

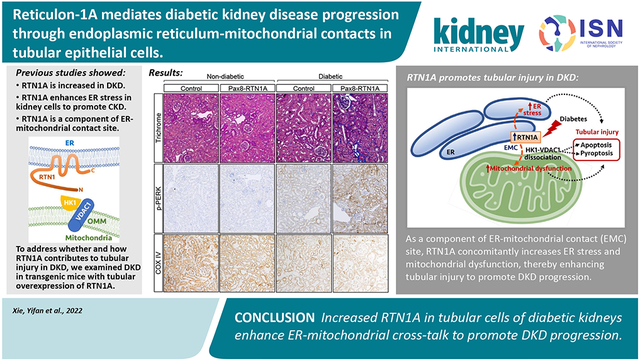
Abstract
Recent epidemiological studies suggest that some patients with diabetes progress to kidney failure without significant albuminuria and glomerular injury, suggesting a critical role of kidney tubular epithelial cell (TEC) injury in diabetic kidney disease (DKD) progression. However, the major risk factors contributing to TEC injury and progression in DKD remain unclear. We previously showed that expression of endoplasmic reticulum-resident protein Reticulon-1A (RTN1A) increased in human DKD, and the increased RTN1A expression promoted TEC injury through endoplasmic reticulum (ER) stress response. Here, we show that TEC-specific RTN1A overexpression worsened DKD in mice, evidenced by enhanced tubular injury, tubulointerstitial fibrosis, and kidney function decline. But RTN1A overexpression did not exacerbate diabetes-induced glomerular injury or albuminuria. Notably, RTN1A overexpression worsened both ER stress and mitochondrial dysfunction in TECs under diabetic conditions by regulation of ER-mitochondria contacts. Mechanistically, ER-bound RTN1A interacted with mitochondrial hexokinase-1 and the voltage-dependent anion channel-1 (VDAC1), interfering with their association. This disengagement of VDAC1 from hexokinase-1 resulted in activation of apoptotic and inflammasome pathways, leading to TEC injury and loss. Thus, our observations highlight the importance of ER-mitochondrial crosstalk in TEC injury and the salient role of RTN1A-mediated ER-mitochondrial contact regulation in DKD progression.
Keywords: Diabetic kidney disease, kidney tubular epithelial cells, endoplasmic reticulum stress, endoplasmic reticulum-mitochondrial contacts
Graphical Abstract
INTRODUCTION
Diabetic kidney disease (DKD) is the most common cause of kidney failure worldwide 1. The current therapeutic regimen provides only partial effects with no ability to forestall the disease progression, suggesting that the key pathogenic mechanisms driving DKD are not adequately inhibited. Histologically, DKD is characterized by glomerular basement membrane (GBM) thickening, mesangial expansion, and podocyte loss at the early disease stages, and diffuse or nodular glomerulosclerosis and tubulointerstitial fibrosis with inflammation at later stages. Notably, renal function of diabetic patients better correlates with the degree of tubulointerstitial injury than that of glomerular lesions 2, and recent evidence indicates that tubular epithelial cell (TEC) injury occurs at early DKD, even preceding the onset of microalbuminuria 3. Moreover, a growing body of evidence indicates that even in normoalbuminuric diabetic patients, DKD can progress to kidney failure and that the disease progression is associated with prominent tubulointerstitial injury and fibrosis instead of glomerular injury 4, 5. While hyperglycemia and increased advanced glycation end products (AGEs) were shown to promote tubular injury and fibrosis in diabetic kidneys 6-8, the molecular and cellular mechanisms of tubular injury in the early stage of DKD and how they contribute to the disease progression remain obscure.
Endoplasmic reticulum (ER) stress and mitochondrial dysfunction are two major cellular mechanisms involved in kidney cell injury in DKD 9-12 The role of ER stress in DKD development and progression is evidenced by studies demonstrating that the exposure of kidney cells to high glucose concentrations induces ER stress-mediated apoptosis 13 and that the genetic or pharmacological intervention of ER stress response attenuates DKD progression in mice 14-16. In humans, the elevated urinary protein excretion is associated with tubular injury and ER stress, and renal expression of ER stress markers in patients with progressive DKD are higher than patients with non-progressive DKD 13, 17, 18. The role of mitochondrial dysfunction in promoting DKD is also well described. Persistent hyperglycemia shifts the metabolic flux of glucose from complete oxidation in mitochondria to the glycolytic pathway, leading to altered metabolic pathways involving pentose phosphate, sorbitol and polyols, and advanced glycation end-products. Activation of these pathways results in increased oxidative stress, inflammation, fibrosis, DNA damage, and vascular changes. However, the relationship between ER stress and mitochondrial dysfunction has not been studied in the context of DKD.
By analyzing the transcriptomic datasets in a mouse model with progressive chronic kidney disease (CKD), we previously demonstrated that the increased expression of Rtn1 (encoding Reticulon-1A, RTN1A) is associated with CKD progression 19 Reticulons localize primarily to the ER membranes, and RTN1A expression was markedly increased in the TECs of the diseased kidneys and inversely correlated with the estimated glomerular filtration rate (eGFR) in diabetic patients 19 Three intronic SNPs of RTN1 were also identified to be associated with diabetic patients with end-stage kidney disease (ESKD) 20. Mechanistically, we demonstrated that beyond its purported role as an ER-membrane shaping protein, RTN1A exacerbates ER stress response and induces apoptosis of tubular cells in kidney disease settings. The knockdown of RTN1A expression in mice attenuated albuminuria and kidney injury in experimental models of type 1 and type 2 diabetic mice 19, 21. The RTN1A knockdown also reduced tubular injury and improved renal function in mice with albumin overload-induced nephropathy 22 and acute kidney injury 23. However, as these studies involved mice with global knockdown of Rtn1a, whether the enhanced tubular expression RTN1A contributed to DKD progression was not clear.
To specifically address the role of RTN1A in TECs, we examined the DKD phenotype in mice with TEC-specific RTN1A overexpression. Induction of mild DKD phenotype with low-dose streptozotocin in the RTN1A transgenic mice surprisingly led to a significant decline in renal function and enhanced tubulointerstitial fibrosis, but without significant change in glomerular injury, thereby mimicking the progressive DKD without overt proteinuria. Similar observations of enhanced tubular injury and worsening of DKD were observed in the transgenic type 1 diabetes model, OVE26. Moreover, as a component of ER-mitochondrial contact site (EMC), increased RTN1A expression augmented the diabetes-induced ER stress and mitochondrial dysfunction in tubular cells in diseased kidneys. We now report for the first time the role and the mechanism of RTN1A-mediated ER-mitochondrial crosstalk in TEC injury and the progression of early DKD.
METHODS
Mouse models
All mouse protocols were approved by the Institutional Animal Care and Use Committee at Icahn School of Medicine at Mount Sinai (Protocol # LA10-0001). The generation of double transgenic mice with tetracycline-responsive RTN1A transgene and Pax8 promoter-driven reverse tetracycline-controlled transactivator (TetO-RTN1A;Pax8-rtTA) in the FVB/NJ background was previously described 23. Streptozotocin-induced diabetic model: Diabetes induction by streptozotocin (STZ) was performed as described 24. Briefly, 8-week-old mice were intraperitoneally administered with low-dose STZ (50 mg/kg) in 50 mM sodium citrate buffer (pH 5.4) for 5 consecutive days. Diabetes was defined as sustained fasting blood glucose above 250 mg/dl at two weeks post-injection. All mice were euthanized after 29 weeks of STZ injection. OVE26 diabetic mouse model: Type 1 diabetic OVE26 transgenic mice [Tg(Cryaa-Tag, Ins2-CALM1)26Ove/PneJ] in the FVB/NJ background were obtained from The Jackson Laboratory, and crossed with TetO-RTN1A;Pax8-rtTA mice to generate OVE26; TetO-RTN1A;Pax8-rtTA transgenic mice. Doxycycline-supplemented chow (625mg/kg, Envigo) was given to transgenic mice starting at 8 weeks of age. Age-matched mice without doxycycline supplementation were used as controls, and all mice were euthanized at 24 weeks of age.
Statistical Analysis
Data expressed as mean±SD. Data were analyzed with an unpaired, two-tailed t-test between two groups, and a nonparametric Mann-Whitney test was applied for tubular injury scores. 2-way ANOVA with Tukey’s post hoc multiple comparison test was applied for analyses between three or more groups. GraphPad Prism (v.9) was used for statistical analysis.
Detailed Methods are provided in the Supplemental Methods section.
RESULTS
TEC-specific RTN1A overexpression worsens tubulointerstitial injury and DKD progression in diabetic mice
To assess the TEC-specific role of RTN1A in the progression of DKD, we utilized the transgenic mice with tetracycline-inducible, TEC-specific RTN1A overexpression (Pax8-rtTA;tetO-RTN1A) in the FVB/NJ strain 23. The administration of chow supplemented with doxycycline (625mg/kg chow) for three weeks led to an average of a four-fold increase in the expression of total RTN1A mRNA (mRtn1a and hRTN1A) in the kidney cortices of transgenic mice by quantitative polymerase chain reaction (qPCR) using a primer pair that simultaneously detects the murine and human transcripts 23 (Supp. Figure 1A). Immunostaining of frozen kidney tissues with RTN1A antibody showed increased RTN1A expression specifically within tubular cells, much of which co-localized with proximal and distal tubular markers, Lotus tetragonolobus agglutinin (LTA) and Dolichos biflorus agglutinin (DBA) (Supp. Figure 1B). We then induced diabetes in 8-week old transgenic mice with low-dose injections of streptozotocin (+STZ), which typically lead to mild to moderate DKD in FVB/NJ strains 25. Non-diabetic control mice (−STZ) received the citrate buffer vehicle. RTN1A expression was subsequently induced at 2 weeks post-STZ injection with Dox-supplemented chow (referred to as Pax8-RTN1A mice), and control transgenic mice without RTN1A induction were given normal chow (referred to as control mice). All mice were euthanized at 29 weeks post-STZ injection. As anticipated, diabetes led to a marked increase in RTN1A expression in the kidneys of control mice, which was further enhanced in the kidneys of Pax8-RTN1A mice (Supp. Figure 1C-E).
The diabetes induction led to similar levels of persistent hyperglycemia in control and Pax8-RTN1A mice (Supp. Figure 1E). However, we observed the worsening of diabetes-induced kidney hypertrophy and albuminuria in the Pax8-RTN1A mice in comparison to the control mice (Figure 1A-B). We also observed a modest elevation in the blood urea nitrogen (BUN) levels in comparison to the control diabetic mice, which became further enhanced in the diabetic Pax8-RTN1A mice (Figure 1C). These results indicated that increased RTN1A tubular expression exacerbated the mild tubular injury noted in the diabetic mice in the FVB/NJ strain. The histologic evaluations of tubular and glomerular injury were consistent with these findings in that mild tubular injuries in the control diabetic mice were further augmented in the diabetic Pax8-RTN1A mice, while diabetes-induced glomerular hypertrophy and mesangial expansion were without notable differences between the diabetic groups (Figure 1D-E). Moreover, although the tubulointerstitial fibrosis is not typically remarkable in the streptozotocin-induced models at this stage, its development was much more evident in the diabetic Pax8-RTN1A kidneys in comparison to the control diabetic kidneys, as shown by Masson's trichrome staining and Collagen 1A immunofluorescence (Figure 2A-C).
Figure 1: TEC-specific RTN1A overexpression accelerated renal function decline in STZ-induced diabetic mice.

(A) Kidney-to-body weight (K/BW) ratio at 29 weeks post-diabetes induction in control and Pax8-RTN1A mice. (B) Total 24-hour urinary albumin excretion at 29 weeks post-diabetes induction. (C) Blood urea nitrogen (BUN) levels at 29 weeks post-diabetes induction. (D) Representative images of periodic acid-Schiff (PAS)-stained kidneys at 100x (top) and 400x magnifications (scale bars: 20μm, top panel, and 10μm, bottom panel). (E) Quantification of average tubular injury score, mesangial matrix fraction (%) and the glomerular area is shown per mouse (n=5 mice per group).*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 between indicated groups by two-way ANOVA with Tukey's post hoc analysis or nonparametric Mann-Whitney test (for tubular injury scores).
Figure 2: TEC-specific RTN1A overexpression accelerated renal fibrosis in STZ-induced diabetic mice.

(A) Masson's trichrome-stained kidney sections of control and Pax8-RTN1A mice. Scale bar, 25μm. (B) Representative image of collagen 1A immunostained kidney sections. DNA is counterstained in blue. Scale bar, 30μm. (C) Quantification of average fold change in collagen 1A+ area is shown per mouse (n=5 mice per group, 10-12 fields evaluated per mouse). *p<0.05, ***p<0.001, and ****p<0.0001 between indicated groups by two-way ANOVA with Tukey's post hoc analysis.
Consistent with the above findings of enhanced tubular injury with RTN1A overexpression in diabetic kidneys, there was a marked increase in kidney injury marker-1 (KIM1) protein and mRNA expression (Figure 3A) and increased cell death, as ascertained by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining in the diabetic Pax8-RTN1A kidneys in comparison to the control diabetic kidneys (Figure 3B). Since increased RTN1A expression exacerbates the ER stress response to promote kidney disease progression 19, 23, we next examined the expression of ER stress markers in the kidneys of control and diabetic mice. While the mRNA expression of C/EBP homologous protein (Chop) and Gadd34 were modestly increased in diabetic mouse kidneys compared to nondiabetic control kidneys, their expression was markedly enhanced in the Pax8-RTN1A diabetic kidneys (Figure 3C). Similarly, the phosphorylation of protein kinase R-like ER kinase (p-PERK) was notably increased in the diabetic Pax8-RTN1A kidney tubules in comparison to all other groups (Figure 3D).
Figure 3: TEC-specific RTN1A overexpression accelerated tubular injury and death in STZ-induced diabetic mice.

(A) Representative image of KIM-1 immunostaining shows significant KIM-1 staining only in the diabetic Pax8-RTN1A mouse kidneys. Scale bar, 30μm. Real-time PCR analysis of Havcr1 in kidney cortices of control and Pax8-RTN1A mice is shown on the right (n=3 mice per group). (B) TUNEL-stained kidney sections. DNA is counterstained in blue. Scale bar, 10μm. Quantification of average TUNEL+ cells per field per mouse is shown on the right (n=5 mice per group, 10-12 fields evaluated per mouse). (C) Real-time PCR analysis of ER stress markers Chop and Gadd34 in kidney cortices of mice (n=3 mice per group). (D) Representative images of phosphorylated PERK (p-PERK) in kidney sections of control and Pax8-RTN1A mice. Scale bar, 20μm. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 between indicated groups by two-way ANOVA with Tukey’s post hoc analysis.
Since streptozotocin poses concerns of kidney cell cytotoxicity, we extended our analysis to the genetic model of type 1 diabetes, OVE26 transgenic mice 26, by generating Pax8-RTN1A;OVE26 triple transgenic mice. As OVE26 mice typically develop significant proteinuria by 8 weeks of age 26, 27, the tubular RTN1A overexpression was induced with doxycycline-supplemented chow in OVE26 mice and nondiabetic wildtype (WT) littermate controls at 8 weeks of age, and all mice were euthanized at 24 weeks of age.
While overt hyperglycemia was observed in all OVE26 mice (Supp. Figure 2A), diabetes-induced kidney hypertrophy and kidney function decline was worsened in the OVE26;Pax8-RTN1A mice than in control OVE26 mice (Figure 4A-C). We also observed a significant increase in tubular injury, apoptosis, and interstitial fibrosis in the OVE26;Pax8-RTN1A mice in comparison to control OVE26 mice (Fig. 4D-E) but without overt changes in the glomerular hypertrophy and mesangial expansion (Supp. Figure 2B). Together with the STZ model, these results highlight the role of RTN1A in the exacerbation of diabetes-induced ER stress response and ensuing tubular injury and fibrosis to promote DKD progression.
Figure 4: TEC-specific RTN1A overexpression accelerated renal function decline and renal fibrosis in OVE26 diabetic mice. Nondiabetic (WT) and diabetic OVE26 mice were evaluated at 24 weeks of age.

(A) Kidney-to-body weight (K/BW) ratio. (B) Total 24-hour urinary albumin excretion. (C) Blood urea nitrogen (BUN) levels. (D) Representative images of periodic acid-Schiff (PAS)-, Masson's trichrome-, and TUNEL-stained kidneys. Scale bars: 30μm. (E) Quantification of average tubular injury score, trichrome-stained fibrosis area (%), and TUNEL+ cells per field is shown per mouse (n=5 mice per group). *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 between indicated groups by two-way ANOVA with Tukey's post hoc analysis or nonparametric Mann-Whitney test (for tubular injury scores).
RTN1A regulates ER-mitochondrial contacts in tubular cells
To further delineate the molecular mechanisms of ER stress induction and tubular injury by RTN1A, we next examined the RTN1A-interacting proteins by mass spectrometry of proteins that co-immunoprecipitated with FLAG-tagged full-length RTN1A in HEK293 cells in comparison to control HEK293 cells (Supp. Table 1). We also examined RTN1A-interacting proteins versus those that immunoprecipitated with RTN1C that lack most of the N-terminal domain (Supp. Figure 3A) and does not participate in ER stress response 19, which resulted in a similar set of pulled-down proteins (data not shown). Surprisingly, proteins that co-immunoprecipitated with RTN1A, but not RTN1C, were predominantly mitochondrial proteins (Table 1). Notably, a recent study identified RTN1A as a component of a protein complex in the ER-mitochondrial contacts (EMCs), also referred to as mitochondria-associated ER membranes 28. EMC is a dynamic contact region, with a reported average of 10 to 30nm in distance between ER and mitochondria 29 Increasing evidence indicates that EMC is an important regulator of mitochondrial homeostasis, apoptosis, and autophagy 30. A recent report also showed that EMC integrity is disrupted in tubular cells in diabetic kidneys 31. Therefore, we also assessed the effects of RTN1A overexpression in EMC in tubular cells. We first performed the in situ proximity ligation assay (PLA) to detect EMCs in the cultured proximal tubular cell line, HK-2, using antibodies against ER-resident protein, inositol 1,4,5-triphosphate receptor-3 (IP3R-3), and mitochondrial voltage-dependent anion channel 1 (VDAC1). Incubation with only a single primary antibody (IP3R3 or VDAC1) served as a negative control. The analysis showed greater PLA signals in HK-2 cells under high glucose conditions in comparison to normal glucose, and these signals were further increased in cells overexpressing RTN1A (Figure 5A). These results suggested that RTN1A may enhance EMCs in tubular cells. To validate these findings in vivo, we next assessed the distance of EMCs between ER and mitochondria on transmission electron microscopy (TEM) images of tubular cells of control and Pax8-RTN1A mice. Examples of measurements are shown in Supp. Figure 2, and the average distance between ER and mitochondria apposition decreased in diabetic mice with RTN1A overexpression (Figure 5B, Suppl. Figure 4), suggesting an increased interaction between ER and mitochondria through RTN1A overexpression. Since the above results indicate that RTN1A overexpression worsens tubular injury in diabetic kidneys, it is likely that RTN1A increases ER-mitochondria crosstalk in kidney injury settings to promote disease progression.
Table 1: List of top 15 RTN1A-interacting proteins that are mitochondria-associated.
Top 15 RTN1A-interacting proteins are shown in the highest ratio of spectra counts between RTN1A-expressing and control (Ctrl) HEK293 cells. Gene names encoding the proteins are indicated in parenthesis.
| Top 15 mitochondria-associated proteins | Accession Number |
Molecular Weight |
Spectra counts (Ctrl) |
Spectra counts (RTN1A) |
Ratio (RTN1A/Ctrl) |
|---|---|---|---|---|---|
| Hexokinase-1 (HK1) | P19367 | 102 kDa | 0 | 25 | 13.5 |
| ATP synthase subunit beta, mitochondrial (ATP5B) | P06576 | 57 kDa | 3 | 43 | 9.0 |
| NADH-ubiquinone oxidoreductase 75kDa subunit, mitochondrial (NDUFS1) | P28331 | 79 kDa | 0 | 12 | 7.0 |
| Cytochrome b-c1 complex subunit 1, mitochondrial (UQCRC1) | P31930 | 53 kDa | 0 | 11 | 6.5 |
| Glycerol kinase (GK) | P32189 | 61 kDa | 0 | 10 | 6.0 |
| Metaxin-2 (MTX2) | O75431 | 30 kDa | 2 | 19 | 5.3 |
| Mitochondrial import receptor subunit TOM40 homolog (TOMM40) | O96008 | 38 kDa | 1 | 13 | 5.0 |
| Calcium-binding mitochondrial carrier protein, Aralar1 (SLC25A12) | O75746 | 75 kDa | 0 | 8 | 5.0 |
| Mitochondrial import receptor subunit TOM22 homolog (TOMM22) | Q9NS69 | 16 kDa | 0 | 7 | 4.5 |
| Voltage-dependent anion-selective channel protein 1 (VDAC1) | P21796 | 31 kDa | 7 | 37 | 4.3 |
| NADH dehydrogenase [ubiquinone] flavoprotein 1, mitochondrial (NDUFV1) | P49821 | 51 kDa | 0 | 6 | 4.0 |
| DnaJ homolog subfamily C member 11 (DNAJC11) | Q9NVH1 | 63 kDa | 0 | 6 | 4.0 |
| Voltage-dependent anion-selective channel protein 2 (VDAC2) | P45880 | 32 kDa | 4 | 20 | 3.7 |
| Cytochrome b-c1 complex subunit 2, mitochondrial (UQCRC2) | P22695 | 48 kDa | 1 | 9 | 3.7 |
| Leucine-rich PPR motif-containing protein, mitochondrial (LRPPRC) | P42704 | 158 kDa | 0 | 5 | 3.5 |
| MICOS complex subunit MIC19 (CHCHD3) | Q9NX63 | 26 kDa | 0 | 5 | 3.5 |
Figure 5: RTN1A overexpression increased the EMC contact sites cultured cells and reduced the distance of EMCs in tubular cells in vivo.

(A) Representative images of in situ proximity ligation assay targeting IP3R3-VDAC1 interaction in control or RTN1A overexpressing HK2 cells that were cultured high mannitol (HM, 25mM+5mM glucose) or high glucose (HG, 30mM) media for 24 hours. PLA red fluorescent dots indicate the location and extent of IP3R3-VDAC1 interaction. Nuclei were stained with DAPI. Each picture is representative of a typical cell staining observed in 10 fields chosen at random. Scale bar, 10μm. Quantification of the PLA signal per cell is shown on the right (average number of dots per cell, n=10 fields per condition). *p<0.05, **p<0.01, and ****p<0.0001 between indicated groups by two-way ANOVA with Tukey’s post hoc analysis. (B) Representative transmission EM images of areas of mitochondria-ER appositions in control and diabetic mice. Examples of distances are indicated. Scale bar, 20μm. (C) Quantification of the relative amount of OMM in contact with ER for each distance category (less than 5, between 6-10, and between 11-20nm). n=10-50 mitochondria in 10 fields per group. ****p<0.0001 compared to all groups by two-way ANOVA with Tukey’s post hoc analysis.
RTN1A worsens tubular injury through ER-mitochondrial cross-talk
Since mitochondrial dysfunction is a key contributor to tubular injury and apoptosis 32-35, we posited that in addition to augmented ER stress response, RTN1A may concomitantly contribute to mitochondrial dysfunction in diabetic kidneys via EMCs. Therefore, we carried out a series of in vitro experiments to test the role of RTN1A in the mitochondrial function of cultured TECs. In the first set of experiments, we assessed the effect of RTN1A overexpression on the mitochondrial function in the immortalized human kidney 2 (HK-2) proximal tubular cells. The overexpression of RTN1A, but not control vector or RTN1C resulted in decreased mitochondrial DNA (mtDNA) copy number (Supp. Figure 3B-C). RTN1A also increased the production of mitochondrial reactive oxygen species (ROS), as measured by mitochondrial superoxide indicator MitoSOX (Supp. Figure 3C). Since we did not detect any remarkable differences between RTN1C and control cells, we used either control or RTN1C overexpressing HK-2 cells interchangeably throughout the in vitro analysis. In the second set of experiments, we examined the effects of ER stress induction in HK-2 cells with or without RTN1A overexpression. Treatment of cells with tunicamycin, a known pharmacological inducer of ER stress, increased the expression of key ER stress sensor, glucose-regulated protein-78 (GRP78), which was further enhanced by RTN1A overexpression (Supp. Figure 5A-B). Interestingly, tunicamycin reduced mtDNA copy number and ATP synthesis (Supp. Figure 5C-D), indicative of reduced mitochondrial function. It also increased mitochondrial ROS (Supp. Figure 5E), while decreasing mitochondrial membrane potential, as detected by a reduction in binding of fluorescent tetramethylrhodamine, methyl ester (TMRM) dye that accumulates in negatively charged polarized mitochondrial membrane (Supp. Figure 5F). In the third set of experiments, we conversely examined the effect of RTN1A knockdown in the tunicamycin-induced ER stress and mitochondrial dysfunction. HK-2 cells were stably transduced with lentivirus expressing either scrambled shRNA (shScr) or shRNA against RTN1A (shRTN1A), as we did previously 19, 22 Cells expressing shRTN1A showed an effective reduction in RTN1A mRNA expression with or without tunicamycin treatment (Supp. Figure 6A). shRTN1A cells also showed reduced expression of GRP78 mRNA and protein levels following the tunicamycin treatment (Supp. Figure 6B-C). Importantly, RTN1A knockdown significantly restored the mitochondrial function in tunicamycin-treated cells (Supp. Figure 6D-G). In the fourth set of experiments, we examined the effects of high glucose-induced ER stress and mitochondrial dysfunction to mimic the diabetic settings in HK-2 cells with or without RTN1A overexpression. Control or RTN1A-overexpressing HK-2 cells were incubated with media with normal glucose (NG), high glucose (HG), or high mannitol (HM) control. High glucose conditions, but not high mannitol, further enhanced RTN1A expression in RTN1A overexpressing HK-2 cells (Figure 6A), which was associated with increased ER stress (Figure 6B) and decreased mitochondrial function (Figure 6C-D). Lastly, we confirmed in primary TECs that overexpression of RTN1A also exacerbated the tunicamycin-induced mitochondrial dysfunction as shown by decreased mitochondrial DNA (mtDNA) copy number (Supp. Figure 7). Taken together, these in vitro experiments indicate that RTN1A enhances ER stress under injury conditions and that this is associated with worsened mitochondrial function.
Figure 6: RTN1A enhances high glucose-induced ER stress and mitochondrial dysfunction in HK2 cells in high glucose conditions.

Control or RTN1A-overexpressing HK2 cells were incubated with media containing normal glucose (NG, 5mM), high mannitol (HM, 25mM+5mM glucose), or high glucose (HG, 30mM) for 24 hours. (A-B) Western blot analysis of RTN1A and densitometric analysis of RTN1A expression normalized to β-actin, shown as fold change to NG control. N.S., not significant. (C) Real-time PCR analysis of GPR78 expression in cells. (D) Average fold change in ATP levels. (E) Average fold change in MitoSOX signals. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 between indicated groups by two-way ANOVA with Tukey's post hoc analysis.
To validate these findings in vivo, we examined the expression of ER stress markers and mitochondrial proteins in kidney cortices of control and Pax8-RTN1A mice. Consistent with elevated ER stress in diabetic kidneys, both RTN1A and CHOP expression was increased in kidneys of diabetic mice compared with nondiabetic mice, but their expression was even further enhanced in diabetic Pax8-RTN1A mouse kidneys (Figure 7A, Supp. Figure 8). These changes were associated with reduced mitochondrial proteins TFAM and COX IV, but with enhanced expression of BAX (Figure 7B, Supp. Figure 8). Moreover, assessment of cytochrome C in the mitochondrial and cytoplasmic fractions showed reduced mitochondrial cytochrome C, but significantly increased cytoplasmic cytochrome C in the diabetic Pax8-RTN1A kidneys in comparison to all other groups (Figure 7C, Supp. Figure 8), suggesting enhanced cytochrome C release in the kidney cells of diabetic Pax8-RTN1A mice. In line with these observations, immunostaining showed reduced COX IV expression and increased mitochondrial fragmentation in the tubular cells of diabetic Pax8-RTN1A mouse kidneys (Figure 7D-F). These data confirm that the induction of RTN1A in TECs in vivo results in heightened ER stress and mitochondrial dysfunction in the diabetic setting, consistent with the role of RTN1A in ER-mitochondria cross-talk in DKD.
Figure 7: TEC-specific RTN1A overexpression exacerbates mitochondrial dysfunction in kidney cells in STZ-induced diabetic mice.

(A) Western blot analysis of RTN1A and CHOP in lysates of kidney cortices of control and diabetic mice. (B) Western blot analysis of BAX, TFAM, and COX IV in kidneys of control and diabetic mice. (C) Western blot analysis of cytochrome C in the mitochondrial (mito) and cytoplasmic (cyto) fractions of lysates of kidney cortices. Mitochondrial COX IV protein was used to normalize mitochondrial protein loading, and β-actin was used to normalize cytoplasmic protein loading. (D) Representative images of COX IV immunostaining in kidneys of mice. Some non-specific nuclear staining of COX IV was also noted in all samples. Scale bar, 30μm. (E) Representative images of transmission electron microscopy of mitochondria in TECs of mice. Scale bar, 2μm. (F) The quantification of the mitochondrial morphologic changes (n=3 mice per group, ***p<0.001 and ****p<0.0001 between indicated groups by two-way ANOVA with Tukey’s post hoc analysis.).
RTN1A interacts with mitochondrial Hexokinase 1 to promote tubular injury
To further delineate the molecular mechanisms of ER stress-mitochondrial cross-talk via RTN1A, we explored the potential function of RTN1A’s interaction with outer mitochondrial membrane (OMM) proteins that were identified in Table 1. Among the top RTN1A-interacting proteins was Hexokinase-1 (HK1), an enzyme that catalyzes the irreversible first step of the glycolytic pathway by phosphorylating glucose to glucose-6-phosphate. Among the mammalian hexokinases, only HK1 is ubiquitously expressed, while HK2 is expressed predominantly in skeletal and cardiac muscle and relatively low expression of HK3 in most tissues 36. Indeed, recently published mouse kidney single-cell transcriptomes 37 indicate that Hexokinase-1 is the predominant HK expressed in the TECs (Supp. Figure 9).
We confirmed the interaction between RTN1A and HK1 by immunoprecipitation in HK-2 proximal tubular cells (Figure 8A). Interestingly, HK1 protein levels were reduced in the kidneys of diabetic Pax8-RTN1A mice (Figure 8B). Importantly, immunostaining of human DKD or control kidney section also showed reduced tubular HK1 expression (Figure 8C). We confirmed in cultured HK-2 cells that RTN1A overexpression led to the reduced HK1 protein expression and that this effect was abrogated by proteasomal degradation inhibitor, PYR41, treatment (Figure 8D), suggesting that the increased RTN1A expression contributes to reduced HK1 protein by proteasomal degradation in TECs.
Figure 8: RTN1A interacts with HK1, and increased RTN1A is associated with reduced HK1 expression in DKD.

(A) Immunoprecipitation (IP) using FLAG-tagged RTN1A overexpressing HK2 cell lysates and western blot analysis using anti-FLAG and anti-HK1 antibodies. Input lysates are shown on the left. (B) Western blot analysis of RTN1A and HK1 expression in kidney cortices of control and diabetic mice. Densitometric analysis is shown on the right (n=3 mice per group). (C) Representative images of HK1 immunostaining in control or DKD biopsy sample. Scale bar, 50μm. (D) Western blot analysis of RTN1A and HK1 expression in HK2 cells with or without PYR41 treatment (25μM for 24 hours).
In addition to its enzymatic function in glycolysis, hexokinases have an important role in cellular homeostasis: mitochondria-associated HK protects against apoptosis caused by members of the BCL2 protein family 38. This is thought to be regulated through HK’s interaction with voltage-dependent anion channel (VDAC), also known as mitochondrial porin, an abundant OMM protein required for the transport of metabolites into and out of the mitochondria 39, 40. HK’s interaction with VDAC competes with VDAC’s association with BCL2 family proteins, thereby suppressing apoptosis 38, 41. Moreover, HK1 dissociated from VDAC triggers the NLRP3 inflammasome assembly and release of interleukin-1β (IL-1β) and IL-18 42, 43. Among the VDAC subtypes (VDAC1-3), VDAC1 is widely expressed in most cell types 39 Interestingly, VDAC1 and VDAC2 were also among the top proteins that interacted with RTN1A in kidney cells (Table 1). Therefore, we posited that RTN1A may disrupt the HK1-VDAC interaction to cell death and inflammation in kidney injury settings.
We first tested the effect of RTN1 overexpression in VDAC1-HK1 interaction by immunoprecipitation in proximal tubular HK-2 cells. As shown in Figure 9A, the overexpression of RTN1A, but not RTN1C, led to the reduced binding between HK1 and VDAC1. We next similarly tested the interaction of HK1 and VDAC1 using purified recombinant proteins. As shown in Figure 9B, increasing the amount of recombinant RTN1A protein led to a proportionate reduction in HK1 that immunoprecipitated with recombinant VDAC1 protein, indicating that RTN1A competes for the interaction between HK1 and VDAC1 and that RTN1A binds to HK1 with higher affinity than with VDAC1. However, increasing amounts of RTN1C did not affect HK1-VDAC1 interaction (Suppl. Figure 10). Moreover, the overexpression of RTN1A, but not that of RTN1C, was associated with increased Caspase 1 expression and increased the release of IL-1β (Figure 9C-D). However, induction of Caspase 1 and IL-1β release by RTN1A was abrogated with overexpression of HK1 (Figure 9E-G), further supporting the observation that increased RTN1A disrupts the availability of HK1 for interaction with VDAC1. Together, our data suggest that RTN1A activates the inflammasome through competitive binding of HK1 with VDAC1, a new mechanism of tubular injury and pyroptosis in DKD (Suppl. Figure 11).
Figure 9: RTN1A competes for HK1 and VDAC1 interaction to activate the inflammasome pathway.

(A) Top: Lysates of HK2 cells overexpressing RTN1A or RTN1C were immunoprecipitated (IP) with anti-HK1 antibody or control IgG and immunoblotted with VDAC1 antibody. Bottom: Input lysates were immunoblotted with VDAC1, RTN1A, and RTN1C antibodies. (B) Immunoprecipitation was performed using purified recombinant proteins (DDK-tagged HK1, His-tagged VDAC1, and Myc-tagged RTN1A). Interaction of HK1 and VDAC1 was assessed with increasing amounts of RTN1A (B) or RTN1C (C) using anti-His antibody and immunoblotted with DDK and Myc antibodies. (C-D) Western blot and densitometric analysis of Caspase-1 in HK2 cells with RTN1A or RTN1C overexpression. (D-G) Western blot and densitometric analysis of Caspase-1 and IL-1β HK2 cells overexpressing RTN1C or RTN1A with or without HK1 overexpression. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 between indicated groups by one-way ANOVA with Tukey's post hoc analysis.
DISCUSSION
Although it is well-recognized that tubular injury and tubulointerstitial fibrosis are associated with late DKD stages 2, whether and how tubular injury occurs in early DKD remains unclear. Animal models of DKD provide limited information on the progressive DKD as observed in human DKD since they typically do not display significant tubular injury or tubulointerstitial fibrosis. The overexpression of RTN1A in TECs results in a marked decline in kidney function accompanied by tubulointerstitial fibrosis that better mimics the progressive human DKD. Moreover, as our previous study showed heightened tubular injury by RTN1A in a non-diabetic context 19, it is likely that RTN1A promotes DKD and CKD in general through the shared mechanisms of tubular injury.
In recent years, the dynamic organelle cross-talk via direct interactions of membrane contact sites has emerged as important regulators of cellular homeostasis 32, 44, 45. In particular, the alteration in contacts between ER and mitochondria via EMCs regulates a variety of intracellular events that control mitochondrial function and regulate autophagy and apoptosis. A recent report demonstrated that the disruption of EMCs aggravates kidney cell injury and DKD in diabetic mice 31. However, how EMCs are regulated in DKD or the role of RTN1A in EMC regulation have not been extensively examined. Our present study demonstrates that RTN1A contributes to the alteration of EMCs in the diabetic kidneys, which were associated with increased ER stress-induced mitochondrial dysfunction. Although the distance of EMCs in the kidney tubular cells has not been previously reported, the average EMCs in neuronal cells were reported to be between 10 to 30nm 29, similar to our findings of 5-20nm in length. However, the distance of EMCs in liver tissue was reported to be between 20-100nm 46, suggesting that the variations may exist between specific cell types and/or among the measurements. Previous studies have also indicated that PERK is found within EMCs and regulates apoptosis through the mitochondrial ROS release 47 Since PERK interacts with RTN1A19, it is likely that RTN1A and PERK interaction is also important for the function of EMCs, which will be explored in future studies. The frequency and distance of EMCs in tubular cells in normal and injured kidneys, and its contribution to mitochondrial-ER crosstalk in kidney disease pathogenesis also warrants in-depth examinations in future studies.
Our study also demonstrates that RTN1A interacts with several mitochondrial OMM proteins, including HK1, and that RTN1A-HK1 interaction disrupts HK1-VDAC1 interaction, thereby releasing VDAC1 to induce inflammasome- and BCL2-mediated pathways 40-42 and leading to HK1 proteasome-mediated degradation. In line with these observations, increased HK activity is shown to protect proximal tubular cells following acute kidney injury by promoting glucose metabolism and/or inhibiting apoptosis through regulation of Bax-mediated mitochondrial membrane injury 48, 49 Thus, our findings underscore a new mechanism of ER-mitochondrial crosstalk in tubular injury in kidney disease (as summarized in Suppl. Figure 11) and further highlight RTN1A as a potential new target of tubular injury and fibrosis in CKD.
Supplementary Material
Translational Statement.
Clinical evidence indicates that in a subset of diabetic patients, diabetic kidney disease (DKD) could progress to kidney failure in the absence of significant glomerular injury. However, the pathological mechanisms of tubular injury in DKD progression are not entirely clear. Previously, we demonstrated that Reticulon-1A (RTN1A), an endoplasmic reticulum (ER) membrane protein, is a risk factor in DKD progression. The present study now demonstrates that RTN1A facilitates ER-to-mitochondrial cross-talk in tubular cells, resulting in a concomitant increase in ER stress and mitochondrial injury in DKD, thus uncovering a mechanism of tubular injury in DKD progression.
Funding:
JCH is supported by NIH/NIDDK R01DK109683, R01DK122980, R01DK129467, P01DK56492, and VA Merit Award I01BX000345; KL is supported by NIH/NIDDK R01DK117913-01 and R01DK129467.
Footnotes
SUPPLEMENTARY MATERIAL: Suppl. Figures 1-10; Supp. Table 1; Supp. Methods and References; Suppl. file of uncropped western blots. Supplementary information is available on Kidney International's website.
DISCLOSURES: The authors have declared that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Collins AJ, Foley RN, Herzog C, et al. US Renal Data System 2010 Annual Data Report. Am J Kidney Dis 57: A8, e1–526. [DOI] [PubMed] [Google Scholar]
- 2.Mauer M, Zinman B, Gardiner R, et al. ACE-I and ARBs in early diabetic nephropathy. Journal of the renin-angiotensin-aldosterone system : JRAAS 2002; 3: 262–269. [DOI] [PubMed] [Google Scholar]
- 3.Bonventre JV. Can we target tubular damage to prevent renal function decline in diabetes? Semin Nephrol 2012; 32: 452–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fioretto P, Mauer M. Histopathology of diabetic nephropathy. Semin Nephrol 2007; 27: 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garofolo M, Russo E, Miccoli R, et al. Albuminuric and non-albuminuric chronic kidney disease in type 1 diabetes: Association with major vascular outcomes risk and all-cause mortality. J Diabetes Complications 2018; 32: 550–557. [DOI] [PubMed] [Google Scholar]
- 6.Lan HY, Mu W, Tomita N, et al. Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J Am Soc Nephrol 2003; 14: 1535–1548. [DOI] [PubMed] [Google Scholar]
- 7.Verzola D, Gandolfo MT, Salvatore F, et al. Testosterone promotes apoptotic damage in human renal tubular cells. Kidney Int 2004; 65: 1252–1261. [DOI] [PubMed] [Google Scholar]
- 8.Saito A, Takeda T, Sato K, et al. Significance of proximal tubular metabolism of advanced glycation end products in kidney diseases. Ann N Y Acad Sci 2005; 1043: 637–643. [DOI] [PubMed] [Google Scholar]
- 9.Cunard R, Sharma K. The endoplasmic reticulum stress response and diabetic kidney disease. Am J Physiol Renal Physiol 2011; 300: F1054–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galvan DL, Green NH, Danesh FR. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int 2017; 92: 1051–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma K. Mitochondrial Dysfunction in the Diabetic Kidney. Advances in experimental medicine and biology 2017; 982: 553–562. [DOI] [PubMed] [Google Scholar]
- 12.Forbes JM, Thorburn DR. Mitochondrial dysfunction in diabetic kidney disease. Nat Rev Nephrol 2018; 14: 291–312. [DOI] [PubMed] [Google Scholar]
- 13.Lindenmeyer MT, Rastaldi MP, Ikehata M, et al. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J Am Soc Nephrol 2008; 19: 2225–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu G, Sun Y, Li Z, et al. Apoptosis induced by endoplasmic reticulum stress involved in diabetic kidney disease. Biochem Biophys Res Commun 2008; 370: 651–656. [DOI] [PubMed] [Google Scholar]
- 15.Wu X, He Y, Jing Y, et al. Albumin overload induces apoptosis in renal tubular epithelial cells through a CHOP-dependent pathway. OMICS 2010; 14: 61–73. [DOI] [PubMed] [Google Scholar]
- 16.Luo ZF, Feng B, Mu J, et al. Effects of 4-phenylbutyric acid on the process and development of diabetic nephropathy induced in rats by streptozotocin: regulation of endoplasmic reticulum stress-oxidative activation. Toxicol Appl Pharmacol 2010; 246: 49–57. [DOI] [PubMed] [Google Scholar]
- 17.Cybulsky AV. Endoplasmic reticulum stress in proteinuric kidney disease. Kidney Int 2010; 77: 187–193. [DOI] [PubMed] [Google Scholar]
- 18.Lee EK, Jeong JU, Chang JW, et al. Activation of AMP-activated protein kinase inhibits albumin-induced endoplasmic reticulum stress and apoptosis through inhibition of reactive oxygen species. Nephron Exp Nephrol 2012; 121: e38–48. [DOI] [PubMed] [Google Scholar]
- 19.Fan Y, Xiao W, Li Z, et al. RTN1 mediates progression of kidney disease by inducing ER stress. Nat Commun 2015; 6: 7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonomo JA, Palmer ND, He JC, et al. Association Analysis of the Reticulon 1 Gene in End-Stage Kidney Disease. American journal of nephrology 2015; 42: 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan Y, Zhang J, Xiao W, et al. Rtn1a-Mediated Endoplasmic Reticulum Stress in Podocyte Injury and Diabetic Nephropathy. Sci Rep 2017; 7: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao W, Fan Y, Wang N, et al. Knockdown of RTN1A attenuates ER stress and kidney injury in albumin overload-induced nephropathy. Am J Physiol Renal Physiol 2016; 310: F409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan Y, Xiao W, Lee K, et al. Inhibition of Reticulon-1A-Mediated Endoplasmic Reticulum Stress in Early AKI Attenuates Renal Fibrosis Development. J Am Soc Nephrol 2017; 28: 2007–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brosius FC 3rd, Alpers CE, Bottinger EP, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol 2009; 20: 2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qi Z, Fujita H, Jin J, et al. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes 2005; 54: 2628–2637. [DOI] [PubMed] [Google Scholar]
- 26.Zheng S, Noonan WT, Metreveli NS, et al. Development of late-stage diabetic nephropathy in OVE26 diabetic mice. Diabetes 2004; 53: 3248–3257. [DOI] [PubMed] [Google Scholar]
- 27.Hong Q, Zhang L, Das B, et al. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho IT, Adelmant G, Lim Y, et al. Ascorbate peroxidase proximity labeling coupled with biochemical fractionation identifies promoters of endoplasmic reticulum-mitochondrial contacts. J Biol Chem 2017; 292: 16382–16392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu L, Wang X, Tong C. Endoplasmic Reticulum-Mitochondria Contact Sites and Neurodegeneration. Front Cell Dev Biol 2020; 8: 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Volkmar N, Christianson JC. Squaring the EMC - how promoting membrane protein biogenesis impacts cellular functions and organismal homeostasis. Journal of cell science 2020; 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang M, Zhao L, Gao P, et al. DsbA-L ameliorates high glucose induced tubular damage through maintaining MAM integrity. EBioMedicine 2019; 43: 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lahiri S, Toulmay A, Prinz WA. Membrane contact sites, gateways for lipid homeostasis. Curr Opin Cell Biol 2015; 33: 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gatta AT, Levine TP. Piecing Together the Patchwork of Contact Sites. Trends Cell Biol 2017; 27: 214–229. [DOI] [PubMed] [Google Scholar]
- 34.Lynes EM, Raturi A, Shenkman M, et al. Palmitoylation is the switch that assigns calnexin to quality control or ER Ca2+ signaling. Journal of cell science 2013; 126: 3893–3903. [DOI] [PubMed] [Google Scholar]
- 35.van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochimica et biophysica acta 2014; 1843: 2253–2262. [DOI] [PubMed] [Google Scholar]
- 36.Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. The Journal of experimental biology 2003; 206: 2049–2057. [DOI] [PubMed] [Google Scholar]
- 37.Park J, Shrestha R, Qiu C, et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 2018; 360: 758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem 2002; 277: 7610–7618. [DOI] [PubMed] [Google Scholar]
- 39.Varughese JT, Buchanan SK, Pitt AS. The Role of Voltage-Dependent Anion Channel in Mitochondrial Dysfunction and Human Disease. Cells 2021; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shoshan-Barmatz V, Golan M. Mitochondrial VDAC1: function in cell life and death and a target for cancer therapy. Current medicinal chemistry 2012; 19: 714–735. [DOI] [PubMed] [Google Scholar]
- 41.Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. Journal of bioenergetics and biomembranes 2008; 40: 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolf AJ, Reyes CN, Liang W, et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 2016; 166: 624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moon JS, Hisata S, Park MA, et al. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep 2015; 12: 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Prinz WA. Bridging the gap: membrane contact sites in signaling, metabolism, and organelle dynamics. J Cell Biol 2014; 205: 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murley A, Nunnari J. The Emerging Network of Mitochondria-Organelle Contacts. Mol Cell 2016; 61: 648–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.D'Eletto M, Rossin F, Occhigrossi L, et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep 2018; 25: 3573–3581 e3574. [DOI] [PubMed] [Google Scholar]
- 47.Verfaillie T, Rubio N, Garg AD, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 2012; 19: 1880–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith JA, Stallons LJ, Schnellmann RG. Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin-induced acute kidney injury. Am J Physiol Renal Physiol 2014; 307: F435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gall JM, Wong V, Pimental DR, et al. Hexokinase regulates Bax-mediated mitochondrial membrane injury following ischemic stress. Kidney Int 2011; 79: 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.