

Summary
Background
Western (WEEV), eastern (EEEV), and Venezuelan (VEEV) equine encephalitis viruses are mosquito-borne pathogens classified as potential biological warfare agents for which there are currently no approved human vaccines or therapies. This clinical trial aimed to evaluate the safety and tolerability of an investigational trivalent virus-like particle (VLP) vaccine, WEVEE VLP, composed of WEEV, EEEV, and VEEV VLPs.
Methods
The WEVEE VLP vaccine was evaluated in a phase 1, randomized, open-label, dose escalation trial at the Hope Clinic of the Emory Vaccine Center at Emory University, Atlanta, GA, USA. Eligible participants were healthy adults aged 18-50. Participants were assigned to a dose group of 6, 30, or 60 mcg and randomized 1:1 to receive the WEVEE VLP vaccine with or without alum adjuvant by intramuscular injection at study day 0 and at week 8. The primary objectives were the safety and tolerability of the vaccine (assessed in all participants who received at least one administration of study product) and the secondary objective was immune response by plaque reduction neutralization test (PRNT) four weeks after second vaccination. This trial is registered at ClinicalTrials.gov, NCT03879603.
Findings
Between April 2 and June 13, 2019, 30 trial participants were enrolled (mean age 32 years; 16 [53%] female, 14 [47%] male). Six groups of five participants each received 6, 30, or 60 mcg vaccine doses with or without adjuvant, and all 30 participants completed study follow-up. Vaccinations were safe and well-tolerated. The most frequently reported symptoms were mild injection-site pain and tenderness (22/30 [73%]) and malaise (15/30 [50%]). Dose-dependent differences in the frequency of pain and tenderness were found between the 6, 30, and 60 mcg groups (p = 0·022); no significant differences were observed between dosing groups for any other reactogenicity symptom. Two adverse events in one trial participant (60 mcg dose with alum) were assessed as possibly related to the study product; both resolved without clinical sequelae. Four weeks following the second vaccine administration neutralizing antibodies were induced in all study groups with the highest response seen against all three vaccine antigens in the 30 mcg + alum group (PRNT80 geometric mean titer: EEEV: 60·8 [95% CI 29·9-124·0]; VEEV: 111·5 [95% CI 49·8-249·8]; WEEV: 187·9 [95% CI 90·0-392·2]. Finally, four weeks following second vaccine administration the majority of trial participants developed an immune response to all three vaccine components (EEEV: 24/29 [83%]; VEEV: 26/29, [90%]; WEEV: 27/29, [93%]; EEEV, VEEV, and WEEV: 22/29, [76%]).
Interpretation
Consistent with phase 1 trials, the primary limitation of this study is the small sample size. The favorable safety profile and neutralizing antibody responses, along with pressing public health need, support further evaluation of this product in advanced phase clinical trials.
Funding
The Vaccine Research Center of the National Institute of Allergy and Infectious Diseases, National Institutes of Health funded the clinical trial. The United States Department of Defense contributed funding for manufacturing of the study product.
Introduction
Western (WEEV), eastern (EEEV), and Venezuelan (VEEV) equine encephalitis viruses are single-stranded RNA alphaviruses that are highly pathogenic to humans and other vertebrates.1 Viral transmission to humans is mediated by more than 20 species of mosquito vectors including Aedes, Coquillettidia, and Culex that are capable of transmitting virus between infected bird or rodent hosts and humans.1–3 These viruses have caused small, but recurrent epizootics in North, South, and Central America over the last several decades. The most recent outbreak documented in Central America occurred in Panama in 2010 with 13 confirmed cases of EEEV, 11 cases of VEEV, and one case of coinfection.4 However, due to overlap in clinical symptoms with other arboviruses such as Dengue, as well as limitations in surveillance and diagnostics, the true disease burden of these viruses it not clearly understood.5 Sporadic outbreaks of EEEV in humans have also occurred throughout the United States, averaging 11 cases per year between 2009-2018.6,7 During 2019, the United States encountered the largest outbreak of EEEV to date with 38 confirmed cases and 15 related deaths predominantly across the northeast.7 The clinical manifestation of VEEV, WEEV, or EEEV infection ranges from mild flu-like symptoms to severe neurological illnesses including fatal encephalitis.1,8 The estimated case fatality rates associated with infection can be low (<1%) for VEEV, moderate (3-15%) for WEEV, and as high as 70% for EEEV.9 The recent outbreaks in the United States and high morbidity and mortality of EEEV demonstrate the importance of developing medical countermeasures against these pathogens.10
Although aerosol transmission does not occur naturally, previous studies and documented accidental infections in laboratory workers have shown that these viruses are highly stable and particularly transmissible in aerosol form. Furthermore, infection acquired by aerosol transmission results in a similar disease as mosquito-borne infection.11–13 These characteristics contribute to the classification of all three alphaviruses as category B bioterrorism agents/priority pathogens by both the United States Centers for Disease Control and Prevention (CDC) and National Institute of Allergy and Infectious Diseases (NIAID), delineating the viruses as potential biological warfare agents that could pose a risk to national security.14
There are currently no therapies or vaccines for VEEV, WEEV, or EEEV approved for human use by the United States Food and Drug Administration (FDA). Results of a phase 1 clinical trial of a DNA vaccine candidate against VEEV, pWRG/VEE, showed the vaccine was safe and induced a durable immune response in some study groups; however, there are no reports of the product in advanced clinical testing or commercial development.15 Beginning in the early 1960s, several candidate vaccines have additionally been utilized under Investigational New Drug (IND) status for at-risk laboratory personnel through the United States Army (US Army) Special Immunization Program.16 The live-attenuated vaccine TC-83 against VEEV induces protective humoral responses but is associated with significant adverse events and reactogenicity in approximately 25% of recipients and has a sub-optimal neutralizing antibody profile in approximately 20% of recipients. Therefore, a formalin-inactivated VEEV vaccine, C-84, has been used as a boost with TC-8317–20. While C-84 is found to be less reactogenic, it provides a suboptimal immune response in TC-83 non-responders.18 Formalin-inactivated vaccines against WEEV (TS-GSD 210) and EEEV (TS-GSD 104) have also been evaluated by the US Army and while safe, demonstrate low rates of seroconversion and limited effectiveness.21,22 Furthermore, sequential vaccination against all three viruses may lead to immune interference.23,24 Together, the limitations associated with these vaccine candidates have thus far hindered further development.
To address the unmet public health and biosecurity need for a prophylactic vaccine, there is an ongoing research effort to develop an effective vaccine against all three viruses that could be utilized by laboratory and military personnel, populations in the Americas at high risk of infection, and as a biodefense agent (NCT04131595; NCT01984983).15 Following the successful development and clinical evaluation of a virus-like particle (VLP) vaccine for Chikungunya virus, the Vaccine Research Center (VRC) developed a trivalent vaccine made up of VEEV, WEEV, and EEEV VLPs.25,26 Preclinical nonhuman primate studies with the western, eastern, and Venezuelan equine encephalitis (WEVEE) VLP vaccine demonstrated that the vaccine was highly immunogenic and offered protection from lethal aerosol challenge against all three viruses without detected immune interference, thus prompting advancement into clinical evaluation.27 Here, we report the safety and immunogenicity results of a phase 1 clinical trial evaluating the WEVEE VLP vaccine in healthy adults.
Methods
Study Design
VRC 313 was a first-in-human phase 1, open-label, dose-escalation clinical trial designed to assess the safety, tolerability, and immunogenicity of a trivalent VLP encephalitis vaccine with or without adjuvant at 6, 30, or 60 mcg administered in two doses eight weeks apart. The study was sponsored by the Vaccine Research Center (VRC), National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), and conducted at the Hope Clinic of the Emory Vaccine Center at Emory University in Atlanta, GA. The clinical trial protocol was reviewed and approved by the Emory University Institutional Review Board (IRB).
Participants
Eligible study participants were healthy adults, aged 18 to 50 years with no prior vaccinations with an investigational alphavirus vaccine. Individuals were recruited through print and electronic media with advertisements reviewed and approved by the Emory IRB. The trial was not designed to achieve an equal distribution of participant ages, and interested participants were enrolled based on the eligibility criteria. To determine eligibility, healthy adults were pre-screened and assessed through clinical laboratory tests, self-reported medical history, and a physical examination. Full inclusion and exclusion criteria are available in the trial protocol. All participants gave written informed consent before enrollment.
Randomization and masking
Once eligibility was assessed, eligible participants were assigned to a dose group and randomized 1:1 to receive the WEVEE VLP vaccine with or without alum using an electronic randomization system generated by the Protocol Statistician. There were two dose-escalation reviews in the study, performed by a Protocol Safety Review Team (PSRT), assessing whether any safety concerns had arisen before escalation to the next dose could occur. No more than one participant was randomized and vaccinated per day for the first three participants at each dose group. Randomization began with the 6 mcg WEVEE VLP dose groups, and the first interim safety review was conducted after the first 3 participants who received a 6 mcg dose of WEVEE VLP had two weeks post-vaccination safety data. After the first dose escalation criteria were met and the 6 mcg dose level was assessed as safe, randomization proceeded for the 30 mcg WEVEE VLP dose groups. The second interim safety review was conducted after the first 3 participants to receive a 30 mcg dose of WEVEE VLP had two weeks of post-vaccination safety data. After the second dose escalation criteria were met and the 30 mcg dose level was assessed as safe, remaining participants were randomized to a 60 mcg WEVEE VLP dose group. Vaccines were administered open label, and the study clinicians and each participant were informed on the assigned vaccine regimen after randomization was complete.
Procedures
The vaccine product, VRC-WEVVVLP073-VP (WEVEE VLP), was developed by the VRC,27 and was generated through construction of eukaryotic expression vectors encoding the structural proteins capsid (C), E1, E2, E3, and 6K from WEEV strain CBA87, EEEV strain PE-6, and VEEV strain TC-83 by methods previously described.27 Briefly, plasmid DNA (raw material) was manufactured by transforming E.coli DH5α cells with respective alphavirus recombinant plasmids, followed by expansion, first in shake flask and then in 100L bioreactors. Plasmids were purified by a combination of filtration and column chromatography methods. Purified plasmids were used to transiently transfect human embryonic kidney 293-derived suspension cell line (HEK293) to express protein that self-assembled to form non-replicating VLPs, which were then harvested from the culture medium. Generated VLPs were purified by filtration and column chromatography and mixed at a 1:1:1 mass ratio. Vaccination doses and regimens were selected based on the preclinical evaluations of WEVEE vaccine in nonhuman primates (NHPs) as well as clinical study outcomes of chikungunya VLP-based vaccine in humans.25–27 The vaccine was filled into single-dose vials at a concentration of 78 mcg/mL. The study adjuvant was an aluminum hydroxide suspension (alum) provided in a sterile, pyrogen-free suspension at a concentration of 5 mg/mL. The alum dose of 500 mcg was mixed in during preparation of each vaccine dose, and the diluent consisted of sterile phosphate buffered saline (PBS). The study products were manufactured under current Good Manufacturing Practices (cGMP) at the VRC Pilot Plant operated by the Vaccine Clinical Material Program, Leidos Biomedical Research, Inc. The study product was administered by intramuscular (IM) injection in the deltoid muscle via needle and syringe at study day 0 and at week 8; all product administrations were monitored by a study clinician. Safety laboratory tests were obtained prior to product administration and throughout the study. Volunteers recorded solicited symptoms for seven days after each product administration and a clinician assessed the site of vaccination on the day of administration, two days after vaccination, and two weeks later. For all dose groups, solicited local reactogenicity including pain/tenderness, redness, and swelling at the injection site, and systemic reactogenicity including fever, malaise, myalgia, joint pain, headache, chills, and nausea, was evaluated using a 7-day diary card. Clinical assessments were also performed at scheduled follow-up study visits throughout the study. Adverse events were graded according to the Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventive Vaccine Clinical Trials (modified from FDA Guidance, September 2007). All adverse events (AEs) were recorded for 28 days after product administration; serious adverse events (SAEs) and new chronic medical conditions were recorded throughout the study.
Outcomes
The primary endpoints were safety, measured by laboratory tests and adverse events, and tolerability, measured by local and systemic reactogenicity. The secondary endpoint was immunogenicity at four weeks after second vaccination, study week 12. WEEV, EEEV, and VEEV neutralizing titers were assessed using a plaque reduction neutralization test (PRNT) at baseline and study weeks 4, 10, 12, 24, and 36 as described previously.27 Details of the assay are provided in the Appendix. The reciprocal of the serum dilution which neutralized 80% of the input virus was reported as PRNT80 values, and a positive response was defined as PRNT80 titers above the limit of detection (PRNT value ≥ 10).
Sample size
Per the trial’s protocol, sample size calculations for safety were expressed in terms of the ability to detect SAEs. Sample sizes were chosen so within each group (n=5), there was over 90% chance to observe at least 1 SAE if the true rate is at least 0.37 and over 90% chance to observe no SAE if the true rate was no more than 0.02.
Statistical analysis
In accordance with the trial’s Statistical Analysis Plan (SAP), the safety population consisted of all participants who received at least one administration of study product summarized according to the actual study product received. The frequencies of maximum severity for any local reactogenicity symptom and any systemic reactogenicity symptom over all vaccinations were compared using Fisher’s exact test for the following groupings: alum and no-alum groups at each dose level; adjuvanted and unadjuvanted participants overall; and by dose level. Quantitative immunogenicity responses, defined as the PRNT80 titers, were summarized using geometric means, and were visually presented on a log-transformed scale. Positive qualitative PRNT80 responses, as defined above, were summarized as the proportion of individuals in each group with a positive response. Responses were analyzed for all participants who completed the full vaccine regimen (product administration at day 0 and week 8). When comparing responses between alum and no-alum groups, quantitative responses were compared using Student’s two-sample t-test with unequal variance. Qualitative responses were compared using Fisher’s exact test. Similarly, when comparing responses between dose groups, quantitative responses were compared on the log scale using Student’s two-sample t-test with unequal variance; qualitative responses were compared using Fisher’s exact test. This trial is registered with ClinicalTrials.gov, NCT03879603.
Role of the funding source
The Vaccine Research Center of the National Institute of Allergy and Infectious Diseases, National Institutes of Health funded the study and its investigators had complete control over study design, data collection, data analysis, data interpretation, and writing of the report. The United States Department of Defense contributed funding for manufacturing of the study product. The corresponding author had access to all the data in the trial and final responsibility for the decision to submit for publication.
Results
Of the 40 individuals screened, 30 participants were enrolled from April 2 through June 13, 2019 (Figure 1). Participants were enrolled in the open-label dose-escalation protocol to receive 6, 30, or 60 mcg of WEVEE VLP and randomized on the product administration day to receive vaccine with or without alum (5 subjects per study group). The second product administration occurred at study week 8, and the final study vaccination occurred on August 9, 2019. The study population comprised 16 women (53%) and 14 men (47%); the mean age was 32 years (range: 21 to 48) (Table 1). One participant in the 6 mcg dose group received only one of the two study vaccinations due to a case of severe neutropenia that was determined to be not related to product administration; all other participants received both scheduled injections for a total of 59 vaccinations. All enrolled participants completed study follow-up. The final study follow-up visit occurred on February 26, 2020 (Figure 1).
Figure 1: VRC 313 Trial CONSORT Diagram.
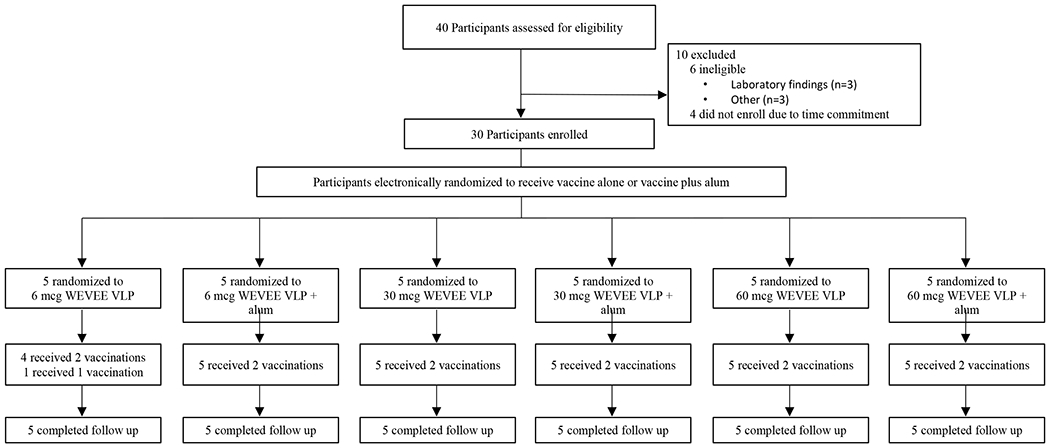
Participants were enrolled according to a dose-escalation protocol and randomized within each dose group to receive two doses of WEVEE VLP with or without alum, eight weeks apart. One participant did not receive the second vaccination due to asymptomatic neutropenia but continued with safety follow-up. All participants completed at least 12 weeks of follow-up.
Table 1:
Summary of Categorical Demographic and Baseline Characteristics by Treatment Group
| 6 mcg WEVEE (n = 5) | 6 mcg WEVEE + alum (n = 5) | 30 mcg WEVEE (n = 5) | 30 mcg WEVEE + alum (n = 5) | 60 mcg WEVEE (n = 5) | 60 mcg WEVEE + alum (n = 5) | All Participants (n = 30) | ||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Variable | Characteristic | n (%) | ||||||
| Sex | Male | 3 (60) | 2 (40) | 3 (60) | 2 (40) | 2 (40) | 2 (40) | 14 (47) |
| Female | 2 (40) | 3 (60) | 2 (40) | 3 (60) | 3 (60) | 3 (60) | 16 (53) | |
| Age | 21-30 | 4 (80) | 3 (60) | 2 (40) | 3 (60) | 2 (40) | 3 (60) | 17 (57) |
| 31-40 | 0 (0) | 2 (40) | 1 (20) | 2 (40) | 1 (20) | 2 (40) | 8 (27) | |
| 41-50 | 1 (20) | 0 (0) | 2 (40) | 0 (0) | 2 (40) | 0 (0) | 5 (17) | |
| Ethnicity | Hispanic or Latino | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (20) | 1 (3) |
| Not Hispanic or Latino | 5 (100) | 5 (100) | 5 (100) | 5 (100) | 5 (100) | 4 (80) | 29 (97) | |
| Race | Asian | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (20) | 1 (3) |
| Black or African American | 2 (40) | 1 (20) | 1 (20) | 0 (0) | 1 (20) | 1 (20) | 6 (20) | |
| White | 3 (60) | 4 (80) | 4 (80) | 5 (100) | 4 (80) | 3 (60) | 23 (77) | |
| BMI | Mean (standard deviation) | 24·4 (3) | 31·6 (10) | 25·9 (3) | 24·7 (8) | 24·5 (4) | 25·0 (4) | 26·0 (6) |
| Education | High school graduate/GED | 0 (0) | 0 (0) | 0 (0) | 1 (20) | 0 (0) | 0 (0) | 1 (3) |
| College/University | 3 (60) | 4 (80) | 3 (60) | 2 (40) | 3 (60) | 3 (60) | 18 (60) | |
| Advanced Degree | 2 (40) | 1 (20) | 2 (40) | 2 (40) | 2 (40) | 2 (40) | 11 (37) | |
Abbreviation: BMI, body mass index, calculated as weight in kilograms divided by height in meters squared.
Vaccinations were safe and well-tolerated with no SAEs or dose-limiting toxicities. All reported local and systemic solicited reactogenicity was mild to moderate in severity. Mild pain and tenderness at the injection site was the most frequently reported symptom, occurring in 22/30 participants (73%); no other local symptoms were reported. Dose-dependent differences in the frequency of pain and tenderness were found between the 6, 30, and 60 mcg groups (p = 0·022). Mild (grade 1) systemic reactogenicity was reported by 17/30 participants (57%), including malaise (n = 15; 50%), myalgia (n = 9; 30%), headache (n = 7; 23%), nausea (n = 6; 20%), joint pain (n = 3; 10%), and chills (n = 1; 3%); no fever was reported. Two out of the 30 participants (7%) reported moderate (grade 2) headache. There were no statistically significant differences in systemic reactogenicity between dosing groups. There were no significant differences in reactogenicity between participants who received alum versus those who did not (Figure 2; Appendix, pages 2-3, Table 1).
Figure 2: Maximum local and systemic solicited reactogenicity.
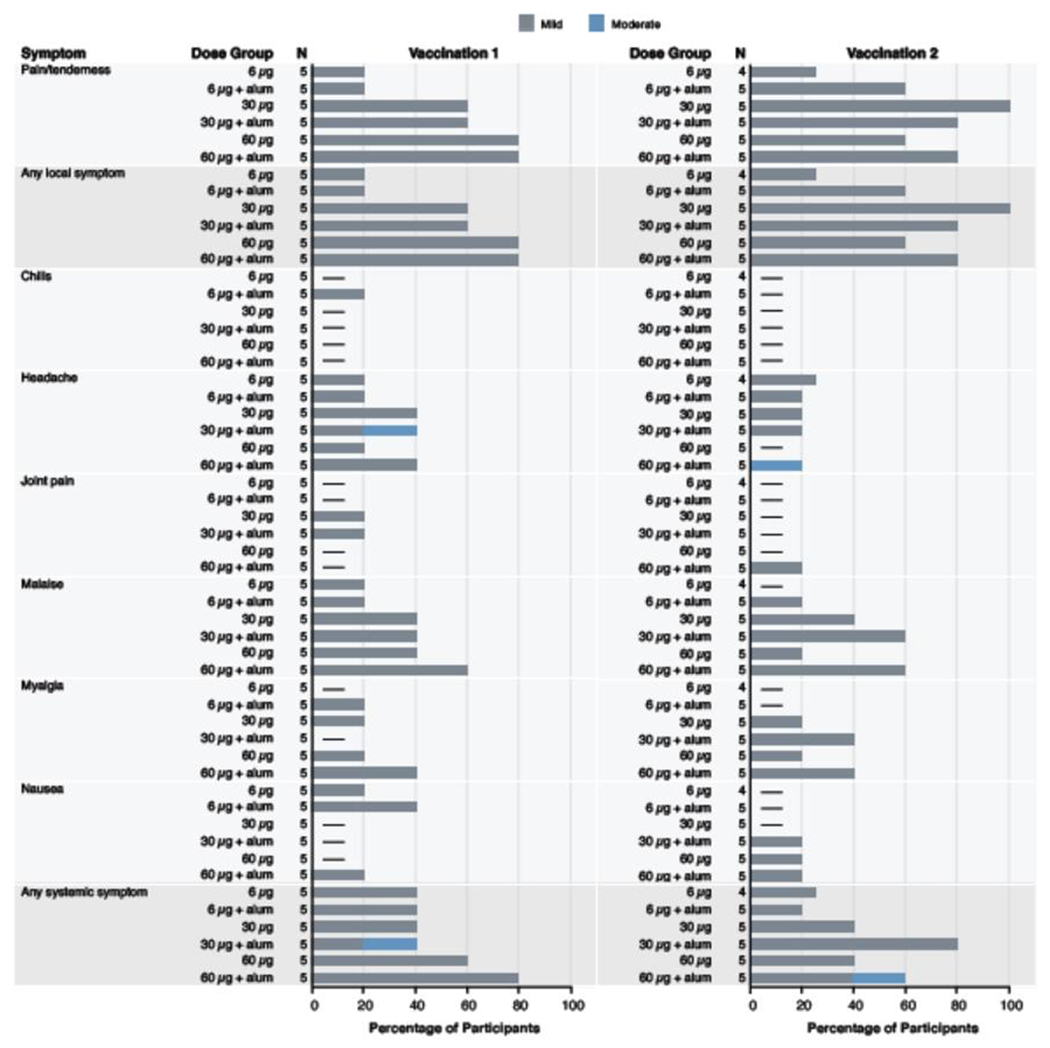
Percent of participants (x-axis) who reported a local or systemic symptom (y-axis) in the seven days following administration of vaccination 1 (study week 0) and vaccination 2 (study week 8). There were no reported local symptoms of redness or swelling, or systemic symptom of fever for any participants following either vaccination, n = 5 for all dose groups except for 6 mcg WEVEE (n = 4) following vaccination 2.
Product administrations were discontinued for one participant receiving a 6 mcg dose without alum who developed severe (grade 3) neutropenia identified 12 days after vaccination, peaked at day 33, and fully resolved 6 months later with no clinical sequelae. Due to the time course and this participant’s pre-existing history of benign neutropenia, this adverse event was evaluated as unrelated to vaccination.
For one participant randomized to receive a 60 mcg dose with alum, two adverse events were assessed as possibly related to the study product. These included mild elevated blood pressure 30 minutes after first vaccination that resolved when assessed 12 days later, and moderate asymptomatic neutropenia at 28 days following the second vaccination that self-resolved 35 days later without clinical sequelae and therefore was not considered of clinical significance.
At baseline, no participants had positive neutralization titers to EEEV, VEEV, or WEEV. By the week 12 primary immunogenicity endpoint (four weeks after the second dose), neutralizing antibodies were induced in 22/29 trial participants (76%) against all three vaccine components (EEEV: 24/29 [83%]; VEEV: 26/29 [90%]; WEEV: 27/29 [93%]) (Figure 3; Appendix, page 7, Table 4). In both the 30 mcg with alum and 60 mcg without alum dosing groups, all 5 participants (100%) in each group had positive neutralization responses to all three vaccine components at week 12 (Appendix, page 7, Table 4). Furthermore, neutralizing antibody responses were durable, induced in comparable magnitude against EEEV, VEEV, and WEEV, and were dose- and adjuvant-dependent (Figure 4; Appendix, pages 5-7, Tables 3 and 4).
Figure 3: Neutralizing Antibodies.
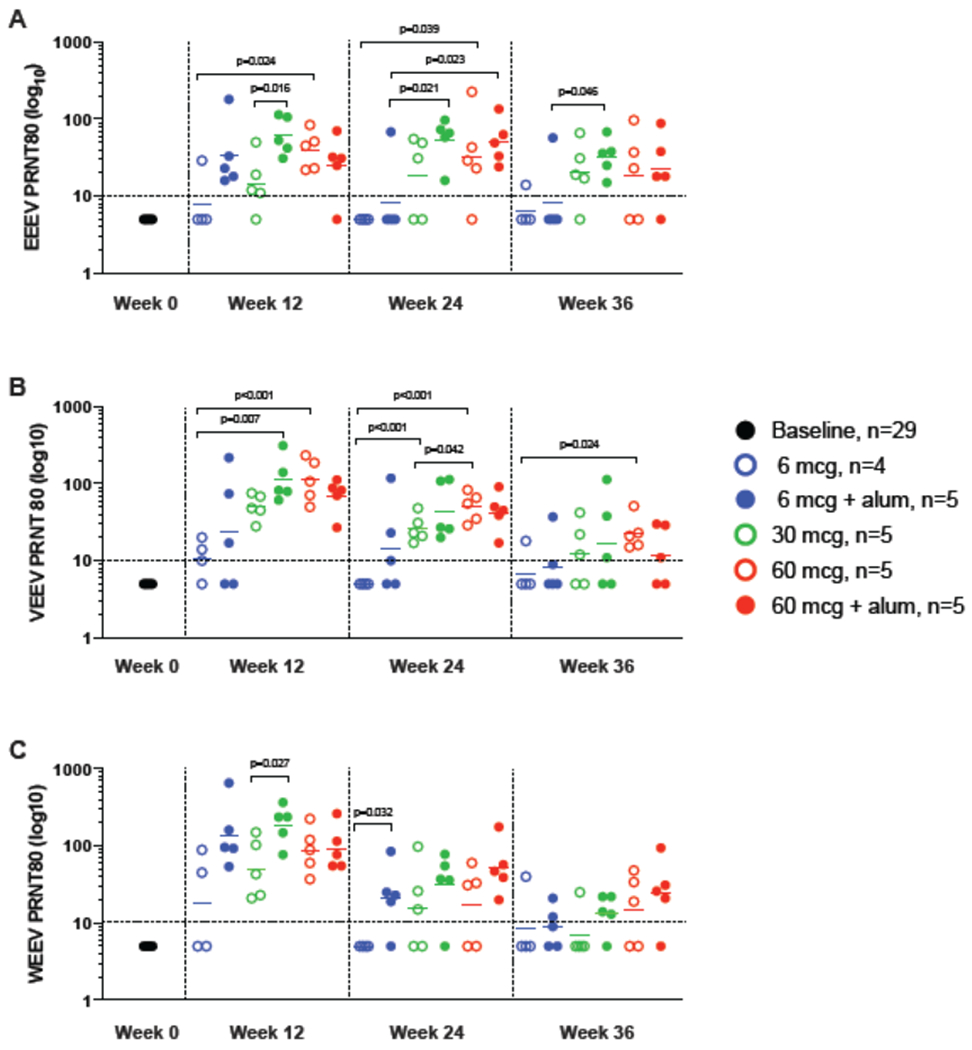
PRNT80 titers for EEEV (Panel A), VEEV (Panel B), and WEEV (Panel C) at baseline (Pre) and at 4, 16, and 24 weeks post the second product administration are shown. Participants received 6, 30, or 60 mcg WEVEE VLP without alum (open circles) or with alum (closed circles). The reciprocal of the serum dilution which neutralizes 80% of the input virus are shown as PRNT80 titers for each participant. A positive response was defined as a PRNT80 value ≥ 10 as indicated by the dotted horizontal line. A two-sample t-test was used to compare groups, and statistically significant p values (<0·05) are shown. Both dose- and alum-dependent responses are observed, and positive responses are seen in nearly all participants in the 30 and 60 mcg dose groups with and without alum. Highest responses are seen in the 30mcg + alum group against all three viruses at week 4 post second vaccine administration.
Figure 4: Durability of Neutralizing Antibody Response.
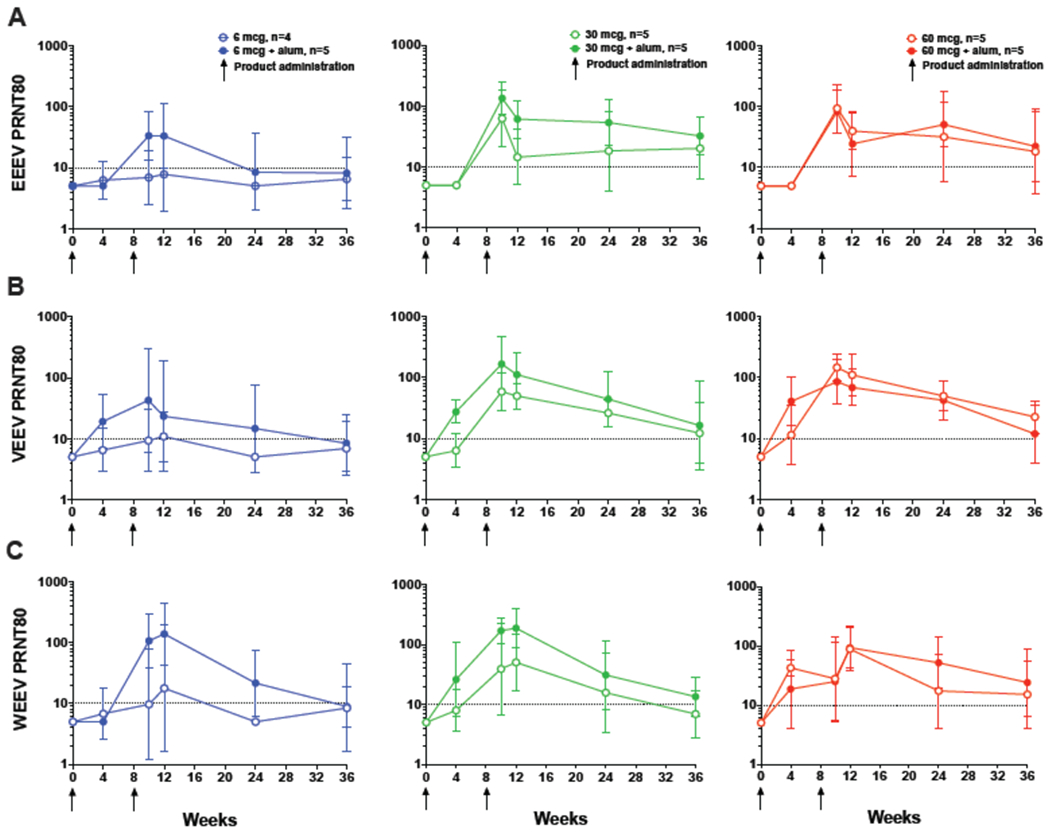
PRNT80 titers for EEEV (Panel A), VEEV (Panel B), and WEEV (Panel C) up to 28 weeks post second product administration. Participants received 6, 30, or 60 mcg WEVEE VLP without alum (open circles) or with alum (closed circles) administered at day 0 and study week 8 indicated by black arrows. The reciprocal of the serum dilution which neutralizes 80% of the input virus is shown as the geometric mean PRNT80 titers by study group, with 95% confidence intervals indicated by error bars. A positive response was defined as a PRNT80 value ≥ 10 as indicated by dotted horizontal line, and n = 5 for all dose groups with the exception of 6 mcg WEVEE (n = 4). Peak neutralizing responses were seen at 2 weeks (EEEV, VEEV) and 4 weeks (WEEV) post second vaccination. The dosing effect is observed out to the last study time point, and an alum-dependent response is seen in the 6 and 30 mcg dosing groups throughout the study time course.
Neutralizing antibody responses to EEEV at week 12 demonstrated a dose- and adjuvant-dependent response profile. The impact of alum was most clearly seen in the 6 and 30 mcg groups; alum did not increase the antibody response in the 60 mcg group. In the 6 mcg group, most participants did not generate neutralizing antibodies unless alum was also delivered, whereas in the 30 mcg dose group addition of alum yielded statistically significant increase in neutralizing antibodies (30 mcg with alum vs 30 mcg without alum GMT: 60·8 [95% CI 29·9-124·0] vs 14·4 [95% CI 5·1-41·2], p = 0·016). There was also increased neutralizing antibody response with increased vaccine dose (6 mcg, 30 mcg, 60 mcg), a difference which was statistically significant between the 6 mcg group and the 60 mcg group (6 mcg vs 60 mcg GMT: 7·8 [95% CI 1·9-31·4] vs 39·6 [95% CI 19·6-80·1], p = 0·024) (Figure 3; Appendix, pages 5-6, Table 3).
Neutralizing antibody responses to VEEV at week 12 followed a similar pattern to EEEV neutralization. The impact of alum was clearly demonstrated in both the 6 and 30 mcg groups; however, no difference in the magnitude of the response was observed in the 60 mcg group. The response also increased with each dosing group, and these increases were statistically significant when comparing the 6 mcg dose to both of the higher doses (6 mcg vs 30 mcg: 10·9 [95% CI 4·3-27·8] vs 49·9 [95% CI 30·7-80·9], p = 0·007; 6 mcg vs 60 mcg: 10·9 [95% CI 4·3-27·8] vs 110·5 [95% CI 49·8-245·5], p<0·001) (Figure 3; Appendix, pages 5-6, Table 3).
The WEEV neutralization responses at week 12 were consistent with those observed for both EEEV and VEEV. The addition of alum increased the magnitude of the response in both the 6 and 30 mcg groups and was statistically significant in the 30 mcg group (30 mcg without alum vs 30 mcg with alum GMT: 50·3 [95% CI 16·8-150·2] vs 187·9 [95% CI 90·0-392·2], p = 0·027). Neutralizing antibody responses increased with increasing dose (Figure 3; Appendix, pages 5-6, Table 3).
Immune responses to all three vaccine components were seen up to 28 weeks post second vaccination at the 36 week time point. The dose effect was observed out to week 36, where neutralizing antibody responses against each virus were seen in the majority of participants in the 30 and 60 mcg groups, but not in the 6 mcg group. At week 36 in the 30 mcg without alum group, 4/5 [80%] participants had a positive response against EEEV, 3/5 [60%] against VEEV, and 1/5 [20%] against WEEV. In the 30 mcg with alum group, 5/5 [100%] participants had a positive response against EEEV, 3/5 [60%] against VEEV, and 4/5 [80%] against WEEV. In the 60 mcg without alum group, 3/5 [60%] participants had a positive response against EEEV, 5/5 [100%] against VEEV, and 3/5 [60%] against WEEV. Finally, in the 60 mcg with alum group, 4/5 [80%] of participants had positive responses against EEEV, 3/5 [60%] against VEEV, and 4/5 [80%] against WEEV (Figures 3 and 4; Appendix, pages 5-6, Table 3). Despite these high responses to each individual vaccine component at the week 36 time point, there was a reduction in the number of participants who responded to all three components simultaneously. For the 30 and 60 mcg dose groups (both with and without alum), 4/5 [80%] – 5/5 [100%] participants responded to all three vaccine components simultaneously at the primary immunogenicity time point at week 12; however, by week 36 these same groups ranged from 1/5 [20%] – 2/5 [40%] participants with neutralizing antibody responses against all three viruses (Appendix, page 7, Table 4). Finally, we observed peak neutralizing antibody responses at either week two (EEEV, VEEV) or four (WEEV) post second vaccination for all groups (Figure 4; Appendix, pages 5-6, Table 3).
Discussion
There remains an unmet public health need for an effective vaccine against the encephalitic alphaviruses WEEV, EEEV, and VEEV, which can cause severe disease, have resulted in recent outbreaks in the United States, and have a potential application as biological warfare agent. There are additionally no antiviral treatments available for any of these viruses.28 This first-in-human trial of a trivalent alphavirus VLP vaccine against WEEV, EEEV, and VEEV demonstrated that both unadjuvanted and alum-adjuvanted vaccine regimens were safe, well-tolerated, and immunogenic against the three antigens in healthy adults.
The magnitude of immune responses induced by the WEVEE VLP vaccine was both adjuvant- and dose-dependent. Significantly higher response rates to all three viruses were observed in study participants who received higher doses of WEVEE VLP vaccine, as well as most participants who received alum. Alum-based adjuvants are safe and well documented to facilitate increased antibody titers and help induce rapid, long-lasting immune responses, and are frequently utilized in first-in-human vaccine safety trials.29 We found that immune responses were potentiated by the addition of alum in the 6 mcg and 30 mcg dose groups; however, this increase was statistically significant in the 30 mcg dose group only (EEEV p = 0·016, WEEV p = 0·027, at week 12). Interestingly, in these two dose groups, the impact of the adjuvant is less pronounced by the final study time point at week 36, a trend most pronounced in the 6 mcg dose group. Due to the robust immunogenicity of VLPs, the 60 mcg dose induced a similar response when administered with or without alum, indicating that the alum adjuvant is dose sparing and most impactful in the groups receiving the lower dose of VLP vaccine.
The correlates of protection against WEEV, EEEV, or VEEV are presumed to be neutralizing antibodies,27 and in this study were measured by plaque reduction neutralization test (PRNT). Prior studies conducted by the US Army on investigational vaccines suggest that for the live-attenuated VEEV vaccine TC-83, a PRNT80 titer of >1:20 may be a protective response.18,19 EEEV and WEEV protective titers are less well defined; however, based on clinical studies of EEEV (TS-GSD 104) and WEEV (TS-GSD 210) vaccines involving subjects with no prior EEEV or WEEV exposure, the threshold titer for protection is considered to be PRNT80 >1:40.21,22 In this context, the WEVEE VLP vaccine induced high seroconversion rates and demonstrated potentially protective immune responses against the three alphaviruses.
VLP vaccines are known to induce durable immune responses,26,30 and here we demonstrate durability of the neutralizing antibody response to all three vaccine components through the final study time point of 28 weeks post second vaccination for most (60-100%) participants in the 30 mcg + alum and both 60 mcg groups. We detected robust response to EEEV, VEEV, and WEEV indicating that consistent with preclinical findings,27 immune interference is unlikely occurring in the WEEVE VLP vaccine as it has in studies in which monovalent alphavirus vaccine candidates TS-GSD 104 (EEEV), TS-GSD 210 (WEEV), and TC-83 (VEEV) were inoculated sequentially or concurrently.23,24
As with many phase 1 trials, the primary limitation of this study is the small sample size within each dosing regimen, limiting the ability to extrapolate detailed differences between dose groups and perform immunogenicity analyses adjusted for multiple comparisons. The favorable safety and immunogenicity elicited by the WEVEE VLP trivalent vaccine in the reported phase 1 trial has prompted formal commercial development of the product. Evaluation in advanced phase clinical trials should be performed to determine optimal dosing and vaccine administration schedule.
Supplementary Material
Research In Context.
Evidence before this study
Western (WEEV), eastern (EEEV), and Venezuelan (VEEV) equine encephalitis viruses are mosquito-borne pathogens classified as potential biological warfare agents for which there are currently no approved human vaccines or therapies. We searched PubMed for research articles from database inception up to October 13, 2021, using the terms “eastern” OR “western” OR “Venezuelan” AND “equine encephalitis virus” AND “vaccin*” AND “clinical trial.” At the time of the search, we found no publications of trials involving the administration of a trivalent vaccine against eastern, western, and Venezuelan equine encephalitis viruses in human participants. We did find publications discussing the phase I trial results of a DNA vaccine candidate against only VEEV. This vaccine, pWRG/VEE, expresses the E3-E2-6K-E1 genes of VEEV, and was found to be safe and induced a durable immune response in some study groups. The phase 1 clinical trial testing pWRG/VEE was completed in 2015 and we could not find any information about advanced phase clinical trial testing or commercial development of this vaccine. There are additionally monovalent vaccines against each of these viruses that have been utilized under Investigational New Drug (IND) status for at-risk laboratory personnel through the United States Army Special Immunization Program. These vaccines include both live-attenuated (TC-83, VEEV) and formalin-inactivated products (C-84, VEEV; TS-GSD 210, WEEV; TS-GSD 104, EEEV). However, suboptimal immunogenicity of the inactivated vaccines, reactogenicity of the live-attenuated vaccine, and potential immune interference following sequential immunization has left a remaining need for novel vaccine platforms against WEEV, EEEV, and VEEV. Following the successful development of a virus-like particle (VLP) vaccine against Chikungunya virus, the Vaccine Research Center has developed the WEVEE VLP vaccine, a trivalent vaccine composed of EEEV, WEEV, and VEEV VLPs. Preclinical studies demonstrated the WEVEE VLP vaccine provided complete protection for all three vaccine components against aerosol challenge in nonhuman primates with no evidence of immune interference. In this phase 1 clinical trial, we evaluated the safety, tolerability, and vaccine-induced antibody response of the WEVEE VLP vaccine in unadjuvanted and alum-adjuvanted regimens in healthy adults.
Added value of this study
To our knowledge, these are the first published results of a trivalent vaccine against eastern, western, and Venezuelan encephalitis viruses in human participants. All vaccine regimens were safe and well-tolerated. Dose- and adjuvant-dependent neutralizing antibody responses were elicited against all three vaccine components four weeks after the second administration and immune responses were durable through the study duration.
Implications of all the available evidence
The results of our trial indicate that the WEVEE VLP vaccine, used both in unadjuvanted and alum-adjuvanted regimens, is safe, well-tolerated, and demonstrated durable immune responses against all three vaccine components in healthy adults, with no evidence of immune interference as observed in studies of sequential administration of monovalent vaccine candidates against equine encephalitis viruses. These results of a trivalent VLP vaccine against eastern, western, and Venezuelan encephalitis viruses support the vaccine’s further development and evaluation in advanced clinical trials.
Acknowledgements
We thank the trial volunteers for their contribution and commitment to vaccine research. We additionally thank our colleagues and collaborators who contributed to this work including: Kevin Newell, Lisa Hoopengardner, Maureen Wilson, and Beth Baseler at Leidos Biomedical Research, Inc.; Jim Albert, Phyllis Renehan, Haley Howell, and Carol Jones at the Emmes Company; and Melicia Gainey at Battelle.
Funding/Support
The clinical trial was fully funded by the Vaccine Research Center, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH). The United States Department of Defense contributed funding for manufacturing of the study product.
VRC 313 Study Team:
Hope Clinic of the Emory Vaccine Center: Ellie Butler, Jean Winter, Jianguo Xu, Amy Sherman, Colleen Kelley, Rameses Fredrick, Nadine Rouphael, Varun Phadke, Cynthia Whitney, Alicarmen Alvarez, Renata Dennis, Rebecca Fineman, Pamela Lankford-Turner, Sha Yi, Lilin Lai; Vaccine Research Center: Gena Burch, Shanker Gupta, Nina Berkowitz, Cristina Carter, Allison Beck, Brenda Larkin, Stephanie Taylor,; Vaccine Research Center Production Program: Mandy Alger, Jessica Bahorich, Amy (Lynch) Chamberlain, Yachen Chang, Rajoshi Chaudhuri, Jonathan Cooper, Jacob Demirji, Fan Yang, Alissa Fernald, Deepika Gollapudi, Janel Holland-Linn, Lisa Kueltzo, James Lee, Jie Liu, Xun Liu, Rachel Mowery, Sarah O’Connell, Erwin Rosales-Zavala, Jason Sands, Xin Wang, Shaojie Weng, Sara Witter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interests
We declare no competing interests.
Data Sharing Statement
Data generated in this study is available as de-identified data on ClinicalTrials.gov, NCT03879603. The study protocol, statistical analysis plan, and informed consent form are available on ClinicalTrials.gov (https://clinicaltrials.gov/ProvidedDocs/03/NCT03879603/Prot_SAP_ICF_000.pdf). Additional de-identified data may be made available upon request to the corresponding author for investigators whose proposed use of the data has been approved by the NIAID IRB.
References
- 1.Ronca SE, Dineley KT, Paessler S. Neurological Sequelae Resulting from Encephalitic Alphavirus Infection. Front Microbiol 2016; 7: 959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arechiga-Ceballos N, Aguilar-Setien A. Alphaviral equine encephalomyelitis (Eastern, Western and Venezuelan). Rev Sci Tech 2015; 34(2): 491–501. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong PM, Andreadis TG. Eastern equine encephalitis virus in mosquitoes and their role as bridge vectors. Emerg Infect Dis 2010; 16(12): 1869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carrera JP, Forrester N, Wang E, et al. Eastern equine encephalitis in Latin America. N Engl J Med 2013; 369(8): 732–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aguilar PV, Estrada-Franco JG, Navarro-Lopez R, Ferro C, Haddow AD, Weaver SC. Endemic Venezuelan equine encephalitis in the Americas: hidden under the dengue umbrella. Future Virol 2011; 6(6): 721–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molaei G, Armstrong PM, Graham AC, Kramer LD, Andreadis TG. Insights into the recent emergence and expansion of eastern equine encephalitis virus in a new focus in the Northern New England USA. Parasit Vectors 2015; 8: 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eastern Equine Encephalatis (EEE), Center for Disease Control and Prevention. 2020. https://www.cdc.gov/easternequineencephalitis/index.html (accessed December 20 2021).
- 8.Zacks MA, Paessler S. Encephalitic alphaviruses. Vet Microbiol 2010; 140(3-4): 281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dupuy LC, Reed DS. Nonhuman primate models of encephalitic alphavirus infection: historical review and future perspectives. Curr Opin Virol 2012; 2(3): 363–7. [DOI] [PubMed] [Google Scholar]
- 10.Morens DM, Folkers GK, Fauci AS. Eastern Equine Encephalitis Virus - Another Emergent Arbovirus in the United States. N Engl J Med 2019; 381(21): 1989–92. [DOI] [PubMed] [Google Scholar]
- 11.Reed DS, Lind CM, Sullivan LJ, Pratt WD, Parker MD. Aerosol infection of cynomolgus macaques with enzootic strains of Venezuelan equine encephalitis viruses. J Infect Dis 2004; 189(6): 1013–7. [DOI] [PubMed] [Google Scholar]
- 12.Hanson RP, Sulkin SE, Beuscher EL, Hammon WM, McKinney RW, Work TH. Arbovirus infections of laboratory workers. Extent of problem emphasizes the need for more effective measures to reduce hazards. Science 1967; 158(3806): 1283–6. [DOI] [PubMed] [Google Scholar]
- 13.Rusnak JM, Dupuy LC, Niemuth NA, Glenn AM, Ward LA. Comparison of Aerosol- and Percutaneous-acquired Venezuelan Equine Encephalitis in Humans and Nonhuman Primates for Suitability in Predicting Clinical Efficacy under the Animal Rule. Comp Med 2018; 68(5): 380–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.NIAID Emerging Infectious Diseases/Pathogens. 2016. https://www.niaid.nih.gov/research/emerging-infectious-diseases-pathogens.
- 15.Hannaman D, Dupuy LC, Ellefsen B, Schmaljohn CS. A Phase 1 clinical trial of a DNA vaccine for Venezuelan equine encephalitis delivered by intramuscular or intradermal electroporation. Vaccine 2016; 34(31): 3607–12. [DOI] [PubMed] [Google Scholar]
- 16.National Research Council (US) Committee on Special Immunizations Program for Laboratory Personnel Engaged in Research on Countermeasures for Select Agents. Protecting the Frontline in Biodefense Research: The Special Immunizations Program. Washington (DC): National Academies Press; (US: ); 2011. 2, History of the Special Immunizations Program and Lessons Learned from Occupational Immunization Against Hazardous Pathogens. https://www.ncbi.nlm.nih.gov/books/NBK209341/. [PubMed] [Google Scholar]
- 17.McKinney RW, Berge TO, Sawyer WD, Tigertt WD, Crozier D. Use of an Attenuated Strain of Venezuelan Equine Encephalomyelitis Virus for Immunization in Man. Am J Trop Med Hyg 1963; 12: 597–603. [DOI] [PubMed] [Google Scholar]
- 18.Pittman PR, Makuch RS, Mangiafico JA, Cannon TL, Gibbs PH, Peters CJ. Long-term duration of detectable neutralizing antibodies after administration of live-attenuated VEE vaccine and following booster vaccination with inactivated VEE vaccine. Vaccine 1996; 14(4): 337–43. [DOI] [PubMed] [Google Scholar]
- 19.Rusnak JM, Kortepeter MG, Aldis J, Boudreau E. Experience in the medical management of potential laboratory exposures to agents of bioterrorism on the basis of risk assessment at the United States Army Medical Research Institute of Infectious Diseases (USAMRIID). J Occup Environ Med 2004; 46(8): 801–11. [DOI] [PubMed] [Google Scholar]
- 20.Berge TO, Gleiser CA, Gochenour WS Jr., Miesse ML, Tigertt WD. Studies on the virus of Venezuelan equine encephalomyelitis. II. Modification by specific immune serum of response of central nervous system of mice. J Immunol 1961; 87: 509–17. [PubMed] [Google Scholar]
- 21.Bartelloni PJ, McKinney RW, Calia FM, Ramsburg HH, Cole FE Jr. Inactivated western equine encephalomyelitis vaccine propagated in chick embryo cell culture. Clinical and serological evaluation in man. Am J Trop Med Hyg 1971; 20(1): 146–9. [DOI] [PubMed] [Google Scholar]
- 22.Bartelloni PJ, McKinney RW, Duffy TP, Cole FE Jr. An inactivated eastern equine encephalomyelitis vaccine propagated in chick-embryo cell culture. II. Clinical and serologic responses in man. Am J Trop Med Hyg 1970; 19(1): 123–6. [DOI] [PubMed] [Google Scholar]
- 23.Pittman PR, Liu CT, Cannon TL, Mangiafico JA, Gibbs PH. Immune interference after sequential alphavirus vaccine vaccinations. Vaccine 2009; 27(36): 4879–82. [DOI] [PubMed] [Google Scholar]
- 24.Reisler RB, Gibbs PH, Danner DK, Boudreau EF. Immune interference in the setting of same-day administration of two similar inactivated alphavirus vaccines: eastern equine and western equine encephalitis. Vaccine 2012; 30(50): 7271–7. [DOI] [PubMed] [Google Scholar]
- 25.Chang LJ, Dowd KA, Mendoza FH, et al. Safety and tolerability of chikungunya virus-like particle vaccine in healthy adults: a phase 1 dose-escalation trial. Lancet 2014; 384(9959): 2046–52. [DOI] [PubMed] [Google Scholar]
- 26.Chen GL, Coates EE, Plummer SH, et al. Effect of a Chikungunya Virus-Like Particle Vaccine on Safety and Tolerability Outcomes: A Randomized Clinical Trial. JAMA 2020; 323(14): 1369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ko SY, Akahata W, Yang ES, et al. A virus-like particle vaccine prevents equine encephalitis virus infection in nonhuman primates. Sci Transi Med 2019; 11(492). [DOI] [PubMed] [Google Scholar]
- 28.Wolfe DN, Heppner DG, Gardner SN, et al. Current strategic thinking for the development of a trivalent alphavirus vaccine for human use. Am J Trop Med Hyg 2014; 91(3): 442–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kool M, Fierens K, Lambrecht BN. Alum adjuvant: some of the tricks of the oldest adjuvant. J Med Microbiol 2012; 61(Pt 7): 927–34. [DOI] [PubMed] [Google Scholar]
- 30.Donaldson B, Lateef Z, Walker GF, Young SL, Ward VK. Virus-like particle vaccines: immunology and formulation for clinical translation. Expert Rev Vaccines 2018; 17(9): 833–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.