

Abstract
A premature truncation of MYBPHL in humans and a loss of Mybphl in mice is associated with dilated cardiomyopathy, atrial and ventricular arrhythmias, and atrial enlargement. MYBPHL encodes myosin binding protein H-like (MyBP-HL). Prior work in mice indirectly identified Mybphl expression in the atria and in small puncta throughout the ventricle. Because of its genetic association with human and mouse cardiac conduction system disease, we evaluated the anatomical localization of MyBP-HL and the consequences of loss of MyBP-HL on conduction system function. Immunofluorescence microscopy of normal adult mouse ventricles identified MyBP-HL-positive ventricular cardiomyocytes that co-localized with the ventricular conduction system marker contactin-2 near the atrioventricular node and in a subset of Purkinje fibers. Mybphl heterozygous ventricles had a marked reduction of MyBP-HL-positive cells compared to controls. Lightsheet microscopy of normal perinatal day 5 mouse hearts showed enrichment of MyBP-HL-positive cells within and immediately adjacent to the contactin-2-positive ventricular conduction system, but this association was not apparent in Mybphl heterozygous hearts. Surface telemetry of Mybphl-null mice revealed atrioventricular block and atrial bigeminy, while intracardiac pacing revealed a shorter atrial relative refractory period and atrial tachycardia. Calcium transient analysis of isolated Mybphl-null atrial cardiomyocytes demonstrated an increased heterogeneity of calcium release and faster rates of calcium release compared to wild type controls. Super-resolution microscopy of Mybphl heterozygous and homozygous null atrial cardiomyocytes showed ryanodine receptor disorganization compared to wild type controls. Abnormal calcium release, shorter atrial refractory period, and atrial dilation seen in Mybphl null, but not wild type control hearts, agree with the observed atrial arrhythmias, bigeminy, and atrial tachycardia, whereas the proximity of MyBP-HL-positive cells with the ventricular conduction system provides insight into how a predominantly atrial expressed gene contributes to ventricular arrhythmias and ventricular dysfunction.
Keywords: Myosin binding protein, MyBP-HL, MYBPHL, Ventricular conduction system, atrial cardiomyocyte, Cardiomyopathy
1. Introduction
Dilated cardiomyopathy (DCM) is a prevalent genetic heart disease characterized by ventricular dilation and systolic dysfunction and is caused by mutations in genes that are involved in a variety of cardiomyocyte functions [5]. While ventricular function has been the major focus of research into DCM, there has been a growing appreciation of the importance of atrial and ventricular conduction system dysfunction as sequelae and modifiers of cardiomyopathies [6, 7]. In DCM, the atria frequently dilate, which is linked with increased occurrence of atrial tachycardia and fibrillation [8–10]. Atrial fibrillation has also been shown to develop concomitantly with remodeling in hypertrophic cardiomyopathy [11]. Progressive atrioventricular block and bundle branch block are comorbidities in dilated cardiomyopathy and are associated with increased mortality [12–14].
The cardiac conduction system is comprised of distinct populations of specialized cardiomyocytes that control the rate and propagation of cardiac action potentials throughout the heart [15, 16]. Cardiomyocytes with distinct gene expression patterns and morphology comprise the sinoatrial node and atrial conduction tract, atrioventricular node, His-bundle, Purkinje fibers, and junctional ventricular zone cells [17]. Single cell sequencing studies have demonstrated that these zones are comprised of several additional subsets of cells, each with their own specific transcriptomic landscape that regulates their function [18]. Most genes that have been identified to cause cardiomyopathy are expressed in throughout the chambers of the heart [5]. Genes that are highly expressed in rare subpopulations of cardiomyocytes have not been well studied in the context of genetic heart disease.
We previously described a premature truncation mutation, MYBPHL R255Stop, in a family with DCM, atrial and ventricular arrhythmias, and atrioventricular conduction dysfunction [1]. MYBPHL encodes the myofilament-associated protein myosin binding protein H-like (MyBP-HL) and is highly expressed in the atria. Using a LacZ reporter, we also identified Mybphl expression in a small subset of ventricular cardiomyocytes, although the precise anatomical distribution was not clear. A Mybphl null allele was characterized in mice and provided evidence that both heterozygous and homozygous disruption of Mybphl produced atrial and ventricular dilation, systolic dysfunction, and premature ventricular contractions [1].
Additional evidence has emerged implicating MYBPHL with conduction system disorder. A genome-wide association meta-analysis identified MYBPHL as a locus associated with prolongation of P-R interval [19]. Single cell RNA-Seq experiments have also identified Mybphl expression within subsets of the mouse conduction system, including the Purkinje cells and Purkinje-to-ventricular transition zone [18]. However, it is unclear how loss of a predominantly atrial myofilament protein could cause ventricular dysfunction and arrhythmia. In the present work, we provide evidence for MyBP-HL expressing cells within and adjacent to components of the ventricular conduction system and provide a rationale by which loss of Mybphl can elicit ventricular rhythm disturbances.
2. Methods
2.1. Animal models
The Mybphl null mouse was obtained from the Knock-Out Mouse Project and the European Mutant Mouse Archive [1, 20]. This mouse carries a null allele of Mybphl where exons 2 – 6 are deleted and replaced with a LacZ reporter. Contactin-2-EGFP bacterial artificial chromosome transgenic mice express enhanced green fluorescent protein in the ventricular conduction system under the control on the contactin-2 promoter [21]. For all terminal time points and tissue collection, animals were humanely sacrificed by cervical dislocation while under inhaled isoflurane anesthesia. All procedures involving mouse models performed in this study were approved by the Institutional Care and Use Committee at Northwestern University and conformed to the ethical treatment of animals standards set out in the Guide for the Care and Use of Laboratory Animals issued by the NIH Office of Laboratory Animal Welfare. Northwestern University’s animal facility maintains the standards required for AAALAC accreditation.
2.2. Cardiomyocyte isolation
Adult mouse cardiomyocytes were isolated as previously reported [22]. Mice were anesthetized with 5% inhaled isoflurane in 100% oxygen and administered 50 U heparin with an intraperitoneal injection 20 minutes prior to sacrifice. The animals were anesthetized with 5% inhaled isoflurane. The thoracic cavity was opened, and the heart and lungs were cut out of the chest with care to preserve the aortic arch. The heart and lungs were immobilized in a 35 mm dish containing Tyrode’s solution with no calcium (143 mM NaCl, 2.5 mM KCl, 25 mM NaHCO3, 2 mM MgCl2, 11 mM glucose, pH 7.4), where the aorta was dissected, cleaned, and bisected at the level of the brachiocephalic artery. A 24-gauge gavage needle (Cadence Science #7900) was fitted to a 1 mL syringe filled with ice cold Tyrode’s solution with no calcium, the aorta was stretched over the ball tip of the catheter and immobilized with a 6–0 silk suture. Care was taken to ensure no air was introduced into the aorta. Retrograde perfusion was initiated via syringe with zero calcium Tyrode’s solution at a rate of 1 mL/minute, after which the gavage catheter and the heart were transferred to a Langendorff system with a constant pressure of 80 mmHg. The Langendorff apparatus contained Tyrode’s solution with no calcium and a modified Krebs-Henseleit buffer (112 mM NaCl, 4.7 mM KCl, 0.6 mM KH2PO4, 40 μM CaCl2, 0.6 mM Na2HPO4, 1.2 mM MgSO4, 30 μM phenol red, 21.4 mM NaHCO3, 10 mM HEPES, and 30 mM taurine, pH 7.4) with 0.15% collagenase type II (Worthington), 0.1% BDM, 0.1% glucose, 1 × insulin-transferrin-selenium (Gibco), and 100 U/mL penicillin/streptomycin added day-of experiment. The buffers were maintained at 37 °C with water-jacketed glassware (Radnoti) and a recirculating water bath. The hearts were perfused with a digestion solution for seven minutes followed by removal of the atria, mincing with fine spring scissors, and trituration using a plastic transfer pipette every five minutes for 40 minutes. Atrial tissue was allowed to settle between the triturations and the supernatant was filtered through a 100 μm cell strainer into a stop buffer (digestion buffer with no collagenase, 40 μM CaCl2, and 1% bovine serum albumin). After complete digestion of the tissue, the cells were centrifuged for 1 minute at 100 × G. The pellet was resuspended in a stop buffer, centrifuged again for 1 minute at 100 × G, and resuspended in a final buffer (stop buffer with no BSA). Ventricular cardiomyocytes were isolated in the same manner except the hearts were digested for 5 minutes and the serial trituration was constant. Glass coverslips (10 mm diameter) were coated with 20 μg/mL laminin (23017–015 Gibco) in phosphate buffered saline (PBS) for one hour. Cardiomyocytes were added in final buffer and allowed to adhere to the coverslips for one hour at 37 °C.
2.3. Immunofluorescence staining of sectioned tissue and isolated cardiomyocytes
Frozen mouse hearts were sectioned at 5 μm thickness and adhered to a Superfrost+ glass slide. Sections were fixed in an ice cold 4% paraformaldehyde solution in PBS for 10 minutes and then rinsed and stored in cold PBS for up to an hour. Coverslips with adhered isolated cardiomyocytes were put into the wells of a 24-well plate. The cells were washed with 500 μL of PBS and incubated in 4% paraformaldehyde for 10 minutes at 4 °C. Tissue or cells were then washed with PBS and then permeabilized in PBS with 0.25% Triton X-100 for 25 minutes at 4 °C. The samples were blocked with a solution of 20% FBS and 0.1% Triton X-100 in PBS for 30 minutes at 4 °C and then incubated in primary antibody in 2% FBS and 0.1% Triton X-100 overnight at 4 °C. Primary antibodies were used against MyBP-HL (Pocono, custom), epitope and antibody validation in Figure 1A, cMyBP-C (Santa Cruz E7), contactin-2 (R&D Systems AF4439) [21], ryanodine receptor 2 (AbCam GR3250452–2). The samples were washed three times in cold PBS containing 0.1% Triton X-100 and incubated with secondary antibodies conjugated with Alexa-488 (Invitrogen A21202) or Alexa-568 (Invitrogen A10042). Samples were again washed in PBS with 0.1% Triton X-100 three times, with the second wash containing 1 μM Hoechst solution (Invitrogen H3570) to visualize nuclei. Sectioned tissue was mounted using VectaShield without DAPI and coverslips were adhered with clear nail polish. Coverslips were mounted on glass slides using ProLong Gold anti-fade reagent (Thermo P10144) and allowed to solidify in a dark dry container overnight.
Figure 1. MyBP-HL is found primarily in the atria.
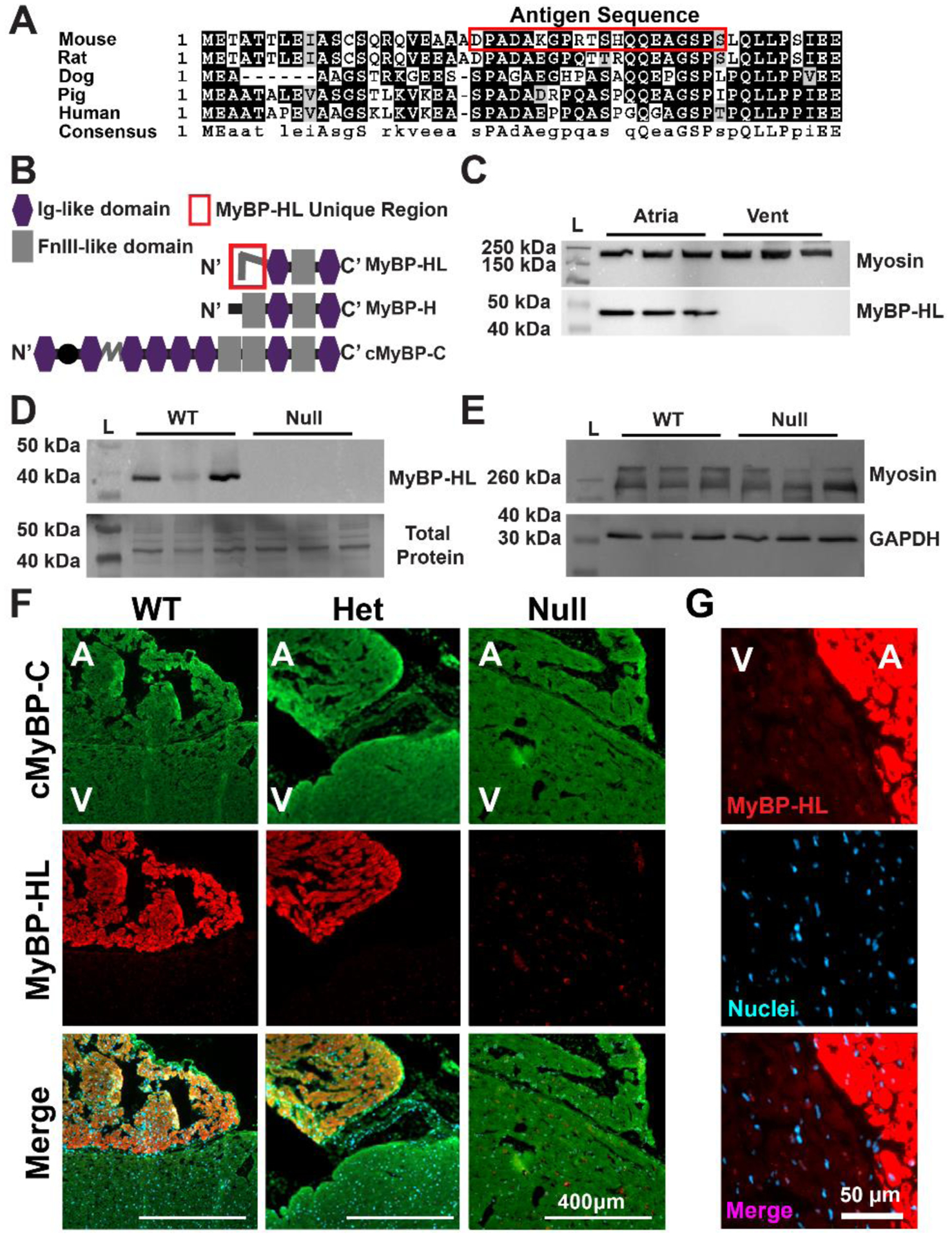
(A) Alignment of the amino terminal residues of MyBP-HL across five species shows similarities between rat and mouse, with less conservation between rodents and larger mammals. (B) Domain structure schematic of MyBP-HL and related myosin binding proteins depicting the MyBP-HL-specific amino-terminal sequence used to generate the antibody. (C) Immunoblotting using the anti-MyBP-HL antibody on protein lysate from mouse atria and ventricles shows atrial expression of MyBP-HL with total sarcomere content represented with myosin immunoblotting. N = 3 hearts, 2 male, 1 female. (D) Immunoblot using total atrial protein from wild type and homozygous Mybphl null mouse hearts showed reactivity with a 40 kDa protein that is absent in Mybphl null atria. N = 3 hearts, 1 male, 2 female per genotype. Total protein content of the membrane shown. N = 3 hearts per genotype. (E) Immunoblotting of the same protein lysates from (D) for total sarcomere content with myosin and GAPDH as a loading control shows similar sarcomere content from WT and Mybphl homozygous null atria. (F) Immunofluorescence microscopy of sectioned frozen mouse hearts immunostained for cMyBP-C found in both atria (A) and ventricle (V), with MyBP-HL localized to the atria in wild type (WT) and heterozygous Mybphl mice. MyBP-HL protein was absent from Mybphl homozygous null hearts. (G) The MyBP-HL antibody shows off-target staining on the nuclear membrane, noticeable in WT ventricle sections.
2.4. Immunofluorescence microscopy
Immunostained fixed tissue sections and isolated cardiomyocytes were imaged on a Zeiss Axio Observer with epifluorescent illumination. Tiled sections of slides were used for quantification of cell density and MyBP-HL positive cardiomyocytes were counted by hand by a blinded operator and normalized as the number of MyBP-HL positive cells per section. Isolated ventricular cardiomyocytes were plated in 35 mm diameter dishes and images were taken of the entire culture area using a 10 × objective, tiled together, and the total cell density per dish was measured using the analyze particles tool in ImageJ. An observer blinded to genotype scanned the stained cells and imaged and counted MyBP-HL expressing ventricular cardiomyocytes. MyBP-HL positive cardiomyocytes were normalized to cell density per individual isolation and plating. Cell morphology was measured by outlining the cells in ImageJ. Sectioned tissue from WT and heterozygous Mybphl null mouse hearts slices were immunostained for MyBP-HL and analyzed by an observer blinded to genotype. MyBP-HL positive ventricular cardiomyocytes were quantified and the prevalence of these cells per section was recorded. Ventricular cardiomyocytes from Cntn2-GFP bacterial artificial chromosome mice [21] were also isolated, counted, and normalized per total cardiomyocytes.
2.5. Structured illumination microscopy
Super-resolution microscopy was performed using a Nikon Structured Illumination Microscope (N-SIM) in the Nikon Imaging Center in Northwestern University’s Center for Advanced Microscopy. The N-SIM was fitted with a 100 × oil objective (NA 1.49) and 488 nm and 561 nm wavelength lasers were used for imaging. All SIM images were acquired and reconstructed using the Nikon Elements software.
2.6. Ryanodine receptor distribution analysis
Reconstructed SIM images were analyzed, and clusters counted by hand by an observer blinded to genotype. For cluster size analysis, images were processed in ImageJ by Otsu thresholding followed by watershed segmentation. The areas of each thresholded cluster was quantified. It should be noted that this is an indirect measurement of cluster size that gives a larger total area for each cluster in comparison to other recent work that has used point-spread function correction and calibration techniques to quantify individual RyR receptors and identify the number of receptors per cluster and cluster size [23, 24].
2.7. Whole-mount immunostaining
Perinatal day 5 mouse hearts were isolated and prepared for whole mount immunostaining using the iDISCO method [25]. Antibodies were first validated by immunostaining sectioned frozen mouse hearts that had undergone the chemical treatments in the iDISCO protocol. For whole mount staining, hearts were collected and rinsed of blood in cold PBS, then shaken overnight in PBS with 4% PFA at 4° C. Hearts were rinsed 3 × 30 min at RT with shaking. Hearts were dehydrated in a methanol/saline series (20%, 40%, 60%, 80%, 2 × 100%) for 1 hour at room temperature for each step, except the last step at 4° C. Samples were incubated overnight at room temperature with shaking in 66% DCM, 33% methanol. Hearts were washed twice in 100% methanol and then transferred to a chilled solution of 5% H2O2 in methanol overnight at 4° C. The hearts were rehydrated with a methanol/saline series (80%, 60%, 40%, 20%) for 1 hour each at room temperature followed by 2 × 1 hour washes in PBS with 0.2% Triton X-100. At this stage, the tubes containing the hearts and solutions were completely devoid of air to avoid sample oxidation. Tissue was permeabilized by incubation with 0.2% Triton X-100, 300 mM glycine, and 20% dimethylsulfoxide in PBS for 2 days at 37° C. The hearts were blocked in 0.2% Triton X-100, 10% dimethylsulfoxide, and 6% donkey serum in PBS for 2 days at 37° C. Primary antibodies were diluted at 1:500 in 0.2% Tween-20, 10 mg/L heparin, 5% dimethylsulfoxide, 3% donkey serum in PBS for 4 days at 37° C. Hearts were then washed 4 – 5 times with 0.2% Tween-20, 10 mg/L heparin in PBS for 30 minutes each, with the last wash overnight. Secondary antibodies were diluted in 0.2% Tween-20, 10 mg/L heparin, 3% donkey serum in PBS for 4 days at 37° C and then washed as after the primary antibody incubation. To optically clear the hearts, the samples were dehydrated in a methanol/saline series (20%, 40%, 60%, 80%, 2 × 100%) for 1 hour at room temperature for each step, or overnight for the final step. Samples were then incubated with 66% DCM/33% methanol at room temperature with shaking for 3 hours followed by 2 × 15-minute washes in 100% DCM. Finally, the tissue was rendered transparent by incubation with dibenzyl ether (Sigma 108014) without shaking at room temperature and stored this way until imaging.
2.8. Lightsheet microscopy and volumetric modeling
A LaVision II lightsheet microscope (LaVision Biotech) fitted with a 6 mm working distance Olympus MVPLAPO 2 × 0.5NA lens was used to determine localization of MyBP-HL and contactin-2 within the perinatal day 5 mouse heart. Hearts were imaged in dibenzyl ether with six light sheets using 561 or 637 nm laser lines to excite Alexa Fluor 568 or 647 secondary antibodies. Hearts were imaged at 1.6 × magnification and the numeric aperture and laser sheet settings were adjusted to provide the optimal illumination and focus across the sample, and images were taken at 5 μm steps.
Image stacks were processed in the Imaris software to create a volumetric representation of the whole heart. The MyBP-HL stained atria were rendered into a surface feature with 3.5 μm surface detail and intensity threshold set at automatic values. This volume was used to mask the MyBP-HL signal from the atria, leaving the ventricular foci. The contactin-2 stained conduction system was rendered as a volumetric surface feature with 3.5 μm surface detail and automatic intensity thresholding. MyBP-HL ventricular foci were rendered as points and were determined by their size (20 μm diameter, 40 μm elongation factor to correct for Z-stack elongation), selected for sphericity >0.083, and automatically thresholded for quality. For all surfaces and spots, regions of signal from outside the heart volume (e.g. reflections from the sample mounting posts) were removed by hand. The distance from each ventricular MyBP-HL rendered spot to the contactin-2 ventricular conduction system surface was measured.
2.9. Implantable telemetry
DSI ETA-F10 biopotential transmitters were implanted subcutaneously in anesthetized mice at 10 – 11 weeks of age and animals were allowed to recover for one week. Conscious ambulatory electrocardiogram recordings were made for one hour at 8:00 am followed by intraperitoneal injection of 1 mg/kg propranolol and continuous recording for the following three hours [26]. Animals recovered for 3 days before a subsequent injection of 20 mg/kg flecainide [27]. The electrocardiogram signals were analyzed using DSI’s Ponemah software. Heart rate variability was visualized using Poincaré plots of the R-R interval graphed against the R-R interval of the subsequent beat (R-R+1) [1].
2.10. Intracardiac pacing
Intracardiac electrophysiological recording and stimulation was performed on mice sedated with inhaled isofluorane. A 1.1-F octapolar catheter (Millar EPR-800) was inserted through the right jugular vein and into the right atria and ventricle in mice sedated with inhaled isoflurane. Animals were positioned prone on a heated table and surface electrocardiograms were measured with limb leads in the table. Protocols for burst pacing of 50 ms cycle length were used to try and elicit bouts of atrial tachycardia or atrial fibrillation [28]. If atrial tachycardia was observed in two or more burst pacing protocols the animal was considered positive. Following baseline recordings, animals were treated with intraperitoneal injection of 4 mg/kg isoproterenol and burst pacing was repeated to assess the occurrence of arrhythmia.
2.11. Confocal calcium imaging
Atrial cardiomyocytes were incubated with 14 μM of the calcium indicator dye Cal520-AM (ATT Bioquest) for 25 minutes. Cells were added to the tissue bath on the stage of an inverted Zeiss LSM 510 (25 × objective, N/A 1.2). The bath was perfused with Tyrode’s solution (143 mM NaCl, 2.5 mM KCl, 25 mM NaHCO3, 2 mM CaCL2, 2 mM MgCl2, 11 mM glucose, pH 7.4) and cells were paced at 1000, 500, 400, 300, and 200 ms cycle lengths [29]. Calcium transients were analyzed using custom software in MatLab 2015b, and WT and null mice were compared at each pacing frequency. Heterogeneity was assessed by segmenting the line-scan calcium transient into 2-micron segments that run the length of the cell, and then is measured as the standard deviation between the segments. Three-dimensional fluorescent intensity graphs were made using the Plotly R Open-Source Graphing Library.
2.12. Data handling and statistics
All data was collected and analyzed by observers blinded to genotype. Data was initially parsed in Microsoft Excel and statistical tests were performed using GraphPad Prism 8. Data were assessed for normalcy using a Bartlett’s test and then evaluated for significance using either a two-tailed Student’s t-test or a repeat measure one-way ANOVA with a Bonferroni’s multiple comparisons post-hoc test. Figure legends provide detail on the group numbers and tests used. Significance was determined a priori as p < 0.05.
3. Results
3.1. MyBP-HL is expressed in discrete foci throughout the ventricle
A polyclonal MyBP-HL antibody was generated that recognizes the amino terminal domain of mouse MyBP-HL (Fig. 1A, B). These 50 N’-terminal amino acids in MyBP-HL are not similar to any region of MyBP-H or MyBP-C. In WT mice, MyBP-HL protein was detected by immunoblot in atrial protein homogenates, but not in ventricle samples (Fig. 1C), consistent with our prior data using a commercial antibody [1]. This antibody detected MyBP-HL protein in wild type (WT) atrial samples but did not detect MyBP-HL protein from Mybphl homozygous null atria (Fig. 1D). Total protein staining of this same membrane illustrates loading of Mybphl null tissue. Immunoblotting for myosin to show total sarcomere content and GAPDH as a control shows similar levels of sarcomere content in both genotypes (Fig. 1E). The atrial expression of Mybphl was confirmed with immunofluorescence microscopy of sectioned hearts from adult WT, heterozygous, and homozygous Mybphl null mice (Fig. 1F). Faint staining can be observed in the Mybphl null mouse using the MyBP-HL antibody. Closer imaging shows a perinuclear staining in ventricular tissue (Fig. 1G). The cause of this off-target binding is unclear, as the MyBP-HL epitope region does not have high homology with other proteins.
Sections of adult mouse hearts were co-stained with antibodies against contactin-2 to mark the ventricular conduction system, including the AV node, His bundles, and Purkinje fibers. Tissue at the base of the ventricular septum in the vicinity of the atrioventricular node showed positive contactin-2 staining that co-stained for MyBP-HL staining (Fig. 2A-i, ii). Other ventricular cardiomyocytes showed positive staining for MyBP-HL, including within contactin-2 positive Purkinje cells in the ventricular sub-endocardium (Fig. 2A-iii), and in cells in the ventricular epicardium that did not co-stain with the ventricular conduction system (Fig. 2A-iv).
Figure 2. MyBP-HL is found in the atrioventricular transition tissue and a subset of the ventricle.
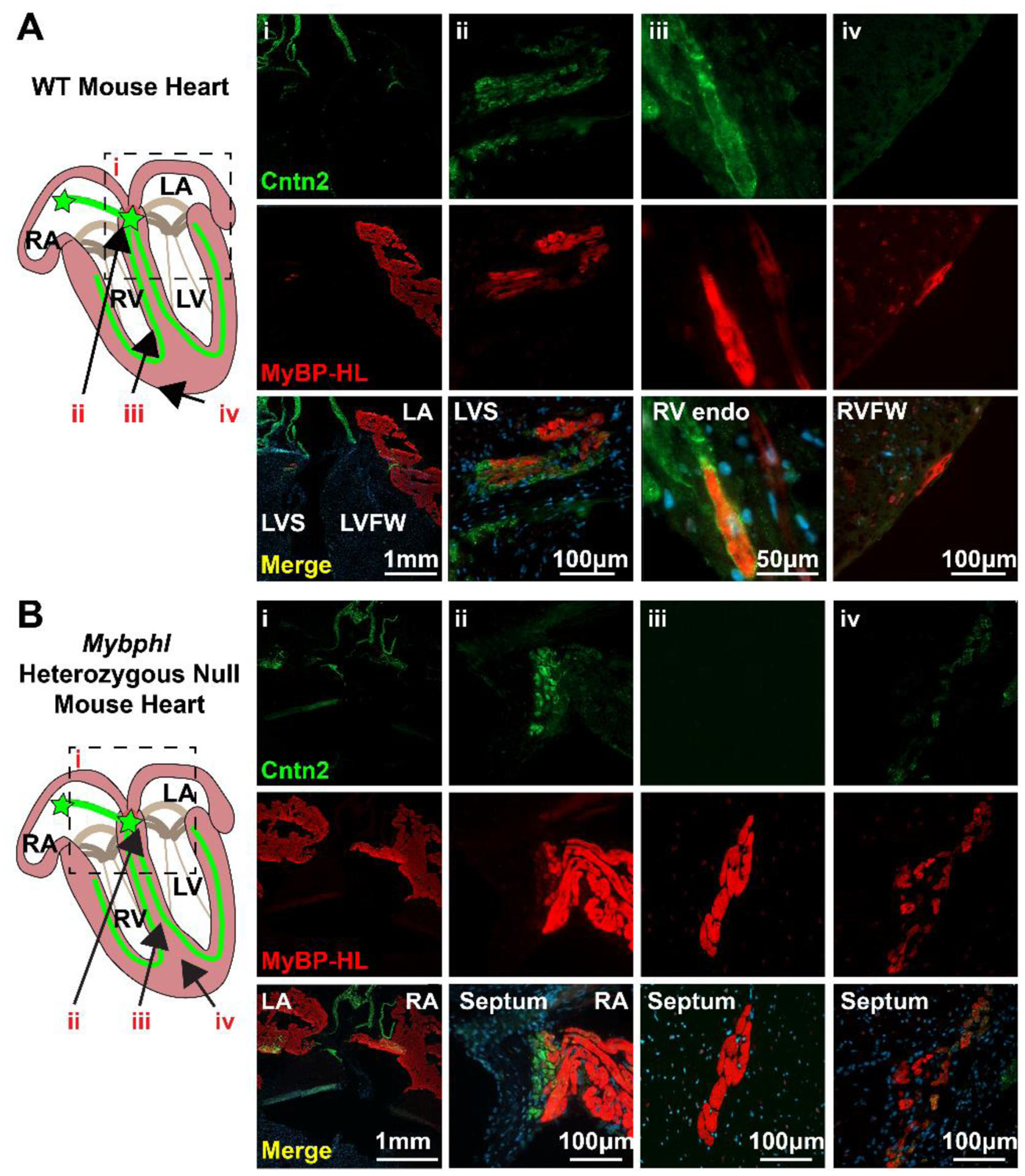
(A) Immunofluorescence microscopy of 5 μm thick frozen sections of wild type mouse hearts stained with MyBP-HL (red) and the ventricular conduction system marker contactin-2 (Cntn2). Panels show: low (i) and high (ii) magnification images of the interventricular septum at the base of the heart where atrial tissue enters the ventricle and MyBP-HL staining overlaps with contactin-2 in the ventricle. (iii) A contactin-2 stained Purkinje fiber in the RV endocardial wall shows co-staining with MyBP-HL in some ventricular conduction system cardiomyocytes. (iv) MyBP-HL-stained ventricular cardiomyocytes on the RV epicardium with no associated contactin-2 staining. (B) Immunofluorescence microscopy of 5 μm thick frozen sections of Mybphl heterozygous mouse hearts stained with MyBP-HL (red) and the ventricular conduction system marker contactin-2 (green). Panels show: low (i) and high (ii) magnification images of the interventricular septum at the base of the heart where atrial tissue enters the ventricle and MyBP-HL staining overlaps with contactin-2 in the ventricle. (iii) A large cluster of MyBP-HL positive cells in the LV septum with no associated contactin-2 staining. (iv) A multicellular cluster of MyBP-HL positive cells in the septum with overlapping contactin-2 staining. Female hearts.
Immunofluorescence microscopy with antibodies against MyBP-HL and contactin-2 on Mybphl heterozygous mouse hearts showed robust MyBP-HL staining in the atria, and MyBP-HL co-staining with contactin-2 in the base of the ventricular septum in the atria to ventricular transition tissue (Fig. 2B-i, ii). In contrast, the pattern of MyBP-HL in the ventricle showed multicellular puncta of MyBP-HL positive cells (Fig. 2B-iii, iv), which was not observed in sections from WT mice. As in the WT sections, heterozygous sections showed that there was a high degree of variability in the association between MyBP-HL positive regions in the ventricle and contactin-2 stained conduction system tissue. Note that the green signal on the top margins of Fig. 2A-i and 2B-i is autofluorescence from the collagen in the vasculature and valves at the base of the heart.
We show MyBP-HL to be predominantly found in the atria, which provides an anatomical basis for the concept that loss of Mybphl could cause atrial dysfunction. Because the phenotype in humans carrying the R255X mutation and the Mybphl null mice include ventricular arrhythmia and dysfunction [1], we wanted to further explore the identity of the Mybphl expressing ventricular cardiomyocytes. Analysis of MyBP-HL localization in sectioned tissue revealed that 67% of the MyBP-HL positive ventricular cardiomyocytes were identified in the right ventricular free wall (Fig. 3A, D). We were able to identify additional multicellular puncta of MyBP-HL positive ventricular cardiomyocytes in Mybphl heterozygous hearts (Fig. 3B). However, when quantifying the number of individual cells, we noted a significant reduction in the number of MyBP-HL positive ventricular cardiomyocytes in heterozygous hearts compared to WT (Fig. 3C), suggesting that partial loss of MyBP-HL can cause alterations in the presence and localization of ventricular MyBP-HL expressing cells.
Figure 3. MyBP-HL heterozygous hearts have reduced numbers of ventricular MyBP-HL foci.
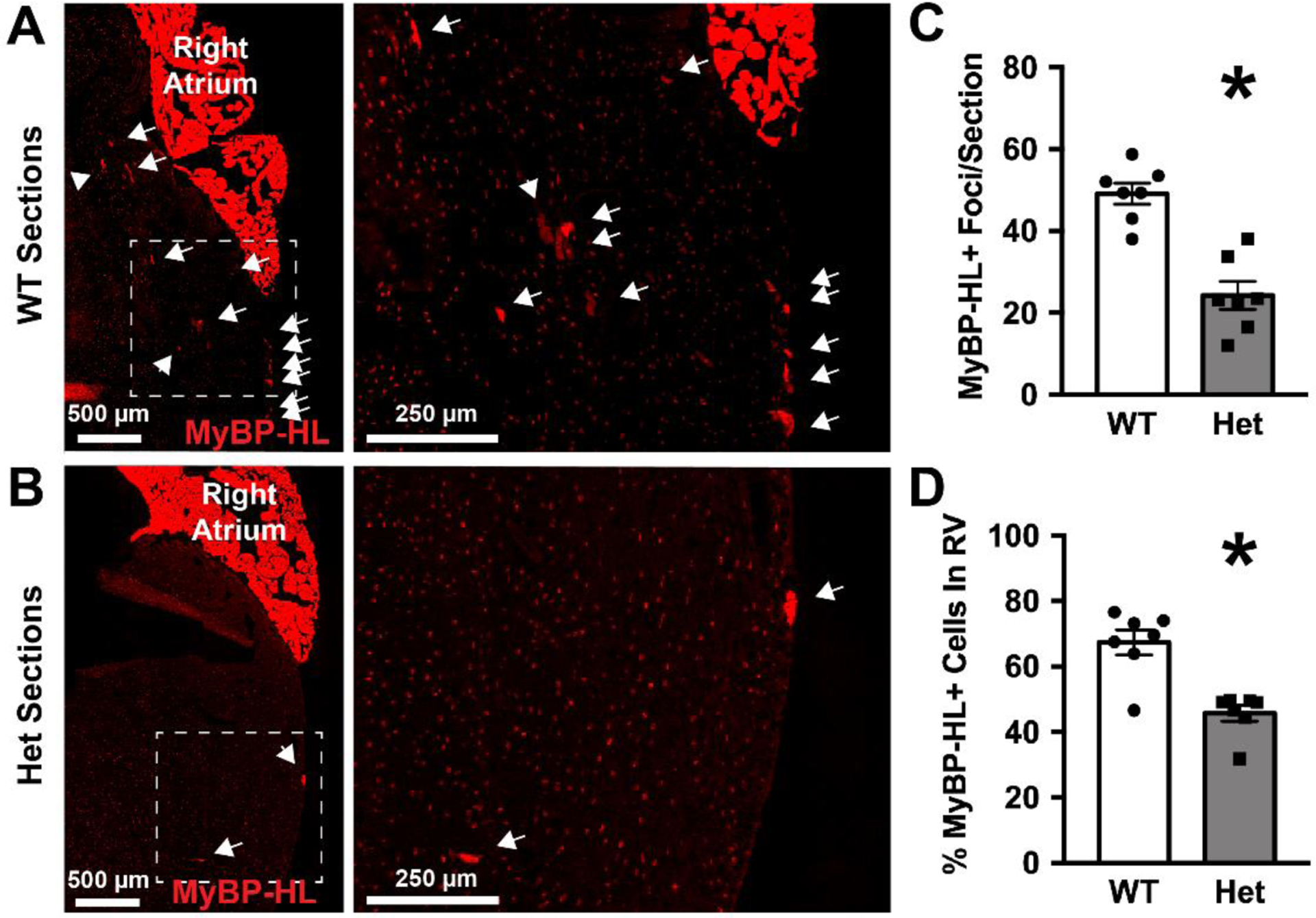
(A) Right ventricular free wall region from a WT heart stained with MyBP-HL (red), with expanded view of the RV free wall showing individual MyBP-HL-positive foci (arrows). (B) Right ventricular free wall region from a Mybphl heterozygous heart stained with MyBP-HL and contactin-2 show fewer and larger MyBP-HL foci. Speckled red background signal in panels A and B are off target staining from this antibody that occurs perinuclearly throughout the heart. (C) Quantification of the numbers of MyBP-HL foci per section. (D) Quantification of the percentage of MyBP-HL foci that were found in the RV free wall. N = average foci count per slide from 4 – 6 sections/slide. N = 7 WT, 7 Het slides. Female hearts. * = P< 0.05 by two-tailed t-test
We isolated individual adult cardiomyocytes from WT and Mybphl heterozygous mouse hearts to evaluate quantity and morphology of ventricular cardiomyocytes. We hypothesized that the MyBP-HL positive ventricular cardiomyocytes would have a Purkinje cell morphology, which is known to be elongated and narrow compared to normal brick-shaped ventricular cardiomyocytes [21, 30, 31]. In WT hearts, we isolated and identified many MyBP-HL positive cardiomyocytes with ventricular conduction cell morphology, but we also found MyBP-HL positive cells with the canonical brick shaped ventricular cardiomyocyte (Fig. 4A). Analysis of cardiomyocyte morphology revealed that ventricular cardiomyocytes that were positive for MyBP-HL were not significantly different from ventricular cardiomyocytes that were MyBP-HL-negative, with no difference in cell length, width, or length/width ratio (Fig. 4C – E). Although the overall size and shape of MyBP-HL positive cells did not differ between heterozygous and WT hearts, there was a significant reduction in the number of MyBP-HL positive ventricular cardiomyocytes normalized to the number of cells isolated, which supports the observations of reduced MyBP-HL ventricular foci in sectioned heterozygous hearts (Fig. 4B).
Figure 4. MyBP-HL expressing ventricular cardiomyocytes have diverse morphologies with fewer MyBP-HL-positive ventricular cells in heterozygous hearts.
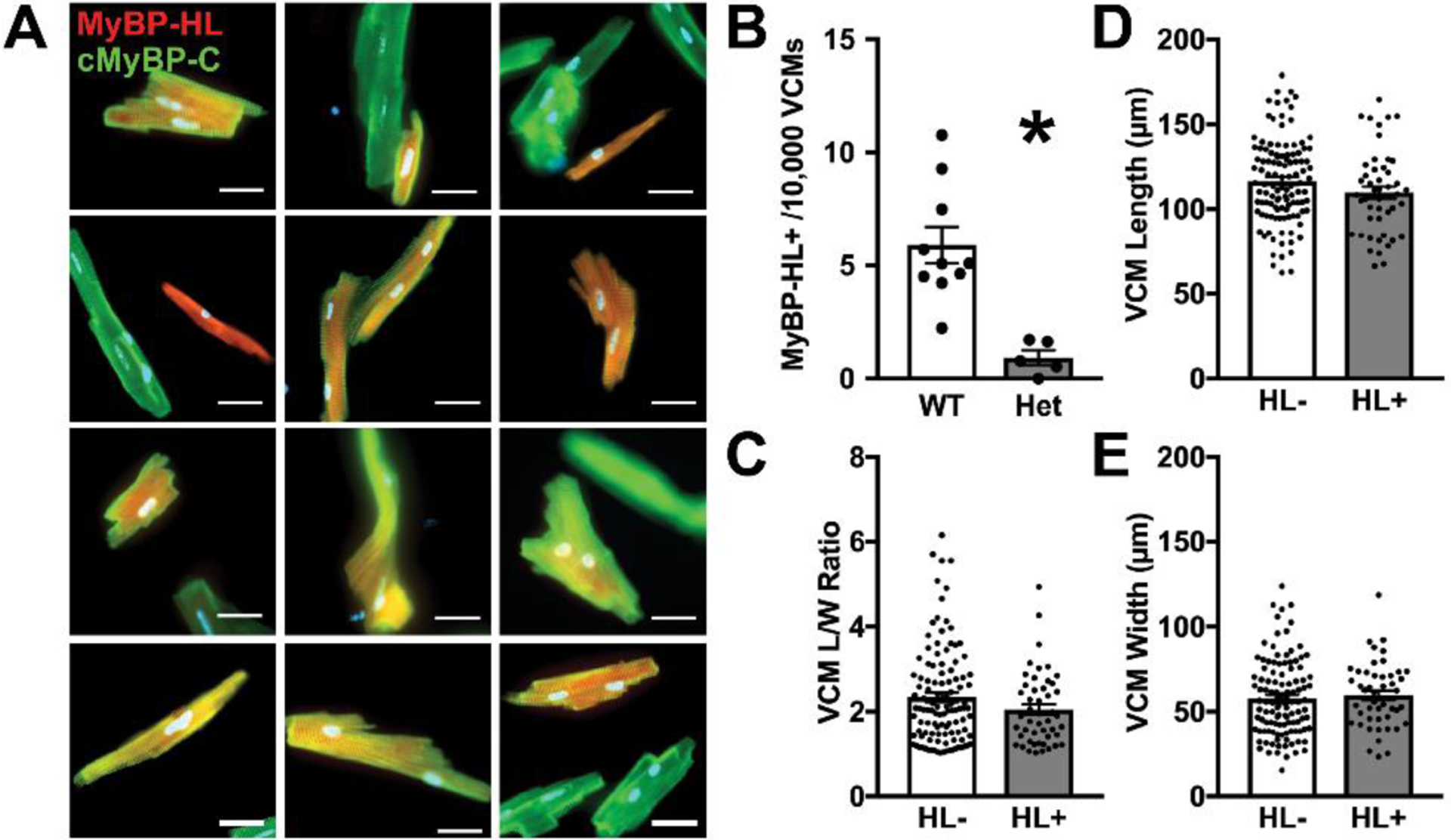
(A) Isolated ventricular cardiomyocytes stained with MyBP-HL (red) and cMyBP-C (green). (B) The number of MyBP-HL positive cells counted in total ventricular cardiomyocyte single cell preparations. N = average frequency in isolations from 10 WT mice, 5 Het mice. * = P< 0.05 by two-tailed t-test. (C – E) The length/width ratio, cell length, and cell width values for isolated MyBP-HL positive ventricular cardiomyocytes compared to MyBP-HL negative ventricular cardiomyocytes. N = 6 WT isolations, 4 male, 2 female; 112 MyBP-HL(−) cells, 48 MyBP-HL(+) ventricular cardiomyocytes.
Our quantification of the number of MyBP-HL positive ventricular cardiomyocytes was surprisingly small, at 5.9 ± 0.16 per 10,000 ventricular cardiomyocytes in WT hearts. We suspected that our isolation of ventricular cells was not yielding representative amounts of conduction system and endocardial cardiomyocytes. Therefore, we quantified the frequency of contactin-2 positive ventricular conduction system cardiomyocytes in our isolations using mice expressing green fluorescent protein under the control of the contactin-2 promoter. Using the same method as we did for the MyBP-HL frequency quantification, we quantified 7.24 ± 0.55 per 10,000 live ventricular cardiomyocytes expressing contactin-2. This is substantially lower than previous quantification of contactin-2 expressing cells in this mouse model measured by fluorescence-activated cell sorting that reported approximately 100 green fluorescent protein positive cells per 10,000 live cardiomyocytes [32]. However, the cells we counted were plated on laminin coated culture dishes for several hours prior to imaging and these steps may have selected for larger non-conduction system ventricular cardiomyocytes. These observations suggest that there may be significantly more MyBP-HL expressing ventricular cardiomyocytes and future generation of a reporter of Mybphl expression would be able to test this hypothesis.
3.2. MyBP-HL positive ventricular cardiomyocytes are enriched near the ventricular conduction system
Therefore, we evaluated the association between MyBP-HL expressing ventricular cardiomyocytes and the ventricular conduction system by immunostaining whole hearts from perinatal day five mice with MyBP-HL and contactin-2, optically clearing the hearts, and imaged with lightsheet microscopy. We assembled Z-stacks from the light sheet images and created three dimensional reconstructions and renderings of the atria, ventricular conduction system, and MyBP-HL ventricular foci (Fig. 5A – C). The atria, which express Mybphl in every cardiomyocyte, showed strong MyBP-HL signal and were used to create an atrial volume rendering to demarcate the atrial to ventricular transition. In the ventricle, MyBP-HL immunostaining was identified and rendered as discrete spots (Fig. 5A). The shortest distance from each MyBP-HL positive ventricular spot to the contactin-2 rendered surface was measured and the percent frequency of the distance were reported with a histogram (Fig. 5B). In WT hearts, MyBP-HL was enriched within 100 μm of the conduction system, whereas this enrichment was not observed in Mybphl heterozygous hearts (Fig. 5B – D). The reduction in ventricular MyBP-HL foci observed in heterozygous hearts (Fig. 3, 4) was supported by this experiment, with heterozygous hearts having red channel spot distribution like the Mybphl null hearts (Fig 5. B – C). The spots observed in the Mybphl null hearts control for staining and rendering artifacts.
Figure 5. Ventricular MyBP-HL puncta are enriched near contactin-2 stained Purkinje fibers.
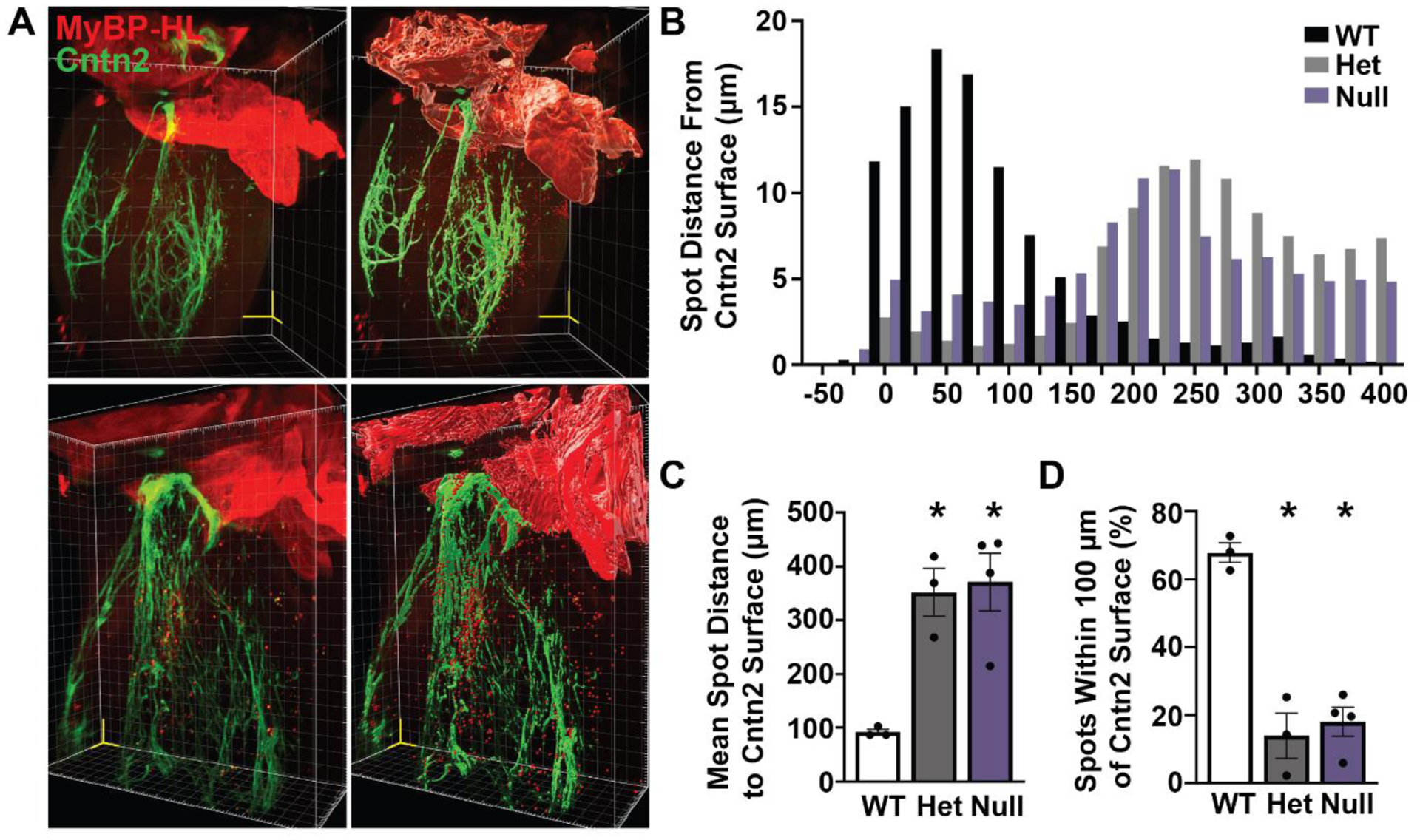
(A) Reconstructed images from 5 μm step size Z-stacks taken using lightsheet microscopy of a WT perinatal day 5 mouse heart immunostained for MyBP-HL (red) and contactin-2 (Cntn2) (green). Yellow scale volume 500 μm/side. Rendering of surface feature details (right panels) of the MyBP-HL stained atria and contactin-2 stained ventricular conduction system, as well as single spot renderings of MyBP-HL positive ventricular foci. (B) A histogram of the percentage of the shortest distance from each MyBP-HL spot to the contactin-2 surface in Mybphl WT, Het, and Null hearts showed enrichment of MyBP-HL spots near the contactin-2 surface in WT mice. (C) The mean shortest distance between MyBP-HL spots and the contactin-2 surface is significantly increased in Het and Null hearts. (D) WT hearts show most MyBP-HL spots are located within 100 μm of the contactin-2 surface. N = 3 WT, 3 Het, 4 Null. Mixed sex litters. * = p > 0.05 by One-Way ANOVA.
3.3. Loss of Mybphl results in atrial and ventricular conduction defects
We previously showed that isoproterenol treatment elicited increased heart rate variability and ventricular arrhythmias in Mybphl hearts [1]. We further evaluated cardiac conduction properties of adult Mybphl mouse hearts using conscious ambulatory telemetry (Fig. 6). Electrocardiogram traces from adult WT and Mybphl homozygous null mice following acute treatment with propranolol showed atrioventricular nodal block, with P waves dissociated from the QRS complexes in Mybphl null mice. Poincaré plots of R-R intervals showed increased heart rate variability compared to the WT control mice (Fig. 6A). Poincaré plots of P-R intervals show the highly variable P-R interval in Mybphl null mice during 1 – 2 second periods of atrioventricular dissociation compared to simultaneous recordings taken in WT mice. Acute treatment with the sodium channel blocker flecainide prolonged the QRS duration and slowed heart rate in both WT and Mybphl null mice (Fig. 6C). Mybphl null mice showed a marked increase in heart rate variability, as seen in the representative electrocardiogram trace and Poincaré plots. At baseline, P wave and QRS duration, and the P-R intervals were similar among WT, Mybphl heterozygous, and homozygous null hearts (Fig. 6D). However, acute propranolol treatment prolonged P wave duration in Mybphl null mice (Fig. 6E). Flecainide increased P wave duration and QRS duration in Mybphl heterozygous mice compared to homozygous null mice (Fig. 6F).
Figure 6. Loss of Mybphl results in atrial and ventricular conduction dysfunction.
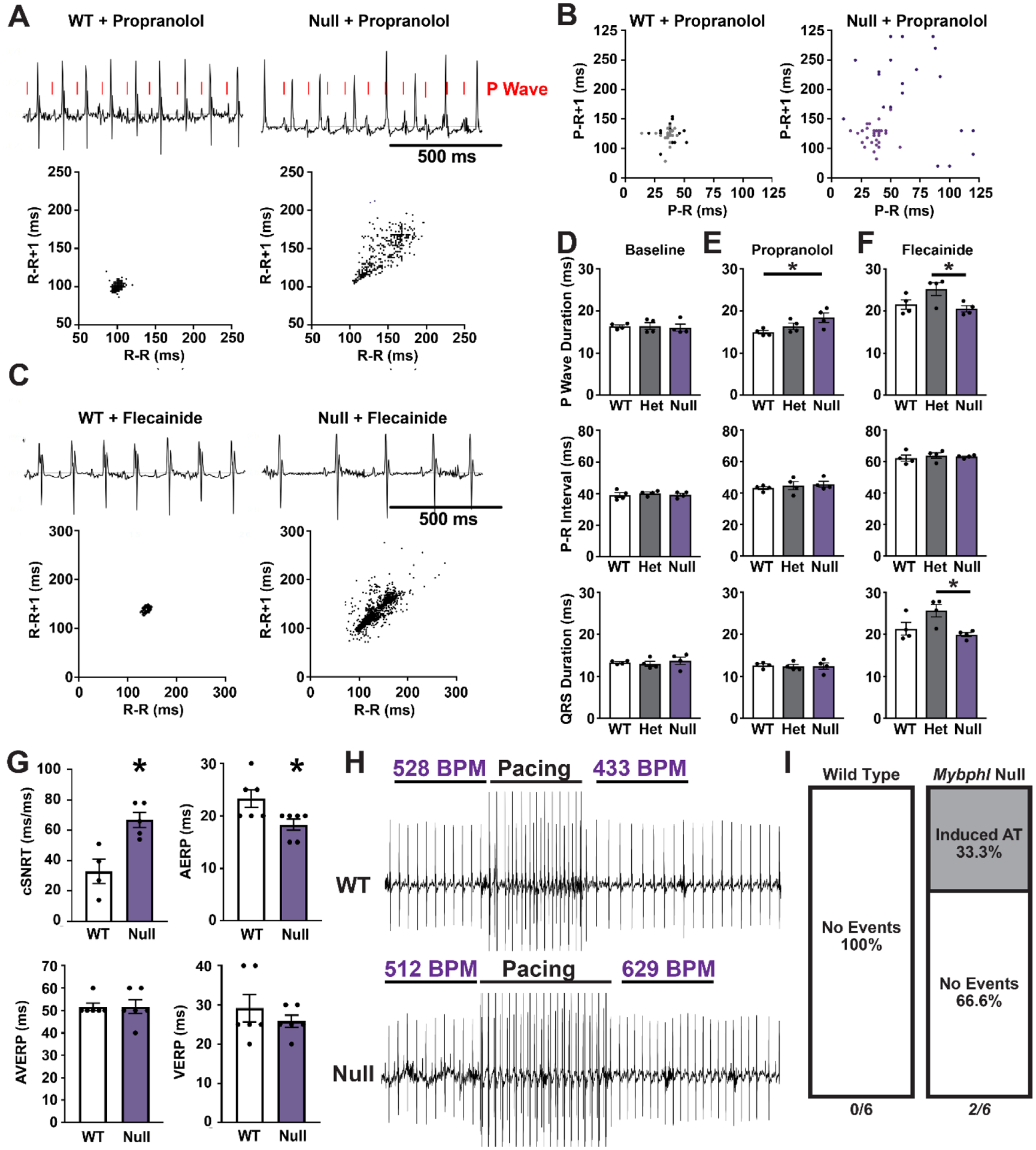
(A) Conscious ambulatory electrocardiogram recordings on WT and Mybphl null mice following acute propranolol injection shows dissociation of the P-waves in null mice. Poincaré plots of the R-R interval show increased heart rate variability (400 consecutive R waves from single representative animals). (B) Poincaré plots of the P-R intervals taken during 1 – 2 second periods of atrioventricular dissociation in Mybphl null mice illustrate the variable P-R interval. Corresponding period of WT traces shown. Data from two mice, (dark and light dots) shown per genotype. Male and female per genotype. (C) Conscious ambulatory telemetry recordings on WT and Mybphl null mice following acute flecainide injection slows heart rates in both groups, with Mybphl null mice exhibiting high R-R variability (plots show 400 consecutive beats from single representative animals). (D) P-wave duration, P-R interval, and QRS duration measurements obtained from conscious ambulatory telemetry from WT, heterozygous, and homozygous Mybphl null mice, at baseline or following either acute propranolol treatment (E) or flecainide treatment (F). Data averaged from two minutes of recording. N = 4 mice per genotype, both with 2 Male, 2 Female. * = P <0.05 by One-Way ANOVA. (G) Durations for corrected sinus node recovery time (cSNRT), atrial effective refractory period (AERP), atrioventricular node effective refractory period (AVERP), and ventricular effective refractory period (VERP) from intracardiac recordings. N = 6 mice per group. * = P< 0.05 by two-tailed t-test. (H, I) Intracardiac programed stimulation at 50 ms cycle lengths elicited periods of atrial tachycardia in 2 out of 6 Mybphl null mice but this programed stimulation was unable to elicit tachycardia in WT mice. Pre- and post-pacing heart rates are marked above the example traces. N = 6 mice per group, all female.
We performed intracardiac programmed electrical stimulation to pace the heart and record electrophysiological parameters. Atrial burst pacing at 50 ms cycle lengths was performed for 1 – 2 seconds in WT and Mybphl null mice (Fig. 6G – I). We observed that Mybphl homozygous null mice had a significantly longer corrected sinus node recovery time following pacing, and that the atrial effective refractory period was shorter in Mybphl homozygous null mice (Fig. 6G). All WT hearts had normal or slower beating rates following cessation of the stimuli, whereas 2/6 Mybphl hearts showed persistent atrial tachycardia for several seconds following burst pacing (Fig. 6H, I). Atrial tachycardia has not able to be elicited in control mice under these conditions in similar experiments [28, 33].
3.4. Atrial cardiomyocytes lacking Mybphl show aberrant calcium handling
Assessing the functional effect of loss of Mybphl at the single cardiomyocyte level was technically prohibitive due to the sparse distribution Mybphl expressing cardiomyocytes in the ventricle, including the atrioventricular node region or the Purkinje fibers. Instead, we isolated atrial cardiomyocytes that all express Mybphl and performed calcium transient analysis to identify any cell intrinsic changes that could contribute to the arrhythmic phenotype observed with loss of Mybphl. Atrial cardiomyocytes isolated from WT mouse hearts showed calcium release events that were spatiotemporally uniform along the length of the cell from 1000 ms to 200 ms cycle length stimuli (Fig. 7A). Due to an increased propensity of triggered waves at shorter cycle lengths, we compared single representative transients at 1000 and 500 ms cycle lengths. Mybphl homozygous null atrial cardiomyocytes showed no difference in peak amplitude of calcium release but did show significantly increased heterogeneity of calcium release along the length of the cell (Fig 7B, C). Analysis of the transients showed no significant difference in time to peak calcium release and no significant difference in the heterogeneity of this parameter (Fig. 7D). The maximum rate of calcium release was significantly higher in Mybphl homozygous null transients compared to WT transients (Fig. 7E) and the heterogeneity in maximum rate of calcium release was also significantly higher in Mybphl null transients (Fig. 7F).
Figure 7. Mybphl heterozygous and homozygous null atrial cardiomyocytes show heterogenous calcium release and aberrant ryanodine receptor organization.
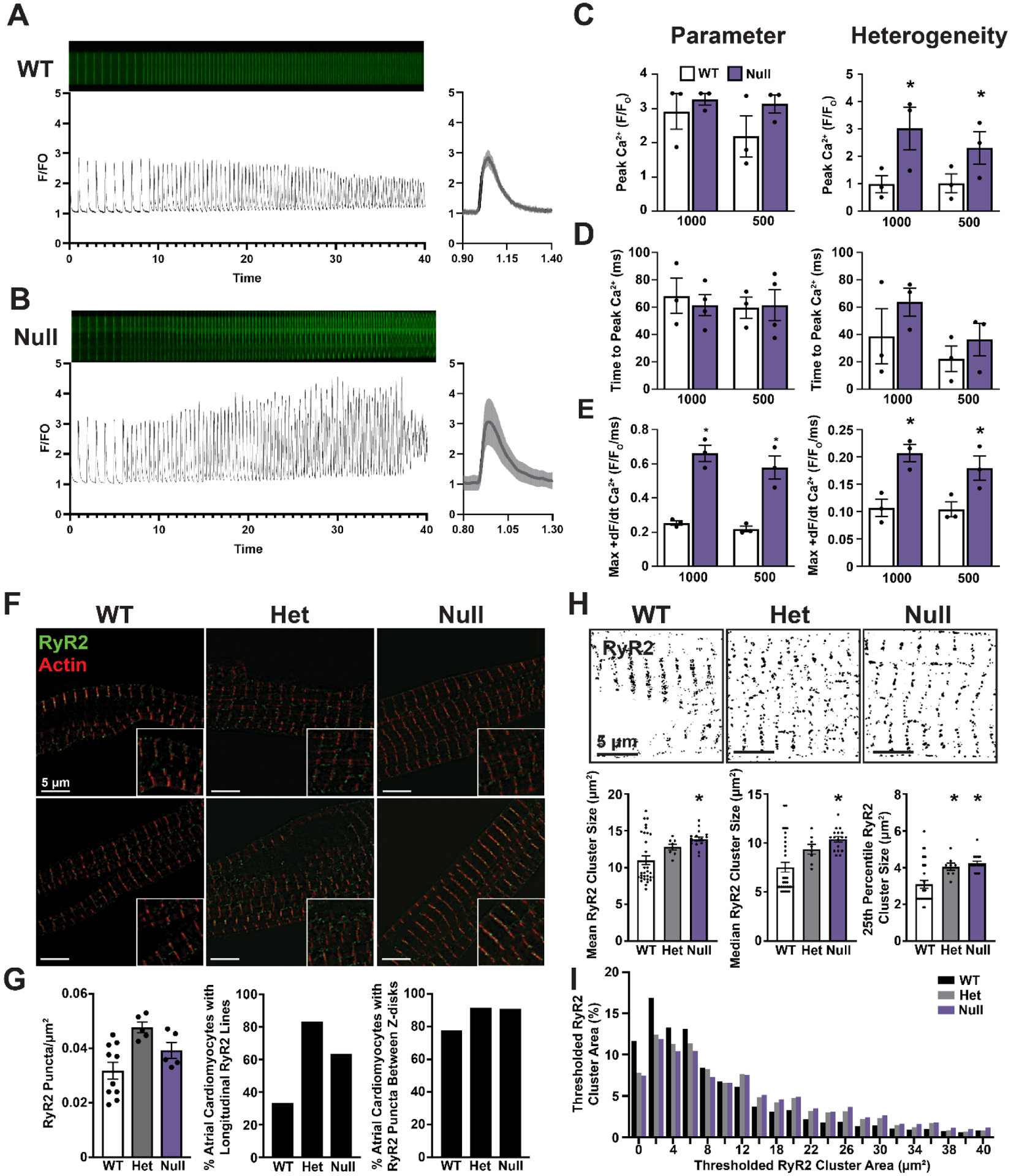
Line scanning confocal images from isolated atrial cardiomyocytes loaded with Cal520 calcium indicator dye and the associated calcium transient profiles from WT (A) and Null (B) groups. Single 1000 ms transient shown with standard deviation across a single cell. (C) Peak calcium transient amplitude at 1000 ms and 500 ms pacing cycle lengths (left) and the heterogeneity index (right) of peak amplitude measured along the length of each cell. N = 3 mice per group, two male, one female WT mice and one male, two female Null mice with averages of 8, 8, and 13 WT cells; 23, 13, and 13 Null cells. (D) Time to peak calcium release at each cycle length (left) and heterogeneity index (right) of time to peak calcium release. (E) The maximal rate of calcium release (+dR/dt) at each frequency and the heterogeneity index of the maximal rate of calcium release. (F) Structured illumination microscopy images of isolated atrial cardiomyocytes stained with anti-ryanodine receptor 2 antibodies and fluorescent conjugated phalloidin to mark actin. (G) Quantification of RyR2 puncta per area, the percent occurrence of cells exhibiting longitudinal lines of RyR2 puncta between myofibers, and the percent of cardiomyocytes with RyR2 puncta outside the peri-Z-disk region. N = Average of all cells from WT, 10; Het 5, Null 5 animals. WT 18 cells, Het 12 cells, Null 11 cells. (H) Otsu thresholding of structured illumination microscopy images and quantification of average RyR2 cluster size, as well as the cluster size of each cell at the 25th percentile and median values. Clusters measured from N = WT 32, Het 8, Null 17 individual cardiomyocytes. (I) Histogram of RyR2 cluster size from thresholded images. N = WT 32, Het 8, Null 17 Individual cardiomyocytes from 2 male, 1 female WT mouse, two female Het mice, and one male, one female null mouse.
3.5. Loss of Mybphl associates with disrupted ryanodine receptor organization
Due to the highly heterogeneous calcium release observed in the Mybphl homozygous null atrial cardiomyocytes, we performed immunostaining and super resolution structured illumination microscopy to visualize ryanodine receptor organization in isolated atrial cardiomyocytes (Fig. 7F). We observed no significant difference in ryanodine receptor cluster density (Fig. 7G) but did note that heterozygous and homozygous Mybphl null cardiomyocytes had more ryanodine receptor clusters that fell between myofibrils and were not organized transversely along the Z-line (Fig. 7G). To quantify cluster sizes, we thresholded the RyR fluorescence signal and measured thresholded cluster area, which showed a significant increase in the mean area in homozygous null Mybphl atrial cardiomyocytes (Fig. 7H). We also noted that clusters tended to be larger in Mybphl heterozygous and homozygous null cells, as noted in the top quartile and median receptor size per cell (Fig. 7H). This is also illustrated by the shoulder on the histogram representing a greater number of larger clusters with thresholded areas over 10 μm2 in Mybphl heterozygous and homozygous null mice (Fig. 7H).
4. Discussion
Our prior report on the association between loss of MYBPHL and the development of atrial and ventricular dysfunction, arrhythmia, and dilated cardiomyopathy provided some evidence that MYBPHL is expressed primarily in the atria [1]. However, we had not deeply explored the anatomical distribution of MYBPHL expression, or effects of the loss of MYBPHL on single cardiomyocyte function. Our results now demonstrate that Mybphl is highly expressed in the atria and is expressed in small amounts in the ventricle, primarily in regions that are within or adjacent to the ventricular conduction system. We provide evidence of atrioventricular dysfunction following loss of Mybphl, which was a major component of the disease in the affected family [1]. We also identified subcellular disarray of ryanodine receptor clusters and increased heterogeneity of calcium release in Mybphl null mice.
4.1. Atrial dilation and arrhythmia susceptibility
MyBP-HL is primarily expressed in the atria, with additional expression in discrete subset of the ventricle. We previously showed that Mybphl homozygous null mice have dilated atria and arrhythmia [1]. We now report that these mice also show a shortened atrial refractory period and increased amplitude and heterogeneity of intracellular calcium release. This is a pathogenic combination of properties that predispose the atria to develop arrhythmia, as atrial dilation and increased pressures have previously been shown to correlate with shortened atrial effective refractory period [34, 35]. Additionally, the clinical features of the family with the MYBPHL R255Stop mutation included significant burden of atrioventricular block [1], which we now observed in Mybphl null mice treated acutely with propranolol, a beta-adrenergic blocker. We also established that Mybphl-expressing ventricular cardiomyocytes are found in superior portions of the atrioventricular node and within and adjacent to the Purkinje fiber network. If cells positioned in these anatomical zones display defects like Mybphl-expressing cells from the atria, this would explain their contribution to atrioventricular conduction defects. However, we have not yet been able to ascertain what cellular or tissue level changes occur in these ventricular cells during the development of disease. Directly validating this hypothesis will require using a combination of reporters to identify the correct cell type that co-expresses ventricular conduction system markers, such as contactin-2, as well as MyBP-HL.
4.2. Intracellular disarray and calcium handling defects
Atrial cardiomyocytes have a less developed t-tubule network that is dysregulated in heart failure, which may lead to the development of subcellular calcium waves that promote arrhythmia [36–38]. The sarcoplasmic reticular of atrial myocytes has been shown to include large axial tubule elements running the length of the cell that facilitates coordinated calcium release in atrial cardiomyocytes [39]. Heterogeneous calcium release, triggered calcium waves, and a higher occurrence of longitudinal interfilamentous RyR clusters in Mybphl null atrial cardiomyocytes are consistent with pathological remodeling seen in other models of atrial dysfunction with transverse-tubule structural defects [36, 40]. We were unable to include assessment of the calcium transient properties in Mybphl heterozygous atrial cardiomyocytes. This is unfortunate, as the Mybphl heterozygous mice demonstrate a phenotype that is similar to the homozygous null mice in many experiments and also phenotypes that are intermediate to the WT and homozygous in others. Importantly, the mechanism of cell-intrinsic dysfunction arising due to the reduction or loss of MyBP-HL may differ between heterozygous and homozygous null cardiomyocytes and warrants further study. Our data also show that Mybphl null cardiomyocytes have increased heterogeneity of calcium release and larger ryanodine receptor areas. We did not assess SR calcium levels, which would inform whether the SR calcium overload occurs in the Mybphl null cardiomyocytes.
Aberrant calcium handling and ryanodine receptor cluster disarray are not expected to be directly regulated by MyBP-HL, which is myofilament associated, but may be a consequence of contractile dysfunction and subcellular disarray observed in other models of myofilament mutations [41, 42]. However, the contribution of MyBP-HL to myofilament structure and function remains to be determined. Isolating ventricular cardiomyocytes that express Mybphl was not technically feasible given their sparsity. We expect that the loss of Mybphl in Mybphl-expressing ventricular conduction system myocytes would result in dysfunction like what we observed in atrial cardiomyocytes. The sarcoplasmic reticulum in Purkinje cells shows minimal transverse tubule network, similar to atrial cardiomyocytes [43–45].
4.3. Cellular heterogeneity
Our prior data showed that Mybphl had a low level of expression in the ventricle [1], but it was unclear whether this was noise, atrial contamination in ventricular tissue preparations, low-level expression in many cells, or high expression in few cells. From our immunostaining data, MyBP-HL is observed to be highly expressed in a small number of cells in specific areas of the ventricle (Fig. 1 – 5). This would result in low and/or highly variable expression levels in RNA-Seq experiments from bulk ventricular myocardium. RNA-Seq data has been published from ventricular cardiomyocytes expressing a green fluorescent reporter driven by the contactin-2 promoter to mark ventricular conduction system cells, allowing ventricular conduction system-enriched tissue to be isolated for analysis [32]. Mybphl was upregulated in ventricular conduction system tissue compared to bulk myocardium, but importantly the relative expression values were highly variable between samples of the Purkinje tissue, with FPKM counts of: 5.7, 0.8, 3.1, 16.1 in total ventricle, and fragments per kilobase of exon per million mapped fragment counts of: 40.0, 130.6, 24.9, 17.4 in Purkinje cell enriched samples [1, 32]. This RNA-Seq data set did not include counts from Mybph, which has previously been suggested to be expressed in Purkinje fibers, although the recent identification of Mybphl as a highly homologous gene may confound some of these experiments [2–4].
Recent experiments using single cell RNA-Seq have shown transcriptional homogeneity within “working” ventricular cardiomyocytes in healthy mouse hearts [46]. Studies of the components of the ventricular conduction system in mice demonstrated the heterogeneity and presence of additional distinct subclusters of cells within the ventricular conduction system [18]. This data set showed Mybphl was differentially regulated in the ventricular transition zone that includes Purkinje cell gene signatures, in the atrioventricular transition zone, and the atrioventricular ring [18]. However, in all these regions, the magnitude of Mybphl expression change was small and may reflect the expression of Mybphl in only a small subpopulation of cells within each of these zones. The localization of MyBP-HL in discrete regions of the ventricle and within and adjacent to the conduction system suggests a nuanced tuning of specific cells.
Assessing the functional consequences of gene expression in highly specific regions of the heart is a fascinating and daunting prospect. An example of cell-scale functional transitions are the junctional cardiomyocytes between the high-speed conducting Purkinje fibers and the bulk myocardium [47, 48]. Cell-level resolution of transcriptional signatures of small groups of specialized cells are revealing the complexity of cardiac regulation, in cardiomyocytes and in non-muscle cell types that comprise the heart [18, 49–51]. Genes identified as highly enriched in small groups of cells, like MyBP-HL, may have an underappreciated contribution to overall heart function, and mutations in these genes may contribute to the development of disease. Future studies on these genes and rare cells may provide fascinating insight into cell level regulation of cardiac physiology.
5. Conclusion
The data we have reported raises several additional questions, most notably regarding the mechanism responsible for how loss of Mybphl results in conduction system dysfunction. We have shown that loss of Mybphl causes subcellular disarray that results in calcium handling abnormalities. We have also shown that Mybphl is localized in regions of the heart that are critical for proper conduction system function. We have not acquired Purkinje fiber morphology at a high enough resolution to determine if any morphological changes to the Purkinje network are present in Mybphl null hearts. As loss of Mybphl in this model is constitutive, it is possible that some defects in anatomical arrangement of the Purkinje fibers may arise during development. Testing a model of conditional deletion of Mybphl in adult mice would determine whether loss of Mybphl during development is required for disease or if MyBP-HL is able to dynamically regulate adult cardiomyocytes such that its absence leads to the observed pathophysiology.
Supplementary Material
Highlights.
MyBP-HL is expressed throughout the mouse atria and in specific ventricular foci
Ventricular cardiomyocytes expressing MyBP-HL occur near the ventricular conduction system
Loss of MyBP-HL results in atrioventricular block and prolonged P-waves
Mybphl null atrial cardiomyocytes exhibit heterogeneous release of calcium
Disclosure of funding
This work was supported by funding from the NIH National Heart Lung and Blood institute grants K99HL141698, R00151698, and F32HL131304 (DYB); HL128075 (EMM); American Heart Association grants ASCD SFRN (EMM), Career Development Award (MRP); Leducq Foundation Transatlantic Network on Editing the Failing Heart (EMM).
Abbreviations:
- MyBP-HL
myosin binding protein-H like
- MYBPHL
human gene encoding MyBP-HL
- Mybphl
mouse gene encoding MyBP-HL
- DCM
dilated cardiomyopathy
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Barefield DY, Puckelwartz MJ, Kim EY, Wilsbacher LD, Vo AH, Waters EA, Earley JU, Hadhazy M, Dellefave-Castillo L, Pesce LL, McNally EM, Experimental Modeling Supports a Role for MyBP-HL as a Novel Myofilament Component in Arrhythmia and Dilated Cardiomyopathy, Circulation 136(16) (2017) 1477–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Alyonycheva T, Cohen-Gould L, Siewert C, Fischman DA, Mikawa T, Skeletal muscle-specific myosin binding protein-H is expressed in Purkinje fibers of the cardiac conduction system, Circ Res 80(5) (1997) 665–72. [DOI] [PubMed] [Google Scholar]
- [3].Mouton J, Loos B, Moolman-Smook JC, Kinnear CJ, Ascribing novel functions to the sarcomeric protein, myosin binding protein H (MyBPH) in cardiac sarcomere contraction, Exp Cell Res 331(2) (2015) 338–51. [DOI] [PubMed] [Google Scholar]
- [4].Mouton JM, van der Merwe L, Goosen A, Revera M, Brink PA, Moolman-Smook JC, Kinnear C, MYBPH acts as modifier of cardiac hypertrophy in hypertrophic cardiomyopathy (HCM) patients, Hum Genet 135(5) (2016) 477–483. [DOI] [PubMed] [Google Scholar]
- [5].McNally EM, Barefield DY, Puckelwartz MJ, The genetic landscape of cardiomyopathy and its role in heart failure, Cell Metab 21(2) (2015) 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tayal U, Newsome S, Buchan R, Whiffin N, Walsh R, Barton PJ, Ware JS, Cook SA, Prasad SK, Truncating Variants in Titin Independently Predict Early Arrhythmias in Patients With Dilated Cardiomyopathy, J Am Coll Cardiol 69(19) (2017) 2466–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lee SP, Ashley EA, Homburger J, Caleshu C, Green EM, Jacoby D, Colan SD, Arteaga-Fernandez E, Day SM, Girolami F, Olivotto I, Michels M, Ho CY, Perez MV, Investigators SH, Incident Atrial Fibrillation Is Associated With MYH7 Sarcomeric Gene Variation in Hypertrophic Cardiomyopathy, Circ Heart Fail 11(9) (2018) e005191. [DOI] [PubMed] [Google Scholar]
- [8].Lakdawala NK, Givertz MM, Dilated cardiomyopathy with conduction disease and arrhythmia, Circulation 122(5) (2010) 527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Patel DA, Lavie CJ, Milani RV, Shah S, Gilliland Y, Clinical implications of left atrial enlargement: a review, Ochsner J 9(4) (2009) 191–6. [PMC free article] [PubMed] [Google Scholar]
- [10].Seko Y, Kato T, Haruna T, Izumi T, Miyamoto S, Nakane E, Inoko M, Association between atrial fibrillation, atrial enlargement, and left ventricular geometric remodeling, Sci Rep 8(1) (2018) 6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, Caleshu CA, Helms AS, Colan SD, Girolami F, Cecchi F, Seidman CE, Sajeev G, Signorovitch J, Green EM, Olivotto I, Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe), Circulation 138(14) (2018) 1387–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Arbustini E, Pilotto A, Repetto A, Grasso M, Negri A, Diegoli M, Campana C, Scelsi L, Baldini E, Gavazzi A, Tavazzi L, Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease, J Am Coll Cardiol 39(6) (2002) 981–90. [DOI] [PubMed] [Google Scholar]
- [13].Brembilla-Perrot B, Alla F, Suty-Selton C, Huttin O, Blangy H, Sadoul N, Chometon F, Groben L, Luporsi JD, Zannad N, Aliot E, Cedano J, Ammar S, Abdelaal A, Juilliere Y, Nonischemic dilated cardiomyopathy: results of noninvasive and invasive evaluation in 310 patients and clinical significance of bundle branch block, Pacing Clin Electrophysiol 31(11) (2008) 1383–90. [DOI] [PubMed] [Google Scholar]
- [14].Aleksova A, Carriere C, Zecchin M, Barbati G, Vitrella G, Di Lenarda A, Sinagra G, New-onset left bundle branch block independently predicts long-term mortality in patients with idiopathic dilated cardiomyopathy: data from the Trieste Heart Muscle Disease Registry, Europace 16(10) (2014) 1450–9. [DOI] [PubMed] [Google Scholar]
- [15].Park DS, Fishman GI, The cardiac conduction system, Circulation 123(8) (2011) 904–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].van Weerd JH, Christoffels VM, The formation and function of the cardiac conduction system, Development 143(2) (2016) 197–210. [DOI] [PubMed] [Google Scholar]
- [17].Munshi NV, Gene regulatory networks in cardiac conduction system development, Circ Res 110(11) (2012) 1525–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Goodyer WR, Beyersdorf BM, Paik DT, Tian L, Li G, Buikema JW, Chirikian O, Choi S, Venkatraman S, Adams EL, Tessier-Lavigne M, Wu JC, Wu SM, Transcriptomic Profiling of the Developing Cardiac Conduction System at Single-Cell Resolution, Circ Res 125(4) (2019) 379–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].van Setten J, Brody JA, Jamshidi Y, Swenson BR, Butler AM, Campbell H, Del Greco FM, Evans DS, Gibson Q, Gudbjartsson DF, Kerr KF, Krijthe BP, Lyytikainen LP, Muller C, Muller-Nurasyid M, Nolte IM, Padmanabhan S, Ritchie MD, Robino A, Smith AV, Steri M, Tanaka T, Teumer A, Trompet S, Ulivi S, Verweij N, Yin X, Arnar DO, Asselbergs FW, Bader JS, Barnard J, Bis J, Blankenberg S, Boerwinkle E, Bradford Y, Buckley BM, Chung MK, Crawford D, den Hoed M, Denny JC, Dominiczak AF, Ehret GB, Eijgelsheim M, Ellinor PT, Felix SB, Franco OH, Franke L, Harris TB, Holm H, Ilaria G, Iorio A, Kahonen M, Kolcic I, Kors JA, Lakatta EG, Launer LJ, Lin H, Lin HJ, Loos RJF, Lubitz SA, Macfarlane PW, Magnani JW, Leach IM, Meitinger T, Mitchell BD, Munzel T, Papanicolaou GJ, Peters A, Pfeufer A, Pramstaller PP, Raitakari OT, Rotter JI, Rudan I, Samani NJ, Schlessinger D, Silva Aldana CT, Sinner MF, Smith JD, Snieder H, Soliman EZ, Spector TD, Stott DJ, Strauch K, Tarasov KV, Thorsteinsdottir U, Uitterlinden AG, Van Wagoner DR, Volker U, Volzke H, Waldenberger M, Jan Westra H, Wild PS, Zeller T, Alonso A, Avery CL, Bandinelli S, Benjamin EJ, Cucca F, Dorr M, Ferrucci L, Gasparini P, Gudnason V, Hayward C, Heckbert SR, Hicks AA, Jukema JW, Kaab S, Lehtimaki T, Liu Y, Munroe PB, Parsa A, Polasek O, Psaty BM, Roden DM, Schnabel RB, Sinagra G, Stefansson K, Stricker BH, van der Harst P, van Duijn CM, Wilson JF, Gharib SA, de Bakker PIW, Isaacs A, Arking DE, Sotoodehnia N, PR interval genome-wide association meta-analysis identifies 50 loci associated with atrial and atrioventricular electrical activity, Nat Commun 9(1) (2018) 2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Austin CP, Battey JF, Bradley A, Bucan M, Capecchi M, Collins FS, Dove WF, Duyk G, Dymecki S, Eppig JT, Grieder FB, Heintz N, Hicks G, Insel TR, Joyner A, Koller BH, Lloyd KC, Magnuson T, Moore MW, Nagy A, Pollock JD, Roses AD, Sands AT, Seed B, Skarnes WC, Snoddy J, Soriano P, Stewart DJ, Stewart F, Stillman B, Varmus H, Varticovski L, Verma IM, Vogt TF, von Melchner H, Witkowski J, Woychik RP, Wurst W, Yancopoulos GD, Young SG, Zambrowicz B, The knockout mouse project, Nat Genet 36(9) (2004) 921–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pallante BA, Giovannone S, Fang-Yu L, Zhang J, Liu N, Kang G, Dun W, Boyden PA, Fishman GI, Contactin-2 expression in the cardiac Purkinje fiber network, Circ Arrhythm Electrophysiol 3(2) (2010) 186–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Aubert G, Barefield DY, Demonbreun AR, Ramratnam M, Fallon KS, Warner JL, Rossi AE, Hadhazy M, Makielski JC, McNally EM, Deletion of Sulfonylurea Receptor 2 in the Adult Myocardium Enhances Cardiac Glucose Uptake and Is Cardioprotective, JACC Basic Transl Sci 4(2) (2019) 251–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Galice S, Xie Y, Yang Y, Sato D, Bers DM, Size Matters: Ryanodine Receptor Cluster Size Affects Arrhythmogenic Sarcoplasmic Reticulum Calcium Release, J Am Heart Assoc 7(13) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hou Y, Jayasinghe I, Crossman DJ, Baddeley D, Soeller C, Nanoscale analysis of ryanodine receptor clusters in dyadic couplings of rat cardiac myocytes, J Mol Cell Cardiol 80 (2015) 45–55. [DOI] [PubMed] [Google Scholar]
- [25].Renier N, Wu Z, Simon DJ, Yang J, Ariel P, Tessier-Lavigne M, iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging, Cell 159(4) (2014) 896–910. [DOI] [PubMed] [Google Scholar]
- [26].Rose RA, Kabir MG, Backx PH, Altered heart rate and sinoatrial node function in mice lacking the cAMP regulator phosphoinositide 3-kinase-gamma, Circ Res 101(12) (2007) 1274–82. [DOI] [PubMed] [Google Scholar]
- [27].Martin CA, Zhang Y, Grace AA, Huang CL, In vivo studies of Scn5a+/− mice modeling Brugada syndrome demonstrate both conduction and repolarization abnormalities, J Electrocardiol 43(5) (2010) 433–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Laforest B, Dai W, Tyan L, Lazarevic S, Shen KM, Gadek M, Broman MT, Weber CR, Moskowitz IP, Atrial fibrillation risk loci interact to modulate Ca2+-dependent atrial rhythm homeostasis, J Clin Invest 129(11) (2019) 4937–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gussak G, Marszalec W, Yoo S, Modi R, O’Callaghan C, Aistrup GL, Cordeiro JM, Goodrow R, Kanaporis G, Blatter LA, Shiferaw Y, Arora R, Zhou J, Burrell AR, Wasserstrom JA, Triggered Ca(2+) Waves Induce Depolarization of Maximum Diastolic Potential and Action Potential Prolongation in Dog Atrial Myocytes, Circ Arrhythm Electrophysiol 13(6) (2020) e008179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Herron TJ, Milstein ML, Anumonwo J, Priori SG, Jalife J, Purkinje cell calcium dysregulation is the cellular mechanism that underlies catecholaminergic polymorphic ventricular tachycardia, Heart Rhythm 7(8) (2010) 1122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ono N, Yamaguchi T, Ishikawa H, Arakawa M, Takahashi N, Saikawa T, Shimada T, Morphological varieties of the Purkinje fiber network in mammalian hearts, as revealed by light and electron microscopy, Arch Histol Cytol 72(3) (2009) 139–49. [DOI] [PubMed] [Google Scholar]
- [32].Kim EE, Shekhar A, Lu J, Lin X, Liu FY, Zhang J, Delmar M, Fishman GI, PCP4 regulates Purkinje cell excitability and cardiac rhythmicity, J Clin Invest 124(11) (2014) 5027–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nadadur RD, Broman MT, Boukens B, Mazurek SR, Yang X, van den Boogaard M, Bekeny J, Gadek M, Ward T, Zhang M, Qiao Y, Martin JF, Seidman CE, Seidman J, Christoffels V, Efimov IR, McNally EM, Weber CR, Moskowitz IP, Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm, Sci Transl Med 8(354) (2016) 354ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ravelli F, Allessie M, Effects of atrial dilatation on refractory period and vulnerability to atrial fibrillation in the isolated Langendorff-perfused rabbit heart, Circulation 96(5) (1997) 1686–95. [DOI] [PubMed] [Google Scholar]
- [35].Neuberger HR, Schotten U, Blaauw Y, Vollmann D, Eijsbouts S, van Hunnik A, Allessie M, Chronic atrial dilation, electrical remodeling, and atrial fibrillation in the goat, J Am Coll Cardiol 47(3) (2006) 644–53. [DOI] [PubMed] [Google Scholar]
- [36].Shiferaw Y, Aistrup GL, Louch WE, Wasserstrom JA, Remodeling Promotes Proarrhythmic Disruption of Calcium Homeostasis in Failing Atrial Myocytes, Biophys J 118(2) (2020) 476–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].McNutt NS, Fawcett DW, The ultrastructure of the cat myocardium. II. Atrial muscle, J Cell Biol 42(1) (1969) 46–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dibb KM, Clarke JD, Eisner DA, Richards MA, Trafford AW, A functional role for transverse (t-) tubules in the atria, J Mol Cell Cardiol 58 (2013) 84–91. [DOI] [PubMed] [Google Scholar]
- [39].Brandenburg S, Kohl T, Williams GS, Gusev K, Wagner E, Rog-Zielinska EA, Hebisch E, Dura M, Didie M, Gotthardt M, Nikolaev VO, Hasenfuss G, Kohl P, Ward CW, Lederer WJ, Lehnart SE, Axial tubule junctions control rapid calcium signaling in atria, J Clin Invest 126(10) (2016) 3999–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Shiferaw Y, Aistrup GL, Wasserstrom JA, Mechanism for Triggered Waves in Atrial Myocytes, Biophys J 113(3) (2017) 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tardiff JC, Hewett TE, Palmer BM, Olsson C, Factor SM, Moore RL, Robbins J, Leinwand LA, Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy, J Clin Invest 104(4) (1999) 469–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Varnava AM, Elliott PM, Baboonian C, Davison F, Davies MJ, McKenna WJ, Hypertrophic cardiomyopathy: histopathological features of sudden death in cardiac troponin T disease, Circulation 104(12) (2001) 1380–4. [DOI] [PubMed] [Google Scholar]
- [43].Legato MJ, Ultrastructure of the atrial, ventricular, and Purkinje cell, with special reference to the genesis of arrhythmias, Circulation 47(1) (1973) 178–89. [DOI] [PubMed] [Google Scholar]
- [44].Sommer JR, Waugh RA, The ultrastructure of the mammalian cardiac muscle cell--with special emphasis on the tubular membrane systems. A review, Am J Pathol 82(1) (1976) 192–232. [PMC free article] [PubMed] [Google Scholar]
- [45].Page E, Tubular systems in Purkinje cells of the cat heart, J Ultrastruct Res 17(1) (1967) 72–83. [DOI] [PubMed] [Google Scholar]
- [46].Yekelchyk M, Guenther S, Preussner J, Braun T, Mono- and multi-nucleated ventricular cardiomyocytes constitute a transcriptionally homogenous cell population, Basic Res Cardiol 114(5) (2019) 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Li ZY, Wang YH, Maldonado C, Kupersmith J, Role of junctional zone cells between Purkinje fibres and ventricular muscle in arrhythmogenesis, Cardiovasc Res 28(8) (1994) 1277–84. [DOI] [PubMed] [Google Scholar]
- [48].Mendez C, Mueller WJ, Merideth J, Moe GK, Interaction of transmembrane potentials in canine Purkinje fibers and at Purkinje fiber-muscle junctions, Circ Res 24(3) (1969) 361–72. [DOI] [PubMed] [Google Scholar]
- [49].Gladka MM, Molenaar B, de Ruiter H, van der Elst S, Tsui H, Versteeg D, Lacraz GPA, Huibers MMH, van Oudenaarden A, van Rooij E, Single-Cell Sequencing of the Healthy and Diseased Heart Reveals Cytoskeleton-Associated Protein 4 as a New Modulator of Fibroblasts Activation, Circulation 138(2) (2018) 166–180. [DOI] [PubMed] [Google Scholar]
- [50].Vigil-Garcia M, Demkes CJ, Eding JEC, Versteeg D, de Ruiter H, Perini I, Kooijman L, Gladka MM, Asselbergs FW, Vink A, Harakalova M, Bossu A, van Veen TAB, Boogerd CJ, van Rooij E, Gene expression profiling of hypertrophic cardiomyocytes identifies new players in pathological remodelling, Cardiovasc Res 117(6) (2021) 1532–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gladka MM, Single-Cell RNA Sequencing of the Adult Mammalian Heart-State-of-the-Art and Future Perspectives, Curr Heart Fail Rep 18(2) (2021) 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.