

Abstract
We have recently developed a new synthetic methodology that provided both N-aryl-5-hydroxytriazoles and N-pyridine-4-alkyl triazoles. A selection of these products was carried through virtual screening towards targets that are contemporary and validated for drug discovery and development. This study determined a number of potential structure target dyads of which N-pyridinium-4-carboxylic-5-alkyl triazole displayed the highest score specificity towards KAT2A. Binding affinity tests of abovementioned triazole and related analogs towards KAT2A confirmed the predictions of the in-silico assay. Finally, we have run in vitro inhibition assays of selected triazoles towards KAT2A; the ensemble of binding and inhibition assays delivered pyridyl-triazoles carboxylates as the prototype of a new class of inhibitors of KAT2A.
Keywords: N-pyridine triazoles, KAT2A inhibitors, virtual screening, acetyl transferases, anti-cancer
1. Introduction
1,2,3-triazoles are commonly recognised as important chemical scaffolds as well as efficient molecules in the area of medicinal chemistry1,2. Given their versatile behaviour in acting as both Lewis acids and bases3,4, triazoles have been used as a core structural motif in a huge variety of drug classes such as: antimicrobial5,6, anti-inflammatory7, antineoplastic8, antiviral9 ,antihypertensive10 or as immunomodulatory agents11.
We have recently reported a novel synthetic pathway that, by reacting β-ketoesters 1 and azides 2, provided 1,2,3-triazoles 3 or 4 (Scheme 1). The reactions employing 2-unsubstituted β-ketoesters were found to provide 5-methyl-1,2,3-triazoles 4; whereas 2-alkyl-substituted β-ketoesters provided 5-hydroxy 1,2,3-triazoles 3 in high yields and as a single regioisomer12.
Scheme 1.
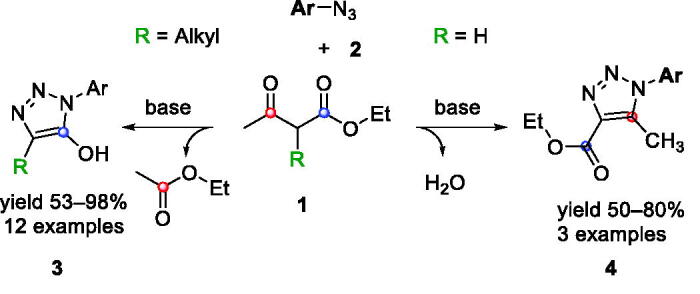
Preparation of triazoles 3 and 4.
As a follow up of this work, we have posed the question of whether or not those classes of new compounds may be useful in medicinal chemistry. Triazoles were repeatedly reported as bioactive compounds1–11 and many drug candidates containing the pyridine ring were equally described. Pyridines are commonly used in medicinal chemistry because of their ability to establish hydrogen bonds either as donors or acceptors, their water solubility, small dimensions and, most importantly, their potential to act as amide bioisosteres13. The latter in particular, makes pyridine pivotal in drug discovery14. Additionally, if compared to the benzene ring15–17, the pyridine unit displays a relevant increased basicity18, an improved aqueous solubility19 and a smaller polar surface; 20; all of these features consent an optimal orientation of the pyridine-containing drugs with the biological target through π-stacking interactions21,22. According to the FDA23, there are more than 95 approved drugs containing the pyridine moiety that are, currently, employed against tuberculosis (i.e., isoniazid24,25 5 and ethionamide26 6), HIV/AIDS (i.e., delavirdine27 7), Alzheimer disease (i.e., tacrine28 8), Raynaud’s syndrome (i.e., nifedipine29 9), hypertension (e.g., nivaldipine30 10) and so on among the others (Figure 1).
Figure 1.
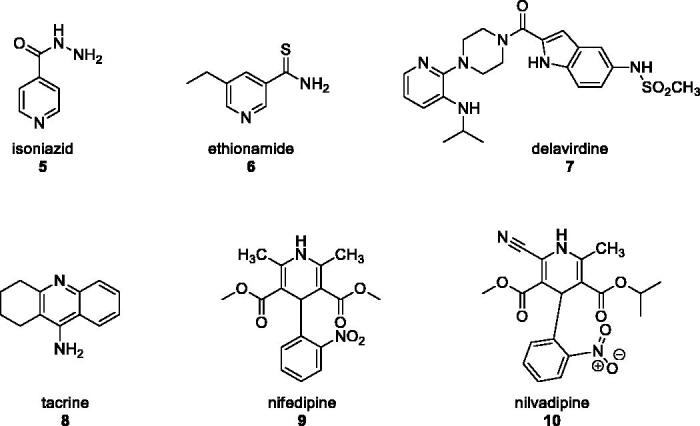
Few examples of approved drugs containing a pyridine unit.
In order to answer the research question on new triazoles’ bioactivity, we submitted a small selection of pyridine-based compounds 11–18 (Figure 2) into a virtual screening study, which identified in N-(pyridin-2-yl)-4-carboxylic-5-methyl triazole 16 (Figure 2) a new chemical template with significant activity and selectivity as KAT2A inhibitor. KAT2A, also known as GCN5 (general control nonderepressible 5), is part of the HAT (histone acetyltransferase) family and, more specifically, belongs to the GCN5-related N-acetyltransferase group31,32. KAT2A promotes the acetylation of the ε-amino group of lysines thus preventing the positive charge on the amino group to impact other proteins interaction. Furthermore, the neutralisation of lysine positive charge leads to a weaker bond with DNA strands allowing transcriptional factors to penetrate DNA itself more easily33. Dysregulation of KATs’ activity is associated with numerous severe tumours such as small-cell lung cancer34 or colon cancer35, inflammatory disorders36, type 2 diabetes37 and so on. So far, only few HATs inhibitors are available and have been approved for therapeutical use (Figure 3). They mainly derive from natural sources such as anacardic acid38 19, garcinol39,40 20 or curcumin41 21 even though synthetic products, bearing thiazole42 22, isothiazole43,44 23 and pyrido/benzothiazolone45 24 units, have been recently introduced and proved to have a higher activity and specificity compared to the natural products.
Figure 2.
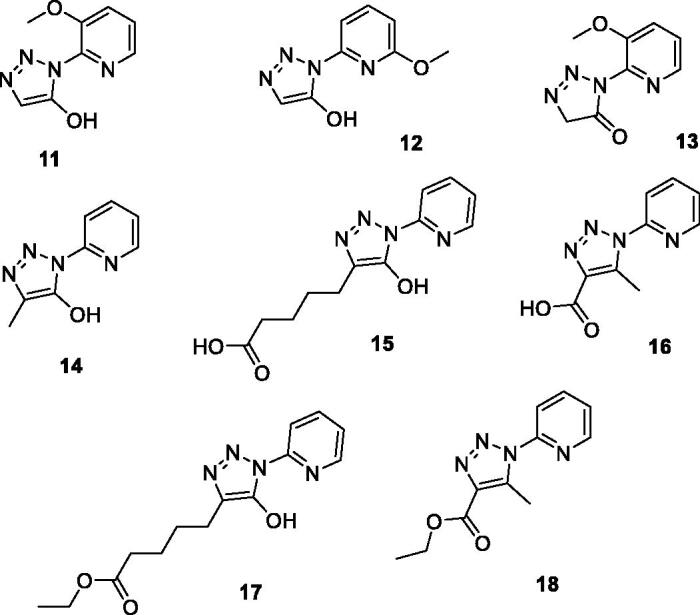
Pyridine-based triazoles tested through docking screening against the BioGPS cavity database.
Figure 3.
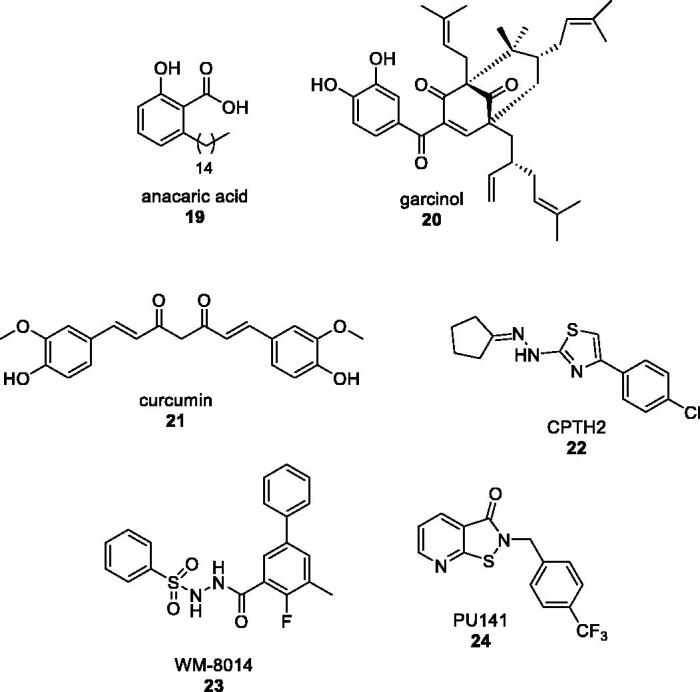
Natural and synthetic HATs inhibitors.
Unfortunately, most of the HATs inhibitors known showed lack of selectivity towards members of the HAT family and, therefore, selectivity for a specific enzyme, for example, KAT2A, is still an outstanding issue.
Herein we describe each stage of our research that led to the identification of compound 16 (Figure 2) as a new KAT2A inhibitor template. This study includes virtual screening, synthesis, binding studies and in vitro testing.
2. Results and discussion
2.1. Virtual screening
Compounds 11–18 were submitted to virtual screening towards a database of proteins with established structure and role in system biology using the BioGPS software in combination with the FLAP algorithm. The BioGPS workflow used for the docking screening of triazoles 11–18 consisted in 5 parts: i) Protein refinement, achieved by using an algorithm known as “Fixpdb” that enables the preparation of the protein structure obtained from the Protein Data Bank (PDB)46; ii) Cavity detection, achieved by using an algorithm known as “Flapsite” that allows for the identification of pocket points located within a distance of 4 Å maximum from the closest protein46; iii) and iv) Cavity characterisation and comparison, achieved by the FLAP algorithm (Fingerprints for Ligands And Proteins) that enables the identification of the potential complementary ligand pharmacophoric features for a protein binding site46,47; v) Data analysis, in which each pocket/target similarity is analysed by using the global scores that attribute to 0 no similarity and to 1 maximum similarity46. This study provided docking results and score distribution of 11–18 versus the protein panel (Table 1)46. Importantly each of the proteins listed in Table 1 is a significant target for medicinal chemistry as for example KAT2A31–37.
Table 1.
Docking scores for triazoles 11–18 found for this dataset of twenty proteins.
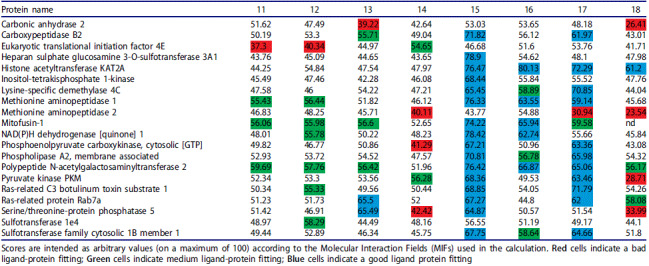
The results collated in Table 1 report the following: (1) numbers are arbitrary scores comprised between 1 and 100 and reflect the fitting of molecules 11–18 to the enzyme pocket; (2) red cells indicate a bad ligand-protein fitting; (3) green cells indicate medium ligand-protein fitting meanwhile (4) blue cells indicate a good ligand protein fitting. The results obtained showed the following: (i) triazoles 11–14, bearing a hydroxyl group at position C5, did not show significant binding scores (Table 1, except for Rab7a48 and Serine/Threonine- protein phosphatase 5 (PP5)49; (ii) long chain acid 15 showed a good fitting with sixteen proteins (Table 1) hence lacked selectivity from the premises and for this reason was discarded. In conclusion, this dataset showed that the presence of a 5-hydroxy, or its ketol form, on the triazole generated a class of compounds that were not useful for further medicinal chemistry investigations with the 20 selected proteins. Equally, compound 17 (Table 1), showed a promiscuous behaviour with up to thirteen proteins and, therefore, was abandoned.
At this point we focussed our attention on compound 16, which docking with KAT2A was reported to be peculiarly higher than any of the other binders 11–15 and 17,18, reaching 80.13 (Table 1)1. Considering that many of the known ligands for KAT2A comprise a carboxylate functionality (Figure 3), we hypothesised that the binding data for compound 16 were dependent on the presence of a carboxylic function at the C4 position which favoured the interaction of triazole 16 with the desired target (Figure 4), whereas triazole 18, bearing an ester function at C4, showed a lower binding score with KAT2A (Table 1). In support of our hypothesis, polar contacts connecting 16 and KAT2A occurred mainly when 16’s OH function of the carboxylic moiety and 16’s N2-N3 atoms of the triazole ring interacted with the NH functions of KAT2A Arg-558 residue (shown with blue and light blue sticks, Figure 4).
Figure 4.

PyMOL outlook of triazole 16 bound to KAT2A active site pocket. Triazole’s 16 nitrogens are coloured in blue, carbons in green and oxygens in red.
In particular, 16’s N2 and N3 atoms of the triazole ring coordinate arginine NH number 20289 (2.2 Å and 2.1 Å distance respectively, Figure 4) while the N3 atom of the triazole ring and the OH function of the carboxylic moiety coordinate arginine NH number 20292 (2.3 Å and 2.5 Å distance respectively, Figure 4).
The low binding-affinity of triazole 18 with all the 20 proteins and the interactions of 16 with KAT2A identified in this work (Figure 4), clearly indicated that this selection of proteins required triazole binders to possess hydrogen-bond donor and acceptors units at C4 and C5, which may have been a discriminator in the selection of targets from the BioGPS database46. In summary, the analysis of the docking studies identified 16 as a potential scaffold to be evaluated as KAT2A inhibitor. For this reason, we proceeded with the synthesis of a small library of triazole 16 analogs 26a-e and 27a-d.
2.2. Synthesis of pyridyl-triazoles 26a-f and 27a-d
A small library of 1,2,3-triazoles 26a-e and 27a-d bearing either a carboxylate or an ester functionality at C4 was therefore prepared (Table 2). We anticipated that the carboxylic functionality was indeed crucial to high binding, however, we also noted that compound 18, i.e., the ethyl ester of compound 16, showed a significant binding for KAT2A. Hence, both carboxylates and their esters were included in the library.
Table 2.
Synthesis of pyridyl-based triazoles 26a-ea.
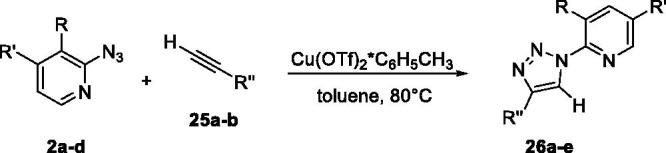
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Azide | R | R’ | Alkyne | R’’ | Product | Yield (%)b |
| 1 | 2a | H | H | 25a | COOEt | 26a | 90 |
| 2 | 2a | H | H | 25b | (CH2)4COOH | 26b | 89 |
| 3 | 2b | OCH3 | H | 25a | COOEt | 26c | 78 |
| 4 | 2c | H | CH3 | 25a | COOEt | 26d | 94 |
| 5 | 2d | H | Cl | 25a | COOEt | 26e | 99 |
aReaction conditions: 2a-d (1 equiv.), 25a,b (1.1 equiv.), Cu(OTf)2*C6H5CH3 (0.1 equiv.), toluene (0.25 M). bIsolated yields.
The synthesis of compounds 26a-e has been carried out following a procedure previously reported12. Pyridine azides 2a-e were reacted with alkynes 25a,b to give triazoles 26a-e via a CuAAc cycloaddition protocol (Table 2)50. Pyridine-based triazoles 26a-d were obtained in high yields (Table 2). Considering that carboxylates were found better ligands by virtual screening, as highlighted in the docking results (Table 1), we then proceeded with the hydrolysis of esters 26a,b and 26d,e to reveal the corresponding acids 27a-d (Table 3). This entailed standard saponification using a solution of potassium hydroxide, which provided 27a-d in good to high yields.
Table 3.
Hydrolysis of triazoles 26a,b-d,e to reveal corresponding carboxylates 27a-da.
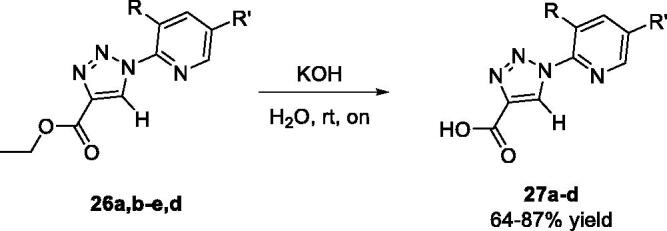
| |||||
|---|---|---|---|---|---|
| Entry | Triazole | R | R’ | Product | Yield (%)b |
| 1 | 26a | H | H | 27a | 87 |
| 2 | 26b | OCH3 | H | 27b | 70 |
| 3 | 26d | H | CH3 | 27c | 71 |
| 4 | 26e | H | Cl | 27d | 64 |
aReaction conditions: 26a,b-d,e (1 equiv.), KOH (1 equiv.), H2O (1 M).
bIsolated yields.
2.3. Preliminary binding studies through fluorescence analysis
The binding properties, alongside the ability of 16 and 27a-d to inhibit the activity of KAT2A were evaluated using a standard protocol based upon the measurement of fluorescence51. KAT2A fluorogenic assay was developed by others to screen for inhibitors of this enzyme52. This test is based on the transfer of an acetyl group from acetyl-CoA (acetyl coenzyme A) to a peptide substrate. After incubation with acetyl-CoA and the inhibitor, the KAT2A enzyme generates acetylated histone H3 peptide and CoASH. The thiol group of CoASH can be detected with fluorescein isothiocyanate isomer I (5-FITC), which is a reliable reaction considering the reactivity towards nucleophiles including amines and sulfhydryl groups present on proteins51. The fluorescein isothiocyanate isomer I (5-FITC) absorption is at wavelength = 495 nm and emission = 525 nm. In the experiment, which relies on the measurement of emission at 525 nm, strong emissions were observed when a poor inhibition of KAT2A was realised; on the contrary, poor emissions at 525 nm indicated high levels of inhibition (Figure 5). Each assay was run in triplicate and at increasing concentrations of 16, 26c and 27a-d (1.5 µM, 5 µM, 10 µM and 15 µM). As shown in Figure 5, compounds 27a and 27 b displayed a good inhibitory activity at as low as 5 µM (coloured in blue) while higher concentrations were required for triazoles 16 and 26c (15 µM, coloured in black, and 10 µM, coloured in red, respectively). Compound 27c also displayed a good inhibition of KAT2A acetylating activity at just 5 µΜ concentration (coloured in blue). A significant divergence was observed for the behaviour of 27d, which holds a p-Cl substitution on the pyridine ring. The results collected indicated that 27d seemed to promote rather than inhibit the enzymatic activity of KAT2A, as highlighted by the increasing level of CoASH produced vs concentration (Figure 5). This result was most unexpected in lieu of the striking similarity between 27d and 27a-c and it should be looked in more details in future work.
Figure 5.

KAT2A fluorescence tests performed on triazoles 16, 26c and 27a-d.
In summary, we have demonstrated that triazoles 16, 26c and 27a-c possess inhibitory activity of KAT2A whereas only 27d proved to behave as an activator. Compounds 16, 26c and 27a-d were then subjected to in vitro assays in a cell model displaying dysfunctional activity of KAT2A to confirm the bioactivity.
2.4. In vitro testing of triazoles 16, 26c and 27a-d
Compounds 16, 26c and 27a-d were tested in U937 cell line (from human myeloid leukaemia, AML) in which KAT2A is known to be overexpressed53. U937 cells were stimulated with different concentrations of compounds 16, 26c and 27a-d, starting from 200 µM. Cell viability was measured by thiazolyl blue tetrazolium bromide (MTT) assay. This assay displayed a slight reduction (10–20%) in cell viability by all the compounds tested (Figure 6).
Figure 6.

MTT assay performed on triazoles 16, 26c and 27a-d. NT = non treated.
To investigate compounds 16, 26c and 27a-d induced inhibition of KAT2A acetylation levels of histone H3K9/14ac were analysed. To this end, U937 cells were treated with a 200 µM concentration of compounds 16, 26c and 27a-d for 24 h. SAHA (suberoylanilide hydroxamic acid), a known histone deacetylase inhibitor, was used as a positive control of acetylation at 5 µM concentration. Histone extraction and subsequent Western Blot (WB) analysis were carried out checking H3K9/14ac acetylation levels. Triazole 26c showed 40% reduction in the acetylation of H3K9/14ac (Figure 7) which was the strongest inhibition value obtained.
Figure 7.

WB analysis (on the left) of 16, 26c and 27a-d showing H3K9/14Ac levels in U937 cells following 24 h treatment at the concentration of 200 µM; 5 µM SAHA treatment was used as a positive control of acetylation. Densitometric analysis of WB is shown on the right.
Considering the results obtained, it is possible that a lateral and flexible chain at C4 on the triazole ring is required to allow a tight interaction of triazoles and KAT2A. This in addition to the presence of a hydrogen-bond acceptor site which is represented by the carboxylate function.
3. Conclusion
In conclusion, we identified a new class of binders/inhibitors of KAT2A which comprises a pyridine and a long chain carboxylate linked via a triazole ring. The study used virtual screening to select a small library of pyridine-based 1,2,3-triazoles, including 26a-e and 27a-d. We then submitted triazoles 26a-e and 27a-d to fluorescence binding assays versus KAT2A enzyme which confirmed the binding abilities of these entities to KAT2A. Fluorescence binding assays revealed that only triazoles 27a-d and 26c interacted with KAT2A, meanwhile their correspondent ester analogs 26a,b,e,f did not show any binding. Finally, we evaluated the in vitro activity of 26c and 27a-d in U937 cell line of human AML and found out 26c to be the most active compound showing a 40% inhibition of KAT2A acetylating activity. It is noteworthy that compound 26c, having a longer chain, displayed the best inhibitory activity in vitro; meanwhile, shorter chain carboxylate, alike 16 or 27, that were demonstrated optimal binders, showed reduced activity compared to 26c. Studies are currently ongoing to (1) determine the structure-activity relationship (SAR) of 26c analogs and (2) improve potency and selectivity of this new template (26c) for KAT2A inhibition at a lower dose concentration.
Supplementary Material
Acknowledgements
The authors acknowledge IRC GOIPG/2018/3165 for support to RP and VALERE: “Vanvitelli per la Ricerca Program: EPInhibitDRUGre (CUP B66J20000680005) and Programma Operativo Nazionale (PON) Ricerca e Innovazione 2014–2020 AIM attrazione e mobilità dei ricercatori” for supporting NDG and GB research about KAT2A biological assays.
Funding Statement
This work was supported by Irish Research Council (GOIPG/2018/3165), Programma Operativo Nazionale Ricerca e Competitività (Ricerca e Innovazione 2014-2020 AIM), and VALERE (EPInhibitDRUGre (CUP B66J20000680005).
Note
Although the dataset predicted 16 as a good binder also for other proteins, the value of these interactions was inferior to a point that selectivity could be, at least in principle, possible.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Angell YL, Burgess K.. Peptidomimetics via copper-catalyzed azide–alkyne cycloadditions. Chem Soc Rev 2007;36:1674–89. [DOI] [PubMed] [Google Scholar]
- 2.Kharb R, Sharma PC, Yar MS, et al. . Pharmacological significance of triazole scaffold. J Enzyme Inhib Med Chem 2011;26:1–21. [DOI] [PubMed] [Google Scholar]
- 3.Schulze B, Schubert US.. Beyond click chemistry-supramolecular interactions of 1,2,3-triazoles. Chem Soc Rev 2014;43:2522–71. [DOI] [PubMed] [Google Scholar]
- 4.Palmer MH, Parsons S.. 4-Methyl-1,2,4-Triazole and 1-Methyl-Tetrazole. Acta Crystallogr Sect C Cryst Struct Commun 1996;52:2818–22. [Google Scholar]
- 5.Sheremet EA, Tomanov RI, Trukhin EV, Berestovitskaya VM.. Synthesis of 4-aryl-5-nitro-1,2,3-triazoles. Russ J Org Chem 2004;40:594–5. [Google Scholar]
- 6.Hafez HN, Abbas HAS, El-Gazzar ARBA.. Synthesis and evaluation of analgesic, anti-inflammatory and ulcerogenic activities of some triazolo- and 2-pyrazolyl-pyrido[2,3-d]-pyrimidines. Acta Pharm 2008;58:359–78. [DOI] [PubMed] [Google Scholar]
- 7.Liu K, Shi W, Cheng P.. The coordination chemistry of Zn(II), Cd(II) and Hg(II) complexes with 1,2,4-triazole derivatives. Dalton Trans 2011;40:8475–90. [DOI] [PubMed] [Google Scholar]
- 8.Passannanti A, Diana P, Barraja P, et al. . Pyrrolo[2,3-d][1,2,3]triazoles as potential antineoplastic agents. Heterocycles 1998;48:1229–35. [Google Scholar]
- 9.Johns BA, Weatherhead JG, Allen SH, et al. . The use of oxadiazole and triazole substituted naphthyridines as HIV-1 integrase inhibitors. Part 1: establishing the pharmacophore. Bioorg Med Chem Lett 2009;19:1802–6. [DOI] [PubMed] [Google Scholar]
- 10.Shalini K, Kumar N, Drabu S, Sharma PK.. Advances in synthetic approach to and antifungal activity of triazoles. Beilstein J Org Chem 2011;7:668–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindstedt R, Ruggiero V, Alessio VD, et al. . Inhibits T cell activation by reducing Nfat nuclear residency. Int J Immunopathol Pharmacol 2009;22:29–42. [DOI] [PubMed] [Google Scholar]
- 12.Pacifico R, Destro D, Gillick-Healy MW, et al. . Preparation of Acidic 5-Hydroxy-1,2,3-triazoles via the Cycloaddition of Aryl Azides with β-Ketoesters. J Org Chem 2021;86:11354–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamada Y. Role of pyridines in medicinal chemistry and design of BACE1 inhibitors possessing a pyridine scaffold. In: PP Pandey, ed. Pyridine. London, UK: IntechOpen; 2018. DOI: 10.5772/intechopen.74719 [DOI] [Google Scholar]
- 14.Asif M. Biological potential and chemical properties of pyridine and piperidine fused pyridazine compounds: pyridopyridazine a versatile nucleus. Asian J Pharm Sci 2016;1:29. [Google Scholar]
- 15.Nobeli I, Price SL, Lommerse JPM, Taylor R.. Hydrogen bonding properties of oxygen and nitrogen acceptors in aromatic heterocycles. J Comput Chem 1997;18:2060–74. [Google Scholar]
- 16.Laurence C, Brameld KA, Graton J, et al. . The pKBHX database: toward a better understanding of hydrogen-bond basicity for medicinal chemists. J Med Chem 2009;52:4073–86. [DOI] [PubMed] [Google Scholar]
- 17.Kenny PW, Montanari CA, Prokopczyk IM, et al. . Hydrogen bond basicity prediction for medicinal chemistry design. J Med Chem 2016;59:4278–88. [DOI] [PubMed] [Google Scholar]
- 18.Eicher T, Hauptmann S, Speicher A.. The chemistry of heterocycles: structures, reactions, synthesis, and applications. Weinheim, Germany: John Wiley & Sons; 2013. [Google Scholar]
- 19.Arnold DS, Plank CA, Erickson EE, Pike FP.. Solubility of benzene in water. J Chem Eng Data 1958;3:253–6. [Google Scholar]
- 20.Ertl P, Rohde B, Selzer P.. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem 2000;43:3714–7. [DOI] [PubMed] [Google Scholar]
- 21.Hohenstein EG, Sherrill CD.. Effects of heteroatoms on aromatic π-π interactions: benzene-pyridine and pyridine dimer. J Phys Chem A 2009;113:878–86. [DOI] [PubMed] [Google Scholar]
- 22.Huber RG, Margreiter MA, Fuchs JE, et al. . Heteroaromatic π-stacking energy landscapes. J Chem Inf Model 2014;54:1371–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vitaku E, Smith DT, Njardarson JT.. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J Med Chem 2014;57:10257–74. [DOI] [PubMed] [Google Scholar]
- 24.Hsu KHK. Thirty years after isoniazid: its impact on tuberculosis in children and adolescents. JAMA 1984;251:1283–5. [DOI] [PubMed] [Google Scholar]
- 25.Pym AS, Domenech P, Honoré N, et al. . Regulation of catalase-peroxidase (KatG) expression, isoniazid sensitivity and virulence by furA of Mycobacterium tuberculosis. Mol. Microbiol 2001;40:879–89. [DOI] [PubMed] [Google Scholar]
- 26.Morlock GP, Metchock B, Sikes D, et al. . ethA, inhA, and katG Loci of ethionamide-resistant clinical mycobacterium tuberculosis isolates. Antimicrob Agents Chemother 2003;47:3799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z, Vince R.. Design and synthesis of dual inhibitors of HIV reverse transcriptase and integrase: introducing a diketoacid functionality into delavirdine. Bioorg Med Chem 2008;16:3587–95. [DOI] [PubMed] [Google Scholar]
- 28.Wang L, Kumar R, Pavlov PF, Winblad B.. Small molecule therapeutics for tauopathy in Alzheimer’s disease: walking on the path of most resistance. Eur J Med Chem 2021;209:112915. [DOI] [PubMed] [Google Scholar]
- 29.Minami J, Numabe A, Andoh N, et al. . Comparison of once-daily nifedipine controlled-release with twice-daily nifedipine retard in the treatment of essential hypertension. Br J Clin Pharmacol 2004;57:632–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang JG, Kario K, Lau T, et al. . Asian Pacific Heart Association. Use of dihydropyridine calcium channel blockers in the management of hypertension in Eastern Asians: a scientific statement from the Asian Pacific Heart Association. Hypertens Res 2011;34:423–30. [DOI] [PubMed] [Google Scholar]
- 31.Voss AK, Thomas T.. Histone lysine and genomic targets of histone acetyltransferases in mammals. BioEssays 2018;40:1800078–16. [DOI] [PubMed] [Google Scholar]
- 32.Ud-Din AIMS, Tikhomirova A, Roujeinikova A.. Structure and functional diversity of GCN5-related n-acetyltransferases (GNAT). Int J Mol Sci 2016;17:1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith BC, Denu JM.. Chemical mechanisms of histone lysine and arginine modifications. Biochim Biophys Acta Gene Regul Mech 2009;1789:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen L, Wei T, Si X, et al. . Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. J Biol Chem 2013;288:14510–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin YW, Jin HJ, Zhao W, et al. . The histone acetyltransferase GCN5 expression is elevated and regulated by c-Myc and E2F1 transcription factors in human colon cancer. Gene Expr 2015;16:187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dekker FJ, Van Den Bosch T, Martin NI.. Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases. Drug Discov Today 2014;19:654–60. [DOI] [PubMed] [Google Scholar]
- 37.Sun C, Wang M, Liu X, et al. . PCAF improves glucose homeostasis by suppressing the gluconeogenic activity of PGC-1α. Cell Reports 2014;9:2250–62. [DOI] [PubMed] [Google Scholar]
- 38.Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK.. Small molecule modulators of histone acetyltransferase p300. J Biol Chem 2003;278:19134–40. [DOI] [PubMed] [Google Scholar]
- 39.Arif M, Pradhan SK, Thanuja GR, et al. . Mechanism of p300 specific histone acetyltransferase inhibition by small molecules. J Med Chem 2009;52:267–77. [DOI] [PubMed] [Google Scholar]
- 40.Balasubramanyam K, Altaf M, Varier RA, et al. . Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J Biol Chem 2004;279:33716–26. [DOI] [PubMed] [Google Scholar]
- 41.Balasubramanyam K, Varier RA, Altaf M, et al. . Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem 2004;279:51163–71. [DOI] [PubMed] [Google Scholar]
- 42.Chimenti F, Bizzarri B, Maccioni E, et al. . Novel histone acetyltransferase inhibitor modulating Gcn5 network: cyclopentylidene-[4-(4’ chlorophenyl)thiazol-2-yl)hydrazone. J Med Chem 2009;52:530–6. [DOI] [PubMed] [Google Scholar]
- 43.Ghizzoni M, Haisma HJ, Dekker FJ.. Reactivity of isothiazolones and isothiazolone-1-oxides in the inhibition of the PCAF histone acetyltransferase. Eur J Med Chem 2009;44:4855–61. [DOI] [PubMed] [Google Scholar]
- 44.Alvarez-Sánchez R, Basketter D, Pease C, Lepoittevin JP.. Studies of chemical selectivity of hapten, reactivity, and skin sensitization potency. Synthesis and studies on the reactivity toward model nucleophiles of the 13C-labeled skin sensitizers, 5-chloro-2-methylisothiazol-3-one (MCI) and 2-methylisothiazol. Chem Res Toxicol 2003;16:627–36. [DOI] [PubMed] [Google Scholar]
- 45.Furdas SD, Shekfeh S, Bissinger EM, et al. . Synthesis and biological testing of novel pyridoisothiazolones as histone acetyltransferase inhibitors. Bioorg Med Chem 2011;19:3678–89. [DOI] [PubMed] [Google Scholar]
- 46.Siragusa L, Cross S, Baroni M, et al. . BioGPS: navigating biological space to predict polypharmacology, off-targeting, and selectivity. Proteins 2015;83:517–32. [DOI] [PubMed] [Google Scholar]
- 47.Baroni M, Cruciani G, Sciabola S, et al. . A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): theory and application. J Chem Inf Model 2007;47:279–94. [DOI] [PubMed] [Google Scholar]
- 48.Davies JP, Cotter PD, Ioannou YA.. Cloning and mapping of human Rab7 and Rab9 cDNA sequences and identification of a Rab9 pseudogene. Genomics 1997;41:131–4. [DOI] [PubMed] [Google Scholar]
- 49.Hinds TD, Sánchez ER.. Protein phosphatase 5. Int J Biochem Cell Biol 2008;40:2358–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB.. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed 2002;41:2596–9. [DOI] [PubMed] [Google Scholar]
- 51.Chaganti LK, Venkatakrishnan N, Bose K.. An efficient method for FITC labelling of proteins using tandem affinity purification. Biosci Rep 2018;38:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.https://westbioscience.com/acetyltransferase/assay-kit/gcn5-fluorogenic-assay-kit-3372.html
- 53.Kikuchi H, Kuribayashi F, Kiwaki N, et al. . GCN5 regulates the superoxide-generating system in leukocytes via controlling Gp91-phox gene expression. J Immunol 2011;186:3015–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.