

Abstract
BACKGROUND AND AIM:
Intrahepatic cholangiocarcinoma (ICC) is a devastating liver cancer with extremely high intra- and inter-tumoral molecular heterogeneity, partly due to its diverse cellular origins. We investigated clinical relevance and the molecular mechanisms underlying hepatocyte (HC)-driven ICC development.
METHODS:
Expression of ICC driver genes in human diseased livers at risk for ICC development were examined. Sleeping beauty and hydrodynamic tail vein injection based Akt-NICD/YAP1 ICC model was used to investigate pathogenetic roles of SOX9 and YAP1 in HC-driven ICC. We identify DNA methyltransferase-1 (DNMT1) as a YAP1 target, which was validated by loss- and gain-of-function studies, and its mechanism addressed by chromatin immunoprecipitation sequencing.
RESULTS:
Co-expression of AKT and NICD/YAP1 in HC yielded ICC which represent 16–30% of clinical ICC. NICD independently regulates SOX9 and YAP1 and deletion of either, significantly delays ICC development. Yap1 or TEAD inhibition, but not Sox9 deletion, impairs HC-to-biliary epithelial cell (BEC) reprogramming. DNMT1 was discovered as a novel downstream effector of YAP1-TEAD complex that directs HC-to-BEC/ICC fate-switch through the repression of HC-specific genes regulated by master regulators for HC differentiation including HNF4α, HNF1α and CEBPα/β. DNMT1 loss prevented Notch/YAP1-dependent HC-driven cholangiocarcinogenesis, and DNMT1 re-expression restored ICC development following TEAD repression. Co-expression of DNMT1 with AKT was sufficient to induce tumor development including ICC. DNMT1 was detected in subset of HCs and dysplastic BECs in cholestatic human livers prone to ICC development.
CONCLUSION:
We identify a novel NOTCH-YAP1/TEAD-DNMT1 axis essential for of HC-to-BEC/ICC conversion, may be relevant in cholestasis-to-ICC pathogenesis in the clinic.
Keywords: liver cancer, bile duct, precision medicine, trans-differentiation, epigenetics
LAY SUMMARY
We identify a subset of intrahepatic bile duct cancers to originate from hepatocytes instead of bile duct cells, and identify the epigenetic molecular basis of this fatal disease.
INTRODUCTION
Intrahepatic cholangiocarcinoma (ICC) has steadily risen in the US, while the five-year survival rate of ICC remains under 10%1. Although FGFR inhibitor and IDH1/2 inhibitors are recently approved as first targeted2,3, the impact of these trials are limited since only ~15% of patients harbor these aberrations and long-term survival benefits are only modest4–6. Thus, there is a dire need for improved understanding of the biology of the disease for developing better therapies.
ICC is traditionally considered to originate from cholangiocytes (biliary epithelial cells; BECs), as tumor cells exhibit luminal structures and express BEC-specific markers such as SOX9 and Cytokeratin (CK)-19. However, a subset of human ICCs have also been theorized to originate from hepatocytes (HCs), based on the frequent detection of human ICC in the pericentral zone of the liver lobule (which is anatomically distinct from the native biliary structure), the high prevalence of mixed HCC-ICC in human patients, and the well-proven occurrence of HC-to-BEC trans-differentiation in various murine models of chronic cholestasis by lineage tracing systems4,7–11. This concept has been supported by several studies demonstrating a rapid onset of HC-derived murine ICC by either thioacetamide (TAA) administration12 or by delivery of combinations of some oncogenes such as myristoylated-Akt (referred hereon as Akt) and Notch intracellular domain (NICD), Akt-YAP1, Akt-Fbxw7ΔF or KRAS G12D-sh-p53 into HCs using the sleeping beauty transposon-transposase and hydrodynamic tail vein injection (SB-HDTVI) technique8,9,11,13,14.
Sox9 and Yap1 are well known biliary-specific Notch target genes or regulators of Notch signaling, and hence critical in BEC maturation and bile duct morphogenesis in murine development15. Expression of YAP1 and SOX9 is prevalent in human ICC and positively correlates with clinical grade16,17. Despite these observations, the underlying molecular mechanisms by which these factors contribute to tumor biology has remained largely elusive.
Herein, we reveal that Akt-NICD-driven HC-derived ICC model shows high genetic similarity to a subset of human ICC cases. We also provide novel mechanistic insights into how NOTCH-YAP1/TEAD signaling governs HC reprogramming into ICC through directing the expression of DNA Methyltransferase 1 (DNMT1), repressing target gene transcription of HC-specific transcription factors including HNF4α and CEBPα/β, allowing cell fate change and eventually neoplastic transformation. Using both loss- and gain-of-function genetic studies, and pharmacological approaches, we demonstrate a heretofore unknown role of DNMT1 downstream of YAP-TEAD activation, in the fate-switch of HC-to-BEC to ICC, thus underscoring the disparate cellular origin of subsets of ICCs, which might have notable biological and therapeutic implications.
Materials and Methods
Animals.
All animal care and experiments were performed in accordance with the Institutional Animal Care and Use Committee at the University of Pittsburgh. Sox9(flox/flox) and Yap1(flox/flox) mice were purchased from Jackson Laboratories for breeding. All transgenic and KO mouse lines were maintained on the immunocompetent C57BL/6 genetic background. All animals ranged from 8–12 weeks (8–12w) in age for analysis and were from either sex.
Patient data.
All human tissue samples were provided by Pittsburgh Liver Research Center’s (PLRC’s) Clinical Biospecimen Repository and Processing Core supported by P30DK120531 under approved Institutional Review Board STUDY19070068. Tissue sections were obtained from archival formalin-fixed paraffin-embedded tissue blocks from 6 patients with healthy liver who underwent biopsy secondary to colon adenocarcinoma, 10 patients with PSC, and 10 patients with NASH (Table S1). The staining was scored as 0 (negative, 0–5 positives hepatocytes observed in a whole section); 1 (positive, <20% hepatocytes positive within a section); and 2 (positive, 20–50% of hepatocytes positive within a section). Scores of 0 are considered negative (NEG), and 1 and 2 are considered positive (POS).
Animal Models of ICC.
The constructs used for mouse SB-HDTVI, including pT3-EF1α, pT3-EF1α- myrAkt-HA (mouse), pT3-EF1α-Myc-N1ICD (mouse), pT3-EF1α-Yap1 S127A (human), pCMV-empty, pCMV-Cre, pT3-EF1α-Sox9 (mouse), pT3-EF1α-HA-myrAkt-sh-Luciferase, pT3-EF1α-HA-myrAkt-Sh-Yap1, pT3-EF1α-myrAkt-HA-Sh-Dnmt1 (mouse), pT3-EF1α-Dnmt1-V5 (mouse), DN-Tead-V5, Fbxw7, Kras G12D, sh-p53 and pCMV-SB were generated or have been described elsewhere8,11,18. All plasmids for in vivo use were purified by Endotoxin Free Maxi Prep kit (Sigma-Aldrich). 6–8w old mice were randomized into groups and subjected to the SB-HTVI as described previously18 and in supplementary information.
5-Azacytidine Treatment.
5-Azacytidine (MCE, HY-10586) was dissolved in DMSO as a 100 mg/ml stock and stored at −80°C in aliquots. Mice were i.p. injected with either 1 mg/kg 5-Azacytidine dissolved in 0.9% saline and 0.9% saline alone as a control.
Immunofluorescence and Immunohistochemistry (IHC).
These were performed as previously described19 (supplementary information). Detailed information on antibodies is provided in Table S2.
RNA sequencing analysis.
For each group (WT, Akt-NICD), 3–5 mice liver samples were processed for RNA-seq analysis. RNA from livers was used to generate library using TruSeq kit from Illumina20,21. We used PLRC’s HiSeq2500 platform and sequenced 200 million reads to accurately quantify genes and transcripts22. Raw sequencing data was analyzed by FastQC for quality control23. Low quality reads or adapter sequences were trimmed out by Trimmomatic24. Sequenced reads were aligned to mouse reference genome mm10 by HISAT2 aligner25. Read counts for each gene were then quantified by HTSeq26. All pipelines were run by default parameter settings. RNA-seq data has been submitted to online database Gene Expression omnibus (GEO) accession ID: GSE200472. Differential expression (DE) analysis was performed to compare WT versus Akt-NICD. Based on the read counts, DE tests were performed by R package ‘DEseq2’27 and top DE genes were selected by absolute fold-change greater than 1 and FDR=0.05.
Bioinformatic comparison between mouse and human models.
To test molecular similarity between Akt-NICD ICC model and human studies, 3 publicly available human ICC datasets (GSE33327, GSE26566 and GSE76297) were analyzed by two comparisons. (1) Gene signature analysis. For microarray data, samples were first pre-processed by quantile normalization to scale the expression in the same level28. Probe-based intensities were then mapped to gene-based expression. If multiple probes were annotated with the same gene, only the probe with the largest interquartile range will be utilized as representative of that gene. After pre-processing, gene expression data were analyzed by R package ‘limma’ test29 and top differentially expressed genes were selected by absolute fold-change greater than 1 and FDR=0.05 (same criteria as the mouse model). Top DE genes from mouse models were converted to human homologous genes by Mouse Genome Database (MGD)30. These genes were then applied into the human studies to check their expression signatures. (2) Signature prediction. Positive prediction of gene signatures tested in this study has been calculated by Nearest Template Prediction (Gene Pattern module)31 as previously described32.
Chromatin immunoprecipitation followed by sequencing (ChIP-Seq) data analysis.
DNMT1 ChIP-seq analysis has been performed by Active Motif, Inc (Carlsbad, CA). Detailed process and antibodies information is provided in supplementary information. DNMT1 ChIP-seq data has been submitted to online database GEO accession ID: GSE200472.
To explore TEAD4 target genes in mouse liver, public ChIP-seq data were obtained from GEO database with accession ID GSE10786033. Library for TEAD4 binding to TetO-YAP1 mice was analyzed. Quality control was first performed on the raw sequencing data using FastQC software and low-quality reads and adapter sequences were removed by tool Trimmomatic24. Surviving reads were next aligned to mouse reference genome mm10 by Burrows-Wheeler Aligner (BWA) software34 and duplicate reads were marked by Picard online tool. Eventually, peaks were called by Model-based analysis of ChIP-Seq (MACS) tool35 to detect local enriched TEAD4 binding sites. Selected binding regions were annotated to mouse genes for downstream analysis.
Searching for transcription factor binding sites.
Validated binding motives for transcription factor TEAD2, TEAD3 and TEAD4 were initially obtained from TRANSFAC database. Find Individual Motif Occurrences (FIMO) tool were applied to scan 5,000 bp upstream sequence of Dnmt1 promoter region for respective motives. Significant matching sequences (p≤0.0001) and their corresponding motives were displayed.
Statistical Analysis.
For all mouse experiments, sample size was pre-determined based on previous literature describing SB-HDTVI-mediated liver carcinogenesis18. Accordingly, littermates were randomized into groups for HDTVI and managed throughout the course of treatment in a non-blinded manner. All subsequent molecular, IHC, and immunofluorescence analysis was performed in a blinded manner. All confidence intervals shown on the bar plots are presented as mean ± standard error of mean (SEM). Differences in mean values of liver volume and liver weight-to body weight ratio (LW/BW) were analyzed by one-way ANOVA assuming normal Gaussian distribution with Geisser Greenhouse posttest correction. For patient data, Fisher’s exact test (two-sided) with post hoc pairwise comparison was utilized to assess statistical significance. p<0.05 was considered significant (*), p<0.01 was considered highly significant (**), p<0.005 was considered extremely significant (***), and so on. All statistical analysis on patient samples is included in the results section and respective p-values included in the pertinent text and figure legends. All statistics were performed using GraphPad Prism 8.0 Software or R software.
RESULTS
p-AKT, and NICD effectors YAP1 and SOX9, are activated in pre-malignant liver diseases.
SB-HDTVI induces HC-driven ICC in Akt-NICD and Akt-YAP1 mouse models8,11. To investigate the relevance of expressing these gene combinations in HCs, we investigated localization of phospho-Ser473-AKT (or p-AKT) and NICD targets SOX9 and YAP115,36, in liver samples of patients with non-alcoholic steatohepatitis (NASH) and primary sclerosing cholangitis (PSC), 2 known risk factors of ICC10,37. We identified significant increase in cytoplasmic and nuclear p-AKT (p=0.024), nuclear SOX9 (p=0.0008) or nuclear YAP1 (p=0.047) in subsets of HCs in all patients (n=20), whereas these markers were rarely detected in HC of healthy controls (Fig.1A, S1A, S1B, Table S1). 35% of cases showed some degree of upregulation of all 3 markers, while 55% showed upregulation of at least 2 of the 3 markers in subsets of HCs (Fig.1B, S1B–E). These results support the relevance of studying SB-HDTVI-driven tumorigenesis in Akt-NICD and Akt-YAP1 murine models.
Figure 1. Upregulation of AKT and NICD targets YAP1 and SOX9 in hepatocytes in patients with risk for ICC.
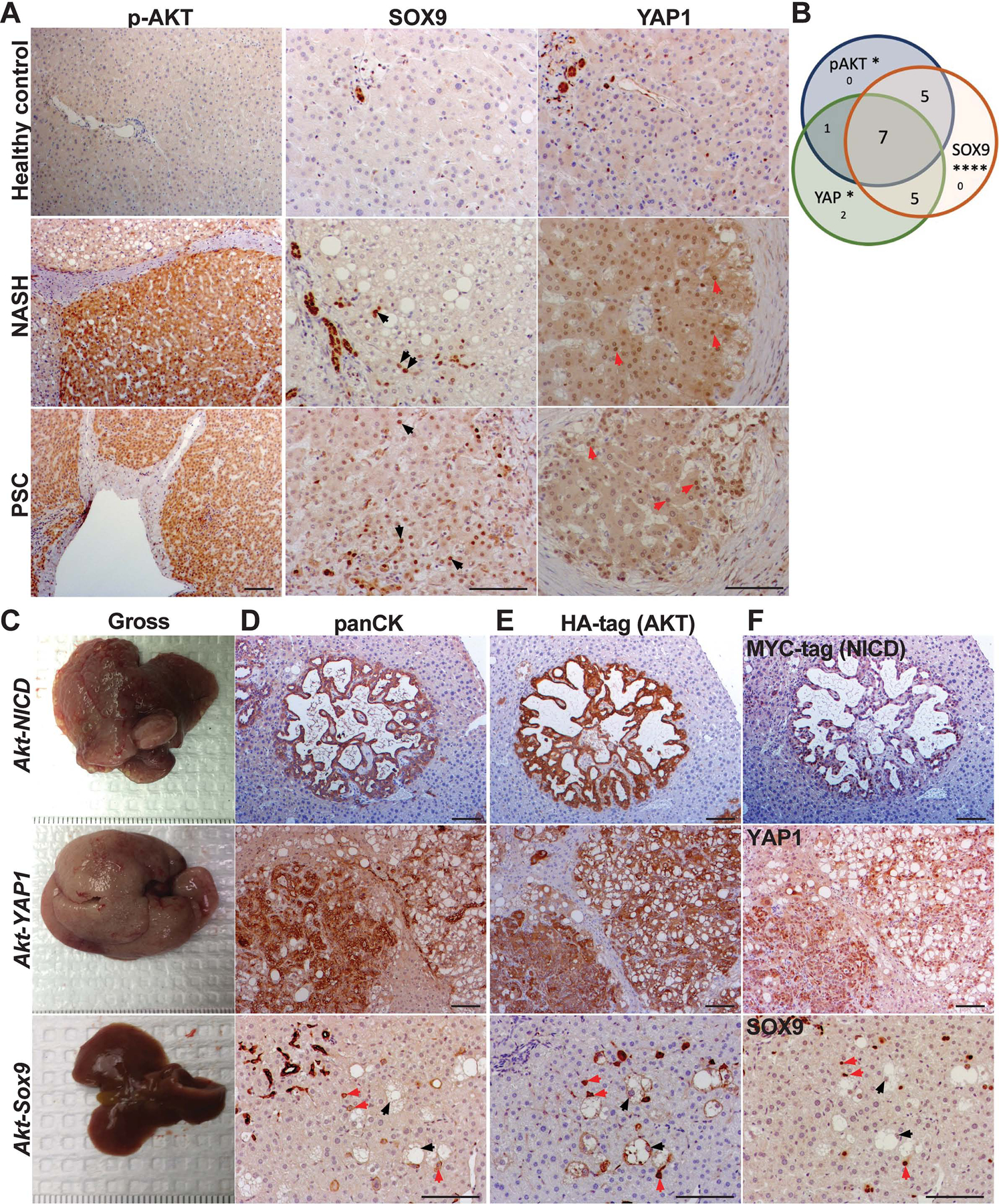
(A) Representative IHC images of liver section from patients with NASH and PSC showing increased p-AKT, SOX9 and YAP1 expression as compared to healthy liver. Black arrows point to YAP1+ hepatocytes; red arrows to SOX9+ hepatocytes; white arrows to p-AKT+ hepatocytes. (B) Venn diagram showing overlap of patient samples with NASH and PSC that exhibited aberrant induction of either p-AKT, SOX9, or YAP1 levels in HCs specifically. (C) Gross images of livers from Akt-NICD (upper panel), Akt-YAP1 (middle panel) and Akt-Sox9 injected mice (lower panel) at 5w post-HDTVI (D) Representative IHC staining for panCK depicts presence of ICC in Akt-NICD model (upper panel), mixed ICC-HCC in Akt-YAP1 model (middle panel) and lack of any tumors with positive staining in normal ducts. Red arrows indicate HA-Akt and Sox9-transfected HCs and black arrows, HA-Akt-transfected but SOX9− foamy hepatocytes in AKT-Sox9 model (lower panel). (E) Representative IHC for HA-tag to identify myr-AKT in serial sections (to D) (upper panel) and mixed ICC/HCC in Akt-YAP1 model (middle panel). Red arrows indicate HA-Akt and Sox9-transfected HCs and black arrows, to HA-Akt-transfected but SOX9− foamy hepatocytes in AKT-Sox9 model (lower panel). (F) Representative IHC on serial sections (to D and E) to identify MYC-tag to identify NICD (upper panel), YAP1 (middle panel) and SOX9 (lower panel) at 5w in Akt-NICD, Akt-YAP1 and AKT-Sox9 livers. Scale bars:100 μm; *p<0.05; ****p<0.0001.
Myristoylated-AKT cooperates with NICD or YAP1 but not SOX9 in inducing hepatic tumors.
Next, we established SB-HDTVI models to investigate the role of NICD effectors YAP1 and SOX9 along with AKT activation, in hepatic tumorigenesis. We observed rapid development of HC-derived ICC 4–5w after HDTVI of Akt (HA-tag) and NICD (Myc-tag) in the Akt-NICD model, while co-delivery of Akt and YAP1-S127A resulted in mixed ICC/HCC in Akt-YAP1 model (Fig.1C–F)8,11. IHC confirmed ICCs to be positive for biliary pan-cytokeratin (panCK), HA-tag (AKT), MYC-tag (NICD), or YAP1 (Fig.1D–F). Given the role for SOX9 downstream of Notch signaling in HC-to-BEC conversion in liver injury36,38,39, we generated Akt-Sox9 model. No tumors were observed at 5w post-injection, either macroscopically or microscopically (Fig.1C–F), and up to 4 months (4m) (not shown). Histologically, HCs transduced with Akt and Sox9 only weakly expressed panCK and exhibited an intermediate morphology between HCs and BECs, remaining as single cells even at 5w (red arrows, Fig.1D–F). Thus, SOX9 is insufficient to drive HC-derived ICC downstream of Notch signaling, despite concurrent AKT activation.
Transcriptomic analysis of HC-derived ICC in Akt-NICD model reveals significant similarity to a subset of human ICCs.
To address the clinical relevance of ICC in the Akt-NICD model, we performed RNA-Seq analysis. When comparing the WT and Akt-NICD livers, 4,284 genes were up-regulated and 2,208 genes were down-regulated by FDR=5% and absolute log2 fold change>1 (Fig.2A) (GEO accession ID: GSE200472). To determine if the mouse model mimics a subtype of patient ICC, a human study GSE33327 of ICC was analyzed40, using similar pipeline. When comparing 6 controls and 149 ICCs categorized as inflammatory (n=57) and proliferative (n=92), 590 upregulated and 781 down-regulated genes were enriched. Another human study (GSE26566), consisting of 104 ICC and 59 surrounding liver samples, was assessed41. By same approach, 3,097 upregulated and 2,876 down-regulated genes were identified. Similarly, a third human study GSE76297 composing 92 non CCA tumor and 91 CCA tumor samples were analyzed, where 759 up and 1,018 down genes were detected. To directly compare mouse and human study, top differentially expressed genes (DEGs) from the Akt-NICD ICC model were converted to human homologues by the Mouse Genome Database42. Top DEGs were then applied to human study GSE33327 and 372 top genes are shown (Fig.2B). These genes could clearly separate normal (orange), inflammation (green) and proliferation (pink) ICC subgroups in patients. Similarly, DEGs from the Akt-NICD mouse model were also applied to GSE26566. In total 1,862 common differentially expressed genes between mouse and human studies that could clearly stratify normal surrounding liver (orange) and ICC (green) (Fig.2C). DEGs from the Akt-NICD mouse model were also applied to the third human study GSE76297. The overlapping 747 DEGs could also separate the non-tumor samples (orange) and ICCs (green) in this dataset (Fig.2D).
Figure 2: RNA-seq analysis of murine Akt-NICD ICC model and comparison with human ICC cases.
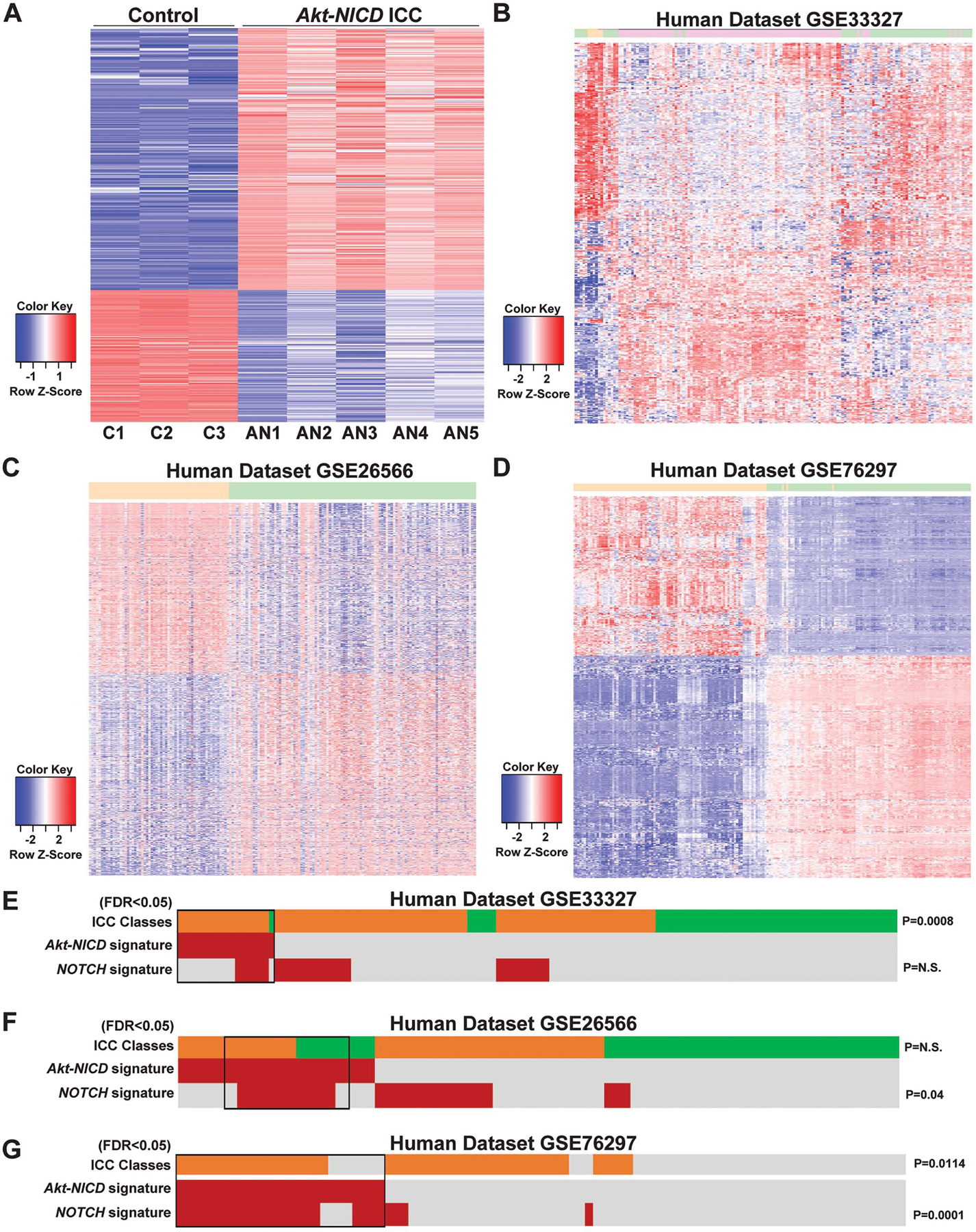
(A) Heatmap for the differentially expressed genes comparing wild-type (WT) liver and Akt-NICD ICC. (B) Heatmap of gene signatures in human GSE33327 study that are selected by mouse model (WT VS Akt-NICD). Orange: normal; Green: inflammation-class; Pink: proliferation-class. (C) Heatmap of gene signatures in human GSE26566 study that are selected by mouse model (WT VS Akt-NICD). Orange: surrounding liver; Green: ICC. (D) Heatmap of gene signatures in human GSE76297 study that are selected by mouse model (WT VS Akt-NICD). Orange: non-tumor; Green: ICC. Nearest Template Prediction (NTP) analysis of three human whole-tumor gene expression datasets GSE33327 (E), GSE26566 (F) and GSE76297 (G) using the Akt-NICD signature generated in this study. Inflammation class is indicated in green; Proliferation class is indicated in orange. In the heatmap, each column represents a patient, and each row represents a different signature; positive prediction of signatures as calculated by NTP is indicated in red and absence in gray. p values that show significant correlation are indicated to the right of the NTP analysis.
Nearest Template Prediction (NTP) analysis was performed using the top 200 DEGs derived from Akt-NICD signature (Table S3). In GSE33327, a small percentage of human patients (13%, 20/149) were positively predicted by the Akt-NICD signature (Fig.2E, red). Patients molecularly similar to Akt-NICD model belonged mostly to the previously described “proliferation class”40 (19/92 vs 1/57, p=0.0008). In the GSE26566 dataset, 28% (26/110) of ICC patients were predicted to be positive by Akt-NICD signature while there was no significant enrichment in proliferation class within this cohort (Fig.2F, orange). In GSE76297, around the same percentage of patients were predicted to be positive by Akt-NICD signature (29%, 26/91); 73% of these patients (19/26), were predicted to belong to the Proliferation class versus 43% (28/65) in the rest of the cohort (Fig.2G, p=0.01). In GSE26566 and GSE76297 but not in GSE33327, the group of human patients resembling the Akt-NICD murine model was significantly enriched in NOTCH signaling (p<0.05)41. Altogether, Akt-NICD model represents a subset of human ICC, possibly suggesting HC origin of ICC in a subset of cases.
Conditional Yap1 or Sox9 deletion significantly delays cholangiocarcinogenesis in the Akt-NICD model.
Given the high nuclear levels of YAP1 and SOX9 in human ICC and a well-known interaction of NOTCH, SOX9 and YAP1 in biliary compartment4,10,11,16,17,36,42, we next investigated the roles of YAP1 and SOX9 in the Akt-NICD model. In addition to Akt-NICD, we co-delivered pCMV-Cre into Yap1flox/flox or Sox9flox/flox mice, to delete Yap1 or Sox9 in transduced HCs, respectively (labeled as Yap1KO or Sox9KO) (Fig.3A). As controls, pCMV-Empty was injected into Yap1flox/flox or Sox9flox/flox mice and for analysis, both groups were combined and called wild-type (WT). All WT mice developed a lethal burden of ICC requiring euthanasia by 5–6w post SB-HDTVI (Fig.3B–F). In contrast, only 1 of 6 Sox9KO became sick due to tumor burden requiring euthanasia at 5w, while the remaining 5 showed no signs of illness, surviving until sacrifice at 38 days (38d). All Akt-NICD Yap1KO mice were healthy at 6w, when they were sacrificed (Fig.3B). Grossly, Akt-NICD Yap1KO or Sox9KO had only rare tumor and significantly lower LW/BW compared to widespread gross disease in WT (Fig.3C–F). Histology verified significantly reduced ICC burden in Yap1KO and Sox9KO livers (Fig.3E–F). Altogether, conditional elimination of either Yap1 or Sox9 significantly diminished HC-derived ICC in the Akt-NICD model.
Figure 3. Tumor-specific Sox9 or Yap1 deletion significantly delays Akt-NICD-mediated hepatocyte-derived ICC development.
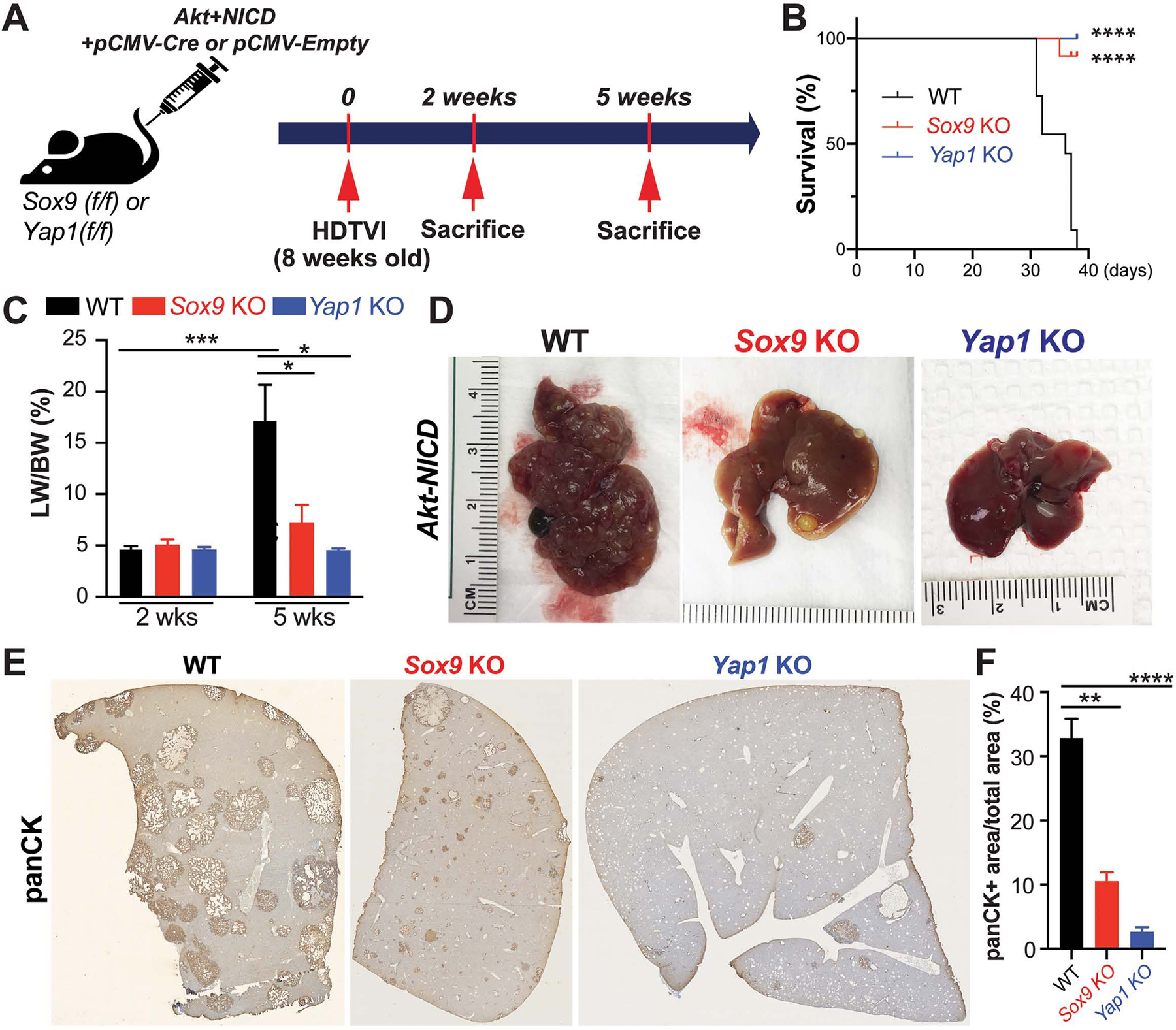
(A) Experimental design illustrating plasmids used for HDTVI, mice used in study and time-points analyzed. (B) Kaplan–Meier curve showing improved survival of Sox9KO and Yap1KO as compared to WT (C) LW/BW ratio depicts comparable low tumor burden in Akt-NICD Sox9KO, Yap1KO and WT mice at 2w but significantly lower in Sox9KO and Yap1KO at 5w. (D) Representative gross images from Akt-NICD WT show multiple large tumors at 5w, with only occasional small tumor seen in Sox9KO and few gross tumor nodules in Yap1KO at the same time. (E) Representative tiled image of panCK staining in Akt-NICD WT mice, Sox9KO and Yap1KO at 5w and quantification (F). Scale bars:100 μm; error bar: standard error of the mean; *p<0.05; **p<0.01; ***, p<0.001; ****p<0.0001.
YAP1, but not SOX9 is essential in Akt-NICD-mediated HC-to-BEC/ICC fate-change.
Given the roles of YAP1 and SOX9 in biliary fate determination42, we next investigated HC-to-BEC reprogramming in the Akt-NICD-transduced HCs in WT, Sox9KO and Yap1KO, 2w post-HDTVI (Fig.4A). At this early stage, Akt-NICD-transduced cells in WT livers begin to display biliary morphology and positivity for SOX9, YAP1 and panCK, and lose HNF4α expression as shown by IHC on serial sections (Fig.4B, red dashed line). These observations were also substantiated by immunofluorescence (Fig.4C). At this stage, Yap1-deleted, Akt-NICD transduced cells continued to exhibit HC morphology and remained HNF4α-positive by IHC and immunofluorescence (Fig.4B, blue dashed line). A subset of Yap1-deleted cells co-expressed SOX9 and HNF4α (Fig.4C, white arrows), and represent a cell-type with incomplete HC-to-BEC reprogramming. Importantly, this intermediate cell population was not detected at 2w in the Akt-NICD WT livers. Sox9-deleted Akt-NICD transduced cells, like WT, showed biliary morphology with positive biliary markers (YAP1 and panCK) and absence of HNF4α, by IHC (Fig.4B, green dashed line) and immunofluorescence (Fig.4C). Altogether, these observations support a unique role of YAP1 in Notch-dependent HC-to-BEC/ICC transdifferentiation.
Figure 4. Role of Yap1 but not Sox9 in hepatocyte-to-biliary reprogramming in Akt-NICD-driven cholangiocarcinogenesis.
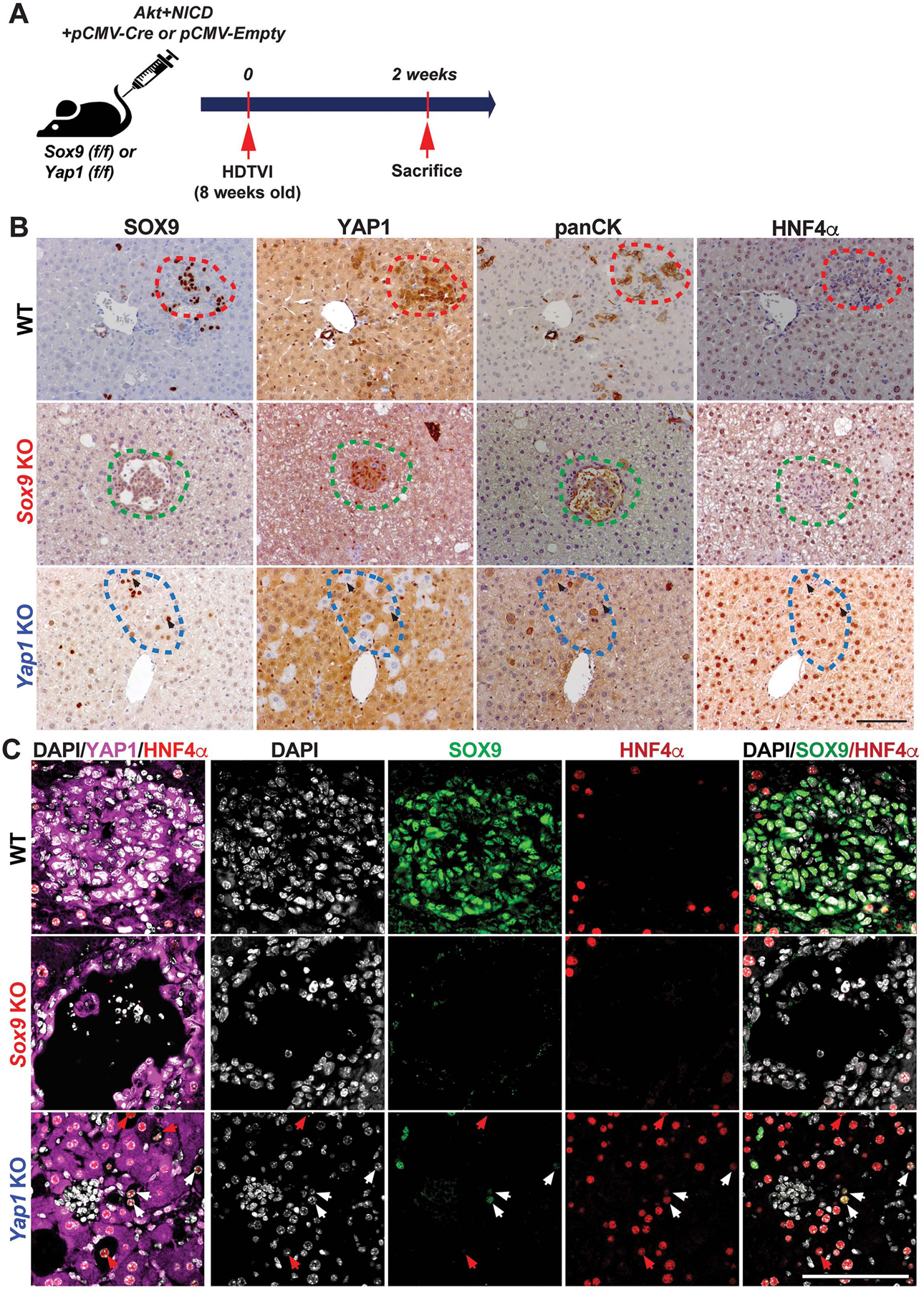
(A) Experimental design illustrating plasmids used for HDTVI, mice used in study and time-points analyzed. (B) Representative IHC staining of WT (red-dashed lines), Sox9KO (green-dashed lines), and Yap1KO livers (blue-dashed lines) at 2w after Akt-NICD injection. (C) Confocal images of IF staining of WT, Sox9KO and Yap1KO livers at 2w verify IHC results in B. Red arrows point to YAP1−;HNF4α+;SOX9− cells, white arrows to YAP1−;HNF4α+;SOX9+ cells. Scale bars:100 μm.
DNMT1 is required for NOTCH-YAP1-driven HC-to-BEC/ICC fate-switch.
Next, we sought to delineate the molecular mechanism by which YAP1 regulates HC-to-BEC/ICC fate-switch under the Notch signaling. A recent study revealed Nuak2 as a key downstream factor for YAP1-driven HCC development by ChIP-seq33. However, Nuak2 expression did not change in Akt-NICD-driven ICC in our RNA-seq data. To assess possible YAP1 target genes in HC-to-BEC reprogramming, we assessed public ChIP-seq data analyzing HC-specific YAP1 over-expression33. We identified 393 potential TEAD4 target genes in YAP1-active HCs. Since, epigenetic regulation including methylation, has been suggested to contribute to cell fate switches43–46, we focused on a master epigenetic regulator DNMT1 (Fig.S2A). We confirmed existence of TEAD2/3/4-specific binding motif in the Dnmt1 promoter (Fig.S2A–B). Based on these observations, we hypothesized that YAP1/TEAD complex upregulates Dnmt1 transcription, and subsequently DNMT1 represses epigenetic targets which maintain HC identity, thus facilitating the conversion of HC-to-BEC/ICC in the Akt-NICD model. Indeed, within 2w of injection of Akt-NICD, the dual transfected cells show high levels of nuclear DNMT1 (Fig.5A).
Figure 5. DNMT1 is required for YAP1-TEAD-driven HC-to-ICC transformation.
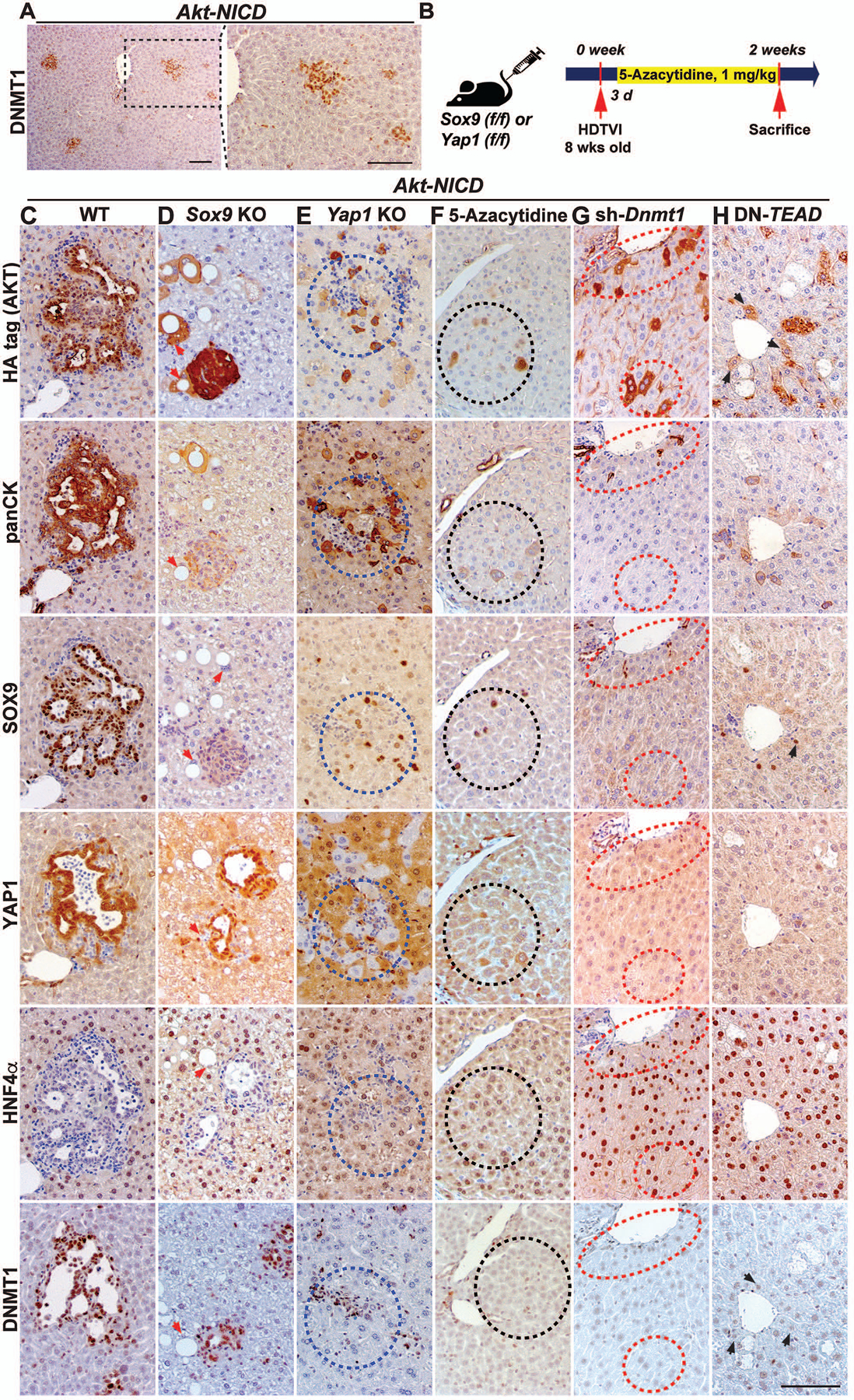
(A) Representative IHC showing DNMT1 induction in Akt-NICD-transfected HCs in the early stage of HC-to-BEC conversion at 2w post-injection. (B) Experimental design illustrating 5-Azacytidine treatment, plasmids used for HDTVI, mice used in study and time-points analyzed. Representative IHC of WT (C) or Sox9KO (D) livers showing Akt-NICD-transfected cells. (E) Yap1KO livers showing Yap1-deleted Akt-NICD-transfected cells with imperfect HC-to-biliary reprogramming (blue-dashed lines). 5-Azacytidine-treated (F), Dnmt1-silenced (G) or TEAD-inhibited livers (H) showing defective HC-to-BEC reprogramming. Akt-NICD-transfected cells (black- or red-dashed lines). Red arrows point to Akt-HA transduced cells. Black arrows point to Akt-HA transduced cells in DN-TEAD livers. Scale bars:100 μm.
Further, we carefully examined the expression pattern of DNMT1 in diverse cell types in the liver using WB, IHC and co-IF analysis (Fig.S2C–F). In addition to ICC cells (Fig.5C), DNMT1 expression was detected in all CD45+ immune cells but not in CD31+ endothelial cells (Fig.S2C, D). Interestingly, only subset of CK19+ BECs express DNMT1 suggesting heterogeneity in DNMT1 activity in adult bile duct (Fig.S2D, arrows). Weak DNMT1 expression is also observed in HCs (Fig.S2C, arrows, Fig.5C) and was dramatically increased in HC-derived ICC, suggesting its likely role in HC-to-ICC transformation (Fig.5C, S2F).
To investigate the role of DNMT1 in HC-driven cholangiocarcinogenesis, we employed both genetic and pharmacologic approaches. We used an FDA-approved DNMT1 specific inhibitor 5-Azacytidine and Akt-sh-Dnmt1 plasmid in the Akt-NICD-ICC model (Fig.5B). Remarkably, both approaches of DNMT1 inhibition completely inhibited Notch-YAP1-dependent initial HC-to-BEC conversion at 2w (Fig.5F, black dashed line; Fig.5G, red dashed line). Upon DNMT1 inhibition, the Akt-NICD-transduced cells continued to display HC morphology retaining HNF4α expression (Fig.5F, black dashed line; Fig.5G, red dashed line). Akt-NICD-transduced HCs in WT or Sox9KO liver converted into BEC/ICC exhibiting BEC morphology, expressing SOX9, YAP1 and panCK, and losing HNF4α (Fig.5C, WT or Fig.5D, Sox9KO). Given the similarities in phenotypes between DNMT1-inhibited and Yap1-deleted Akt-NICD-ICC, the presence of well-defined TEAD4 binding on Dnmt1 genomic locus (Fig.S2A), and the presence of DNMT1 in early HC-derived BEC/ICC (Fig.5A), we next compared DNMT1 expression in Akt-NICD-WT, Akt-NICD-Yap1KO and Akt-NICD-Sox9KO liver, 2w post-injection. In Akt-NICD injected WT or Sox9KO liver, DNMT1 was detected in HA-tag+;YAP1+ ICC (Fig.5C, WT or Fig.5D, Sox9KO), while HA-tag+;YAP1−;HNF4α+ cells which exhibited impaired HC-to-ICC conversion, were negative for DNMT1 (Fig.5E, blue dash line). This suggested YAP1-dependent regulation of DNMT1 in Notch-driven HC-to-BEC/ICC conversion. Likewise, DNMT1 was not detected in any of HCs transduced with Sox9 alone at 3w after injection (Fig.S3A, red dashed line). Akt singular transduced foamy cells were DNMT1− (Fig.5D, red arrows), whereas HA-tag+;SOX9− cells strongly expressed DNMT1 (Fig.5D, Sox9KO), suggesting AKT and SOX9 are not involved in DNMT1 induction. Given the strong DNMT1 expression in Akt-NICD-dependent HC-derived-ICC, we also examined DNMT1 expression in other SB-HDTVI or chemical-induced HC-derived ICC models with distinct molecular drivers. We found strong DNMT1 expression in ICCs at 5w in the Akt+Fbxw7ΔF (Fig.S3B), Kras G12D+sh-p53 (Fig.S3C) and in the ICC component of the Akt-Yap-driven mixed ICC/HCC model (Fig.S3E). We also observed strong DNMT1 expression in ICCs that develop after 24w treatment with TAA (300mg/ml) (Fig.S3D). Thus, we observed strong DNMT1 expression in all examined HC-derived ICC models, suggesting its critical role in HC-to-ICC transformation irrespective of molecular drivers.
NICD-YAP drives HC-to-ICC transformation through TEAD-dependent DNMT1 induction.
Although both YAP1 and DNMT1 inhibition prevents Akt-NICD-driven HC-to-ICC conversion at 2w post-injection, Yap1-deleted-mice eventually develop YAP1−;DNMT1+ ICC at 5w post injection (Fig.3, S4A–B), suggesting an existence of YAP1-independent Dnmt1 compensation. Indeed, TAZ, with 47% amino acid sequence identity to YAP1 and encoded by a paralogue, is known for such redundancy, and simultaneous deletion of Yap1 and Taz potently represses Akt-NICD-ICC47,48. Given that transcription factor TEAD1/2/3/4 possess a similar DNA binding motif49 and is required by TAZ or YAP1 to regulate target gene expression, we next asked if using truncated-mutant lacking DNA binding domain or dominant negative (DN)-TEAD, could impair Akt-NICD-driven ICC development. Given that DN-TEAD expression represses transcriptional activity of all TEADs and of YAP1-TEAD/TAZ-TEAD complex49–51, we used DN-Tead2 expression plasmid along with Akt-NICD to attain tumor-specific co-expression (Fig.5H). As controls, we injected NICD, pT3-Ef1α-Empty and Akt plasmid to develop wild-type Akt-NICD-ICC (Fig.5C). Consistent with previous results, Akt-NICD-transduced HCs yielded SOX9+;YAP1+;panCK+;HNF4α− ICC with biliary morphology (Fig.5C). Upon DN-TEAD co-delivery, the Akt-NICD-transduced cells continued to display HC morphology maintaining HNF4α expression, indicating a block in HC-to-BEC trans-differentiation (Fig.5H, black arrows), similar to what was observed with YAP1 or DNMT1 inhibition at same stage (Fig.5E–G). Intriguingly, we found that over-expression of NICD alone induced fate-switch of HC into SOX9+;YAP1+;panCK+;DNMT1+;HNF4α− BEC at 3w post-injection, although these foci were notably fewer than NICD-Akt (Fig.S4C–D). NICD and DN-TEAD co-transduced cells retained HC morphology and HNF4α expression and never acquired SOX9, panCK or DNMT1 expression (Fig.S4C–D), indicating that NOTCH-YAP1/TEAD cascade drives HC-to-BEC fate conversion and ICC development through DNMT1.
Pharmacologic DNMT1 inhibition induces molecular phenotype switch of Akt-YAP1-driven mixed ICC/HCC to HCC.
Previously, it has been reported that co-delivery of Akt and YAP1, instead of NICD, induced HC-derived mixed ICC/HCC in a Notch-dependent manner11. Tumor-specific Notch2 deletion switched the tumor type in the Akt-YAP1 model from ICC-dominant but mixed ICC/HCC to a benign hepatocellular adenoma-like tumor at the expense of ICC. Given that DNMT1 is strongly induced only in Akt-YAP1-mediated ICC nodules (SOX9+) whereas DNMT1 expression is weak or absent in HCC regions (Fig.S3E), we sought to investigate the impact of pharmacologic DNMT1 inhibition in the Akt-YAP1 model. Akt and YAP1 S127A plasmids were co-delivered by SB-HDTVI into WT and 5-Azacytidine was administered i.p. to inhibit DNMT1 (Fig.6A). Mice were sacrificed at 2w post-injection to evaluate HC-to-BEC/ICC fate conversion. Macroscopically, there was no significant difference in gross tumor burden (Fig.6B) or LW/BW ratio between 5-Azacytidine- and vehicle-treated liver (Fig.6C). Microscopically, the vehicle-treated Akt-YAP1 livers showed many intensely SOX9-positive ICC nodules scattered throughout a lobe (Fig.6D). These tumors were mixed and showed ICC component, which was SOX9+;YAP1+;panCK+ and HCC component with nuclear HNF4α (Fig.6E). 5-Azacytidine group showed diffuse HA-positive tumor foci which were SOX9− but HNF4α+ and of HC morphology (Fig.6D–E). The tumors were HCCs, which lacked panCK, expressed HNF4α and showed scattered and small subset of YAP1+ cells (Fig.6E). Also, we assessed cell death by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining at 3 weeks post Akt-YAP1 delivery and found comparable number of TUNEL+ cells in both of vehicle- or 5-azacytidine-treated livers (Fig.S3F). Importantly, most of TUNEL+ cells retain HC morphology (Fig.S3F arrows), indicating that absence of ICC tumor in 5-Azacytidine-treated liver is not due to enhanced cell death of HC-derived BEC/ICC. Altogether, these data show that DNMT1 is required for YAP1-driven HC-to-BEC/ICC fate switch while its loss does not impair Akt-YAP1-driven HCC development.
Figure 6: Pharmacologic DNMT1 inhibition switches the fate of Akt-YAP1-transduced hepatocytes from mixed ICC/HCC to HCC at the expense of ICC.
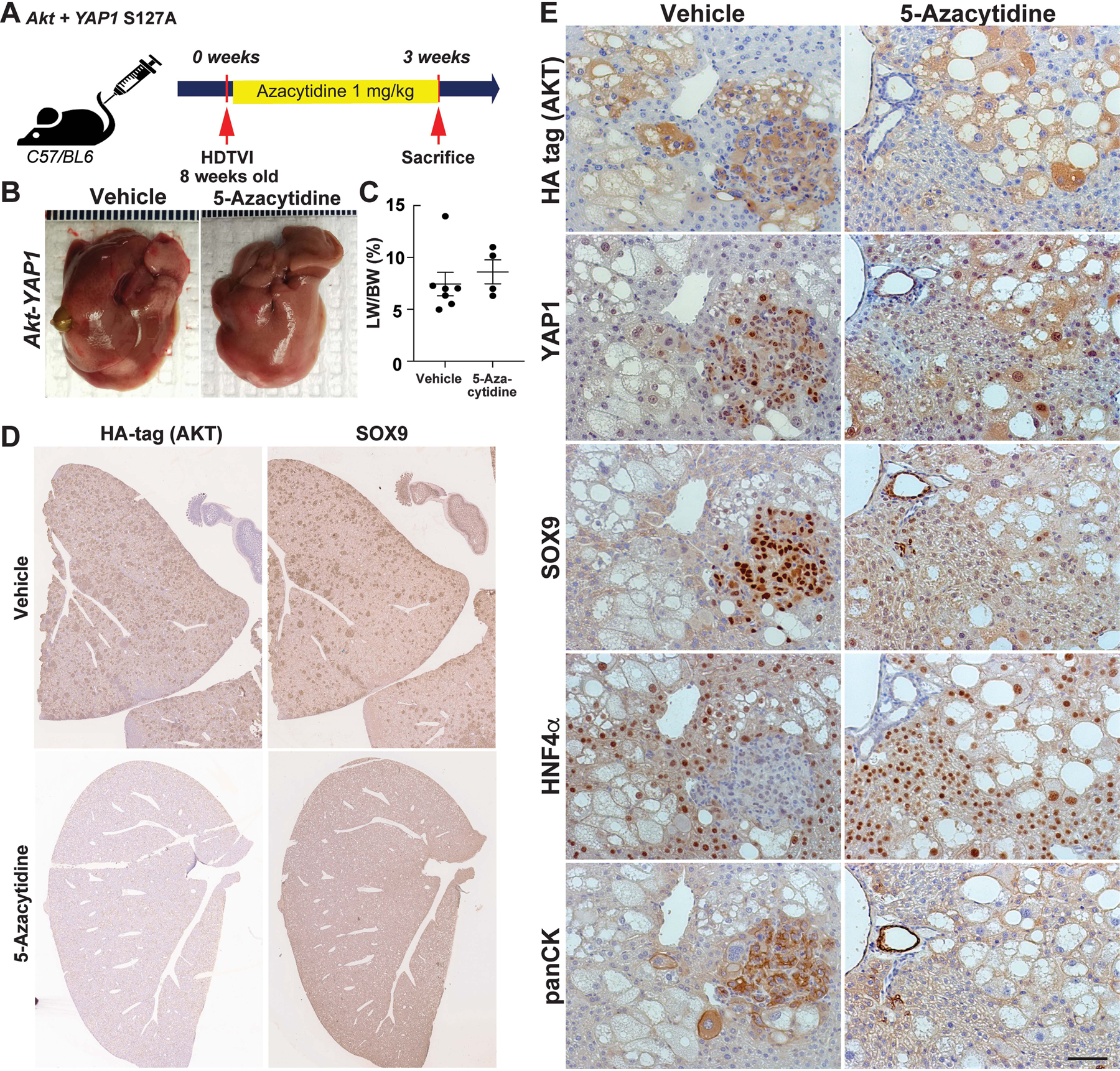
(A) Experimental design illustrating 5-Azacytidine treatment, plasmids used for HDTVI, mice used in study and time-points analyzed. (B) Representative gross images show comparable Akt-YAP1 tumor development between Vehicle or 5-Azacytidine-treated livers at 3w post-injection (C) LW/BW ratio depicts comparable tumor burden in Vehicle or 5-Azacytidine-treated animals at 3w. (D) Representative tiled image of HA-tag (AKT) and SOX9 staining in ICC nodules of the mixed ICC/HCC lesions in vehicle- or 5-Azacytidine-treated livers. (E) Representative IHC images of 3w vehicle-treated livers bearing Akt-YAP1-driven ICC component and HCC positive for HNF4α. In 5-Azacytidine-treated livers, no ICC but only HCC was detected.
DNMT1 or TEAD inhibition permanently abrogate Akt-NICD-driven ICC.
Given that DNMT1 or TEAD inhibition completely blocked NICD-YAP1-dependent HC-to-ICC conversion at 2w post-HDTVI in the Akt-NICD/YAP1 model (Fig.5E–G), we next asked if this abrogation of ICC development is permanent, unlike after Yap1 deletion in which tumors were evident later (Fig.3, Fig.S4B). We injected Akt-NICD along with DN-TEAD expression plasmid, or treated with 5-Azacytidine until sacrifice, and assessed these animals at 5w post-HDTVI (Fig.7A). Akt-NICD-injected mice developed lethal ICC requiring euthanasia at 5w while both DNMT1- and TEAD-inhibited mice were asymptomatic, normal LW/BW ratios and no gross tumors (Fig.7B–C). Histology verified presence of none to a rare microscopic HA-tag (AKT)+ tumor foci in 5-Azacytidine-treated (Fig.S4E–F) or TEAD-repressed group (Fig.7D). These data show an indispensable role of TEAD-DNMT1 axis in Akt-NICD-dependent HC-driven ICC development, and also suggests that singular targeting of either is sufficient to eliminate Akt-NICD-ICC without relapse due to any redundant compensatory mechanism, unlike targeting YAP1 alone.
Figure 7. NICD-YAP1/TEAD-DNMT1 axis drives HC-to-ICC transformation.
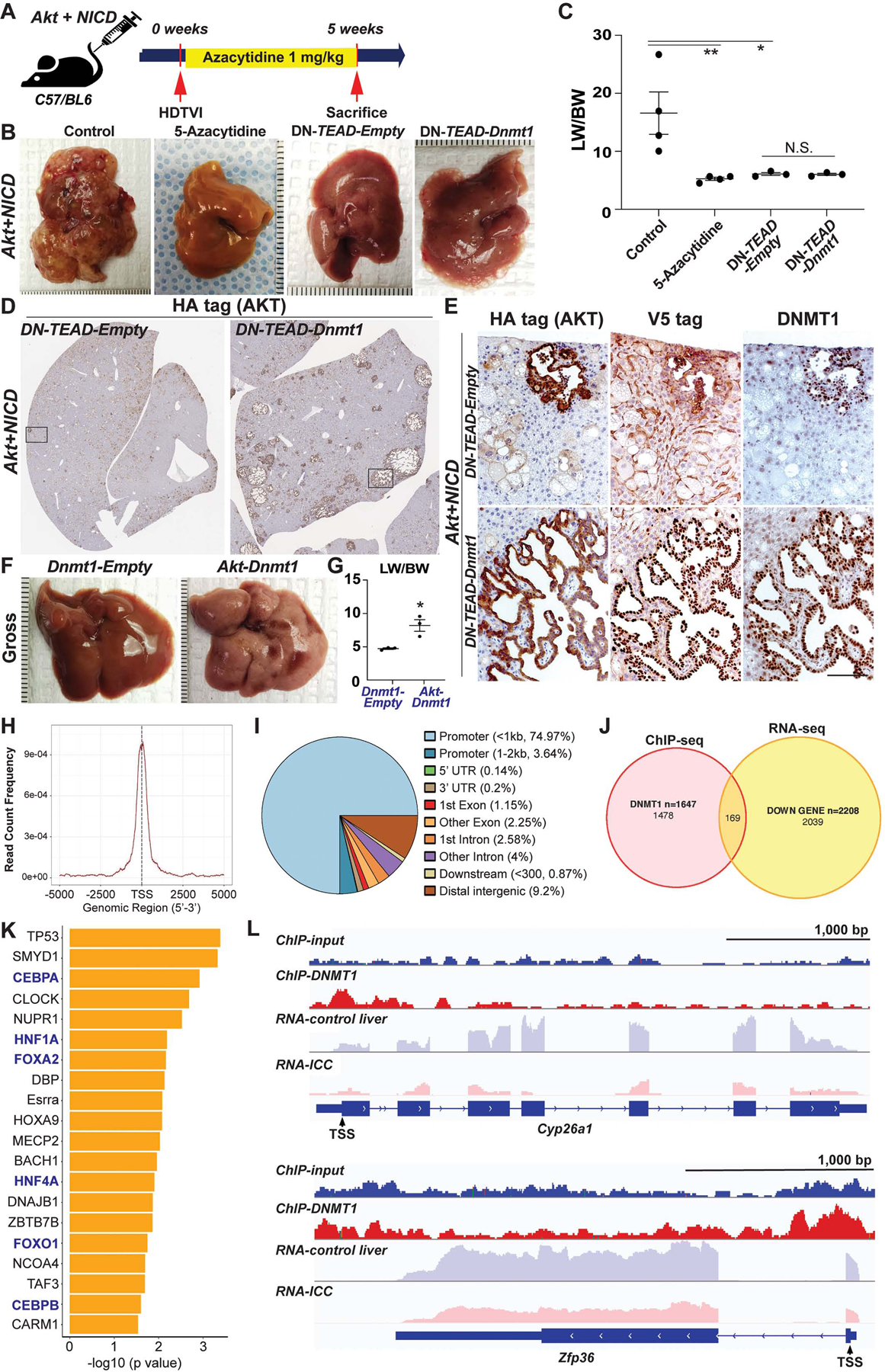
(A) Experimental design illustrating 5-Azacytidine treatment, plasmids used for HDTVI, mice used in study and time-points analyzed. (B) Representative gross images from Akt-NICD control, 5-Azacytidine-treated or DN-TEAD injected livers at 5w. (C) LW/BW ratio depicts comparable low tumor burden in 5-Azacytidine-treated, DN-TEAD and DN-TEAD-Dnmt1 co-injected animals at 5w. (D) Representative tiled image of HA-tag (AKT) staining showing near-complete abrogation of tumor by TEAD repression whereas widespread cystic ICC nodules were detected in Dnmt1 re-expressed livers. (E) IHC of squared region (D panel) showing HA-tag+;V5-tag (DNMT1)+;endogenous DNMT1+ ICC nodules in Dnmt1 re-expressed liver (bottom). Few tumor nodules observed in Akt-NICD; DN-TEAD were V5-tag− but positive for HA-tag and endogenous DNMT1 (top). Representative gross images (F) and LW/BW (G) ratio from Akt-Dnmt1-injected group shows tumor burden within entire liver lobes while no gross tumor nodule was observed in Dnmt1-Empty-injected livers. (H) Enrichment of DNMT1 ChIP-Seq binding within +/− 2.5kb from transcription start site (TSS) region. (I) Annotation of 2,174 DNMT1 ChIP-Seq peaks relative to Ref Seq transcript annotations showing robust enrichment in promoter region. (J) Venn diagrams showing the overlap of DNMT1 binding (ChIP-seq) and decreased RNA expression (RNA-seq) in Akt-NICD-ICC. (K) IPA-URA analysis results displaying top 20 predicted upstream transcriptional regulators for 169 genes shown in (J). (L) Visualization of DNMT1 ChIP-Seq peaks in correlation with transcripts mapping to representative genes within each regulatory group.
Pharmacologic DNMT1 inhibition does not lower fully developed ICC tumor burden.
Given that 5-Azacytidine treatment completely prevents Akt-NICD-ICC development, we next investigated the therapeutic effect of pharmacologic DNMT1 inhibition on fully developed ICC representing advanced tumor stage. Akt and NICD plasmids were co-delivered by SB-HDTVI into WT and 5-Azacytidine was administered i.p. to inhibit DNMT1 at 2w post injection when ICC became visible. Mice were sacrificed at 5w post-injection to evaluate ICC tumor burden (Fig.S5A). There was no significant difference in macroscopic and microscopic tumor burden (Fig.S5B, E), LW/BW ratio (Fig.S5C) or survival (Fig.S5D) between 5-Azacytidine- and vehicle-treated animals. Altogether, these data show that DNMT1 is required for Akt-NICD-driven HC-to-ICC transformation but dispensable for maintenance of advanced ICC.
Dnmt1 re-expression restores ICC development in Akt-NICD-driven-TEAD-inhibited tumors.
Next, to conclusively determine DNMT1 as a functional Notch-YAP1/TEAD downstream effector in HC-driven ICC, we delivered Dnmt1 expression plasmid along with Akt-NICD and DN-TEAD. As a control, pT3-Ef1α-Empty was injected. Re-expression of Dnmt1 restored ICC development in Akt-NICD model despite TEAD inhibition as observed by gross and histological analysis (Fig.7B–E). LW/BW of DN-TEAD-Dnmt1 liver was still comparable to DN-TEAD livers at this stage (Fig.7C) suggesting that DNMT1 only partially rescues Akt-NICD-ICC formation blocked by TEAD repression. Microscopically, widespread ICC nodules in DN-TEAD-Dnmt1-livers were HA-tag (AKT)+;V5-tag (DNMT1)+;endogenous DNMT1+ whereas the majority of Akt-transduced cells (HA-tag+) in DN-TEAD-livers were V5-tag (DNMT1)−;DNMT1− with HC morphology indicating impaired HC-to-BEC/ICC conversion (Fig.7E). Interestingly, few ICC nodules in DN-TEAD-livers were V5-tag (DNMT1)− but endogenous DNMT1+ showing its indispensable role in HC-to-ICC transformation (Fig.7E). Altogether, these data demonstrate Notch-YAP1/TEAD-DNMT1 axis as being crucial and indispensable for HC-driven cholangiocarcinogenesis.
DNMT1 represses the transcription of HC-specific genes during HC-to-ICC transformation.
To address the mechanism by which DNMT1 downstream of NICD/YAP1 axis promotes HC-to-ICC reprogramming, we performed ChIP-Seq for DNMT1 in murine Akt-NICD-ICC liver together with input control (pulled, N=5) to identify direct DNMT1-targets. DNMT1 ChIP-seq captured a total of 2,174 peaks in Akt-NICD-ICC genome and majority (78.64%) were found at <2.5kb promoter regions, suggesting its main role in transcriptional regulation (Fig.7H, I). Gene annotation analysis of DNMT1 peaks identified 1,647 DNMT1 bound genes in Akt-NICD-ICC liver (Fig.7J and GSE200472). These DNMT1-bound and regulated genes were then cross-referenced to data obtained from DNMT1 ChIP-Seq and RNA-seq analysis generated using corresponding Akt-NICD-ICC sample (GSE200472). Given the well-known role of DNMT1 as a transcriptional repressor, we selected 169 of overlapped genes between the 1,647 DNMT1 bound genes in ChIP-seq (Akt-NICD-ICC) and 2,039 of decreased genes in Akt-NICD-ICC transcriptome compared to healthy control liver (RNA-seq). List of 169 genes is shown in Table S4. Next, we explored tentative upstream regulators for expression of selected 169 genes using Ingenuity Pathway Analysis Upstream Regulator Analysis (IPA-URA) tool52. IPA-URA analysis predicted a total of 320 upstream regulators denoted in Table S5. Considering majority of DNMT1 binding peaks were found in <2.5kb promoter region (Fig.7H, I), we next examined transcriptional regulators and selectively displayed top 20 annotated transcription factors from the list (Fig.7K). The prominent top upstream transcription regulators included multiple critical factors associated with HC differentiation and maturation functions such as HNF4α, HNF1α and CEBPα/β (blue letters, Fig.7K). To visualize DNMT1 binding and relative transcription of representative genes determined by IPA-URA analysis, we selectively depicted peak patterns of Cyp26a1 and Zfp36 which are known to be regulated by HNF4α, HNF1α, FOXA2 and/or TP53. Peak analysis shows robust DNMT1 binding in the promoter regions along with notable decrease of overall transcription, implying that promoter-bound DNMT1 repressed RNA transcription of Cyp26a1 and Zfp36 in Akt-NICD-ICC (Fig.7L). Overall, our data suggests that DNMT1 is required for fate conversion of Akt-NICD-transfected HCs into biliary lineage by inhibiting expression of genes related to HC function and identity, regulated by HC-specific transcription factors.
Co-expression of Dnmt1 and Akt in HC provokes mixed HCC/ICC.
Given the critical role of DNMT1 in HC-driven ICC development, we next tested if co-expression of Dnmt1, instead of NICD or YAP1, along with Akt, is sufficient to induce tumors. Co-expression of Akt-Dnmt1 in HC led to liver cancer with significantly high LW/BW at 5w, while Dnmt1 alone group showed normal LW/BW and no gross or microscopic tumors (Fig.7F,G). Microscopically, majority of Dnmt1-transduced cells (V5-tag+;DNMT1+) were panCK−;HNF4α+ HCC-like tumors while a small number of V5-tag+;DNMT1+;panCK+;HNF4α− ICC nodules were also observed in Akt-Dnmt1 livers (Fig.S6). Histology showed mixed ICC/HCC foci similar to Akt-YAP1-driven tumors, although fewer ICC nodules were evident in Akt-Dnmt1 model (Fig.1, 6 and S6B), further supporting the essential roles for Dnmt1 in liver cancer development downstream of YAP1.
Lastly, to lend clinical credibility to this observation, we re-evaluated liver sections from NASH and PSC patients (Fig.1) to examine DNMT1 expression. We observed enhanced and widespread nuclear DNMT1 expression in HCs in PSC but less so in NASH or healthy controls livers (Fig.S7A–B). Two of the 9 PSC explanted livers exhibited bile duct dysplasia and ICC-like regions (SOX9+) associated with inflammation and stromal expansion (Fig.S4C–D). We found most SOX9+ HCs and duct-like cells also expressed nuclear DNMT1 while subsets of these cells were also p-AKT positive (Fig.S4C–D). These data suggest that DNMT1 and/or AKT co-activation in early neoplastic ICC in high-risk cases like PSC, may be driving the process of HC-to-BEC metaplasia. Altogether, co-expression of Dnmt1 with Akt in HC was sufficient to induce mixed HCC/ICC in mice and may have notable translational implications.
Discussion
Our study addresses heterogeneity in cellular origin of ICC. Active cell fate-transition between HC and BEC demonstrating the plasticity of hepatic epithelial during hepatobiliary injury is well-recognized39, while this is an important tier of liver repair, such reprogramming entails chromatin remodeling which may increase temporal susceptibility to a stochastic event. Accumulation of additional hits over time could result in neoplastic transformation53,54. Identifying drivers of reprogramming is thus of high relevance. We identified the presence of p-AKT as well as NICD targets YAP1 and SOX9 in livers of patients with well-known risk of ICC development. A subgroup of PSC and NASH patients showed increased expression of these HC-to-BEC reprogramming mediators in a subset of HCs. Both PSC and NASH are associated with risk of ICC development55,56. In conditions like PSC where BECs are the afflicted cell type, upregulation of biliary markers in HCs may be a reparative response, whereas the upregulation of biliary markers in in HCs in NASH may be an adaptive response to lipid accumulation. Aberrant expression of these factors in subsets of HCs may facilitate their de-differentiation and may be the first hit towards transformation, while additional mutagenic signals may be emanating from the adverse microenvironment of ongoing injury/inflammation due to the disease process. Interestingly, we found DNMT1-positive dysplastic bile ducts/ICC-like cells in 2/9 PSC patient livers. While there was no visible bile duct around them, we observed DNMT1+ and SOX9+ HCs partially connected to these pre-neoplastic ductal cells surrounded by stroma. DNMT1 was only rarely seen in NASH cases suggesting an alternative ICC pathogenesis. Further studies are needed to directly investigate the fate of YAP1 and DNMT1 positive HCs in primary biliary injury models as the tumor cell source.
Although the cystic histology of ICC in the Akt-NICD model is not commonly observed in human ICC, we believe this to be due to SB-HDTVI technique which delivers DNA into a subset of HCs located around zone-3 of a lobule. It is likely that these cystic patterns emerge due to spatial constraints faced by these ‘transfected’ cells as they proliferate under the “inside-out” oncogenic signals. In fact, molecular analysis of Akt-NICD tumors showed significant genetic similarity to ICCs seen in patients. Using a list of signature genes from the Akt-NICD model, transcriptomic and NTP analysis provided class prediction with confidence when computed in patient gene-expression data datasets32. Akt-NICD model signature predicted ICC in 13% of cases in GSE33327, 12% in GSE26566 and 30% in GSE76297. Most of these cases belonged to the “proliferation class” of ICC in all datasets as defined in an earlier study40. In addition, in all 3 datasets, the group of human patients resembling the Akt-NICD murine model were enriched in NOTCH signaling (p<0.05)41 and in 1/3 studies in the stem cell-like signature of human ICC57.
In Akt-NICD ICC, YAP1, but not SOX9, is essential for completion of HC-to-BEC reprogramming. YAP1 has been shown to be critical for biliary lineage commitment15,58 as well as malignant HC-to-BEC trans-differentiation through the regulation of Notch-SOX9 cascade in the absence of Notch activity42. However, in Akt-NICD ICC development, we reveal a novel molecular mechanism: YAP1-dependent Dnmt1 expression, where DNMT1 provokes HC-to-BEC conversion when Notch signaling is active. This aspect is also confirmed in Akt-YAP1 mixed HCC/ICC model. DNMT1 inhibition eliminates ICC whereas Akt-YAP1-transduced HCs remain as HNF4α+ HCCs, implying a specific role of DNMT1 in YAP1-mediated HC-to-BEC fate transition but being dispensable in oncogenic transformation.
There have been several studies demonstrating the critical roles for DNMT in liver cancer development and maintenance45,59–61; most of studies showed that pharmacologic DNMT inhibition leads to hypomethylation of the promoters of various tumor suppressors such as p16INK4a, FoxM1, TP53, PTEN, and others62, which typically induced death of malignant cells. At the same time, like other epigenetic regulators19,39,63, DNMT1 was among the list of suggested regulators of various cell fate switches43–46,64. We provide unique and direct evidence that DNMT1 inhibition impaired Akt-NICD/YAP1-mediated HC-to-BEC conversion, thereby completely abrogating HC-derived murine ICC development. Importantly, through integration of DNMT1 ChIP-seq and transcriptome of Akt-NICD-ICC, we uncovered the function of DNMT1 in repressing transcription of multiple genes crucial for maintaining HC differentiation and function. This novel observation suggests that DNMT1 plays permissive role in conversion of HC into biliary lineage by repressing HC identity. The unique role of DNMT1 in fate conversion in our model may also explain the absence of therapeutic effect of DNMT1 inhibition on advanced ICC and supports the notion that HC-to-ICC fate conversion may not be actively contributing to tumor maintenance but only to tumor initiation. There are also likely additional targets of the Notch-YAP1/TEAD axis contributing to HC-to-ICC transformation, requiring further studies.
Supplementary Material
WHAT YOU NEED TO KNOW.
Background and Context:
Intrahepatic biliary cancers remain a fatal disease due to poor understanding of their pathogenesis, and while subsets of these tumors are thought to originate from hepatocytes, the mechanism is unknown.
New Findings:
We identify a novel Notch-Yap/Tead-DNMT1 axis that suppresses key genes involved in hepatocyte identity that allow reprogramming of this cell type into biliary and in turn biliary cancer fate.
Limitations:
While biliary tumors in our model represent subsets of human biliary tumors by in silico analysis, further validation studies are important, along with uncovering additional determinants of cell fate switch.
Impact:
The identified targetable epigenetic regulator of fate switch of hepatocytes to bile duct cancers may allow development of biomarkers and novel chemoprophylactic therapies in high-risk patients like primary sclerosing cholangitis.
Grant support:
This work was supported in part by NIH grants R01DK101426, R01DK116993, R01CA204586, R01CA251155, R01CA250227, and Endowed Chair for Experimental Pathology to S.P.M., and by NIH grant 1P30DK120531 to Pittsburgh Liver Research Center (PLRC) and by R01CA258449 to S.K and PLRC Pilot & Feasibility grant PF 2019-05 to S.K.
Abbreviations used in this paper:
- ICC
intrahepatic cholangiocarcinoma
- FGFR
fibroblast growth factor receptor
- IDH1/2
isocitrate dehydrogenase 1/2
- BECs
biliary epithelial cells
- HCs
hepatocytes
- SOX9
SRY-box transcription factor 9
- CK
cytokeratin
- HCC
hepatocellular carcinoma
- TAA
thioacetamide
- NICD
Notch intracellular domain
- YAP1
yes-associated protein 1
- Fbxw7
F-box/WD repeat-containing protein 7
- SB-HDTVI
sleeping beauty transposon-transposase and hydrodynamic tail vein injection
- DNMT1
DNA methyltransferase 1
- HNF4α
hepatocyte nuclear factor 4 alpha
- CEBPA/B
CCAAT/enhancer-binding protein alpha/beta
- IACUC
Institutional Animal Care and Use Committee
- PSC
primary sclerosing cholangitis
- NASH
non-alcoholic steatohepatitis
- RNA-seq
RNA-sequencing
- IHC
immunohistochemistry
- GEO
gene expression omnibus
- MGD
mouse genome database
- ChIP-seq
Chromatin immunoprecipitation followed by sequencing
- BWA
Burrows-Wheeler Aligner
- MACS
Model-based analysis of ChIP-Seq
- FIMO
Find Individual Motif Occurrences
- SEM
standard error of mean
- DEG
differentially expressed gene
- NTP
Nearest Template Prediction
- WT
wild-type
- KO
knockout
- LW/BW
liver weight-to body weight ratio
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- TSS
transcription start site
- IPA-URA
Ingenuity Pathway Analysis Upstream Regulator Analysis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: There are no financial conflict of interests to declare relevant to the current manuscript for any of the authors.
Transcript profiling: The RNA-seq and ChIP-seq data has been submitted to online database GEO accession ID: GSE200472.
Data Transparency Statement:
Data and analytic methods have been included in the methods section. Study materials will be made available to other researchers, if requested under an appropriate Materials Transfer Agreement.
References
- 1.National Cancer Institute NIoH. Cancer Stat Facts: Liver and Intrahepatic Bile Duct Cancer, 2018.
- 2.Dabney RS, Khalife M, Shahid K, et al. Molecular pathways and targeted therapy in cholangiocarcinoma. Clin Adv Hematol Oncol 2019;17:630–637. [PubMed] [Google Scholar]
- 3.Lamarca A, Barriuso J, McNamara MG, et al. Molecular targeted therapies: Ready for “prime time” in biliary tract cancer. J Hepatol 2020. [DOI] [PubMed] [Google Scholar]
- 4.Banales JM, Cardinale V, Carpino G, et al. Expert consensus document: Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol 2016;13:261–80. [DOI] [PubMed] [Google Scholar]
- 5.Cai Y, Cheng N, Ye H, et al. The current management of cholangiocarcinoma: A comparison of current guidelines. Biosci Trends 2016;10:92–102. [DOI] [PubMed] [Google Scholar]
- 6.Lamarca A, Hubner RA, David Ryder W, et al. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol 2014;25:2328–38. [DOI] [PubMed] [Google Scholar]
- 7.Abou-Alfa GK, Andersen JB, Chapman W, et al. Advances in cholangiocarcinoma research: report from the third Cholangiocarcinoma Foundation Annual Conference. J Gastrointest Oncol 2016;7:819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan B, Malato Y, Calvisi DF, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest 2012;122:2911–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hill MA, Alexander WB, Guo B, et al. Kras and Tp53 Mutations Cause Cholangiocyte- and Hepatocyte-Derived Cholangiocarcinoma. Cancer Res 2018;78:4445–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet 2014;383:2168–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Dong M, Xu Z, et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018;37:3229–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sekiya S, Suzuki A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J Clin Invest 2012;122:3914–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Dell MR, Huang JL, Whitney-Miller CL, et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res 2012;72:1557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Wang H, Peters M, et al. Loss of Fbxw7 synergizes with activated Akt signaling to promote c-Myc dependent cholangiocarcinogenesis. J Hepatol 2019;71:742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lemaigre FP. Development of the Intrahepatic and Extrahepatic Biliary Tract: A Framework for Understanding Congenital Diseases. Annu Rev Pathol 2019. [DOI] [PubMed] [Google Scholar]
- 16.Wu H, Liu Y, Jiang XW, et al. Clinicopathological and prognostic significance of Yes-associated protein expression in hepatocellular carcinoma and hepatic cholangiocarcinoma. Tumour Biol 2016;37:13499–13508. [DOI] [PubMed] [Google Scholar]
- 17.Yuan X, Li J, Coulouarn C, et al. SOX9 expression decreases survival of patients with intrahepatic cholangiocarcinoma by conferring chemoresistance. Br J Cancer 2018;119:1358–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tao J, Zhang R, Singh S, et al. Targeting beta-catenin in hepatocellular cancers induced by coexpression of mutant beta-catenin and K-Ras in mice. Hepatology 2017;65:1581–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ko S, Russell JO, Tian J, et al. Hdac1 Regulates Differentiation of Bipotent Liver Progenitor Cells During Regeneration via Sox9b and Cdk8. Gastroenterology 2019;156:187–202 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen ZH, Yu YP, Tao J, et al. MAN2A1-FER Fusion Gene Is Expressed by Human Liver and Other Tumor Types and Has Oncogenic Activity in Mice. Gastroenterology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen ZH, Yu YP, Zuo ZH, et al. Targeting genomic rearrangements in tumor cells through Cas9-mediated insertion of a suicide gene. Nat Biotechnol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conesa A, Madrigal P, Tarazona S, et al. A survey of best practices for RNA-seq data analysis. Genome Biol 2016;17:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andrews S. FastQC- a quality control tool for high throughput sequence data. 2010.
- 24.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015;12:357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 2014;15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolstad BM, Irizarry RA, Astrand M, et al. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003;19:185–93. [DOI] [PubMed] [Google Scholar]
- 29.Smyth GK. Limma: linear models for microarray data. In Bioinformatics and computational biology solutions using R and Bioconductor 2005:397–420. [Google Scholar]
- 30.Bult CJ, Blake JA, Smith CL, et al. Mouse Genome Database (MGD) 2019. Nucleic Acids Research 2019;47:D801–D806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reich M, Liefeld T, Gould J, et al. GenePattern 2.0. Nat Genet 2006;38:500–1. [DOI] [PubMed] [Google Scholar]
- 32.Hoshida Y. Nearest template prediction: a single-sample-based flexible class prediction with confidence assessment. PLoS One 2010;5:e15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan WC, Pepe-Mooney B, Galli GG, et al. NUAK2 is a critical YAP target in liver cancer. Nat Commun 2018;9:4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zong Y, Panikkar A, Xu J, et al. Notch signaling controls liver development by regulating biliary differentiation. Development 2009;136:1727–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bragazzi MC, Ridola L, Safarikia S, et al. New insights into cholangiocarcinoma: multiple stems and related cell lineages of origin. Ann Gastroenterol 2018;31:42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han X, Wang Y, Pu W, et al. Lineage Tracing Reveals the Bipotency of SOX9(+) Hepatocytes during Liver Regeneration. Stem Cell Reports 2019;12:624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ko S, Russell JO, Molina LM, et al. Liver Progenitors and Adult Cell Plasticity in Hepatic Injury and Repair: Knowns and Unknowns. Annu Rev Pathol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sia D, Hoshida Y, Villanueva A, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013;144:829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villanueva A, Alsinet C, Yanger K, et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology 2012;143:1660–1669 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yimlamai D, Christodoulou C, Galli GG, et al. Hippo pathway activity influences liver cell fate. Cell 2014;157:1324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aloia L, McKie MA, Vernaz G, et al. Epigenetic remodelling licences adult cholangiocytes for organoid formation and liver regeneration. Nat Cell Biol 2019;21:1321–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDaniel K, Meng F, Wu N, et al. Forkhead box A2 regulates biliary heterogeneity and senescence during cholestatic liver injury in micedouble dagger. Hepatology 2017;65:544–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Merino-Azpitarte M, Lozano E, Perugorria MJ, et al. SOX17 regulates cholangiocyte differentiation and acts as a tumor suppressor in cholangiocarcinoma. J Hepatol 2017;67:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu R, Kim KY, Jung YW, et al. Dnmt1 regulates the myogenic lineage specification of muscle stem cells. Sci Rep 2016;6:35355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moya IM, Castaldo SA, Van den Mooter L, et al. Peritumoral activation of the Hippo pathway effectors YAP and TAZ suppresses liver cancer in mice. Science 2019;366:1029–1034. [DOI] [PubMed] [Google Scholar]
- 48.Pocaterra A, Romani P, Dupont S. YAP/TAZ functions and their regulation at a glance. J Cell Sci 2020;133. [DOI] [PubMed] [Google Scholar]
- 49.Zhang S, Wang J, Wang H, et al. Hippo Cascade Controls Lineage Commitment of Liver Tumors in Mice and Humans. Am J Pathol 2018;188:995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu-Chittenden Y, Huang B, Shim JS, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 2012;26:1300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J, Liu P, Tao J, et al. TEA Domain Transcription Factor 4 Is the Major Mediator of Yes-Associated Protein Oncogenic Activity in Mouse and Human Hepatoblastoma. Am J Pathol 2019;189:1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X, Long J, He T, et al. Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer’s disease. Sci Rep 2015;5:12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nebbioso A, Tambaro FP, Dell’Aversana C, et al. Cancer epigenetics: Moving forward. PLoS Genet 2018;14:e1007362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sandhu DS, Shire AM, Roberts LR. Epigenetic DNA hypermethylation in cholangiocarcinoma: potential roles in pathogenesis, diagnosis and identification of treatment targets. Liver Int 2008;28:12–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rizvi S, Eaton JE, Gores GJ. Primary Sclerosing Cholangitis as a Premalignant Biliary Tract Disease: Surveillance and Management. Clin Gastroenterol Hepatol 2015;13:2152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wongjarupong N, Assavapongpaiboon B, Susantitaphong P, et al. Non-alcoholic fatty liver disease as a risk factor for cholangiocarcinoma: a systematic review and meta-analysis. BMC Gastroenterol 2017;17:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oishi N, Kumar MR, Roessler S, et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology 2012;56:1792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee DH, Park JO, Kim TS, et al. LATS-YAP/TAZ controls lineage specification by regulating TGFbeta signaling and Hnf4alpha expression during liver development. Nat Commun 2016;7:11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Braconi C, Huang N, Patel T. MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology 2010;51:881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakamura K, Nakabayashi K, Htet Aung K, et al. DNA methyltransferase inhibitor zebularine induces human cholangiocarcinoma cell death through alteration of DNA methylation status. PLoS One 2015;10:e0120545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Rourke CJ, Lafuente-Barquero J, Andersen JB. Epigenome Remodeling in Cholangiocarcinoma. Trends Cancer 2019;5:335–350. [DOI] [PubMed] [Google Scholar]
- 62.Loaeza-Loaeza J, Beltran AS, Hernandez-Sotelo D. DNMTs and Impact of CpG Content, Transcription Factors, Consensus Motifs, lncRNAs, and Histone Marks on DNA Methylation. Genes (Basel) 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cho YD, Ryoo HM. Trans-differentiation via Epigenetics: A New Paradigm in the Bone Regeneration. J Bone Metab 2018;25:9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Page A, Paoli P, Moran Salvador E, et al. Hepatic stellate cell transdifferentiation involves genome-wide remodeling of the DNA methylation landscape. J Hepatol 2016;64:661–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data and analytic methods have been included in the methods section. Study materials will be made available to other researchers, if requested under an appropriate Materials Transfer Agreement.