

Abstract
Huntington’s disease (HD) is a heritable, fatal neurodegenerative disorder caused by a mutation in the Huntingtin gene. It is characterized by chorea, as well as cognitive and psychiatric symptoms. Histopathologically, there is a massive loss of striatal projection neurons and less but significant loss in other areas throughout the cortico-basal ganglia-thalamocortical (CBGTC) loop. The mutant huntingtin protein has been implicated in numerous functions, including an important role in synaptic transmission. Most studies on anatomical and physiological alterations in HD have focused on striatum and cerebral cortex. However, based on recent CBGTC projectome evidence, the need to study other pathways has become increasingly clear. In this review, we examine the current status of our knowledge of morphological and electrophysiological alterations of those pathways in animal models of HD. Based on recent studies, there is accumulating evidence that synaptic disconnection, particularly along excitatory pathways, is pervasive and almost universal in HD, thus supporting a critical role of the huntingtin protein in synaptic transmission.
Keywords: Huntington’s disease, Genetic models, Basal ganglia, Synaptic activity, Disconnection
1. Introduction
Huntington’s Disease (HD) is an inherited, ultimately fatal neurodegenerative disease caused by a mutation in the Huntingtin (HTT) gene, containing an elongated CAG (glutamine) chain (The Huntington’s Disease Collaborative Research Group, 1993). When the number of CAG triplet repeats exceeds 36, patients will inexorably develop the disease. The main motor symptom of HD is chorea (abnormal dance-like movements), along with psychiatric and cognitive abnormalities. Other movement disorders also occur including; impairment in coordination of voluntary movements such as speech, swallowing, and balance (Duff et al., 2007; Paulsen et al., 2008). As HD progresses, predominant motor symptoms evolve into bradykinesia and rigidity. Histopathologically, HD primarily affects the basal ganglia, specifically the striatal medium-sized spiny neurons (MSNs), also known as striatal projection neurons (SPNs). Neurons in other brain regions, including cortical pyramidal neurons (CPNs) and thalamic neurons, also are lost (Vonsattel et al., 1985; Waldvogel et al., 2015). In the striatum, various interneuron types are spared, except for the parvalbumin (PV)-expressing fast-spiking interneurons (FSIs) (Ferrante et al., 1987; Harrington and Kowall, 1991; Reiner et al., 2013). In human motor cortex there is selective loss of calbindin-D28k (CB) interneurons in cases with motor disorder but not mood disorder. Whereas in the anterior cingulate cortex there is loss of CB, calretinin, and PV interneurons in cases with mood disorder but not motor disorder (Kim et al., 2014).
The huntingtin protein (HTT) is critical for normal brain synaptic transmission. Thus, HTT acts as a scaffold protein and is linked to diverse processes such as transport of vesicles and brain-derived neurotrophic factor (BDNF) (Saudou and Humbert, 2016; Zuccato and Cattaneo, 2009). Studies in HD animal models have shown altered exocytosis of presynaptic vesicles and Ca2+ influx (Joshi et al., 2009; Morton et al., 2001). This has led to the idea that HD is not just a neurodegenerative disease but it is actually a synaptopathy (Cepeda and Levine, 2020; Li et al., 2003; Zieger and Choquet, 2021). Indeed, targeted deletion of normal HTT from striatal MSNs disrupts synaptic connectivity to output structures (Burrus et al., 2020).
The normal HTT protein is also critical during development, as lack of the protein is lethal (Duyao et al., 1995; Nasir et al., 1995; Saudou and Humbert, 2016; Zeitlin et al., 1995). Inactivation of HTT by RNA interference or deletion of the gene affects spindle orientation and cell fate of cortical progenitors in the ventricular zone of mouse embryos (Godin et al., 2010; Molina-Calavita et al., 2014). In addition, mutant (m)HTT protein presence during embryonic development alters neuronal migration, orientation and final positioning (Barnat et al., 2017; Osmand et al., 2016). Interestingly, expression of mHTT during early development is sufficient to cause a permanent HD phenotype even when mHTT expression is terminated by postnatal day 21 (Molero et al., 2016). In HD patients, cortical thinning and loss of white matter are common in pre-manifest HD subjects (Aylward, 2007; Reading et al., 2005; Rosas et al., 2005; Rosas et al., 2006; Waldvogel et al., 2015). Prodromal HD patients exhibit small intracranial volumes, suggesting mHTT can cause abnormal brain development (Nopoulos et al., 2011). Our own studies demonstrated the frequent occurrence of dysplastic lesions in the cortex of symptomatic and presymptomatic HD mice (Cepeda et al., 2019). Aberrant cortical development is likely to affect synaptic transmission along multiple pathways in the cortico-basal ganglia-thalamocortical (CBGTC) loop. The question can be raised as to whether there is a node in the CBGTC loop where synaptic pathology begins or if it starts simultaneously in several regions. Only examination of areas beyond the corticostriatal pathway during brain development, as well as during presymptomatic and symptomatic stages of HD will allow answering those questions. However, in order to uncover the functional alterations along the CBGTC loop, an intimate knowledge of the anatomical pathways of this circuit is required. The development of new and more advanced anatomical tracing techniques, e.g., anterograde and retrograde viruses, is beginning to provide a more comprehensive picture, albeit of increasing complexity, of a growing number of parallel and convergent pathways constituting the CBGTC loop. Knowledge of how these pathways are affected in HD is a conditio sine qua non for a better understanding of underlying pathological mechanisms. This review focuses primarily on synaptic mechanisms, more detailed information on the molecular and cellular mechanisms of HD neuropathology, including receptor alterations of MSNs and CPNs in mouse models, can be found in a number of comprehensive reviews (Mackay et al., 2018; Plotkin and Surmeier, 2015; Raymond et al., 2011). We apologize for not being able to cite the already abundant and excellent literature on this subject matter.
2. A new vista of the cortico-basal ganglia-thalamocortical loop
The classical view of the CBGTC loop postulates the existence of parallel pathways, each carrying different types of information. Very succinctly, information flows through this loop in three parallel channels; associative, limbic and sensorimotor through the dorsomedial, ventromedial and lateral two quadrants of the striatum, respectively (Alexander et al., 1986; Aoki et al., 2019; Haber, 2003; Mandelbaum et al., 2019; Parent and Hazrati, 1994; Wallace et al., 2017). However, this scheme is insufficient to explain the multiple functions performed by the basal ganglia (Plotkin and Goldberg, 2019). Likewise, the segmentation of the striatum itself has increased considerably. Thus, a mesoscale mouse corticostriatal projectome from the entire cerebral cortex to the dorsal striatum identified 29 distinct functional striatal domains based on the topography of cortical inputs (Hintiryan et al., 2016).
In a recent paper, the same group provided a detailed map of the multi-synaptic output pathways arising from these striatal domains. The authors identified 14 substantia nigra pars reticulata (SNr), 36 external globus pallidus (GPe), and 6 parafascicular and ventromedial thalamic nuclei domains, as well as a direct cortico-SNr projection. They also demonstrated that multiple striatal domains of the direct pathway display a greater convergence (i.e., more direct pathway inputs consolidate in the same output structure, SNr/GPi) than that of the more parallel striatopallidal indirect pathway, where different striatal domains remain separate in the GPe. Importantly, thalamic domains relay this output back to the originating corticostriatal neurons of each subnetwork forming a veritable closed loop (Foster et al., 2021).
3. Mouse models of HD
The introduction of genetic rodent models of HD, in particular mouse models, has allowed examination of the evolution of symptoms as well as the localization of neuronal and synaptic abnormalities (Brooks and Dunnett, 2015; Levine et al., 2004). The current mouse models can replicate some characteristics of HD to different degrees. No one model, however, can replicate the symptomatology and histopathology of human HD (Levine et al., 2004). Differences in mouse models are due to the expression of either truncated or full-length human mHTT. We will describe the most common transgenic and knock-in (KI) HD mouse models discussed in this review. The first transgenic mouse model of HD, and the most extensively studied, is the R6/2 model. This model carries exon 1 of the human gene, which contains the N-terminal 171 amino acids and therefore can cause acute neuronal toxicity. The model produces a severe, fast-progressing phenotype similar to juvenile HD (Mangiarini et al., 1996). However, these mice do not express the mHTT in its natural genomic and protein context, which could lead to the loss of any potential post-translational modifications and protein interactions occurring in human HD. Additionally, it has been shown that mHTT fragments generated by intracellular cleavage in human HD and full-length transgenic models have a different subcellular localization than pre-cleaved truncated mHTT in R6/2 mice (Warby et al., 2008). This again, raises doubts about the model’s capability to recapitulate all aspects of disease pathology.
The most widely studied full-length transgenic mouse models are the yeast artificial chromosome (YAC) and the bacterial artificial chromosome (BAC) HD models, which express human mHTT (Gray et al., 2008; Hodgson et al., 1999). The YAC128 model exhibits age-dependent striatal, followed by cortical atrophy and mimics human disease progression by displaying first a hyperkinetic and later a hypokinetic phenotype (Slow et al., 2003). The BACHD model carries 97 mixed CAACAG repeats. It shows reduced cortical and striatal volume and progressive motor impairments, consistent with the findings in the YAC128 mice (Gray et al., 2008). Although similar in terms of deficits in motor learning and coordination, depressive-like symptoms, and striatal volume loss, the two models also show differences. For example, while YAC128 mice exhibit widespread accumulation of mHTT aggregates in striatum, these aggregates are absent in BACHD mice. In addition, DARPP-32, enkephalin, and dopamine (DA) D1 and D2 receptor mRNAs are significantly decreased in YAC128 but not BACHD mice (Pouladi et al., 2012).
The knock-in mouse models express mHTT in the most realistic genomic context, since the human polyQ sequence is inserted into the endogenous mouse HTT gene (Shelbourne et al., 1999). The Q140 model contains the chimeric mouse/human exon 1 with 140 CAG repeats. This model displays early selective atrophy in the striatum and cortex as well as behavioral symptoms at 4 months of age. A recent KI model derived from the Q140 is the Q175, which is now widely used because of its breeding stability and replication of adult-onset progressive motor symptoms and brain pathology (Smith et al., 2014). Although each model shows relatively different characteristics of the HD progression in humans, the major findings on electrophysiological and morphological changes have been quite consistent across multiple lines (Cummings et al., 2009).
Most basic research has focused on examining the effect and the underlying mechanisms of neuronal and synaptic dysfunction in the striatum and cortex (Cepeda et al., 2010; Cummings et al., 2009; Heikkinen et al., 2012; Miller et al., 2008; Rebec et al., 2006; Walker et al., 2008). All studies have confirmed a progressive disconnection along the corticostriatal pathway (Cepeda et al., 2003). Based on the new and more detailed corticostriatal connectome, a recent study examined the specific corticostriatal connections primarily affected in symptomatic Q175 HD mice. The authors examined projections from the upper limb motor regions to the intermediate level of the caudate putamen (CPi). They showed that while primary motor cortex projections were unaffected, secondary motor cortex projections to the CPi were significantly reduced (Hintiryan et al., 2016). The specific loss of projections from the secondary but not the primary motor cortex suggests a pathway-specific loss instead of a complete corticostriatal connectome loss.
4. Striatal output structures
Striatal output structures are innervated by projection MSNs giving rise to two major pathways: direct and indirect. Direct pathway MSNs, expressing DA D1 receptors and substance P, project to the internal globus pallidus (GPi)/SNr. Indirect MSNs, expressing DA D2 receptors and enkephalin, project to the GPe, which in turn projects to the subthalamic nucleus (STN) before reaching their final destination in the SNr (Gerfen, 1992). A third pathway, the hyperdirect pathway, arises from CPNs that project directly to the STN. The hyperdirect pathway is able to bypass the indirect pathway, with the STN projection neurons exciting the SNr to suppress thalamocortical activity, thus increasing inhibition of the motor neurons and suppressing movement (DeLong and Wichmann, 2007) (Fig. 1). An imbalance in basal ganglia connectivity plays an important role in the pathogenesis of HD. Thus, while motor signs are associated with altered connectivity in the indirect pathway, apathy seems more associated with changes in the direct pathway (Nair et al., 2021). Although both the direct and indirect pathways are affected as disease progresses, it has long been held that, at least in human patients, indirect pathway MSNs are affected earlier compared to those in the direct pathway (Albin et al., 1992; Reiner et al., 1988). This could mean that indirect pathway MSNs are more vulnerable to the HD mutation. In mouse models, cell loss is less evident and occurs very late (Turmaine et al., 2000). For example, in Q175 mice, no loss of striatal MSNs was seen up to 18 months of age, although enkephalin expression was reduced (Deng et al., 2021). Interestingly, and probably as a compensatory mechanism, enkephalin and substance P terminals were more abundant in GPe and SNr/GPi respectively at both 6 and 18 months. Electrophysiological studies revealed functional alterations more selectively on direct pathway MSNs in early stages of disease in two HD mouse models, YAC128 and BACHD, with an increase in spontaneous excitatory post-synaptic currents (sEPSCs), whereas in late stages both direct and indirect pathway MSNs were affected (Andre et al., 2011). It is thus expected that alterations in striatal MSNs lead to faulty communication with their output regions. Although few, recent studies highlight changes in these output regions (Barry et al., 2018; Perez-Rosello et al., 2019), as well as in other nodes of the CBGTC loop (Fig. 2).
Fig. 1.
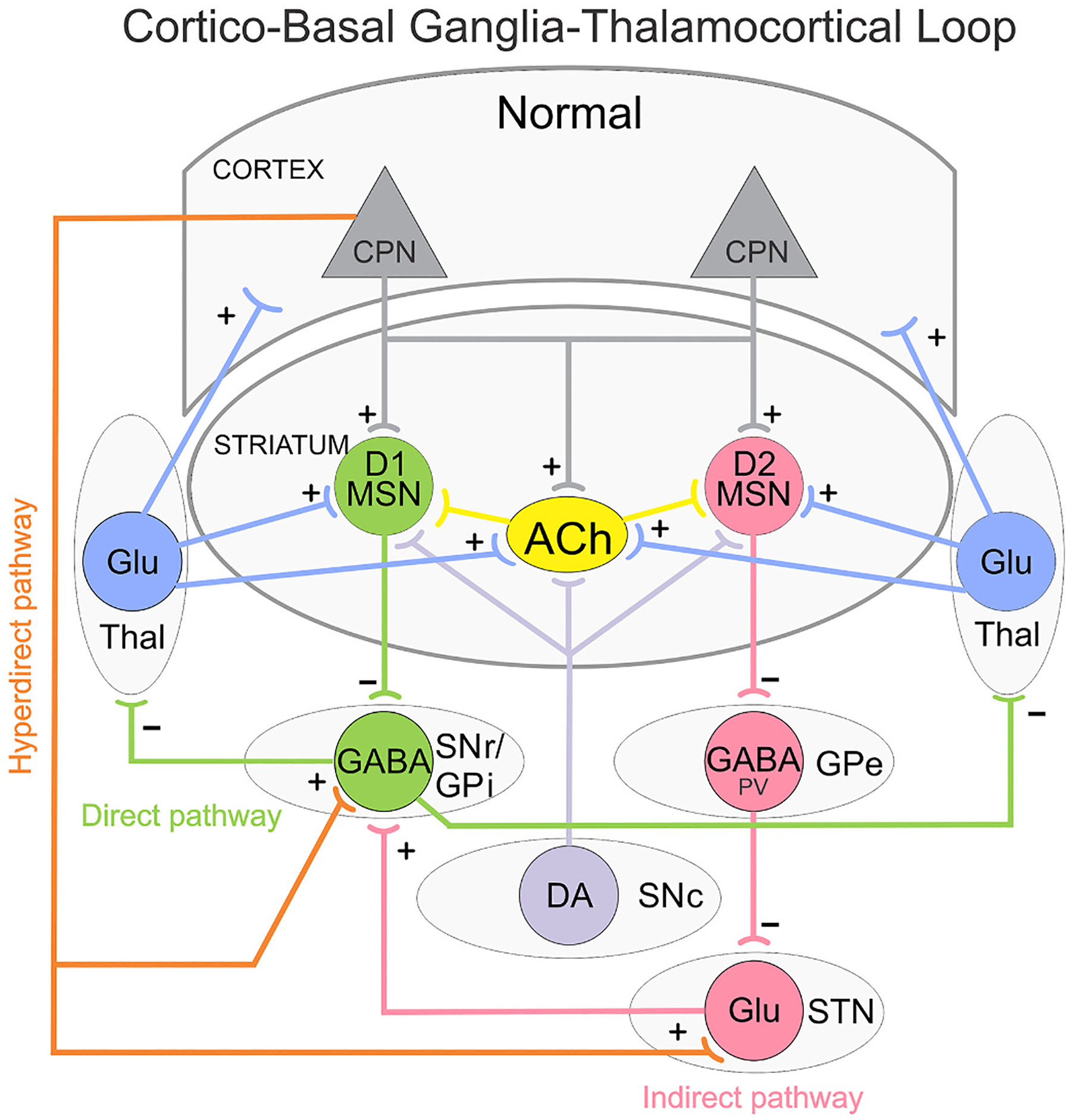
Simplified diagram of the cortico-basal ganglia-thalamocortical (CBGTC) loop. Striatal medium-sized spiny neurons (MSNs) expressing dopamine (DA) D1 receptors receive glutamatergic (Glu) inputs (+) from cortical pyramidal neurons (CPNs) and project to the substantia nigra pars reticulata (SNr) as well as the internal segment of the globus pallidus (GPi). This is called the direct pathway and promotes movement. MSNs expressing DA D2 receptors also receive inputs from CPNs and project to the external segment of the globus pallidus (GPe). The GPe, in turn, projects to the subthalamic nucleus (STN) and this to the SNr/GPi. This is known as the indirect pathway and counteracts movement. GABA (−) outputs from SNr/GPi project to the thalamus (Thal), which in turn, sends Glu inputs (+) back to the cortex thus completing the CBGTC loop. Both D1 and D2 MSNs also receive afferents from the substantia nigra pars compacta (SNc) and Thal. A hyperdirect pathway connects the CPNs of the cortex directly to the STN. In addition, a newly described pathway from CPNs to SNr GABAergic neurons is illustrated. Several classes of interneurons, including acetylcholine (ACh) interneurons, exert control over the MSNs. For the sake of simplicity, GABAergic interneurons and some additional synaptic pathways are not illustrated.
Fig. 2.
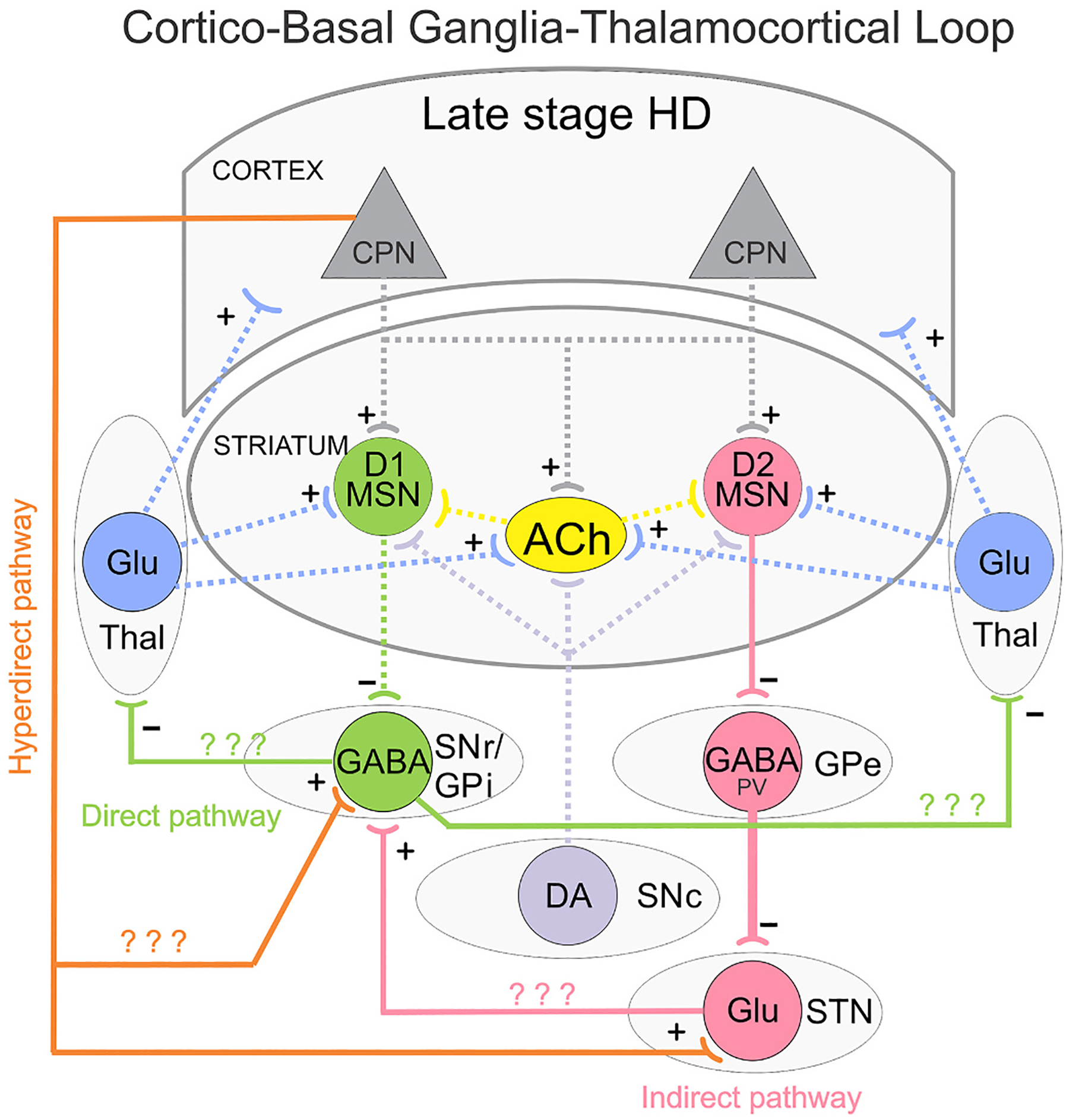
Simplified diagram of the CBGTC loop in late stage HD. Glu inputs (+) from CPNs are reduced onto both direct (D1) and indirect (D2) pathway MSNs. GABA (−) outputs from D1 MSNs are reduced onto SNr/GPi neurons, dis-inhibiting these neurons and promoting hypokinesia. GABA (−) outputs from D2 MSNs are unchanged or decreased, but reduced GABA reuptake disrupts firing patterns of PV-expressing GABAergic neurons in the GPe and facilitate bursting activity. This, in conjunction with reduced cortical input through the hyperdirect pathway, may reduce STN firing. Question marks indicate that alterations of these pathways in HD remain unknown or studies are inconclusive.
The Indirect Pathway: The GP, both GPe and GPi, are important nodes in the CBGTC loop and have been the targets for ablation and neuromodulation, i.e., deep brain stimulation (DBS), to improve motor disorders, including HD (Bonomo et al., 2021; Cif and Hariz, 2017; Reiner, 2004; Wojtecki et al., 2016). As the first relay station of the indirect pathway and considering that striatal enkephalin-containing neurons appear more susceptible than direct pathway MSNs, it is important to know how this nucleus is altered in HD.
GPe:
There are three main cellular types in the GPe that have been characterized morphologically and electrophysiologically (Cooper and Stanford, 2000; Hegeman et al., 2016; Kita and Kitai, 1991). The most abundant are the prototypical (PV-expressing) neurons, that project to the STN, and the arkypallidal (Npas- or FoxP2-expressing) neurons that project back to the striatum. Interestingly, it is the arkypallidal, not the prototypical GPe neurons that are lost in symptomatic Q175 mice at 18 months (Deng et al., 2021). We examined passive and active membrane properties of GPe neurons in R6/2 model mice. As HD progresses GPe neurons in this model showed an increase in cell membrane capacitance and input resistance (Akopian et al., 2016). R6/2 GPe neurons also showed alterations in firing patterns; increased interspike interval variation and number of bursts. Optogenetically-evoked responses of indirect pathway MSNs onto GPe neurons, in both R6/2 and YAC128 HD mice, showed an increase in decay time and area while amplitude remained unchanged (Barry et al., 2018). In contrast, another study reported no change in decay time but a significant increase in amplitude of optically-evoked GABA responses (Perez-Rosello et al., 2019). However, the underlying mechanism for this change was not explained as both pre- and post-synaptic mechanisms were invoked. A more recent study in our laboratory determined that the increase in decay time of the optogenetically-evoked responses of GPe neurons is probably caused by alterations in expression of GABA transporter-3 (GAT-3). In agreement, a GAT-3 inhibitor, SNAP5114, caused an increase in decay time in WT but not R6/2 neurons (Barry et al., 2020). Overall, there were no significant differences in frequency of spontaneous inhibitory post-synaptic currents (sIPSCs) or inter-event interval (IEI) in symptomatic R6/2 (2 months), presymptomatic (2 months) or symptomatic (12 months) YAC128 GPe neurons compared to WT littermates (Barry et al., 2018). Although the kinetics of sIPSCs of GPe neurons showed no significant differences between R6/2 and WT mice, there was a non-significant increase in decay time in the R6/2 neurons (Barry et al., 2018).
STN:
There is a significant loss of STN neurons in symptomatic Q175 mice (Atherton et al., 2016; Deng et al., 2021). Furthermore, the neuronal activity of the STN is altered in early stages of HD in two HD mouse models, BACHD and Q175, exhibiting prolonged N-methyl-Daspartate (NMDA) receptor mediated synaptic currents due to a reduction in glutamate uptake (Atherton et al., 2016). In symptomatic YAC128 mice, a progressive age-dependent disconnection of the hyperdirect pathway, similar to that seen in the corticostriatal pathway, was observed (Callahan and Abercrombie, 2015a). Thus, cortical entrainment of STN activity was disrupted and there was an increase in the proportion of STN neurons that were less phase-locked to cortical activity. In addition, evoked responses of STN neurons to cortical stimulation as well as spontaneous firing of STN neurons were decreased. In R6/2 mice, cortical entrainment of STN neural activity also was disrupted, leading the authors to conclude that miscommunication between cortex and STN may be a mechanism contributing to disordered motor control in HD (Callahan and Abercrombie, 2015b).
A recent study examined neuronal activity throughout different regions of the indirect pathway using in vivo and ex vivo electrophysiological recordings in Q175 mice at early symptomatic stages (6–12 months). Compared with WT mice, D2-MSNs were hypoactive, leading to hyperactivity of prototypic PV-positive GPe neurons. In contrast, arkypallidal GPe neurons and STN neurons were hypoactive. Notably, the hypoactivity of STN and arkypallidal GPe neurons was partially alleviated by optogenetic inhibition of prototypic GPe neurons (Callahan et al., 2021). Another recent study identified a subnetwork of direct pathway MSNs specifically targeting arkypallidal Npas-positive neurons in the GPe (Cui et al., 2021). Its role in HD pathogenesis remains to be determined.
The direct pathway: As HD progresses changes occur in the direct pathway output structures [SNr and substantia nigra pars compacta (SNc)] both in terms of intrinsic and synaptic properties. SNr neurons of the CAG140 HD model were shown to have increased burst rates compared to their WT littermates during early development of symptoms (Murphy-Nakhnikian et al., 2012). In R6/2 mice, SNr neurons were shown to have decreased cell membrane capacitance and increased membrane input resistance indicating decreases in somatic area and dendritic processes (Barry et al., 2018). This is similar to alterations found in the cortex and striatum of symptomatic R6/2 mice (Klapstein et al., 2001). Most SNr neurons fire spontaneously with a rhythmic pattern. In R6/2 mice there was an increase in low frequency firing cells compared to WT littermates (Barry et al., 2018). Using optogenetic techniques, we also showed a decrease in amplitude of the evoked synaptic response of direct pathway MSN terminals onto SNr neurons. Decreased amplitude of evoked responses in the YAC128 HD mouse model also was observed (Barry et al., 2018). Compared to results from GPe neurons, there were significant decreases of sIPSCs frequency and amplitude in symptomatic R6/2 (2 months) SNr neurons compared to WT littermates (Barry et al., 2018). sIPSCs amplitude was also decreased in symptomatic (12 months) YAC128, but unchanged in presymptomatic (2 months) compared to WT littermates. The decreased amplitude of optogenetically evoked GABAergic responses corroborated the reduction in frequency and amplitude of the sIPSCs (Barry et al., 2018).
5. The SNc-striatum DA pathway
The SNc plays an important role in HD pathophysiology. Indeed, biphasic changes in striatal DA content, increased in the early stage but decreased in the late stage, reflect clinical symptoms, i.e., hyperkinesia followed by bradykinesia (Cepeda et al., 2014). In addition, regions with early HD pathology all receive dense DA inputs (Menalled et al., 2003). Although DA SNc neurons do not appear to be lost in HD, they undergo functional changes. Electrophysiological studies in R6/1 mice, a model similar to the R6/2 but with slow progression, demonstrated that, although DA neurons do not show changes in basic membrane properties, there is a reduction of small-conductance Ca2+-activated K+ channels (SK3, responsible for the slow afterhyperpolarization), leading to hyperexcitability and concomitant increases in DA release followed by drastic reductions in DA availability in late stages of the disease (Dallerac et al., 2015). Consistent with this observation, studies have shown progressive loss of DA in the striatum of HD animal models (Johnson et al., 2006; Ortiz et al., 2011; Ortiz et al., 2012), which could explain bradykinesia in the late stages. The origin of early increases in DA is unknown but anatomical studies have suggested that early degeneration of striosomal MSNs may produce hyperactivity of the nigrostriatal DA pathway, causing chorea (Hedreen and Folstein, 1995). In addition, changes in cortical input onto SNc neurons could also contribute to biphasic changes in DA release.
6. The thalamostriatal pathway: morphological and electrophysiological findings
Aside from the cortex, the striatum also receives considerable excitatory inputs from the thalamus, which end mainly on the spines and dendrites of MSNs and cholinergic interneurons (Doig et al., 2010; Lapper and Bolam, 1992; Smith et al., 2004). The thalamic projection is topographically organized and arises predominantly from the centromedian (CM) and parafascicular (Pf) nuclei (Berendse and Groenewegen, 1990; Wall et al., 2013).
Tracing studies indicated that direct and indirect pathway MSNs in mice receive inputs from relatively equal numbers of thalamic neurons (Wall et al., 2013). In Q140 mice it was shown that thalamostriatal projections are reduced compared to WT mice (Deng et al., 2013). Using VGLUT2 (vesicular glutamate transporter 2) immunohistochemistry to detect thalamostriatal terminals, they observed a significant reduction in both axodendritic and axospinous thalamostriatal terminals that was already evident in the dorsolateral striatum at 1 month of age and continued up to 12 months. Consistent with the early loss of thalamostriatal projections in Q140 mice, striatal expression of proteins critical to thalamostriatal synapse formation, such as the semaphorin 3E receptor Plexin-D1 signaling complex (Ding et al., 2011), were significantly reduced early in the lifespan of several genetic mouse models of HD (Kuhn et al., 2007). A follow up study from Reiner’s group compared cortical and thalamic inputs to the direct and indirect pathway MSNs in 4 and 12 month-old Q140 mice. The authors found that the loss of corticostriatal terminals at 12 months of age was preferential for D1-positive spines. In contrast, thalamostriatal terminal loss was comparable for spines from both pathways (Deng et al., 2014). They concluded that a differential thalamic and cortical input loss to MSNs is an early event in HD and that the loss of cortical input onto direct pathway neurons may contribute to pre-manifest motor slowing. These early and consistent changes suggest the abnormal development of basal ganglia connectivity.
Functional studies using optogenetic stimulation of the thalamostriatal pathway highlighted overall increases in glutamate release probability and decreases in AMPAR/NMDAR current ratios (Kolodziejczyk and Raymond, 2016; Parievsky et al., 2017). Parievsky et al. found increased decay time in both AMPAR and NMDAR currents, potentially as a compensatory mechanism to counter the reduced number of excitatory synaptic contacts and therefore, reduced peak amplitude. A higher relative proportion of NMDARs at thalamostriatal synapses, longer decay times of NMDAR-mediated currents, and increased glutamate release probability, all support an important role of thalamostriatal projections in excitotoxicity. Both studies also found increased activation of extrasynaptic NMDARs, which is associated with cell death signaling (Hardingham and Bading, 2010), in different HD mouse models. Notably, Kolodziejczyk and Raymond observed this occurrence as early as 14 days in vitro in their YAC128 thalamostriatal co-culture system (Kolodziejczyk and Raymond, 2016). This further established the possible role of thalamic afferents in HD pathology, as well as suggested that these early changes are possibly regulated by an impaired receptor transport or distribution mechanism in HD.
There is little data on the abnormalities of thalamic input to striatal interneurons in both animal models and human HD. One potential cell type prone to dysfunction is the cholinergic interneuron, because it receives excitatory inputs directly from the thalamus in both rodents and primates (Doig et al., 2010; Lapper and Bolam, 1992). Cholinergic interneurons project to both direct and indirect pathway MSNs, and modulate the responsivity of these neurons at corticostriatal synapses (Ding et al., 2010; Pisani et al., 2007; Smith et al., 2011). A study in R6/2 mice showed that neurons of the Pf nucleus, the main source of thalamostriatal afferents providing trophic support to cholinergic interneurons, degenerate at an early stage (Crevier-Sorbo et al., 2020). Using ChAT immunolabeling to identify cholinergic interneurons, significant changes in the VGLUT2 labeled thalamic input to striatal cholinergic interneurons were observed in 1 and 4 month-old Q140 mice (Deng and Reiner, 2016). The dendrites of Q140 cholinergic interneurons were significantly fewer and shorter as early as 1 month of age, and there were fewer dendritic arborizations from individual interneurons as well, consistent with another report (Holley et al., 2015). In this study, we also reported enhanced inhibitory inputs to cholinergic interneurons in R6/2 mice, which would reduce their activity and pacemaking ability. Overall, these results show that the abundance of thalamic input to striatal cholinergic interneurons is reduced early on in Q140 mice. In support, it has been demonstrated that the responsiveness of striatal cholinergic interneurons to thalamic inputs in presymptomatic Q175 mice also is reduced (Tanimura et al., 2016). To our knowledge, no published information from studies of mouse models is available on how HD may affect thalamic inputs to other striatal interneuron types.
7. The thalamocortical pathway: morphological and electrophysiological findings
The role of the thalamus and the thalamocortical projection in HD has been understudied, even though some of the earliest sensory, attention, and cognitive deficits are clearly associated with thalamocortical circuits. In HD, it is hypothesized that the early loss of indirect pathway MSNs results in disinhibition of the motor thalamocortical pathway (Albin et al., 1992; Deng et al., 2004; Reiner et al., 1988), although evidence from mouse models suggests the disinhibition may also be due to alterations in the direct pathway (Andre et al., 2011). In addition, CPNs that synapse on thalamic nuclei neurons are preferentially lost (Hedreen et al., 1991), suggesting potential reciprocal disruption in the circuitry.
Using patch clamp electrophysiology in brain slices, preliminary studies in our lab described altered membrane properties in thalamic neurons of R6/2 mice. Even at the presymptomatic stage both ventral anterior lateral (VAL) and ventral posteromedial (VPM) neurons start to show alterations in either membrane properties (VAL) or somatic size (VPM). These changes become more pronounced in the symptomatic stage leading to decreased capacitance, increased input resistance and faster decay time constants. This suggests signs of degeneration in both regions. CPNs in the barrel cortex of symptomatic R6/2 mice also exhibited altered membrane properties indicative of degeneration. When optically stimulating these thalamocortical terminals, smaller IPSCs and EPSCs were observed in CPNs, which suggests a possible disconnection between the thalamus and the cortex, especially between the thalamus and GABAergic interneurons of the barrel cortex in the case of IPSCs (Holley et al., 2019). In a more recent study, we utilized high-density silicone probes to examine the effects of altered thalamocortical pathway in HD in more physiological conditions. Early symptomatic Q175 mice were trained to perform a simple cued-response behavioral task. It consisted of licking for milk in response to an audible cue delivered via a solenoid valve actuation. Responses to the reward stimuli, i.e., licking, were measured and analyzed for various brain regions (Shobe et al., 2021). We observed delayed cortical (both CPNs and FSIs) and thalamic responses to reward stimuli, as well as impaired thalamocortical coherence (Shobe et al., 2021). Specifically, we found an increase in delta band power in the cortex of Q175 mice, which is consistent with many HD studies in humans (Hunter et al., 2010; Leuchter et al., 2017; Painold et al., 2010) and is suggestive of impaired thalamocortical communication. As the thalamus does not receive inputs directly from the striatum, but through SNr/GPi, we believe that cell dysfunction in the striatum is unlikely the culprit of the thalamocortical changes found in this study. Instead, early cognitive and behavioral changes are primarily due to altered thalamocortical activity, which subsequently affects the corticostriatal pathway.
8. Therapies to rescue synaptic pathology
Based on our knowledge of synaptic pathology, which therapies are more suitable to treat HD symptoms? Classically, therapies for HD primarily target the symptom of chorea with a variety of treatments including: DA antagonists, DA-depleting agents [tetrabenazine (TBZ)], glutamate antagonists, benzodiazepines, cannabinoids, lithium and deep brain stimulation to name a few (Armstrong et al., 2012; Bagchi, 1983; Paleacu, 2007; Pidgeon and Rickards, 2013). Currently TBZ, a vesicular monoamine transporter 2 inhibitor, is one of the very few FDA approved drugs for HD (Huntington Study Group, 2006). YAC128 mice in the early stages of the disease show increased stereotypies that are decreased by TBZ treatment, supporting increased DA tone in direct pathway neurons (Andre et al., 2011). TBZ helps control chorea but has negligible effects on other HD symptoms such as psychiatric disturbances.
New therapies use Zinc-finger transcription repressors and CRISPRCas9 to target HTT DNA (Estevez-Fraga et al., 2020). Others use genetargeting strategies specifically for the mHTT gene and/or mHTT protein (Leavitt and Tabrizi, 2020). However, recent studies by large pharmaceutical companies using antisense oligonucleotides (ASOs) to lower mHTT in HD patients have stalled. A phase I/II trial by Roche, using the drug tominersen, showed significantly lower levels of mHTT in patients’ cerebrospinal fluid without significant side effects. However, the phase III trial of tominersen was halted as the drug had less efficacy than placebo, and when given more frequently, led to a worse outcome. An independent committee of experts reviewed the data, leading to an early termination of the trial due to the drug’s potential benefits not outweighing the risks (Kwon, 2021). Other studies examined whether selective phosphodiesterase (PDE) inhibitors could improve pathogenesis of the disease in HD rodent models. Indeed, R6/2 mice treated with a highly selective PDE10A inhibitor, TP-10, ameliorated behavioral deficits and brain pathology by increasing striatal and cortical levels of phosphorylated cAMP response element-binding protein (CREB) and BDNF (Beaumont et al., 2016; Giampa et al., 2010). A randomized, double-blind, placebo controlled, phase II clinical trial (Amaryllis) used the PDE10A inhibitor PF-02545920 to examine the efficacy on motor function along with the safety and tolerability of the drug. The trial used the Unified-Huntington’s-Disease-Rating-scale Total-Motor-Score (UHDRS-TMS), UHDRS-Total-Maximum-Chorea score and Clinical-Global-Impression of Improvement as measurements of changes in HD patients who were separated into three groups (5 mg PF-02545920, 20 mg PF-02545920 or placebo). Unfortunately, the results showed no benefits of the drug compared to placebo (Delnomdedieu et al., 2018). Similarly, a selective PDE9A inhibitor, PF-04447943, rescued corticostriatal transmission in BACHD transgenic rats (Chakroborty et al., 2020). In contrast, no significant beneficial role for SCH-51866, a PDE1/5 inhibitor, was observed in either motor or cognitive behavioral tasks (Beaumont et al., 2014). Another strategy aimed at restoring synaptic corticostriatal communication used an ampakine, Cx614, known to facilitate glutamate release and increase BDNF, which is reduced in HD (Park, 2018; Zuccato et al., 2005). There was an augmentation of synaptic activity in both WT and R62 mice, but this effect was reduced in symptomatic mice (Cepeda et al., 2010). Recently, optogenetic activation of cortical regions projecting to the striatum, in particular the secondary motor cortex (M2), was reported to be beneficial to treat some motor symptoms and rescue synaptic deficits caused by the corticostriatal disconnection in R6/2 mice (Fernandez-Garcia et al., 2020). This stimulation could restore levels of trophic factors such as BDNF.
Neural Stem Cells:
As mentioned before, in rodent models of HD neuronal loss is only mild or occurs very late in disease progression. Thus, symptoms are caused principally by cell dysfunction, in particular abnormal synaptic communication (Levine et al., 2004). In contrast, in human patients clinical symptoms occur as a combination of functional and structural changes. The question is, how can we replace the massive loss of neurons and reconstruct synaptic microcircuits in striatum and other brain regions? Experimental models are beginning to provide an answer (Bjorklund and Parmar, 2020). Implantation of multiple stem cell lines has been used to treat HD symptoms in animal models and were recently reviewed (Holley et al., 2018). These include adult multipotent stem cells (e.g., adipose- and bone marrow-derived mesenchymal stem cells), pluripotent stem cells and, more recently, embryonically-derived neural stem cells (NSCs), which can differentiate into neurons, astrocytes, and oligodendrocytes (Park et al., 2021). We used ESI-017-derived human (h)NSCs for transplantation studies in HD mice. ESI-017 is one of the six clinical-grade human embryonic stem cell lines generated from supernumerary embryos and approved for clinical application. We demonstrated that ESI-017-derived hNSCs survive, make synaptic contacts, and are electrophysiologically active in striatum and cerebral cortex of R6/2 mice. Further, they partially rescue some MSN membrane properties, attenuate epileptiform activity, and improve some behavioral symptoms (Reidling et al., 2018). These differentiated stem cells also increase BDNF and reduce aberrant accumulation of mHTT in the striatum of transplanted animals. Thus, hNSCs have the potential of restoring corticostriatal connectivity in HD. Two other strategies have been attempted that promise restoration of basal ganglia connectivity. One consists of grafting differentiated human striatal progenitors. In a rat model of HD, these progenitors undergo maturation and integrate into host striatal circuits, extend projections to target output regions and receive synaptic contacts. Notably, transplanted rats show significant improvement in sensory-motor tasks up to 2 months after transplant (Besusso et al., 2020). The other strategy used an in vivo cell conversion technology to reprogram striatal astrocytes into GABAergic neurons through AAV-mediated ectopic expression of NeuroD1 and Dlx2 transcription factors. The striatal astrocyteconverted neurons showed action potentials and synaptic events, and projected their axons to the appropriate target regions. Behavioral analyses of treated R6/2 mice showed a significant extension of life span and improvement of motor deficits (Wu et al., 2020). Our more recent study demonstrated that 8-month implantation of hNSCs into the striatum of Q175 HD mice ameliorates behavioral deficits, increases BDNF and reduces mHTT. More importantly, in the context of synaptic pathology, electrophysiological and morphological studies demonstrated that hNSCs differentiate into diverse neuronal populations, including MSN- and interneuron-like cells. Remarkably, hNSCs receive synaptic inputs, innervate host neurons, and improve membrane and synaptic properties (Holley et al., 2020).
9. Conclusions
Almost 20 years ago, we postulated the idea that in HD there is a progressive disconnection along the corticostriatal pathway (Cepeda et al., 2003). This can now be generalized to other excitatory pathways within the CBGTC loop, supporting an important role of HTT in synaptic transmission. Nonetheless, a comprehensive hodology of synaptic dysfunction throughout the CBGTC loop is still incomplete and future advances in the understanding of HD mechanisms should concentrate on finding early changes not only in the corticostriatal projection but also in other pathways. In particular, alterations in thalamostriatal and thalamocortical pathways, that seem to precede and set the stage for corticostriatal pathway dysfunction, are beginning to be unraveled. The GPi/SNr-thalamic projection also should be studied.
In addition, investigators should try to tease apart an underlying cause from a compensatory mechanism. For example, late decreases in glutamate or DA release could very well be a homeostatic adaptation for early increases. Similarly, increased abundance of striatal terminals in output regions could be a compensatory consequence of reduced MSN output. It is important to realize that with disease progression both degenerative and regenerative morphological changes occur pari passu (Ferrante et al., 1991; Graveland et al., 1985). In fact, axonal regeneration and sprouting have been proposed as a potential therapeutic target in neurodegenerative disorders (Marshall and Farah, 2021). Based on the above findings, it appears that the striatum itself is not the main or sole instigator of neuropathological changes in HD. We need to look at other nodes in the CBGTC loop to find the answer. Either an altered cortical input due to aberrant development or a combination of thalamocortical and thalamostriatal disrupted connectivity, appears to be the primum movens of synaptic pathology.
Another pressing question is how early synaptic dysfunction can be identified. Is the faulty cortical architecture reported in some animal models (Cepeda et al., 2019) the trigger of a cascade of morphological and functional changes in the CBGTC loop? Based on the fact that mHTT is already present during the embryonic period and leads to aberrant brain development, we may speculate that some of the early changes in morphology and synaptic activity could represent preemptive adaptations aimed at delaying disease manifestations.
Acknowledgments
The authors would like to acknowledge Dr. Sandra M. Holley for her comments on the manuscript. This work was funded by USPHS grants NS096994 (MSL) and NS111316 (CC). Also by the Cell, Circuits and Systems Analysis Core supported by USPHS grant P50HD103557.
References
- Akopian G, Barry J, Cepeda C, Levine MS, 2016. Altered membrane properties and firing patterns of external globus pallidus neurons in the R6/2 mouse model of Huntington’s disease. J. Neurosci. Res 94, 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Reiner A, Anderson KD, Dure LS, Handelin B, et al. , 1992. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington’s disease. Ann. Neurol 31, 425–430. [DOI] [PubMed] [Google Scholar]
- Alexander GE, DeLong MR, Strick PL, 1986. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu. Rev. Neurosci 9, 357–381. [DOI] [PubMed] [Google Scholar]
- Andre VM, Cepeda C, Fisher YE, Huynh M, Bardakjian N, et al. , 2011. Differential electrophysiological changes in striatal output neurons in Huntington’s disease. J. Neurosci 31, 1170–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki S, Smith JB, Li H, Yan X, Igarashi M, et al. , 2019. An open cortico-basal ganglia loop allows limbic control over motor output via the nigrothalamic pathway. eLife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong MJ, Miyasaki JM, Academy, American, of N., 2012. Evidence-based guideline: pharmacologic treatment of chorea in Huntington disease: report of the guideline development subcommittee of the American Academy of Neurology. Neurology 79, 597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton JF, McIver EL, Mullen MR, Wokosin DL, Surmeier DJ, Bevan MD, 2016. Early dysfunction and progressive degeneration of the subthalamic nucleus in mouse models of Huntington’s disease. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward EH, 2007. Change in MRI striatal volumes as a biomarker in preclinical Huntington’s disease. Brain Res. Bull 72, 152–158. [DOI] [PubMed] [Google Scholar]
- Bagchi SP, 1983. Differential interactions of phencyclidine with tetrabenazine and reserpine affecting intraneuronal dopamine. Biochem. Pharmacol 32, 2851–2856. [DOI] [PubMed] [Google Scholar]
- Barnat M, Le Friec J, Benstaali C, Humbert S, 2017. Huntingtin-mediated multipolar-bipolar transition of newborn cortical neurons is critical for their postnatal neuronal morphology. Neuron 93, 99–114. [DOI] [PubMed] [Google Scholar]
- Barry J, Akopian G, Cepeda C, Levine MS, 2018. Striatal direct and indirect pathway output structures are differentially altered in mouse models of Huntington’s disease. J. Neurosci 38, 4678–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry J, Sarafian TA, Watson JB, Cepeda C, Levine MS, 2020. Mechanisms underlying the enhancement of gamma-aminobutyric acid responses in the external globus pallidus of R6/2 Huntington’s disease model mice. J. Neurosci. Res 98, 2349–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont V, Park L, Rassoulpour A, Dijkman U, Heikkinen T, et al. , 2014. The PDE1/5 inhibitor SCH-51866 does not modify disease progression in the R6/2 mouse model of Huntington’s disease. PLoS Curr. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont V, Zhong S, Lin H, Xu W, Bradaia A, et al. , 2016. Phosphodiesterase 10A inhibition improves Cortico-basal ganglia function in Huntington’s disease models. Neuron 92, 1220–1237. [DOI] [PubMed] [Google Scholar]
- Berendse HW, Groenewegen HJ, 1990. Organization of the thalamostriatal projections in the rat, with special emphasis on the ventral striatum. J. Comp. Neurol 299, 187–228. [DOI] [PubMed] [Google Scholar]
- Besusso D, Schellino R, Boido M, Belloli S, Parolisi R, et al. , 2020. Stem Cell-Derived Human Striatal Progenitors Innervate Striatal Targets and Alleviate Sensorimotor Deficit in a Rat Model of Huntington Disease (Stem cell reports). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund A, Parmar M, 2020. Neuronal replacement as a tool for basal ganglia circuitry repair: 40 years in perspective. Front. Cell. Neurosci 14, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomo R, Elia AE, Bonomo G, Romito LM, Mariotti C, et al. , 2021. Deep brain stimulation in Huntington’s disease: a literature review. Neurol. Sci 42, 4447–4457. [DOI] [PubMed] [Google Scholar]
- Brooks SP, Dunnett SB, 2015. Mouse models of Huntington’s disease. Curr. Top. Behav. Neurosci 22, 101–133. [DOI] [PubMed] [Google Scholar]
- Burrus CJ, McKinstry SU, Kim N, Ozlu MI, Santoki AV, et al. , 2020. Striatal projection neurons require huntingtin for synaptic connectivity and survival. Cell Rep. 30 (642–57), e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan JW, Abercrombie ED, 2015a. Age-dependent alterations in the cortical entrainment of subthalamic nucleus neurons in the YAC128 mouse model of Huntington’s disease. Neurobiol. Dis 78, 88–99. [DOI] [PubMed] [Google Scholar]
- Callahan JW, Abercrombie ED, 2015b. Relationship between subthalamic nucleus neuronal activity and electrocorticogram is altered in the R6/2 mouse model of Huntington’s disease. J. Physiol 593, 3727–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan JW, Wokosin DL, Bevan MD, 2021. Dysregulation of the Basal Ganglia Indirect Pathway Prior to Cell Loss in the Q175 Mouse Model of Huntington’s Disease BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Levine MS, 2020. Synaptic dysfunction in Huntington’s disease: lessons from genetic animal models. Neuroscientist, 1073858420972662, Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Calvert CR, Hernandez-Echeagaray E, Nguyen OK, et al. , 2003. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington’s disease. J. Neurosci 23, 961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Cummings DM, Hickey MA, Kleiman-Weiner M, Chen JY, et al. , 2010. Rescuing the Corticostriatal Synaptic Disconnection in the R6/2 Mouse Model of Huntington’s Disease: Exercise, Adenosine Receptors and Ampakines. PLoS currents 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Murphy KP, Parent M, Levine MS, 2014. The role of dopamine in huntington’s disease. Prog. Brain Res 211, 235–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Oikonomou KD, Cummings D, Barry J, Yazon VW, et al. , 2019. Developmental origins of cortical hyperexcitability in Huntington’s disease: review and new observations. J. Neurosci. Res 97, 1624–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Manfredsson FP, Dec AM, Campbell PW, Stutzmann GE, et al. , 2020. Phosphodiesterase 9A inhibition facilitates Corticostriatal transmission in wild-type and transgenic rats that model Huntington’s disease. Front. Neurosci 14, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cif L, Hariz M, 2017. Seventy years with the Globus pallidus: pallidal surgery for movement disorders between 1947 and 2017. Mov. Disord 32, 972–982. [DOI] [PubMed] [Google Scholar]
- Cooper AJ, Stanford IM, 2000. Electrophysiological and morphological characteristics of three subtypes of rat globus pallidus neurone in vitro. J. Physiol 527 (Pt 2), 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crevier-Sorbo G, Rymar VV, Crevier-Sorbo R, Sadikot AF, 2020. Thalamostriatal degeneration contributes to dystonia and cholinergic interneuron dysfunction in a mouse model of Huntington’s disease. Acta Neuropathol. Commun 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Du X, Chang IYM, Pamukcu A, Lilascharoen V, et al. , 2021. Striatal direct pathway targets Npas1(+) Pallidal neurons. J. Neurosci 41, 3966–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DM, Andre VM, Uzgil BO, Gee SM, Fisher YE, et al. , 2009. Alterations in cortical excitation and inhibition in genetic mouse models of Huntington’s disease. J. Neurosci 29, 10371–10386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallerac GM, Levasseur G, Vatsavayai SC, Milnerwood AJ, Cummings DM, et al. , 2015. Dysfunctional dopaminergic Neurones in mouse models of Huntington’s disease: a role for SK3 channels. Neurodegener. Dis 15, 93–108. [DOI] [PubMed] [Google Scholar]
- Delnomdedieu M, Tan Y, Ogden A, Berger Z, Reilmann R, 2018. A randomized, double-blind, placebo-controlled phase ii efficacy and safety study of the PDE10A inhibitor PF-02545920 in Huntington disease (amaryllis). J. Neurol. Neurosurg. Psychiatry 89, A99.3–A100. [Google Scholar]
- DeLong MR, Wichmann T, 2007. Circuits and circuit disorders of the basal ganglia. Arch. Neurol 64, 20–24. [DOI] [PubMed] [Google Scholar]
- Deng YP, Reiner A, 2016. Cholinergic interneurons in the Q140 knock-in mouse model of Huntington’s disease: reductions in dendritic branching and thalamostriatal input. J. Comp. Neurol 524, 3518–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng YP, Albin RL, Penney JB, Young AB, Anderson KD, Reiner A, 2004. Differential loss of striatal projection systems in Huntington’s disease: a quantitative immunohistochemical study. J. Chem. Neuroanat 27, 143–164. [DOI] [PubMed] [Google Scholar]
- Deng YP, Wong T, Bricker-Anthony C, Deng B, Reiner A, 2013. Loss of corticostriatal and thalamostriatal synaptic terminals precedes striatal projection neuron pathology in heterozygous Q140 Huntington’s disease mice. Neurobiol. Dis 60, 89–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng YP, Wong T, Wan JY, Reiner A, 2014. Differential loss of thalamostriatal and corticostriatal input to striatal projection neuron types prior to overt motor symptoms in the Q140 knock-in mouse model of Huntington’s disease. Front. Syst. Neurosci 8, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Wang H, Joni M, Sekhri R, Reiner A, 2021. Progression of basal ganglia pathology in heterozygous Q175 knock-in Huntington’s disease mice. J. Comp. Neurol 529, 1327–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JB, Guzman JN, Peterson JD, Goldberg JA, Surmeier DJ, 2010. Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron 67, 294–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JB, Oh WJ, Sabatini BL, Gu C, 2011. Semaphorin 3E-Plexin-D1 signaling controls pathway-specific synapse formation in the striatum. Nat. Neurosci 15, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig NM, Moss J, Bolam JP, 2010. Cortical and thalamic innervation of direct and indirect pathway medium-sized spiny neurons in mouse striatum. J. Neurosci 30, 14610–14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Paulsen JS, Beglinger LJ, Langbehn DR, Stout JC, Predict HDIotHSG., 2007. Psychiatric symptoms in Huntington’s disease before diagnosis: the predict-HD study. Biol. Psychiatry 62, 1341–1346. [DOI] [PubMed] [Google Scholar]
- Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, et al. , 1995. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 269, 407–410. [DOI] [PubMed] [Google Scholar]
- Estevez-Fraga C, Flower MD, Tabrizi SJ, 2020. Therapeutic strategies for Huntington’s disease. Curr. Opin. Neurol 33, 508–518. [DOI] [PubMed] [Google Scholar]
- Fernandez-Garcia S, Conde-Berriozabal S, Garcia-Garcia E, Gort-Paniello C, Bernal-Casas D, et al. , 2020. M2 cortex-dorsolateral striatum stimulation reverses motor symptoms and synaptic deficits in Huntington’s disease. eLife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Beal MF, Martin JB, Bird ED, Richardson EP Jr., 1987. Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol 46, 12–27. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Richardson EP Jr., 1991. Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J. Neurosci 11, 3877–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster NN, Barry J, Korobkova L, Garcia L, Gao L, et al. , 2021. The mouse cortico-basal ganglia-thalamic network. Nature 598, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, 1992. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 15, 133–139. [DOI] [PubMed] [Google Scholar]
- Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR, 2010. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington’s disease. PLoS One 5, e13417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin JD, Colombo K, Molina-Calavita M, Keryer G, Zala D, et al. , 2010. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron 67, 392–406. [DOI] [PubMed] [Google Scholar]
- Graveland GA, Williams RS, DiFiglia M, 1985. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 227, 770–773. [DOI] [PubMed] [Google Scholar]
- Gray M, Shirasaki DI, Cepeda C, Andre VM, Wilburn B, et al. , 2008. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci 28, 6182–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, 2003. The primate basal ganglia: parallel and integrative networks. J. Chem. Neuroanat 26, 317–330. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H, 2010. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci 11, 682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington KM, Kowall NW, 1991. Parvalbumin immunoreactive neurons resist degeneration in Huntington’s disease striatum. J. Neuropathol. Exp. Neurol 50, 309. [Google Scholar]
- Hedreen JC, Folstein SE, 1995. Early loss of neostriatal striosome neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol 54, 105–120. [DOI] [PubMed] [Google Scholar]
- Hedreen JC, Peyser CE, Folstein SE, Ross CA, 1991. Neuronal loss in layers V and VI of cerebral cortex in Huntington’s disease. Neurosci. Lett 133, 257–261. [DOI] [PubMed] [Google Scholar]
- Hegeman DJ, Hong ES, Hernandez VM, Chan CS, 2016. The external Globus pallidus: Progress and perspectives. Eur. J. Neurosci 43, 1239–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen T, Lehtimaki K, Vartiainen N, Puolivali J, Hendricks SJ, et al. , 2012. Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington’s disease. PLoS One 7, e50717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintiryan H, Foster NN, Bowman I, Bay M, Song MY, et al. , 2016. The mouse cortico-striatal projectome. Nat. Neurosci 19, 1100–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, et al. , 1999. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 23, 181–192. [DOI] [PubMed] [Google Scholar]
- Holley SM, Joshi PR, Parievsky A, Galvan L, Chen JY, et al. , 2015. Enhanced GABAergic Inputs Contribute to Functional Alterations of Cholinergic Interneurons in the R6/2 Mouse Model of Huntington’s Disease. ENEURO.: 0008–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley SM, Kamdjou T, Reidling JC, Fury B, Coleal-Bergum D, et al. , 2018. Therapeutic effects of stem cells in rodent models of Huntington’s disease: review and electrophysiological findings. CNS Neurosci. Ther 24, 329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley SM, Galvan L, Kamdjou T, Dong A, Levine MS, Cepeda C, 2019. Major contribution of somatostatin-expressing interneurons and cannabinoid receptors to increased GABA synaptic activity in the striatum of Huntington’s disease mice. Front. Synaptic Neurosci 11, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter A, Bordelon Y, Cook I, Leuchter A, 2010. QEEG measures in Huntington’s disease: a pilot study. PLoS Curr. 2, RRN1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington Study Group, 2006. Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology 66, 366–372. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Rajan V, Miller CE, Wightman RM, 2006. Dopamine release is severely compromised in the R6/2 mouse model of Huntington’s disease. J. Neurochem 97, 737–746. [DOI] [PubMed] [Google Scholar]
- Joshi PR, Wu NP, Andre VM, Cummings DM, Cepeda C, et al. , 2009. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J. Neurosci 29, 2414–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EH, Thu DC, Tippett LJ, Oorschot DE, Hogg VM, et al. , 2014. Cortical interneuron loss and symptom heterogeneity in Huntington disease. Ann. Neurol 75, 717–727. [DOI] [PubMed] [Google Scholar]
- Kita H, Kitai ST, 1991. Intracellular study of rat globus pallidus neurons: membrane properties and responses to neostriatal, subthalamic and nigral stimulation. Brain Res. 564, 296–305. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, et al. , 2001. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington’s disease transgenic mice. J. Neurophysiol 86, 2667–2677. [DOI] [PubMed] [Google Scholar]
- Kolodziejczyk K, Raymond LA, 2016. Differential changes in thalamic and cortical excitatory synapses onto striatal spiny projection neurons in a Huntington disease mouse model. Neurobiol. Dis 86, 62–74. [DOI] [PubMed] [Google Scholar]
- Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, et al. , 2007. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol. Genet 16, 1845–1861. [DOI] [PubMed] [Google Scholar]
- Kwon D, 2021. Failure of genetic therapies for Huntington’s devastates community. Nature 593, 180. [DOI] [PubMed] [Google Scholar]
- Lapper SR, Bolam JP, 1992. Input from the frontal cortex and the parafascicular nucleus to cholinergic interneurons in the dorsal striatum of the rat. Neuroscience 51, 533–545. [DOI] [PubMed] [Google Scholar]
- Leavitt BR, Tabrizi SJ, 2020. Antisense oligonucleotides for neurodegeneration. Science 367, 1428–1429. [DOI] [PubMed] [Google Scholar]
- Leuchter MK, Donzis EJ, Cepeda C, Hunter AM, Estrada-Sanchez AM, et al. , 2017. Quantitative electroencephalographic biomarkers in preclinical and human studies of Huntington’s disease: are they fit-for-purpose for treatment development? Front. Neurol 8, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MS, Cepeda C, Hickey MA, Fleming SM, Chesselet MF, 2004. Genetic mouse models of Huntington’s and Parkinson’s diseases: illuminating but imperfect. Trends Neurosci. 27, 691–697. [DOI] [PubMed] [Google Scholar]
- Li JY, Plomann M, Brundin P, 2003. Huntington’s disease: a synaptopathy? Trends Mol. Med 9, 414–420. [DOI] [PubMed] [Google Scholar]
- Mackay JP, Nassrallah WB, Raymond LA, 2018. Cause or compensation?-altered neuronal ca(2+) handling in Huntington’s disease. CNS Neurosci. Ther 24, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelbaum G, Taranda J, Haynes TM, Hochbaum DR, Huang KW, et al. , 2019. Distinct cortical-thalamic-striatal circuits through the Parafascicular nucleus. Neuron 102 (636–52), e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, et al. , 1996. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506. [DOI] [PubMed] [Google Scholar]
- Marshall KL, Farah MH, 2021. Axonal regeneration and sprouting as a potential therapeutic target for nervous system disorders. Neural Regen. Res 16, 1901–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF, 2003. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J. Comp. Neurol 465, 11–26. [DOI] [PubMed] [Google Scholar]
- Miller BR, Walker AG, Shah AS, Barton SJ, Rebec GV, 2008. Dysregulated information processing by medium spiny neurons in striatum of freely behaving mouse models of Huntington’s disease. J. Neurophysiol 100, 2205–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molero AE, Arteaga-Bracho EE, Chen CH, Gulinello M, Winchester ML, et al. , 2016. Selective expression of mutant huntingtin during development recapitulates characteristic features of Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A 113, 5736–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Calavita M, Barnat M, Elias S, Aparicio E, Piel M, Humbert S, 2014. Mutant huntingtin affects cortical progenitor cell division and development of the mouse neocortex. J. Neurosci 34, 10034–10040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ, Faull RL, Edwardson JM, 2001. Abnormalities in the synaptic vesicle fusion machinery in Huntington’s disease. Brain Res. Bull 56, 111–117. [DOI] [PubMed] [Google Scholar]
- Murphy-Nakhnikian A, Dorner JL, Fischer BI, Bower-Bir ND, Rebec GV, 2012. Abnormal burst patterns of single neurons recorded in the substantia nigra reticulata of behaving 140 CAG Huntington’s disease mice. Neurosci. Lett 512, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair A, Razi A, Gregory S, Rutledge RR, Rees G, Tabrizi SJ, 2021. Imbalanced basal ganglia connectivity is associated with motor deficits and apathy in Huntington’s disease. Brain. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, et al. , 1995. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 81, 811–823. [DOI] [PubMed] [Google Scholar]
- Nopoulos PC, Aylward EH, Ross CA, Mills JA, Langbehn DR, et al. , 2011. Smaller intracranial volume in prodromal Huntington’s disease: evidence for abnormal neurodevelopment. Brain 134, 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz AN, Kurth BJ, Osterhaus GL, Johnson MA, 2011. Impaired dopamine release and uptake in R6/1 Huntington’s disease model mice. Neurosci. Lett 492, 11–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz AN, Osterhaus GL, Lauderdale K, Mahoney L, Fowler SC, et al. , 2012. Motor function and dopamine release measurements in transgenic Huntington’s disease model rats. Brain Res. 1450, 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmand AP, Bichell TJ, Bowman AB, Bates GP, 2016. Embryonic mutant huntingtin aggregate formation in mouse models of Huntington’s disease. J. Huntington’s Dis 5, 343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painold A, Anderer P, Holl AK, Letmaier M, Saletu-Zyhlarz GM, et al. , 2010. Comparative EEG mapping studies in Huntington’s disease patients and controls. J. Neural Transm. (Vienna) 117, 1307–1318. [DOI] [PubMed] [Google Scholar]
- Paleacu D, 2007. Tetrabenazine in the treatment of Huntington’s disease. Neuropsychiatr. Dis. Treat 3, 545–551. [PMC free article] [PubMed] [Google Scholar]
- Parent A, Hazrati LN, 1994. Multiple striatal representation in primate substantia nigra. J. Comp. Neurol 344, 305–320. [DOI] [PubMed] [Google Scholar]
- Parievsky A, Moore C, Kamdjou T, Cepeda C, Meshul CK, Levine MS, 2017. Differential electrophysiological and morphological alterations of thalamostriatal and corticostriatal projections in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis 108, 29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, 2018. Cortical axonal secretion of BDNF in the striatum is disrupted in the mutant-huntingtin Knock-in mouse model of Huntington’s disease. Exp. Neurobiol 27, 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Jeon J, Choi J, Kim JY, Kim HS, et al. , 2021. Human iPSC-derived neural precursor cells differentiate into multiple cell types to delay disease progression following transplantation into YAC128 Huntington’s disease mouse model. Cell Prolif. 54, e13082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, et al. , 2008. Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J. Neurol. Neurosurg. Psychiatry 79, 874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Rosello T, Gelman S, Tombaugh G, Cachope R, Beaumont V, Surmeier DJ, 2019. Enhanced striatopallidal gamma-aminobutyric acid (GABA)a receptor transmission in mouse models of huntington’s disease. Mov. Disord 34, 684–696. [DOI] [PubMed] [Google Scholar]
- Pidgeon C, Rickards H, 2013. The pathophysiology and pharmacological treatment of Huntington disease. Behav. Neurol 26, 245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Bernardi G, Ding J, Surmeier DJ, 2007. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 30, 545–553. [DOI] [PubMed] [Google Scholar]
- Plotkin JL, Goldberg JA, 2019. Thinking outside the box (and arrow): current themes in striatal dysfunction in movement disorders. Neuroscientist 25, 359–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JL, Surmeier DJ, 2015. Corticostriatal synaptic adaptations in Huntington’s disease. Curr. Opin. Neurobiol 33, 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouladi MA, Stanek LM, Xie Y, Franciosi S, Southwell AL, et al. , 2012. Marked differences in neurochemistry and aggregates despite similar behavioural and neuropathological features of Huntington disease in the full-length BACHD and YAC128 mice. Hum. Mol. Genet 21, 2219–2232. [DOI] [PubMed] [Google Scholar]
- Raymond LA, Andre VM, Cepeda C, Gladding CM, Milnerwood AJ, Levine MS, 2011. Pathophysiology of Huntington’s disease: time-dependent alterations in synaptic and receptor function. Neuroscience 198, 252–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reading SA, Yassa MA, Bakker A, Dziorny AC, Gourley LM, et al. , 2005. Regional white matter change in pre-symptomatic Huntington’s disease: a diffusion tensor imaging study. Psychiatry Res. 140, 55–62. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Conroy SK, Barton SJ, 2006. Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: variations with behavioral state and repeated ascorbate treatment. Neuroscience 137, 327–336. [DOI] [PubMed] [Google Scholar]
- Reidling JC, Relano-Gines A, Holley SM, Ochaba J, Moore C, et al. , 2018. Human neural stem cell transplantation rescues functional deficits in R6/2 and Q140 Huntington’s disease mice. Stem Cell Rep. 10, 58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, 2004. Can lesions of GPe correct HD deficits? Exp. Neurol 186, 1–5. [DOI] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D’Amato CJ, Penney JB, Young AB, 1988. Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. U. S. A 85, 5733–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Shelby E, Wang H, Demarch Z, Deng Y, et al. , 2013. Striatal parvalbuminergic neurons are lost in Huntington’s disease: implications for dystonia. Mov. Disord 28, 1691–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas HD, Hevelone ND, Zaleta AK, Greve DN, Salat DH, Fischl B, 2005. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology 65, 745–747. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Tuch DS, Hevelone ND, Zaleta AK, Vangel M, et al. , 2006. Diffusion tensor imaging in presymptomatic and early Huntington’s disease: selective white matter pathology and its relationship to clinical measures. Mov. Disord 21, 1317–1325. [DOI] [PubMed] [Google Scholar]
- Saudou F, Humbert S, 2016. The biology of huntingtin. Neuron 89, 910–926. [DOI] [PubMed] [Google Scholar]
- Shelbourne PF, Killeen N, Hevner RF, Johnston HM, Tecott L, et al. , 1999. A Huntington’s disease CAG expansion at the murine Hdh locus is unstable and associated with behavioural abnormalities in mice. Hum. Mol. Genet 8, 763–774. [DOI] [PubMed] [Google Scholar]
- Shobe JL, Donzis EJ, Lee K, Chopra S, Masmanidis SC, et al. , 2021. Early impairment of thalamocortical circuit activity and coherence in a mouse model of Huntington’s disease. Neurobiol. Dis 157, 105447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, et al. , 2003. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet 12, 1555–1567. [DOI] [PubMed] [Google Scholar]
- Smith Y, Raju DV, Pare JF, Sidibe M, 2004. The thalamostriatal system: a highly specific network of the basal ganglia circuitry. Trends Neurosci. 27, 520–527. [DOI] [PubMed] [Google Scholar]
- Smith Y, Surmeier DJ, Redgrave P, Kimura M, 2011. Thalamic contributions to basal ganglia-related behavioral switching and reinforcement. J. Neurosci 31, 16102–16106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Syed A, Lukacsovich T, Purcell J, Barbaro BA, et al. , 2014. A potent and selective Sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington’s disease. Hum. Mol. Genet 23, 2995–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura A, Lim SA, Aceves Buendia JJ, Goldberg JA, Surmeier DJ, 2016. Cholinergic interneurons amplify Corticostriatal synaptic responses in the Q175 model of Huntington’s disease. Front. Syst. Neurosci 10, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington’s Disease Collaborative Research Group, 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983. [DOI] [PubMed] [Google Scholar]
- Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW, 2000. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A 97, 8093–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr., 1985. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol 44, 559–577. [DOI] [PubMed] [Google Scholar]
- Waldvogel HJ, Kim EH, Tippett LJ, Vonsattel JP, Faull RL, 2015. The neuropathology of Huntington’s disease. Curr. Top. Behav. Neurosci 22, 33–80. [DOI] [PubMed] [Google Scholar]
- Walker AG, Miller BR, Fritsch JN, Barton SJ, Rebec GV, 2008. Altered information processing in the prefrontal cortex of Huntington’s disease mouse models. J. Neurosci 28, 8973–8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall NR, De La Parra M, Callaway EM, Kreitzer AC, 2013. Differential innervation of direct- and indirect-pathway striatal projection neurons. Neuron 79, 347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace ML, Saunders A, Huang KW, Philson AC, Goldman M, et al. , 2017. Genetically distinct parallel pathways in the Entopeduncular nucleus for limbic and sensorimotor output of the basal ganglia. Neuron 94, 138–152 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warby SC, Doty CN, Graham RK, Carroll JB, Yang YZ, et al. , 2008. Activated caspase-6 and caspase-6-cleaved fragments of huntingtin specifically colocalize in the nucleus. Hum. Mol. Genet 17, 2390–2404. [DOI] [PubMed] [Google Scholar]
- Wojtecki L, Groiss SJ, Hartmann CJ, Elben S, Omlor S, et al. , 2016. Deep brain stimulation in Huntington’s disease-preliminary evidence on pathophysiology, efficacy and safety. Brain Sci. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Parry M, Hou XY, Liu MH, Wang H, et al. , 2020. Gene therapy conversion of striatal astrocytes into GABAergic neurons in mouse models of Huntington’s disease. Nat. Commun 11, 1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A, 1995. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet 11, 155–163. [DOI] [PubMed] [Google Scholar]
- Zieger HL, Choquet D, 2021. Nanoscale synapse organization and dysfunction in neurodevelopmental disorders. Neurobiol. Dis 158, 105453. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E, 2009. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol 5, 311–322. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Liber D, Ramos C, Tarditi A, Rigamonti D, et al. , 2005. Progressive loss of BDNF in a mouse model of Huntington’s disease and rescue by BDNF delivery. Pharmacol. Res 52, 133–139. [DOI] [PubMed] [Google Scholar]