

Abstract
Background & Aims:
YAP aberrant activation is implicated in intrahepatic cholangiocarcinoma (iCCA). TEAD mediated transcriptional regulation is the primary signaling event downstream of YAP. The role of Wnt/β-Catenin signaling in cholangiocarcinogenesis remains undetermined. Here, we investigated the possible molecular interplay between YAP and β-Catenin cascades in iCCA.
Methods:
Activated Akt (Myr-Akt) was co-expressed with Yap (YapS127A) or Tead2VP16 via hydrodynamic tail vein injection into the mouse livers. Tumor growth was monitored, liver tissues were collected and analyzed using histopathologic and molecular analysis. Yap, β-Catenin, and TEAD interaction in iCCAs was investigated through co-immunoprecipitation. Conditional Ctnnb1 KO mice were utilized to determine β-Catenin function in murine iCCA models. RNA sequencing (RNASeq) was performed to analyze the genes regulated by YAP and/or β-Catenin. Immunostaining of total and non-phosphorylated/activated β-Catenin staining was performed in mouse and human iCCAs.
Results:
We discovered that TEAD factors are required for YAP-dependent iCCA development. However, transcriptional activation of TEADs did not fully recapitulate YAP’s activities in promoting cholangiocarcinogenesis. Notably, β-Catenin physically interacted with YAP in human and mouse iCCA. Ctnnb1 ablation strongly suppressed human iCCA cell growth and Yap-dependent cholangiocarcinogenesis. Furthermore, RNASeq analysis revealed that YAP/TAZ regulate a set of genes significantly overlapping with those controlled by β-Catenin. Importantly, activated/non-phosphorylated β-Catenin was detected in over 80% of human iCCAs.
Conclusion:
YAP induces cholangiocarcinogenesis via TEAD-dependent transcriptional activation and interaction with β-Catenin. β-Catenin binds to YAP in iCCA and is required for YAP full transcriptional activity, revealing the functional crosstalk between YAP and β-Catenin pathways in cholangiocarcinogenesis.
Keywords: Hippo/YAP, β-Catenin, TEADs, Intrahepatic cholangiocarcinoma
Lay summary:
YAP drives cholangiocarcinogenesis via TEAD mediated transcriptional activation and functional crosstalk with β-Catenin. β-Catenin physically interacts with Yap in cholangiocarcinoma and is required for YAP’s oncogenic activity.
Graphical Abstract
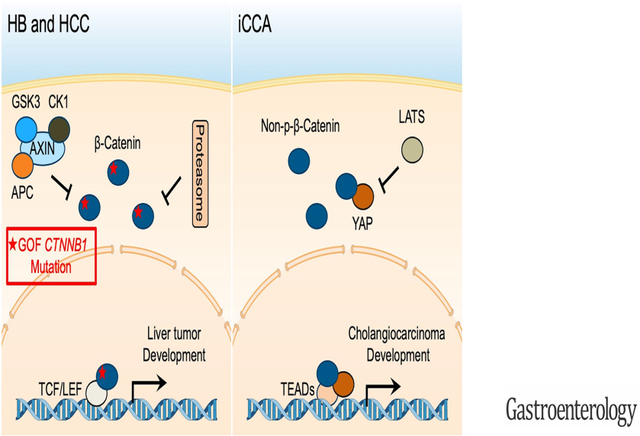
Introduction
Cholangiocarcinomas (CCAs) are highly heterogeneous hepatobiliary malignant tumors with features of cholangiocyte differentiation. They can be subdivided into three subtypes according to the anatomical location: intrahepatic (iCCA), perihilar (pCCA), and distal (dCCA) 1. iCCA is the second most common type of primary liver tumor after hepatocellular carcinoma (HCC), accounting for ~10–15% of all hepatobiliary malignancies 2, 3. iCCA incidence is increasing globally 1. Due to limited therapeutic options, there is an urgent need to uncover signaling pathways underlying iCCA tumorigenesis to develop novel and effective therapies against this deadly malignancy.
Hippo is a tumor suppressor pathway inactivated in several tumor types, including iCCA 4, 5. YES-associated protein (YAP) acts as a transcriptional coactivator downstream of Hippo. YAP exerts its functions predominantly by interacting with the TEA domain (TEAD)-containing transcription factors and additional proteins (Fig. S1) 6. Structurally, YAP is a multidomain protein, possessing an N-terminal proline-rich domain, the TEAD binding domain (TBD), WW domain(s), the SH3-binding motif, the transcriptional activation domain (TAD), and a C-terminal PDZ-binding motif 7. TBD and TAD are essential for YAP transcriptional activities, whereas the other domains mediate YAP-protein interactions (Fig. S1). YAP is critical for cancer initiation, progression, and metastasis 8. Studies have demonstrated increased YAP nuclear expression in human iCCA specimens, and activated YAP levels correlate with a worse prognosis in iCCA patients 1. The oncogenic role of YAP in cholangiocarcinogenesis has been further substantiated in vivo. Indeed, the co-expression of activated forms of Akt (myr-Akt) and Yap (YapS127A) induces iCCA formation in mice (Akt/Yap)9.
The Wnt/β-Catenin pathway is an evolutionarily conserved pathway with a critical role in regulating liver homeostasis, regeneration, and carcinogenesis. Activated Wnt/β-Catenin is implicated in hepatoblastoma (HB) and HCC development, as gain of function (GOF) mutations in the CTNNB1 gene, encoding β-Catenin, occur in these two tumor entities. CTNNB1 mutations are instead infrequent in iCCA 10. A few studies have reported the activation of the Wnt/β-Catenin signaling in human iCCA 11, and the precise function(s) of this pathway in cholangiocarcinogenesis remains poorly understood.
Using the Akt/Yap induced iCCA model, we investigated the molecular mechanisms whereby YAP drives cholangiocarcinogenesis. The present data suggest that YAP induces iCCA development via TEAD mediated transcriptional activation and crosstalk with β-Catenin.
Materials and Methods
Constructs and Reagents
The plasmids used for in vitro and in vivo studies are detailed in Sup. Table 1. All plasmids were extracted using the Endotoxin Free Prep Kit (Sigma-Aldrich, St. Louis, MO, USA). Detailed cloning information is available in Supporting Materials and Methods.
Cell lines and cell Culture
Human iCCA cell lines RBE, HUCCT-1 and KKU156, mouse iCCA cell lines M1-1a, M4-2a, and M4-2b (kindly provided by Dr. Gregory J. Gores of Mayo Clinic, Rochester, MN), and human embryonic kidney cell line HEK-293FT were used in this study. All cells were cultured in Dulbecco’s modified Eagle medium (DMEM) (Sigma-Aldrich, St. Louis, MO) with 10% fetal bovine serum (FBS) (Sigma-Aldrich) and 1% penicillin-streptomycin (PS) (Sigma-Aldrich), and incubated at 37°C, 5% CO2.
siRNA transfection
Cells were plated in a 6-well culture plate. When cells reached 60% confluence, cells were transfected with siRNA (siCon, siYAP, siTAZ, or siYAP/TAZ) and Lipofectamine RNAiMAX Transfection Reagent (ThermoFisher Scientific) diluted in Opti-MEM (Gibco, Grand Island, NY). siCon, siYAP (cat AM16708, assay ID 17374), siTAZ (cat AM16708, assay ID 23134) were purchased from ThermoFisher Scientific. Detail dosage and procedures were conducted according to the manufacturer’s protocol. After 48 hours of transfection, cells were harvested for RNA analysis.
Lentivirus packaging and cell transduction
Early passaged HEK-293FT cells were cultured in a DMEM medium without PS before producing lentiviral particles. HEK-293FT cells were plated in a 10 cm dish and incubated at 37°C, 5% CO2. When cells reached 60–70% confluence, cells were co-transfected with plasmids mixture (9.2 μg psPAX2 + 2.8 μg pMD2.G + 12 μg Plenti-Gene) and 30 μl Lipofectamine 2000 reagents (Invitrogen) diluted in 500μl Opti-MEM. Cells were incubated at 37°C, 5% CO2 for 48 or 72 hours. Lentiviral supernatant was harvested, spun at 500 g for 3 minutes, and filtered through a 0.45-mm PES filter (Millipore, Bedford, MA). RBE, HUCCT-1, and KKU156 cells were infected with virus and fresh culture medium at the volume ratio 1:1. Then, 48 hours later, cells were treated with puromycin-containing media (2 μg/ml for RBE, 1.0 μg/ml for HUCCT-1, and 1.5 μg/ml for KKU156) to select for cells with stable expression of the target gene with the puromycin resistance. Stable selected cells were used for IF, qRT-PCR, co-IP, WB, colony formation assay, and RNA Sequencing Analysis. Detailed protocols and information are available in Supporting Materials and Methods.
Human Tissue Samples
For Co-immunoprecipitation (co-IP) studies, human CCA tissues were obtained following curative surgical resections from patients with CCA at the West China Hospital (Sichuan University, Chengdu, China). Additional human CCA and corresponding non-tumorous liver tissues were retrieved from the Clinical Biospecimen Repository and Processing Core, Pittsburgh Liver Research Center (Pittsburgh, PA). For the CTNNB1 expression and β-Catenin activation studies, human iCCA and corresponding surrounding non-tumorous liver tissues were collected at the Universities of Greifswald (Greifswald, Germany) and Regensburg (Regensburg, Germany). Institutional Review Board approval was obtained at the local Ethical Committee of the Sichuan University (Chengdu, China, approval code: 2013–210), University of Pittsburgh (approval codes: 20040276 and 19070068), and the Medical Universities of Greifswald (approval code: BB 67/10) and Regensburg (approval code: 17-1015-101), in compliance with the Helsinki Declaration. Written informed consent was obtained from all individuals.
Mouse experiments
Wildtype FVB/N mice and Ctnnb1flox/flox mice (in the FVB/N genetic background) were from the Jackson Laboratory (Sacramento, CA). Yapflox/flox mice (in the FVB/N genetic background) were kindly provided by Dr. Eric Olson from the University of Texas Southwestern Medical Center (Dallas, TX). At five to seven weeks of age, mice of the same gender were subjected to hydrodynamic tail vein injection in parallel, as described in our previous study 12, to induce iCCA formation. The dosages of plasmids used for iCCA murine models are shown in Sup. Table 2. Mice were maintained and monitored continually following the Committee for Animal Research protocols at the University of California, San Francisco (San Francisco, CA). Mouse liver tissues were harvested for H&E, IHC, IF, co-IP, WB, and qRT-PCR analysis. Detailed protocols and information are available in Supporting Materials and Methods.
Statistical Analysis
Data were analyzed using the Prism 6.0 software (GraphPad, San Diego, CA). Comparisons between the two groups were performed using the Mann-Whitney U test for non-parametric data or the Unpaired t-test for parametric data. Survival curves were analyzed using the Log-rank (Mantel-Cox) test. Data are presented as mean ± SD. P < .05 was considered statistically significant.
Results
TEAD mediated transcriptional activation is required for YAP driven cholangiocarcinogenesis
YAP primarily functions via TEAD mediated transcriptional activation. Based on The Cancer Genome Atlas (TCGA) CHOL dataset, the expression levels of TEAD1-4 were all significantly increased in human iCCAs compared to non-neoplastic surrounding livers (Fig. S2). Furthermore, we consistently observed nuclear accumulation of TEAD in human iCCA cell lines (Fig. S3–S5). Thus, we hypothesized that YAP/TEAD mediated transcriptional activation is sufficient and necessary for YAP-induced cholangiocarcinogenesis. To test this hypothesis, we utilized the Akt/Yap-induced murine iCCA model 9. Specifically, mice were subjected to hydrodynamic injection of activated Akt together with either activated YapS127A (Akt/Yap) or YapS127AS94A, a Yap mutant form unable to bind to TEADs (Akt/YapS94A) (Fig. 1A). Notably, the prevention of YAP/TEAD interaction completely inhibited liver tumor formation in mice. Indeed, Akt/YapS94A mice appeared healthy by 21 weeks post-injection, while all Akt/Yap mice were moribund due to a high tumor burden within 11 weeks after injection (Fig. 1B). Numerous highly proliferative iCCAs occupied most liver parenchyma in Akt/Yap mice (Fig. 1C). In striking contrast, liver tissues from Akt/YapS94A mice were normal, with few Ki67 positive proliferating cells, at the same time point (Fig. 1C). Even by 21 weeks post-injection, no tumor nodules were detectable on Akt/YapS94A livers. Histologically, only AKT-induced hepatic steatosis 13 was appreciable in those livers (Fig. S6).
Figure 1. TEADs is required for Yap-driven iCCA formation in mice.
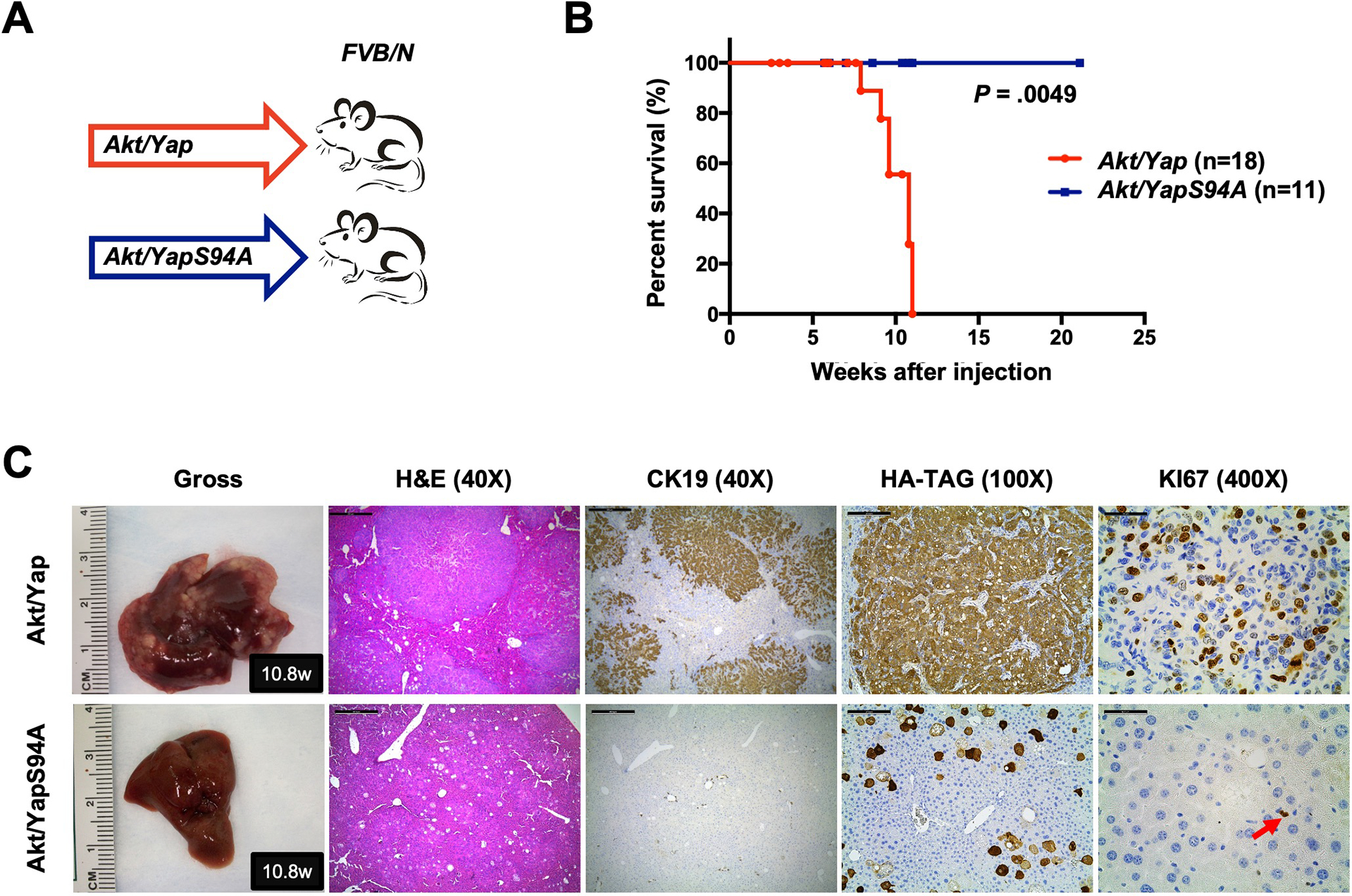
(A) Study design. FVB/N mice were subjected to HTVi of either Akt/Yap (n=18) or Akt/YapS94A (n = 11) plasmids. (B) Mouse survival curves. (C) Representative gross images, H&E, and immunohistochemistry for CK19, HA-TAG and KI67 of liver sections from Akt/Yap (M10.8w.p.i) and Akt/YapS94A (F10.8w.p.i) mice. The red arrow indicates KI67 positive stained nuclei. Scale bar: 500 μm (40x), 100 μm (100x), and 50 μm (400x). Abbreviations: H&E, hematoxylin and eosin staining; w.p.i, weeks post-injection.
Next, we determined whether inhibiting Yap/TEAD interaction could suppress iCCA growth. For in vivo experiments, we co-expressed Akt/Yap oncogenes together with the dominant-negative form of TEAD2 (dnTEAD2) (Akt/Yap/dnTEAD2), or the competitive TEAD binding protein VGLL4 (Akt/Yap/VGLL4) in mice. pT3-EF1α empty vector was co-injected with Akt/Yap as control (Akt/Yap/pT3) (Fig. S7A). Notably, Yap/TEAD interaction inhibition strongly hampered Akt/Yap-driven iCCA formation (Fig. S7B–S7C), leading to prolonged mouse survival and decreased iCCA tumor burden. Consistently, in human and mouse iCCA cell lines, overexpression of dnTEAD2 decreased YAP target genes’ expression, including CTGF and CYR61, and strongly inhibited iCCA cell growth (Fig. S8).
Altogether, the data demonstrate the requirement of TEAD for YAP-induced cholangiocarcinogenesis.
TEAD dependent transcriptional activation does not fully recapitulate YAP activation during cholangiocarcinogenesis
Using the Tead2VP16 construct, we demonstrated recently that TEAD mediated transcription fully recapitulates Yap activities during Yap/β-Catenin induced hepatoblastoma (HB) development 14. To determine whether Yap/TEAD mediated transcriptional activation is sufficient to drive Yap-driven iCCA formation in vivo, activated Akt and Tead2VP16 plasmids were co-injected into mice (Akt/Tead2VP16) (Fig. 2A). It is important to note that Tead2VP16 fusion induces TEAD-mediated transcription independent of the endogenous TEAD proteins. Intriguingly, Akt/Tead2VP16 co-expression drove iCCA formation in mice, although tumor development required a significantly longer latency than Akt/Yap mice (Fig. 2B–2C). Tumor lesions were appreciable in Akt/Yap liver as early as 2.5 weeks post-injection (Fig. S9A and S9C). At this time point, no iCCA lesions developed in Akt/Tead2VP16 mice (Fig. S9A and S9C). In the Akt/Tead2VP16 model, small iCCA lesions were detected ~8 weeks post-injection, when numerous iCCA lesions occupied most of the liver parenchyma in Akt/Yap mice (Fig. S9B–S9C). Consistently, a significantly smaller CK19 positive area and a lower tumor cell proliferation characterized Akt/Tead2VP16 mouse livers (Fig. 2D–2E). The data imply that TEAD mediated transcriptional activity is not entirely equivalent to YAP activation during iCCA development.
Figure 2. TEAD mediated transcriptional activation is not sufficient to induce Yap-dependent cholangiocarcinogenesis.
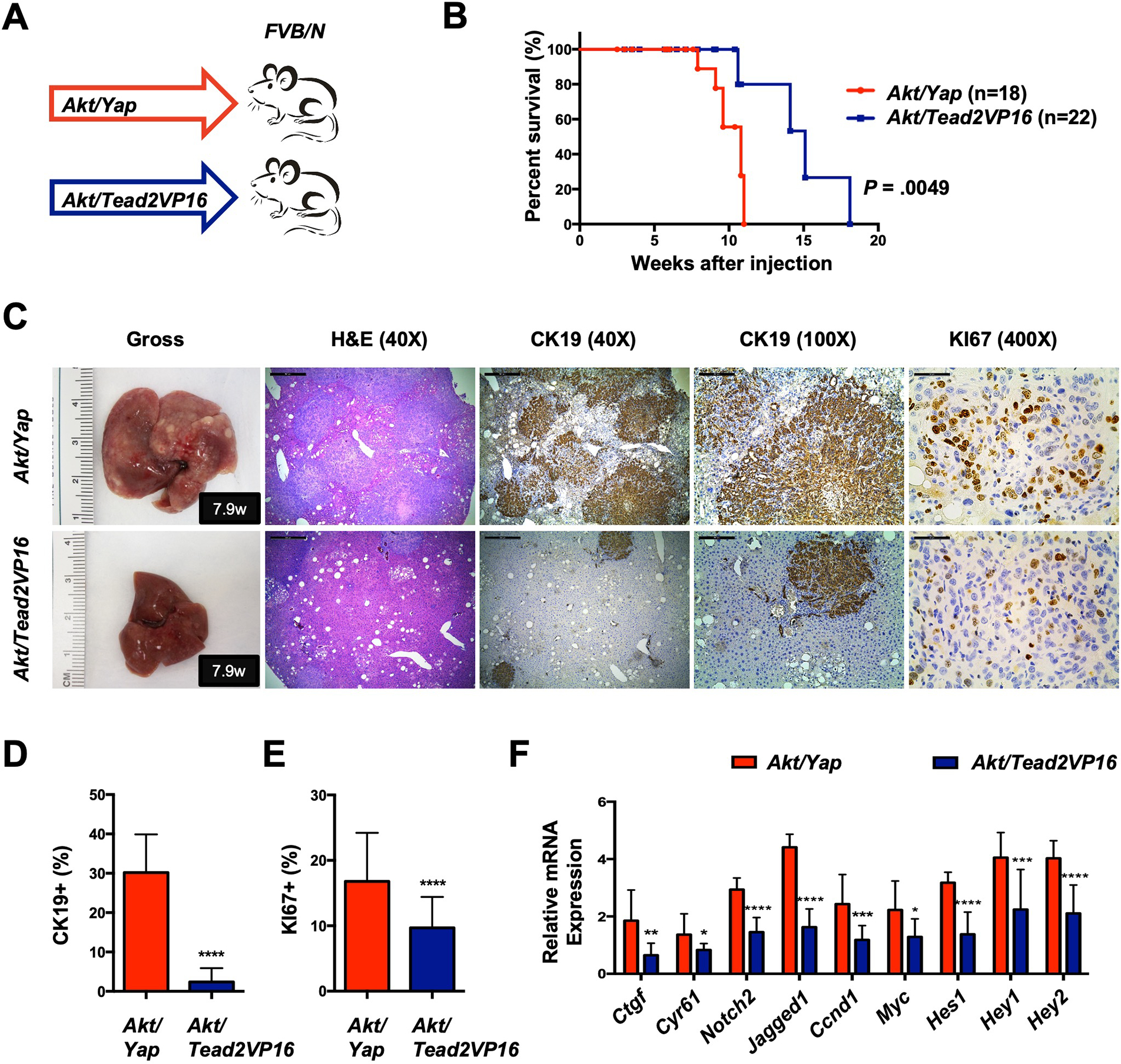
(A) Study design. FVB/N mice were subjected to HTVi of either Akt/Yap (n=18) or Akt/Tead2VP16 (n = 22) plasmids. Akt/Yap, Akt/YapS94A (Fig.1B), and Akt/ead2VP16 mice were generated in parallel. (B) Mouse survival curves. (C) Representative gross images, H&E, and immunohistochemistry for CK19 and KI67 of liver sections from Akt/Yap (M7.9w.p.i) and Akt/Tead2VP16 (M7.9w.p.i) mice. (D) Analysis of CK19-positive areas in Akt/Yap and Akt/Tead2VP16 mouse liver tissues at 2.5–11 w.p.i. (E) Quantification of the KI67 positive staining in liver sections from the depicted mice. (F) mRNA expression of Yap and Notch targets. Data were analyzed by the Mann-Whitney test. Statistical significance: *P < .05, **P < .01, ***P < .001, ****P < .0001. Scale bar: 500 μm (40x), 100 μm (100x) and 50 μm (400x). Abbreviations: H&E, hematoxylin and eosin staining; w.p.i, weeks post-injection.
To investigate the molecular mechanisms underlying this phenotype, we performed molecular analyses of iCCA lesions from Akt/Yap and Akt/Tead2VP16 mice. Similar AKT/mTOR activation levels were detected in the iCCA lesions from the two mouse models (Fig. S10A–S10B). Intriguingly, most Yap target genes, such as Ctgf, Cyr61, Notch2, and Jag1, and downstream Notch pathway genes, such as Hes1, Hey1, and Hey2, were expressed at lower levels in Akt/Tead2VP16 iCCA lesions (Fig. 2F), suggesting a lower level of Yap transcriptional activity in Akt/Tead2VP16 driven iCCA. Consistently, RNA sequencing (RNASeq) analysis of the Akt/Yap and Akt/Tead2VP16 iCCA transcriptome revealed that multiple YAP target genes 15 were expressed at lower levels in Akt/Tead2VP16 iCCA (Fig. S10C, Supplementary Dataset). Furthermore, the lower expression of NOTCH2 and JAGGED1 was confirmed at the protein level (Fig. S10A–S10B). In addition, Ccnd1 and c-Myc, known to be regulated by multiple pathways, also exhibited lower expression levels in Akt/Tead2VP16 iCCA lesions (Fig. 2F). Besides, Western blotting revealed lower levels of PCNA and SURVIVIN in Akt/Tead2VP16 iCCAs (Fig. S10A–S10B), supporting lower cell proliferation in this model.
In summary, these findings imply that in addition to TEAD mediated transcriptional activation, other mechanisms support the full activation of YAP and its oncogenic properties in cholangiocarcinogenesis.
Yap promotes cholangiocarcinogenesis independent of WW, SH3, and PDZ domains
Yap interacts with various proteins through the WW, SH3, and PDZ domains (Fig. S1). We hypothesized that one of these domains might be required for the full activation of Yap in iCCA. Therefore, we generated different recombinant Yap constructs by deleting WW, SH3, or PDZ domains (YapΔWW, YapΔSH3, or YapΔPDZ). The expressions of the resulting proteins were confirmed by Western Blotting (Fig. S11A). Each construct was co-injected into mice associated with activated Akt, with Akt/Yap as the control (Fig. S11B). Surprisingly, all the truncated Yap forms effectively induced iCCA in mice (Fig. S11C–S11D). Ablation of SH3 did not affect iCCA formation, as Akt/YapΔSH3 and Akt/Yap mice developed cholangiocellular tumors with similar latency. Notably, deletion of WW or PDZ significantly accelerated tumor progression (Fig. S11D–S11E), suggesting that WW and PDZ domains negatively regulate Yap-mediated iCCA development.
Overall, the findings suggest that the major protein interaction domains, including WW, SH3, and PDZ, are dispensable or negatively regulate Yap-driven cholangiocarcinogenesis.
Yap physically interacts with β-Catenin in cholangiocarcinoma
RNASeq on Akt/Yap iCCA and Akt/Tead2VP16 iCCA lesions identified 1354 genes differentially regulated in the two models (Fig. S12A, Supplementary Dataset). We focused on genes downregulated in Akt/Tead2VP16 iCCA. Pathway analyses revealed that the Wnt signaling was enriched in the downregulated genes (Fig. S12B), suggesting the possible involvement of Wnt/β-Catenin cascade in YAP-driven cholangiocarcinogenesis. Previous studies indicate that YAP binds to β-Catenin in various cell types 16–19. Therefore, we hypothesized that YAP/β-Catenin interaction might be necessary for Yap full activation in iCCA.
First, we determined whether YAP physically binds to β-Catenin in human and mouse iCCAs. For this purpose, the RBE and KKU156 cell lines were transfected with a Flag-tagged YapS127A plasmid. Immunoprecipitation (IP) of YAP followed by immunoblotting for β-Catenin revealed that YAP directly interacts with β-Catenin in RBE and KKU156 cells (Fig. 3A). Similarly, direct binding of YAP and β-Catenin was detected in Akt/Yap mouse iCCA lesions (Fig. 3B), as well as human iCCAs collected at two different medical centers (Fig. 3C and Fig. S13A). Furthermore, additional experiments demonstrated that β-Catenin could also co-IP with TEADs, suggesting that β-Catenin, YAP, and TEADs form a complex in iCCA cells (Fig. S13B).
Figure 3. Yap physically interacts with β-Catenin in iCCA.
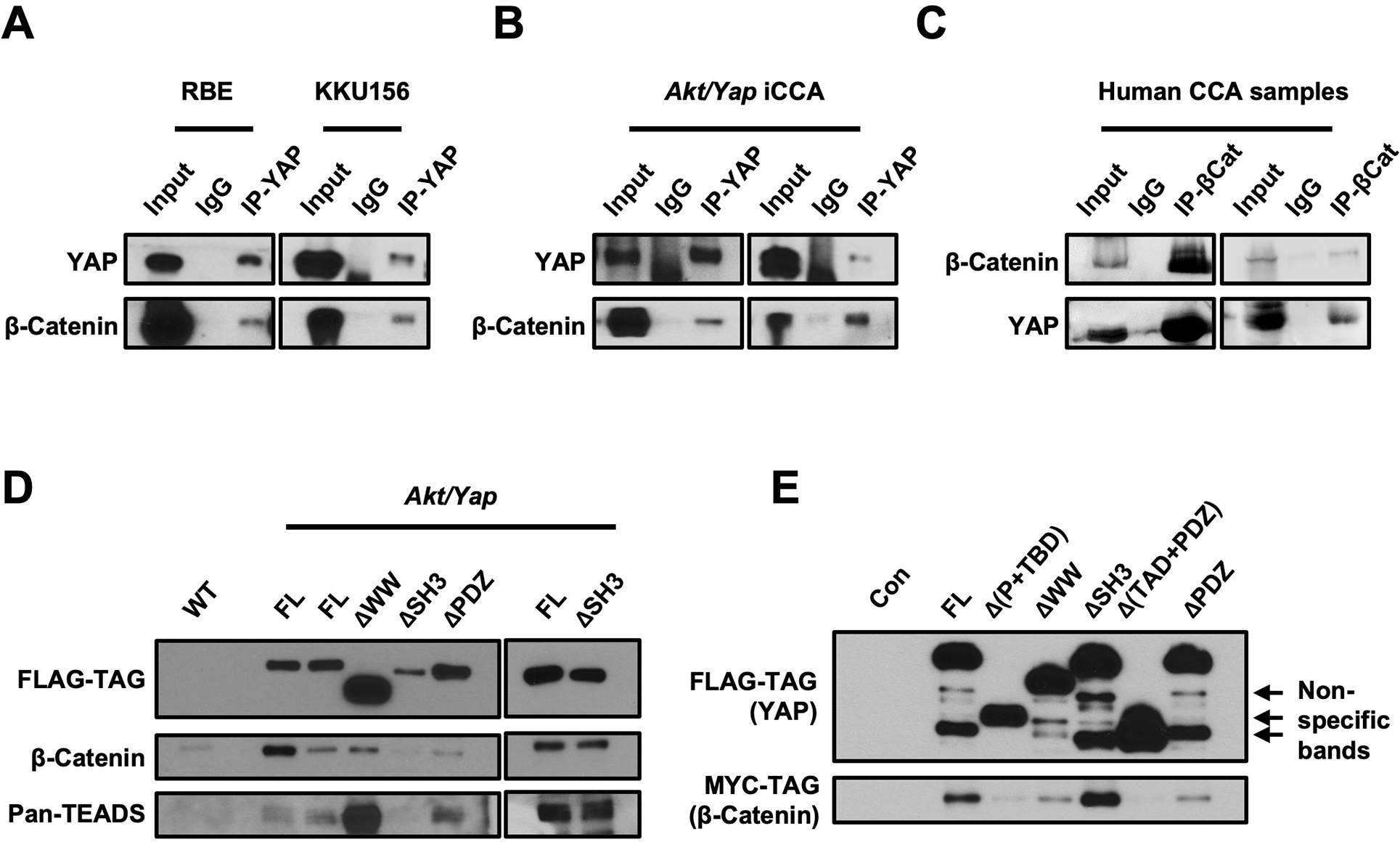
Co-immunoprecipitation assays to detect β-Catenin and Yap interaction. (A, B) Lysates from human CCA cells transfected with the pT3-EF1αH-FLAG-YapS127A plasmid (A) or Akt/Yap iCCA tumors (B) were subjected to immunoprecipitation with an anti-Yap antibody. (C) Lysates from human CCA samples were subjected to immunoprecipitation with an anti-β-Catenin antibody. (D, E) Lysates from Akt/Yap (FL), Akt/YapΔWW, Akt/YapΔSH3, Akt/YapΔPDZ tumors (D), or 293FT cells transfected with truncated Flag-Yap and Myc-β-Catenin plasmids (E) were subjected to immunoprecipitation with anti-FLAG beads. FVB/N mouse livers (WT) (D) and untreated 293FT cells (E) were used as the controls.
Next, we analyzed the cellular localization of YAP and β-Catenin in human and mouse iCCA cells using immunofluorescence. In human iCCA cell lines, pronounced nuclear and weak cytoplasmic immunoreactivity for YAP was detected, whereas β-Catenin showed predominantly membranous staining. However, a small subset of RBE and KKU156 cells exhibited a positive β-Catenin nuclear staining. In these cells, β-Catenin co-localized with Yap (Fig. S14–S15). Similar results were obtained in Akt/Yap iCCA lesions (Fig. S16A). Isolating the cytoplasmic and nuclear extracts and performing co-IP revealed that YAP and β-Catenin interacted in both cytoplasmic and nuclear fractions (Fig. S16B). Consistently, immunohistochemical staining of YAP and β-Catenin in human iCCA sections revealed the simultaneous nuclear and cytoplasmic localization of YAP and β-catenin as well as activated/non-phosphorylated β-Catenin (non-p-β-Catenin) (Fig. S17).
Finally, we investigated the functional domains allowing the YAP interaction with β-Catenin. In mouse iCCAs induced by various YAP deletions, including YapΔWW, YapΔSH3, and YapΔPDZ (Fig. S11), we could readily detect the binding of β-Catenin and TEAD with these YAP truncated forms (Fig. 3D). We made additional truncated forms of Yap constructs, including YapΔTBD and YapΔ(TAD+PDZ) (Fig. S18). When transfected into the cells, β-Catenin successfully bound to most truncated Yap forms, including YapΔPDZ, except for YapΔ(TAD+PDZ) (Fig. 3E). The results demonstrate that Yap interacts with β-Catenin via its transcriptional activation domain in iCCA.
YAP/TAZ and β-Catenin regulate an overlapping set of genes in human iCCA cells
Based on these observations, we hypothesized that β-Catenin interacts with YAP in iCCA cells. Once associated, this complex might regulate the expression of a set of target genes in cholangiocarcinogenesis. To test this hypothesis, we silenced β-Catenin in RBE human iCCA cells using shβ-Catenin (shβCat) (Fig. S19) and performed RNASeq. We identified 3338 genes downregulated and 3259 genes upregulated by shβCat, respectively (Fig. S20A, Supplementary Dataset). Knockdown of β-Catenin in iCCA cells decreased mRNA levels of CTNNB1 and its targets AXIN2, c-MYC, and CCND1, but not many other canonical Wnt target genes, such as GLUL, TBX3, TCF7, LEF1, RNF43, TIAM1, MMP7, and CD44 (Fig. S20B). Consistently, further analysis of the canonical Wnt/β-Catenin pathway genes revealed that this gene expression signature was not enriched in the genes downregulated by shβCat (Fig. S20C). The results suggest that β-Catenin might regulate distinct pathways instead of the canonical Wnt cascade in iCCA. The analysis of the Kyoto Encyclopedia of Genes and Genomes (KEGG) revealed that pathways downregulated by shβCat were involved in RNA transport, DNA replication, cell cycle, and p53 signaling (Fig. S21).
Next, we specifically searched for canonical Hippo/YAP/TAZ targets to assess whether they are regulated by β-Catenin in iCCA cells. Notably, silencing of β-Catenin triggered consistent downregulation of Hippo/YAP/TAZ effectors, including CTGF, NOTCH2, ABCB1, TXN, CRIM1, CCDC80, AREG, and ASAP1 (Fig.4A), most of which are TEADs-dependent YAP target genes. The results support our hypothesis that an intact β-Catenin might be necessary for YAP’s full activation in iCCA.
Figure 4. Genomic profiling reveals the crosstalk between YAP and β-Catenin signaling cascade in iCCA.
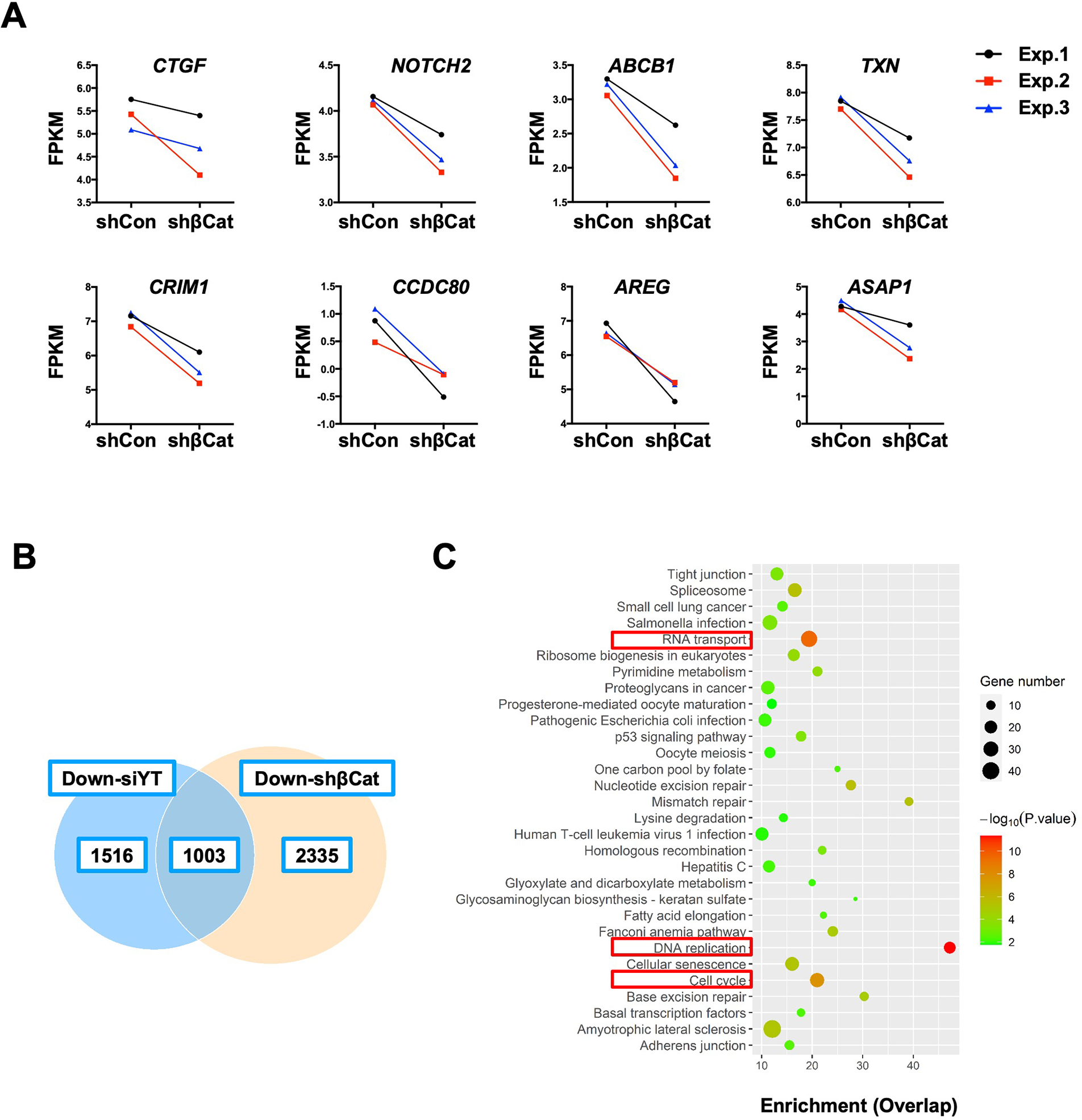
(A) Fragments per kilobase of transcript per million mapped reads (FPKM) results of YAP target genes expression. (B) The overlapping downregulated genes in the shβCat and siYT RBE cell lines. (C) KEGG analysis of downregulated genes in both shβCat and siYT RBE cell lines. P-value ranked the genes. Red inserts indicate overlapping pathways related to tumorigenesis.
To further substantiate this observation, we analyzed genes regulated by Hippo or β-Catenin in human iCCA cells globally. Studies have shown that YAP and its paralog TAZ may play redundant roles in regulating target gene expression in tumors 20. Indeed, knockdown YAP alone only led to a moderate reduction of its target gene expression, while simultaneous depletion of YAP and TAZ resulted in robust suppression of CTGF and CYR61 levels in human iCCA cells (Fig. S22). Therefore, we performed RNAseq following YAP/TAZ knockdown (siYT) in RBE cells (Fig. S23). RNASeq identified 2519 genes downregulated, including YAP1 and WWTR1 (which encodes TAZ), and 1994 genes upregulated by siYT, respectively, in RBE cells (Fig. S24A and Supplementary Dataset). Among the genes downregulated by siYZ are the canonical Hippo/YAP/TAZ target genes, such as CTGF, NOTCH2, ABCB1, TXN, CRIM1, and CCDC80 (Fig. S24B and Supplementary Dataset). KEGG analysis revealed that the pathways downregulated by siYT include RNA transport, p53 signaling, DNA replication, and cell cycle pathways (Fig. S25). Further investigation showed that 1003 genes were commonly downregulated in siYZ- and shβCat-treated RBE cells (Fig.4B). These overlapping genes belong to RNA transport, DNA replication, and cell cycle categories (Fig. 4C) and have prominent roles during tumorigenesis. In all siYZ downregulated genes, 39.8% were downregulated by shβCat, whereas only 8.3% were induced by shβCat. The difference was statistically significant (P < .01).
Altogether, YAP/TAZ and β-Catenin regulate an overlapping set of genes with critical roles in tumorigenesis. Thus, β-Catenin might significantly contribute to the full activation in iCCA.
β-Catenin is critical for Yap dependent iCCA development in mice
Our data suggest that an intact β-Catenin signaling is necessary to fully activate YAP and cholangiocarcinogenesis. In human iCCA cells, β-catenin knockdown significantly inhibited cell growth and YAP target gene expression (Fig. S26). If the hypothesis is correct, one would predict that ablation of Ctnnb1 would significantly suppress Yap-driven iCCA formation in mice. For this purpose, we utilized conditional Ctnnb1 KO mice (Ctnnb1flox/flox). Ctnnb1flox/flox mice were hydrodynamically injected with Akt and Yap plasmids together with the pCVM/Cre construct (Akt/Yap/Cre). This strategy allowed the deletion of Ctnnb1 while co-expressing Akt and Yap oncogenes in the same set of mouse hepatocytes. Additional Ctnnb1flox/flox mice were injected with Akt, Yap plasmids and the pCVM empty vector (Akt/Yap/pCMV) as a control (Fig. 5A). Ablation of endogenous β-Catenin strongly delayed tumor development in mice (Fig. 5B and S27A). Specifically, all Akt/Yap/pCMV mice became moribund by 7 to 10 weeks post-injection due to a high tumor burden, similar to Akt/Yap injected wildtype mice (Sup. Table 3). In striking contrast, all Akt/Yap/Cre mice appeared healthy, and no liver tumor developed (Sup. Table 3). When mice were harvested at around 21 weeks post-injection, most mice showed only a few small tumor nodules on the liver (Fig. 5C and Sup. Table 3). To exclude that the co-injection of Cre interferes with transformation efficiency, wildtype FVB/N mice were co-injected with Akt, Yap, and Cre plasmids. Akt/Yap/Cre mice in wildtype background became moribund by 7 to 10 weeks post-injection, similar to Akt/Yap/pCMV injected Ctnnb1flox/flox mice (Fig.S27B).
Figure 5. Deletion of β-Catenin significantly inhibits Akt/Yap iCCA development in mice.
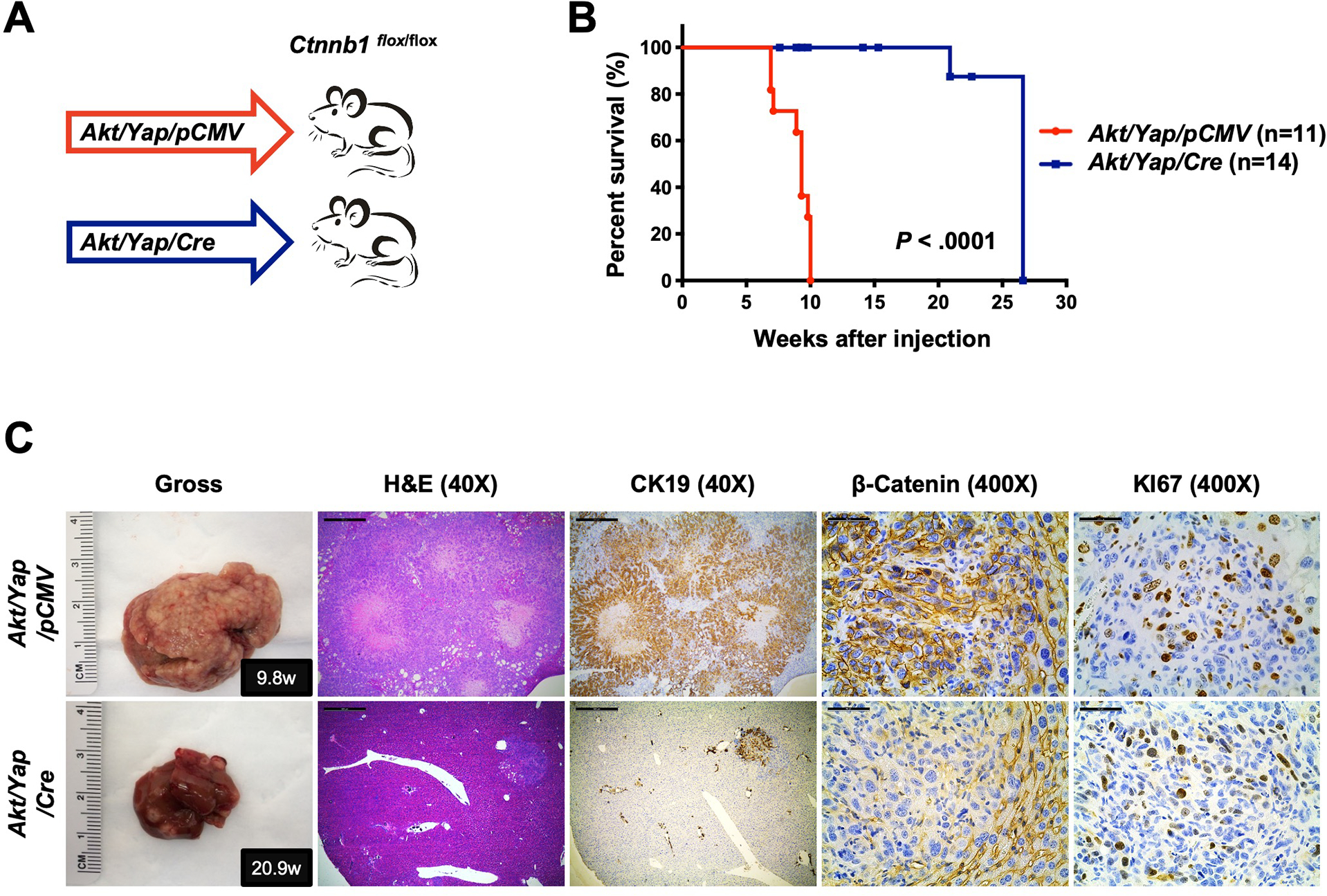
(A) Study design. Ctnnb1flox/flox mice were subjected to HTVi of either Akt/Yap/pCMV (control, n = 11) or Akt/Yap/Cre (n = 14) plasmids. (B) Mouse survival curves. (C) Representative gross images, H&E, and immunohistochemistry for CK19, β-Catenin, and KI67 of liver sections from Akt/Yap/pCMV (M9.8w.p.i) and Akt/Yap/Cre (M20.9w.p.i) mice. Data were analyzed using the Mann-Whitney test. Scale bar: 500 μm (40x) and 50 μm (400x). Abbreviations: H&E, hematoxylin and eosin staining; w.p.i, weeks post-injection.
Histological evaluation revealed that ablation of β-Catenin did not change the tumor phenotype, as tumors developing in Akt/Yap/pCMV and Akt/Yap/Cre mice shared similar histological features (Fig.5C, Fig. S27C, and Sup. Table 3). All tumors stained positive for CK19 and KI67 (Fig.5C and S27C). IHC and Western blotting confirmed AKT and YAP overexpression in Akt/Yap/pCMV and Akt/Yap/Cre lesions (Fig. S27C and S28A). All Akt/Yap/pCMV iCCAs demonstrated positive β-Catenin staining, whereas all Akt/Yap/Cre lesions showed only sporadic β-catenin staining (Fig. 5C and S28B). Using double immunofluorescence (IF) staining, we confirmed that the β-Catenin positive cells within the iCCA lesions were either fibroblasts (VIMENTIN (+)) or endothelial cells (CD31 (+)), whereas all tumor cells (CK19 (+)) lost β-Catenin expression (Fig. S28B–S28E). The results indicate that ablation of β-Catenin strongly suppresses the initiation of Akt/Yap-driven iCCA development in vivo, but does not affect iCCA differentiation program.
To further substantiate our conclusion, we injected Akt and Yap plasmids together with AXIN2, a known β-Catenin inhibitor, into mice (Akt/Yap/Axin2). Akt/Yap/pT3 mice were used as the control (Fig.S27D). As expected, β-Catenin inhibition via AXIN2 overexpression strongly inhibited Akt/Yap-driven iCCA formation (Fig.S27E).
Notch signaling is a critical pathway downstream of Yap in Akt/Yap-driven iCCA development, as blocking the Notch cascade suppressed Akt/Yap-dependent cholangiocarcinogenesis 21. Co-expression of activated Akt and Notch1 (Nicd) led to rapid iCCA formation (Akt/Nicd) 22. While Yap expression could be detected in Akt/Nicd iCCA lesions, Yap ablation only mildly delayed Akt/Nicd iCCA development (Fig. 6A–6C and S29A). Thus, if β-Catenin modulates Yap activity, we predicted that β-Catenin has a limited role in Akt/Nicd iCCA formation. To test this hypothesis, we hydrodynamically injected Akt/Nicd/pCMV or Akt/Nicd/Cre plasmids into Ctnnb1flox/flox mice (Fig. 6D). Distinct from Akt/Yap mice, where ablation of Ctnnb1 strongly suppressed tumor development (Fig. 5B), deletion of β-Catenin only mildly delayed Akt/Nicd iCCA formation (Fig. 6E), phenocopying Yap deletion in this iCCA model (Fig. 6B). Importantly, histological and biochemical analysis confirmed that all Akt/Nicd/Cre tumor cells were β-Catenin negative (Fig. 6F and S29B), indicating that Akt/Nicd readily induces iCCA formation in the Ctnnb1 KO genetic background. As Tead2V16 mimics TEAD mediated transcription, Akt/Tead2V16 drives iCCA formation in a largely Yap-independent and endogenous TEAD-independent manner. Consistently, we found that ablation of Ctnnb1 only mildly affected Akt/Tead2V16 driven iCCA pathogenesis (Fig. S30).
Figure 6. Deletion of β-Catenin mildly delays Akt/Nicd driven iCCA.
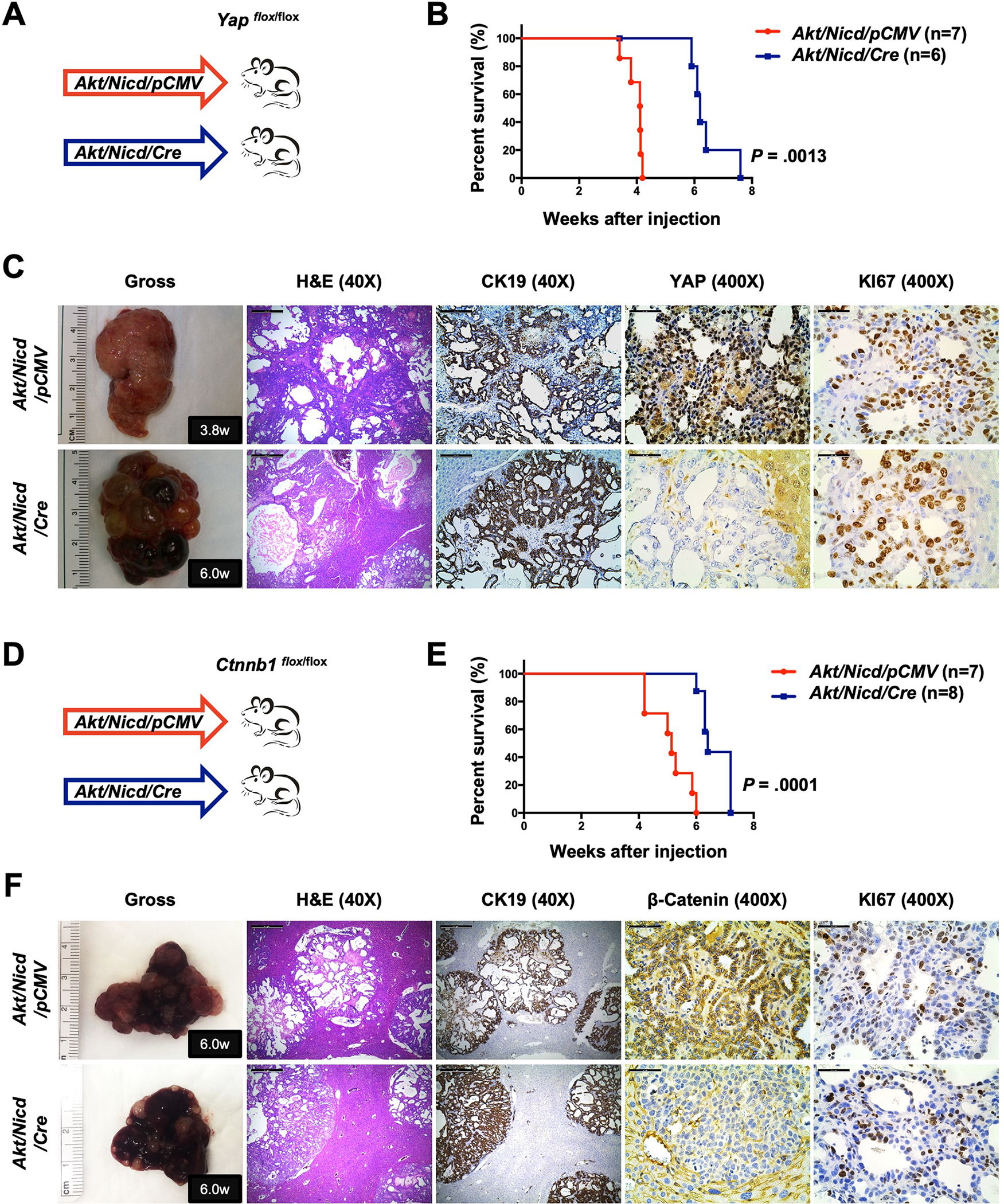
(A) Study design. Yapflox/flox conditional knockout mice were subjected to HTVi of either Akt/Nicd/pCMV (control, n = 7) or Akt/Nicd/Cre (n = 6) plasmids. (B) Mouse survival curves. (C) Representative gross images, H&E, and immunohistochemistry for CK19, YAP, and KI67 of liver sections from Yapflox/flox Akt/Nicd/pCMV (M3.8w.p.i) and Yapflox/flox Akt/Nicd/Cre (M6.0w.p.i) mice. (D) Study design. Ctnnb1 flox/flox mice were subjected to HTVi of either Akt/Nicd/pCMV (control, n = 7) or Akt/Nicd/Cre (n = 8) plasmids. (E) Mouse survival curves. (F) Representative gross images, H&E, and immunohistochemistry for CK19, β-Catenin, and KI67 of liver sections from Ctnnb1flox/flox Akt/Nicd/pCMV (M6.0w.p.i) and Ctnnb1flox/flox Akt/Nicd/Cre (M6.0w.p.i) mice. Scale bar: 500 μm (40x) and 50 μm (400x). Abbreviations: H&E, hematoxylin and eosin staining; w.p.i, weeks post-injection.
To further validate that intact β-catenin is indispensable for Yap-dependent iCCA development, we selected another mouse iCCA model induced by Akt, Jag1, and Fak co-expression (Akt/Jag1/Fak). Akt/Jag1/Fak combination induces iCCA formation in a Yap-dependent manner 23. Thus, we hydrodynamically injected Akt/Jag1/Fak/pCMV or Akt/Jag1/Fak/Cre plasmids into Ctnnb1flox/flox mice (Fig. 7A). Notably, ablation of Ctnnb1 strongly suppressed Akt/Jag1/Fak induced iCCA formation, recapitulating the results from the Akt/Yap iCCA model (Fig. 5B and 7B). Akt/Jag1/Fak/pCMV livers were almost entirely occupied by numerous large iCCAs at 11 weeks post-injection when Akt/Jag1/Fak/Cre livers were virtually normal (Fig. S31). Even 35 weeks post-injection, Akt/Jag1/Fak/Cre mice were still alive (Fig. 7B), and, upon dissection, only small tumor nodules were detected. Histologically, the lesions were iCCA, characterized by the loss of β-catenin expression in tumor cells and low proliferation (Fig. 7C). These data indicate that β-Catenin deletion significantly delays Yap-dependent Akt/Jag1/Fak iCCA development.
Figure 7. Deletion of β-Catenin strongly suppresses Akt/Jag1/Fak-induced iCCA.
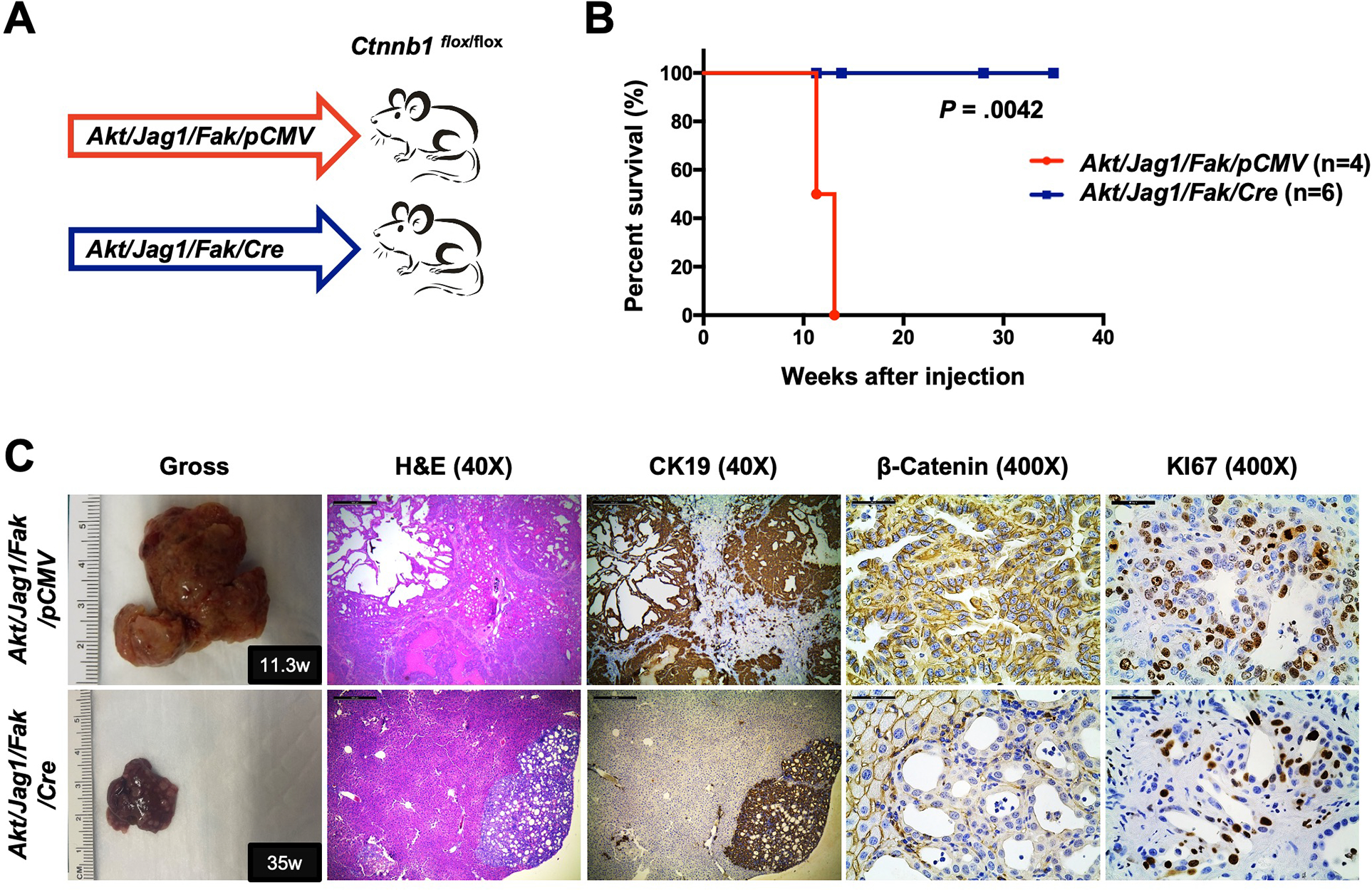
(A) Study design. Ctnnb1flox/flox mice were subjected to HTVi of either Akt/Jag1/Fak/pCMV (control, n = 4) or Akt/Jag1/Fak/Cre (n = 6) plasmids. (B) Mouse survival curves. (C) Representative gross images, H&E, and immunohistochemistry for CK19, β-Catenin, and KI67 of liver sections from Akt/Jag1/Fak/pCMV (F11.3w.p.i) and Akt/Jag1/Fak/Cre (F35w.p.i) mice. Scale bar: 500 μm (40x) and 50 μm (400x). Abbreviations: H&E, hematoxylin and eosin staining; w.p.i, weeks post-injection.
Overall, the present findings indicate that functional β-catenin is indispensable for Yap-dependent iCCA development.
Inactivation of Hippo and activation of the Wnt/β-catenin pathway in human iCCA
Inactivation of the Hippo cascade and consequent upregulation of YAP/TAZ are prominent molecular events in human iCCA 4, 5. This finding was validated here using genes downregulated in siYT cells. Notably, this gene list was enriched in genes upregulated in iCCA samples using the TCGA CHOL dataset and the NCI human iCCA dataset 24, 25 (Fig. S32A). Furthermore, KEGG pathway analysis revealed that genes downregulated in siYT cells and upregulated in human iCCA samples are predominantly involved in cell cycle and DNA replication (Fig.S32B–S32C).
However, whether the β-Catenin pathway is activated in human iCCA remains unclear as genetic events leading to β-Catenin activation, such as mutations of CTNNB1 or AXIN1, are rarely found in human iCCA. Analysis of the TCGA CHOL dataset revealed that the canonical target genes of β-Catenin, such as GLUL, TBX3, SP5, LGR5, LECT2, OAT, HADH, and PLOD2, are not upregulated in human iCCAs (Fig. S33). These data suggest the lack of activation of the canonical Wnt/β-Catenin signaling in iCCA. Interestingly, increased CTNNB1, multiple WNT ligands, and FZDs mRNA expression was observed in human iCCA based on the TCGA database (Fig. S34–S35) and in Akt/Yap iCCAs (Fig. S36). The increased CTNNB1 mRNA expression in iCCA was further validated using our human iCCA collection (n=58; Fig. S37A, Sup. Table 4). We also found that human iCCA patients with high CTNNB1 mRNA expression display a poor prognosis (Fig.S37B). Furthermore, this association remained strongly significant after multivariate Cox regression analysis (Supplementary Material), suggesting CTNNB1 mRNA levels as an independent prognostic factor for iCCA.
Next, we analyzed whether the genes downregulated in shβCat RBE cells were enriched in human iCCA samples. Notably, 28.7% of shβCat-downregulated genes were upregulated in iCCA based on the NCI dataset, and this was statistically significant (P < .001) (Fig. S38A). KEGG analysis of these overlapping genes revealed that they are associated with cell cycle and DNA replication (Fig. S38B). Similar results were obtained using the TCGA CHOL dataset (Fig. S38A, S38C). Altogether, these data support the activation of β-Catenin in human iCCA.
To further investigate this important question, we analyzed a collection of human iCCA (n=225) by immunohistochemistry (Fig. S39). In nontumorous surrounding liver tissues, weak staining for β-catenin was observed in hepatocytes, whereas a more robust membranous and cytoplasmic staining characterized the biliary cells. In contrast, strongly homogeneous membranous and cytoplasmic immunoreactivity for total and activated/non-phosphorylated β-catenin was predominant in iCCA samples (182/225; 80.9% and 191/225; 84.8%, respectively). In addition, intense and homogeneous nuclear staining was detected in 7 samples (3.11%; Fig. S40), while 12 (5.3%) iCCA exhibited uniform or sparse areas of intense β-catenin cytoplasmic immunoreactivity containing scattered tumor cells with nuclear β-catenin accumulation (Fig. S40).
Consistent with human iCCA data, overexpression of non-phosphorylated β-Catenin was detected in all Akt/Yap, Akt/Nicd, and Akt/Jag1/Fak iCCA tumors when compared with normal livers (Fig.S41–S42).
Altogether, these results demonstrate the increased non-p-β-Catenin expression regardless of the patients’ clinicopathological data, implying that activation of β-Catenin is a general phenomenon in human iCCA. The data highlight the increased expression and activation of β-Catenin independent of CTNNB1 mutations in iCCA.
Discussion
A major conclusion of the current study is the discovery of β-Catenin activation in human and mouse iCCAs. As CTNNB1 mutations are rare in iCCA, it is most likely that β-Catenin is activated via the Wnt ligands. Indeed, we observed increased expression of multiple Wnt ligands and FZD receptors in human iCCA samples and mouse Akt/Yap iCCA lesions (Fig.S34–S36). We further analyzed the single cell RNASeq data from mouse iCCA data set 26. Multiple Wnt ligands could be found to be expressed in cancer-associated fibroblasts (CAFs) (Fig. S43). These data together indicate that tumor microenvironment might be the major sources of Wnt ligands in iCCA; and the activation of β-Catenin in iCCA is likely Wnt-ligand dependent. Nevertheless, additional studies, for example, by deleting the Wnt co-receptors Lrp5/6 10 in iCCA, are necessary to demonstrate the requirement of the Wnt pathway in cholangiocarcinogenesis. Previous studies revealed that β-Catenin is predominantly localized in the membrane of human iCCA cells, suggesting the lack of β-Catenin activation. However, several studies have suggested that IHC staining is not a sensitive technique to address β-Catenin activation status in subsets of liver tumors as can be seen by lack of clear nuclear β-Catenin but an increase of its target genes like GLUL, TBX3, and AXIN2 27. Therefore, a more reliable approach to determine the Wnt/β-Catenin pathway activation status in the absence of CTNNB1 mutations is evaluating activated/non-phosphorylated β-Catenin levels using either IHC or Western blotting. Using activated/non-phosphorylated β-Catenin levels as a surrogate marker of β-Catenin activation, our data support a broad activation of β-Catenin in human and mouse iCCA samples.
In addition, the current study revealed that β-Catenin is a YAP/TEAD partner required for YAP-dependent cholangiocarcinogenesis. Since β-Catenin is known to function via TCF4 mediated transcriptional regulation, we asked whether TCF4 is the major β-Catenin partner in iCCA. We thus performed RNASeq on RBE cells with overexpression of the dominant-negative form of TCF4 (dnTCF4) (Fig. S44A), which blocks β-Catenin/TCF4 mediated gene expression28. We found that overexpression of dnTCF4 downregulated multiple canonical Wnt/β-Catenin target genes, including AXIN2, RNF43, MMP7, and TIAM1 (Fig. S44B). However, dnTCF4 only led to the downregulation of 167 genes and upregulation of 143 genes in RBE cells (Fig. S44C, Supplementary Dataset), significantly less than those modulated by β-Catenin or YAP/TAZ (Fig. S20A and S24A). Importantly, KEGG analysis revealed that pathways downregulated by dnTCF4 were different from that by shβCat (Fig. S45). These data indicate that the primary function of β-Catenin in regulating iCCA is independent of TCF4. The results might also explain why the canonical Wnt/β-Catenin pathway, regulated by the β-Catenin/TCF4 complex, is not activated in human iCCAs.
To further investigate how β-Catenin modulates YAP-dependent cholangiocarcinogenesis, we performed RNASeq studies of Akt/Yap iCCAs in wildtype mice and Ctnnb1 KO background (Fig. S46A, Supplementary Dataset). Pathway analysis revealed that the Ras and MAPK signaling pathways were significantly downregulated in β-Catenin (−) Akt/Yap iCCA lesions (Fig. S46B). Moreover, several genes controlling the Ras/MAPK signaling, including ERBB2, SHC1, GRB2, HRAS, NRAS, KRAS, ARAF, BRAF, RAF1, MAP2K1, and MAPK1, were downregulated in RBE shβCat cells (Fig. S47. Consistent with RNASeq results, a lower level of phosphorylated/activated (p-)Erk1/2 characterized β-Catenin (−) Akt/Yap iCCA lesions (Fig. S48A–S48B). Furthermore, silencing of β-Catenin led to diminished p-Erk1/2 in human iCCA cell lines (Fig. S48C). These data suggest that, in addition to supporting Yap activity, β-Catenin contributes to iCCA tumorigenesis through the Ras/MAPK cascade. Moreover, RNASeq analysis revealed decreased expression of cholangiocarcinoma stem cell markers, such as Cd24, Cd44, and Cd133 29, in β-Catenin (−) Akt/Yap iCCAs (Fig. S49), suggesting that β-Catenin might also regulate cholangiocarcinogenesis by inducing stemness features in cancer cells.
One of the interesting observations of our study is that WW and PDZ domains negatively regulate Yap-mediated iCCA development. As these domains mediate YAP-protein interactions, the specific proteins that mediate these inhibitory effects on Yap-driven cholangiocarcinogenesis should be determined. Previous studies suggest that PRGP2 and ARID1A sequester YAP via its WW domain and attenuate its transcriptional activity 30, 31. In addition, zona occludens proteins have been identified as YAP/TAZ negative regulators via the PDZ domain 32. It would be of interest to determine whether these proteins interact with YAP in iCCA via the WW or PDZ domain and how they function to inhibit iCCA development. These studies will provide novel insight into how YAP activity is regulated in iCCA and possible new targets for iCCA treatment.
Supplementary Material
What You Need to Know:
BACKGROUND AND CONTEXT:
YES-associated protein (YAP) aberrant activation is a predominant oncogenic event in human intrahepatic cholangiocarcinoma. However, the precise molecular mechanisms by which YAP induces cholangiocarcinoma development remain unclear.
NEW FINDINGS:
YAP drives cholangiocarcinogenesis via TEAD mediated transcriptional activation and interaction with β-Catenin. β-Catenin is activated/non-phosphorylated in most human cholangiocarcinoma and is required for YAP-dependent cholangiocarcinoma formation.
LIMITATIONS:
As CTNNB1 mutations are rare in human cholangiocarcinoma, the molecular mechanisms leading to the activation of β-Catenin in cholangiocarcinoma remain undetermined.
IMPACT:
Our findings provide novel mechanisms by which the non-mutant form of β-Catenin contributes to cholangiocarcinoma formation.
Acknowledgments
We deeply thank Dr. Gregory J. Gores (Mayo Clinic, Rochester, MN) for sharing the M1-1a, M4-2a, and M4-2b mouse iCCA cell lines; Dr. Zhiqiang Lin (Masonic Medical Research Institute, Utica, NY) for sharing the AAV9.VGLL4-GFP plasmid.
Grant support
This study was funded by R01CA19606 (to XC) and R01CA228483 (to XC and RFS); R01CA204586 and R01CA250227 (to XC and SPM), P30DK026743 (to XC by UCSF Liver Center), P30DK120531 (to SPM by the Pittsburgh Liver Research Center), and R01CA258449 (to SK).
Abbreviations
- CCA
cholangiocarcinoma
- dnTEAD2
dominant-negative form of TEAD2
- HB
hepatoblastoma
- HCC
hepatocellular carcinoma
- iCCA
intrahepatic cholangiocarcinoma
- TAD
transcriptional activation domain
- TBD
TEAD binding domain
- TCGA
The Cancer Genome Atlas
- TEADs
TEA domain (TEAD)-containing transcription factors
- YAP
YES-associated protein
Footnotes
Disclosures
The authors declare no potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability statement
The data that support the findings of this study are included within the article and supplementary materials.
References:
- 1.Banales JM, Marin JJG, Lamarca A, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol 2020;17:557–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 3.Cheng R, Du Q, Ye J, et al. Prognostic value of site-specific metastases for patients with advanced intrahepatic cholangiocarcinoma: A SEER database analysis. Medicine (Baltimore) 2019;98:e18191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen-Lefebvre AT, Selzner N, Wrana JL, et al. The hippo pathway: A master regulator of liver metabolism, regeneration, and disease. FASEB J 2021;35:e21570. [DOI] [PubMed] [Google Scholar]
- 5.Sugihara T, Isomoto H, Gores G, et al. YAP and the Hippo pathway in cholangiocarcinoma. J Gastroenterol 2019;54:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev 2016;30:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen YA, Lu CY, Cheng TY, et al. WW Domain-Containing Proteins YAP and TAZ in the Hippo Pathway as Key Regulators in Stemness Maintenance, Tissue Homeostasis, and Tumorigenesis. Front Oncol 2019;9:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pocaterra A, Romani P, Dupont S. YAP/TAZ functions and their regulation at a glance. J Cell Sci 2020;133. [DOI] [PubMed] [Google Scholar]
- 9.Zhang S, Song X, Cao D, Xu Z, et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J Hepatol 2017;67:1194–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perugorria MJ, Olaizola P, Labiano I, et al. Wnt-beta-catenin signalling in liver development, health and disease. Nat Rev Gastroenterol Hepatol 2019;16:121–136. [DOI] [PubMed] [Google Scholar]
- 11.Wang W, Smits R, Hao H, et al. Wnt/beta-Catenin Signaling in Liver Cancers. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol 2014;184:912–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calvisi DF, Wang C, Ho C, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011;140:1071–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Liu P, Tao J, et al. TEA Domain Transcription Factor 4 Is the Major Mediator of Yes-Associated Protein Oncogenic Activity in Mouse and Human Hepatoblastoma. Am J Pathol 2019;189:1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H, Liu CY, Zha ZY, et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem 2009;284:13355–13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azzolin L, Panciera T, Soligo S, et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 2014;158:157–70. [DOI] [PubMed] [Google Scholar]
- 17.Deng F, Peng L, Li Z, et al. YAP triggers the Wnt/beta-catenin signalling pathway and promotes enterocyte self-renewal, regeneration and tumorigenesis after DSS-induced injury. Cell Death Dis 2018;9:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan JX, Xiong L, Zhao K, et al. YAP promotes osteogenesis and suppresses adipogenic differentiation by regulating beta-catenin signaling. Bone Res 2018;6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imajo M, Miyatake K, Iimura A, et al. A molecular mechanism that links Hippo signalling to the inhibition of Wnt/beta-catenin signalling. EMBO J 2012;31:1109–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, Wang J, Zhang S, et al. Distinct and Overlapping Roles of Hippo Effectors YAP and TAZ During Human and Mouse Hepatocarcinogenesis. Cell Mol Gastroenterol Hepatol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Dong M, Xu Z, et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018;37:3229–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan B, Malato Y, Calvisi DF, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest 2012;122:2911–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song X, Xu H, Wang P, et al. Focal adhesion kinase (FAK) promotes cholangiocarcinoma development and progression via YAP activation. J Hepatol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farshidfar F, Zheng S, Gingras MC, et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep 2017;18:2780–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andersen JB, Spee B, Blechacz BR, et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012;142:1021–1031 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Affo S, Nair A, Brundu F, et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 2021;39:866–882 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiao Y, Xu M, Tao J, et al. Oncogenic potential of N-terminal deletion and S45Y mutant beta-catenin in promoting hepatocellular carcinoma development in mice. BMC Cancer 2018;18:1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tao J, Zhang R, Singh S, et al. Targeting beta-catenin in hepatocellular cancers induced by coexpression of mutant beta-catenin and K-Ras in mice. Hepatology 2017;65:1581–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu HJ, Chu PY. Role of Cancer Stem Cells in Cholangiocarcinoma and Therapeutic Implications. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulman JD, Harris JE, Xie L, et al. Proline-rich Gla protein 2 is a cell-surface vitamin K-dependent protein that binds to the transcriptional coactivator Yes-associated protein. Proc Natl Acad Sci U S A 2007;104:8767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang L, Azzolin L, Di Biagio D, et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 2018;563:265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Remue E, Meerschaert K, Oka T, et al. TAZ interacts with zonula occludens-1 and –2 proteins in a PDZ-1 dependent manner. FEBS Lett 2010;584:4175–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are included within the article and supplementary materials.