

Abstract
Objective
The aim of the current study is to understand why some individuals avoid developing Parkinson disease (PD) despite being at relatively high genetic risk, using the largest datasets of individual‐level genetic data available.
Methods
We calculated polygenic risk score to identify controls and matched PD cases with the highest burden of genetic risk for PD in the discovery cohort (International Parkinson's Disease Genomics Consortium, 7,204 PD cases and 9,412 controls) and validation cohorts (Comprehensive Unbiased Risk Factor Assessment for Genetics and Environment in Parkinson's Disease, 8,968 cases and 7,598 controls; UK Biobank, 2,639 PD cases and 14,301 controls; Accelerating Medicines Partnership–Parkinson's Disease Initiative, 2,248 cases and 2,817 controls). A genome‐wide association study meta‐analysis was performed on these individuals to understand genetic variation associated with resistance to disease. We further constructed a polygenic resilience score, and performed multimarker analysis of genomic annotation (MAGMA) gene‐based analyses and functional enrichment analyses.
Results
A higher polygenic resilience score was associated with a lower risk for PD (β = −0.054, standard error [SE] = 0.022, p = 0.013). Although no single locus reached genome‐wide significance, MAGMA gene‐based analyses nominated TBCA as a putative gene. Furthermore, we estimated the narrow‐sense heritability associated with resilience to PD (h 2 = 0.081, SE = 0.035, p = 0.0003). Subsequent functional enrichment analysis highlighted histone methylation as a potential pathway harboring resilience alleles that could mitigate the effects of PD risk loci.
Interpretation
The present study represents a novel and comprehensive assessment of heritable genetic variation contributing to PD resistance. We show that a genetic resilience score can modify the penetrance of PD genetic risk factors and therefore protect individuals carrying a high‐risk genetic burden from developing PD. ANN NEUROL 2022;92:270–278
Parkinson disease (PD) is the second most common neurodegenerative disorder. 1 Although there are monogenic forms of PD, the majority of individuals with PD do not harbor pathogenic mutations and are often said to have "sporadic" PD. 2 Over the past decade, significant progress has been made in understanding the genetic architecture of sporadic PD by conducting genome‐wide association studies (GWASs). The latest PD GWAS meta‐analysis has robustly identified 90 independent risk signals that can explain between 16 and 36% of the heritable risk of PD in individuals of European ancestry. 3 The genetic risk burden, or "polygenic risk score" (PRS; ie, a weighted sum of the PD risk alleles an individual carries), has been shown to correlate with PD susceptibility, age at onset, and progression in independent cohorts. 4 , 5 , 6
In the PD field, PRS has been extensively applied in an effort to distinguish PD cases from controls. Optimized PRS analyses are able to differentiate disease status with 56.9% sensitivity and 63.2% specificity when estimated alone, and with 83.4% sensitivity and 90.3% specificity when the score is combined with family history, olfactory function, age, and gender. 7 Furthermore, PRS has been successfully applied to explore novel functional pathways in PD, 8 to study gene–environment interactions, 9 to estimate potential shared genetic etiologies, 10 and as a disease penetrance modifier in LRRK2 and GBA mutation carriers11, 12 all toward the implementation of personalized medicine. 13
In addition, polygenic scores and GWASs provide an opportunity to research genetic factors that confer resilience. In the context of genetics, resilience is defined as heritable variation that promotes resistance to disease by reducing the penetrance of risk loci. The first polygenic resilience score study on a complex genetic disorder has been recently published. 14 The authors found that a polygenic resilience score managed to differentiate high‐risk controls from equal‐risk schizophrenia cases. Furthermore, the Resilience Project by Chen et al found that 13 of 589,306 healthy adults were genetically resilient to highly penetrant forms of genetic childhood disorders. 15 Studies that focus on resilience genetic factors in both monogenic and polygenic forms of disease 16 , 17 are therefore crucial to shed light on disease mechanisms that may be more amenable to therapeutic intervention.
Resilience is not simply the inverse of risk, which refers to "protective variants" (ie, the alternate alleles at each risk‐associated locus that have a higher frequency in controls than in cases). 14 On the contrary, resilience alleles are thought to mitigate the effects of the risk loci and reduce the likelihood of the disorder in higher risk groups.
A priority in elucidating PD etiology lies in defining cumulative risk; however, very little is known about genetic factors that enhance resistance to PD development. Why some people avoid illness despite being at elevated risk remains unexplored in the field. The current study aims to explore the genetic architecture of resilience in PD. Here, we conduct the first GWAS of resilience to polygenic PD risk and construct a polygenic resilience score that could decrease susceptibility to PD risk variants. Finally, we explore functional enrichment of resilient variants by performing pathway analyses and expression enrichment across tissues and cell types.
Subjects and Methods
Demographic and Cohort Characteristics, Quality Control Procedures, and Study Design
Figure 1 summarizes the workflow and data used in this project. To assess PD risk, we obtained summary statistics defining risk allele weights from Chang et al's PD GWAS meta‐analysis, 18 including 26,035 PD cases and 403,190 controls of European ancestry. There were 7,909,453 imputed single nucleotide polymorphisms (SNPs) tested for association with PD in this study. Individual‐level genotyping data not included in Chang et al and from the International Parkinson's Disease Genomics Consortium (IPDGC) 3 were used as a discovery cohort containing 7,204 PD cases and 9,412 controls, all unrelated and of European ancestry (Supplementary Table 1). Additional details of the IPDGC cohort and detailed quality control (QC) and imputation methods are further described elsewhere. 3
FIGURE 1.
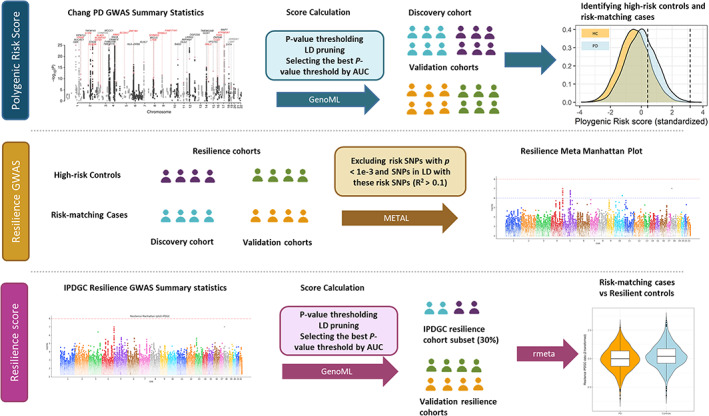
Workflow and rationale summary. AUC = area under curve; GWAS = genome‐wide association study; HC = healthy control; IPDGC = International Parkinson's Disease Genomics Consortium; LD = linkage disequilibrium; PD = Parkinson disease; SNP = single nucleotide polymorphism.
Imputed individual‐level genotyping data from the Comprehensive Unbiased Risk Factor Assessment for Genetics and Environment in Parkinson's Disease (COURAGE‐PD) and UK Biobank (UKBB), and whole genome sequencing data from the Accelerating Medicines Partnership–Parkinson's Disease Initiative (AMP‐PD; v2.5; www.amp-pd.org) were used as validation cohorts. All participants were of European ancestry. The COURAGE‐PD cohort consists of 8,968 cases and 7,598 controls genotyped on Neurochip. 19 Demographic and clinical characteristics of COURAGE‐PD are described in Supplementary Table 2. Imputation was performed in a cloned Michigan Imputation Server (MIS) (https://193.196.20.138:8080/) at the deNBI cloud (https://denbi.uni-tuebingen.de/). The Haplotype Reference Consortium Release 1.1 (HRC) data usage request was approved by the Sanger Institute (dataset ID: EGAD00001002729). The datasets were prepared in accordance with the reference panel criteria for the MIS (https://imputationserver.sph.umich.edu/index.html). The HRC/1000G imputation preparation and checking tool (https://www.well.ox.ac.uk) was used to check for Ref/Alt allele assignments, incorrect strands, deviation from allele frequency, and palindromic SNPs. Later, post‐QC data were phased using Eagle v2.4 in our MIS. Imputation of autosomal variants was performed separately for each dataset using the HRC reference panel and the GRCh37/hg19 assembly with an R 2 filter of 0.3. Finally, imputed data were hard‐called using R 2 of 0.8 on PLINK 2.0. 20
The UKBB cohort was composed of 2,639 unrelated PD cases and 14,301 unrelated controls (UKBB data v2) with recruitment age > 60 years and without medical history of neurological diseases (PD field code: 131023; European ethnic grouping field code: 22006). Additional details on this cohort, along with QC procedures, are described in Bycroft et al. 21 Finally, the AMP‐PD dataset (v2) was composed of 2,248 cases and 2,817 controls of European ancestry, unrelated and without any PD known genetic cause, with an average age at onset of 61.3 years in cases and an average age of 69.3 years in controls (Supplementary Table 3). Additional and detailed cohort characteristics, as well as QC methods, can be found at https://amp-pd.org/whole-genome-data.
PRS Calculation to Identify High‐Risk Resilient Controls and Risk‐Matching PD Cases
PRS analyses required 2 key input data sets: (1) reference data—published GWAS summary statistics including variants and effect sizes, for which Chang et al 18 PD data were used; and (2) target data—genotyping, imputed individual‐level data, for which nonoverlapping IPDGC, UKBB, AMP‐PD, and COURAGE‐PD cohorts were used. The reference and target datasets used were independent from each other because sample overlap could cause substantial inflation between PRS and disease status association in the target dataset. Because the IPDGC and UKBB cohorts are included in the Nalls et al3 summary statistics, we used the Chang GWAS summary statistics to avoid spurious results.
We applied supervised machine learning to select the p value threshold of independent variants that best predicted PD risk in the IPDGC dataset. We used GenoML, an open‐source python package that automates machine learning workflows for genomics (https://genoml.com/). Source code and documentation are available at https://genoml.com/ and on GitHub (https://github.com/GenoML/genoml2). The process we used for selecting the best p value threshold mirrored that in Makarious et al. 22 We ran the discrete supervised learning workflow for munging, followed by training on a series of p value thresholds taken from Chang et al 18 PD GWAS summary statistics, which included each incremental order of magnitude ranging from 0.01 to 1 × 10−8. Each model included SNPs as well as sex, age, and 20 principal components (PCs) to account for population stratification. In the following paragraph, we summarize the GenoML workflow carried out to establish our optimal p value threshold.
For each p value threshold, we first performed data munging that included feature selection via extraTreesClassifier (up to 500 trees), linkage disequilibrium (LD) pruning (R 2 > 0.1 within 1MB sliding windows), and normalization (z‐scaling of features including sex, age and PCs). Feature selection was performed using the extremely randomized trees classifier algorithm (extraTrees) on combined data modalities to remove redundant feature contributions that could overfit the model, and to optimize the information content from the features and limit artificial inflation in predictive accuracy that might be introduced by including such a large number of features before filtering. By removing redundant features using correlation‐based pruning and an extraTrees classifier as a data‐munging step, the potential for overfitting is limited, and it also makes models that are likely to be more conservative and generalizable.
We then completed all available algorithms in the package, which we trained on 70% of the samples and tested on the randomly selected remaining 30% of the sample (under default settings). Briefly, the GenoML workflow consists of the top dozen algorithms stemming from standard linear models used in genetic prediction analyses, employing tree‐based methods (boosting), kernel‐based methods (k‐nearest neighbors, support vectors, discriminant analysis, and random forests), and deep learning (perceptron and gradient descent). For each p value threshold, we selected the model that produced the highest area under curve (AUC) and then compared across p value thresholded models. The p value threshold with the highest AUC was set at 1e‐3. We selected this threshold for our PRS construction that followed. A total of 1,060 variants were used to construct PRS using the 1e‐3 p value threshold. The Chang GWAS only identified 41 significant loci of the current 90 PD risk regions. This model included variants nominated by GenoML with p < 1e‐3 to identify high‐risk cases and equally high‐risk controls through PRS analyses capturing current risk loci considered subtop hits in the Chang et al study. PRS was computed using PLINK v1.9 20 and was standardized using z‐score scaling. A logistic regression model, adjusted by age, sex, and 10 PCs, was used to examine the correlation between PRS and PD status. We then ranked subjects by PRS and categorized the controls that had a PRS above the 75th percentile as "PD resilient." 14 PD cases whose PRS was between the 75th percentile and the maximum PRS for controls were retained as the comparison group. This method detected 2,353 high‐risk resilient controls and 3,011 risk‐matching cases in the IPDGC cohort (Supplementary Table 4).
PD Resilience GWAS and Polygenic Resilience Score Calculation
Discovery Phase Analyses
To avoid potential bias affecting our analyses, we excluded SNPs with a p ≤ 1e‐3 in Chang et al18 summary statistics in addition to variants in LD with these SNPs at R 2 > 0.1. This step avoided rediscovering risk variants, ensuring that any resilience genetic variants derived from our analysis were independent from known risk loci. We also excluded variants in the major histocompatibility complex region (hg19, chr6:28477797–33,448,354), due to inter‐region variability and extensive LD. A minor allele frequency threshold of 0.05 was applied to further filter the inclusion of variants due to power concerns. A GWAS for PD resilience was conducted including 2,353 high‐risk resilient controls and 3,011 equal‐risk cases generated from the IPDGC cohort using PLINK v2.0. 20
To calculate a polygenic resilience score, we randomly split the 2,353 high‐risk resilient controls and 3,011 equal‐risk cases in a 70–30% ratio. The GenoML pipeline described above was applied to select the best p value threshold of independent variants predicting resilience. A total of 239 variants with a p value < 1e‐3 were used to construct a polygenic resilience profile, which was calculated using the “‐‐score” function in PLINK v1.9. Risk allele dosages were counted (giving a dose of 2 if homozygous for the risk allele, 1 if heterozygous, and 0 if homozygous for the reference allele). All SNPs were weighted by the log odds ratios obtained from the resilience GWAS using the 70% data subset, with a greater weight given to alleles with higher risk estimates. Polygenic resilience scores were converted to z scores for easier interpretation. A logistic regression model was used to explore the resilience scores and resilience status after adjusting for age, sex, and 10 PCs. For easier interpretation, beta values are reported relating to an increasing dosage of alleles conferring resilience to PD (meaning that as the resilience score increases, the risk of PD decreases).
Finally, Pearson correlation coefficient was applied to explore the linear correlation between risk and resilience scores in four separate groups: (1) PD cases and controls, (2) resilient controls and risk‐matching cases.
Replication Phase and Meta‐Analysis
PRSs were calculated in the validation cohorts (COURAGE‐PD, UKBB, and AMP‐PD) using weights derived from the Chang et al GWAS summary statistics and mimicking the pipeline used in the discovery phase (IPDGC dataset). A logistic regression model, adjusted by age, sex, and 10 PCs, was used to examine the association between PRS and PD status within each cohort. We applied the 75th percentile threshold method to identify resilient controls and equal‐risk cases.
A resilience GWAS was conducted on these 3 validation cohorts following the same criteria described above. We then meta‐analyzed GWAS summary statistics from all 4 cohorts (IPDGC, COURAGE‐PD, UKBB, and AMP‐PD) using the METAL package. 23 Briefly, results from the 4 GWAS analyses were combined in a fixed‐effect meta‐analysis to obtain the overall effects. Resilience scores were calculated in the validation cohorts using weights derived from the IPDGC resilience GWAS conducted in 70% of the data. A logistic regression model was used to explore the resilience scores and resilience status after adjusting for age, sex, and 10 PCs within each cohort. Similarly, we then performed a fixed‐effect meta‐analysis with the R package rmeta using effect sizes and standard errors for resilience scores obtained from the 4 cohorts to evaluate the aggregate predictive capacity of the resilience scores.
Heritability Analyses
SNP‐based heritability estimates associated with resilience to PD were calculated using LD score regression. 24 This approach involves running regression analyses to examine the relationship between LD scores and the test statistics of the SNPs from the GWAS. Here, the LD score for an SNP is the sum of LD R 2 measured with all other SNPs.
Functional Enrichment of Resilience SNPs
To conduct a gene‐based GWAS and assess expression enrichment across tissues and cell types, we uploaded meta‐GWAS summary statistics to the Functional Mapping and Annotation of Genome Wide Association Studies (FUMA) webserver (https://fuma.ctglab.nl/). Gene‐based GWAS was computed by multimarker analysis of genomic annotation (MAGMA) implemented in FUMA. In the MAGMA gene‐based GWAS, SNPs are mapped to 16,956 protein‐coding genes, and the resulting SNP p values are combined into a gene test statistic using the SNP‐wise mean model. MAGMA gene set pathway analyses using the full distribution of SNP p values were performed for curated gene sets and Gene Ontology terms obtained from MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/). To determine the tissues and cell types most relevant to PD resilience, summary statistics were analyzed using MAGMA gene property tests to compare enrichment of the average gene expression per tissue using GTEx v8 (54 tissues; https://gtexportal.org). Bonferroni correction was performed for all tested gene sets. In addition, single‐cell RNA sequencing data from the Dropviz 25 dataset (spanning 88 possible tissues and cell type combinations) were queried for cell enrichment analyses using FUMA.
Results
PRS Identifies High‐Risk Resilient Controls and Matched‐Risk PD Cases
Using the largest datasets of individual‐level genetic data available for PD to date, and in concordance with previous PD genetic studies, PRS could significantly detect an association with PD status in all 4 tested cohorts (Supplementary Fig 1). The regression model indicated that a higher PRS per standard deviation of genetic risk was significantly associated with PD risk in the IPDGC (1,060 variants, β = 0.354, standard error [SE] = 0.020, p = 4.19e‐70), COURAGE‐PD (1,034 variants, β = 0.240, SE = 0.017, p = 2.95e‐45), AMP‐PD (977 variants, β = 0.283, SE = 0.030, p = 3.97e‐21), and UKBB cohorts (802 variants, β = 0.215, SE = 0.022, p = 1.47e‐22).
These analyses enabled the identification of high‐risk controls and risk‐matching cases. We detected 2,353 resilient controls and 3,011 risk‐matched cases in IPDGC, 1,900 resilient controls and 3,102 cases in COURAGE‐PD, 3,576 resilient controls and 847 cases in UKBB, and 705 resilient controls and 798 cases in AMP‐PD data (Supplementary Table 4).
GWAS and Meta‐Analysis of Resilience in PD Provide Insights into the Genetic Architecture of Resistance to Disease
A GWAS approach followed by meta‐analysis was implemented to explore genetic variants associated with resilience to PD in high‐risk controls and equally high‐risk cases (λIPDGC = 1.03, λCOURAGE−PD = 1.01, λUKBB = 1.02, λAMP−PD = 1.01). We performed a fixed‐effect meta‐analysis of GWAS summary statistics from all 4 cohorts. The resilience meta‐GWAS included a total of 8,534 resilient controls and 7,758 risk‐matching cases. We compared the genome‐wide resilience p values with an expected (ie, null) distribution of p values, revealing that the observed values fit closely with expected values as shown in the quantile–quantile (Q‐Q) plot (λmeta = 1.065; Supplementary Fig 2).
We performed power calculations using the GAS power calculator 26 at our meta‐analysis sample size to confirm we could achieve >90% statistical power at genome‐wide significance for a variant with a minor allele frequency of 20%, a genotype relative risk of 1.2, and a disease prevalence of 0.5%. When using a more stringent threshold (10% of the data), our ability to reach genome‐wide significance drops to 17% of statistical power (Supplementary Fig 3).
No significant loci were found to be linked with resilience to PD at a genome‐wide level (Fig 2). However, our analyses nominated 4 independent subtop resilience signals that require further validation at p < 1.0 × 10−6 (Supplementary Table 5). We observed suggestive associations with rs62325099 (β = −0.328, SE = 0.061, p = 1.03e‐07) near LINC01262, rs2652202 (β = 0.127, SE = 0.024, p = 1.64e‐07) near TBCA, rs12245509 (β = −0.248, SE = 0.049, p = 5.21e‐07) near LINC01375, and rs292289 (β = −0.301, SE = 0.056, p = 9.85e‐08) near C18orf42. The effect sizes of the 4 subtop SNPs on PD risk in the Nalls et al GWAS 3 are also shown in Supplementary Table 5. Expanding future studies will identify new loci and improve the AUC for a genetic predictor of resilience to PD.
FIGURE 2.
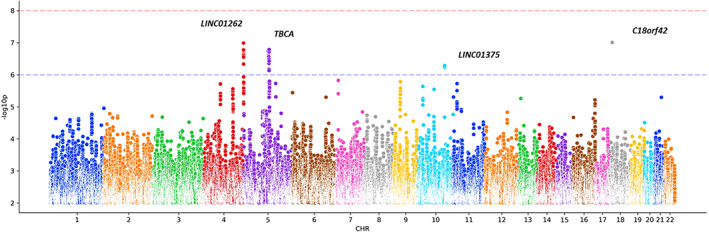
Manhattan plot showing genome‐wide association results conferring resilience to Parkinson disease. CHR = Chromosome.
Narrow‐sense heritability analyses revealed that the proportion of resilience to PD explained by genetic factors was 9.6% (h 2 = 0.081, SE = 0.035, p = 0.0003).
Gene‐Based MAGMA Analyses Nominate TBCA as a Potential Gene Involved in Resilience to PD
After performing MAGMA annotation and gene mapping, we conducted a gene‐based association analysis using all SNPs in the GWAS. Our results nominated TBCA (top lead SNP: rs2652202, p = 1.64e‐07) as significantly associated with PD resilience (FUMA Bonferroni adjusted p value, 0.05/16,956 = 2.95e‐6; Supplementary Fig 4A and B). The Q‐Q plot for the gene‐based GWAS is shown in Supplementary Figure 4C.
Polygenic Resilience Score Modulates the Effect of PD Genetic Risk Factors
The associations between resilience scores and PD status per high‐risk cohort are represented in Figure 3. The regression model indicated that a lower resilience score was significantly associated with PD risk in the IPDGC (239 variants, β = −0.097, SE = 0.052, p = 0.060) and COURAGE‐PD cohorts (227 variants, β = −0.080, SE = 0.031, p = 0.011). We were not able to identify a significant association in the AMP‐PD (216 variants, β = 0.002, SE = 0.060, p = 0.979) and UKBB (232 variants, β = 0.010, SE = 0.048, p = 0.838) cohorts, likely due to the limited sample size of the resilient controls and equally high‐risk cases (Supplementary Fig 5). Meta‐analysis results of the 4 cohorts showed that a higher resilience score was associated with a lower risk of PD (β = −0.054, SE = 0.022, p = 0.013, I 2 = 0.262; see Fig 3).
FIGURE 3.
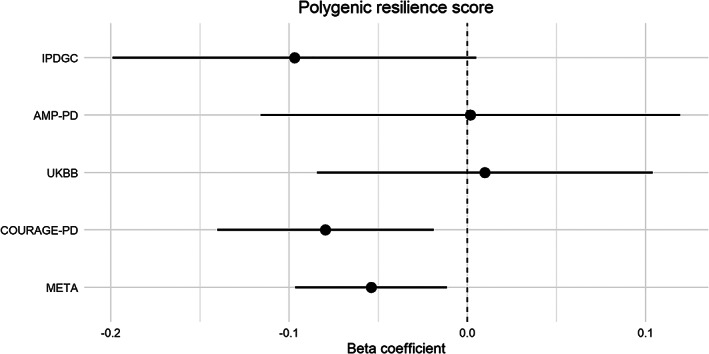
Forest plot depicting the effect (β coefficient) of polygenic resilience score on Parkinson disease risk (95% confidence interval) across cohorts. AMP‐PD = Accelerating Medicines Partnership–Parkinson's Disease Initiative; COURAGE‐PD = Comprehensive Unbiased Risk Factor Assessment for Genetics and Environment in Parkinson's Disease; IPDGC = International Parkinson's Disease Genomics Consortium; UKBB = UK Biobank.
In the full IPDGC cohort, risk and resilience scores were positively correlated in controls (Pearson r = 0.132, 95% confidence interval [CI] = 0.111–0.151, p = 2.2e‐16) and negatively correlated in PD cases (Pearson r = −0.092, 95% CI = −0.115 to −0.069, p = 2.2e‐16; Supplementary Fig 6). However, risk and resilience scores were not correlated in high‐risk controls and cases in the IPDGC cohort and not correlated in all groups within validation cohorts (Supplementary Table 6).
Functional Enrichment Pathway Analyses Highlight Molecular Processes Harboring Resilience Alleles
Based on MAGMA gene set pathway analyses using GWAS summary statistics, we identified one significant enriched pathway (GO_bp: go_histone_h3_k9_dimethylation, β = 0.024, SE = 0.292, Bonferroni adjusted p = 0.021). The top 10 biological pathways are shown in Supplementary Table 7. Results of FUMA analysis for tissue and cell type‐specific expression enrichment are shown in Supplementary Figures 7 and 8. We did not find any significant tissues and cell types associated with resilience to PD after false discovery rate correction. The top 10 nominated tissues are brain‐derived, and the top 5 nominated brain cell types are neuronal.
Code Availability
Analysis code is available at https://github.com/ipdgc/IPDGC-Trainees/blob/master/RESILIENCE.md.
Discussion
Despite success at unraveling genetic risk factors associated with PD, our understanding of the heritable variation that promotes resistance to PD risk is widely unknown. Using the largest genetic PD cohorts available to date, we aimed to explore the genetic architecture of resilience to PD with the goal of studying genetic variation that helps unaffected individuals cope with a relatively large genetic burden of disease‐associated variants. To our knowledge, there are no previous reports in the PD field where a similar approach has been implemented.
We performed a meta‐analysis of GWASs including 4 datasets of European ancestry totaling 7,758 cases and 8,534 controls. Although no variants reached genome‐wide significance, we observed 4 subtop loci: TBCA, LINC01262, LINC01375, and C18orf42. Gene‐based GWAS analyses also highlighted TBCA as a potential gene involved in the resilience to PD. TBCA is thought to play a relevant role in modulating the stability and polymerization of microtubules. Substantial evidence supports the view that altered microtubule dynamics underlies or contributes to neurodegenerative disorders. Expression of the tubulin chaperone TBCA has been found to be altered in PD dementia patients, suggesting that defects in synaptic transmission and axonal function are early events in the pathogenesis of PD. 27 Interestingly, two long intergenic non‐protein coding RNA genes (lincRNAs; LINC01262 and LINC01375) were nominated as potential subtop hits. Recent studies have shown that lincRNAs might alter the expression of PD‐linked genes, such as PINK1, LRRK2, and SNCA. 28 Future studies are needed to explore the regulatory role of these two lincRNAs in PD. In addition to these two loci, C18orf42 encodes a protein kinase A (PKA) binding protein and is expressed preferentially in neural tissues. 29 It has been shown that the loss of PKA signaling regulates mitochondrial function and neuronal development, contributing to the etiology of PD. 30
A novel aspect of this study is that a lower polygenic resilience score constructed from the resilience GWAS (conducted in 70% of the IPDGC data) was significantly associated with PD among high‐risk individuals and that this is a cumulative protective score specific to samples in the highest quartile of generalized genetic risk from previous publications. The current study suggests that polygenic resilience score modifies the risk of PD in the top quartile of individuals carrying the highest burden of known genetic risk factors. Our results show a significant positive correlation between risk and resilience scores in the discovery IPDGC control cohort, which validates the notion that, as risk score increases, so too must the resilience score for an at‐risk individual to remain unaffected. In concordance, we observe a negative correlation between risk and resilience scores in the discovery IPDGC cases cohort. We assume the limitation that no significant correlations between risk and resilience scores were observed in the replication cohorts and the high‐risk subset of cases or controls within IPDGC, probably due to a lack of statistical power. In the PD field, extensive research has been done in terms of risk, but few studies have focused on resilience. A study conducted by Iwaki et al 11 found that lower PRS constructed from 89 PD risk variants was associated with a lower penetrance of disease in LRRK2 G2019S carriers, especially in younger individuals. In a similar context, Blauwendraat et al 12 found that PRS modifies risk for disease and reduces age at onset in GBA carriers. Although we would have liked to further explore LRRK2, GBA, and additional PD known risk loci in the context of resilience, the sample size required to draw meaningful conclusions in carriers versus noncarriers limited this genome‐wide power‐hungry approach. We were not able to assess resilience in specific carriers of LRRK2 and GBA mutations. Exploring resilience to PD in LRRK2 and GBA carriers through gene–gene interaction analyses, where the penetrance of a risk variant could change based on the effect of a resilience variant, presents an excellent opportunity to understand the complexity of disease, which in turn is crucial to developing predictive and preventive approaches. We encourage other researchers to expand on this pilot study. Additionally, although the current study sought to explore resilience from a mere genetics perspective, it should be pointed out that disease penetrance can be largely affected by environmental factors, which we did not account for. Further studies focused on analyzing gene–environment interactions and their role in resilience are warranted. It is hoped that large‐scale, collaborative, and multicenter research will help plug current knowledge gaps in the near future. Altogether, identification of factors influencing the penetrance of disease in high‐risk burden carriers could be relevant to identify protective mechanisms against illness. Interestingly, our heritability estimates revealed a substantial contribution of genetic factors to the genetic architecture of resilience to PD. In the context of neurodegenerative diseases, Dumitrescu et al have recently reported that the narrow‐sense heritability of resilience in Alzheimer disease (AD) ranges between 19 and 67%. 16 Notably, the authors highlight a putative role of vascular risk, metabolism, and mental health in protection from the cognitive consequences of neuropathology in AD.
Overall, our genome‐wide enrichment pathway analysis implicated the histone h3‐k9 dimethylation (H3K9me2) pathway in the resilience to PD. Interestingly, histone methylation is a crucial epigenetic mechanism regulating gene expression. Sugeno et al 31 reported that overexpression of α‐synuclein in transgenic drosophila and in inducible human neuroblastoma SH‐SY5Y cells led to enhanced histone H3K9me2, which eventually impaired synaptic activity. Histone methylation H3K9me2 is also significantly elevated in the prefrontal cortex and hippocampus of late stage familial AD mice, which links to the epigenetic regulation of reduced glutamate receptor transcription. 32 Interestingly, Belzil et al 33 found that reduced C9orf72 mRNA levels in amyotrophic lateral sclerosis and frontotemporal dementia patients was caused by histone H3K9me3. Future studies are warranted to investigate how specific histone methylation mechanisms regulate synaptic and other pathophysiological changes identified in PD patients. Although we did not find any significant enrichment for tissues or cell types associated with resilience, likely due to limited sample size, our analyses suggest the possibility of resilience alleles being enriched for expression in brain and neuronal cell types known to be involved in disease etiology. 3
Finally, although this is the most comprehensive genetic analysis of resilience in PD, some limitations should be acknowledged in this work. Although the largest available individual level PD genetics cohorts were explored, the sample size of high‐risk individuals was still limited, and we remained underpowered to detect genome‐wide single variant effects. We defined individuals with PRS above the 75th percentile to the maximum of the control group as high‐risk individuals. Future work including larger sample sizes containing non‐European individuals, and stricter cutoff (90th percentile to the maximum) are needed to further delineate PD resilience. Additionally, we are aware of the limitation that the current study only focused on European individuals. The genetic architecture of resilience in PD should further be explored in ancestrally diverse populations. Prior to running the machine learning model, PC analyses were conducted similarly among the 3 datasets, where population outliers deviating 6 standard deviations from the population mean for European ancestry were removed. In addition, all SNP minor allele dosages were adjusted for PCs 1–10 to account for population substructure, allowing the model to be built using genotype dosages adjusted for population substructure. However, we cannot predict how this model might perform on other populations, and future analyses should be conducted as new data in different populations become available. Our approach was designed to identify resilience SNPs that are LD‐independent of risk SNPs based on liberal definitions of risk (p < 0.001) and of LD (R 2 < 0.1 with a risk‐conferring variant) so that we avoided detecting additional risk SNPs. We assume the limitation that biologically, it is expected that resilience SNPs can reduce the penetrance of nearby risk SNPs, even those within the same gene or LD block. Future conditional association analysis in much larger datasets could be an accurate approach to test whether resilience signals are more likely to colocalize with loci harboring risk variants. In our study, we only explored resilience variants that can confer resistance to disease. We encourage other researchers to study how resilience variants may affect the age at onset or disease progression.
In conclusion, the present study represents a step forward in understanding genetic factors contributing to PD resistance. We performed the first GWAS of PD resilience and conducted comprehensive follow‐up analyses highlighting novel pathways contributing to PD resilience. We showed that our resilience score can modify the penetrance of known and unknown PD genetic risk factors and therefore protect individuals carrying a high‐risk genetic burden from developing PD. Here, we present a pipeline that can serve as a foundational publicly available resource for continued investigation of a crucial scientific question as new data are generated.
Author Contributions
H.Li. and S.B.‐C. contributed to the conception and design of the study. H.Li., S.B.‐C., M.D., C.B., M.B.M., H.Le., J.J.K., C.S., M.S., M.N., A.S., and T.G. contributed to the acquisition and analysis of the data. H.Li., S.B.‐C., M.D., C.B., M.B.M., A.N., B.M.J., I.F., M.S., M.N., A.S., and T.G. contributed to drafting a significant portion of the manuscript or figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Supplementary Table 1. Demographic and clinical characteristics of the IPDGC cohort.
APPENDIX S1. Supporting Information
Acknowledgments
This research was supported by the NIH Intramural Research Program (National Institute on Aging, National Institute of Neurological Disorders and Stroke; project numbers 1ZIA‐NS003154, Z01‐AG000949‐02, and Z01‐ES10198).
We would like to thank all of the subjects who donated their time and biological samples to be a part of this study. We also would like to thank all members of the IPDGC. For a complete overview of members, acknowledgments, and funding, please see http://pdgenetics.org/partners. We would also like to thank the COURAGE‐PD Consortium and the AMP‐PD (see Supplementary Information for additional details).
Contributor Information
Thomas Gasser, Email: thomas.gasser@uni-tuebingen.de.
Sara Bandres‐Ciga, Email: sarabandres@gmail.com.
International Parkinson's Disease Genomics Consortium; Comprehensive Unbiased Risk Factor Assessment for Genetics and Environment in Parkinson's Disease Consortium:
Sulev Koks, George D Mellick, Walter Pirker, Alexander Zimprich, Anthony E Lang, Ekaterina Rogaeva, Pille Taba, Alexis Brice, Marie‐Christine Chartier‐Harlin, Jean‐Christophe Corvol, Cloé Domenighetti, Alexis Elbaz, Suzanne Lesage, Eugenie Mutez, Pierre‐Emmanuel Sugier, Ashwin Ashok Kumar Sreelatha, Sandeep Grover, Kathrin Brockmann, Angela B Deutschländer, Thomas Gasser, Jens Krüger, Peter Lichtner, Milena Radivojkov‐Blagojevic, Claudia Schulte, Manu Sharma, Efthimos Dardiotis, Georges M Hadjigeorgiou, Athina Maria Simitsi, Leonidas Stefanis, Grazia Annesi, Laura Brighina, Carlo Ferrarese, Simona Petrucci, Gianni Pezzoli, Andrea Quattrone, Letizia Straniero, Monica Gagliardi, Enza Maria Valente, Anna Zecchinelli, Nobutaka Hattori, Akiyoshi Nakayama, Hirotaka Matsuo, Kenya Nishioka, Dheeraj Bobbili, Rejko Kruger, Zied Landoulsi, Patrick May, Lukas Pavelka, Bastiaan R Bloem, Bart PC van de Warrenburg, Jan Aasly, Lasse Pihlstrøm, Mathias Toft, Joaquim J Ferreira, Leonor Correia Guedes, Soraya Bardien, Jonathan Carr, Sun Ju Chung, Yun Joong Kim, Monica Diez‐Fairen, Mario Ezquerra, Pau Pastor, Eduardo Tolosa, Andrea C Belin, Nancy L Pedersen, Andreas Puschmann, Caroline Ran, Emil Y Rödström, Karin Wirdefeldt, Carl E Clarke, Karen E Morrison, Manuela Tan, Connor Edsall, Matt J Farrer, Dimitri Krainc, Andrew B Singleton, Lena F Burbulla, and Dena G Hernandez
References
- 1. Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nat Rev Dis Primer 2017;3:1–21. [DOI] [PubMed] [Google Scholar]
- 2. Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 2009;18:R48–R59. [DOI] [PubMed] [Google Scholar]
- 3. Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta‐analysis of genome‐wide association studies. Lancet Neurol 2019;18:1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nalls MA, Escott‐Price V, Williams NM, et al. Genetic risk and age in Parkinson's disease: continuum not stratum. Mov Disord Soc 2015;30:850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Paul KC, Schulz J, Bronstein JM, et al. Association of polygenic risk score with cognitive decline and motor progression in Parkinson disease. JAMA Neurol 2018;75:360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Duncan L, Shen H, Gelaye B, et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat Commun 2019;10:3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nalls MA, McLean CY, Rick J, et al. Diagnosis of Parkinson's disease on the basis of clinical and genetic classification: a population‐based modelling study. Lancet Neurol 2015;14:1002–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bandres‐Ciga S, Saez‐Atienzar S, Kim JJ, et al. Large‐scale pathway specific polygenic risk and transcriptomic community network analysis identifies novel functional pathways in Parkinson disease. Acta Neuropathol 2020;140:341–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jacobs BM, Belete D, Bestwick J, et al. Parkinson's disease determinants, prediction and gene‐environment interactions in the UK Biobank. J Neurol Neurosurg Psychiatry 2020;91:1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chia R, Sabir MS, Bandres‐Ciga S, et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat Genet 2021;53:294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iwaki H, Blauwendraat C, Makarious MB, et al. Penetrance of Parkinson's disease in LRRK2 p.G2019S carriers is modified by a polygenic risk score. Mov Disord 2020;35:774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blauwendraat C, Reed X, Krohn L, et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson's disease and Lewy body dementia. Brain 2020;143:234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reed X, Schumacher‐Schuh A, Hu J, Bandres‐Ciga S. Advancing personalized medicine in common forms of Parkinson's disease through genetics: current therapeutics and the future of individualized management. J Pers Med 2021;11:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hess JL, Tylee DS, Mattheisen M, et al. A polygenic resilience score moderates the genetic risk for schizophrenia. Mol Psychiatry 2021;26:800–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen R, Shi L, Hakenberg J, et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotechnol 2016;34:531–538. [DOI] [PubMed] [Google Scholar]
- 16. Dumitrescu L, Mahoney ER, Mukherjee S, et al. Genetic variants and functional pathways associated with resilience to Alzheimer's disease. Brain 2020;143:2561–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khera AV, Chaffin M, Aragam KG, et al. Genome‐wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chang D, Nalls MA, Hallgrímsdóttir IB, et al. A meta‐analysis of genome‐wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet 2017;49:1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blauwendraat C, Faghri F, Pihlstrom L, et al. NeuroChip, an updated version of the NeuroX genotyping platform to rapidly screen for variants associated with neurological diseases. Neurobiol Aging 2017;57:247.e9–247.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chang CC, Chow CC, Tellier LC, et al. Second‐generation PLINK: rising to the challenge of larger and richer datasets. GigaSci 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Makarious MB, Leonard HL, Vitale D, et al. Multi‐modality machine learning predicting Parkinson's disease. NPJ Parkinsons Dis 2022;8:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinforma Oxf Engl 2010;26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bulik‐Sullivan BK, Loh P‐R, Finucane HK, et al. LD score regression distinguishes confounding from polygenicity in genome‐wide association studies. Nat Genet 2015;47:291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saunders A, Macosko EZ, Wysoker A, et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 2018;174:1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnson JL, Abecasis GR. GAS power calculator: web‐based power calculator for genetic association studies. bioRxiv 2017; 10.1101/164343 [preprint]. https://www.biorxiv.org/content/10.1101/164343v1 [Google Scholar]
- 27. Doi S, Fujioka N, Ohtsuka S, et al. Regulation of the tubulin polymerization‐promoting protein by Ca2+/S100 proteins. Cell Calcium 2021;96:102404. [DOI] [PubMed] [Google Scholar]
- 28. Elkouris M, Kouroupi G, Vourvoukelis A, et al. Long non‐coding RNAs associated with neurodegeneration‐linked genes are reduced in Parkinson's disease patients. Front Cell Neurosci 2019;13:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fukuda M, Aizawa Y. Hypothetical gene C18orf42 encodes a novel protein kinase A‐binding protein. Genes Cells 2015;20:267–280. [DOI] [PubMed] [Google Scholar]
- 30. Dagda RK, Das BT. Role of protein kinase a in regulating mitochondrial function and neuronal development: implications to neurodegenerative diseases. Rev Neurosci 2015;26:359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sugeno N, Jäckel S, Voigt A, et al. α‐Synuclein enhances histone H3 lysine‐9 dimethylation and H3K9me2‐dependent transcriptional responses. Sci Rep 2016;6:36328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng Y, Liu A, Wang Z‐J, et al. Inhibition of EHMT1/2 rescues synaptic and cognitive functions for Alzheimer's disease. Brain J Neurol 2019;142:787–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Belzil VV, Bauer PO, Prudencio M, et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol 2013;126:895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Demographic and clinical characteristics of the IPDGC cohort.
APPENDIX S1. Supporting Information