

Abstract
We report here “sandwich” diimine-copper (I) catalysts for C(sp3)-H bond functionalization. Reactions of alkanes and ethers with trimethylsilyldiazomethane, ethyl diazoacetate, and trifluoromethyldiazomethane have been demonstrated. We also report C(sp3)-H bond methylation, benzylation, and diphenylmethylation by diazomethane, phenyldiazomethanes, and diphenyldiazomethane. These reactions are rare examples of base-metal catalyzed, intermolecular C(sp3)-H functionalizations by employing unactivated diazocompounds. Electrophilicity and unique steric environment of “sandwich”-copper catalysts are likely reasons for their catalytic efficiency.
Keywords: C-H activation, carbenes, copper, diazo compounds, diimines
Graphical Abstract
A new catalyst platform for C(sp3)-H bond functionalization has been discovered. “Sandwich” diimine-copper (I) complexes catalyze reactions of alkane, ether, and amine C-H bonds with a large panel of diazo compounds. Additionally, the first metal-catalyzed C(sp3)-H methylation by CH2N2 is disclosed. The electrophilicity and extreme steric bulk of these catalysts are likely reasons for their efficiency.
Introduction
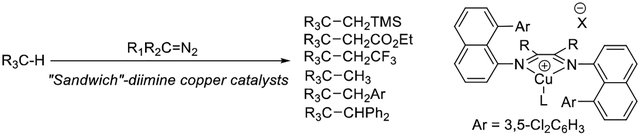
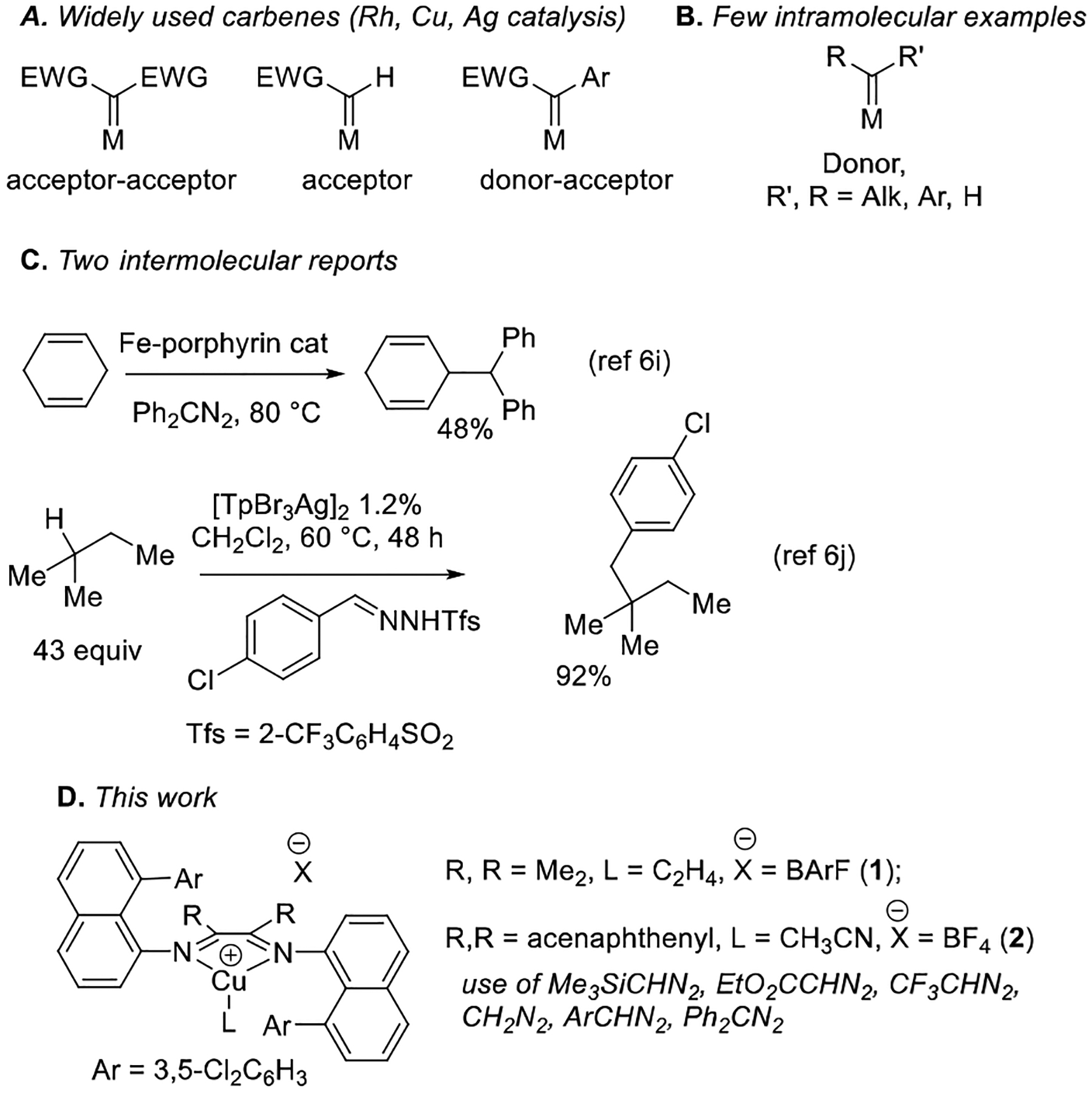
Selective C(sp3)-H bond functionalization is a challenging transformation that has attracted significant current interest.[1] Among metal-catalyzed reactions, most examples involve formation of a metal-carbon bond-containing intermediate that reacts with another reagent affording the final product. Alternatively, C-H functionalizations involving metal carbenes have been extensively employed for alkane functionalization.[2] These reactions do not involve a discrete alkylmetal intermediate. Instead, a transient metal carbene reacts with a C-H bond to afford the final product and regenerated catalyst. Currently, dirhodium complexes are the most synthetically useful catalysts, allowing for highly enantioselective transformations. These catalysts have been used in total syntheses of complex natural products.[3,4] Group 11 metal complexes have been extensively used for alkane functionalization as well.[2,5] Notable examples include modified tris(pyrazolyl)borate-copper, silver, and gold catalysts pioneered by the group of Díaz-Requejo and Pérez.[2e,f,g,5b,c] However, both rhodium- and group 11 metal-catalyzed C-H insertions are typically limited to either acceptor or donor-acceptor carbene precursors (Scheme 1A). In contrast, use of donor carbenes in C-H functionalization is rare and, in most cases, restricted to intramolecular reactions (Scheme 1B), while intermolecular insertions are particularly scarce (Scheme 1C).[6] The first intermolecular examples were reported by the Che group.[6i] Benzylic and allylic C-H bonds react with diphenyldiazomethane under iron porphyrin catalysis, affording modest yields of products. A recent paper from the Bi group introduces a convenient method for benzylation of alkane C-H bonds catalyzed by a highly electrophilic perbrominated tris(pyrazolylborate)-Ag complex.[6j] The reactions are high-yielding and very general with respect to the substitution on the carbene aryl group. Non-activated (not allylic, benzylic, or α to heteroatom) C(sp3)-H bonds are readily functionalized.
Scheme 1.
Metal carbene C-H functionalization.
Review of earlier work shows that general intermolecular C-H functionalization with donor carbenes catalyzed by a base metal complex has not been reported. Furthermore, it appears that efficient metal-catalyzed C-H insertions by diazomethane and trimethylsilyldiazomethane are not known, and trifluoromethyldiazomethane has been employed only for functionalization of activated benzylic C(sp3)-H bonds.[7] We report here that “sandwich” diimine-copper catalysts 1 and 2 allow for a general, intermolecular C(sp3)-H functionalization by donor, donor-acceptor, and acceptor diazocompounds. Importantly, diazomethane, trifluoromethyldiazomethane, and trimethylsilyldiazomethane can be employed in C(sp3)-H bond functionalization.
Results and Discussion
In 2016, we disclosed the synthesis of “sandwich” diimine-copper ethylene complex 1.[8] Related “sandwich” diimine-palladium and nickel catalysts are capable of olefin living polymerization, attesting to high axial steric hindrance exhibited by this ligand platform.[9] We were interested in exploring 1 and related complexes as carbene C-H insertion catalysts. Potential advantages relative to other catalysts include their electrophilicity and extreme steric environment, resulting in a highly reactive, cationic copper carbene intermediate that may be less sensitive to dimerization leading to alkene and other side reactions.[10]
Reactions with Trimethylsilyldiazomethane.
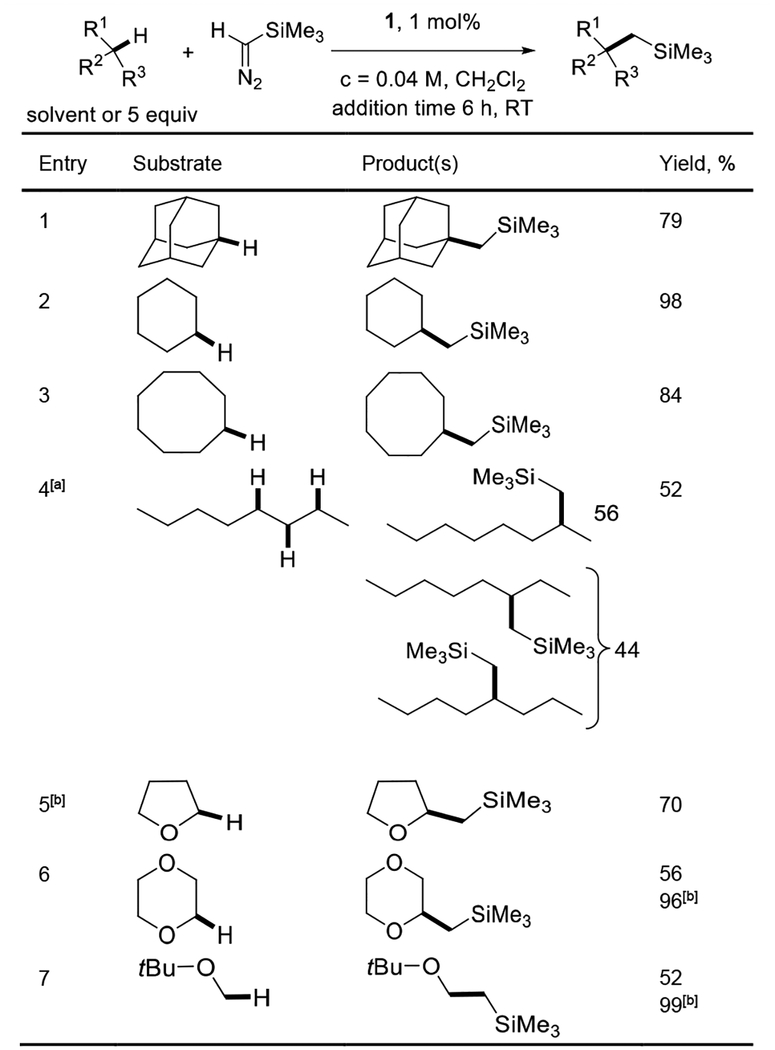
We examined the challenging reaction of trimethylsilyldiazomethane with dioxane (Table 1).[11] Both “sandwich” diimine-copper ethylene and acetonitrile complexes 1 and 2 gave nearly quantitative conversion to the C-H insertion product 3 (entries 1 and 2). Other diimine-copper complexes, such as the phenyl derivative 4 and even highly electrophilic, nitro-substituted 5 afforded lower conversions (entries 3 and 4). Control experiments with NaBArF (entry 5) and copper(tetrakis)acetonitrile complex gave at best trace conversion to product (entries 5 and 6). Other catalysts that facilitate C-H insertion reactions, such as dirhodium tetraacetate, Rh2(esp)4, [12] and tris(3,4,5-tribromopyrazolyl)borate copper(I) acetonitrile complex gave very low conversions to 3 (entries 7–9). Thus, the best results were obtained with “sandwich” diimine-copper complexes which were used for further investigations.
Table 1.
Catalyst screening.
| ||
|---|---|---|
| Entry | Catalyst (mol%) | Yield, % |
| 1 | 1 (1) | >99 |
| 2 | 2 (1) | >99 |
| 3 | 4 (1) | 61 |
| 4 | 5 (1) | 72 |
| 5 | NaBArF (5) | nr |
| 6 | Cu(MeCN)4PF6 (5), NaBArF (6) | 2.5 |
| 7 | Rh2(esp)4 (5) | 12 |
| 8 | Rh2(OAc)4 (15) | nr |
| 9 | TpBr3Cu(MeCN) (1) | 2.5 |
Catalyst, CH2Cl2 (2.5 mL), dioxane (4.0 mL), add TMSCHN2 (0.5 mmol, 1.0 equiv, in hexanes + 6.0 mL dioxane) in 6 hours at RT (20–23 °C). Yields determined by 1H NMR spectroscopy with an internal standard. Abbreviations: NaBArF = NaB[3,5-(CF3)2C6H3]4. Rh2(esp)4 = Bis[Rh(α,α,α′,α′-tetramethyl-1,3-benzenedipropionic acid)]. TpBr3Cu(MeCN) – tris(3,4,5-tribromopyrazolyl)borate Cu(I) acetonitrile complex.
“Sandwich”-copper complex 1 is sufficiently active to catalyze insertions of Me3SiCHN2 into unactivated C(sp3)-H bonds even at low 1% loading (Table 2). Adamantane gave product in 79% yield (entry 1). Cyclohexane and cyclooctane afforded the insertion products in 98 and 84% yields, respectively (entries 2 and 3). n-Octane reacted to give a mixture of three insertion products in a combined 52% yield (entry 4). Tetrahydrofuran (entry 5), dioxane (entry 6), and t-butyl methyl ether (entry 7) are reactive and products were formed in high yields, while isolated yields are somewhat lower due to volatility of products. Trimethylsilyl moiety can be chemoselectively converted to a hydroxy group by using I(OCOCF3)3 reagent.[13]
Table 2.
Reactions with trimethylsilyldiazomethane.
|
Catalyst (1 mol%), entries 1 and 3: CH2Cl2 (6.5 mL), substrate (2.5 mmol, 5.0 equiv), add Me3SiCHN2 (0.5 mmol, 1.0 equiv, in hexanes + 6.5 mL CH2Cl2) in 6 hours at RT. Rest of entries: substrate used as solvent (10 mL, 128–246 equiv). Yields are isolated yields. [a] Isolated as a mixture of isomers. [b] Yield determined by 1H NMR spectroscopy with an internal standard.
Reactions with Ethyl Diazoacetate.
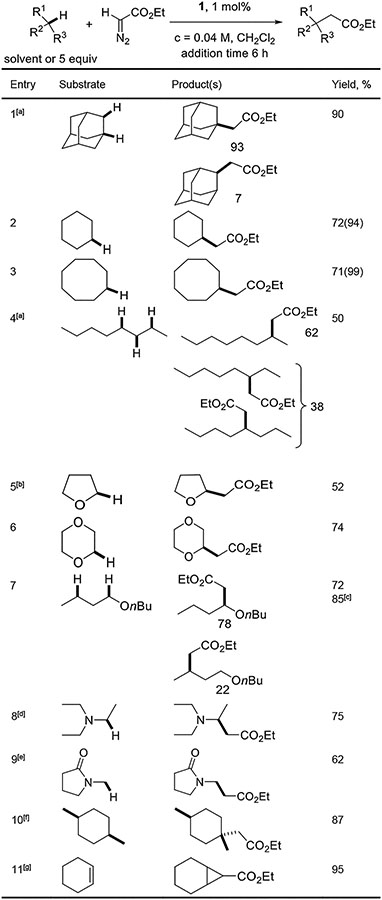
The reactions of ethyl diazoacetate with a number of substrates were explored next (Table 3). Adamantane gave a combined 90% yield of two isomers, with the tertiary C-H insertion product predominating (entry 1). Lower selectivity compared with that of entry 1, Table 2 can be rationalized by higher electrophilicity and reactivity of carbene derived from ethyl diazoacetate. Cyclohexane and cyclooctane afforded products in excellent yields (entries 2, 3). n- Octane reacted to give three insertion products in 50% isolated yield, with preferential functionalization at 2-position (entry 4). Reactions with ethers were successful as well. Tetrahydrofuran gave product in 52% yield (entry 5), while challenging substrate dioxane underwent C-H insertion in 74% yield (entry 6).[11] Di-n-butyl ether gave a mixture of two isomeric products in 82% NMR and 72% isolated yield, with major product arising from insertion α to oxygen (entry 7). Amines are reactive as well, and C-H insertion into α-C-H bond of triethylamine gave product in 75% isolated yield at 70 °C (entry 8). N-Methylpyrrolidone afforded product arising from insertion into methyl group in 62% yield (entry 9). Use of cis-1,4-dimethylcyclohexane substrate resulted in formation of one product diastereomer in 87% yield, with preservation of stereochemistry at reactive site (entry 10). Reaction with cyclohexene (entry 11) afforded a mixture of cyclopropanation and C-H insertion products in ca. 10:1 ratio and 95% combined yield.
Table 3.
Reactions with ethyl diazoacetate.
|
Catalyst (1 mol%), entries 1, 3, 4: CH2Cl2 (6.5 mL), substrate (2.5 mmol, 5.0 equiv), add ethyl diazoacetate (0.5 mmol, 1.0 equiv, in 6.5 mL CH2Cl2) in 6 hours at RT. Entries 2, 6, 7, 10: substrate used as solvent (10 mL, 118–234 equiv). Yields are isolated yields. [a] Isolated as a mixture of isomers. [b] Scale: 1 mmol, THF solvent (20 mL, 246 mmol). [c] Yield determined by 1H NMR spectroscopy with an internal standard. [d] Catalyst 2 (3 mol%), Et3N (15 mL, 108 mmol), CHCl3 (15 mL), add ethyl diazoacetate (10 mmol) at 70 °C in 3 h. [e] Catalyst 2 (3 mol%), NMP (5 mL, 52 mmol), CHCl3 (5 mL), add ethyl diazoacetate (1 mmol in CHCl3) at 60 °C in 12 h. [f] cis-1,4-Dimethylcyclohexane (7 equiv), catalyst 2 (2 mol%). Contains less than 5% of another product arising from insertion in a secondary C-H bond. [g] Major product. About 10/1 ratio of cyclopropanation/C-H insertion observed.
Reactions with Trifluoromethyldiazomethane.
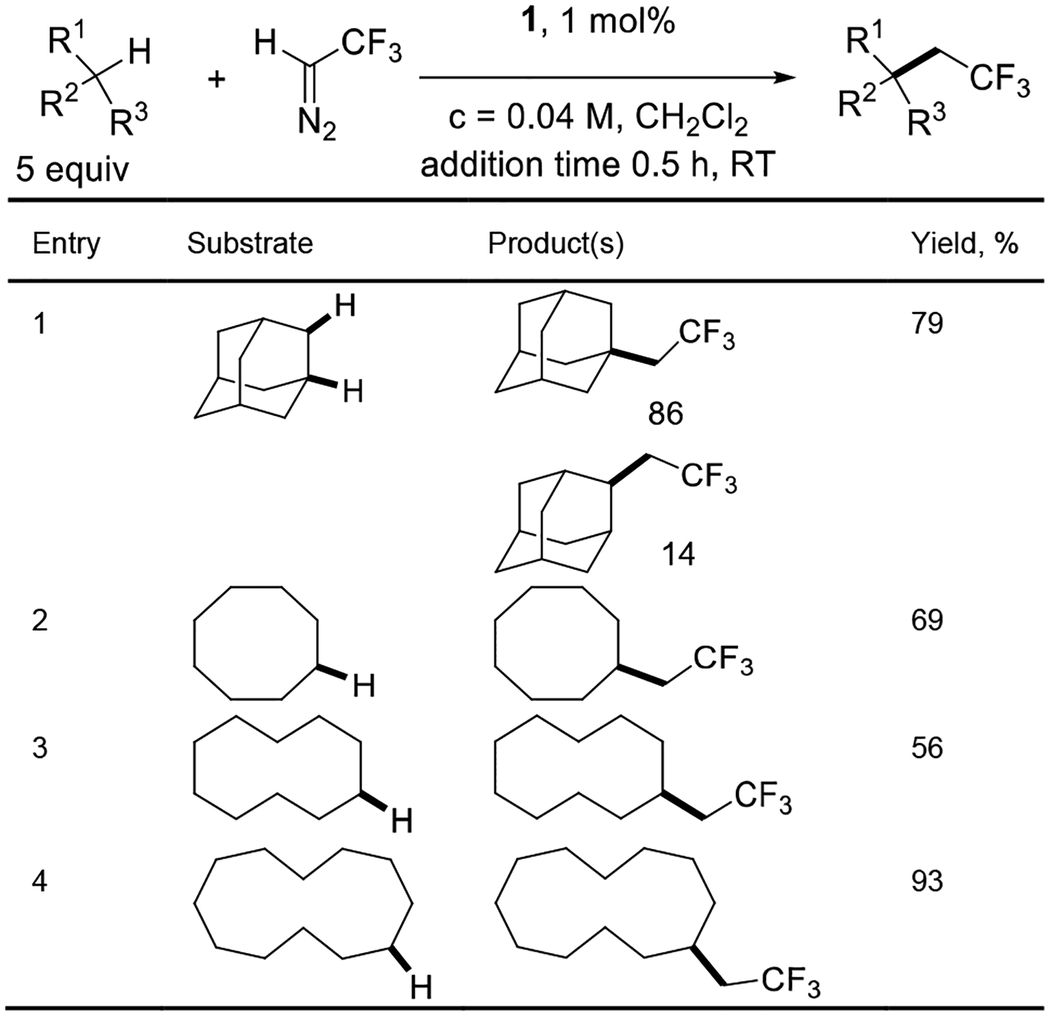
Carbon-hydrogen bond functionalization with trifluoromethyldiazomethane was successful as well (Table 4). Adamantane gave a mixture of insertion into secondary and tertiary C-H bonds in ca. 6/1 ratio in 79% combined yield (entry 1). Cyclooctane, cyclodecane, and cyclododecane all reacted to give acceptable to excellent yields of products (entries 2–4). Metal carbene trifluoroethylation of unactivated (not benzylic or α to heteroatom) C(sp3)-H bonds has not been reported, while reactions with heteroatom-hydrogen bonds are well-precedented.[7,14]
Table 4.
Trifluoromethyldiazomethane reactions.
|
Catalyst (1 mol%), CH2Cl2 (12.5 mL), substrate (2.5 mmol, 5.0 equiv), add CF3CHN2 (0.5 mmol, 1.0 equiv, 1.47 mL of 0.340 M solution in CH2Cl2) in 0.5 hours at RT, stir 1 hour. Yields determined by 19F NMR with internal standard.
Reactions with Phenyl Methyldiazoacetate, Diazomethane, and Aryldiazomethanes.
A number of other diazocompound C-H insertions were explored to determine generality of this methodology (Scheme 2). Phenyl methyl diazoacetate reacts with adamantane to give 77% yield of 6. Methylation of adamantane gave a mixture of mono- and dimethylation products 7 and 8 in modest conversion. New methods for C-H methylation are of particular interest, as methylation of drugs often leads to pharmaceutically interesting products.[15] This appears to be the first instance of metal-catalyzed C(sp3)-H functionalization by diazomethane.[16] Benzylation and diphenylmethylation of THF gave 9 and 10 in 51 and 31% yields, respectively. Reaction of adamantane with p—nitrophenyldiazomethane afforded 11 in 75% yield.
Scheme 2.
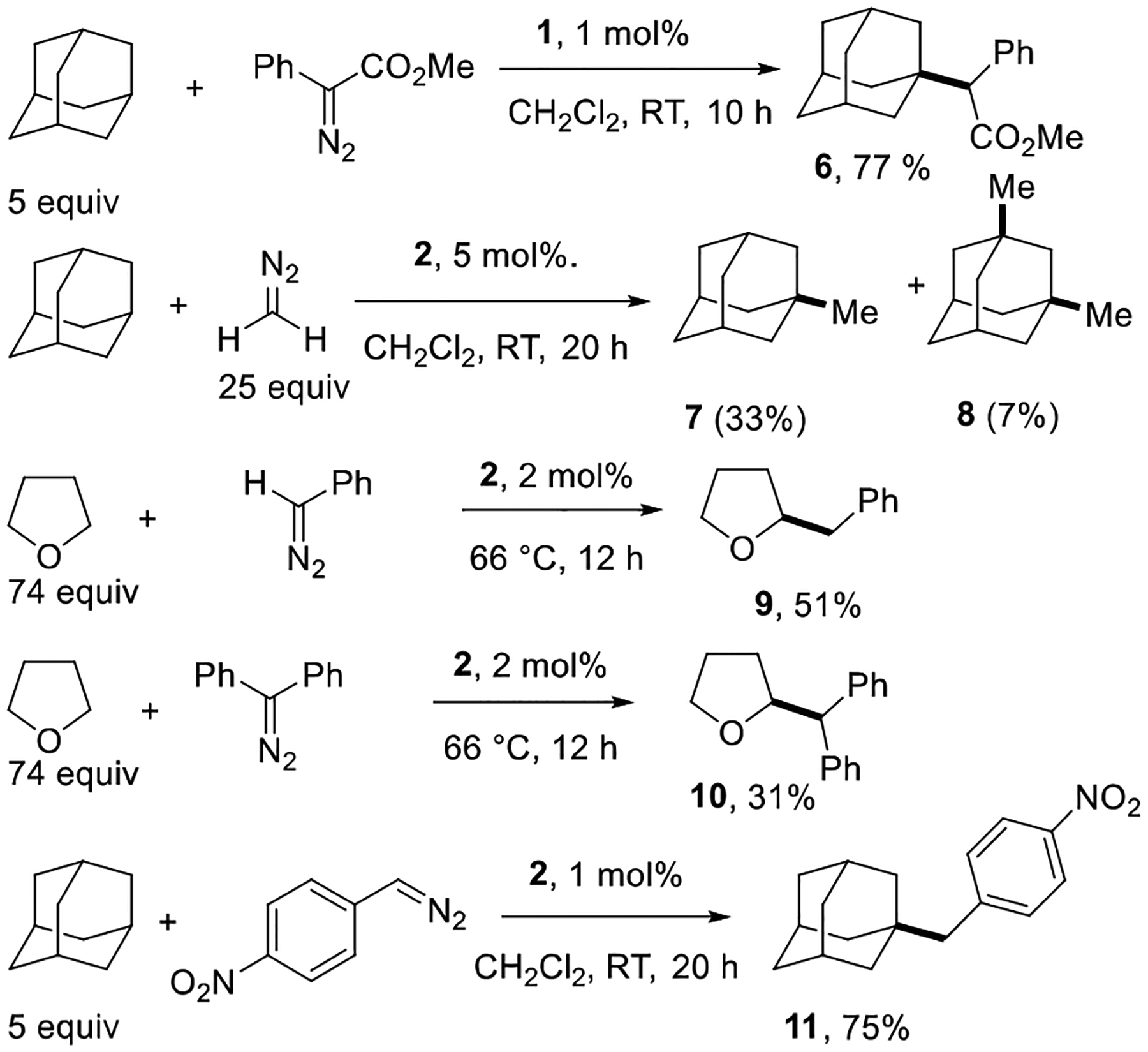
Reactions with other diazocompounds.
Mechanistic Considerations.
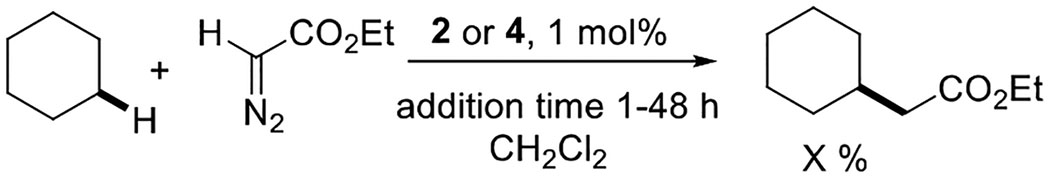
The key features of the new catalytic system are the high reactivity of the intermediate copper carbene, short diazo compound addition times for more reactive substrates, and ability to employ relatively low excess (5 equivalents) of hydrocarbon. Specifically, catalysts 1 and 2 perform better than highly electrophilic nitroaryl-substituted complex 5 (Table 1). Structurally characterized copper carbenes L2Cu=CR2 possess geometry where the L2Cu plane is perpendicular to the Cu=CR2 plane.[10] Extensive axial shielding provided by two 3,5-dichloroaryl moieties in a “sandwich” orientation may force the distortion of the carbene moiety, rising the ground state energy of the intermediate, thus increasing its reactivity. Structurally characterized “sandwich” diimine-NiBr2 complexes show deviations from the expected tetrahedral geometry.[9a,b] Steric bulk of ligands may prevent formation of bridging dicopper carbenes, increasing concentration of the active monomeric species.[10a] Increase of C-H insertion yield relative to carbene coupling product alkene has been observed if catalyst loading is decreased, suggesting that in some cases carbene coupling may be second-order in catalyst.[17a,b] Isolated copper carbene complexes form alkene if heated in a bimolecular reaction.[10a] In this scenario, bulky ligands around carbene should minimize formation of alkene while the formation of C-H insertion product would be less impacted. Alternatively, reaction of metallocarbene with diazo compound may form the alkene byproduct.[17c] In this case, the relationship between ligand bulk and alkene byproduct formation is less clear. Increased yields of Si-H insertion products were observed if bulkier ligands on rhodium were used. This was attributed to steric interactions blocking approach of the diazo compound to the intermediate rhodium carbene.[17d] We examined the reaction of cyclohexane with ethyl diazoacetate catalyzed by “sandwich” catalyst 2 and simple phenyl-substituted complex 4. Ethyl diazoacetate in CH2Cl2 was added to cyclohexane/CH2Cl2 solution of the catalyst over the designated time (Table 5). For 2, the yield plateaus between three and six hours addition time. For the less bulky 4, yield keeps increasing with addition time and does not plateau even after 48 hours. These results show that steric bulk around the reactive copper center plays an important role in enhancing the efficiency of C-H insertion reactions.
Table 5.
Comparison of catalysts 2 and 4.
| |||||||
|---|---|---|---|---|---|---|---|
| Catalyst | 1 h | 2 h | 3 h | 6 h | 12 h | 24 h | 48 h |
| 2 | 45 % | 66 % | 85 % | 92 % | 93 % | 95 % | 95 % |
| 4 | 14 % | 24 % | 34 % | 44 % | 54 % | 64 % | 83 % |
Catalyst (1 mol%), CH2Cl2 (12.5 mL) + cyclohexane (10 mL), add ethyl diazoacetate (1.0 mmol, 1.0 equiv) in CH2Cl2 (1.0 mL) in 1–48 hours at RT. Yields determined by 1H-NMR analysis with an internal standard.
Conclusion
In conclusion, we have shown that “sandwich” diimine-copper (I) complexes are efficient catalysts for C(sp3)-H functionalization. These complexes display unusually high reactivity allowing for the first general use of a base metal catalyst in carbene C(sp3)-H functionalization. Importantly, this chemistry substantially expands the scope of diazocompounds employed for C(sp3)-H functionalization. Relatively short addition times and, for less volatile substrates, 5 equivalents of C-H coupling component can be employed.
Supplementary Material
Acknowledgements
This research was supported by the Welch Foundation (Chair E-0044) and NIGMS (Grant R01GM077635). We thank Dr. Hendrich A. Chiong for initial experiments with “sandwich”-copper catalysts.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].(a) Baudoin O. Angew. Chem., Int. Ed 2020, 59, 17798–17809. [DOI] [PubMed] [Google Scholar]; (b) Gandeepan P, Müller T, Zell D, Cera G, Warratz S, Ackermann L, Chem. Rev 2019, 119, 2192–2452. [DOI] [PubMed] [Google Scholar]; (c) Das J, Guin S, Maiti D, Chem. Sei 2020, 11, 10887–10909. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen Z, Rong M-Y, Nie J, Zhu X-F, Shi B-F, Ma J-A, Chem. Soc. Rev 2019, 48, 4921–4942. [DOI] [PubMed] [Google Scholar]; (e) Chu JCK, Rovis T, Angew. Chem., Int. Ed 2018, 57, 62–101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Hartwig JF, Larsen MA, ACS Cent. Sci 2016, 2, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Tang X, Jia X, Huang Z, Chem. Sci 2018, 9, 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Saint-Denis TG, Zhu R-Y, Chen G, Wu Q-F, Yu J-Q, Science 2018, 359, eaao4798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Gutekunst WR, Baran PS, Chem. Soc. Rev 2011, 40, 1976–1991. [DOI] [PubMed] [Google Scholar]
- [2].(a) Xia Y, Qiu D, Wang J, Chem. Rev 2017, 117 13810–13889. [DOI] [PubMed] [Google Scholar]; (b) Damiano C, Sonzini P, Gallo E, Chem. Soc. Rev 2020, 49, 4867–4905. [DOI] [PubMed] [Google Scholar]; (c) Slattery CN, Ford A, Maguire AR, Tetrahedron 2010, 66, 66816705. [Google Scholar]; (d) Doyle MP, Duffy R, Ratnikov M, Zhou L, Chem. Rev 2010, 110, 704–724. [DOI] [PubMed] [Google Scholar]; (e) Díaz-Requejo MM, Pérez PJ, Chem. Rev 2008, 108, 3379–3394. [DOI] [PubMed] [Google Scholar]; (f) Díaz-Requejo MM, Pérez PJ, Eur. J. Inorg. Chem 2020, 879–885. [Google Scholar]; (g) Caballero A, Díaz-Requejo MM, Fructos MR, Olmos A, Urbano J, Pérez PJ, Dalton Trans 2015, 44, 20295–20307. [DOI] [PubMed] [Google Scholar]; (h) Zhao X, Zhang Y, Wang J, Chem. Commun 2012, 48, 10162–10173. [DOI] [PubMed] [Google Scholar]; (i) Kirmse W, Angew. Chem. Int. Ed 2003, 42, 1088–1093. [DOI] [PubMed] [Google Scholar]
- [3].(a) Davies HML, J. Org. Chem 2019, 84, 12722–12745. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davies HML, Denton JR, Chem. Soc. Rev 2009, 38, 3061–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shih J-L, Chen P-A, May JA, Beilstein J Org. Chem 2016, 12, 985–999. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) DeAngelis A, Panish R, Fox JM, Acc. Chem. Res 2016, 49, 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].(a) Selected examples:Fu J, Ren Z, Bacsa J, Musaev DG, Davies HML, Nature 2018, 564, 395–399. [DOI] [PubMed] [Google Scholar]; (b) Lu P, Mailyan A, Gu Z, Guptill DM, Wang H, Davies HML, Zakarian A, J. Am. Chem. Soc 2014, 136, 17738–17749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].(a) Scott LT, DeCicco GJ, J. Am. Chem. Soc 1974, 96, 322–323. [Google Scholar]; (b) Díaz-Requejo MM, Belderraín TR, Nicasio MC, Trofimenko S, Pérez PJ, J. Am. Chem. Soc 2002, 124, 896–897. [DOI] [PubMed] [Google Scholar]; (c) Gava R, Olmos A, Noverges B, Varea T, Álvarez E, Belderrain TR, Caballero A, Asensio G, Pérez PJ, ACS Catal 2015, 5, 3726–3730. [Google Scholar]; (d) Flores JA, Badarinarayana V, Singh S, Lovely CJ, Dias HVR, Dalton Trans 2009, 7648–7652. [DOI] [PubMed] [Google Scholar]
- [6].(a) Zhu D, Chen L, Fan H, Yaoa Q, Zhu S, Chem. Soc. Rev 2020, 49, 908–950. [DOI] [PubMed] [Google Scholar]; (b) Bergstrom BD, Nickerson LA, Shaw JT, Souza LW, Angew. Chem. Int. Ed 2021, 60, 6864–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Morton D, Blakey SB, ChemCatChem 2015, 7, 577–578. [Google Scholar]; (d) Soldi C, Lamb KN, Squitieri RA, Gonzalez-Lopez M, Di Maso MJ, Shaw JT, J. Am. Chem. Soc 2014, 136, 15142–15145. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Reddy AR, Zhou C-Y, Guo Z, Wei J, Che CM, Angew. Chem., Int. Ed 2014, 53, 14175–14180. [DOI] [PubMed] [Google Scholar]; (f) Wen X, Wang Y, Zhang XP, Chem. Sci 2018, 5082–5086. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Souza LW, Squitieri RA, Dimirjian CA, Hodur BM, Nickerson LA, Penrod CN, Cordova J, Fettinger JC, Shaw JT, Angew. Chem., Int. Ed 2018, 57, 15213–15216. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zhu D, Ma J, Luo K, Fu H, Zhang L, Zhu S, Angew. Chem., int. Ed 2016, 55, 8452–8456. [DOI] [PubMed] [Google Scholar]; (i) Wang H-X, Wan Q, Low K-H, Zhou C-Y, Huang J-S, Zhang J-L, Che C-M, Chem. Sci 2020, 11, 2243–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Liu Z, Cao S, Yu W, Wu J, Yi F, Anderson EA, Bi X, Chem 2020, 6, 2110–2124. [Google Scholar]
- [7].(a) Zhang J, Huang X, Zhang RK, Arnold FH, J. Am. Chem. Soc 2019, 141, 9798–9802. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Atherton JH, Fields R, J. Chem. Soc C 1968, 2276–2278. [Google Scholar]; (c) Mykhailiuk PK, Chem. Rev 2020, 120, 12718–12755. [DOI] [PubMed] [Google Scholar]; (d) Hyde S, Veliks J, Liegault B, Grassi D, Taillefer M, Gouverneur V, Angew. Chem., Int. Ed 2016, 55, 3785–3789. [DOI] [PubMed] [Google Scholar]
- [8].Klimovica K, Kirschbaum K, Daugulis O, Organometallics 2016, 35, 2938–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].(a) Zhang D, Nadres ET, Brookhart M, Daugulis O, Organometallics 2013, 32, 5136–5143. [Google Scholar]; (b) Vaidya T, Klimovica K, LaPointe A, Keresztes I, Lobkovsky EB, Daugulis O, Coates G, J. Am. Chem. Soc 2014, 136, 7213–7216. [DOI] [PubMed] [Google Scholar]; (c) Allen KE, Campos J, Daugulis O, Brookhart M, ACS Catalysis 2015, 5, 456–464. [Google Scholar]
- [10].(a) Dai X, Warren TH, J. Am. Chem. Soc 2004, 126, 10085–10094. [DOI] [PubMed] [Google Scholar]; (b) Straub B, Hofmann P, Angew. Chem., Int. Ed 2001, 40, 1288–1290. [DOI] [PubMed] [Google Scholar]; (c) Barluenga J, López LA, Löber O, Tomás M, García-Granda S, Alvarez-Rúa C, Borge J, Angew. Chem., Int. Ed 2001, 40, 3392–3394. [DOI] [PubMed] [Google Scholar]; (c) Hofmann P, Shishov IV, Rominger F, Inorg. Chem 2008, 47, 11755–11762. [DOI] [PubMed] [Google Scholar]
- [11]. Reaction of a more electrophilic carbene source ethyl diazoacetate with dioxane catalyzed by a hydrotris(3-mesityl)pyrazolylborate Cu complex gave C-H insertion product in 20% yield. Please see ref. 5b.
- [12].Espino CG, Fiori KW, Kim M, Du Bois J, J. Am. Chem. Soc 2004, 126, 15378–15379. [DOI] [PubMed] [Google Scholar]
- [13].Matsuoka K, Komami N, Kojima M, Mita T, Suzuki K, Maeda S, Yoshino T, Matsunaga S, J. Am. Chem. Soc 2021, 143, 103–108. [DOI] [PubMed] [Google Scholar]
- [14]. For enantioselective, engineered cytochrome P450 enzyme-catalyzed amine α-C-H trifluoroethylation please see ref. 7a.
- [15].Barreiro EJ, Kümmerle AE, Fraga CAM, Chem. Rev 2011, 111, 5215–5246. [DOI] [PubMed] [Google Scholar]
- [16].Non-selective photochemical insertions of methylene into C-H bonds are known. Addition of transition metal, such as rhodium, shuts down C-H insertion reactivity in favor of ylide formation.; (a) Meerwein H, Rathjen H, Werner H, Chem. Ber 1942, 75, 1610–1622. [Google Scholar]; (b) Kirmse W, Chiem PV, Tetrahedron Lett 1985, 197–200. [Google Scholar]; (c) Olah GA, Doggweiler H, Felberg JD, J. Org. Chem 1984, 49, 2116–2120. [Google Scholar]
- [17].(a) Caballero A, Díaz-Requejo MM, Trofimenko S, Belderrain TR, Pérez PJ, Eur. J. Inorg. Chem 2007, 2848–2852. [DOI] [PubMed] [Google Scholar]; Reactions of TMSCHN2 with dioxane give higher yields if lower catalyst loadings are employed. Please see Table S-2 in Supporting Information.; (c) Besora M, Braga AAC, Sameera WMC, Urbano J, Fructos MR, Pérez PJ, Maseras FA, J. Organomet. Chem 2015, 784, 2–12. [Google Scholar]; (d) Jagannathan JR, Fettinger JC, Shaw JT, Franz AK, J. Am. Chem. Soc 2020, 142, 11674–11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.