

Summary.
The mechanisms of volatile anesthetic action remain among the most perplexing mysteries of medicine. Across phylogeny, volatile anesthetics selectively inhibit mitochondrial complex I and they also depress presynaptic excitatory signaling. To explore how these effects are linked, we studied isoflurane effects on presynaptic vesicle cycling and ATP levels in hippocampal cultured neurons from wildtype and complex I mutant (Ndufs4(KO)) mice. To bypass complex I, we measured isoflurane effects on anesthetic sensitivity in mice expressing NADH dehydrogenase (NDi1). Endocytosis in physiologic concentrations of glucose was delayed by effective behavioral concentrations of isoflurane in both wildtype (τ (unexposed) 44.8+/−24.2s; τ (exposed) 116.1+/−28.1s; p<0.01) and Ndufs4(KO) cultures( τ (unexposed) 67.6+/−16.0s; τ (exposed) 128.4+/−42.9s; p=0.028). Increasing glucose, to enhance glycolysis and increase ATP production, led to maintenance of both ATP levels and endocytosis (τ (unexposed) 28.0+/−14.4; τ (exposed) 38.2+/−5.7; reducing glucose worsened ATP levels and depressed endocytosis (τ (unexposed) 85.4+/−69.3; τ (exposed) >1000; p<0.001). The block in recycling occurred at the level of reuptake of synaptic vesicles into the presynaptic cell. Expression of NDi1 in wildtype mice caused behavioral resistance to isoflurane for tail clamp response (EC50 Ndi1(−) 1.27+/−0.14%; Ndi1(+) 1.55+/−0.13%) and halothane (EC50 Ndi1(−) 1.20+/−0.11%; Ndi1(+) 1.46+/−0.10%); expression of NDi1 in neurons improved hippocampal function, alleviated inhibition of presynaptic recycling and increased ATP levels during isoflurane exposure. The clear alignment of cell culture data to in vivo phenotypes of both isoflurane sensitive and resistant mice indicates that inhibition of mitochondrial complex I is a primary mechanism of action of volatile anesthetics.
Keywords: Volatile anesthetics, mitochondria, complex I, ATP, presynapse, endocytosis, exocytosis, mouse
ETOC Blurb
Jung et al. demonstrate that in neuronal cultures isoflurane decreases presynaptic ATP levels and inhibits presynaptic endocytosis after intense neuronal stimulation. Both effects result from inhibition of mitochondrial complex I. ATP dependent failure of presynaptic endocytosis is one primary mechanism of action of isoflurane.
Introduction.
For over 170 years, the mechanisms of action for volatile anesthetics (VAs) have remained among the most perplexing mysteries of medicine. Unlike the situation with parenteral anesthetics 1,2, the molecular mechanisms by which VAs function have not been clearly identified 3–6. Several molecular targets have been proposed for VAs 7,8. However, animal models that have tested the roles of most targets by mutating candidate proteins have either failed to show an effect or shown only modest changes in VA sensitivity 9,10.
In contrast, disruption of complex I of the mitochondrial electron transport chain (ETC) causes hypersensitivity to VAs across the animal kingdom, including humans, nematodes, flies, and mice 11–15. Previous work in isolated nematode 16–18 and mammalian mitochondria 19–21 revealed that, in contrast to ETC complexes II-IV, complex I was significantly inhibited by VAs. In mitochondria isolated from mitochondrial mutants hypersensitive to volatile anesthetics, complex I function was also hypersensitive to these agents 18,22,23. Complex I is the entry site for electron transport from NADH to oxygen 24,25. It is the rate limiting step in the electron transport 26 indicating that inhibition of complex I may have dramatic effects on energy production.
Mice in which the complex I protein NDUFS4 is knocked out show the largest increase reported in VA sensitivity in a mammal 13. The EC50s for isoflurane and halothane are one-third that of controls 13 despite the mice displaying no overt disease at this age (≤P30) 27. The evolutionary dependence of VA sensitivity on complex I implies that an ancient mechanism is at hand, linking mitochondrial function to synaptic silencing in the presence of VAs.
Several laboratories have shown that presynaptic function of excitatory neurons is inhibited by VAs 28–33. Our studies with wildtype and Ndufs4(KO) mice showed that mitochondrial function in glutamatergic neurons determined VA sensitivity to tail clamp and loss of righting reflex 23,33. Furthermore, presynaptic frequencies of excitatory signaling were inhibited by VAs at concentrations approximating the whole animal EC50s of wildtype and Ndufs4(KO) mice 23,33. The sensitivity of presynaptic frequencies to isoflurane increased when neurons were made dependent on mitochondrial respiration.
Synaptic neurotransmitter recycling, in particular endocytosis, is uniquely dependent on ATP availability 34,35. Since VAs are known potent inhibitors of mitochondrial complex I 18–21, its inhibition may underlie the presynaptic changes caused by VAs. We hypothesized that VAs acutely inhibit mitochondrial ATP production which, in turn, decreases presynaptic ATP and disrupts endocytosis. If this relationship exists, then both presynaptic ATP levels and endocytosis should be sensitive to approximately equal concentrations of VA. These phenomena should occur at concentrations of isoflurane that match the whole animal sensitivities of both wild type and mutant animals.
We hypothesized that isoflurane would inhibit endocytosis and ATP production at a concentration approximating the whole animal EC95 of wild type. In addition, a similar inhibition should be seen in Ndufs4(KO) at lower concentrations of isoflurane matching the EC95 of the mutant. To establish that complex I is a primary component controlling VA sensitivity, a change in complex I leading to resistance would be invaluable. The recent availability of a genetic model that can bypass complex I (NDi1)36,37 as an electron donor allows direct analysis of the mitochondrial effects of isoflurane on both whole animal behavior and endocytosis.
The goals of this work are to establish the importance of complex I in anesthetic response and to decipher the molecular mechanism by which mitochondrial inhibition by VAs disrupts synaptic function. The results indicate that VAs cause neuronal quiescence by inhibiting mitochondrial complex I, creating an acute, reversible energy failure in the presynaptic cell that inhibits synaptic vesicle endocytosis. We propose inhibition of complex I as a primary mechanism of action for volatile anesthetics.
Results.
Baseline Energetics
Ndufs4(KO) mice lack a subunit of complex I of the mitochondrial electron transport chain (Figure S1A). Multiple studies have shown that mitochondria from Ndufs4(KO) mice have a lower maximal complex I-dependent function than do wildtype controls 23,27,38. However, the unstimulated baseline metabolic rates of CNS cells isolated from the two genotypes have not been reported. We measured baseline and uncoupled respiratory rates in wildtype controls and Ndufs4(KO) hippocampal cells cultured in 1.5mM glucose plus 10mM pyruvate and found them to be indistinguishable (Figure S1B). When neurons are unstimulated, respiration devoted to ATP synthesis was identical between the two genotypes.
Synaptic Function in Physiologic Conditions
In contrast to spontaneous firing, presynaptic activities of CA1 hippocampal neurons were previously shown to be depressed following strong electrical stimulation in both Ndufs4(KO) and wildtype controls at concentrations correlating with those affecting behavior in whole animals 23,33. The depression in both genotypes was profoundly worsened in the presence of isoflurane. We questioned which processes at the presynapse might explain this difference. We used VGlut1-pHluorin, which quenches in an acidic environment, i.e. within synaptic vesicles, and fluoresces in a neutral milieu such as the extracellular space (Figure 1A). It is commonly used to trace synaptic vesicle recycling (Figure 1B) 30,39,40
Figure 1.
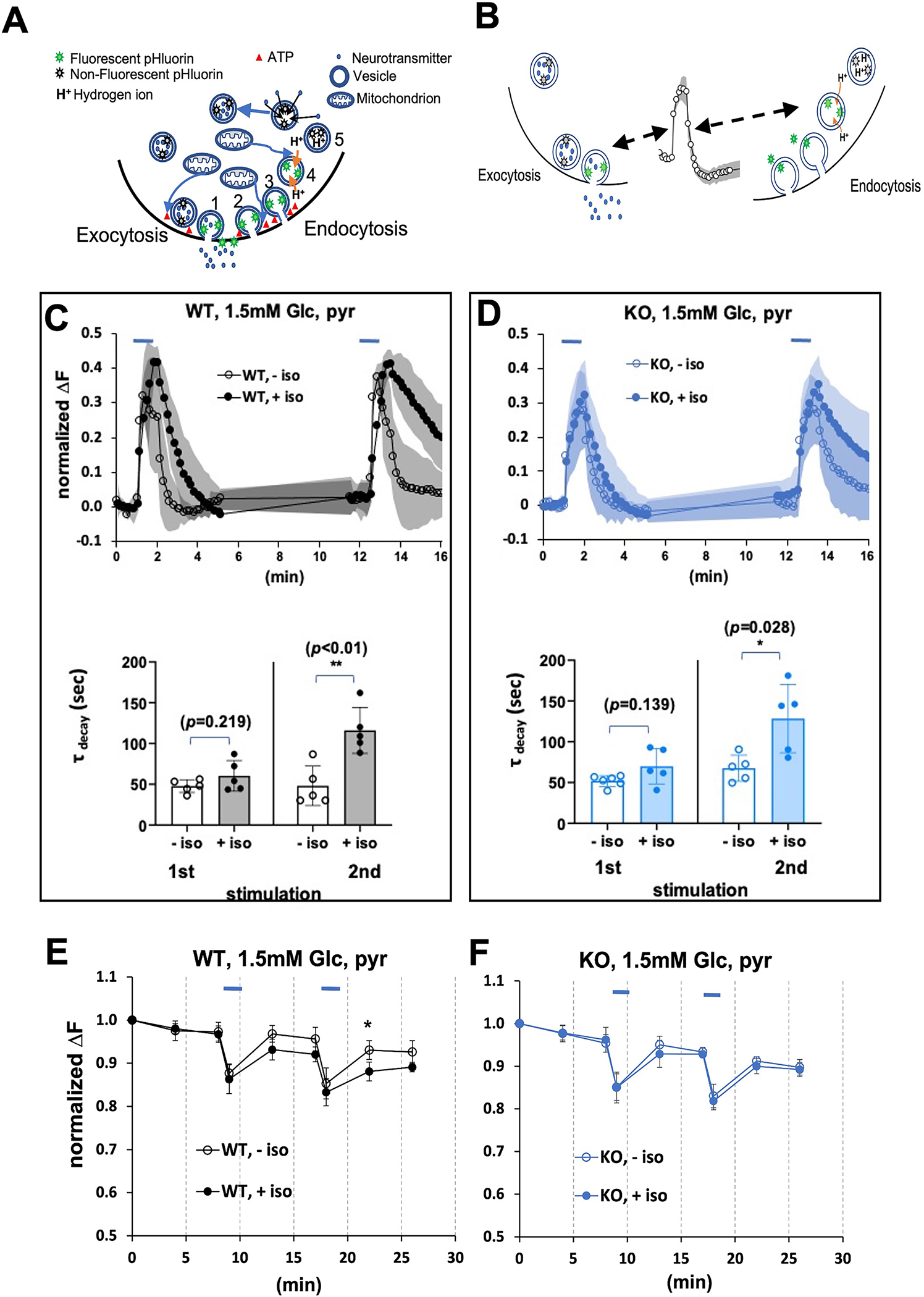
A. A simplified depiction of pHluorin fluorescence to trace synaptic vesicle recycling. Synaptic transmission at individual boutons is the physiological result of exocytosis (release, 1 in figure) of neurotransmitter into the synaptic cleft. This is followed by endocytosis (reuptake of vesicles, 2 in figure) and vesicle scission (3 in figure) into the presynaptic neuron. Following completion of endocytosis, hydrogen ions are pumped into the vesicles (4 in figure) which allows neurotransmitter to re-enter the vesicles (5 in figure). VGLUT1-pHluorin does not fluoresce (grey stars) in an acidic environment, i.e. within the acidified synaptic vesicle, and fluoresces (green stars) when exposed to the neutral extracellular milieu 39. Docking of vesicles, reuptake of vesicles and re-acidification of vesicles are all ATP-(red triangles) dependent processes. B. Fluorescence of VGLUT1-pHluorin during synaptic transmission. Synaptic transmission was assessed in neurons expressing a pH-sensitive green fluorescent protein (GFP) targeted to synaptic vesicles (VGLUT1-pHluorin, diagram green stars) as in 1b. The rise in fluorescence during electrical stimulation primarily reflects exocytosis (pHluorin fluoresces upon exposure to the pH-neutral extracellular space), while the downstroke reflects endocytosis and synaptic vesicle reacidification (pHluorin loses fluorescence in an acidic environment). C,D. Exocytosis and endocytosis following electrical field stimulation (10Hz*60s, blue bars) of hippocampal cultures in 1.5mM glucose, supplemented with pyruvate. In figures 2–5,7 the mean change in fluorescence (ΔF) is always normalized to the size of the total pool of VGLUT-pHluorin in each bouton (determined by measuring the fluorescence after application of 50 mM NH4Cl following the second stimulation). C. Upper panel. Wild type cells electrically stimulated in the absence (black open circles) or presence (black filled circles) of 0.74mM isoflurane. D. Upper panel. Ndufs4(KO) cells electrically stimulated in the absence (blue open circles) or presence (blue filled circles) of 0.25mM isoflurane. Lower panels. In this and subsequent figures, tau is the exponential constant for a first order fit for the decay curve. Upon repeated stimulation, tau increased (endocytosis was delayed) in both genotypes with isoflurane exposure at their respective whole animal 2XEC50s. The colors (black, wildtype; blue, Ndufs4(KO)) and symbol designations (blue bar, electrical stimulus: open circles, (−) isoflurane; filled circles, (+) isoflurane) are the same in figures 2–6. E. Synaptic transmission decreases ATP at the WT synapse in 1.5 mM glucose. Hippocampal cultures expressing the ATP FRET sensor and mCherry-synaptophysin (to identify synaptic boutons) were subjected to electrical field stimulation as in c. in the presence of 1.5mM glucose supplemented with 10mM pyruvate. Individual synaptic boutons were imaged, and the ATP FRET signal was quantified 54. Electrical stimulation produces a decrease in ATP that fails to fully recover in wildtype with or without isoflurane (isoflurane, 0.74mM). However, the decrease in ATP is greater in the presence of isoflurane. F. Synaptic transmission decreases ATP at the Ndufs4(KO) synapse in 1.5 mM glucose. Electrical stimulation produces a decrease in ATP that fails to fully recover in Ndufs4(KO) with or without isoflurane. (isoflurane, 0.25mM). ATP levels recover to ~90% of baseline over 5–7 minutes following stimulation. In this and all subsequent figures, * indicates p<0.05, ** indicates p<0.01, and *** indicates p<0.001 when comparing results with and without isoflurane.
We first measured stimulated synaptic vesicle recycling in primary cultures of hippocampal cells from wildtype mice in the presence of approximately physiologic concentrations of glucose (1.5mM) 41,42 (Figure 1C, upper left panel). For these and subsequent experiments, we supplemented cells with the mitochondrial specific substrate, pyruvate, to support mitochondrial function as we changed the potential impact of glycolysis. In the absence of isoflurane, we measured rapid exocytosis (rise time of VGLUT1-pHluorin fluorescence) and endocytosis (return to baseline following stimulation). In the presence of isoflurane (0.74mM; approximately 2XEC50 corresponding to about 3% isoflurane in air. In all cases, EC50 refers to the concentrations preventing a response to tail clamp), in wildtype neurons exocytosis was not affected compared to unexposed neurons. Upon repeat stimulation there was a delay in return to baseline of the pHluorin signal (τ (unexposed) 44.8 ± 24.2s (n=5); τ (exposed) 116.1 ± 28.1s (n=5); p<0.01) indicating a defect in endocytosis (Figure 1c, lower left panel).
If inhibition of mitochondrial complex I function was the mechanism by which endocytosis was affected in the wild type, then we hypothesized that lower isoflurane concentrations would achieve the same effect in Ndufs4(KO) neurons since complex I function is already compromised. We exposed cultures from Ndufs4(KO) to their behavioral (tail clamp) 2XEC50 of isoflurane (0.25mM corresponding to ~0.8% isoflurane) and repeated the paradigm (Figure 1D, upper panel). There was a defect in endocytosis in the mutant in 0.25mM isoflurane (Figure 1D, lower panel, τ (unexposed) 67.6+/−16.0s (n=5); τ (exposed) 128.4+/− 42.9s (n=5); p=0.028). In the KO neurons at 0.25mM isoflurane, endocytosis resembled that of the wildtype at 0.74mM isoflurane. No defect in endocytosis was seen in the wildtype neurons at 0.25mM isoflurane (not shown). We compared the pHluorin response with FRET analysis to determine whether ATP levels differed between the two genotypes. At their respective 2XEC50s, we observed a decrease in ATP levels in both genotypes during stimulation, which did not fully recover once stimulation ceased (Figures 1E,F). There was a significant difference in ATP levels in the wildtype between cultures exposed and unexposed to isoflurane following the second stimulation (t=22min). The difference between ATP levels in exposed and unexposed neurons was not seen in the mutant although levels of ATP were significantly decreased in both compared to baseline (p<0.01). Since ATP levels were decreased and endocytosis was delayed when isoflurane was applied to the cultures, we next tested conditions which would support or limit ancillary ATP production by glycolysis to isolate the effects of isoflurane on mitochondrial function and endocytosis.
Synaptic Function in High Glucose
To determine if decreased ATP levels contribute to the mechanism underlying isoflurane inhibition of endocytosis, we tested the effects of increased ATP availability. To increase ATP availability in the presence of mitochondrial inhibition by isoflurane, we increased the glucose concentration beyond normal physiologic levels to 30mM glucose. At this concentration, increased glycolytic capacity should exceed physiologic levels 34. We then measured stimulated synaptic vesicle recycling in the wildtype in the presence and absence of isoflurane (Figure 2A, upper panel). In the absence of isoflurane, in the wildtype both exocytosis and endocytosis were brisk as in 1.5mM glucose. In the presence of isoflurane (0.74mM) the inhibition of endocytosis seen after the second stimulation in physiologic glucose (1.5mM) was rescued by 30mM glucose (Figure 2A, lower panel), τ (unexposed) 28.0+/− 14.4 (n=11); τ (exposed) 38.2+/− 5.7 (n=9); p=0.061).
Figure 2. Synaptic Function in High Glucose.
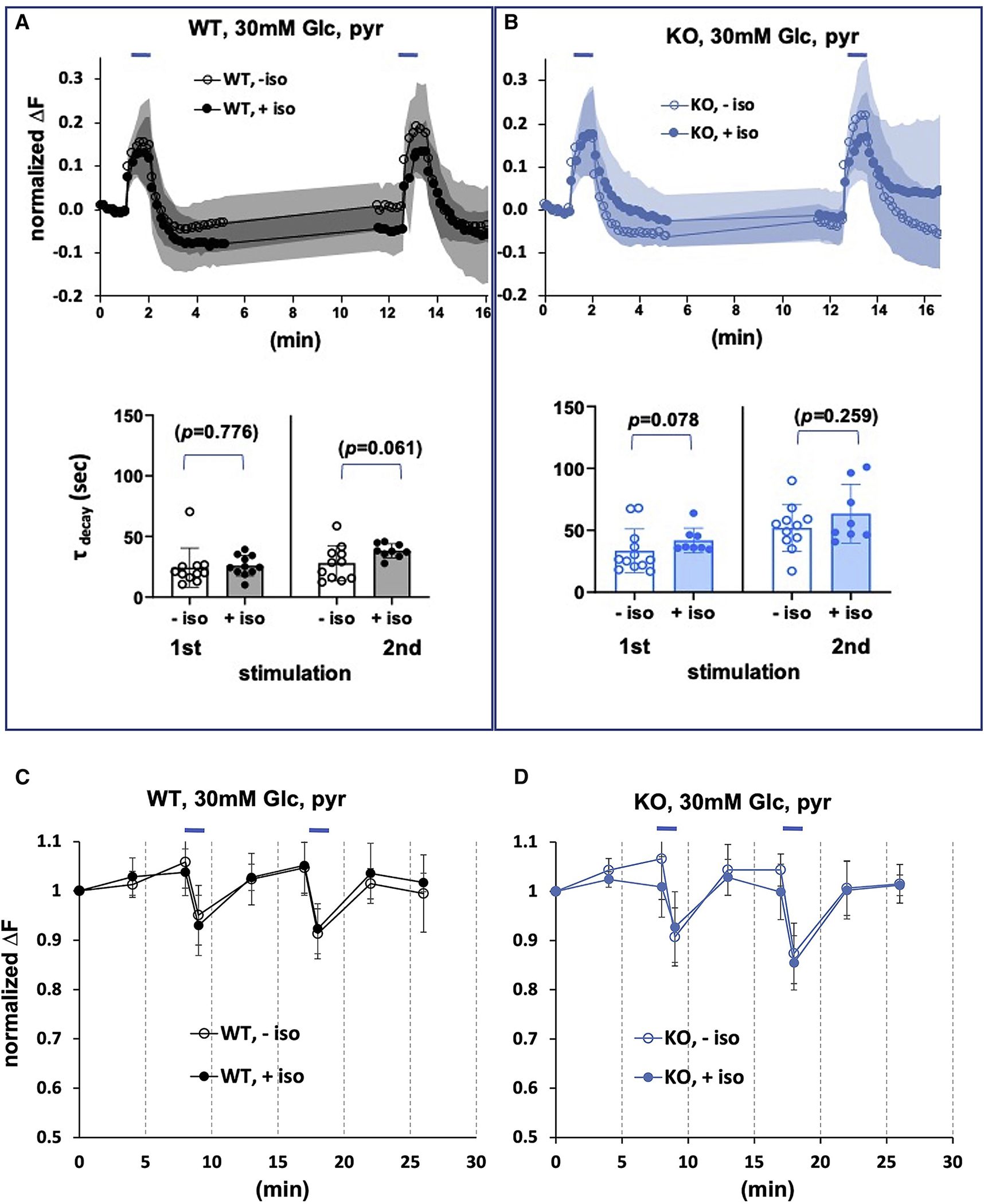
A. Exocytosis and endocytosis following electrical field stimulation of WT hippocampal cultures in the presence of 30mM glucose, supplemented with pyruvate. Upper panel. Wild type cells electrically stimulated (10Hz*60s, blue bars) in the absence or presence of isoflurane (0.74mM). B. Exocytosis and endocytosis of Ndufs4(KO) hippocampal cultures in the presence of 30mM glucose, supplemented with pyruvate. Upper panel. Ndufs4(KO) cells electrically stimulated (10Hz*60s, blue bars) in the absence or presence of isoflurane (0.25mM). Lower panels. There are no increases in tau for either genotype upon isoflurane exposure after the second stimulus (compare to results in 1.5mM glucose (Figures 1c,d). C,D. Synaptic ATP fully recovers in 30mM glucose following stimulation in the presence of isoflurane. Hippocampal cultures expressing the ATP FRET sensor as described in Figure 1E. C. Electrical stimulation produces a transient decrease in FRET signal (ATP) that fully recovers in wildtype cultures in absence or presence of isoflurane (0.74mM). D. Electrical stimulation produces a transient decrease in FRET signal (ATP) that fully recovers in Ndufs4(KO) cultures in absence or presence of isoflurane (0.25mM).
Rescuing the decreased ATP levels seen in Figure 1 might also alleviate the endocytosis defect seen in the KO neurons. In the absence of isoflurane, both exocytosis and endocytosis were normal in the KO (Figure 2B, upper panel). At 0.25mM isoflurane, there was no failure in endocytosis in the mutant, as evidenced by the return of fluorescence to baseline after both the first and second stimulation (Figure 2B, lower panel, τ (unexposed) 52.0+/− 18.8 (n=11); τ (exposed) 63.4+/− 23.8 (n=8); p=0.259). We examined ATP levels with FRET analysis to determine whether ATP levels differed between the exposed and unexposed neurons in either genotype under these conditions. At their respective 2XEC50s, ATP levels decreased in both genotypes during stimulation, but fully recovered (unlike at 1.5mM glucose, Figure 2b) (Figures 2C,D).
Synaptic and Mitochondrial Function with Restricted Glucose
If ATP production by glycolysis supports recycling in the face of mitochondrial inhibition by isoflurane, restriction of glycolysis to endogenous reserves should augment the endocytosis defect. We removed glucose from the perfusate to restrict glycolysis to endogenous intracellular stores (with added pyruvate to support mitochondrial function). Under these conditions, in the absence of isoflurane, a normal first peak (both exocytosis and endocytosis) was seen in response to electrical stimulation in the wildtype cells (Figure 3A, upper panel), while in the second peak, endocytosis was delayed. When isoflurane was added at the wildtype EC95 (0.74mM) to the neuronal cultures, endocytosis failed completely (Figure 3A, lower panel, First peak, τ (unexposed) 48+/− 23 (n=11); τ (exposed) 223+/− 147 (n=12); p<0.001; Second peak τ (unexposed) 85.4+/− 69.3 (n=10); τ (exposed) >1000 (n=10); p<0.001). A similar pattern was seen when the wildtype was exposed to ~1.4XEC50, (0.5mM isoflurane, corresponding to about 2% isoflurane) (Figure 3A, upper and lower panels). The pattern seen in the KO at 0.25mM isoflurane was also similar to that seen in the wildtype both at 0.5mM and 0.74mM isoflurane (Figure 3B, upper and lower panels). There was a defect in endocytosis in the absence of isoflurane in the second peak and a complete failure of endocytosis in the presence of isoflurane (Figure 3B, lower panel, First peak, τ (unexposed) 54+/− 43 (n=13); τ (exposed) 400+/− 260 (n=10); p<0.001; Second peak τ (unexposed) 316+/− 218 (n=13); τ (exposed) >1000 (n=10); p<0.001). No defect in endocytosis was seen in the wildtype neurons at 0.25mM isoflurane (not shown).
Figure 3. Synaptic and Mitochondrial Function following electrical field stimulation of hippocampal cultures with no exogenous glucose, supplemented with pyruvate.
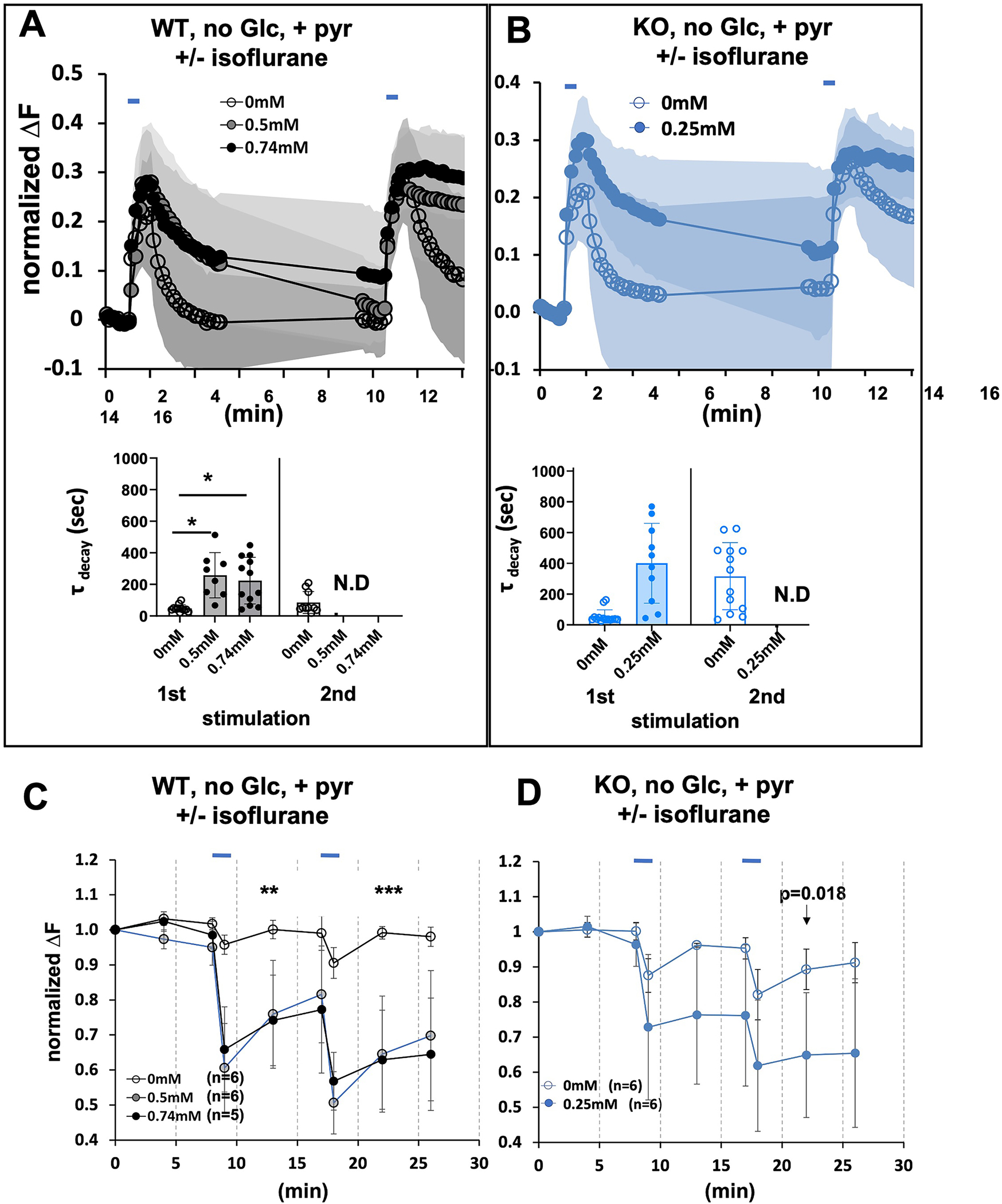
A. Upper Panel. Wild type cells electrically stimulated in the absence or presence of isoflurane (0.5mM and 0.74mM). B. Upper Panel. Ndufs4(KO) cells stimulated in the absence or presence of isoflurane (0.25mM). Lower A,B panels. There is an increase in decay time after the first stimulation in isoflurane for all isoflurane exposed cultures. Tau could not be calculated after the second stimulation for either (complete failure of endocytosis) genotype in the presence of isoflurane (complete failure of endocytosis). C,D. The presence of isoflurane decreases the ATP FRET signal at the synapse following stimulation. Hippocampal cultures, in the absence of exogenous glucose but with added pyruvate to support mitochondrial respiration as described in Figure 2. C. Electrical stimulation of wildtype cells in the absence of isoflurane produces a reversible decrease in ATP. In the presence of isoflurane (0.5mM and 0.74mM) the FRET signal dramatically decreases and fails to recover. D. Electrical stimulation of Ndufs4(KO) cells in the absence of isoflurane produces a modest decrease in ATP. In the presence of isoflurane (0.25mM) the FRET signal dramatically decreases and fails to recover.
As noted in the Methods, imaging cultures at a rate of once per 3 seconds (0.3 Hz) led to complete a failure of the fluorescent signal to return to baseline following the second stimulus, independent of experimental isoflurane treatment. Since the focus of these experiments is to isolate the effects of isoflurane on endocytosis in the face of significant neuronal activity, we used 10 second sampling (0.1Hz). However, for comparison the effects of isoflurane on endocytosis are shown for both 0.1Hz and 0.3Hz sampling following a single stimulus in Figure S2 and Figure S3 (See also STAR Methods). Decay times of this single peak are reported for both acquisition rates. These data show similar failure to return to baseline during the first peak using both acquisition rates. Since our acquisition rates differs from prior studies in rat cultures, our values for decay times are for direct comparison of neuronal activity with and without isoflurane and not for comparison to decay times with other protocols.
In the absence of isoflurane, FRET analysis showed a decrease in ATP in both genotypes (Figure 3C,D) during stimulations, followed by complete (wildtype) or near complete recovery of the ATP driven FRET signal (KO). When isoflurane was added (0.5mM and 0.74mM wildtype; 0.25mM KO) to the neuronal cultures with no exogenous glucose, the FRET signals reflecting ATP levels were dramatically decreased in both the wildtype and KO cultures (Figure 3C,D) and failed to recover.
Synaptic Functioning with Inhibited Glycolysis
We determined whether residual glucose/glycogen allows glycolysis to play a role in the initial (first peak) synaptic response in the presence of isoflurane. Therefore, we pretreated the wildtype controls and Ndufs4(KO) neuronal cultures with the glycolysis inhibitors, 2-deoxy-d-glucose (2DG) and iodoacetic acid (IAA) (Figure 4A,B upper panels). Under these conditions, in the absence of isoflurane, in the first stimulation endocytosis was inhibited in wildtype controls in the presence and absence of isoflurane, and completely failed in the second stimulation (Figure 4A, lower panel, First peak, τ (unexposed) 367+/− 367 (n=10); τ (exposed) 694+/− 334 (n=8); p=0.069). A similar pattern was seen in Ndufs4(KO) at 0.25mM isoflurane (Figure 4B, lower panel, First peak, τ (unexposed) 301+/− 314 (n=8); τ (exposed) 802+/− 461 (n=7); p=0.027). There was no decrease in the upslope in fluorescence during the first peak in either genotype in isoflurane, indicating no defect in exocytosis (Data not shown). Compared to stimulation in the absence of glucose, but with no glycolytic inhibitors, the addition of 2DG and IAA in isoflurane caused a failure of endocytosis following the first stimulation, followed by complete failure of exocytosis in the second stimulation for both mutant and wild type. FRET analysis showed a profound decrease in presynaptic ATP levels in both genotypes in the presence of isoflurane at their respective 2XEC50s (Figures 4C,D).
Figure 4. Synaptic Functioning with glycolytic blockers supplemented with pyruvate.
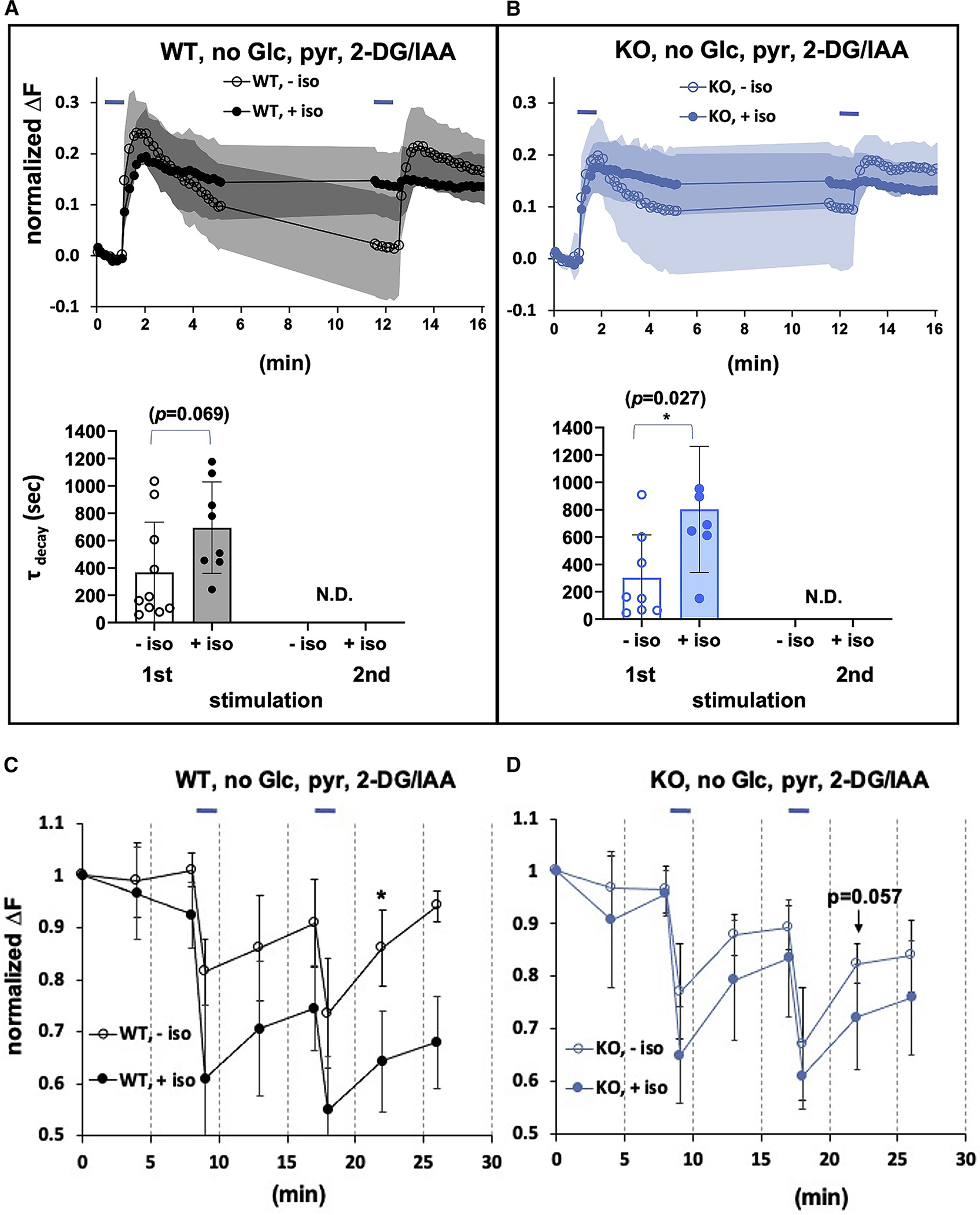
A. Upper panel. Wild type cells, electrically stimulated in the absence (or presence) of isoflurane (0.74mM) with glycolysis inhibited, B. Upper panel. Ndufs4(KO) cells electrically stimulated in the absence or presence of isoflurane (0.25mM) with glycolysis inhibited. Lower A,B panels. Decay time tau is not calculable in the presence of isoflurane following the second stimulation, denoting a complete failure of endocytosis in both genotypes. C,D. The presence of isoflurane decreases the ATP FRET signal at the synapse following stimulation. Hippocampal cultures, with glycolysis inhibited but with added pyruvate to support mitochondrial respiration as described in Figure 2c. C. Electrical stimulation of wildtype cells in the absence of isoflurane with glycolysis inhibited produces a reversible decrease in ATP. In the presence of isoflurane (0.74mM) the FRET signal dramatically decreases and fails to recover. D. Electrical stimulation of Ndufs4(KO) cells in the absence of isoflurane with glycolysis inhibited produces a modest decrease in ATP. In the presence of isoflurane (0.25mM) the FRET signal dramatically decreases and fails to recover.
Recovery from Anesthetic Inhibition
A characteristic of volatile anesthetic action is recovery when the anesthetic is removed. To determine whether the inhibition of endocytosis was reversible, we first exposed mutant and control neuronal cultures to isoflurane (at their respective 2XEC50s) in the absence of glucose (as in Figure 3). We then removed isoflurane from the culture and supplied glucose to allow glycolysis, as well as mitochondrial function, to proceed. In both wildtype and KO, we observed recovery of both endocytosis and ATP levels in the second peak consistent with recovery from the anesthetic effect on mitochondrial and synaptic functions (Figures 5A,B).
Figure 5. Recovery from Anesthetic Inhibition.
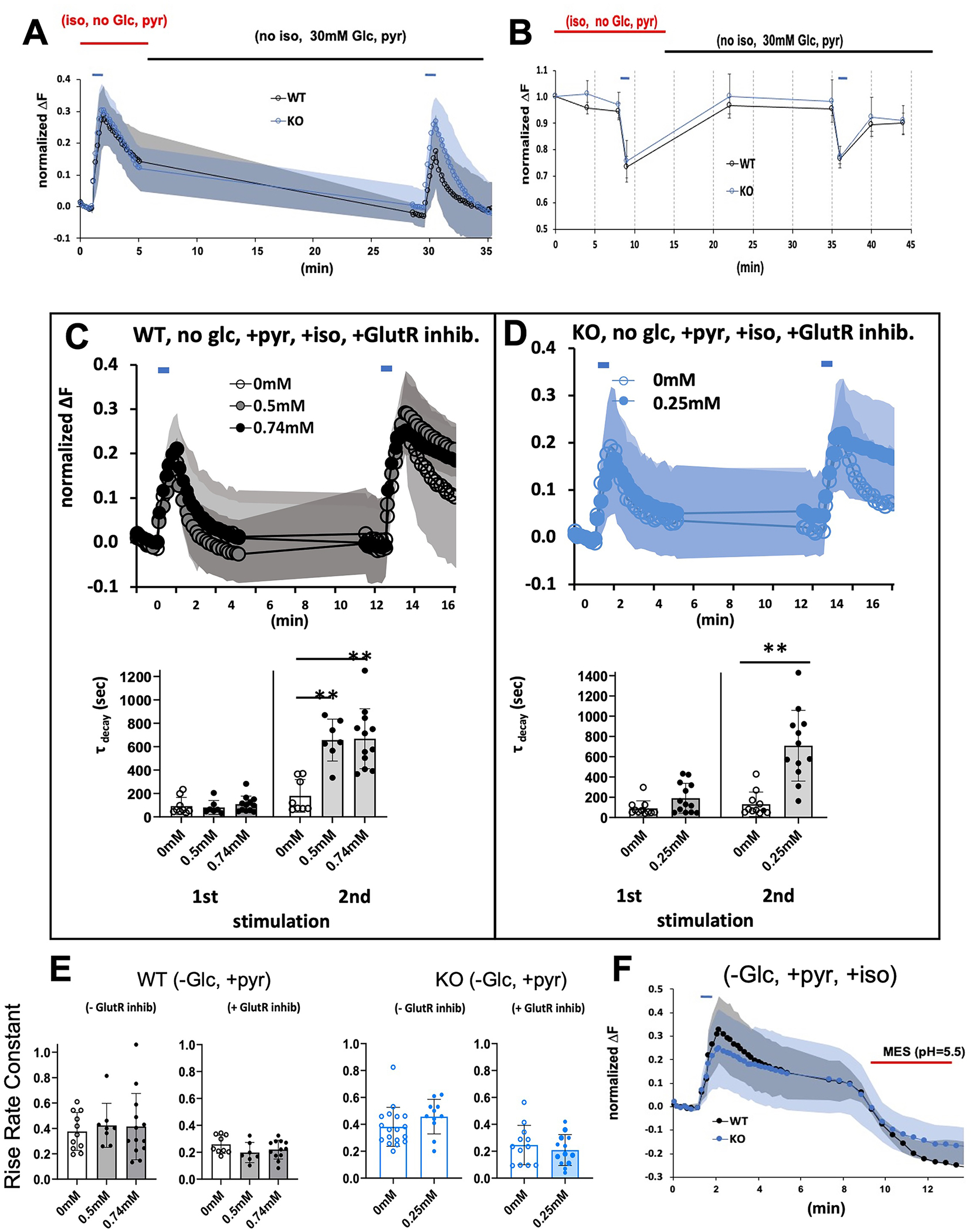
A. Recovery of synaptic recycling following stimulation and removal of isoflurane. Stimulation was as in Figures 1–4. Wildtype and KO cells were exposed to isoflurane at their 2XEC50s in the absence of glucose but with pyruvate (as in Figure 3). Following the first stimulation, isoflurane was removed, and 30mM glucose was added. Endocytosis was re-established following removal of isoflurane and addition of glucose in both genotypes showing reversibility of the anesthetic effects. B. Recovery of ATP levels following stimulation and removal of isoflurane. Stimulation was as in Figures 1–4. Wildtype and KO cells were exposed to isoflurane at their EC95s in the absence of glucose but with pyruvate (as in Figure 3). Following the first stimulation, isoflurane was removed, and 30mM glucose was added. ATP levels recovered to baseline establishing reversibility of the anesthetic effects. C,D. Synaptic Functioning in presence of glutamatergic blockade. To decrease stimulation input, glutamatergic receptors were blocked with antagonists, 6,7-dinitroquinoxaline-2,3-dione (10uM) and 3-(2-Carboxypiperazin-4-yl)propyl-1-phosphonic acid (10uM). C. Wild type cells in 0.5mM or 0.74mM isoflurane in 0mM glucose supplemented with pyruvate. D. Ndufs4(KO) cells in 0.25mM isoflurane in 0mM glucose supplemented with pyruvate. In the presence of isoflurane, endocytosis was markedly defective following the second stimulation. Lower C,D panels. Decay times calculated for WT and KO cells in presence and absence of isoflurane and glutamatergic blockade. Corresponding ATP levels for the WT experiments are shown in Figure S4. E. Histograms showing the effects of isoflurane (WT, 0.5mM and 0.74mM; KO, 0.25mM) on rise time coefficients in the absence or presence of glutamatergic blockade. Left panel represents wildtype cells, right graph represents Ndufs4(KO) cells. F. Stimulating wild type cells in the presence of 0.74mM isoflurane or KO cells in 0.25mM isoflurane in Tyrode buffer (pH 7.4) blocked endocytosis as evidenced by failure of fluorescence to return to baseline. Addition of MES (25 mM, pH 5.5; red bar) to either culture, which acidifies the extracellular compartment, rapidly quenched fluorescence, indicating that pHluorin was exposed to the mild acid.
Synaptic Functioning with Glutamatergic Blockade
Previous work from the Hemmings laboratory indicated that 0.7mM isoflurane inhibited presynaptic calcium influx and exocytosis but did not affect endocytosis 28. Two aspects of their protocol may underlie the differences from those seen here. First, the authors used culture media containing 30mM glucose which would obscure the effects of mitochondrial inhibition (Figure 2). Second, we used intense stimulation to model input from surgical pain while they used a single stimulus with less overall intensity than we employed. For comparison with those and other earlier studies, we decreased the excitatory input in our experiments by using inhibitors of glutamatergic receptors to eliminate network amplification. In addition, glutamatergic inhibition removes the effects of asynchronous stimulation to more definitively reflect input from a single source of stimulation and make failure of the pHluorin signal to return to baseline easier to interpret as endocytosis. In wildtype controls at 0mM, 0.5mM and 0.74mM isoflurane (Figure 5C, upper panel) and in Ndufs4(KO) at 0mM and 0.25mM (Figure 5D, upper panel), defective endocytosis was apparent following the second peak (Figure 5C,D lower panels). This contrasts with results shown for similar conditions without inhibitor (Figure 3). With the addition of glutamatergic blockade, we noted a slower rise in VGLUT1-pHluorin fluorescence consistent with a decrease in rise time coefficients (exocytosis) in both genotypes in the absence of isoflurane (Figure 5E), but which was not changed further by the addition of isoflurane. The change in decay time was correlated by a decrease in ATP levels at both isoflurane concentrations following the second stimulation when glutamatergic blockade was included (Figure S4).
Site of Endocytosis Inhibition
Both dynamin-directed closure of vesicles and reestablishment of the pH gradient within enclosed vesicles are energy-dependent steps that define endocytosis 34,43. To determine which step is susceptible to the anesthetic effects on ATP levels, we inhibited endocytosis as in Figure 3 (no external glucose, isoflurane at their respective 2XEC50s for both genotypes), and then washed the cells in a mildly acidic buffer (MES buffer, pH 5.5, see Methods). If the pHluorin is enclosed in vesicles, and the endocytosis is inhibited at the re-establishment of the pH gradient, then the acidic buffer will not reach the pHluorin and the fluorescence will persist. If the pHluorin remains exposed to the surface of the cell, we would expect the buffer to quench the fluorescence. Treatment of both KO and wildtype cells with MES buffer, when endocytosis was inhibited by isoflurane, led to complete quenching of the VGLUT1-pHluorin fluorescence, consistent with blockade of reuptake of synaptic vesicles at the cell surface (Figure 5F).
Behavioral Resistance to Volatile Anesthetics
Inhibition of mitochondrial complex I prevents TCA cycle activity and the production of ATP from the ETC. If inhibition of complex I with resulting failure of synaptic recycling is a primary effect of VAs leading to the anesthetized state, then restoration of complex I function might lead to resistance of the whole animal response to VAs. NDi1 is a yeast NADH dehydrogenase single protein that functions in a manner similar to mammalian complex I. NDi1 can accept electrons from NADH and donate them to complex III via coenzyme Q; however, NDi1 does not pump protons to contribute to ATP production 36,37. Therefore, NDi1 expression in the presence of complex I inhibition would restore the NAD+/NADH ratio allowing the TCA cycle to proceed, and it would also partially restore the generation of ATP. Importantly, NDi1 is resistant to canonical mitochondrial complex I inhibitors such as rotenone and piericidien A 36,37. A compelling test of our model that complex I is a primary VA target that controls anesthetic sensitivity would be to show VA resistance in a wildtype mouse expressing the NDi1 protein.
We tested NDi1 expressing mice for sensitivity to the VAs, isoflurane and halothane. Compared to WT mice, NDi1 expressing mice showed a 20–25% resistance (reported as Effect Size, the fractional change in EC50) (Figure 6A) to each anesthetic for both loss of righting reflex (sedation) and movement in response to a tail clamp (the classic endpoint for determining MAC). This resistance is consistent with the hypothesis that mitochondrial inhibition is a primary mechanism for the action of volatile anesthetics. We then constructed and tested the double mutant, NDi1;Ndufs4(KO) and tested that strain for sensitivity to isoflurane and halothane. As seen in the wildtype background, NDi1 caused a 20–30% resistance for both loss of righting reflex and tail clamp to both isoflurane and halothane (Figure 6B).
Figure 6. Effects of NDi1 on sensitivity to volatile anesthetics.
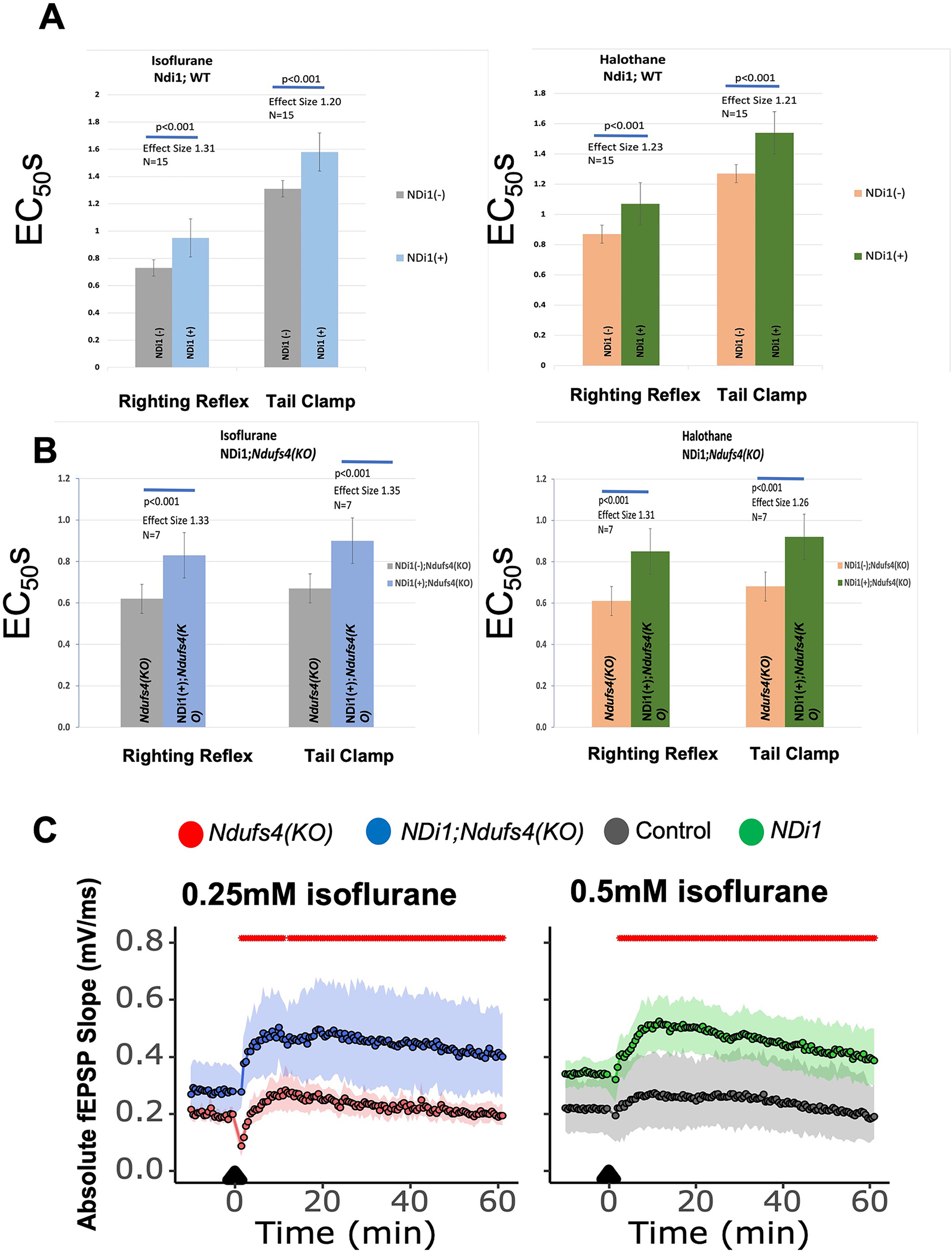
A. EC50s for Loss of Righting Reflex (LORR) (isoflurane, EC50,LORR Ndi1(−) 0.73+/−0.03%; Ndi1(+) 0.95+/−0.08%; n=15; p<0.001) and (halothane, EC50,LORR Ndi1(−) 0.88+/−0.07%; Ndi1(+) 1.12+/−0.11%; n=15; p<0.001) and nonmovement to tail clamp (TC) (isoflurane, EC50,TC Ndi1(−) 1.31+/−0.08%; Ndi1(+) 1.58+/−0.12%; n=15; p<0.001) and (halothane, EC50,TC Ndi1(−) 1.27+/−0.09%; Ndi1(+) 1.54+/−0.11%; n=15; p<0.001) for wildtype and NDi1(+) mice. Left panel is the response to isoflurane, right panel is the response to halothane. Effect sizes are the percent change of the Ndi1 responses compared to the wildtype responses. B. EC50s for Loss of Righting Reflex (LORR) exposed to isoflurane (EC50,LORR Ndi1(−),Ndufs4(KO) 0.62+/−0.10%; Ndi1(+),Ndufs4(KO) 0.83+/−0.16%; n=7; p<0.001) and halothane (EC50,LORR Ndi1(−),Ndufs4(KO) 0.61+/−0.06%; Ndi1(+),Ndufs4(KO) 0.85+/−0.12%; n=7; p<0.001) and nonmovement to tail clamp (TC) for Ndufs4(KO) and NDi1;Ndufs4(KO) mice (isoflurane, EC50,TC Ndi1(−),Ndufs4(KO) 0.67+/−0.07%; Ndi1(+),Ndufs4(KO) 0.90+/−0.11%; n=7; p<0.001) and (halothane, EC50,TC Ndi1(−),Ndufs4(KO) 0.68+/−0.01%; Ndi1(+),Ndufs4(KO) 0.92+/−0.06%; n=7; p<0.001). Left panel is the response to isoflurane, right panel is the response to halothane. Effect sizes are the percent change of the NDi1;Ndufs4(KO) responses compared to the Ndufs4(KO) responses. C. NDi1 increases fEPSPs in the presence of isoflurane in hippocampal slices from both Ndufs4(KO) (left panel) and wildtype (right panel) mice. Hippocampal slices from Ndufs4(KO) and NDi1;Ndufs4(KO) (left panel) are exposed to 0.25mM isoflurane; those from wildtype and NDi1 (right panel) are exposed to 0.5mM isoflurane. NDi1-containing slices recover more quickly, and show pronounced long term potentiation compared to those without NDi1 (Ndufs4(KO): n=6, NDi1;Ndufs4(KO): n=12, wildtype: n = 6, NDi1: n = 12). Red marks at top indicate that NDi1 differs from wildtype and NDi1;Ndufs4(KO) differs from Ndufs4(KO), p<0.05 at all points.
Field Recordings with NDi1
Using field recordings following high frequency stimulation (HFS) in the hippocampus, we previously showed that presynaptic function in the wildtype was depressed by isoflurane in a manner that indicated defective recycling of neurotransmitter, i.e. the function of VA exposed neurons did not recover from strong stimulation as quickly as in unexposed neurons 23,33. We repeated those experiments in hippocampal slices where NDi1 was expressed. In the absence or presence of isoflurane, the fiber volley (FV) amplitudes were not changed from the baseline, pre-HFS levels (data not shown). The dependence of CA1 field excitatory postsynaptic potentials (fEPSP) on input of CA3 presynaptic fibre volleys (FVs) was similar in NDi1 and control slices in the absence of isoflurane (data not shown). We then measured the responses of control and NDi1 slices to high frequency stimulation (HFS) in the presence of isoflurane at the EC50 for wildtype animals (0.5mM) (Figure 6C). fEPSPs evoked between 2 minutes and 5 minutes post-HFS were characteristically depressed 44, but recovered more quickly in NDi1 slices than in control slices (Figure 6C). Subsequent fEPSPs, from 5.5 minutes post-HFS up until 1-hour post-HFS, remained significantly elevated in NDi1 slices compared to those from wildtype animals (red marks above tracings show those points significantly different between genotypes, p<0.05). A similar improvement was seen in recovery from HFS in slices from NDi1;Ndufs4(KO) animals compared to Ndufs4(KO) (Figure 6C).
We also examined whether the fEPSPs of NDi1 slices deviated from that of control slices during HFS trains. In both genotypes, slices pre-exposed to 0.5mM isoflurane developed larger responses in the second pulse than in the first pulse in each train of stimulation, but were not different between genotypes (data not shown). The results indicated the improved recovery in NDi1 slices was consistent with improved recycling.
Synaptic and Mitochondrial Function in NDi1
The presence of NDi1 is associated with behavioral resistance to isoflurane (Figure 6A,B) and with improved neuronal function in the presence of isoflurane (Figure 6C). We postulated that the presence of the yeast NADH dehydrogenase would rescue the isoflurane-induced decrease in presynaptic ATP levels and would result in an alleviation of the isoflurane-induced failure of presynaptic endocytosis. We repeated the protocol seen in Figure 3 (no glucose, 0.74mM isoflurane) in hippocampal neurons from NDi1 containing animals. We found a complete rescue of the endocytosis failure (Figure 7A,B). Ndi1 also rescued the decrease in ATP in the presence of isoflurane (Figure 7C,D). Both results correlate with the behavioral resistance of the NDi1 mouse.
Figure 7. Effects of NDi1 on physiologic responses to volatile anesthetics. Effects of NDi1 on Synaptic function during neuronal stimulation.
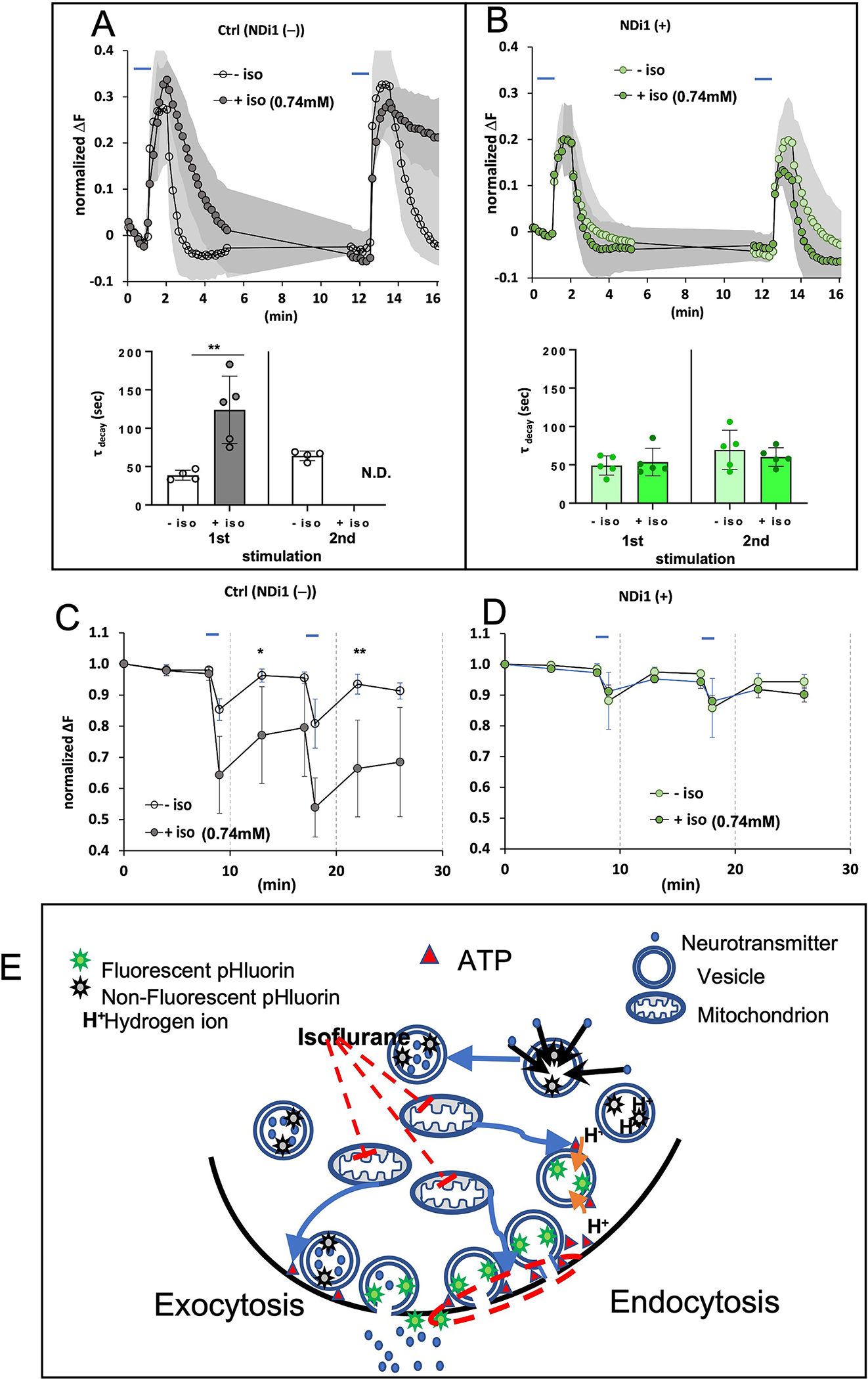
Exocytosis and endocytosis following electrical field stimulation of hippocampal cultures with no exogenous glucose, supplemented with pyruvate. In b. and c. control studies with wildtype cultures are from the siblings of the NDi1 animals and were repeated at the time of the NDi1 studies. A. Upper panel. Wild type cells electrically stimulated in the absence or presence of isoflurane (0.74mM). This protocol was identical to that in Figure 3A for comparison to the right panel of data from NDi1(+).B. Upper panel. NDi1(+) cells stimulated in the absence or presence of isoflurane (0.74mM). Note the recovery of endocytosis after first or second stimulation even in the presence of isoflurane (green filled symbols). (Ndi1(+) cells, First peak, τ (unexposed) 49+/−13 (n=5); τ (exposed) 54+/−18 (n=5); N.S.; Second peak τ (unexposed) 70+/−26 (n=5); τ (exposed) 60+/−12 (n=5); N.S.). A,B Lower panels. The tau values for peak decay (endocytosis) corresponding to the pHluorin signals in the upper panels. C,D. Effects of NDi1 on ATP levels during neuronal stimulation. Hippocampal cultures as described in Figure 3. C. There was a difference in ATP levels in the wildtype cultures exposed and unexposed to isoflurane. Electrical stimulation of wildtype cells in the absence of isoflurane produces a reversible decrease in ATP. In the presence of isoflurane (0.74mM) the FRET signal dramatically decreases and fails to recover. D. ATP levels completely recover following electrical stimulation of NDi1 cells in the presence or absence of isoflurane (0.74mM). Ndi1 rescued the decrease in ATP in the presence of isoflurane (Right panel, Second peak ATP fraction (unexposed) 0.94+/−0.03 (n=5); (exposed) 0.92+/−0.03 (n=5); N.S.). E. Mechanism of isoflurane-induced block of endocytosis. Model for the action of isoflurane on synaptic silencing. Isoflurane inhibits mitochondrial complex I causing decreased oxidative phosphorylation and a fall in presynaptic ATP (Figure S1). Endocytosis is blocked at or before the vesicle scission step upon exposure to isoflurane (step 3 in Figure 1A). The red dashed circle in this figure shows the step inhibited by loss of ATP, the result of inhibition of mitochondrial complex I by isoflurane.
Discussion.
Across the animal kingdom, organisms with mitochondrial complex I defects display hypersensitive responses to VAs both in behavior and presynaptic function 3,11–14. Complex I is the only putative molecular target of VAs for which this holds true, and therefore is a candidate to be a universal target for the anesthetic effects of these drugs. The resistance to volatile anesthetics both at the behavioral level and the neuronal level in NDi1 expressing mice further indicates that mitochondrial inhibition is a primary target in the whole animal response to VAs.
Pathak et al., showed that mitochondrial function was crucial to endocytosis at the presynapse 34. Given the genetic data showing hypersensitivity in complex I mutants 11–14 and the sensitivity of complex I to volatile anesthetics 18–21, we hypothesized that VAs caused neuronal silencing (the anesthetic state) by creating an acute mitochondrial failure at the presynapse. The data presented here, in combination with prior whole animal 11,13,14 and electrophysiologic results 23,33, offer an answer to the question….how do volatile anesthetics inhibit strong neuronal signaling? The sensitivity of whole animal behaviors to isoflurane is transduced through complex I function. Isoflurane exposure reduces presynaptic ATP levels, leading to a failure in recycling of synaptic vesicles in the face of strong neuronal stimulation (Figure 7E).
The finding that respiration is identical between the wildtype and Ndufs4(KO) in unstimulated neurons (Figure S1B) suggests that ATP levels at rest are the same in those genotypes which in turn makes changes in FRET signal easier to compare. The measured respiration is, of course, from neurons in culture and not specifically from synapses where ATP demand is undoubtedly higher 34,35. However, the respiration rates at rest were similar between genotypes indicating that one can approximate that both genotypes (wildtype and Ndufs4(KO)) start at a similar baseline ATP level in the boutons. This corroborated our earlier synaptic findings in CA1 cells in glutamatergic neurons from brain slices which showed no differences at baseline between wildtype and Ndufs4(KO) neurons in spontaneous synaptic activities 23,33. However, isoflurane concentrations that anesthetized only Ndufs4(KO) mice (0.25mM) also decreased the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) only in Ndufs4(KO) CA1 neurons 23,33. Isoflurane concentrations effective in control mice (0.5mM) decreased sEPSC frequencies in both control and Ndufs4(KO) CA1 pyramidal cells 33. Follow-up studies indicated that the pattern of inhibition was most consistent with inhibition of neurotransmitter recycling 23. The reductions in endocytosis and ATP levels seen here also occur at concentrations that match the whole animal EC95s for isoflurane. ATP levels correlate well with failure of endocytosis, which occurs at reuptake of synaptic vesicles at the presynaptic cell surface. When ATP production is supplemented by elevated glucose levels supporting glycolysis or by a transgenic form of NADH dehydrogenase, successful endocytosis is seen with repeated stimulations. Inhibition of glycolysis, with resulting decreased ATP levels, does the opposite; synaptic function becomes more sensitive to isoflurane. Furthermore, preexisting mitochondrial dysfunction, as seen in Ndufs4(KO) cells, leads to a significant hypersensitivity to the effects of isoflurane of both endocytosis and ATP levels. Thus, the sensitivity of endocytosis to isoflurane can be driven in either direction (decreased or increased sensitivity) by increasing or decreasing the availability of ATP from glycolysis or mitochondrial respiration.
Both glycolytic and mitochondrial production of ATP support synaptic endocytosis 34,45. Consistent with the dependence of VA sensitivity on ATP production, two recent studies showed that inhibition of glycolysis or glycogenolysis can increase VA sensitivity while augmentation of glycolysis causes a mild resistance 46,47. Glycolytic activity secondarily impacts the inhibition by isoflurane on the ability of mitochondria to meet the increased demand for ATP production in response to stimulated neuronal activity. Our results support the importance of glycolysis although our use of chemical blockade must be interpreted with caution. Iodoacetate has additional effects beyond inhibition of glycolysis, specifically inhibition of other cysteine peptidases48.
One unresolved aspect of our results is seen in Figure 1. A limitation of our study is that we do not know the concentration of glucose that best reflects in vivo conditions. Even at 1.5mM glucose, while isoflurane has a significant effect on endocytosis, only modest differences were seen in FRET signals in the wildtype. In addition, while similar falls in ATP levels were seen in Ndufs4(KO) neurons, there was no additional difference caused by isoflurane. However, improvement of even this modest change in ATP by the transgenic NDi1 or increased glucose allows a complete recovery of endocytosis and, in the case of the transgenic animal, a relative resistance in sensitivity to isoflurane. Thus, the importance of both mitochondrial complex I and ATP levels are clearly indicated. Possible explanations of the discordance of endocytosis and ATP levels in Ndufs4(KO) are that endocytosis is more affected by ATP produced in the mitochondria compared to by glycolysis or that a second effect of volatile anesthetics, in concert with the presence of modestly decreased ATP levels, affects endocytosis. An example of such a possibility is a decreased sensitivity to ATP levels in dynamin, the GTP-requiring protein involved in closure of synaptic vesicles during endocytosis. Experiments to discern between these possibilities are being undertaken.
It is also important to note that inhibition of complex I will decrease the NAD+/NADH ratio which theoretically could play a role in sensitivity. This point is accentuated by the fact that the presence of NDi1, which will both partially alleviate the NAD+/NADH imbalance and improve ATP production, leads to resistance. However, high concentrations of glucose favoring glycolysis will increase ATP but not alleviate the NAD+/NADH imbalance. Since high glucose restored endocytosis, we interpret that it is ATP levels rather than NAD+/NADH ratios that are important for the anesthetic effects.
The metabolic effects of volatile anesthetics were noted first in the 1970s as a possible important mechanism of their action 20,49–51. In mammals it was clear that volatile anesthetics specifically inhibited complex I function in isolated mitochondria. However, failure to find a widespread decrease in ATP in CNS homogenates then52 and more recently53, led to the conclusion that ATP levels were not of primary importance in causing the anesthetized state. Despite these findings, the potential roles of mitochondrial function in the response to volatile anesthetics received occasional consideration 21. Beginning in 2001, genetic data accumulated indicating that behavioral sensitivity to the anesthetizing effects of VAs was dependent on complex I activity 11–14,22. The mouse knockout of NDUFS4, a complex I subunit 27, was a major advancement in the field for it allowed behavior of cells in culture to be interpreted considering the behavior of the animal in volatile anesthetics. The whole animal EC50 for Ndufs4(KO) in vivo is profoundly lower than that of wildtype, the greatest hypersensitivity reported for a mammal. Ex vivo exposure of cells of each genotype to concentrations that matched the vulnerability of the parent animal led to cellular responses that were basically superimposable between mutant and KO at equipotent doses of isoflurane. The availability of this model then allowed interpretation of electrophysiologic characterization of synaptic function in neurons 13.
Examination of the prior data from the Hemmings et al. 28 does show a delayed return to baseline of VGLUT1-pHluorin fluorescence following stimulation. We interpret this to indicate a defect in endocytosis also existed in their paradigm. In addition, in those experiments the cells were perfused with concentrations of glucose that would favor glycolysis as an energy source and possibly limit the effects of VAs on endocytosis because of maintenance of ATP levels. When we dampened the electrical stimulus of the neuronal cultures in this study using glutamatergic blockade, endocytosis continued to be inhibited by isoflurane in both genotypes.
We do not yet know how expression of the yeast NADH dehydrogenase NDi1 confers anesthetic resistance in our model. If behavior in a volatile anesthetic is simply dependent on resistance of the protein to these agents, then we would predict that both wild type and Ndufs4(KO) mice expressing NDi1 would have the same absolute EC50s in VAs, rather than an identical relative shift. Since this single protein does not pump protons, it may be that the loss of ATP production relative to the native complex is insufficient to compensate fully for anesthetic inhibition of the native complex I. It also may be that the yeast protein is so resistant to VAs that it reveals anesthetic effects on a second target with a higher EC50 than complex I. These speculative hypotheses await further testing.
In conclusion, these results show that presynaptic endocytosis following intense neuronal stimulation is inhibited by isoflurane via inhibition of mitochondrial complex I with a resulting fall in ATP at the presynapse. Complex I inhibition emerges as a candidate to be the primary mechanism of action of isoflurane by ATP dependent failure of presynaptic endocytosis.
STAR Methods.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Philip G. Morgan (pgm4@uw.edu).
Materials availability
Nestin-Cre, NDi1-LSL;Ndufs4(KO) mice were generated by breeding for this manuscript. Ndufs4(KO) mice as available for Jackson Laboratory (JAX:027058). Nestin-Cre, NDi1-LSL mice were the gift of Navdeep Chandel (Northwestern University) and only available from that source.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of Seattle Children’s Research Institute. Mice were housed at 22°C with a 12-hour light-dark cycle and maintained on a standard rodent diet. Food and water were available ad libitum. For all behavioral studies, approximate equal numbers of each sex were studied.
NDi1-LSL mice were a kind gift from Dr. Navdeep Chandel (Northwestern University, IL, USA) and have been previously described36,37. Briefly, Cre-mediated removal of the Lox-STOP-Lox (LSL) cassette allows for expression of the yeast NDI1 protein. Cre expression for removal of the stop codon in Ndi1 was driven by the nestin promoter which limits expression to brain as described. 37 Mice with a floxed allele of the Ndufs4 gene (Ndufs4lox/lox) were the kind gift of Richard Palmiter (University of Washington)27. Ndufs4(KO) mice were global knockouts and were also previously described. 27,33
For behavioral experiments, mice expressing Ndi1 were generated by crossing mice heterozygous for Nestin-Cre and heterozygous for Ndufs4 to mice homozygous for Ndi1-LSL and heterozygous for Ndufs4. All progeny were either heterozygous for Nestin-Cre and Ndi1-LSL or null for Nestin-Cre and heterozygous for Ndi1-LSL. Progeny homozygous for Ndufs4 were used for anesthetic experiments for the effects of Ndi1 on Ndufs4. Progeny with one or no Ndufs4 alleles were used for higher concentration (0.5mM or 0.74mM isoflurane) experiments for the effects of Ndi1 on wildtype mice.
In the field recording experiments, mice expressing Ndi1 were generated by crossing mice homozygous for Nestin-Cre and heterozygous for Ndufs4 to mice homozygous for Ndi1-LSL and heterozygous for Ndufs4. All progeny were heterozygous for Nestin-Cre and Ndi1-LSL. Progeny homozygous for Ndufs4 were used for 0.25mM isoflurane experiments, and progeny with one or no Ndufs4 alleles were used for 0.5mM isoflurane experiments. Mice for comparison at each isoflurane concentration were generated by crossing mice heterozygous for Ndufs4 and did not have alleles of Nestin-Cre or Ndi1-LSL. pHluorin and Fret experiments were done as described below but with hippocampal neurons from Ndi1-LSL mice. (pCAG as control or pCAG-Cre for Ndi1(+) were co-transfected.)
Constructs.
VGlut1-pHluorin and mCherry-synaptophysin in pCAGGS were kind gifts of Dr. Robert Edwards (UCSF)55, and AT1.03YEMK of Hiromi Imamura (Kyoto University, Japan) 54. AT1.03YEMK was placed in pCAGGS by the Nakamura laboratory (Gladstone Institutes, USA). The use of VGLUT1-pHluorin, mCherry-synaptophysin 34,39,56–58 and AT1.03YEMK have been described 54. VGlut1-pHluorin quenches in an acidic environment, i.e. within synaptic vesicles, and fluoresces in a neutral milieu such as the extracellular space (Figure 1A). It is commonly used to trace synaptic vesicle recycling (Figure 1B) 30,39,40.
Cells.
Primary hippocampal cells from floxed Ndufs4 mice were prepared as previously described 40. Briefly, hippocampi were isolated from P0 or P1 mouse pups and cultures were immediately transfected using nucleofection. For VGLUT1-pHluorin experiments, the constructs pCAG-VGlut-pHluorin, pCAG-mCherry-Synaptophysin and pCAG were transfected to be used as wildtype controls. A pCAG-Cre construct was used to establish Ndufs4 (KO) cultures. For ATP FRET experiments, pCAG-AT1.03YEMK, pCAG-mCherry-Synaptophysin, and pCAG as wildtype control or pCAG-Cre for Ndufs4 (KO) were co-transfected.
Hippocampal neurons were maintained at 37°C in Neurobasal media (Thermo Fisher Scientific, USA) supplemented with 1% heat-inactivated fetal bovine serum (Thermo Fisher Scientific, USA), 1X GlutaMax, 15 mM NaCl, 1X B27 (Thermo Fisher Scientific, USA) and 1X Primocin (Invitrogen, USA). 5-Fluoro-2-deoxyuridine (10uM) and uridine (10uM) were added at division 5 to prevent glial growth. Primocin was used since it is known to not affect neurons strongly and has been used in other studies of GABAA receptors without problems 59.
METHOD DETAILS
Live cell imaging
Live imaging was performed as described 33. Briefly, mouse hippocampal neurons were imaged 10–14 days in vitro (DIV 10–14) after transfection, a standard range for mouse 34,60–62. Live-cell images were acquired using an Axio ZEISS Observer microscope with a 40X objective lens coupled with an Axio ZEISS camera and LED illumination. Isoflurane was applied as previously described at 30°C 23.
Coverslips containing transfected neurons were mounted in a laminar-flow perfusion chamber in modified Tyrode’s solution (pH 7.4; in mM: 127 NaCl, 10 HEPES-NaOH, 2.5 KCl, 2 MgCl2, and 2 CaCl2, at 30°C with either 0, 1.5mM, or 30 mM glucose with 10 mM pyruvate, unless otherwise specified. The total size of the synaptic vesicle pool was determined by application of Tyrode’s solution with 50mM NH4Cl at the end of each run of VGLUT1-pHluorin experiments. MES solution (25mM, pH 5.5) was prepared by replacing HEPES with 2-(N-Morpholino) ethanesulfonic acid as previously described34. To inhibit endogenous network activity, glutamate receptor antagonists, 6,7-dinitroquinoxaline-2,3-dione (DNQX, 10uM) and 3-(2-Carboxypiperazin-4-yl)propyl-1-phosphonic acid (CPP, 10uM), were used63. Field stimulations were elicited at 10Hz for 60s using an A385 stimulus isolator and an A310 Accupulser signal generator (World Precision Instruments, USA).
For VGLUT1-pHluorin experiments, images were acquired using 475/20 nm excitation and 524/46 nm band-pass emission filters using an acquisition rate of once ever 10 seconds (0.1Hz), as done by Pathak et al.34 Acquisition rates of once per 3 seconds (0.3 Hz), a standard for many studies in rats39,60,64 resulted in a failure in return to baseline following the second stimulation (Figure S2). This failure was due to fluorescent imaging per se, rather than electrical stimulation (Figure S3). Since the failure in endocytosis was clearly demonstrated in both the first and second peaks at both acquisition rates (0.3Hz and 0.1Hz), we used 0.1Hz to be able to interpret endocytosis rates after a second stimulation, a key test in our paradigm. The areas of pHluorin fluorescence were regarded as synaptic boutons if they co-localized with mCherry-synaptophysin and increased green fluorescence upon superfusion of 50 mM ammonium Tyrode’s solution. To determine the fluorescence response to stimulation over time, background-subtracted changes in fluorescence at each time point were normalized to the total amount of fluorescence as determined by application of Tyrode’s solution with 50mM NH4Cl. The averaged baseline fluorescence intensity determined from the six frames before the first stimulation was set to zero 34.
For ATP FRET experiments, sequential images were obtained through FRET channels (430/26 excitation, 562/45 emission), CFP (430/26 excitation, 485/30 emission) and YFP (511/67 excitation, 562/45 emission). The FRET/donor ratio was calculated for each bouton as described65, where FRET = IFRET/ICFP, where IX is the background-corrected fluorescence intensity (Avg IntensityFRET Channel – Avg Intensitybackground) measured in a given channel at each time point. For relative changes in FRET, values were normalized to the first time point in each bouton.
For both FRET and pHluorin fluorescent measurements, at least 20 synaptic boutons per coverslip were averaged and the means from 5 to 15 coverslips from at least two independent cultures were averaged. Images were analyzed using a custom-written Python script or ImageJ with Time Series Analyzer plugin. Circular regions of interest (~2um) corresponding to responding synaptic boutons were selected and fluorescence response to stimulation over time was determined 66. Fluorescence decay time constants (tau) and rise rate constants were obtained by fitting with a single exponential function.
Anesthetic sensitivity
Mice between 23–29 days old were anesthetized with either halothane or isoflurane, while their temperature was maintained by a water filled heating pad, following the techniques of Sonner67,68. For mice exposed to both anesthetics, the order of anesthetic treatment was randomized and at least 48 hours were provided between exposures. Anesthetic endpoints were loss of righting reflex (LORR) or nonmovement during a non-crushing tail clamp (TC). The concentrations of halothane and isoflurane were monitored using a calibrated inline analyzer.
Field recordings
Field recordings were performed and analysed as previously described 23,33. Briefly, the stimulating electrode was positioned in the area of Schaffer collateral fibres, and the recording electrode was placed in CA1 stratum radiatum to record fEPSPs (field excitatory postsynaptic potentials). Fibres were stimulated every 30s for 10 minutes for baseline activity, and for at least 60min following high frequency stimulation (HFS). fEPSPs during and following HFS were normalized to their average values for each recording during the final ten minutes prior to HFS. HFS consisted of 3 one-second trains of 100Hz, delivered at 20s intervals. The isoflurane-containing solution was superfused for 40min prior to HFS, and for the duration of the experiment. The synaptic input/output curve was constructed by varying stimulation amplitude in the 100–500 μA range.
Respiratory Capacity of Primary Hippocampal Cells
The progeny of Ndufs4(KO) heterozygotes were genotyped on the day of birth, completed within 3 hours as previously described33. Hippocampal cells from homozygous Ndufs4(KO) pups and from wildtype littermates were cultured as described above with the following modifications: 1. No transfection was done. 2. 75,000 cells were plated in each of the experimental wells of a 24-well Seahorse cell culture plate, pretreated with overnight incubation at 37°C of 120ug poly-L-lysine (Sigma, P2636), followed by 1.2ug laminin (Gibco, 23017–015) in borate buffer (0.1M Boric Acid; 0.1M Borax (decahydrate); pH 8.5). 3. The final growth medium (neurobasal with FuDR and uracil) was exchanged every 3 days.
On day 10 in vitro the respiratory capacity of the cells was measured with a Seahorse XF24 Flux Analyzer (Seahorse, Agilent, USA) (Figure S1). Prior to loading the cell plate into the instrument, the growth medium was replaced with respiration medium and cells were incubated for at least 1 hour at 37°C in room air, as previously described69. The share of basal OCR that was linked to mitochondrial ATP production was calculated as the difference between initial respiration and oligomycin-inhibited OCR. The difference between initial OCR and FCCP-uncoupled OCR estimates the “spare respiratory capacity” which cells may utilize when energy demand increases. OCR was normalized to either the number of nuclei (DAPI fluorescence) in a random sample image of each well, or to protein content of the wells.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Methods
All histograms and optical curves are presented as the Means +/− Standard Deviations (SDs); greyed areas represent the SDs, regions of darker grey represent overlap in the SDs. Oxygen consumption curves are presented as Means +/− Standard Errors of the Mean. Decay values (τ) for the pHluorin signals are reported for both first and second stimulation in all cases. Since exocytosis rise times were unaffected the values are not reported (other than for glutamatergic inhibition experiments) but the graphs are shown. For FRET optical signals, graphs are shown but values not reported. Statistical significance was determined by Student t test (unpaired, two tailed) or a one-way ANOVA with subsequent Tukey’s post hoc analysis (if necessary). Significance was defined as p < 0.05. In all figures, * indicates p<0.05, ** indicates p<0.01, and *** indicates p<0.001. For the FRET curves, only the points 4 minutes following each electrical stimulation were compared, in order to correlate to the pHluorin data. Rise times and decay times for pHluorin experiments were well fit to simple exponential curves.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| None | ||
| Bacterial and virus strains | ||
| pCAG-mCherry-synaptophysin | Robert Edwards (UCSF)55 | N/A |
| pCAG-VGlut1-pHluorin | Robert Edwards (UCSF)55 | N/A |
| AT1.03YEMK | Hiromi Imamura (Kyoto University, Japan)54 | N/A |
| pCAG-AT1.03YEMK | Ken Nakamura (Gladstone, CA, USA) | N/A |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| 6,7-dinitroquinoxaline-2,3-dione (DNQX, 10uM) 63 | Alomone Laboratories | Cat #:D130 |
| 3-(2-Carboxypiperazin-4-yl)propyl-1-phosphonic acid (CPP, 10uM) 63 | Tocris | Cat#: 0130 |
| Critical commercial assays | ||
| Deposited data | ||
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Nestin-Cre, NDi1-LSL mice | Navdeep Chandel (Northwestern University, IL, USA) 37 | N/A |
| Ndufs4lox/lox mice | Richard Palmiter (University of Washington)27 | B6.129S4-Ndufs4tm1.1Rpa/J JAX:027058 |
| Nestin-Cre, NDi1-LSL; Ndufs4(KO) mice | This manuscript | N/A |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and algorithms | ||
| Python script | In Lab | |
| ImageJ with Time Series Analyzer | NIH | Public Domain |
| Other | ||
Highlights.
Isoflurane inhibits mitochondrial complex I and decreases presynaptic ATP levels.
The fall in ATP levels accompanies a failure in endocytosis at the presynapse.
NDi1 bypasses complex I and rescues isoflurane’s effects on ATP and endocytosis.
NDi1 causes resistance to the behavioral effects of volatile anesthetics.
Acknowledgements.
The authors are indebted to the laboratories of Drs. Robert Edwards (UCSF, San Francisco, USA) and Susan Voglmaier (UCSF, San Francisco, USA) who provided significant initial technical assistance and sharing of pHluorin constructs, and to Dr. Navdeep Chandel (Northwestern University, Chicago, IL) who kindly shared the NDi1-LSL mouse and guided us in its use. We also thank Dr. Hiromi Imamura (Kyoto University, Japan) for sharing the FRET constructs. In addition, we thank Drs. Hugh Hemmings (Weill Cornell Medical College, NYC, USA) and Kira Spencer for their open and insightful discussions. Finally, the authors thank Dr. Beatrice Predoi for her continuous excellent technical assistance.
Support. PGM, MMS, JMR, PIZ and SJ were each supported in part by NIH Grants R01 GM122899 and R35GM139566. KN and DH were each supported in part by NIH Grant R01 NS091902. CRR was supported in part by NIH Grant NHLBI T32 HL076139–11.
Footnotes
Declarations of Interest. The authors declare no competing interests.
Inclusion and Diversity. We worked to ensure sex balance in our non-human (rodent) subjects.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Guitchounts G, Stewart DS, and Forman SA (2012). Two etomidate sites in alpha1beta2gamma2 gamma-aminobutyric acid type A receptors contribute equally and noncooperatively to modulation of channel gating. Anesthesiology 116, 1235–1244. 10.1097/ALN.0b013e3182567df3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruesch D, Neumann E, Wulf H, and Forman SA (2012). An allosteric coagonist model for propofol effects on alpha1beta2gamma2L gamma-aminobutyric acid type A receptors. Anesthesiology 116, 47–55. 10.1097/ALN.0b013e31823d0c36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falk MJ, Kayser EB, Morgan PG, and Sedensky MM (2006). Mitochondrial complex I function modulates volatile anesthetic sensitivity in C. elegans. Curr Biol 16, 1641–1645. 10.1016/j.cub.2006.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franks NP, and Lieb WR (1994). Molecular and cellular mechanisms of general anaesthesia. Nature 367, 607–614. 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- 5.Humphrey JA, Sedensky MM, and Morgan PG (2002). Understanding anesthesia: making genetic sense of the absence of senses. Human molecular genetics 11, 1241–1249. [DOI] [PubMed] [Google Scholar]
- 6.Campagna JA, Miller KW, and Forman SA (2003). Mechanisms of actions of inhaled anesthetics. The New England journal of medicine 348, 2110–2124. 10.1056/NEJMra021261. [DOI] [PubMed] [Google Scholar]
- 7.Franks NP, and Lieb WR (1982). Molecular mechanisms of general anaesthesia. Nature 300, 487–493. [DOI] [PubMed] [Google Scholar]
- 8.Pavel MA, Petersen EN, Wang H, Lerner RA, and Hansen SB (2020). Studies on the mechanism of general anesthesia. Proc Natl Acad Sci U S A. 10.1073/pnas.2004259117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao M, Sonner JM, Jurd R, Rudolph U, Borghese CM, Harris RA, Laster MJ, and Eger EI 2nd (2005). Beta3-containing gamma-aminobutyric acidA receptors are not major targets for the amnesic and immobilizing actions of isoflurane. Anesthesia and analgesia 101, 412–418, table of contents. 10.1213/01.ANE.0000154196.86587.35. [DOI] [PubMed] [Google Scholar]
- 10.Sonner JM, Werner DF, Elsen FP, Xing Y, Liao M, Harris RA, Harrison NL, Fanselow MS, Eger EI 2nd, and Homanics GE (2007). Effect of isoflurane and other potent inhaled anesthetics on minimum alveolar concentration, learning, and the righting reflex in mice engineered to express alpha1 gamma-aminobutyric acid type A receptors unresponsive to isoflurane. Anesthesiology 106, 107–113. [DOI] [PubMed] [Google Scholar]
- 11.Kayser EB, Morgan PG, and Sedensky MM (1999). GAS-1: a mitochondrial protein controls sensitivity to volatile anesthetics in the nematode Caenorhabditis elegans. Anesthesiology 90, 545–554. [DOI] [PubMed] [Google Scholar]
- 12.Morgan PG, Hoppel CL, and Sedensky MM (2002). Mitochondrial defects and anesthetic sensitivity. Anesthesiology 96, 1268–1270. [DOI] [PubMed] [Google Scholar]
- 13.Quintana A, Morgan PG, Kruse SE, Palmiter RD, and Sedensky MM (2012). Altered anesthetic sensitivity of mice lacking Ndufs4, a subunit of mitochondrial complex I. PLoS One 7, e42904. 10.1371/journal.pone.0042904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olufs ZPG, Loewen CA, Ganetzky B, Wassarman DA, and Perouansky M (2018). Genetic variability affects absolute and relative potencies and kinetics of the anesthetics isoflurane and sevoflurane in Drosophila melanogaster. Sci Rep 8, 2348. 10.1038/s41598-018-20720-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsieh VC, Niezgoda J, Sedensky MM, Hoppel CL, and Morgan PG (2021). Anesthetic Hypersensitivity in a Case-Controlled Series of Patients With Mitochondrial Disease. Anesth Analg. 10.1213/ANE.0000000000005430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suthammarak W, Morgan PG, and Sedensky MM (2010). Mutations in mitochondrial complex III uniquely affect complex I in Caenorhabditis elegans. J Biol Chem 285, 40724–40731. 10.1074/jbc.M110.159608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suthammarak W, Yang YY, Morgan PG, and Sedensky MM (2009). Complex I function is defective in complex IV-deficient Caenorhabditis elegans. The Journal of biological chemistry 284, 6425–6435. 10.1074/jbc.M805733200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kayser EB, Suthammarak W, Morgan PG, and Sedensky MM (2011). Isoflurane selectively inhibits distal mitochondrial complex I in Caenorhabditis elegans. Anesth Analg 112, 1321–1329. 10.1213/ANE.0b013e3182121d37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen PJ (1973). Effect of anesthetics on mitochondrial function. Anesthesiology 39, 153–164. 10.1097/00000542-197308000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Harris RA, Munroe J, Farmer B, Kim KC, and Jenkins P (1971). Action of halothane upon mitochondrial respiration. Arch Biochem Biophys 142, 435–444. [DOI] [PubMed] [Google Scholar]
- 21.Hanley PJ, Ray J, Brandt U, and Daut J (2002). Halothane, isoflurane and sevoflurane inhibit NADH:ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J Physiol 544, 687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kayser EB, Morgan PG, Hoppel CL, and Sedensky MM (2001). Mitochondrial expression and function of GAS-1 in Caenorhabditis elegans. J Biol Chem 276, 20551–20558. 10.1074/jbc.M011066200. [DOI] [PubMed] [Google Scholar]
- 23.Zimin PI, Woods CB, Kayser EB, Ramirez JM, Morgan PG, and Sedensky MM (2018). Isoflurane disrupts excitatory neurotransmitter dynamics via inhibition of mitochondrial complex I. Br J Anaesth 120, 1019–1032. 10.1016/j.bja.2018.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lenaz G, Fato R, Genova ML, Bergamini C, Bianchi C, and Biondi A (2006). Mitochondrial Complex I: structural and functional aspects. Biochimica et biophysica acta 1757, 1406–1420. 10.1016/j.bbabio.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 25.Lenaz G, and Genova ML (2012). Supramolecular organisation of the mitochondrial respiratory chain: a new challenge for the mechanism and control of oxidative phosphorylation. Advances in experimental medicine and biology 748, 107–144. 10.1007/978-1-4614-3573-0_5. [DOI] [PubMed] [Google Scholar]
- 26.Telford JE, Kilbride SM, and Davey GP (2009). Complex I is rate-limiting for oxygen consumption in the nerve terminal. J Biol Chem 284, 9109–9114. 10.1074/jbc.M809101200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, and Palmiter RD (2008). Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab 7, 312–320. 10.1016/j.cmet.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baumgart JP, Zhou ZY, Hara M, Cook DC, Hoppa MB, Ryan TA, and Hemmings HC Jr. (2015). Isoflurane inhibits synaptic vesicle exocytosis through reduced Ca2+ influx, not Ca2+-exocytosis coupling. Proc Natl Acad Sci U S A 112, 11959–11964. 10.1073/pnas.1500525112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie Z, McMillan K, Pike CM, Cahill AL, Herring BE, Wang Q, and Fox AP (2013). Interaction of anesthetics with neurotransmitter release machinery proteins. J Neurophysiol 109, 758–767. 10.1152/jn.00666.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hemmings HC Jr., Yan W, Westphalen RI, and Ryan TA (2005). The general anesthetic isoflurane depresses synaptic vesicle exocytosis. Mol Pharmacol 67, 1591–1599. 10.1124/mol.104.003210. [DOI] [PubMed] [Google Scholar]
- 31.Winegar BD, and MacIver MB (2006). Isoflurane depresses hippocampal CA1 glutamate nerve terminals without inhibiting fiber volleys. BMC Neurosci 7, 5. 10.1186/1471-2202-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacIver MB (2014). Anesthetic agent-specific effects on synaptic inhibition. Anesth Analg 119, 558–569. 10.1213/ANE.0000000000000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zimin PI, Woods CB, Quintana A, Ramirez JM, Morgan PG, and Sedensky MM (2016). Glutamatergic Neurotransmission Links Sensitivity to Volatile Anesthetics with Mitochondrial Function. Curr Biol 26, 2194–2201. 10.1016/j.cub.2016.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pathak D, Shields LY, Mendelsohn BA, Haddad D, Lin W, Gerencser AA, Kim H, Brand MD, Edwards RH, and Nakamura K (2015). The role of mitochondrially derived ATP in synaptic vesicle recycling. J Biol Chem 290, 22325–22336. 10.1074/jbc.M115.656405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rangaraju V, Calloway N, and Ryan TA (2014). Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. 10.1016/j.cell.2013.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Vries S, and Grivell LA (1988). Purification and characterization of a rotenone-insensitive NADH:Q6 oxidoreductase from mitochondria of Saccharomyces cerevisiae. Eur J Biochem 176, 377–384. 10.1111/j.1432-1033.1988.tb14292.x. [DOI] [PubMed] [Google Scholar]
- 37.McElroy GS, Reczek CR, Reyfman PA, Mithal DS, Horbinski CM, and Chandel NS (2020). NAD+ Regeneration Rescues Lifespan, but Not Ataxia, in a Mouse Model of Brain Mitochondrial Complex I Dysfunction. Cell Metab 32, 301–308 e306. 10.1016/j.cmet.2020.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, et al. (2013). mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342, 1524–1528. 10.1126/science.1244360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sankaranarayanan S, De Angelis D, Rothman JE, and Ryan TA (2000). The use of pHluorins for optical measurements of presynaptic activity. Biophys J 79, 2199–2208. 10.1016/S0006-3495(00)76468-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Santos MS, Park CK, Dobry Y, and Voglmaier SM (2017). VGLUT2 Trafficking Is Differentially Regulated by Adaptor Proteins AP-1 and AP-3. Front Cell Neurosci 11, 324. 10.3389/fncel.2017.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McNay EC, Fries TM, and Gold PE (2000). Decreases in rat extracellular hippocampal glucose concentration associated with cognitive demand during a spatial task. Proc Natl Acad Sci U S A 97, 2881–2885. 10.1073/pnas.050583697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rex A, Bert B, Fink H, and Voigt JP (2009). Stimulus-dependent changes of extracellular glucose in the rat hippocampus determined by in vivo microdialysis. Physiol Behav 98, 467–473. 10.1016/j.physbeh.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 43.Budzinski KL, Zeigler M, Fujimoto BS, Bajjalieh SM, and Chiu DT (2011). Measurements of the acidification kinetics of single SynaptopHluorin vesicles. Biophys J 101, 1580–1589. 10.1016/j.bpj.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leung LS, and Fu XW (1994). Factors affecting paired-pulse facilitation in hippocampal CA1 neurons in vitro. Brain Res 650, 75–84. [DOI] [PubMed] [Google Scholar]
- 45.Jang S, Nelson JC, Bend EG, Rodriguez-Laureano L, Tueros FG, Cartagenova L, Underwood K, Jorgensen EM, and Colon-Ramos DA (2016). Glycolytic Enzymes Localize to Synapses under Energy Stress to Support Synaptic Function. Neuron 90, 278–291. 10.1016/j.neuron.2016.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang H, Xu Z, Wu A, Dong Y, Zhang Y, Yue Y, and Xie Z (2015). 2-deoxy-D-glucose enhances anesthetic effects in mice. Anesth Analg 120, 312–319. 10.1213/ANE.0000000000000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan Z, Zhang Z, Zhao S, Zhu Y, Guo D, Yang B, Zhuo L, Han J, Wang R, Fang Z, et al. (2020). Dynamic Variations in Brain Glycogen are Involved in Modulating Isoflurane Anesthesia in Mice. Neurosci Bull 36, 1513–1523. 10.1007/s12264-020-00587-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szabo S, Nagy L, and Plebani M (1992). Glutathione, protein sulfhydryls and cysteine proteases in gastric mucosal injury and protection. Clin Chim Acta 206, 95–105. 10.1016/0009-8981(92)90010-n. [DOI] [PubMed] [Google Scholar]
- 49.Nahrwold ML, Lecky JH, and Cohen PJ (1974). The effect of halothane on mitochondrial permeability to NADH. Life sciences 15, 1261–1265. [DOI] [PubMed] [Google Scholar]
- 50.Nahrwold ML, Clark CR, and Cohen PJ (1974). Is depression of mitochondrial respiration a predictor of in-vivo anesthetic activity? Anesthesiology 40, 566–570. 10.1097/00000542-197406000-00010. [DOI] [PubMed] [Google Scholar]
- 51.Brunner EA, Cheng SC, and Berman ML (1975). Effects of anesthesia on intermediary metabolism. Annu Rev Med 26, 391–401. 10.1146/annurev.me.26.020175.002135. [DOI] [PubMed] [Google Scholar]
- 52.Newberg LA, Milde JH, and Michenfelder JD (1983). The cerebral metabolic effects of isoflurane at and above concentrations that suppress cortical electrical activity. Anesthesiology 59, 23–28. 10.1097/00000542-198307000-00005. [DOI] [PubMed] [Google Scholar]
- 53.Berndt N, Kovacs R, Schoknecht K, Rosner J, Reiffurth C, Maechler M, Holzhutter HG, Dreier JP, Spies C, and Liotta A (2021). Low neuronal metabolism during isoflurane-induced burst suppression is related to synaptic inhibition while neurovascular coupling and mitochondrial function remain intact. J Cereb Blood Flow Metab 41, 2640–2655. 10.1177/0271678X211010353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Imamura H, Nhat KP, Togawa H, Saito K, Iino R, Kato-Yamada Y, Nagai T, and Noji H (2009). Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci U S A 106, 15651–15656. 10.1073/pnas.0904764106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Foss SM, Dobryy YL, Park CK, Hires SA, Shaner NC, Tsien RY, Osborne LC, and Voglmaier SM (2011). Concurrent imaging of synaptic vesicle recycling and calcium dynamics. Front Mol Neurosci 4, 34. 10.3389/fnmol.2011.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Voglmaier SM, Kam K, Yang H, Fortin DL, Hua Z, Nicoll RA, and Edwards RH (2006). Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron 51, 71–84. 10.1016/j.neuron.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 57.Hua Z, Leal-Ortiz S, Foss SM, Waites CL, Garner CC, Voglmaier SM, and Edwards RH (2011). v-SNARE composition distinguishes synaptic vesicle pools. Neuron 71, 474–487. 10.1016/j.neuron.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakamura K, Nemani VM, Wallender EK, Kaehlcke K, Ott M, and Edwards RH (2008). Optical reporters for the conformation of alpha-synuclein reveal a specific interaction with mitochondria. J Neurosci 28, 12305–12317. 10.1523/JNEUROSCI.3088-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Telias M, Segal M, and Ben-Yosef D (2016). Immature Responses to GABA in Fragile X Neurons Derived from Human Embryonic Stem Cells. Front Cell Neurosci 10, 121. 10.3389/fncel.2016.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Linghu C, Johnson SL, Valdes PA, Shemesh OA, Park WM, Park D, Piatkevich KD, Wassie AT, Liu Y, An B, et al. (2020). Spatial Multiplexing of Fluorescent Reporters for Imaging Signaling Network Dynamics. Cell 183, 1682–1698 e1624. 10.1016/j.cell.2020.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tantama M, Martinez-Francois JR, Mongeon R, and Yellen G (2013). Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun 4, 2550. 10.1038/ncomms3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harb A, Vogel N, Shaib A, Becherer U, Bruns D, and Mohrmann R (2021). Auxiliary Subunits Regulate the Dendritic Turnover of AMPA Receptors in Mouse Hippocampal Neurons. Front Mol Neurosci 14, 728498. 10.3389/fnmol.2021.728498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gemba-Nishimura A, Inoue T, Nakamura S, Nakayama K, Mochizuki A, Shintani S, and Yoshimura S (2010). Properties of synaptic transmission from the reticular formation dorsal to the facial nucleus to trigeminal motoneurons during early postnatal development in rats. Neuroscience 166, 1008–1022. 10.1016/j.neuroscience.2009.12.065. [DOI] [PubMed] [Google Scholar]
- 64.Foss SM, Li H, Santos MS, Edwards RH, and Voglmaier SM (2013). Multiple dileucine-like motifs direct VGLUT1 trafficking. J Neurosci 33, 10647–10660. 10.1523/JNEUROSCI.5662-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shields LYM, B. A., Nakamura K (2017). Measuring ATP in Axons with FRET”. In book: Techniques to Investigate Mitochondrial Function in Neurons. Neuromethods 123, 115–131. [Google Scholar]
- 66.Ariel P, and Ryan TA (2010). Optical mapping of release properties in synapses. Front Neural Circuits 4. 10.3389/fncir.2010.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sonner JM, Gong D, and Eger EI 2nd (2000). Naturally occurring variability in anesthetic potency among inbred mouse strains. Anesth Analg 91, 720–726. 10.1097/00000539-200009000-00042. [DOI] [PubMed] [Google Scholar]
- 68.Sonner JM, Gong D, Li J, Eger EI 2nd, and Laster MJ (1999). Mouse strain modestly influences minimum alveolar anesthetic concentration and convulsivity of inhaled compounds. Anesthesia and analgesia 89, 1030–1034. [DOI] [PubMed] [Google Scholar]
- 69.Kayser EB, Sedensky MM, and Morgan PG (2016). Region-Specific Defects of Respiratory Capacities in the Ndufs4(KO) Mouse Brain. PLoS One 11, e0148219. 10.1371/journal.pone.0148219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.