

Abstract
Genome-wide unbiased identification of double stranded breaks enabled by sequencing (GUIDE-seq) is a sensitive, unbiased, genome-wide method for defining the activity of genome editing nucleases in living cells. GUIDE-seq is based on the principle of efficient integration of an end-protected double-stranded oligodeoxynucleotide tag into sites of nuclease-induced DNA double stranded breaks, followed by amplification of tag-containing genomic DNA molecules and high-throughput sequencing. Here we describe a detailed GUIDE-seq protocol including cell transfection, library preparation, sequencing, and bioinformatic analysis. The entire protocol including cell culture can be completed in 9 days. Once tag-integrated genomic DNA is isolated, library preparation, sequencing and analysis can be performed in 3 days. The result is a genome-wide catalogue of off-target sites ranked by nuclease activity as measured by GUIDE-seq read counts. GUIDE-seq is one of the most sensitive cell-based methods for defining genome-wide off-target activity and has been broadly adopted for research and therapeutic use.
Keywords: genome editing, gene editing, off-target, CRISPR-Cas, specificity, engineered nucleases
INTRODUCTION
Genome editing nucleases are a widely used technology for modifying the genome of living cells with many important applications in biology and medicine. There are four major classes of engineered nuclease: zinc fingers nucleases (ZFNs), meganucleases, transcription activator-like effector nucleases (TALENs) and CRISPR-Cas nucleases1. In particular, CRISPR-Cas nucleases have been broadly adopted, due to the ease with which they can be programmed to target new sites simply by providing a new guide RNA (gRNA)2.
In mammalian cells, engineered nuclease-induced DNA double-stranded breaks (DSBs) are commonly repaired by competing endogenous cellular DNA repair pathways: error-prone nonhomologous end-joining (NHEJ) or relatively precise homology-directed repair (HDR). NHEJ is often utilized to introduce indel mutations for gene disruption while HDR can mediate precise gene correction.
Genome editing nucleases can have unintended genome-wide ‘off-target’ activity in a wide variety of cell types and organisms3–5. For basic research, one concern about off-target activity is that they may confound the interpretation of genome editing experiments. For clinical applications, there is a risk that off-target mutations in functional genomic regions may result in cells with increased proliferative capacity and oncogenic potential6. Thus, sensitive, unbiased, genome-wide methods are critical for defining the global activity of genome editors.
Development of GUIDE-seq.
We developed GUIDE-seq based on our initial discovery that blunt, phosphorylated, and end-protected double-stranded oligodeoxynucleotides (dsODN) tags could be efficiently integrated into sites of nuclease-induced DSBs. We found that terminal phosphorothioate linkages were key for achieving high-frequency tag integration presumably by resisting cellular exonuclease activity7. 3’-end protection is more important than 5’-end protection for achieving efficient integration8 and we recommend the use of the 3’-only end-protected dsODN tags in this protocol. To map dsODN integration sites genome-wide, we adapted anchored multiplex PCR sequencing (AMP-seq) to specifically amplify genomic DNA molecules containing the dsODN tag9.
Overview of the workflow.
Here, we present a detailed working protocol for performing GUIDE-seq. Using CRISPR-Cas9 nuclease as an example, first Cas9 and gRNA are delivered into cultured cells as mammalian expression plasmids or as a ribonucleoprotein (RNP) complex along with a short dsODN tag. After delivery of RNP and dsODN tag, cells are typically cultured for 3 days prior to harvesting for GUIDE-seq library preparation.
Library preparation requires several straightforward steps (Fig. 1). Genomic DNA is isolated and randomly fragmented, followed by end-repair, A-tailing, and ligation of a single-tailed high-throughput sequencing adapter containing a randomized 8-bp unique molecular index (UMI). Next, two rounds of PCR are performed with primers complementary to the ligated single-tailed NGS adaptor and the dsODN tag. (Fig. 2).
Figure 1. Schematic workflow of GUIDE-seq method.
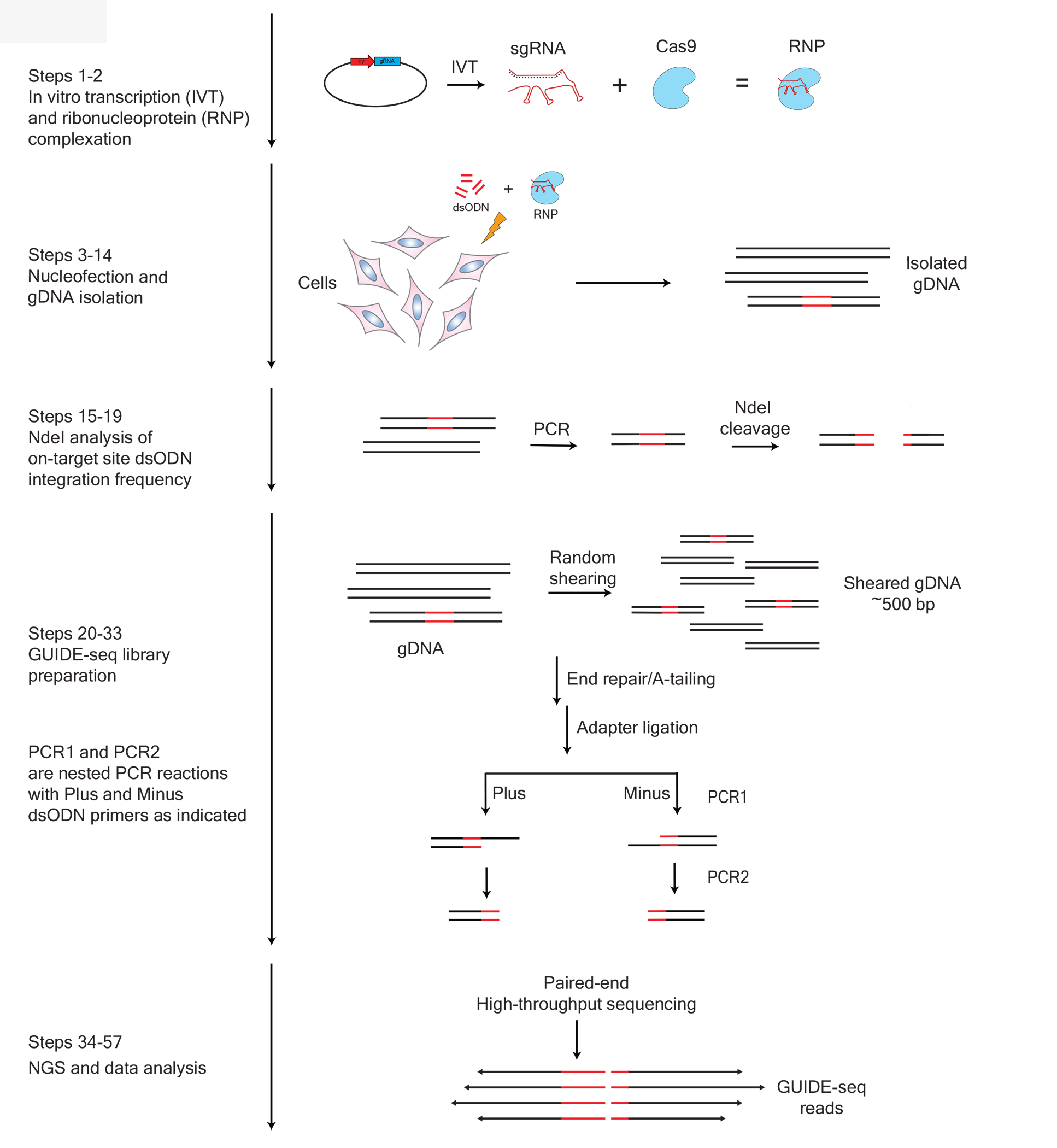
The GUIDE-seq procedure is divided into 5 sections with the protocol step numbers as shown on the left: 1) In vitro transcription (IVT) and ribonucleoprotein (RNP) complexation, 2) nucleofection, cell culturing and genomic DNA (gDNA) isolation; 3) NdeI restriction and indel analysis, 4) GUIDE-seq library preparation, and 5) next-generation sequencing and data analysis. Original figure.
Figure 2. Principle of GUIDE-seq library preparation and paired-end analysis.
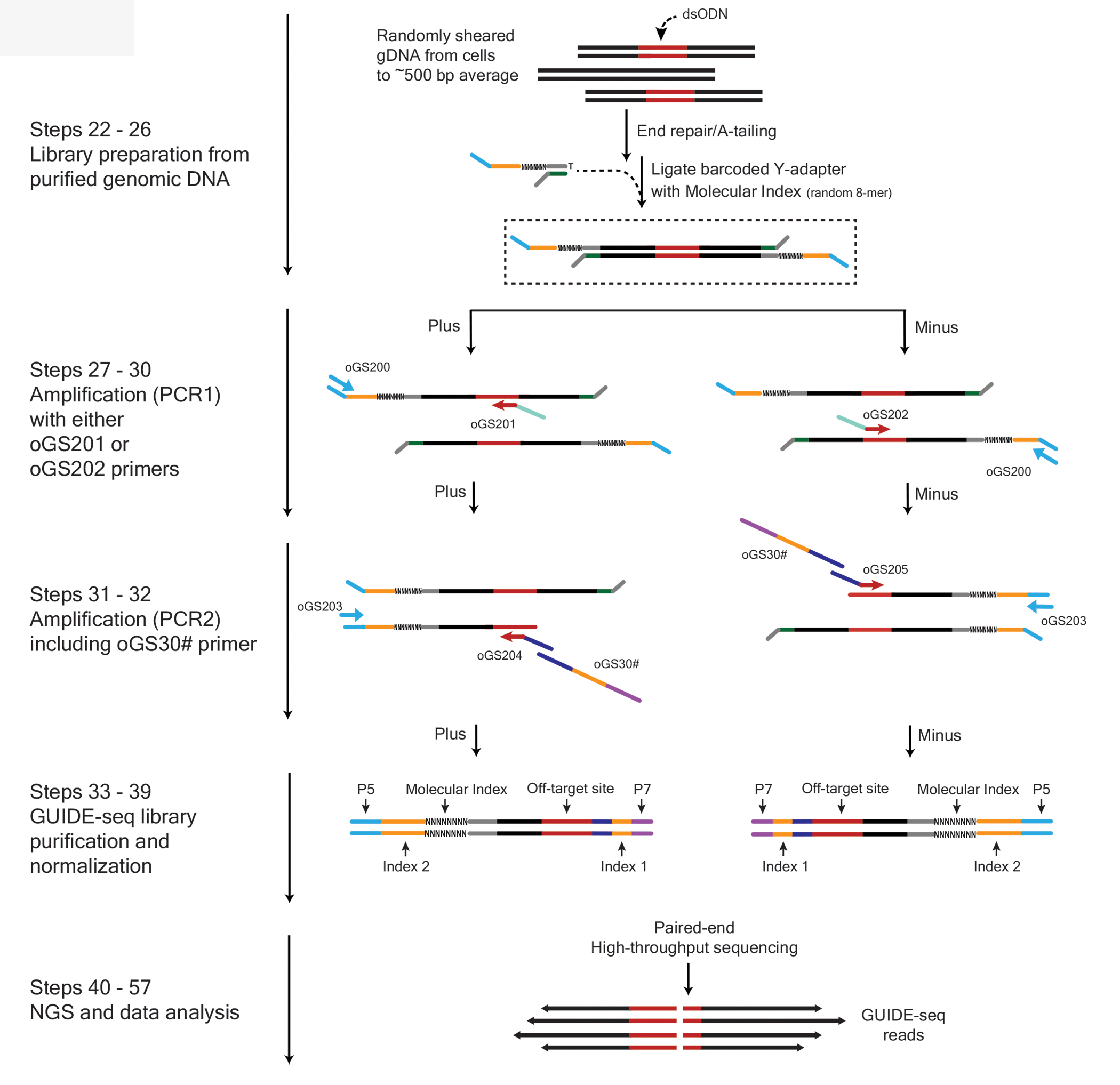
(i) Genomic DNA is sonicated to average size of 500 bp, followed by a standard library generation steps (end repair, A-tailing and adapter ligation); (ii) PCR1 with primer complementary to the ligated adapter on one side and either sense D or antisense R primer complementary to dsODN sequence on the other. The opposite strands dsODN primers are used in separate PCR reactions; (iii) Nested PCR2 includes an additional primer containing p7 index and complementary to the dsODN primer on its 3-prime. i5 index encoded in amplified part of the adapter. Each sample has unique combination of i5 and i7 indices, allowing for parallel sequence and analysis of numerous samples; (iv) library normalization is required for balanced representation of samples and opposite strands for each sample; (v) Library sequencing and analysis, the reads are grouped by unique combination of p5 (blue) and p7 (purple) indices, indexes are marked yellow, dsODN sequence is red. Bidirectionally mapped indicate DSB locations. Original figure.
The UMI introduced with the ligated adapter enables correction of any PCR bias introduced during amplification, allowing accurate quantification of editing activity. Tag-specific amplified sequencing reads are consolidated based on their UMI, mapped to a reference genome, and on- and off-target sites are identified by characteristic bi-directional mapping read signatures.
Comparison to alternative methods.
A number of different techniques have been developed for defining the genome-wide activity of genome-editing nucleases4,8,10–16. These methods fall into two broad categories: cell-based and biochemical methods. In general, cellular methods are the most direct while biochemical methods are the most sensitive.
GUIDE-seq is one of the most sensitive cellular genome-wide activity detection methods. IDLV (integrase defective lentivirus) capture, the first method for unbiased genome-wide identification of nuclease DSBs, is based on similar principle as GUIDE-seq but is less sensitive and has higher background due to residual integrase activity of IDLV vectors12.
As GUIDE-seq is based on tagging nuclease-induced DSBs with a short dsODNs, the location and frequency of tags associated with DSB repair events can be tracked even after the DNA repair event has occurred. In this way, GUIDE-seq read counts reflect the ongoing history of cellular DSB repair events. Other cellular methods such as BLESS, BLISS, and DISCOVER-seq detect transient DSBs as they are occurring but become insensitive to sites of off-target mutation after DSB repair is complete. These methods are complementary to GUIDE-seq as they can be used to define the kinetics of off-target activity.
Complementary biochemical methods such as Digenome-seq, CIRCLE-seq, and CHANGE-seq operate on the principle of detecting nuclease-induced DSBs from purified genomic DNA. These methods have a lower apparent validation rate than GUIDE-seq, but it remains unclear whether this is due to false positives that result from high protein:DNA concentrations used in the in vitro system that may not be achievable in cells or due to false negatives that arise from the lower limits of detection of standard targeted sequencing (~0.1%). These types of methods are recommended when the highest sensitivity is required or in cell types that are sensitive to being transfected with dsODN tags.
Advantages.
Advantages of GUIDE-seq are that it is highly sensitive for a cell-based method, allows detection of cell-type specific cut sites, is sequencing efficient, unbiased, and quantitative. GUIDE-seq can detect off-target sites with associated mutation frequencies of 0.1% and below. GUIDE-seq requires only 2–5 million reads per sample so it can be easily run on benchtop sequencers such as an Illumina MiSeq or MiniSeq systems. GUIDE-seq is an unbiased method that does not make a priori assumptions about sequence characteristics of potential off-target sites. All sites that fit the criteria of bidirectional mapping GUIDE-seq reads are reported, and the subset of sites with a user-specified number of mismatches are further labelled as high-priority off-target sites. GUIDE-seq read counts are strongly correlated with indel mutation frequencies in cells edited in the absence of dsODN tag and, therefore, are a good proxy for genome-wide off-target activity8. Because it is a direct cell-based method, GUIDE-seq has a very high validation rate compared to in vitro methods such as CHANGE-seq based on the principle of selective sequencing of nuclease-cleaved genomic DNA.
Limitations.
The primary limitation of GUIDE-seq is that it requires transfection with dsODN tags that may not be tolerated by all cell types. Some cell types such as human hematopoietic stem cells or iPS cells may have a robust DNA damage response and undergo apoptosis in response to high levels of free DNA ends. The use of ‘surrogate’ cell types that are more compatible for GUIDE-seq analysis may have limitations because there would not account for cell-type specific effects. If GUIDE-seq cannot be specifically optimized for a cell type of interest, it may be reasonable to try a sensitive biochemical method for defining genome-wide activity such as CHANGE-seq8. Finally, although theoretically possible, to our knowledge, in vivo delivery of dsODNs for GUIDE-seq analysis has not been reported in the published literature.
Extended applications.
GUIDE-seq or similar methods can also be used to characterize nucleases beyond broadly adopted Cas9, such as meganucleases17, ZFNs18, TALENs, and CRISPR-Cas nucleases that generate overhangs, such as Cas12a19. Interestingly, we found that blunt end-protected dsODN tags integrate relatively efficiently into both blunt DSBs and those with 5’-overhangs. For enzymes with 3’ overhangs such as meganucleases, others have found that using an end-protected dsODN with randomized 3’ overhangs may be helpful.
GUIDE-seq can be utilized to validate candidate sites that were detected in silico or by in vitro assays such as CHANGE-seq, Digenome-seq and DISCOVER-seq in cell types such as human primary T-cells that are tolerant to transfection with GUIDE-seq dsODN tags8.
Variations of GUIDE-seq that utilize Tn5 tagmentation for library preparation or a longer dsODN tag have also been recently described15,20. An adapted version of GUIDE-seq has been used to discover off-target sites in a mouse cell line that may also be unintentionally modified in mice in vivo3. While we describe the standard GUIDE-seq approach that has been extensively validated here, we anticipate that we and others will continue to improve and adapt the method for future needs.
Experimental design
Optimization of dsODN incorporation.
Here we describe GUIDE-seq protocols for analysis of genome-wide genome editing activity in U2OS and human primary T-cells. To successfully optimize GUIDE-seq for a new cell type, the critical step is to optimize the integration frequencies of dsODN when delivered alongside with Cas9 and gRNA. This optimization is performed by a simple titration of the amount of transfected dsODN to simultaneously maximize integration frequencies while preserving cell viability and recovery. On average, dsODN integrations should represent approximately one third of all nuclease-induced mutations with integration rates greater than 5%4. For example, if we observed 30% indel mutations at a particular target, we would anticipate a GUIDE-seq dsODN integration rate of 10%. 10% dsODN integrations would exceed our absolute integration rate guideline of >5% for successful GUIDE-seq experiments. Failed GUIDE-seq experiments are most often associated with low tag integration frequencies.
For quick validation and optimization of dsODN tag integration, the GUIDE-seq dsODN tag contains a NdeI restriction enzyme site, allowing for the tag detection by NdeI restriction digestion of PCR amplicons. For the NdeI restriction analysis, it is important to design PCR primers generating an amplicon from 300 to 500 bp that does not contain additional NdeI sites, and the position of the Cas9 cut site is located asymmetrically. Indel rate and dsODN integration can also be evaluated by targeted amplicon sequencing analysis. A third alternative for estimating the dsODN incorporation rate for Cas9 is the ICE tool for HDR efficiency (https://ice.synthego.com).
Delivery of editing components and dsODN.
We have been most successful in delivering Cas9 and gRNA as plasmids or RNP alongside with dsODN using electroporation by Lonza Nucleofection. Others have also reported successful dsODN delivery into HEK293 cells by Mirus TransIT-X2 transfection reagent21. Many cationic lipids are not capable of delivering small DNA molecules to cells, so they will need to be tested on a cell-type specific basis. Therefore, we generally recommend use of electroporation-based methods to deliver editing components and dsODN as a starting point for optimizing GUIDE-seq in new cell types.
Transfection of approximately 600,000 to 900,000 cells will usually ensure that sufficient genomic DNA can be isolated to run the GUIDE-seq protocol. This can be achieved with one larger transfection or by pooling multiple smaller transfections together.
Required controls.
To exclude random DSBs not associated with Cas9 activity, control genomic DNA from cells transfected with dsODN only is required. Ideally, GUIDE-seq requires a dsODN-only transfected control for each cell type and independent set of samples. This control is required to utilize the standard GUIDE-seq analytical pipeline.
Assignment of sample barcodes.
Each GUIDE-seq sample should be assigned a unique combination of indexing barcodes added during adapter ligation (Step 24) and PCR2 (Step 31).
Library quantification and sequencing.
Completed libraries should be quantified by qPCR. It is convenient to make six serial 1:10 dilutions to find the optimal concentration range for subsequent pooling.
Sequencing depth.
GUIDE-seq runs should be sequenced to a depth of at least 2 – 5 million reads per sample, achieving on-target read counts above 500 to maximize sensitivity.
Overage.
For GUIDE-seq transfections and for all enzymatic reactions, include 10–20% overage to account for pipetting losses.
MATERIALS
Biological Materials
-
U2OS cell line RRID CVCL_0042 (https://web.expasy.org/cellosaurus/CVCL_0042) (ATCC cat. no. HTB-96)
!CAUTION Cell lines used in your research should be regularly checked to ensure they are authentic and are not infected with mycoplasma.
Human primary CD4+/CD8+ T cells (Key Biologics)
Reagents
gRNA preparation
RNAse ZAP solution (Fisher Scientific cat. no. AM9780)
Plasmid pCRL1 for T7 in vitro transcription of gRNA (Addgene cat. no. 153997)
HindIII-HF restriction enzyme (New England Biolabs cat. no. R3104S)
HiScribe T7 RNA Synthesis Kit (New England Biolabs cat. no. E2050S)
MEGAclear Transcription Clean-Up Kit (Fisher Scientific cat. no. AM1908)
RNA High Sensitivity Screen Tape (Agilent cat. no 5067-5579)
RNA High Sensitivity Sample Buffer (Agilent cat. no. 5067-5580)
Mammalian Cell culture and nucleofection
24 Well Cell Culture Plate, Flat Bottom with Lid, Tissue Culture Treated, Non-Pyrogenic, Polystyrene (Corning Life Sciences cat. no. 3524)
Non-Tissue Culture Treated Plate, 24 Well, Flat Bottom with Low Evaporation Lid (Corning Life Sciences cat. no. 351147)
Non-Tissue Culture Treated Plate, 96 Well, Flat Bottom with Low Evaporation Lid (Corning Life Sciences cat. no. 351172)
Cas9 expressing construct pSQT817 (Addgene cat. no. 53373)
gRNA expression construct, backbone BPK1520 (Addgene cat. no. 65777)
SE nucleofection kit (Lonza cat. no. V4SC-1096)
Advanced DMEM (Fisher Scientific cat. no. 12491023)
PBS (Fisher Scientific cat. no. 10010049)
Disposable Hemocytometer (Fisher Scientific cat. no. 22600100)
Trypan Blue (Fisher Scientific cat. no. 15250061)
FBS, Fetal bovine serum (Fisher Scientific cat. no. 10-082-147)
Trypsin-EDTA 0.05% (Fisher Scientific cat. no. 25300120)
Penicillin/Streptomycin (Fisher Scientific cat. no. 15-070-063)
GlutaMax 100X (Fisher Scientific cat. no. 35-050-061)
96 wells plate V-shape (Fisher Scientific cat. no. 720096)
X-vivo 15 Culture Media (Lonza BEBP04-744Q)
P3 nucleofection kit (Lonza V4SP-3096) !CRITICAL Protocol for T-cell transfection has been optimized with this specific reagent
Human serum (Fisher Scientific cat. no. MT35060CI)
IL-15 (Miltenyi Biotec cat. no. 170-076-114)
IL-7 (Miltenyi Biotec cat. no. 170-076-111)
Steriflip Sterile Disposable Vacuum Filter Units (Fisher Scientific cat. no. SCGP00525)
T cell TransAct beads (Miltenyi Biotec cat. no. 130-019-011)
SpCas9 NLS recombinant protein (New England Biolabs cat. no. M0646)
Genomic DNA isolation
96-well Clear V-Bottom 2 mL Polypropylene Deep Well Plate (Corning Life Sciences cat. no. 3960)
Agencourt DNAdvance purification kit (Beckman Coulter cat. no. A48705)
Lysis LBH (Beckman Coulter), supplied with Agencourt DNAdvance purification kit
Proteinase K (Beckman Coulter), supplied with Agencourt DNAdvance purification kit
Pre-Bind BBA (Beckman Coulter), supplied with Agencourt DNAdvance purification kit
Bind BBE (Beckman Coulter), supplied with Agencourt DNAdvance purification kit
Elution EBA (Beckman Coulter), supplied with Agencourt DNAdvance purification kit
Dithiothreitol DTT solution 1M (Sigma cat. no. 43816)
Qubit dsDNA BR Kit (Fisher Scientific cat. no. Q32850) !CRITICAL This assay can distinguish dsDNA versus ssDNA and is essential for accurate dsDNA quantification.
Qubit Assay Tubes (Fisher Scientific cat. no. Q32856)
DNA shearing
microTUBE AFA Pre-Slit Snap-Cap 6×16mm (Covaris cat. no. 520045)
1X TE buffer (Fisher Scientific cat. no. AM9848)
GUIDE-seq library preparation and next-generation sequencing
Sodium Chloride 5M (VWR cat. no. 97062-858)
Tris-HCl 1M pH 8.0 (Fisher Scientific cat. no. AM9855G)
EDTA 0.5M pH 8.0 (Fisher Scientific cat. no. PAV4233)
UltraPure DNase/RNase-Free Distilled Water (Fisher Scientific cat. no. 10977015)
Ethanol (Sigma, cat. no. E7023) !CAUTION Flammable, keep away from open flame.
1X TE buffer (Fisher Scientific cat. no. AM9848)
NdeI restriction enzyme (New England Biolabs cat. no. R0111S)
Agencourt AMPure XP PCR purification kit (Beckman Coulter cat. no. A63881)
End-repair Mix (Enzymatics cat. no. Y9140-LC-L)
T4 DNA ligase (Enzymatics cat. no. L6030-LC-L)
25 mM dNTP solution mix (Enzymatics cat. no. N2050L)
Taq polymerase (non-hot start) (Fisher Scientific cat. no. 10342-053) !CRITICAL: non-hot start Taq is critical for the procedure
10X PCR Rxn Buffer (-MgCl2) (Fisher Scientific), supplied with Taq polymerase
Platinum Taq polymerase (Fisher Scientific cat. no. 10966-026)
10X PCR Rxn Buffer (-MgCl2) (Fisher Scientific), supplied with Platinum Taq polymerase
50mM MgCl2 (Fisher Scientific), supplied with Platinum Taq polymerase
Adhesive PCR plate seals (Fisher Scientific cat. no. AB0558)
Tetramethylammonium chloride (TMAC) 5M (Sigma cat. no. T3411-500ML) !CAUTION Toxic if swallowed or in contact with skin
DNA High Sensitivity D1000 Screen Tape (Agilent cat. no. 5067-5582)
DNA High Sensitivity D1000 Ladder (Agilent cat. no. 5067-5586)
DNA High Sensitivity D1000 Sample Buffer (Agilent cat. no. 5067-56020)
Optical tube 8x Strip (Agilent cat. no. 401428)
Optical cap 8x Strip (Agilent cat. no. 401425)
KAPA Library Quantification Kit (Illumina) Universal qPCR Mix (Kapa cat. no. KK4824)
Hard-Shell 96 well PCR plates, thin wall (Bio-Rad cat. no. HSP9601)
Microseal Adhesive qPCR film (Bio-Rad cat. no. MSB1001)
Phusion Hot Start Flex 2X Mix (New England Biolabs cat. no. M0536L)
MiSeq reagent kit v3 600 cycles (Illumina cat. no. MS102-3003)
Phix control V3 kit (Illumina cat. no. FC-110-3001)
Tween-20 (Sigma cat. no. P7949)
Custom DNA oligonucleotides from Table 1 are synthesized at 250 nmol scale; oligonucleotides from Table 2 and Table 3 are synthesized at 100 nmol. All are sourced from Integrated DNA Technologies.
Table 1.
Sequences of dsODN complementary oligonucleotides.
| Oligo (Name) | sequence 5’ to 3’ | Purpose |
|---|---|---|
| oGS1 (dsODN FWD) | /5Phos/GTTTAATTGAGTTGTCATATGTTAATAACGGT*A*T | use for dsODN annealing |
| oGS2 (dsODN REV) | /5Phos/ATACCGTTATTAACATATGACAACTCAATTAA*A*C | use for dsODN annealing |
/5Phos/ indicates 5’ phosphorylation
designates phosphorothioate linkage. S
Table 2.
oGS100 and oGS101–116 are adapter oligonucleotides used in ligation step.
| Oligo (Name) | sequence 5’ to 3’ | Purpose |
|---|---|---|
| oGS100 (Common) | /5Phos/GATCGGAAGAGC*C*A | To anneal with A## barcoded oligo |
| oGS101 (A01) | AATGATACGGCGACCACCGAGATCTACACTAGATCGCNNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS102 (A02) | AATGATACGGCGACCACCGAGATCTACACCTCTCTATNNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS103 (A03) | AATGATACGGCGACCACCGAGATCTACACTATCCTCTNNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS104 (A04) | AATGATACGGCGACCACCGAGATCTACACAGAGTAGANNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS105 (A05) | AATGATACGGCGACCACCGAGATCTACACGTAAGGAGNNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS106 (A06) | AATGATACGGCGACCACCGAGATCTACACACTGCATANNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS107 (A07) | AATGATACGGCGACCACCGAGATCTACACAAGGAGTANNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS108 (A08) | AATGATACGGCGACCACCGAGATCTACACCTAAGCCTNNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS109 (A09) | AATGATACGGCGACCACCGAGATCTACACGACATTGTNNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS110 (A10) | AATGATACGGCGACCACCGAGATCTACACACTGATGGNNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS111 (A11) | AATGATACGGCGACCACCGAGATCTACACGTACCTAGNNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS112 (A12) | AATGATACGGCGACCACCGAGATCTACACCAGAGCTANNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS113 (A13) | AATGATACGGCGACCACCGAGATCTACACCATAGTGANNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS114 (A14) | AATGATACGGCGACCACCGAGATCTACACTACCTAGTNNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS115 (A15) | AATGATACGGCGACCACCGAGATCTACACCGCGATATNNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
| oGS116 (A16) | AATGATACGGCGACCACCGAGATCTACACTGGATTGTNNWNNWNNACACTCTTTCCCTACACGACGCTCTTCCGATC*T | Single-tailed unique barcode oligo |
Adapters contain 8-mer molecular indexes NNWNNWNN (where N is any nucleotide; W is either A or T) and i5 index (highlighted in bold and underline).
designates phosphorothioate linkage.
Table 3.
Primers for two sequential PCR reactions and custom primers for Illumina NGS sequencing. oGS200 and oGS203 are common primers for both direction-specific reactions in PCR1 and PCR2 respectively. oGS201, oGS202 and oGS204, oGS205 are direction specific primers for PCR1 and PCR2 respectively. PCR2 contains additional primers: oGS301 through oGS308. These primers contain i7 Illumina index highlighted in bold and added one per reaction. These primers are used to introduce the index in PCR2 as they anneal to region common for both oGS204 and oGS205. oGS309 and oGS310 are sequencing primers, refer to the text for details.
| Oligo (Name) | sequence 5’ to 3’ | Purpose |
|---|---|---|
| oGS200 (P5_PCR1) | AATGATACGGCGACCACCGAGATCTA | PCR1 common primer |
| oGS201 (P_PCR1) | GGATCTCGACGCTCTCCCTATACCGTTATTAACATATGACA | Plus dsODN primer for PCR1 |
| oGS202 (M_PCR1) | GGATCTCGACGCTCTCCCTGTTTAATTGAGTTGTCATATGTTAATAAC | Minus dsODN primer for PCR1 |
| oGS203 (P5_PCR2) | AATGATACGGCGACCACCGAGATCTACAC | PCR2 common primer |
| oGS204 (P_PCR2) | CCTCTCTATGGGCAGTCGGTGATACATATGACAACTCAATTAAAC | Plus dsODN primer for PCR2 |
| oGS205 (M_PCR2) | CCTCTCTATGGGCAGTCGGTGATTTGAGTTGTCATATGTTAATAACGGTA | Minus dsODN primer for PCR2 |
| oGS301 (P701_PCR2) | CAAGCAGAAGACGGCATACGAGATTCGCCTTAGTGACTGGAGTCCTCTCTATGGGCAGTCGGTGA | i7 index PCR2 |
| oGS302 (P702_PCR2) | CAAGCAGAAGACGGCATACGAGATCTAGTACGGTGACTGGAGTCCTCTCTATGGGCAGTCGGTGA | i7 index PCR2 |
| oGS303 (P703_PCR2) | CAAGCAGAAGACGGCATACGAGATTTCTGCCTGTGACTGGAGTCCTCTCTATGGGCAGTCGGTGA | i7 index PCR2 |
| oGS304 (P704_PCR2) | CAAGCAGAAGACGGCATACGAGATGCTCAGGAGTGACTGGAGTCCTCTCTATGGGCAGTCGGTGA | i7 index PCR2 |
| oGS305 (P705_PCR2) | CAAGCAGAAGACGGCATACGAGATAGGAGTCCGTGACTGGAGTCCTCTCTATGGGCAGTCGGTGA | i7 index PCR2 |
| oGS306 (P706_PCR2) | CAAGCAGAAGACGGCATACGAGATCATGCCTAGTGACTGGAGTCCTCTCTATGGGCAGTCGGTGA | i7 index PCR2 |
| oGS307 (P707_PCR2) | CAAGCAGAAGACGGCATACGAGATGTAGAGAGGTGACTGGAGTCCTCTCTATGGGCAGTCGGTGA | i7 index PCR2 |
| oGS308 (P708_PCR2) | CAAGCAGAAGACGGCATACGAGATCCTCTCTGGTGACTGGAGTCCTCTCTATGGGCAGTCGGTGA | i7 index PCR2 |
| oGS309 (Index1_SEQ) | ATCACCGACTGCCCATAGAGAGGACTCCAGTCAC | Index 1 sequencing primer |
| oGS310 (Read2_SEQ) | GTGACTGGAGTCCTCTCTATGGGCAGTCGGTGAT | Read 2 sequencing primer |
Equipment
Universal magnet plate (Alpaqua cat. no. A00400)
Qubit Fluorometer 3.0 (Fisher Scientific cat. no. q33216)
Acoustic Focused Ultrasonicator (Covaris cat. no. E220)
Nucleofector 4-D (Lonza cat. no. AAF-1002X)
Veriti 96-well thermocycler or equivalent (Fisher Scientific cat. no. 4375786)
CFX96 Real-Time PCR thermocycler or equivalent (Bio-Rad cat. no. 1855195)
Incubating Microplate Shaker (Fisher Scientific cat. no. 02217759)
Eppendorf 5424 micro-centrifuge or equivalent (Eppendorf cat. no. 022620401)
Eppendorf 5920 centrifuge or equivalent (Eppendorf cat. no. 2231000383)
4200 TapeStation Instrument and accessories (Agilent cat. no. G2991AA)
Microtiter shaker IKA MS3 (Fisher Scientific cat. no. 1476321)
MiSeq (Illumina cat. no. SY-410-1003)
Standard inverted microscope
Tissue culture biosafety level BSL2 cabinet
Cell culture CO2 incubator
Software
Python v.2.7 or 3: https://www.python.org/downloads/
Burrows–Wheeler Aligner (BWA): https://github.com/lh3/bwa
Bedtools: http://bedtools.readthedocs.io/en/latest/content/installation.html
GUIDE-seq: https://github.com/tsailabSJ/guideseq
REAGENT SETUP
Ethanol, 70% Prepare fresh 70% (vol/vol) ethanol for purifications.
! CAUTION Ethanol is highly flammable.
Bind BBE.
Reagent is from Agencourt DNAdvance purification kit. Equilibrate at room temperature (24°C) and vortex vigorously immediately before use. When not in use store at 4°C.
Culture media for U2OS cells.
Supplement Advanced DMEM with FBS to 10% (vol/vol) final concentration, antibiotics (Penicillin/Streptomycin) and GlutaMAX - each from 100X stocks to 1X final dilutions (i.e. 5 ml per 500 ml of DMEM). Store at 4 °C up to 3 months.
Culture media for T-cells.
Supplement X-vivo 15 media with HSA to 5% (vol/vol) (or 20% (vol/vol) where indicated), filter sterilize and add IL-15 with IL-7 to the final concentration 10 ng/ml each. Prepare fresh every time.
10X STE solution.
The composition of the 10X STE stock solution is 500 mM NaCl, 100 mM Tris-HCl (pH 8.0) and 10 mM EDTA. To prepare 10 ml of 10X STE stock solution, combine 1 ml of 5 M NaCl, 1 ml of 1M Tris HCl (pH 8.0) and 0.2 ml of 0.5M EDTA, and bring the total volume to 10 ml with molecular biology grade water. Store at room temperature for at least 1 year.
GUIDE-seq oligonucleotides.
Re-suspend the lyophilized GUIDE-seq oligos oGS1 and oGS2 in TE buffer at a concentration of 250 μM each (Table 1). Mix 40 μl of each oligo, 10 μl water, and 10 μl of 10X STE buffer and anneal with the following thermocycler program:
| Step | Temperature | Time |
|---|---|---|
| Denaturation | 95°C | 5 min |
| 70 cycles | −0.1°C /cycle | 30 s |
| End | 4°C | Hold |
Annealed oligonucleotides can be stored at −20°C for up to 3 months in aliquots of 20 μl.
Library adapter.
Anneal the MiSeq common adapter oGS100 with one of the oGS101 to oGS116 barcoded oligos for each sample (Table 2). Re-suspend lyophilized oligos in TE buffer to the concentration of 100 μM. Combine 10 μl of each oligo and bring the total volume to 100 μl with final buffer composition 1X STE. Anneal using thermocycler program as above. Annealed oligonucleotides aliquoted by 10 μl and stored at −20°C for up to 3 months.
KAPA qPCR kit.
Prepare for use by adding supplemented primers to 2X master mix according to manufacturer’s instructions. The reagent can be stored at −20°C for several months.
PROCEDURE
Preparation of gRNA ● TIMING 1 w
1| Obtain gRNA either as synthetic chemically modified gRNA22 (option A) or by in vitro transcription reaction with T7 driven template such as targets cloned into pCRL1 or equivalent (option B).
(A). Synthetic gRNA.
Synthetic gRNA is commercially available from several sources (e.g., IDT,Synthego, or Trilink).
(B). In vitro transcribed gRNA.
As template for gRNA synthesis, clone gRNA target sequence into pCRL1 plasmid as previously described23 and verify by sequencing. Use RNAse-free grade water and reagents. Clean pipettors with RNAse ZAP spray.
-
iLinearize the template plasmid using a unique enzyme immediately downstream of gRNA scaffold sequence. Digest up to 5 μg of plasmid, use HindIII-HF restriction enzyme:
Component Volume (μl) Final concentration CutSmart 10X buffer 5 1X gRNA plasmid (3–5 μg) Variable up to 0.1 μg /μl HindIII-HF (20 U/μl) 4 1.6 U/μl Nuclease-free H2O to 50 Total 50 -
ii
Incubate at 37 °C for 3 h and heat inactivate at 80 °C for 20 min.
-
iii
Purify the DNA with Agencourt AMPure XP beads following the manufacturer’s protocol. Briefly, add 90 μl beads suspension (1.8X volume of sample) and mix well by pipetting up and down 10 times. Incubate at room temperature for 5 min and separate beads on magnetic plate for 5 min. Aspirate supernatant without disturbing the pellet. Add 200 μl of 70% ethanol, incubate 30 s and then aspirate. Repeat 70% ethanol wash and air dry the pellet for 3 min. Remove the plate from the magnetic rack and re-suspend the beads in 15 μl TE buffer by pipetting up and down 10 times. Incubate at room temperature for 2 min. Move the plate back to the magnetic rack, allow beads to settle for 1 min, and carefully transfer supernatant to a new plate.
▲ CRITICAL STEP Maintain an RNase-free environment, use RNase-free water and plastic consumables. Pipette accurately to avoid cross contamination from bead purification. Do not over-dry beads to prevent loss of yield and samples cross-contamination.
-
ivSet up the in vitro transcription with the HiScribe T7 RNA Synthesis Kit:
Component Volume (μl) Final concentration HindIII Digested linearized plasmid DNA (1 μg) variable 0.05 μg/μl NTP Buffer Mix 10 10 mM each NTP T7 RNA Polymerase 2 Nuclease-free H2O to 20 Total 20 -
v
Incubate the reaction at 37°C for 16 h.
-
vi
Add 2 μl of DNase I (2 U/μl).
-
vii
Incubate at 37°C for an additional 15 min.
-
viii
Purify the in vitro synthesized gRNA with MEGAclear purification kit according to the manufacturer’s instructions.
-
ix
Elute with pre-heated (95 °C) 50 μl elution solution and collect gRNA by spinning for 1 min at 15 000 × g. To increase the yield, repeat elution with the flow through from first elution.
-
x
Dilute an aliquot of sample 10X before measuring to ensure that it is within the linear range.
-
xi
Quantify the RNA using a NanoDrop spectrophotometer. Choose RNA mode to use correct extinction coefficient.
-
xii
Check gRNA integrity on Agilent TapeStation using RNA High Sensitivity Screen Tape and Buffer according to the manufacturer’s instructions.
PAUSE POINT Make 5 μl aliquots and freeze at −80 °C. gRNA aliquots can be stored at −80°C for several months.
▲ CRITICAL STEP Measuring gRNA concentration in undiluted samples may result in underestimated concentration, resulting in suboptimal genome editing and increased cell toxicity.
Culture, nucleofect, and harvest cells ● TIMING 7 d
2| Here we provide two alternative protocols for nucleofection of U2OS cells with SpCas9 and gRNA expression plasmids (option A), primary CD4+/CD8+ T cells with reconstituted Cas9:gRNA RNP (option B), as well as a guideline for a optimizing a new cell line (option C).
A). U2OS cells
Culture U2OS cells under standard conditions (37 °C, 5% CO2) in complete Advanced DMEM (refer to reagent set up).
-
i
Plate cells at 40% confluency in 100 mm dishes one day before nucleofection.
▲ CRITICAL STEP Culturing cells for a longer time before nucleofection may result in lower cell viability and editing efficiency after transfection.
-
ii
Prior to transfection, pre-equilibrate a 24-well recovery plate(s) with 500 μl of complete DMEM media in each well by incubating in 37 °C, 5% CO2 incubator.
-
iii
Collect cells for nucleofection. Wash cells with PBS, add up to 2 ml per 100 mm dish of 0.05% trypsin to fully cover the cell’s monolayer, and incubate for 5 min in 37 °C, 5% CO2 incubator.
-
iv
Re-suspend cells in the same volume of media used for culturing and count with hemocytometer.
-
v
Collect cells needed for nucleofections by centrifugation for 1 min at 1200 × g, room temperature. For each nucleofection, 2 × 105 cells are resuspended in 20 μl of SE buffer with 100 pmol of dsODN (i.e. 1 μl of annealed dsODN, refer to Reagent setup). We recommend making a single pool of cells resuspended in SE buffer with dsODN for all nucleofections. For instance, for 10 samples resuspend 2 × 106 cells in 200 μl of SE buffer and add 10 μl of annealed dsODN.
-
vi
In 96-well plate, combine 20 μl of cell suspension with dsODN, 500 ng of Cas9 expression plasmid SQT817 and 250 ng gRNA expression plasmid. Control samples should include cell suspension with dsODN only.
▲ CRITICAL STEP Keep volume of DNA added to 2 μl or less for optimal nucleofection efficiency. Control samples should be included into each experiment to monitor random dsODN incorporation as well as cytotoxicity of nucleofection procedure.
-
vii
From the plate, mix and transfer 20 μl of the mixture to the Lonza 4-D cuvette.
-
viii
Nucleofect the samples using DN-100 pulse code on a Lonza 4-D nucleofector following the manufacturer’s instructions.
-
ix
Incubate at room temperature for 10 minutes.
▲ CRITICAL STEP Do not omit room temperature incubation as transfection efficiency can drop significantly.
-
x
Add 80 μl media from the pre-incubated 24-well plate (prepared at step ii) to the nucleofection cuvette and gently re-suspend cells 2–3 times.
-
xi
Transfer the wells content back to the plate and return the plate to the CO2 incubator for 72 hours before harvesting.
B). CD4+/CD8+ T cells
Thaw, then activate and culture CD4+/CD8+ T cells from frozen stock before nucleofection by following the steps below. We plate cells in non-TC treated 24-well plates.
Thaw cells and pipette into 50 ml tube, slowly add 10 ml of pre-warmed X-Vivo 15 media without supplements.
Spin for 10 minutes at 300 × g at room temperature, remove supernatant, and resuspend cells at 1×106 cells/ml in T cell media prepared as described in the reagent set up.
Activate T cells with TransAct polymeric nanomatrix by adding the TransAct reagent directly to the cell culture at the specific dilution ratio specified by the manufacturer (1:100) and culture in a non-TC 24-well plate for 72 hours.
Prior to nucleofection, pre-equilibrate a non-TC 96-well recovery plate(s) with 250 μl per well of filter sterilized complete X-vivo 15 media with 20% HSA and 10 ng/ml of IL-7 and IL-15 in 37°C, 5% CO2 incubator.
For each GUIDE-seq sample, setup 3 nucleofections for pooling to ensure that sufficient tag-integrated cells are available for the analysis. For each nucleofection, use 75 pmol of the Cas9 protein and 3-fold molar excess of gRNA in a final total volume of 5 μl. Prepare RNP by combining the Cas9 protein and gRNA accounting for the required molar amount of each nucleofection, pooling and overage. Incubate at room temperature for 20 minutes.
Pool and count the cells with hemocytometer.
Collect cells needed for nucleofection by centrifugation 10 min at 300 × g, room temperature. For each nucleofection, 3×105 cells are resuspended in 20 μl of P3 buffer with 100 pmol of dsODN (i.e. 1 μl of annealed dsODN, refer to Reagent Setup). We recommend making a single pool of cells resuspended in P3 buffer with dsODN for all nucleofections. For example, for 10 nucleofections resuspend 3×106 cells in 200 μl of P3 buffer and add 10 μl of annealed dsODN.
In non-TC 96 well plate, for each nucleofection combine 20 μl of the cell suspension with dsODN from step (vi) and 5 μl of the RNP complex from step (iv). Control samples should include cell suspension with dsODN only.
From the plate, for each nucleofection mix and transfer 20 μl of the mixture to one well 16 well Lonza 4-D strip. Nucleofect the samples using EO-115 pulse code on a Lonza 4-D nucleofector following the manufacturer’s instructions.
Incubate at room temperature for 7 minutes.
Add 100 μl of media from the plate prepared in step (ii) to the well.
Transfer 120 μl of the content of well back to the plate, culture in CO2 incubator for 72 hours before harvesting. Pool 3 nucleofections for each sample when the cells are harvested for genomic DNA isolation.
C). Optimize for new cell type
Screen for an optimal nucleofection program with an expression plasmid of a fluorescent protein.
Further optimize efficiency of editing using well-characterized target by varying number of cells per nucleofection and amount of RNP.
Using optimal editing conditions, titrate the amount of GUIDE-seq dsODN per nucleofection. 5 to 200 pmol is a good starting range for small scale nucleofections in 20 ul.
Analyze the rate of tag integrations compared to overall indel mutation frequencies.
Choose a GUIDE-seq dsODN amount that results in integration rates above 5% with integrations optimally representing 1/3 of overall indel mutations observed.
▲ CRITICAL STEP For highest nucleofection efficiency the volume of added RNP should be less than 5 μl.
Genomic DNA isolation ● TIMING 3 h
3| 3 days after nucleofection, isolate genomic DNA with Agencourt DNAdvance kit according to manufacturer’s instructions:
Prepare lysis mixture for required number of wells:
| Component | Volume (μl) |
|---|---|
| Lysis LBH | 188 |
| 1M DTT | 5 |
| Proteinase K | 7 |
| Total | 200 |
4| Aspirate media from the plate and directly add 200 μl of lysis mixture to wells of the 24-well plate.
5| Seal the plate with PCR film and incubate at 55 °C for 2–16 hours, with gentle 250 rpm shaking.
6| Transfer all recovered volume of the lysate to Costar 96 deep-well block.
7| Add 100 μl Pre-Bind PBBA and pipette 10 times to mix.
8| Add 170 μl Bind BBE (refer to REAGENT SET UP), pipette 15 times to mix and incubate at room temperature for 5 minutes.
9| Incubate on universal magnet plate for 5 minutes at room temperature to separate the magnetic beads. Aspirate the supernatant without disturbing the beads.
10| Remove the 96-well block from the magnet. Add 340 μl of freshly made 70% ethanol to each well. Re-suspend beads by pipetting 20 times or until fully suspended.
11| Return block to the magnetic plate for 1 minute or until beads collected, discard the supernatant.
12| Repeat the ethanol wash steps 10–11 two more times for a total of three ethanol washes.
13| Elute by resuspending pellet in 200 μl Elution EBA, pipette 10 times then separate beads on magnetic plate for 5 min. Collect 190 μl of the supernatant.
? TROUBLESHOOTING
14| Measure DNA concentration with Qubit Broad Range kit on Fluorometer 3.0 according to manufacturer’s instructions.
▲ CRITICAL STEP Use a fluorescent quantification method like Qubit to avoid overestimating double-stranded DNA concentration.
? TROUBLESHOOTING
■ PAUSE POINT At this point samples can be stored at −20 °C.
Estimating dsODN integration frequency ● TIMING 4 h
15| Assemble PCR reactions for each experimental sample (gDNA from nuclease and dsODN-treated cells from step 13) and corresponding control sample (gDNA from cells transfected only with dsODN).
Please refer to the experimental design section of the introduction for general considerations for primer design:
| Component | Volume (μl) | Final concentration |
|---|---|---|
| 2X Phusion Mix | 25 | 1X |
| Forward primer (10 μM) | 2.5 | 0.5 μM |
| Reverse primer (10 μM) | 2.5 | 0.5 μM |
| Genomic DNA (100 ng) from Step 13 | Variable | 2 ng/μl |
| Nuclease-free water | to 50 | |
| Total | 50 | |
16| Run standard thermocycler program (make adjustments as needed for primer-specific conditions):
| Step | Temperature | Time |
|---|---|---|
| Denaturation | 98°C | 5 min |
| 98°C | 15 s | |
| 35 cycles | 68°C | 20 s |
| 72°C | 30 s | |
| Extension | 72°C | 5 min |
| End | 4°C | Hold |
17| Purify PCR product using 1.8X volume (90 μl) of Agencourt AMPure XP beads according to the manufacturer’s instructions as described in n Step 1(B)(iii). Elute in 30 μl of TE buffer.
18| Analyze tag integration frequency by performing (A) NdeI assay or (B) targeted sequencing.
A). NdeI assay
- Digest 200 ng of PCR product in volume of 20 μl with 10 units NdeI enzyme at 37 °C for 1hr.
Component Volume (μl) Final concentration 10X CutSmart buffer 2.0 1X NdeI 20U/ μl 1.0 1 U/μl PCR product (200 ng) from Step 17 Variable 10 ng/μl Nuclease-free water to 20 Total 20 Purify NdeI-treated DNA using 1.8X volume (36 μl) of Agencourt AMPure XP beads according to manufacturer’s instructions as described in Step 1(B)(iii). Elute in 20 μl of TE buffer.
Dilute 5 μl of each sample with TE buffer and analyze on Agilent TapeStation using D1000 High Sensitivity Screen Tape, Ladder and Buffer according to the manufacturer’s instructions. Determine fraction of DNA cleaved in each sample, using software provided with the instrument. Example is shown in Fig. 3.
Figure 3. Quality control steps in the GUIDE-seq protocol.
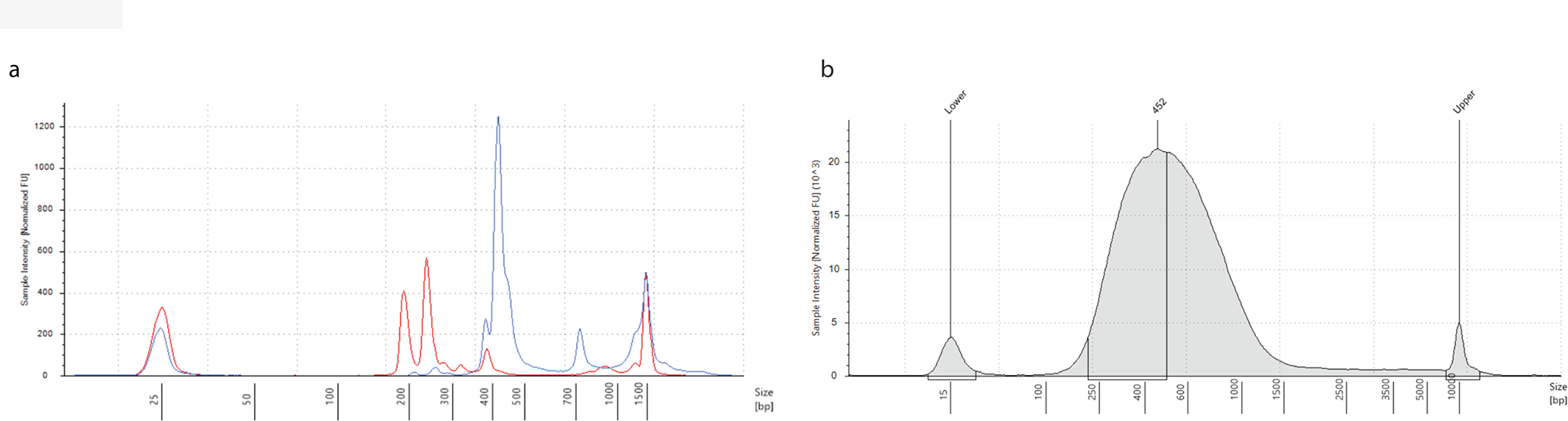
(a) NdeI analysis of dsODN incorporation on Agilent TapeStation. Profile of uncut PCR amplicon (blue) is superimposed with profile of the same PCR product digested with NdeI (red). Positions of the molecular size marker are indicated at the bottom. The targeted PCR is encompassing B2M site 6 as described by Lazzarotto et al.8. (b) Agilent TapeStation analysis of DNA shearing distribution that shows a peak with median size centered around ~450 bp. Original figure.
B). Targeted sequencing analysis
Design primers for NGS analysis by applying guidelines from the Experimental design section.
Proceed with dual-barcoded two-step PCR Amplicon sequencing following Illumina protocols for multiplex amplicon sequencing.
Analyze sequence data using publicly available tools such as CRISPRESSO224: https://github.com/pinellolab/CRISPResso2
19| Check that the percentage of tag integration is more than 5% and represents greater than 30% of detectable mutagenesis frequencies.
▲ CRITICAL STEP GUIDE-seq assay sensitivity is dependent on efficient tag integration.
? TROUBLESHOOTING
GUIDE-seq library preparation ● TIMING 8 h
20| Load 400 ng of gDNA into Covaris DNAmicroTUBE tube in a total volume of 130 μl with TE buffer and shear gDNA to average size of 500 bp using the following Covaris E220 settings: peak incident power 105 W, duty factor 5%, cycles per burst 200, treatment time 80 s. Manufacturer’s recommended settings for shearing should be followed if using a different model instrument. Example of sheared DNA is shown in Fig. 3.
▲ CRITICAL STEP Shear in 130 μl volume to prevent formation of air bubbles that may interfere with the process.
21| Purify sheared DNA using 1X volume (130 μl) of Agencourt AMPure XP beads according to manufacturer’s instructions as described in Step 1(B)(iii). Elute in 15 μl of TE buffer.
22| Assemble single-tube end repair/A-tailing reaction:
| Component | Volume (μl) |
|---|---|
| dNTP mix (5 mM each) | 1 |
| 10X ligation buffer | 2.5 |
| End repair mix | 2 |
| PCR Rxn Buffer (−MgCl2) (10X) | 2 |
| Taq polymerase (non-hot start) | 0.5 |
| Nuclease-free water | 0.5 |
| DNA sample from step 21 | 14 |
| Total | 22.5 |
23| Perform thermocycler program for end repair/A-tailing:
| Step | Temperature | Time |
|---|---|---|
| 1 | 12°C | 15 min |
| 2 | 37°C | 15 min |
| 3 | 72°C | 15 min |
24| Add 1 μl of annealed Y-adapter (oGS100 with one of the oGS101-116 see Table 2 and REAGENT SETUP), providing sample-specific barcode from 01 to 16 and 2 μl of T4 DNA ligase to the reaction mix from the previous step:
| Component | Volume (μl) |
|---|---|
| Reaction from step 22 | 22.5 |
| Y-adapter | 1 |
| T4 ligase | 2 |
| Total volume | 25.5 |
▲ CRITICAL STEP Each test or control sample should have a unique i5/i7 sample index combination. The i5 sample index is added in this step by selecting one of annealed adapters (oGS101 through 116) that contains the index sequence highlighted in bold (Table 2). The i7 sample index is added in step 31.
25| Perform ligation in thermocycler as follows:
| STEP | TEMPERATURE | TIME |
|---|---|---|
| 1 | 16°C | 30 min |
| 2 | 22°C | 30 min |
| End | 4°C | Hold |
26| Purify ligated product using 0.9X volumes (23 μl) of Agencourt AMPure XP beads according to the manufacturer’s instructions as described in Step 1(B)(iii). Elute in 22 μl of TE buffer.
27| Split adapter ligated product for two PCR1 reactions, 10 μl each, label one as Plus the other Minus.
▲ CRITICAL STEP From this point forward, each sample is processed with parallel Plus (P) and Minus (M) reactions. The products from Plus PCR1 are used for Plus PCR2, and Minus PCR1 for Minus PCR2. It is critical to keep track of the P and M reactions throughout two subsequent PCR reactions and to keep them separate.
28| Assemble Plus and Minus PCR1 reactions for each sample. The only difference is the sequence of tag-specific primers (highlighted in bold):
| Plus PCR1 | Minus PCR1 | ||
|---|---|---|---|
| Component | Volume (μl) | Component | Volume (μl) |
| Nuclease-free water | 11.9 | Nuclease-free water | 11.9 |
| Platinum Taq PCR Rxn Buffer (−MgCl2) (10X) | 3 | Platinum Taq PCR Rxn Buffer (−MgCl2) (10X) | 3 |
| dNTP mix (10 mM) | 0.6 | dNTP mix (10 mM) | 0.6 |
| MgCl2 (50 mM) | 1.2 | MgCl2 (50 mM) | 1.2 |
| oGS201 primer (10μM) | 1 | oGS202 primer (10μM) | 1 |
| TMAC (500 mM) | 1.5 | TMAC (500 mM) | 1.5 |
| oGS200 primer (10 μM) | 0.5 | oGS200 primer (10 μM) | 0.5 |
| Platinum Taq polymerase | 0.3 | Platinum Taq polymerase | 0.3 |
| Template DNA (from step 26) | 10 | Template DNA (from step 26) | 10 |
| Total | 30 | 30 | |
29| Run the PCR1 thermocycler program. 70°C Δ-1.0C/cycle in 15 cycles step designates decreasing the temperature for one degree per cycle starting with 70°C:
| Step | Temperature | Time |
|---|---|---|
| Denaturation | 95°C | 5 min |
| 95°C | 30 s | |
| 15 cycles: | 70°C, Δ-1°C/cycle | 2 min |
| 72°C | 30 s | |
| 95°C | 30 s | |
| 10 cycles: | 55°C | 1 min |
| 72°C | 30 s | |
| extension | 72°C | 5 min |
| End | 4°C | Hold |
30| Keep Plus and Minus PCR1 reactions separate. Purify DNA from each reaction using 1.2X volumes (36 μl) of Agencourt AMPure XP beads according to manufacturer’s instructions as described in Step 1(B)(iii). Elute in 15 μl TE buffer and keep the beads.
▲ CRITICAL STEP For higher yield and library complexity, keep the magnetic beads in the sample for the next step.
31| Assemble PCR2 reactions using PCR1 products as template. The Plus and Minus reactions remain separated. The same oGS30# primer (where # is 1 through 8, refer to Table 3) primer added to Plus and Minus reactions, oGS203 primer is universal for both:
| Plus PCR2 | Minus PCR2 | ||
|---|---|---|---|
| Component | Volume (μl) | Component | Volume (μl) |
| Nuclease-free water | 5.4 | Nuclease-free water | 5.4 |
| PCR Rxn Buffer (−MgCl2) (10X) | 3 | PCR Rxn Buffer (−MgCl2) (10X) | 3 |
| dNTP mix (10 mM) | 0.6 | dNTP mix (10 mM) | 0.6 |
| MgCl2 (50 mM) | 1.2 | MgCl2 (50 mM) | 1.2 |
| oGS204 (10 μM) | 1 | oGS205 (10 μM) | 1 |
| TMAC (500 mM) | 1.5 | TMAC (500 mM) | 1.5 |
| oGS203 (10 μM) | 0.5 | oGS203 (10 μM) | 0.5 |
| oGS30# (10 μM) | 1.5 | oGS30# (10 μM) | 1.5 |
| Platinum Taq polymerase | 0.3 | Platinum Taq polymerase | 0.3 |
| Template DNA from step 30 | 10 | Template DNA from step 30 | 10 |
| Total | 30 | 30 | |
▲ CRITICAL STEP Each sample should have a unique combination of i5/i7 indexes, where both P and M reactions of each sample have identical i5/i7 combination. The i5 index, encoded in the Y-adapter, was added earlier at step 24. i7 index is added in the current step, when the same i7 primer (one of oGS301-308) is added to both P and M reactions. Thus, similar to PCR1, the only difference between the P and M reactions for each sample is presence of either P_PCR2 (oGS204) or M_PCR2 (oGS205) primers:
32| Run the PCR program:
| Step | Temperature | Time |
|---|---|---|
| Denaturation | 95°C | 5 min |
| 95°C | 30 s | |
| 15 cycles: | 70°C, Δ-1°C/cycle | 2 min |
| 72°C | 30 s | |
| 95°C | 30 s | |
| 10 cycles: | 55°C | 1 min |
| 72°C | 30 s | |
| extension | 72°C | 5 min |
| End | 4°C | Hold |
33| Purify product of the PCR2 reaction using 0.7X volumes (21 μl) of Agencourt AMPure XP beads according to the manufacturer’s instructions as described in Step 1(B)(iii). Elute in 30 μl of TE buffer.
▲ CRITICAL STEP Keep all PCR products separate, do not pool together until after quantification.
■ PAUSE POINT Samples can be stored at −20°C at this point.
Library quantification and high-throughput sequencing ● TIMING 24 h
34| Prepare 10-fold serial dilutions of the libraries from 10−1 to 10-6. Typically, the 10−5 dilution range is optimal for quantification by qPCR. It is convenient to prepare 50 μl dilutions to leave sufficient material for sample pooling and sequencing. The samples, no template control (NTC), and standards are commonly used in triplicates for each qPCR run.
■ PAUSE POINT Samples can be stored at −20°C at this point.
35| Assemble SYBR qPCR reactions (in triplicates) in 96 well plate with reconstituted 2X KAPA master mix (see “Reagent setup”):
| Component | Volume (μl) |
|---|---|
| Water | 4 |
| Template | 4 |
| KAPA qPCR MM with primer (2X) | 12 |
| Total | 20 |
36| Perform SYBR qPCR reaction: in the plate layout mark standards, indicate corresponding concentrations. Concentrations are expressed as number of molecules per μl.
37| Run qPCR program:
| Step | Temperature | Time |
|---|---|---|
| Initial denaturation | 95°C | 5 min |
| 95°C | 30 s | |
| 35 cycles | 60°C | 45 s |
| Plate read | ||
| Melt curve | 65°C – 95°C Δ0.5°C | 5 s |
38| qPCR data will be expressed both as a table with concentration as defined in step 36, as well as standard curve plot with marked individual samples (Y axis is Cq, X axis is logarithmic scale of standards quantity). When standards are expressed as number of molecules per reaction, the unknown samples concentration will be automatically calculated in the same units. On the plot, choose a sample dilution that falls well within the middle of standards range. Use the table below for Kapa standards to generate data in molecules per microliter:
| Standard | Concentration (pM) | dsDNA molecules/uL |
|---|---|---|
| 1 | 20 | 12000000 |
| 2 | 2 | 1200000 |
| 3 | 0.2 | 120000 |
| 4 | 0.02 | 12000 |
| 5 | 0.002 | 1200 |
| 6 | 0.0002 | 120 |
39| Based on calculated concentrations for each library component, pool equal number of molecules of the individual components to a total of 4.5×109 molecules in the volume of 5 μl, resulting in a final library concentration of 1.5 nM, required for MiSeq sequencing. Adjustment for average library fragment size is not typically required.
? TROUBLESHOOTING
40| One hour before sequencing, bring the reagent cartridge to room temperature in a water bath to thaw all the buffers and mix well.
41| Prepare 4 nM PhiX control using 10 mM Tris HCl 0.1% Tween-20 (vol/vol) as diluent: 2 μl of 10 nM PhiX stock plus 3 μl of 10mM Tris HCl 0.1% Tween.
42| Denature 5 μl of DNA library by adding 5 μl of 0.2 N NaOH (prepared fresh), vortex, collect by brief centrifugation, incubate 5 min at room temperature. In a separate tube, denature 5 μl of 4 nM PhiX prepared in step 41.
43| Add 990 μl of HT1 buffer from the kit to each of the two tubes containing denatured library and PhiX control. The concentration of diluted library is now 7.5 pM and diluted PhiX control is now 20 pM.
44| Further dilute PhiX with HT1 buffer to 12.5 pM by adding 225 μl HT1 buffer to 375 μl of 20pM PhiX.
45| Add 50 μl of the diluted PhiX control (12.5 pM PhiX) to 1 ml of diluted library (7.5 pM) from step 43.
46| In the sequencing cartridge, use separate fresh P1000 pipet tips to pierce the foil sealing wells 13, 14 and 17.
47| Add 3 μl of 100 μΜ Index1 (oGS309 sequencing primer from Table 3) to well 13; 3 μl of 100 μΜ Read2 (oGS310 sequencing primer from Table 3) to well 14. Mix each well carefully using 1 ml serological pipette. Add 600 μl of the library to well 17.
▲ CRITICAL STEP Double check the custom primers added - if omitted, the sequencing will still be performed but without the correct reads.
48| For NGS analysis, GUIDE-seq specific options need to be defined in the Sample Sheet: add blank row, fill in sample ID with “1”, two adapters with N701 and N501. Save and open as text. Enter AAAAAAAA after N501 sequence and save. See example sample sheet (Supplementary File).
▲ CRITICAL STEP In the sample sheet do not check the boxes to use custom sequencing primers. Custom primers used in this protocol are spiked into the standard wells to enable reading of the PhiX control along with library.
49| Load and sequence the library, using a MiSeq Reagent Kit v3 and MiSeq system according to the manufacturer’s instructions or alternative Illumina sequencer of choice.
Post-run analysis ● TIMING 2 h
53| Install GUIDE-seq software pipeline25.
The GUIDE-seq code is compatible with both Python 2 and Python 3. It requires Burrows-Wheeler Aligner (BWA), bedtools, and several other Python packages (see https://github.com/tsailabSJ/guideseq#dependencies for details). Thus, the recommended way to install guide-seq pipeline is via conda.
conda create -n guideseq -c conda-forge -c bioconda -c anaconda -c tsailabSJ guide_seq
source activate guideseq
guideseq.py -h
54| Setup reference genome
Download the appropriate reference genome in FASTA format.
For example, human genome reference Hg19 can be obtained here:
http://www.broadinstitute.org/ftp/pub/seq/references/Homo_sapiens_assembly19.fasta
Please make an index of the reference genome once it is downloaded, for example:
bwa index Homo_sapiens_assembly19.fasta
55| Generate a manifest file (.yaml) referencing the program dependencies and samples FASTQ files paths. A detailed example and description of this file is at https://github.com/tsailabSJ/guideseq#write_manifest
Briefly, create text file with .yaml extension. In the file, follow the example in the link above, indicate the reference genome file, analysis folder and path of bwa and bedtools command. Next, indicate the path for undemultiplexed fastq files and the barcode and target sequences for each experiment. Read 2 FASTQ files must be reverse complemented if sequenced on a NextSeq or MiniSeq before running the analysis pipeline using a tool such as seqtk (https://github.com/lh3/seqtk).
56| Run the analysis with the command line:
guideseq.py all -m manifest_name.yaml
57| To check results please follow the description on GitHub:
https://github.com/tsailabSJ/guideseq#pipeline_output
? TROUBLESHOOTING
| Step | Problem | Possible Reason | Possible solution |
|---|---|---|---|
| 13 | Beads clumping during gDNA purification. Beads present in eluted sample. | Cold reagents or excessive amount of DNA | Place beads on magnet while pipetting for next step. Use binding beads at 24°C. Consider using less of starting material |
| 14 | Amount of DNA lower than 400 ng of DNA is required for the assay. | High rate of cell death after nucleofection | U2OS cells: trypsinize and plate cells one day before transfection to maximize numbers of dividing cells and minimize toxicity. |
| CD4/CD8 T cells: recover with 20% HSA and pool cells from two or more technical replicates if necessary. | |||
| 19 | dsODN incorporation rate is less than 5% | Low efficiency of integration | Optimize amount of dsODN used in the procedure. Titrate between 5 and 100 pmol for 200,000 cells. |
| Low activity of Cas9 | Optimize editing activity using a control gRNA with validated high efficiency of Cas9 cutting | ||
| 39 | Volume is too large to combine library in 5 μl | Low yield of the library | Use undiluted library components and reduce the volume by concentrating in speed-vac. |
| 57 | High-frequency unmatched off-targets observed | Low-level cross contamination of gRNA or gRNA expressing plasmid | Ensure that gRNA and gRNA expressions plasmid are purified carefully to minimize cross contamination. |
● TIMING
Step 1, gRNA synthesis: 1 w
Step 2, cell culture and nucleofection: 1 w
Steps 3–14, Genomic DNA isolation: 4–16 h
Steps 15–19, Estimate dsODN integration frequency: 4 h
Steps 20–33, GUIDE-seq library preparation: 1 d
Steps 34–52, Library quantification and sequencing: 2 d
Steps 53–57, Post run analysis: 2 h
Note that physical random shearing of DNA for GUIDE-seq library preparation is performed sequentially on a Covaris E220 shearing instrument that can handle 96 samples at a time. Time should be allotted on a per-sample basis for this step, in contrast to other library preparation steps where samples can be processed in parallel with multichannel pipettors or automated liquid handling.
Anticipated Results
3 days after nucleofection U2OS cells should reach near confluency. Trypsinizing and re-plating cells 24 h before nucleofection is critical; it should not be omitted as doing otherwise will result in suboptimal growth and DNA recovery. Purified genomic DNA concentration is expected to be 6–10 μg. CD4+/CD8+ T cells tend to be more sensitive to dsODN toxicity than U2OS cells, which might lead to lower genomic DNA yield (see step 14 Troubleshooting).
Optimized dsODN integration frequency is expected be greater than 5% and at least one third of the total indel mutagenesis rate by Cas9:gRNA as measured by NdeI restriction digest assay or targeted sequencing (Fig. 3A). Control DNA from unedited cells should always be analyzed and processed within each set of samples throughout the library preparation. In addition to dsODN incorporation by NdeI digest, another helpful quality control is estimate of DNA fragmentation size on TapeStation (Fig. 3B).
After high-throughput sequencing data is analyzed through GUIDE-seq pipeline, the results are stored in an output folder with separate subfolders for individual steps.
These two folders contain the main GUIDE-seq output.
identified contains text files created with each off-target site listed on a separate line. It is most useful to examine the identified files, sorted by target and GUIDE-seq read count (bi.sum.mi).
visualization contains final output product as *.svg image file for each target depicting on- and off-target sites and number of reads for each. This is a vector image file with *.svg extension.
filtered contains text files that list DSB sites that are identified as background based on comparison with unedited control samples.
The remaining folders contain intermediate reads and alignments that can be consulted for further analysis.
demultiplexed contains four sequence reads (forward, reversed, index1, index2) for each sample.
umitagged contains sequences with UMI tags added to each read for consolidation of duplicate reads.
consolidated contains consolidated reads in FASTQ format.
aligned contains consolidated read sequences mapped to the genome.
Examples of the visualization output is shown in Fig. 4 for two targets with low (Fig. 4A) and high (Fig. 4B) specificity. The Manhattan plot below each visualization output represents number of reads for on and off targets as distributed across chromosomal locations. The number of off-target sites detected may vary significantly for different gRNAs and this can be used to pick highly specific targets.
Figure 4. GUIDE-seq example output.
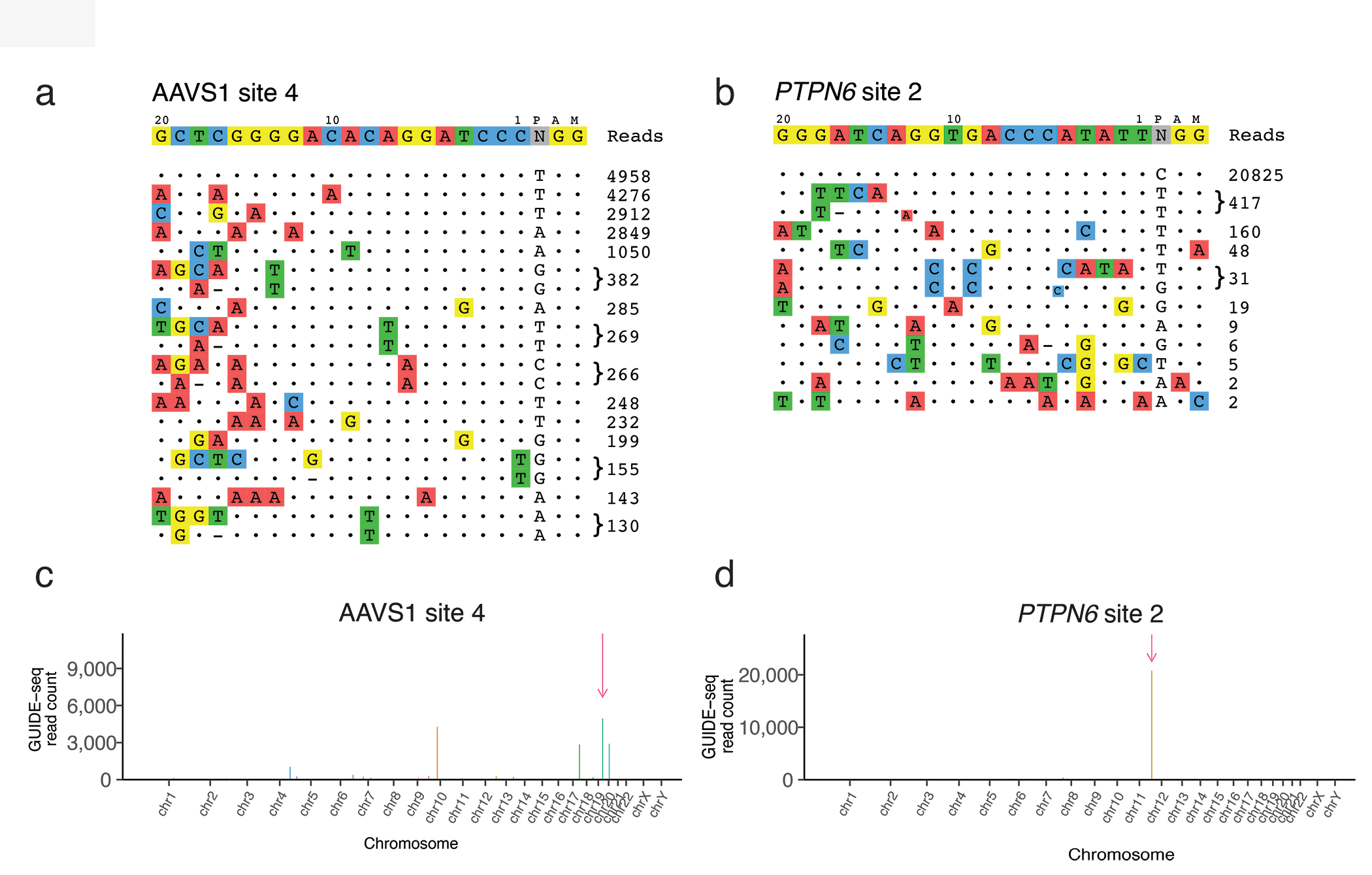
GUIDE-seq on-target and off-target detailed visualizations are shown from previously published experiments7 (a) low-specificity gRNA targeting AAVS1 site 4, (b) high-specificity gRNA targeting PTPN6 site 2. Each row represents an on-target or off-target site and GUIDE-seq read counts corresponding to that site are shown to the right. A dot represents matches with respect to the intended target sequence, and colored nucleotides indicate the sequence of mismatches. Visualizations are ordered by GUIDE-seq read counts. Manhattan plots for (c) AAVS1 site 4 and (d) PTPN6 corresponding to the same GUIDE-seq data are shown; x-axis represents chromosomal location and y-axis represents GUIDE-seq read counts. Original figure.
Supplementary Material
Acknowledgments
This work was supported by St. Jude Children’s Research Hospital and ALSAC, National Institutes of Health (NIH) Office Of The Director (OD) Somatic Cell Genome Editing (SCGE) initiative grant U01AI157189 (to S.Q.T.), St. Jude Children’s Research Hospital Collaborative Research Consortium on Novel Gene Therapies for Sickle Cell Disease (SCD), the Doris Duke Charitable Foundation (2017093), an (NIH) Directors Pioneer Award (DP1 GM105378), NIH R01 GM088040, NIH R01 AR063070, and the Jim and Ann Orr Massachusetts General Hospital (MGH) Research Scholar Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Code availability statement
Open-source GUIDE-seq analysis code is available here:
CFI
S.Q.T. is a co-inventor on patents covering the GUIDE-seq method. S.Q.T. is a member of the scientific advisory board of Kromatid. M.J.A. and J.K.J. hold equity in SeQure Dx, Inc. J.K.J. holds equity in Chroma Medicine. J.K.J. has financial interests in Beam Therapeutics, Editas Medicine, Excelsior Genomics, Pairwise Plants, Poseida Therapeutics, Transposagen Biopharmaceuticals, and Verve Therapeutics (f/k/a Endcadia). J.K.J.’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. J.K.J. is a co-inventor on various patents and patent applications that describe gene editing and epigenetic editing technologies, including the GUIDE-seq method.
Data Availability Statement
Sequencing data from experiments originally described in Lazzarotto et al.8 are available in NCBI Sequence Read Archive PRJNA625995.
References
- 1.Maeder ML & Gersbach CA Genome-editing Technologies for Gene and Cell Therapy. Mol Ther 24, 430–446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang H, Russa ML & Qi LS CRISPR/Cas9 in Genome Editing and Beyond. Annu Rev Biochem 85, 1–38 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Anderson KR et al. CRISPR off-target analysis in genetically engineered rats and mice. Nature Methods 1 (2018) doi: 10.1038/s41592-018-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai SQ et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology 33, 187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie K & Yang Y RNA-Guided Genome Editing in Plants Using a CRISPR–Cas System. Molecular Plant 6, 1975–1983 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Cheng Y & Tsai SQ Illuminating the genome-wide activity of genome editors for safe and effective therapeutics. Genome Biol 19, 226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Putney S, Benkovic S & Schimmel P A DNA fragment with an alpha-phosphorothioate nucleotide at one end is asymmetrically blocked from digestion by exonuclease III and can be replicated in vivo. Proc National Acad Sci 78, 7350–7354 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lazzarotto CR et al. CHANGE-seq reveals genetic and epigenetic effects on CRISPR–Cas9 genome-wide activity. Nat Biotechnol 38, 1317–1327 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng Z et al. Anchored multiplex PCR for targeted next-generation sequencing. Nature Medicine 20, 1479–1484 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Tsai SQ et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nature Methods 14, 607–614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wienert B et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Sci New York N Y 364, 286–289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X et al. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nature Biotechnology 33, 175–178 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Kim D et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature Methods 12, 237–243 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Yan WX et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nature Communications 8, 15058 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmid-Burgk JL et al. Highly Parallel Profiling of Cas9 Variant Specificity. Mol Cell 78, 794–800.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crosetto N et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nature Methods 10, 361–365 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L et al. Meganuclease targeting of PCSK9 in macaque liver leads to stable reduction in serum cholesterol. Nat Biotechnol 36, 717–725 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Miller JC et al. Enhancing gene editing specificity by attenuating DNA cleavage kinetics. Nat Biotechnol 37, 945–952 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Kleinstiver B et al. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nature Biotechnology 34, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nobles CL et al. iGUIDE: an improved pipeline for analyzing CRISPR cleavage specificity. Genome Biol 20, 14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walton RT, Christie KA, Whittaker MN & Kleinstiver BP Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science eaba8853 (2020) doi: 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hendel A et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology 33, 985–989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lazzarotto CR et al. Defining CRISPR–Cas9 genome-wide nuclease activities with CIRCLE-seq. Nature Protocols 13, 2615–2642 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiume M et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nature Biotechnology 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai SQ, Topkar VV, Joung KJ & Aryee MJ Open-source guideseq software for analysis of GUIDE-seq data. Nature Biotechnology 34, 483–483 (2016). [DOI] [PubMed] [Google Scholar]
Key references using this protocol:
- Tsai SQ et al. Nature Biotechnology 33, 187 (2015). doi: 10.1038/nbt.3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzarotto CR et al. Nat Biotechnology 38, 1317 (2020). doi: 10.1038/s41587-020-0555-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver PB et al. Nat Biotechnology 34 869 (2016). doi: 10.1038/nbt.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data from experiments originally described in Lazzarotto et al.8 are available in NCBI Sequence Read Archive PRJNA625995.