D. Dose titration of BAF312 attenuates the initial heart rate reducing effect in healthy subjects. Br J Clin Pharmacol. 2013;75(3):831‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lott D, Lehr T, Dingemanse J, Krause A. Modeling tolerance development for the effect on heart rate of the selective S1P1 receptor modulator Ponesimod. Clin Pharmacol Ther. 2018;103(6):1083‐1092. [DOI] [PubMed] [Google Scholar]
- 25. Brossard P, Scherz M, Halabi A, Maatouk H, Krause A, Dingemanse J. Multiple‐dose tolerability, pharmacokinetics, and pharmacodynamics of ponesimod, an S1P1 receptor modulator: favorable impact of dose up‐titration. J Clin Pharmacol. 2014;54(2):179‐188. [DOI] [PubMed] [Google Scholar]
- 26. Winkler MS, Nierhaus A, Poppe A, Greiwe G, Gräler MH, Daum G. Sphingosine‐1‐phosphate: a potential biomarker and therapeutic target for endothelial dysfunction and sepsis? Shock. 2017;47(6):666‐672. [DOI] [PubMed] [Google Scholar]
- 27. Sattler K, Gräler M, Keul P, et al. Defects of high‐density lipoproteins in coronary artery disease caused by low Sphingosine‐1‐phosphate content: correction by Sphingosine‐1‐phosphate‐loading. J am Coll Cardiol. 2015;66(13):1470‐1485. [DOI] [PubMed] [Google Scholar]
- 28. Knapp M, Lisowska A, Zabielski P, Musiał W, Baranowski M. Sustained decrease in plasma sphingosine‐1‐phosphate concentration and its accumulation in blood cells in acute myocardial infarction. Prostaglandins Other Lipid Mediat. 2013;106:53‐61. [DOI] [PubMed] [Google Scholar]
- 29. Jing XD, Wei XM, Deng SB, Du JL, Liu YJ, She Q. The relationship between the high‐density lipoprotein (HDL)‐associated sphingosine‐1‐phosphate (S1P) and coronary in‐stent restenosis. Clin Chim Acta. 2015;446:248‐252. [DOI] [PubMed] [Google Scholar]
- 30. Punsawad C, Viriyavejakul P. Reduction in serum sphingosine 1‐phosphate concentration in malaria. PLoS One. 2017;12(6):e0180631. 10.1371/journal.pone.0180631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kovarik JM, Schmouder B, Barilla D, Wang Y, Kraus G. Single dose FTY720 pharmocokinetics, food effect and pharmacological responses in healthy subjects. Br J Clin Pharmacol. 2004;57(5):586‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boehler M, Juif PE, Hoch M, Dingemanse J. Absolute bioavailability of Ponesimod, a selective S1P1 receptor modulator, in healthy male subjects. Eur J Drug Metab Pharmacokinet. 2017;42(1):129‐134. [DOI] [PubMed] [Google Scholar]
- 33. Tran JQ, Hartung JP, Peach RJ, et al. Results from the first‐in‐human study with Ozanimod, a novel, selective sphingosine‐1‐phosphate receptor modulator. J Clin Pharmacol. 2017;57(8):988‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Plasma concentration–time profiles of SAR247799 in healthy subjects following administration of single doses (A) and following multiple doses on day 1 (B) or day 14 (C).
FIGURE S2 Trough concentrations of SAR247799 after repeated oral once daily administrations (0.5–15 mg) in fasted conditions
TABLE S1 Baseline lymphocyte count in each dose group of single ascending dose (SAD) and multiple ascending dose (MAD) study parts.
Data Availability Statement
All relevant data are provided within the paper. Qualified researchers may request access to patient level data and related study documents including the clinical study report, blank case report form, statistical analysis plan and dataset specifications. Patient level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi's data sharing criteria, eligible studies and process for requesting access can be found at www.clinicalstudydatarequest.com.