

Abstract
Background:
Rescue treatment for delayed cerebral ischemia (DCI) after subarachnoid hemorrhage can include induced hypertension (iHTN) and, in refractory cases, endovascular approaches, of which selective, continuous intraarterial nimodipine (IAN) is one variant. The combination of iHTN and IAN can dramatically increase vasopressor demand. In case of unsustainable doses, iHTN is often prioritized over IAN. However, evidence in this regard is largely lacking. We investigated the effects of a classical (iHTN+IAN) and modified (IANonly) treatment protocol for refractory DCI in an observational study.
Methods:
Rescue treatment for DCI was initiated with iHTN (target >180mmHg systolic) and escalated to IAN in refractory cases. Until 07/2018, both iHTN and IAN were offered in cases refractory to iHTN alone. After protocol modification, iHTN target was preemptively lowered to >120mmHg when IAN was initiated (IANonly). Primary outcome was noradrenaline demand. Secondary outcomes included noradrenaline-associated complications, brain tissue oxygenation (ptiO2), DCI-related infarction and favorable 6-month outcome (Glasgow Outcome Scale 4–5).
Results:
N=29 and n=20 patients were treated according to the classical and modified protocol, respectively. Protocol modification resulted in a significant reduction of noradrenaline demand (iHTN+IAN 0.70±0.54μg/kg/min, IANonly 0.26±0.20μg/kg/min, p<0.0001) and minor complications (15.0% versus 48.3%, unadjusted OR 0.19 (95% CI 0.05–0.79), p<0.05) with comparable rates of major complications (20.0% versus 20.7%, OR 0.96 (0.23–3.95), p=0.95). Incidence of DCI-related infarction (45.0% versus 41.1%, OR 1.16 (0.37–3.66), p=0.80) and favorable clinical outcome (55.6% versus 40.0%, OR 1.88 (0.55–6.39), p=0.32) were similar. PtiO2 was significantly higher with IANonly (26.6±12.8mmHg, 39.6±15.4mmHg, p<0.01).
Conclusions:
Assuming the potential of iHTN to be exhausted in case of refractory hypoperfusion, additional IAN may serve as a last-resort measure to bridge hypoperfusion in the DCI phase. With close monitoring, preemptive lowering of pressure target after induction of IAN may be a safe alternative to alleviate total noradrenaline load and potentially reduce complication rate.
Graphical Abstract
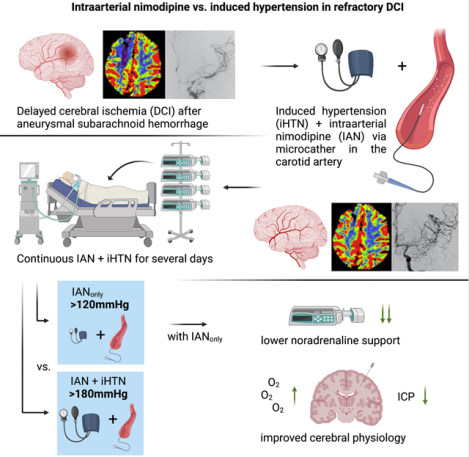
Introduction
Delayed cerebral ischemia (DCI) is one of the known key contributors to cognitive impairment after aneurysmal subarachnoid hemorrhage1. The currently recommended treatment modalities for DCI are largely aimed at maximizing supply to underserved territories, regardless of the exact underlying pathomechanism(s). If DCI occurs, vasopressor-induced hypertension (iHTN) is a common, albeit critically debated first-line approach to improve perfusion2. Further escalation in case of refractory DCI may ultimately include local application of vasodilators as second-line therapy. However, there is still a lack of robust evidence regarding the guidance of rescue treatment for DCI, resulting in highly heterogenous treatment algorithms3,4. Controlled trials for endovascular rescue treatment are not available and unlikely to be performed due to the ethical concern of withholding treatment at this critical point5. The latest international guidelines generally recommend iHTN as primary rescue treatment and intraarterial vasodilation as adjunct with weaker recommendation but are unable to provide clear directive due to the paucity of evidence5–8. Short-term, repetitive intraarterial spasmolysis in the angiography suite is a common form of endovascular rescue treatment3. Leaving the intraarterial catheter(s) in place and continuing vasodilation in the intensive care unit is a less frequently practiced method, accompanied by a separate risk profile but with the opportunity to bridge the oftentimes prolonged DCI phase9,10. Centers implementing continuous endovascular rescue treatment may frequently observe an inherent conflict of mechanisms when adding intraarterial vasodilation on top of iHTN. Secondary systemic distribution of a locally applied vasodilator often increases vasopressor demand to counteract this effect peripherally11. Escalation of vasopressor support may facilitate dangerous complications up to cardiopulmonary exhaustion and multi-organ failure due to impairment of microcirculatory perfusion, requiring reduction of rescue treatment to curb vasopressor demand12. A balance must be found between the risk of cerebral infarction and a critical compromise of peripheral microcirculation. There is, however, currently no evidence on the prioritization of iHTN over intraarterial vasodilation, nor on the safety and efficacy of measures to reduce vasopressor demand in either scenario.
We have previously documented our standard operating procedure with continuous intraarterial nimodipine (IAN) in case of DCI refractory to iHTN and found promising effects on cerebral hypoxia and disturbed local metabolism9,13. Nevertheless, we also encountered critical vasopressor increases which classically triggered reduction of IAN while iHTN was maintained. In search for an alternative algorithm with lower vasopressor demand but non-inferior efficacy, we hypothesized that the effect of iHTN on DCI may be exhausted in cases with refractory hypoperfusion. We modified our treatment protocol accordingly, henceforth lowering our pressure-target preemptively when IAN was initiated after failure of iHTN. The aim of this prospective observational study was to compare our classical (iHTN+IAN) and modified (IANonly) treatment protocols in patients refractory to iHTN alone, with the hypothesis that protocol modification would lead to lower vasopressor demand without a compromise in rescue treatment efficacy.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Patient cohort
Data of patients ≥18 years who were treated at our institution for verified aSAH between January 2014 to December 2020 were collected into a prospective databank. Patients with DCI deemed refractory to iHTN were included into this observational study. Written, informed consent for databank inclusion and data analysis was obtained by a legal representative if capacity for informed decision making was absent. Approval was granted by a local ethics committee (EK062/14). Reporting follows the STROBE criteria14.
Classical and modified DCI treatment protocols
Figure 1 details our treatment algorithm with a classical protocol (iHTN+IAN) followed by protocol modification in 07/2018 (IANonly). All aSAH patients were cared for in an intensive care unit specialized on care of neurosurgical patients after securing the ruptured aneurysm. Fluid balance is targeted at −500 to 0ml daily and further fluid management is aligned with the KDIGO guideline15. All patients received oral nimodipine prophylaxis with 360mg/day unless contraindicated. Awake patients were monitored for DCI clinically, while unconscious and/or analgosedated patients or those at high risk for DCI (Hunt and Hess grade ≥3, or grade 1–2 with modified Fisher grade ≥3, unless the hemorrhage is deemed fatal) received invasive neuromonitoring probes consisting of a combined ptiO2/ICP-probe (Neurovent PTO, Raumedic, Helmbrechts, Germany) and adjacent cerebral microdialysis (71 High Cut-Off Brain Microdialysis Catheter, μdialysis, Stockholm, Sweden). Probes were placed into the watershed area between anterior and middle cerebral artery territories on the side of the offending aneurysm, or in the non-dominant hemisphere for midline aneurysms. Transcranial doppler sonography was used on an as needed basis.
Figure 1.
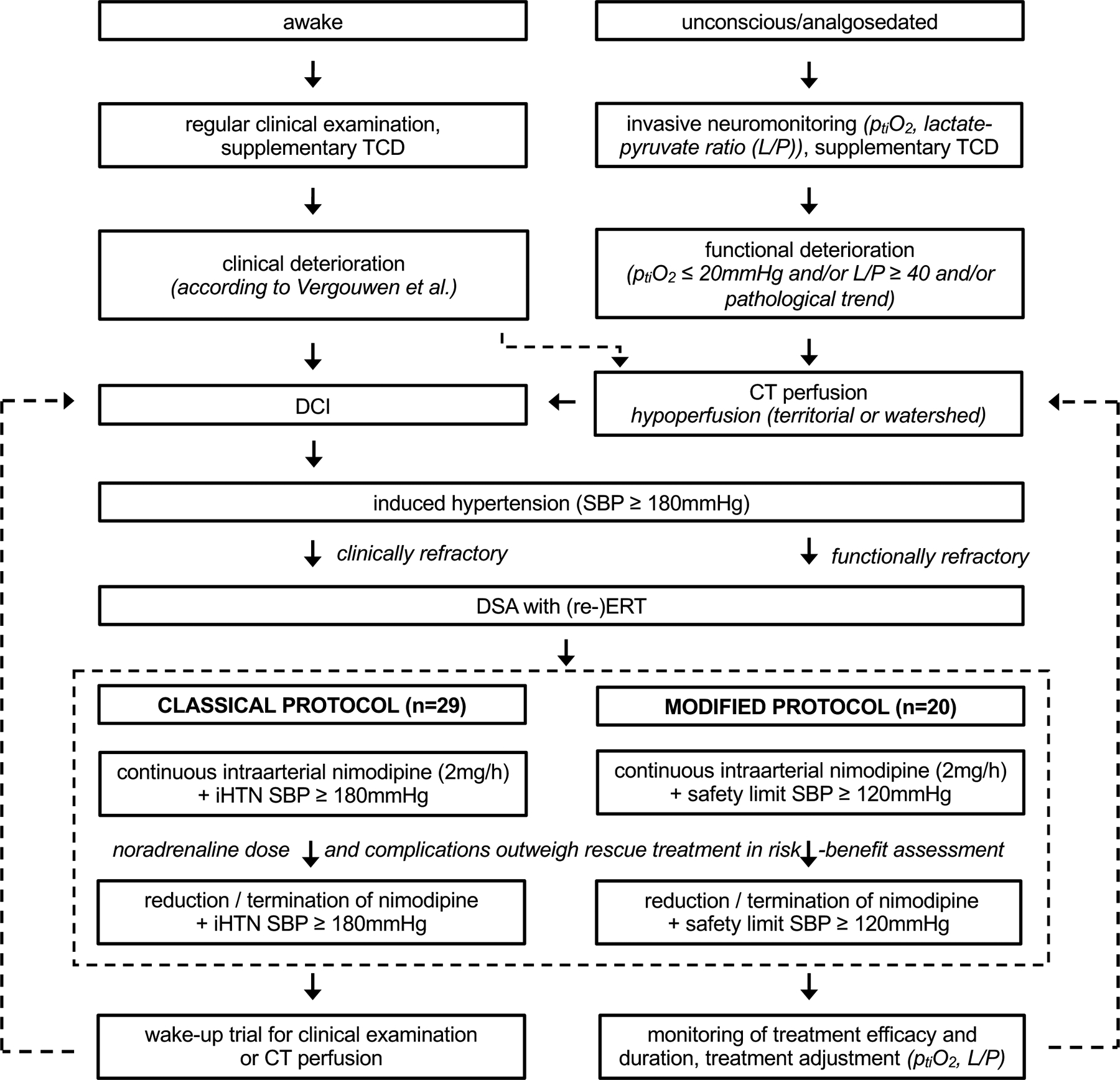
Classical (iHTN+IAN) and modified (IANonly) treatment protocols.
DCI: delayed cerebral ischemia; ERT: endovascular rescue treatment; iHTN+IAN: patients treated according to the classical protocol; IANonly: patients treated according to the modified protocol; TCD: transcranial doppler ultrasonography; SBP: systolic blood pressure.
In awake patients, DCI was diagnosed according to the definition by Vergouwen et al.16. If clinical examination was not available, functional deterioration as observed by invasive neuromonitoring (individual worsening of ptiO2 or lactate/pyruvate ratio) was used to trigger CT perfusion, in which case territorial or watershed perfusion deficit (time to drain >10s, mean transit time >6.7s) was included to also diagnose DCI. The term “DCI” therefore refers to either the first clinical deterioration or to CT perfusion deficit in this cohort. Of note, during IAN, where patients are analgosedated (see below), only repeated CT perfusion scans can indicate if DCI is still ongoing. Infarction on native CT imaging is termed DCI related infarction (if deemed unrelated to embolic infarction).
After DCI diagnosis, iHTN served as first-line treatment with systolic blood pressure >180mmHg by intravenous noradrenaline infusion in both protocols. If clinical deficit or hypoperfusion on CT perfusion persisted or aggravated under iHTN, digital subtraction angiography was conducted. Moderate vasoconstriction and perfusion delay of contrast agent were criteria to consider additional, continuous intraarterial nimodipine application (IAN). For this purpose, microcatheters were placed into one or both internal carotid arteries and/or one vertebral artery depending on the affected regions. Infusion amounted to a maximum cumulative dose of 2mg/h. IAN was accompanied by deep analgosedation (Riker Sedation Agitation Scale 1–2 (unarousable or very sedated) by infusion of propofol, midazolam, ketamine and/or clonidine together with sufentanil), inhibition of platelet-aggregation by tirofiban (Aggrastat®, Correvio, Heathrow, United Kingdom), and minimal patient handling.
Under the classical protocol, iHTN was maintained with a pressure target >180mmHg after initiation of IAN (iHTN+IAN). In case of critical escalation of noradrenaline demand (>0.5μg/kg/min), a risk-benefit assessment was made by the treating team whether to maintain or reduce rescue treatment. Without present or anticipated noradrenaline-related complications and persistent indication for iHTN and IAN in CT perfusion, treatment was continued despite noradrenaline dose >0.5μg/kg/min. Otherwise, IAN dose was reduced to 0.5–1mg/h or terminated completely while pressure target remained >180mmHg. If this did not lower vasopressor demand sufficiently, pressure target was also lowered (up to a safety limit >120mmHg). After protocol modification, rescue treatment was escalated comparably (iHTN >180mmHg as first-line, IAN as second line), but the pressure target was preemptively lowered to >120mmHg systolic blood pressure with the initiation of IAN. To ensure safety of this modification to our approach, CT perfusion was performed immediately after the induction of IAN under normotensive blood pressure regime to document sufficient reversal of hypoperfusion. Reduction of rescue treatment was evaluated analogously to the classical protocol (risk-benefit assessment with noradrenaline >0.5μg/kg/min) and, if noradrenaline-related complications (see below) were present or anticipated, IAN dosage was reduced in a step-wise fashion. Rescue treatment was maintained until resolution of DCI as shown by neuromonitoring and CT perfusion studies, overall condition of the patient and timing within DCI phase. In case of microcatheter thrombosis, the catheter was removed in the angiography suite and timely CT perfusion was conducted. If perfusion re-deterioration was observed on CT perfusion imaging following thrombosis or elective nimodipine dose reduction, microcatheters were replaced again and/or nimodipine treatment was re-escalated. In patients with iHTN+IAN, the pressure target was also re-escalated >180mmHg with microcatheter replacement, while it remained >120mmHg in patients with IANonly.
Outcome parameters
Baseline demographic parameters were recorded, as were details of the clinical course and neuromonitoring data. The primary outcome parameter was the mean noradrenaline demand for the whole duration of combined iHTN+IAN treatment or IANonly, respectively. Breaks between IAN applications >24hours were excluded from mean value calculations. Secondary outcome parameters were focused on the safety of noradrenaline application and the efficacy of cerebral rescue treatment.
Safety We recorded the number of additional treatment de-escalations to curb noradrenaline demand (reduction of pressure target or IAN dose, according to each protocol). We compared the frequency of side effects that may be caused or aggravated by prolonged, high noradrenaline infusion. Side effects were stratified into minor complications with optional treatment reduction (peripheral limb malperfusion visible as cooled, blanched or marbled extremities; new cardiac arrhythmia) and major complications with obligatory interruption of rescue treatment (new cardiac insufficiency diagnosed by transthoracic echocardiography, cardiogenic shock or myocardial infarction with or without ST-elevation; paralytic ileus or diffuse ischemic gastrointestinal injury; secondary sclerosing cholangitis in critically ill patients). The number of CT perfusion scans during IAN (including for diagnosis of DCI) were documented per group. Complications associated with indwelling microcatheters were also documented (thrombosed microcatheter or visible thrombi surrounding the catheter in CT angiography or digital subtraction angiography, necessary re-angiography with catheter replacement, thromboembolic cerebral infarction or venous or arterial thromboembolism diagnosed during IAN or up to one week after catheter removal).
Efficacy To evaluate the efficacy of cerebral rescue treatment, we calculated mean brain tissue oxygenation (ptiO2) and ICP during treatment, recorded the number of patients with ≥1 necessary treatment re-escalation (with or without previous reduction of pressure target or IAN dose, any time CT perfusion indicated that the current strategy was insufficient, according to each protocol), the occurrence of DCI related cerebral infarction (consistent with current or previously hypoperfused territory, absence of thrombi)16, clinical outcome 6 months after discharge with Glasgow Outcome Scale 4–5 defined as favorable outcome, as well as duration of treatment and hospitalization.
Statistical analysis
Statistical analyses were conducted with IBM SPSS Statistics 26 (SPSS Inc., Chicago, IL, USA). Graphical elements were created using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, USA). Continuous parameters are shown as mean ± standard deviation, discrete variables as n (percentage). Continuous data were tested for normality distribution with the Kolmogorow-Smirnov normality test. Unpaired t-test or Mann-Whitney U test assessed differences between groups, as appropriate. Fisher’s exact test was used for comparisons of discrete demographic variables. Binary outcome parameters were compared in a univariate logistic regression analysis and an unadjusted odds ratio (OR) with 95% confidence interval (CI) was calculated. Two-sided p-values <0.05 were considered statistically significant. Missing data were not imputed.
Results
Out of n=243 patients with SAH treated at our institution in the respective time frame, n=49 (20.2%) developed DCI refractory to iHTN. 29 patients were treated with the iHTN+IAN and 20 with IANonly. Baseline demographic and bleeding characteristics were comparable between both groups (Table 1). IHTN was initiated 5.4±2.7 and 8.1±3.4 days after hemorrhage (p<0.01), followed by IAN 2.6±2.7 and 1.8±2.3 days afterwards (p=0.36) in patients with iHTN+IAN and IANonly, respectively. N=10 (34.5%) patients with iHTN+IAN had one repeat endovascular intervention with microcatheter replacement (after elective removal or thrombosed catheter), n=1 (3.5%) had three repeat interventions. Of patients with IANonly, n=4 (20.0%) had one and n=1 (5.0%) had three repeat intervention. 21 patients with iHTN+IAN (72.4%) and 11 patients with IANonly (55.0%) had a ptiO2/ICP-probe. Nimodipine dose during total treatment duration was similar (iHTN+IAN 19.0±7.7μg/kg/h, IANonly 19.8±7.7μg/kg/h, p=0.61), but systolic blood pressure was significantly lower with IANonly (iHTN+IAN 185±15.0mmHg, IANonly 154.8±15.0mmHg, p<0.0001).
Table 1.
Demography and bleeding characteristics.
| iHTN+IAN | IANonly | p-value | |
|---|---|---|---|
| n | 29 | 20 | |
| age, years | 53.3±7.7 | 53.2±10.4 | 0.99 |
| female sex, n (%) | 20 (69.0%) | 17 (85.0%) | 0.31 |
| body mass index | 26.3±5.2 | 25.8±4.8 | 0.96 |
| arterial hypertension, n (%) | 12 (41.4%) | 8 (40.0%) | 1.00 |
| smoking, n (%) | 10 (34.5%) | 9 (45.0%) | 0.56 |
| Hunt & Hess grade 4–5, n (%) | 5 (17.2%) | 6 (30.0%) | 0.32 |
| modified Fisher grade 3–4, n (%) | 17 (58.6%) | 15 (75.0%) | 0.36 |
| aneurysm in the anterior circulation, n (%) | 24 (82.8%) | 20 (100%) | 0.07 |
| endovascular, n (%) | 16 (55.2%) | 11 (55.0%) |
Unpaired t-test or Mann-Whitney U test assessed differences between groups, as appropriate. Fisher’s exact test was used for comparisons of discrete variables. iHTN+IAN: patients treated according to the classical protocol; IANonly: patients treated according to the modified protocol.
Safety
Noradrenaline demand was significantly lower in patients with IANonly (iHTN+IAN 0.70±0.54μg/kg/min, IANonly 0.26±0.20μg/kg/min, p<0.0001) (Figure 2). The proportion of patients developing minor complications was lower with IANonly (iHTN+IAN n=14 (48.3%), IANonly n=3 (15.0%), OR 0.19 (95% CI 0.05–0.79), p<0.05), while the frequency of major complications was unchanged (iHTN+IAN n=6 (20.7%), IANonly n=4 (20.0%), OR 0.96 (95% CI 0.23–3.95), p=0.95) (Table 2). The proportion of patients with any further coerced treatment de-escalations due to high vasopressor demand or complications was also comparable (iHTN+IAN n=15 (51.7%), IANonly n=7 (35.0%), OR 0.50 (95% CI 0.16–1.62), p=0.25) but the noradrenaline dose at which treatment was de-escalated was significantly lower after protocol modification (iHTN+IAN 1.31±0.63μg/kg/min, IANonly 0.73±0.24μg/kg/min, p<0.01). Patients with iHTN+IAN received a mean number of 4.7±2.5 CT perfusion scans during IAN, while patients with IANonly received 6.1±3.3 scans (p=0.12). Complications ascribable to intraarterial microcatheters were not different between groups (thrombosed microcatheter or thrombi surrounding the catheter in imaging iHTN+IAN n=6 (20.7%), IANonly n=5 (25.0%), OR 1.28 (95% CI 0.33–4.95), p=0.72; replacement of microcatheter due to previous catheter thrombosis iHTN+IAN n=5 (17.2%), IANonly n=3 (15.0%), OR 0.85 (95% CI 0.18–4.03), p=0.84; thromboembolic cerebral infarction iHTN+IAN n=3 (10.3%), IANonly n=1 (5.0%), OR 0.46 (95% CI 0.04–4.73), p=0.51; venous or arterial thromboembolism iHTN+IAN n=3 (10.3%), IANonly n=6 (30.0%), OR 3.71 (95% CI 0.80–17.16), p=0.09).
Figure 2.
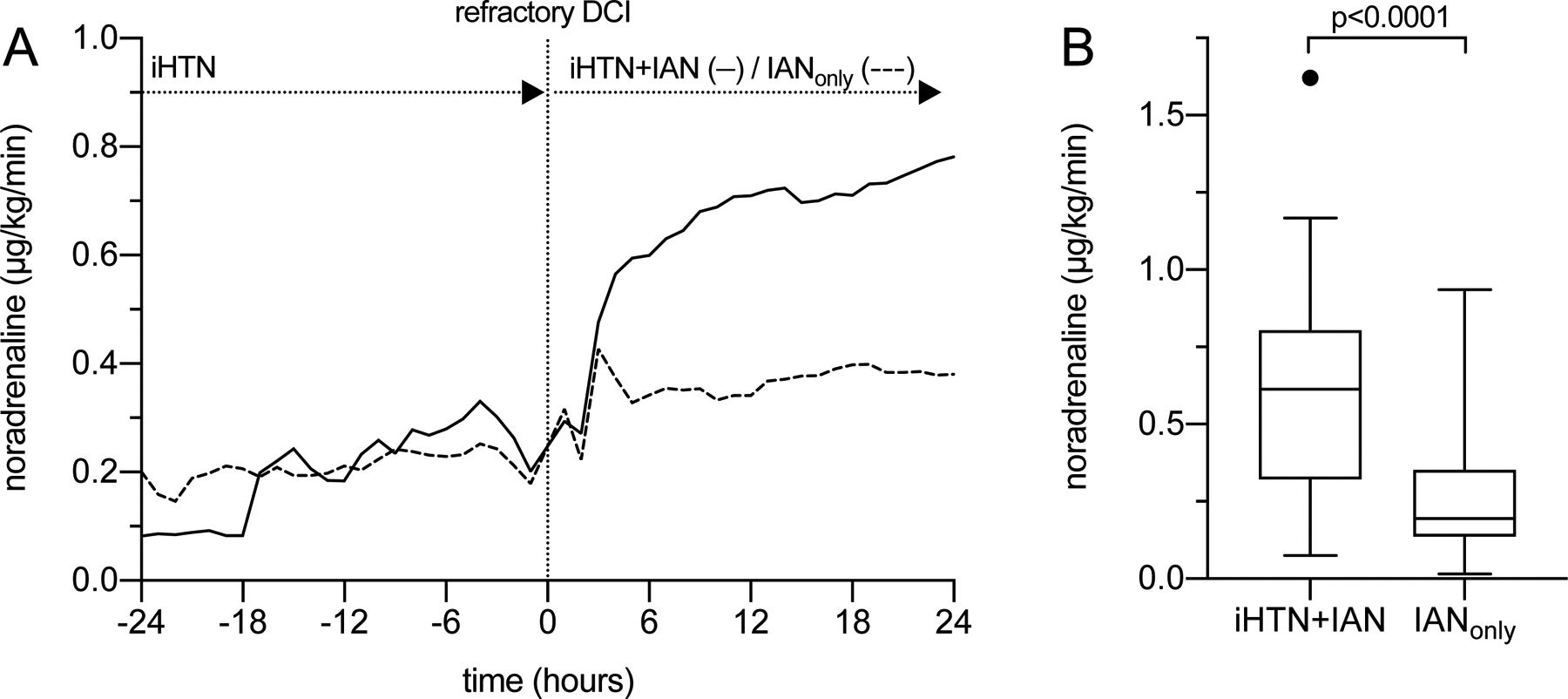
Primary outcome (noradrenaline demand).
(A) Noradrenaline requirements were strongly increased 24 hours after initiation of IAN in patients treated with iHTN+IAN (0.23±0.32 to 0.67±0.56μg/kg/min, Wilcoxon rank sum test, p<0.0001). With IANonly, demand was still elevated after initiation of IAN (0.21±0.17 to 0.36±0.30μg/kg/min, p<0.05) but the increase was more moderate than with iHTN+IAN (p<0.05). (B) Mean noradrenaline demand displayed as Tukey boxplot. The line in the middle of the box represents the median value, the box edges represent 25th and 75th percentiles, the whiskers represent all other values up to 1.5 times the interquartile range, outliers are shown as dots. Noradrenaline demand of the total treatment duration was significantly reduced with IANonly (iHTN+IAN 0.70±0.54μg/kg/min, IANonly 0.26±0.20μg/kg/min, p<0.0001).
Table 2.
Secondary outcome parameters.
| iHTN+IAN | IANonly | p-value | OR (95% CI) | |
|---|---|---|---|---|
| safety (noradrenaline) | ||||
| coerced treatment de-escalations | ||||
| n (%) patients with ≥1 de-escalation | 15 (51.7%) | 7 (35.0%) | 0.25 | 0.50 (0.16–1.62) |
| reduction (IAN or iHTN), n | 34 | 11 | ||
| termination (IAN or iHTN), n | 4 | 1 | ||
| noradrenaline dose triggering de-escalation, μg/kg/min | 1.31±0.63 | 0.73±0.24 | <0.01* | |
| minor complications | ||||
| n (%) patients with ≥1 minor | 14 (48.3%) | 3 (15.0%) | <0.05* | 0.19 (0.05–0.79) |
| complication | 13 | 3 | ||
| peripheral hypoperfusion, n | 3 | 1 | ||
| cardiac arrhythmia, n | ||||
| major complications | ||||
| n (%) patients with ≥1 major complication | 6 (20.7%) | 4 (20.0%) | 0.95 | 0.96 (0.23–3.95) |
| cardiac insufficiency, n | 1 | 0 | ||
| myocardial infarction, n | 0 | 0 | ||
| paralytic ileus or gut ischemia, n | 2 | 3 | ||
| acute kidney failure, n | 3 | 0 | ||
| SSC-CIP, n | 0 | 1 | ||
| CT perfusion images during IAN, n | 4.7±2.5 | 6.1±3.3 | 0.12 | |
| efficacy (rescue treatment) | ||||
| mean ptiO2, mmHg | 26.6±15.3 | 39.6±15.4 | <0.01* | |
| mean ICP, mmHg | 12.0±5.0 | 8.1±2.3 | <0.01* | |
| ≥1 treatment re-escalation, n (%) | 17 (58.6%) | 13 (65.0%) | 0.65 | 1.31 (0.40–4.26) |
| duration of IAN, days | 7.6±4.7 | 11.5±6.2 | <0.05* | |
| duration of iHTN, days | 17.7±7.2 | 11.6±8.9 | <0.01* | |
| hospitalization, days | 38.8±17.5 | 42.7±10.0 | 0.38 | |
| DCI related infarction | ||||
| n (%) patients with ≥1 infarction | 12 (41.1%) | 9 (45.0%) | 0.80 | 1.16 (0.37–3.66) |
| infarctions before iHTN, n | 0 | 3 | ||
| infarctions during iHTN, before IAN, n | 7 | 5 | ||
| infarctions during IAN, n | 5 | 1 | ||
| infarctions after IAN, n | 2 | 3 | ||
| Glasgow Outcome Scale 4–5 after 6 months, n (%) | 10 (40.0%) | 10 (55.6%) | 0.32 | 1.88 (0.55–6.39) |
Unpaired t-test or Mann-Whitney U test assessed differences between groups, as appropriate. Binary logistic regression analysis was used for comparisons of discrete variables.
significant p-value; DCI: delayed cerebral ischemia; ICP: intracranial pressure; iHTN+IAN: patients treated according to the classical protocol; IANonly: patients treated according to the modified protocol; SSC-CIP: secondary sclerosing cholangitis in critically ill patients.
Efficacy
In patients with IANonly, mean ptiO2 during the total treatment duration was significantly higher (iHTN+IAN 26.6±15.3mmHg, IANonly 39.6±15.4mmHg, p<0.01) (Figure 3A). ICP was lower with IANonly (iHTN+IAN 12.0±5.0mmHg, IANonly 8.1±2.3mmHg, p<0.01) (Figure 3B). Re-escalations of treatment (re-increase of nimodipine dose or pressure target) (iHTN+IAN n=17 (58.6%), IANonly n=13 (65.0%), OR 1.31 (95% CI 0.40–4.26), p=0.65) were observed in a comparable number of cases. With IANonly, the total duration of IAN was longer (iHTN+IAN 7.6±4.7 days, IANonly 11.5±6.2 days, p<0.05). Hospitalization time was similar (iHTN+IAN 38.8±17.5 days, IANonly 42.7±10.0 days, p=0.38). The overall occurrence of DCI related infarction was comparable (iHTN+IAN n=12 (41.1%), IANonly n=9 (45.0%), OR 1.16 (95% CI 0.37–3.66), p=0.80) as was clinical outcome 6 months after discharge with favorable Glasgow Outcome Scale in n=10 (40.0%) patients with iHTN+IAN and in n=10 (55.6%) patients with IANonly (OR 1.88 (95% CI 0.55–6.39), p=0.32). Four patients with IHTN+IAN and two patients with IANonly were lost to follow-up.
Figure 3.
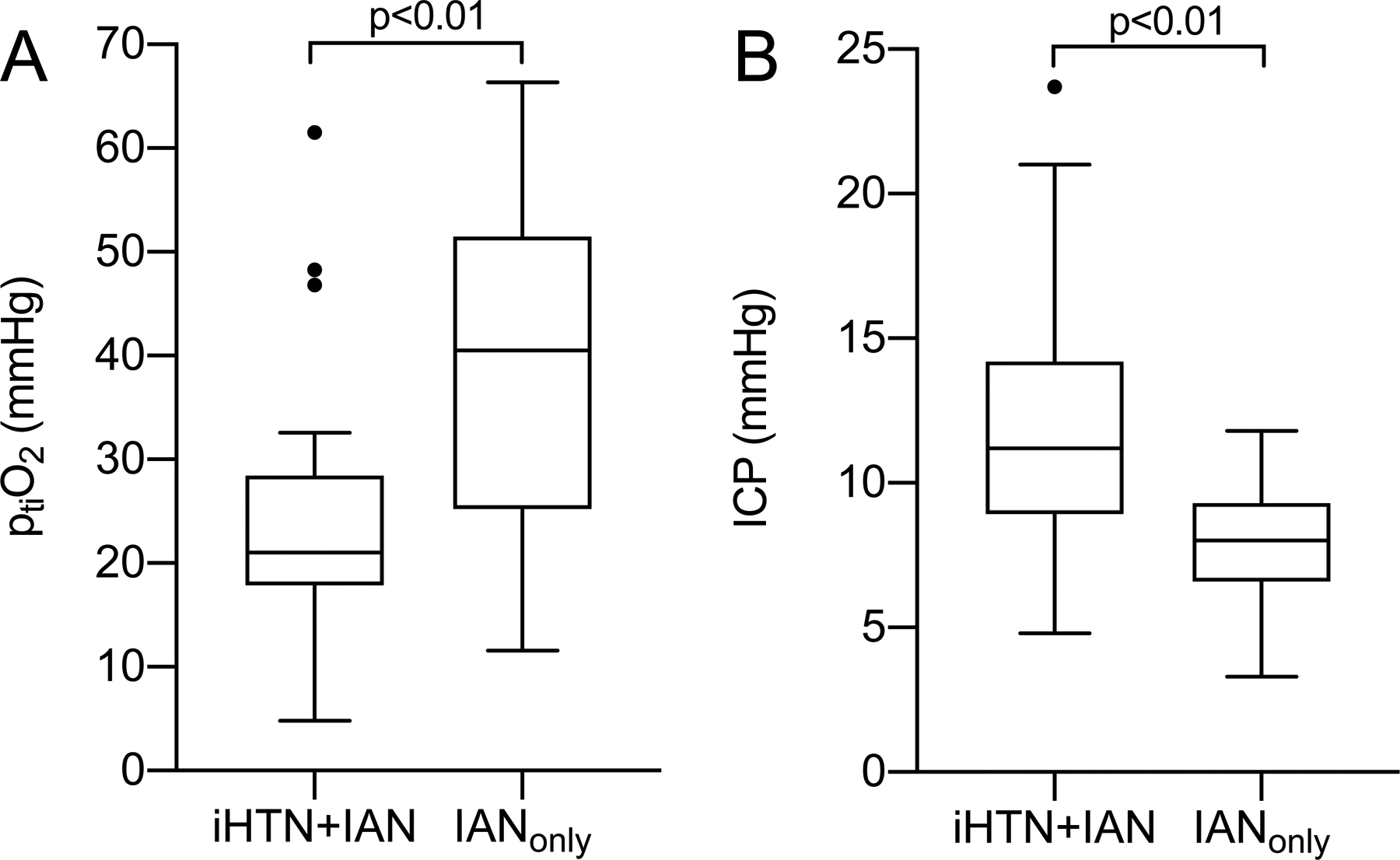
Brain tissue oxygenation and intracranial pressure.
(A) Brain tissue oxygenation (ptiO2) and (B) intracranial pressure (ICP) calculated as means over the total treatment duration (neuromonitoring iHTN+IAN n=21 (72.4%), IANonly n=11 (55.0%)) with significantly higher ptiO2 and lower ICP with IANonly.
Discussion
We recently changed our treatment algorithm in the context of refractory DCI and shifted emphasis of treatment towards intraarterial vasodilation with preemptive lowering of target pressure towards normotension. Protocol modification proved effective in terms of our primary outcome as mean noradrenaline demand was significantly reduced. This is particularly important as many patients in both groups exceeded our internal noradrenaline threshold of >0.5μg/kg/min, beyond which treatment reduction is discussed in a risk-benefit analysis of rescue treatment versus potential complications. Without present or anticipated complications, rescue treatment was oftentimes prioritized over absolute noradrenaline demand. Minor complications of noradrenaline treatment were less frequent but severe complications may nevertheless occur. In a stable situation with IAN and without infections, iHTN target may be a driving factor of noradrenaline demand and reduce minor complications if noradrenaline demand is consistently low (IANonly) rather than moderately elevated (iHTN+IAN). Major complications occurred with severe escalations of noradrenaline demand. It was our observation that severe escalations of vasopressor demand were triggered by concomitant infections, common in the critical care setting and unrelated to iHTN target, which may account for the lack of difference between both groups. Early, rigorous treatment of infection may therefore be equally important to avoid major complications as reducing baseline vasopressor demand by protocol adjustment.
Protocol modification was also associated with a longer total duration of IAN, and thus prolonged need for platelet aggregation inhibition, analgosedation and minimal handling. Continuous intraarterial spasmolysis in awake patients has been reported in few selected cases with focal neurological deficit only, while the majority of patients is either not eligible for wake-up trial or has a high risk of failure due to lack of orientation and manageability with an intraarterial microcatheter in place17. Thromboembolic complications were overall comparable but extracranial thromboses were more frequent with the new protocol occurring in about a third of cases, although this effect was not statistically significant. Of note, platelet aggregation inhibition may benefit cerebral perfusion on top of nimodipine’s vasodilating effect by preventing and/or treating cerebral microthrombosis as part of the presumed pathophysiology of DCI18. These potentially synergistic effects cannot be separated in our cohort but platelet aggregation inhibition has been emerging in recent years from a by-product of endovascular catheterization to being investigated as a separate form of DCI treatment19.
We also evaluated the efficacy of rescue treatment with IANonly as compared to iHTN+IAN and expected no difference. Respective variables confirmed similar treatment outcomes, including comparable rates of DCI related infarction and clinical outcome. Interestingly, intracranial pressure was lower and brain tissue oxygenation was significantly higher in patients with IANonly, suggesting that lower doses of noradrenaline and/or less aggressive hypertension may even have a beneficial effect on cerebral physiology. ICP is typically stabilized by cerebral autoregulation and should be largely unaffected by fluctuations in blood pressure. Higher concentrations of noradrenaline in the iHTN+IAN group could even be hypothesized to lead to lower ICP by noradrenaline-mediated vasoconstriction (analogously to the effects of ICP treatment by hyperventilation). However, impairment of autoregulation is considered part of the pathophysiology of DCI and may additionally be severely disturbed by intraarterial nimodipine infusion as shown by Hockel et al.20. Assuming that autoregulation could be similarly disturbed in this severely compromised patient cohort during nimodipine infusion, ICP may react more passively to blood pressure variations21. This may in turn lead to lower ICP with much lower blood pressure in patients treated with IANonly (ICP increase with high blood pressure during disturbed autoregulation > ICP decrease through noradrenaline).
Higher brain tissue oxygen values may result from improved oxygen delivery. Noradrenaline activates α−1 adrenergic receptors on blood vessels with vasoconstriction and blood pressure increase as part of the flight-reflex. Although the role of noradrenaline in cerebral α−1 activation is controversial, the current understanding is that this activation is much less pronounced in cerebral blood vessels under normal circumstances, preventing unwanted cerebral vasoconstriction. There is mounting evidence that the usually inert cerebral α−1 response to noradrenaline may be altered during aSAH with higher catecholamine sensitivity after brain hemorrhage, hypertension or hypoxia22. At the same time, it has been shown that α−1 mediated vasoconstriction of cerebral arterioles is stronger with increasing doses of noradrenaline and additionally altered with analgosedation23,24. While this reflex may avoid hyperperfusion during episodes of extreme adrenergic activation, it could counteract the intended effects of hemodynamic augmentation with vasopressors. Conversely, high noradrenaline doses may also impede nimodipine-mediated vasodilation. Intracellular Ca2+ is released as part of the activated α−1 cascade to initiate constriction of the smooth muscle cell layer in response to noradrenaline, counteracting nimodipine as a Ca2+-channel blocker in the same cellular compartment. Thus, by reducing pressure target and noradrenaline doses with IANonly, ptiO2 may increase through less pronounced adrenergic activation despite lower cerebral perfusion pressure. In our cohort with the modified protocol, this did not translate into a significant clinical benefit as measured by GOS after 6 months, although any effect would likely not be detectable due to the limited sample size; more subtle advantages, however, may only become apparent at a later follow-up or with detailed neuropsychological testing, as these deficits are frequently reported in SAH patients but are not regularly assessed.
The need to advance rescue treatment for DCI is pressing as convincing evidence for treatment escalation beyond iHTN is still lacking, resulting in highly divergent treatment algorithms for DCI, guided by clinical experience and opinion3; our own treatment protocol and modification are no exemption to this. At most, there is consensus to reduce oral nimodipine during episodes of hypotension5. Excessive increase of vasopressor requirements is a frequent observation with continuous intraarterial vasodilation and has triggered the development of alternative delivery strategies, including local application of vasodilating agents into the cerebrospinal fluid system10. Examples are nimodipine microparticles delivered via external ventricular drain (NEWTON trial) or nicardipine pellets placed onto brain cortex surgically (NicaPlant) but superiority of such approaches could not be demonstrated recently25,26. Prophylactic alternatives to oral nimodipine such as cilostazol may have similar effects on cerebral infarction but do not prevent the necessity for effective rescue strategies as measures of last resort27. A variety of intravenous or intraarterial, short- or longer-term applications of several vasodilators (e.g. nimodipine, nicardipine, milrinone, verapamil) is being utilized as rescue treatment without dominant evidence for any one approach that may justify introducing it as global standard treatment. Intravenous approaches lack the risk of catheterization but supposedly reach lower local concentrations of the vasodilating agent with a similar risk of hypotension and increased vasopressor demand28. Whether vasopressor support is lower with short-term intraarterial spasmolysis is plausible but has not been investigated so far. However, short-term spasmolysis may have to be repeated more frequently with the risk of infarction in between procedures and additional interventional risk29. With an established protocol and effective interdisciplinary collaboration, we believe that continuous intraarterial vasodilation with nimodipine is appropriate as treatment for refractory DCI due to the oftentimes prolonged disease course. Recognizing the limitations and risk profile, our study supports that iHTN can be reduced to a safety limit with nimodipine infusion with beneficial effects on vasopressor demand and cerebral physiology.
Limitations
The sample size, lack of neuromonitoring data in some patients and of a randomization protocol are the main limitations of our study. The study is not powered to detect differences in clinical outcome. Our diagnostic and treatment protocol was created in this form in 2014, with the introduction of invasive neuromonitoring and an extended DCI definition (CT perfusion deficit additional to clinical deterioration). All patients were treated according to this protocol, but the cohort with iHTN+IAN was collected before the IANonly cohort. We could not identify reasons for the later time point of DCI diagnosis in patients with IANonly, particularly contrary to the trend of earlier DCI diagnosis with invasive neuromonitoring in our center30. We are not aware of other diagnostic or treatment changes during this time, but further confounders cannot be completely excluded.
Conclusion
Aggressive rescue treatment for DCI with iHTN and IAN may require excessive vasopressor support. Preemptive lowering of pressure target after induction of IAN can dramatically reduce vasopressor requirements and improve cerebral oxygenation while lowering intracranial pressure. Our protocol modification may serve as incentive for centers active in endovascular rescue treatment to review options of protocol change to reduce vasopressor dosages.
Supplementary Material
Acknowledgements
The graphical abstract was created with biorender.com.
Disclosures
Marcel Aries is supported by the non-profit organization BrainBattle Foundation (HersenStrijd). Dr. Park discloses a grant by the National Institutes of Health (R21NS113055), which was not used for the funding of this study. Dr Wiesmann reports compensation from Stryker for consultant services.
Non-Standard Abbreviations and Acronyms:
- aSAH
aneurysmal subarachnoid hemorrhage
- DCI
delayed cerebral ischemia
- IAN
continuous intraarterial nimodipine
- ICP
intracranial pressure
- iHTN
induced hypertension
- ptiO2
brain tissue oxygenation
References
- 1.Springer MV, Schmidt JM, Wartenberg KE, Frontera JA, Badjatia N, Mayer SA. Predictors of global cognitive impairment 1 year after subarachnoid hemorrhage. Neurosurgery. 2009;65:1043–1050; discussion 1050–1041. doi: 10.1227/01.NEU.0000359317.15269.20 [DOI] [PubMed] [Google Scholar]
- 2.Gathier CS, Dankbaar JW, van der Jagt M, Verweij BH, Oldenbeuving AW, Rinkel GJ, van den Bergh WM, Slooter AJ, Group HS. Effects of Induced Hypertension on Cerebral Perfusion in Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage: A Randomized Clinical Trial. Stroke. 2015;46:3277–3281. doi: 10.1161/STROKEAHA.115.010537 [DOI] [PubMed] [Google Scholar]
- 3.Hollingworth M, Chen PR, Goddard AJ, Coulthard A, Soderman M, Bulsara KR. Results of an International Survey on the Investigation and Endovascular Management of Cerebral Vasospasm and Delayed Cerebral Ischemia. World Neurosurg. 2015;83:1120–1126 e1121. doi: 10.1016/j.wneu.2015.01.036 [DOI] [PubMed] [Google Scholar]
- 4.Brown RJ, Kumar A, McCullough LD, Butler K. A survey of blood pressure parameters after aneurysmal subarachnoid hemorrhage. Int J Neurosci. 2017;127:51–58. doi: 10.3109/00207454.2016.1138952 [DOI] [PubMed] [Google Scholar]
- 5.Diringer MN, Bleck TP, Claude Hemphill J, 3rd, Menon D, Shutter L, Vespa P, Bruder N, Connolly ES, Jr., Citerio G, Gress D, et al. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care. 2011;15:211–240. doi: 10.1007/s12028-011-9605-9 [DOI] [PubMed] [Google Scholar]
- 6.Connolly ES, Jr., Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, Hoh BL, Kirkness CJ, Naidech AM, Ogilvy CS, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/american Stroke Association. Stroke. 2012;43:1711–1737. doi: 10.1161/STR.0b013e3182587839 [DOI] [PubMed] [Google Scholar]
- 7.Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G, European Stroke O. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis. 2013;35:93–112. doi: 10.1159/000346087 [DOI] [PubMed] [Google Scholar]
- 8.Kimball MM, Velat GJ, Hoh BL, Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid H. Critical care guidelines on the endovascular management of cerebral vasospasm. Neurocrit Care. 2011;15:336–341. doi: 10.1007/s12028-011-9600-1 [DOI] [PubMed] [Google Scholar]
- 9.Weiss M, Conzen C, Mueller M, Wiesmann M, Clusmann H, Albanna W, Schubert GA. Endovascular Rescue Treatment for Delayed Cerebral Ischemia After Subarachnoid Hemorrhage Is Safe and Effective. Front Neurol. 2019;10:136. doi: 10.3389/fneur.2019.00136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kieninger M, Flessa J, Lindenberg N, Bele S, Redel A, Schneiker A, Schuierer G, Wendl C, Graf B, Silbereisen V. Side Effects of Long-Term Continuous Intra-arterial Nimodipine Infusion in Patients with Severe Refractory Cerebral Vasospasm after Subarachnoid Hemorrhage. Neurocrit Care. 2018;28:65–76. doi: 10.1007/s12028-017-0428-1 [DOI] [PubMed] [Google Scholar]
- 11.Kieninger M, Gruber M, Knott I, Dettmer K, Oefner PJ, Bele S, Wendl C, Tuemmler S, Graf B, Eissnert C. Incidence of Arterial Hypotension in Patients Receiving Peroral or Continuous Intra-arterial Nimodipine After Aneurysmal or Perimesencephalic Subarachnoid Hemorrhage. Neurocrit Care. 2019. doi: 10.1007/s12028-019-00676-w [DOI] [PubMed] [Google Scholar]
- 12.Gathier CS, van den Bergh WM, van der Jagt M, Verweij BH, Dankbaar JW, Muller MC, Oldenbeuving AW, Rinkel GJE, Slooter AJC, Group HS. Induced Hypertension for Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage: A Randomized Clinical Trial. Stroke. 2017. doi: 10.1161/STROKEAHA.117.017956 [DOI] [PubMed] [Google Scholar]
- 13.Albanna W, Weiss M, Muller M, Brockmann MA, Rieg A, Conzen C, Clusmann H, Hollig A, Schubert GA. Endovascular Rescue Therapies for Refractory Vasospasm After Subarachnoid Hemorrhage: A Prospective Evaluation Study Using Multimodal, Continuous Event Neuromonitoring. Neurosurgery. 2017. doi: 10.1093/neuros/nyw132 [DOI] [PubMed] [Google Scholar]
- 14.von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP, Initiative S. Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. BMJ. 2007;335:806–808. doi: 10.1136/bmj.39335.541782.AD [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney inter, Suppl. 2012;2: 1–138. [Google Scholar]
- 16.Vergouwen MD, Vermeulen M, van Gijn J, Rinkel GJ, Wijdicks EF, Muizelaar JP, Mendelow AD, Juvela S, Yonas H, Terbrugge KG, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke. 2010;41:2391–2395. doi: 10.1161/STROKEAHA.110.589275 [DOI] [PubMed] [Google Scholar]
- 17.Pala A, Schneider M, Brand C, Pedro MT, Ozpeynirci Y, Schmitz B, Wirtz CR, Kapapa T, Konig R, Braun M. The evolution of invasive cerebral vasospasm treatment in patients with spontaneous subarachnoid hemorrhage and delayed cerebral ischemia-continuous selective intracarotid nimodipine therapy in awake patients without sedation. Neurosurg Rev. 2018. doi: 10.1007/s10143-018-0986-5 [DOI] [PubMed] [Google Scholar]
- 18.Snyder MH, Ironside N, Kumar JS, Doan KT, Kellogg RT, Provencio JJ, Starke RM, Park MS, Ding D, Chen CJ. Antiplatelet therapy and delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. J Neurosurg. 2021:1–13. doi: 10.3171/2021.7.JNS211239 [DOI] [PubMed] [Google Scholar]
- 19.Ditz C, Machner B, Schacht H, Neumann A, Schramm P, Tronnier VM, Kuchler J. Effects of post-interventional antiplatelet therapy on angiographic vasospasm, delayed cerebral ischemia, and clinical outcome after aneurysmal subarachnoid hemorrhage: a single-center experience. Neurosurg Rev. 2021;44:2899–2912. doi: 10.1007/s10143-021-01477-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hockel K, Diedler J, Steiner J, Birkenhauer U, Ernemann U, Schuhmann MU. Effect of Intra-Arterial and Intravenous Nimodipine Therapy of Cerebral Vasospasm After Subarachnoid Hemorrhage on Cerebrovascular Reactivity and Oxygenation. World Neurosurg. 2017;101:372–378. doi: 10.1016/j.wneu.2017.02.014 [DOI] [PubMed] [Google Scholar]
- 21.Bouma GJ, Muizelaar JP, Bandoh K, Marmarou A. Blood pressure and intracranial pressure-volume dynamics in severe head injury: relationship with cerebral blood flow. J Neurosurg. 1992;77:15–19. doi: 10.3171/jns.1992.77.1.0015 [DOI] [PubMed] [Google Scholar]
- 22.Purkayastha S, Raven PB. The functional role of the alpha-1 adrenergic receptors in cerebral blood flow regulation. Indian J Pharmacol. 2011;43:502–506. doi: 10.4103/0253-7613.84950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King BD, Sokoloff L, Wechsler RL. The effects of l-epinephrine and l-norepinephrine upon cerebral circulation and metabolism in man. J Clin Invest. 1952;31:273–279. doi: 10.1172/JCI102603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Myburgh JA, Upton RN, Grant C, Martinez A. The cerebrovascular effects of adrenaline, noradrenaline and dopamine infusions under propofol and isoflurane anaesthesia in sheep. Anaesth Intensive Care. 2002;30:725–733. doi: 10.1177/0310057X0203000602 [DOI] [PubMed] [Google Scholar]
- 25.Carlson AP, Hanggi D, Wong GK, Etminan N, Mayer SA, Aldrich F, Diringer MN, Schmutzhard E, Faleck HJ, Ng D, et al. Single-Dose Intraventricular Nimodipine Microparticles Versus Oral Nimodipine for Aneurysmal Subarachnoid Hemorrhage. Stroke. 2020;51:1142–1149. doi: 10.1161/STROKEAHA.119.027396 [DOI] [PubMed] [Google Scholar]
- 26.Akbik F, Waddel H, Jaja BNR, Macdonald RL, Moore R, Samuels OB, Sadan O. Nicardipine Prolonged Release Implants for Prevention of Delayed Cerebral Ischemia after Aneurysmal Subarachnoid Hemorrhage: A Meta-Analysis. J Stroke Cerebrovasc Dis. 2021;30:106020. doi: 10.1016/j.jstrokecerebrovasdis.2021.106020 [DOI] [PubMed] [Google Scholar]
- 27.Senbokuya N, Kinouchi H, Kanemaru K, Ohashi Y, Fukamachi A, Yagi S, Shimizu T, Furuya K, Uchida M, Takeuchi N, et al. Effects of cilostazol on cerebral vasospasm after aneurysmal subarachnoid hemorrhage: a multicenter prospective, randomized, open-label blinded end point trial. J Neurosurg. 2013;118:121–130. doi: 10.3171/2012.9.JNS12492 [DOI] [PubMed] [Google Scholar]
- 28.Seker F, Hesser J, Brockmann MA, Neumaier-Probst E, Groden C, Schubert R, Brockmann C. Pharmacokinetic Modeling of Intra-arterial Nimodipine Therapy for Subarachnoid Hemorrhage-Related Cerebral Vasospasm. Clin Neuroradiol. 2017;27:199–203. doi: 10.1007/s00062-015-0464-1 [DOI] [PubMed] [Google Scholar]
- 29.Andereggen L, Beck J, Z’Graggen WJ, Schroth G, Andres RH, Murek M, Haenggi M, Reinert M, Raabe A, Gralla J. Feasibility and Safety of Repeat Instant Endovascular Interventions in Patients with Refractory Cerebral Vasospasms. AJNR Am J Neuroradiol. 2017;38:561–567. doi: 10.3174/ajnr.A5024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veldeman M, Albanna W, Weiss M, Conzen C, Schmidt TP, Schulze-Steinen H, Wiesmann M, Clusmann H, Schubert GA. Invasive neuromonitoring with an extended definition of delayed cerebral ischemia is associated with improved outcome after poor-grade subarachnoid hemorrhage. J Neurosurg. 2020:1–8. doi: 10.3171/2020.3.JNS20375 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.