

Abstract
Since the emergence of the pandemic of the coronavirus disease (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the discovery of antiviral phytoconstituents from medicinal plants against SARS-CoV-2 has been comprehensively researched. In this study, thirty-three plants belonging to seventeen different families used traditionally in Saudi Arabia were tested in vitro for their ability to inhibit the SARS-CoV-2 main protease (MPRO). Major constituents of the bio-active extracts were isolated and tested for their inhibition potential against this enzyme; in addition, their antiviral activity against the SARS-CoV-2 Egyptian strain was assessed. Further, the thermodynamic stability of the best active compounds was studied through focused comparative insights for the active metabolites regarding ligand–target binding characteristics at the molecular level. Additionally, the obtained computational findings provided useful directions for future drug optimization and development. The results revealed that Psiadia punctulata, Aframomum melegueta, and Nigella sativa extracts showed a high percentage of inhibition of 66.4, 58.7, and 31.5%, against SARS-CoV-2 MPRO, respectively. The major isolated constituents of these plants were identified as gardenins A and B (from P. punctulata), 6-gingerol and 6-paradol (from A. melegueta), and thymoquinone (from N. sativa). These compounds are the first to be tested invitro against SARS-CoV-2 MPRO. Among the isolated compounds, only thymoquinone (THY), gardenin A (GDA), 6-gingerol (GNG), and 6-paradol (PAD) inhibited the SARS-CoV-2 MPRO enzyme with inhibition percentages of 63.21, 73.80, 65.2, and 71.8%, respectively. In vitro assessment of SARS-CoV-2 (hCoV-19/Egypt/NRC-03/2020 (accession number on GSAID: EPI_ISL_430820) revealed a strong-to-low antiviral activity of the isolated compounds. THY showed relatively high cytotoxicity and was anti-SARS-CoV-2, while PAD demonstrated a cytotoxic effect on the tested VERO cells with a selectivity index of CC50/IC50 = 1.33 and CC50/IC50 = 0.6, respectively. Moreover, GNG had moderate activity at non-cytotoxic concentrations in vitro with a selectivity index of CC50/IC50 = 101.3/43.45 = 2.3. Meanwhile, GDA showed weak activity with a selectivity index of CC50/IC50 = 246.5/83.77 = 2.9. The thermodynamic stability of top-active compounds revealed preferential stability and SARS-CoV-2 MPRO binding affinity for PAD through molecular-docking-coupled molecular dynamics simulation. The obtained results suggest the treating potential of these plants and/or their active metabolites for COVID-19. However, further in-vivo and clinical investigations are required to establish the potential preventive and treatment effectiveness of these plants and/or their bio-active compounds in COVID-19.
Keywords: SARS-CoV-2 main protease, coronavirus, SARS-CoV-2 Egyptian strain, thymoquinone, gardenin A, 6-gingerol, 6-paradol
1. Introduction
In December 2019, in Wuhan, China, there was an ongoing epidemic of pneumonia relevant to the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1]. There are six targets for SARS-CoV-2 that play a crucial role in the virus life cycle; the papain-like protease [2], the main protease (MPRO) [3], RNA-dependent RNA polymerase (RdRp) [4], non-structural protein (Nsp15) [5], spike protein [6], and angiotensin-converting enzyme (ACE2) [7].
The genomes of the SARS- and MERS-CoVs include two open reading frames, ORF1a and ORF1b, which are, respectively, translated by host ribosomes into two viral polyproteins, pp1a and pp1ab. Two cysteine proteases, a 3C-like protease (3CLpro) and a papain-like protease (PLpro), are encoded by ORF1a. While PLpro cleaves the polyprotein’s initial three cleavage sites, 3CLpro cleaves the other 11 sites, releasing a total of 16 non-structural proteins (nsp) for both SARS-CoV and MERS-CoV [8]. Targeting the main protease (3CL) as a primary enzyme mediated in the replication and transcription of the virus is a promising tool for the discovery of new antiviral drugs. The SARS-CoV-2 3CL (PDB ID: 6LU7) has a 96% resemblance to SARS-CoV 3CL. Since proteases are promising targets for SARS-CoV-2 replication inhibition, and amino acids Thr24, Thr26, and Asn119 3CL can play a part in binding with antiviral drugs, most of the medications currently available for the use of SARS-CoV-2 specifically work on the main protease (3CL) [9]. Natural products are a good source of bioactive antiviral compounds [10]. In this regard, diverse plants are proven to have a substantial traditional role in treating respiratory disorders and are a good source of bioactive antiviral metabolites. A number of plant-based natural compounds are under investigation, are in preclinical trials, or are in clinical trials. For example, (+)-Calanolide A from Calophyllum langigerum and SP-303 from the latex of Croton lechleri are under clinical investigation [11]. Moreover, different phyto-constituents were reported to be potent inhibitors against SARS-CoV-2, such as baicalin, ivermectin, and artemisinin. Their activity depends on the targeting of viral protease, virus entry, replication, and release from the infected cells [12]. It was also reported that thymoquinone, rosmarinic acid, ellagic acid, and thymol prevent virus entry. Meanwhile, quercetin and caffeic acid were reported as 3CL protease inhibitors [13]. To accelerate the discovery of antiviral hits against SARS-CoV-2, molecular docking for thousands of known phyto-constituents was performed on the main protease by utilizing computer-aided programs [14,15,16,17]. The compounds with high activity in virtual screening encourage researchers to test them in vitro on SARS-CoV-2 MPRO. These studies afforded many active compounds against the main protease, such as neoechinulin A from Red-Sea-derived Aspergillus fumigatus MR2012 [16], naringenin [17], and cnicin from Carduus benedictus [18]. Moreover, thirty-eight African medicinal plants traditionally used as an antiviral and immunomodulator and for treating COVID-19 symptoms were reviewed previously to open the door for drug discovery from medicinal plants [19].
In this work, in-house crude plant extracts were tested in vitro for their ability to inhibit the SARS-CoV-2 main protease (MPRO). Major constituents of the bio-active extracts were isolated and tested for their inhibitory capacity versus this enzyme as well as against the SARS-CoV-2 Egyptian strain. The most active compounds were further inspected by utilizing molecular docking and molecular dynamics to comprehend specific amino acids’ participation with the inhibitor at the active sites and to assess the thermodynamic stability.
2. Materials and Methods
2.1. Plant Material
Thirty-three plants belonging to seventeen different families were collected from Al-Taif and Al-Baha governorates between March and May 2020. Aframomum melegueta (AM-1307) was purchased from a local Saudi market. Identification of all plant samples was confirmed by members of plant taxonomy at College of Science, Jeddah University, Saudi Arabia. Voucher specimens were deposited at the herbarium of the Department of Natural Products and Alternative Medicine, College of Pharmacy, King Abdulaziz University, Jeddah, Saudi Arabia.
2.2. Preparation of the Crude Plant Extracts
Fifty grams of each dried plant material were extracted with methanol (3 × 200 mL) till exhaustion. The collected extracts were concentrated under vacuum and kept for biological study.
2.3. Isolation of Major Active Constituents
2.3.1. Isolation of 6-Gingerol and 6-Paradol
Dried pulverized seeds (500 g) of A. melegueta (Roscoe) K. Schum (AM-1307) were extracted with MeOH until exhaustion. The total MeOH extract was evaporated under vacuum to give 30 g of dark brown residue that was suspended in the least amount of water and partitioned with chloroform (CHCl3); the pooled fractions were concentrated to yield 20 g dried extract. The chloroform fraction was chromatographed on SiO2 CC (silica gel column chromatography) and gradiently eluted with n-hexane-EtOAc (5–80% v/v) to obtain ten subfractions (1–10). Subfractions 3 and 7 contained two major spots. They were separately chromatographed on SiO2 CC using n-hexane-EtOAc (9:1 v/v) and n-hexane-EtOAc (7:3 v/v) to yield 6-paradol and 6-gingerol, respectively.
2.3.2. Isolation of Gardenins A and B
Dried aerial parts of P. punctulata (500 g) (PP-1065) were extracted with methanol (2.5 L × 4). The obtained extract was evaporated to afford brown residue (50 g). This residue was suspended in water and partitioned with CHCl3. The collected fractions were concentrated to give 15 g of chloroform fraction that was then chromatographed on SiO2 CC using n-hexane-EtOAc gradient to obtain 4 major subfractions (1–4). Subfraction 1 contained two major spots that were submitted to SiO2 CC (30 g, 50 × 2 cm, n-hexane:EtOAc 95:5) to give gardenins A and B.
2.3.3. Isolation of Thymoquinone
A total of 200 g Nigella sativa (NS-0801) dried pulverized seeds (250 G) was extracted with MeOH. The concentrated total extract (20 g) was suspended in water and partitioned with CHCl3. SiO2 CC of chloroform fraction (15 g) using n-hexane-EtOAc (97:3 v/v) afforded thymoquinone.
2.4. Identification of Isolated Compounds
The isolated compounds were identified utilizing spectroscopic data (e.g., 1H and 13C) or co-TLC in addition to comparison with the published data [20,21,22]. The spectral data of isolated compounds are represented in the Supplementary Materials (Figures S1–S10).
2.5. In Vitro Screening
MPRO Protease, Untagged (SARS-CoV-2) Assay Kit, Catalog #: 78042-1, BPS Bioscience, Inc., San Diego, CA, USA), was used to investigate enzyme-inhibitory activities in vitro [23]. The inhibition was carried out using a fluorescent substrate containing the cleavage site (↓) of SARS-CoV-2 Mpro (Dabcyl-KTSAVLQ↓SGFRKM-E (Edans), MPRO protease (SARS-CoV-2 MPRO Protease), GenBank Accession No. YP 009725301, a.a. 1–306 (full length), expressed in E. coli expression system, MW 77.5 kDa, with a buffer containing 20 mM Tris, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 7.3, and GC376, an MPRO protease inhibitor with a molecular weight of 507.5 Da. The fluorescence signal of the Edans generated due to the MPRO Protease cleavage of the substrate was monitored using an Flx800 fluorescence spectrophotometer at an emission wavelength of 460 nm and an excitation wavelength of 360 nm in the FRET-based cleavage assay (BioTek, Winooski, VT, USA). Initially, 30 µL of diluted SARS-CoV-2 MPRO protease was pipetted onto a 96-well plate containing 10 µL of pre-pipetted test compounds at a final concentration of 15 ng. The mixture was incubated for 30 min at room temperature (RT) with moderate shaking. The reaction was then started by adding the substrate (10 µL) dissolved in the reaction buffer to a final volume of 50 µL, at a concentration of 40 M, then incubated at RT for 4 h with gentle shaking. The plates were then sealed with tape. A microtiter plate-reading fluorimeter capable of excitation at 360 nm and detection of emission at 460 nm was used to assess fluorescence intensity.
2.6. MTT Cytotoxicity Assay
Half-maximal cytotoxic concentration (CC50) was assessed using 3-(4, 5-dimethylthiazol -2-yl)-2, 5-diphenyltetrazolium bromide (MTT) method in VERO-E6 cells, as previously described [17,24]. Tested compounds were prepared as a stock solution in 10% DMSO in double-distilled water, and final concentration was obtained by dilution with DMEM (Dulbecco’s Modified Eagle’s Medium).
2.7. Inhibitory Concentration 50 (IC50) Determination
IC50 of isolated compounds was assessed using hCoV-19/Egypt/NRC-03/2020 (accession number on GSAID: EPI_ISL_430820) that was adsorbed on Vero-E6 cells, as previously reported [25].
2.8. Molecular Modeling Investigation
Molecular docking was performed via MOE2019 software, as previously reported [26]. In brief, ligands were sketched and minimized through 0.0001 kcal/mol.Å2 gradient applying MMFF94s forcefields with standard ionization at pH 7.40. Crystalline structures of the SARS-Cov2 MPRO proteins (PDB entries; 6W63 or 7CBT) were utilized for the non-covalent or covalent ligand docking protocols, respectively [27]. The PDB entry 7CBT was co-crystallized with the covalent inhibitor, GC376, which was used as positive control reference. However, the 6W63 PDB entry in complex with the non-covalent inhibitor, X77, was adopted for molecular docking of the isolated natural compounds against an MPRO non-covalent reference inhibitor. Both proteins were prepared through standard preparation settings of 3D_protonation under physiological pH and Amber14:EHT forcefields. Covalent docking of GC376 at MPRO was completed through acetalization reaction between the activated GC376 reactive aldehydic group and the catalytic Cys145 sulfhydryl group forming the hemi-thioacetal adduct [28]. Non-covalent docking within the MPRO putative active site was completed through rigid docking protocol via Triangular-Matcher method, initially scored via London_dG, and final re-scoring with Generalized-Born solvation IV/Weighted-Surface Area dG throughout the post-placement refinement stage.
Adopting the rigid docking protocol was rationalized since several reported analyses of the MPRO substrate-binding site considered it of limited flexibility (<2.00 Å RMSD) [29,30]. Additionally, aligning the MPRO at apo (PDB: 6M03) and holo states with either non-covalent (PDB: 6W63 or 5R7Z) or covalent binders (PDB: 7JYC, 6XHO, or 7CBT) depicted great superimposition at the alpha-carbon RMSD at 0.395 Å, 0.377 Å, 0.288 Å, 0.519 Å, and 0.503 Å, respectively. The latter indicated a non-presentable difference between both MPRO states with non-relevant impact of local-ligand-induced-fitting on MPRO-holo structures, at least within macromolecular crystallized states [31]. Validation of both docking protocols was performed through re-docking (self-docking) the co-crystallized ligands within the target’s canonical binding site [32]. Ligand’s pose prediction was completed by ranking the MOE-docking scores, RMSDs below a 2.00 Å threshold, and depicted relevant residue-wise binding interactions, as reported within literature. PyMol2.0.6 was used for pose visual inspection and binding interaction analyses [33].
GROMACS-2019 software package under CHARMM36m force field for protein and CHARMM-General Force Field program (Param-Chem project; https://cgenff.umaryland.edu/ accessed on 13 December 2021) for ligands was used to conduct the explicit molecular dynamics simulations [34,35]. Ligand–protein model was solvated within TIP3P cubic box under periodic boundary conditions with 10 Å marginal distances [36]. Protein residues were assigned at their standard ionization states (pH 7.4), while the entire system’s net charge was neutralized via potassium and chloride ions [34]. Constructed systems were minimized through 5 ps under the steepest descent algorithm [35], and they were then equilibrated for 100 ps under NVT ensemble (303.15 K) followed by 100 ps NPT ensemble (1 atm. pressure and 303.15 K) [37]. The production stage involved 100 ns MD simulation runs under NPT ensemble while using the Particle Mesh Ewald algorithm for computing the long-range electrostatic interactions [38]. Covalent bond lengths were modeled under LINCS with 2 fs integration time step size [39]. Both Coulomb’s and van der Waals’s non-bonded interactions were truncated at 10 Å using the Verlet cut-off scheme [40]. The binding-free energy between the ligand and protein, as well as the residue-wise contributions within the binding-free energy calculations, were estimated via MM/PBSA on representative frames for the whole-MD simulation runs (100 ns) [41].
2.9. Statistical Analysis
IC50s were estimated by Graph-pad-Prism 8®. One-way ANOVA followed by Tukey’s test was utilized for calculation of significant differences between means.
3. Results and Discussion
3.1. SARS-CoV-2 MPRO Inhibitory Activity of Plant Extracts and Major Isolated Constituents
Thirty-three plant extracts were screened for their inhibitory activity against SARS-CoV-2 MPRO using a FRET assay at 100 ug/mL, and GC376 was used as a positive control. The results (Table 1, Figure 1) reveal the activity of Psiadia punctulata, Echinops macrochaetus, Abutilon pannosum, Lavandula dentata, Cometes abyssinica, Aframomum melegueta, and Nigella sativa extracts with percentage inhibitions of 66.4, 8.13, 3.2, 4.43, 0.83, 58.7, and 31.5%, respectively.
Table 1.
Inhibitory activity of the tested plant extracts against SARS-CoV-2 viral main protease (SARS-CoV-2 MPRO).
| Family | Sample No. | Plant Name | Specimen Number | % Inhibition |
|---|---|---|---|---|
| Acanthaceae | 1 | Barleria trispinosa (Forssk.) Vahl | BT-1003 | - |
| Amaranthaceae | 2 | Traganum nudatum Delile | TN-0554 | - |
| Apocynaceae | 3 | Caralluma russelliana (Courbai ex Brongn.) Cufod. | CR-1168 | - |
| 4 | Leptadenia pyrotechnica (Forssk.) Decne. | LP-0840 | - | |
| 5 | Rhazya stricta Decne. | RS-1014 | - | |
| Asparagaceae | 6 | Dracaena cinnabari Balf.f. Resin | DC-1140 | - |
| Asteraceae | 7 | Conyza pyrrhopappa Sch.Bip. ex A.Rich. | CP-1060 | - |
| 8 | Echinops macrochaetus Fresen. | EM-0535 | 8.13 | |
| 9 | Lactuca serriola L. | LS-1050 | - | |
| 10 | Psiadia punctulata Vatke | PP-1065 | 66.45 | |
| 11 | Pulicaria arabica (L.) Cass. | PA-0733 | - | |
| 12 | Saussurea lappa (Decne.) Sch.Bip | SL-1050 | - | |
| 13 | Tagetes minuta L. | TM-3015 | - | |
| 14 | Verbesina encelioides (Cav.) Benth. & Hook. f. ex A.Gray | VE-1053 | - | |
| Brassicaceae | 15 | Lepidium sativum L. | LS-1040 | - |
| Capparaceae | 16 | Maerua crassifolia Forssk. | MC-1034 | - |
| Caryophyllaceae | 17 | Cometes abyssinica R. Br. Ex Wall. | CA-1315 | 0.83 |
| Cleomaceae | 18 | Cleome viscosa L. | CV-0541 | - |
| Convolvulaceae | 19 | Convolvulus glomeratus Choisy. | CG-0440 | - |
| Fabaceae | 20 | Crotalaria emarginella Vatke | CE-1119 | - |
| 21 | Pithecellobium dulce (Roxb.) Benth. | PD-1020 | - | |
| 22 | Parkinsonia aculeata L. | PA-1166 | - | |
| 23 | Tephrosia nubica (Bioss.) Baker | TN-1120 | - | |
| Lamiaceae | 24 | Lavandula dentata L | LD-1090 | 4.43 |
| 25 | Origanum majorana L. | OM-1239 | - | |
| 26 | Phlomis floccosa D.Don | PF-1140 | - | |
| Malvaceae | 27 | Abutilon pannosum (G.Forst.) Schltdl. | AP- 1041 | 3.2 |
| 28 | Triumfetta flavescens Hochst. ex A.Rich. | TF-1142 | - | |
| Peraceae | 29 | Clutia myricoides Jaub. & Spach | CM-1088 | - |
| Ranunculaceae | 30 | Nigella sativa L. seeds | NS-0801 | 31.5 |
| Solanaceae | 31 | Solanum surattense Burm. f. | SS-1141 | - |
| 32 | Withania somnifera (L.) Dunal | WS-1154 | - | |
| Zingiberaceae | 33 | Aframomum melegueta K. Schum | AM-1307 | 58.7 |
Figure 1.

Plant extracts with significant inhibitory activity against the viral protease (SARS-CoV-2 MPRO) GC376; positive control, *** significantly different at p < 0.0001, ** significantly different at p < 0.001.
The potent extracts with significant inhibition of the viral protease (Figure 1), including P. punctulate, A. melegueta, and N. sativa, were subjected to different chromatographic procedures to isolate their major constituents.
Gardenins A and B were isolated from P. punctulata. Meanwhile, 6-gingerol and 6-paradol were isolated from A. melegueta, and thymoquinone was isolated from N. sativa seeds. The isolated compounds were also tested for their inhibitory activity against SARS-CoV-2 MPRO using a FRET assay.
The results (Table 2) reveal the ability of thymoquinone, gardenin A, 6-gingerol, and 6-paradol to inhibit the SARS-CoV-2 MPRO enzyme with inhibition percentages of 63.21, 73.80, 65.2, and 71.8%, respectively.
Table 2.
Percentage inhibition of the isolated compounds from bioactive extracts against viral protease (SARS-CoV-2 MPRO).
| Compound Name | Structure | % Inhibition |
|---|---|---|
| Thymoquinone |
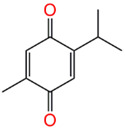
|
63.21 |
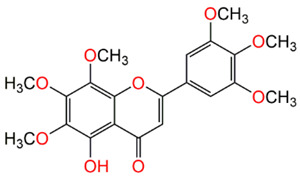
| Gardenin B |
|
1.00 |
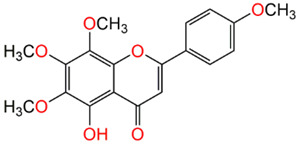
| Gardenin A |
|
73.80 |
| 6-Gingerol |
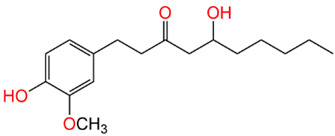
|
65.2% |
| 6-Paradol |
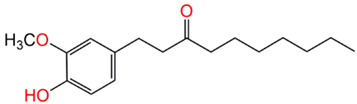
|
71.8% |
Moreover, their IC50 was assessed relative to GC376 (Standard MPRO Protease enzyme inhibitor) on the SARS-COV-2 viral main protease (Table 3 and Figure 2). 6-paradol showed the highest potency (IC50 0.1682 µM), followed by gardenin A, 6-gingerol, and thymoquinone, with an IC50 of 5.964, 9.327, and 10.26µM, respectively.
Table 3.
IC50 of isolated compounds compared to GC376 (Standard MPRO protease enzyme inhibitor) on SARS-CoV-2 viral main protease.
| Compound Name | IC50 (µM) |
|---|---|
| Thymoquinone | 10.26 |
| Gardenin A | 5.964 |
| 6-Gingerol | 9.327 |
| 6-Paradol | 0.1682 |
| GC376 (positive control) | 0.0012 |
Figure 2.


Inhibition of MPRO protease enzyme activity by isolated compounds.
3.2. Antiviral Activity of Isolated Compounds on the SARS-CoV-2 Viral Main Protease Inhibitors
Based on the obtained IC50 (Table 3), the active compounds (thymoquinone, 6-gingerol, 6-paradol, and gardenin A) were tested for their anti-viral activity on SARS-CoV-2 (hCoV-19/Egypt/NRC-03/2020 (accession number on GSAID: EPI_ISL_430820). The results (Figure 3) indicated that tested compounds displayed strong-to-low (5 µg/mL < IC50 < 100 µg/mL) antiviral activity against SARS-CoV-2.
Figure 3.

Dose-inhibition curves for bioactive compounds, where IC50 values were calculated using nonlinear regression analysis of GraphPad Prism software (version 5.01) by plotting log inhibitor versus normalized response.
Major constituents of Nigella sativa were screened virtually against 3CLpro (the main protease) and NSP15 (nonstructural protein 15 or exonuclease). The results showed that dithymoquinone has promising binding activity against these two targets, which suggests this molecule is a potential therapeutic molecule against COVID-19 [42]. Thymoquinone (THY) is one of the major constituents (28–57%) of black seed (N. sativa) oil. It has different biological activities, including cytotoxic, anti-inflammatory, anti-microbial, anti-viral, anti-oxidant, immune-stimulant, and anticoagulant effects [43]. Moreover, it has the ability to reduce the levels of pro-inflammatory mediators (ILs 2, 4, 6, and 12) and increases INF-γ [44]. THY is able to increase IgG1 and IgG2a levels as well as improve pulmonary function tests in restrictive respiratory disorders [44]. Previous in silico studies reported the ability of THY to inhibit the SARS-CoV-2 protease [45] as well as ACE2 [46].
Moreover, it has the ability to block the binding of the viral S-protein to the cellular receptor ACE2 of designed coronavirus pseudoparticles, thus blocking viral entry into the host cell. Our results proved, for the first time, the ability of THY to inhibit the SARS-CoV-2 viral main protease in vitro with IC50 10.26 µM. THY showed relatively high cytotoxicity and strong anti-SARS-CoV-2 activity with a low selectivity index (CC50/IC50 = 8.2/6.14 = 1.33).
From a clinical point of view, THY was safe at a dose of 5 mg/day to 2600 mg/day without any toxic symptoms [47]. Pharmacokinetic studies were performed after oral and IV administration, and they showed that THY has rapid-elimination properties and relatively slower absorption following oral administration [48].
6-Gingerol (GNG) and 6-paradol (PAD) are the major constituents of Aframomum melegueta seeds. The seed is traditionally used for respiratory tract infections, tuberculosis, and coughs [19]. Previous reports proved the anti-inflammatory and immunomodulatory properties of the seed [19]. Previous in silico studies and molecular dynamics showed that GNG has binding affinities of −5.60, −5.40, and −5.37 (kcal/mol) against Cathepsin K, the COVID-19 main protease, and the SARS-CoV 3 C-like protease, respectively [49]. Meanwhile, it showed low potency against SARS-CoV-2 infection with IC50 > 100 μM (CC50 > 100 μM) [50]. In this study, for the first time, GNG was tested on the SARS-CoV-2 viral main protease in vitro with IC50 9.327 µM. Although previous results showed a low potency of GNG on the virus [50], our data showed moderate activity against the SARS-CoV-2 virus at non-cytotoxic concentrations in vitro with a significant selectivity index (CC50/IC50 = 101.3/43.45 = 2.3).
Meanwhile, PAD was not tested before, in silico or in vitro, against the six targets for SARS-CoV-2. Although the obtained results showed the highest potency (IC50 0.1682 µM) against the SARS-CoV-2 viral main protease, it showed a cytotoxic effect on the tested VERO cells with a selectivity index of CC50/IC50 = 155.1/281 = 0.6, which indicated the unsuitability of this method to detect its activity.
Pharmacokinetic studies proved low levels of toxicity and high levels of tolerability of GNG in humans at doses up to 2.0 g daily, with only mild gastrointestinal complaints being reported [51]. GNG is well-absorbed after oral administration and detected in blood as glucuronide and sulfate conjugates [49,51].
Gardenin A (GDA) was not tested before for its activity on SARS-CoV-2. It showed week activity on SARS-CoV-2 virus at non-cytotoxic concentrations in vitro with a significant selectivity index (CC50/IC50 = 246.5/83.77 = 2.9).
3.3. Molecular Modeling of the In-Vitro-Active Compounds
An initial molecular docking study was conducted to investigate the differential binding poses of the in-vitro-active metabolites towards the SARS-Cov2 MPRO active site. The computational study was also beneficial for identifying the key residues involved in ligand–target binding interactions. The above in vitro reference inhibitor, GC376, in complex with MPRO (PDB: 7CBT, reported bioassay IC50 = 0.026 μM), was also adopted for the presented in silico study as a positive control. Typically, GC376 is a bispeptidyl disulfite adduct salt that is well-recognized as a broad-spectrum prodrug exerting strong inhibitory activities against coronaviruses and picornaviruses [52,53,54]. However, this positive control inhibitor has been reported to exert its MPRO inhibition through covalent interaction within the target catalytic site [28,55]. Therefore it was highly rationalized to utilize another crystallized target (PDB: 6W63) bounded to a non-covalent MPRO inhibitor, X77 (reported bioassay IC50 = 2.3 μM), as a second positive control to mimic the predicted binding interactions of our isolated plant metabolites. The latter non-proteinomimetic MPRO inhibitor is an imidazole-carboxamide-based small molecule exhibiting potent broad-spectrum activity against coronaviruses [27]. In this regard, investigating the docking binding energies (kCal/mol) and MPRO residue-wise binding interactions of the isolated compounds in relation to both covalent and non-covalent reference ligands highlight the key structural activity features required for MPRO inhibition.
Throughout the molecular docking protocols, docked ligands showed relevant anchoring at the MPRO binding site (Figure 4A). The SARS-CoV-2 protease is of similar topology as any MPRO protease enzyme, where the substrate-binding site comprises four important subsites, S1′, S2, S3, and S4, correlating to the peptide-based substrate residues (P1′, P2, P3, and P4, respective) [56]. Both 6-paradol (PAD) and 6-gingerol (GNG) depicted common conformation/orientation, with their substituted aromatic scaffold being settled at the S1 subsite while their tail was extended across the other subsites, at the end reaching towards the S3 subsite. The flavonoid-based ligand, gardenin A (GDA), predicted extended and almost linear conformation across several MPRO subsites due to its inherited structural rigidity related to its chromone ring. This ligand showed the deepest anchoring towards the S3 subsite via its tris-methoxy-substituted phenyl ring. The small-sized monoterpene molecule, thymoquinone (THY), exhibited significant orientation at the MPRO pocket, being almost limited to the S1′ and S1 subsites. Limited orientation to Mpro-pocket was also reported for small molecules docked at the Mpro pocket, which was correlated with their modest Mpro-binding affinities [57]. Both reference inhibitors illustrated significant accommodation of the MPRO pocket, with X77 being the one depicting the most extended conformation, reaching up to all four MPRO subsites via its four ring/alkyl scaffolds. On the other hand, GC376 showed an enclosed conformation, with its two terminal rings deeply anchored at the S1 subsite, while its peptidomimetic linker adopted a C-shaped twist to allow significant contact with other subsites. Nevertheless, the anchoring of GC376 was more limited at S2 and S3 subsites compared to the other MPRO subsites. Validation of the obtained binding modes was confirmed through self-docking (re-docking) studies where small root-mean-square deviations (RMSDs) were depicted for the positive controls in relation to their co-crystallized heavy atoms (0.920 Å and 1.779 Å for X77 and GC376, respectively) (Supplementary Materials, Figure S11). The latter ensured the validity and accuracy of the adopted docking protocols and algorithms in predicting the best ligand binding mode while ensuring the biological significance of these furnished poses and their respective energies [58].
Figure 4.


Ligand/MPRO binding modes and interactions. (A) Surface rendition of SARS-CoV-2 MPRO, with an overlay of docked isolated compounds (yellow lines) and potent reference (blue or orange sticks for X77 or GC376, respectively). The protein is colored in dark and light gray colors for protomer A and B, respectively, while the target binding subsites are shown in red, magenta, green, and cyan for S1′, S1, S2, and S3 subsites, respectively; (B) Docked binding modes of investigated compounds (sticks), where residues (lines) only located within 5Å radius of bound ligands are displayed, labeled by sequence numbers, and colored based on respective subsite location. Polar interactions (hydrogen bonds) are shown as dashed black lines.
The anchoring of docked ligands within the MPRO pocket was mediated through binding interactions with several key pocket residues (Table 4). Only PAD managed to depict relevant hydrogen bonding with both S1′ catalytic dyads, His 41 and Cys145, via the ligand’s carbonyl group at its aliphatic tail (Figure 4B). The latter suggested significant PAD inhibition activity of the MPRO enzymatic machinery. It has been reported that these residues correspond to strong ligand–target binding where several natural Mpro-potent inhibitors are reported, such as residue-wise binding [35,59,60,61]. Further stability of PAD at MPRO was mediated through polar contacts with the Asn142 sidechain and Phe140 mainchain via the ligand’s oxygen functionalities on its aromatic scaffold. Although GNG shared close structural similarity to PAD, the earlier ligand failed to exhibit polar contacts with the catalytic residue Cys145 sidechain while keeping its aromatic head deeply anchored at the S1 subsite. Hydrogen bond pairing for the GNG’s free hydroxyl groups with His41 and His163 sidechains was shown instead. Depicting these binding interactions could be due to the presence of extra OH at the GNG’s aliphatic linker. The latter extra functionality might impose steric hindrances on surrounding residues as well as polar attraction forces better than the linker carbonyl, causing the latter to be at the solvent side while forcing the GNG’s head deeper into the S1 subsite. The latter differential binding mode was translated into higher docking binding energies for PAD over GNG (−6.3012 vs. −5.8539 kCal/mol). Regarding the flavonoid ligand, GDA, only two hydrogen bindings were depicted for the chromone oxygen functionalities towards the His41 and Ser144 mainchains. Due to the ligand’s limited maneuvers, GDA showed no significant polar contacts with the hydrophilic residues lining the S3 subsite; this could be correlated to a docking binding energy (−5.5162 kCal/mol) comparable to GNG.
Table 4.
Ligand-target binding interaction parameters for the docked ligands and positive controls within MPRO binding site.
| Compound | Docking Energy (Kcal/mol) | H-Bond Interactions (Distance Å; Angle °) |
Hydrophobic Interactions/van der Waal with Side Chain Carbons | π-Interaction (Distance Å) |
|---|---|---|---|---|
| PAD | −6.3012 | His41 (3.2 Å; 121.6°), Cys145 (3.4 Å; 125.7°), Phe140 (1.9 Å; 129.5°), Asn142 (1.4 Å; 149.7°) |
Met49, His164, Met165, Leu167/Asp187 (Cβ,Cδ), Gln189 (Cβ,Cδ) | Phe140 (π-H) (3.7 Å) |
| GNG | −5.8539 | His41 (3.4 Å; 120.4°), His163 (3.0 Å; 161.0°) |
Met49, Phe140, Met165, His164/Asp187 (Cβ), Gln189 (Cβ) | — |
| GDA | −5.5162 | His41 (3.1 Å; 123.9°), Ser144 (2.6 Å; 141.1°) |
Met165, Leu167, His164/Glu166 (Cβ,Cδ) | Phe140 (π-H) (5.2 Å) His163 (π-π) (5.3 Å) |
| THY | −4.2899 | Gly143 (2.4 Å; 164.4°), Glu166 (1.8 Å; 132.3°) |
Phe140, Met165, His164 | His163 (π-H) (4.5 Å) |
| X77 | −8.5790 | Gly143 (2.4 Å; 145.6°), Gly143 (2.7 Å; 139.5°), His163 (3.0 Å; 142.7°), Glu166 (2.1 Å; 161.9°) |
Met49, Phe140, Leu141, His163, Met165, Leu167/ Asn142 (Cβ,Cδ), Asp187 (Cβ,Cδ), Gln189 (Cβ,Cδ) |
His164 (π-π) (5.1 Å) |
| GC376 | −9.0396 | Phe140 (1.7 Å; 139.1°) Gly143 (2.5 Å; 138.3°), Cys145 (2.3 Å; 158.3°), His164 (2.3 Å; 169.6°), His163 (2.6 Å; 130.6°), Glu166 (2.6 Å; 130.6°) |
Met49, Phe140, Leu141, His163, Met165, Leu167/ Asn142 (Cβ,Cδ), Asp187 (Cδ), Glu166 (Cβ,Cδ) |
— |
Limited docking of THY at S1′ and S1 subsites with only two polar interactions with Gly143 and Glu166 mainchains was reasonably translated to the ligand’s lowest docking energy (−4.2899 kCal/mol) among all docked ligands. Moving towards the positive control ligands, both X77 and GC376 exhibited the most extended polar contacts with several pocket residues, including His164, Cys145, Gly143, His163, Phe140, and/or Glu166, comprising a large number of MPRO subsites. The latter was translated to high docking energies of −8.5790 and −9.0396 kCal/mol for X77 and GC376, respectively. Notably, GC376 exhibited double-bonding with the catalytic Cys145, where the ligand furnished the hemi-thioacetal adduct with the residue’s SH group while depicting polar hydrogen bonding with its NH mainchain. The above docking highlights the key role of the Glu166 mainchain NH group in stabilizing both drug-like molecules (THY/X77) and the proteinomimetic ligand (GC376) at the MPRO pocket. The highlighted Glu166 role came in great agreement with several reported studies [3,30,57,59,62].
The contribution of hydrophobic interactions within the depicted docking binding scores was further evaluated through an investigation of ligand-MPRO van der Waals and π-mediated interactions. Interestingly, the top-docked ligands showed a wider range of non-polar contacts with several hydrophobic residues, including Met49, Phe140, Leu141, His163, His164, Met165, and/or Leu167. Several reports illustrated the significant role of the latter hydrophobic contacts, where His41, Met49, and Asp187 at S2 subsite as well as Met165 and Gln189 from subsite S3 served as the hydrophobic grip for pinning ligands at the MPRO target pocket [3,59,62,63]. Furthermore, almost all ligands except THY showed a significant van der Waals interaction with the carbon sidechains of lining residues (Glu166, Asp187, and Gln189), and this was more extended for PAD and both positive controls. Despite being polar or even charged at physiological pH, the latter residues could manage to achieve non-polar contacts via their sidechain Cβ and/or Cδ atoms, as previously reported in the current literature [30,63]. Finally, ligand stability was further mediated through π-driven non-polar interactions for the PAD, THY, and X77 molecules towards Phe140, His163, and/or His164 sidechains. Despite incorporating aromatic scaffolds within their molecular structures, both GNG and GC376 failed to depict relevant π-interactions due to unfavorable proximity/orientation towards the aromatic lining residues.
A molecular dynamic simulation was further performed to gain more insights regarding the differential thermodynamic behavior of the investigated ligand-MPRO complexes at near-physiological conditions. Moreover, molecular dynamics studies would provide a scientific-based approach to validate the creditability of the obtained molecular docking binding interactions [64]. The stability of the simulated ligand-MPRO complexes was illustrated by monitoring the RMSD trajectories of the combined ligand–target complex across the whole 100 ns explicit molecular dynamics simulation runs (Figure 5). Typically, RMSD measures the molecular deviation from the initial reference structure and provides a good indication of molecular stability and simulation validity. High target RMSDs confer instability and significant conformational changes [65] and correlate with weak ligand/target affinity, being incapable of accommodating the ligands within the protein’s pocket across the simulation timeframe [66].
Figure 5.

Stability analysis of the ligand-MPRO complex across the 100 ns explicit molecular dynamics simulation runs. The generated RMSD trajectories (Å) are represented across the simulation timeframes (ns).
Herein, steady RMSD tones were illustrated for PAD and X77-bound target complexes, reaching their respective dynamic equilibration plateaus after the initial 20 ns until the end of the simulation run. These depicted RMSDs were around average values of 2.58 ± 0.32 Å and 3.02 ± 0.23 Å for X77 and PAD, respectively, and were maintained for more than half of the molecular dynamic simulation runs (>75 ns). This thermodynamic behavior indicated the significant convergence and stability of these bounded MPRO proteins as well as the adequacy of minimization/equilibration stages prior to the simulation production run, requiring no further simulation time extensions. These convergence and stability findings were consistent with reported studies investigating small molecule’s affinity to the SARS-CoV-2 Mpro target through molecular docking and dynamics simulations. The depicted PAD and X77-related RMSD tones were as steady as those for investigated top-active anti-Mpro beta-blocker agents, angiotensin II receptor blockers, marine polyketides, scalarane sesterterpenes, and natural flavonoid aglycones [60,61,67,68,69]. The latter confirmed the thermodynamic stability of PAD at the Mpro pocket.
Regarding GNG- and GDA-bound complexes, the RMSDs were steady for the first half of the simulation run, and then high fluctuations were depicted, conferring great ligand-pocket instability and poor accommodation within the active site. The highest RMSD trajectory fluctuations were assigned to the monoterpene THY complex, where at early frames, the RMSDs spiked high up to ~6.00 Å and continued until the end of the simulation run. Higher RMSD fluctuations were reported as significant for the conformational shift during dynamic simulations as well as non-confinement for poor Mpro-binding ligands, as seen in studies by Al-Karmalawy and his research groups [70,71]. Moving towards the covalently bound GC376 complex, steady low RMSD trajectories (2.70 ± 0.21 Å) were depicted until 50 ns, where the tones slightly rose to ~4.00 Å, where they showed limited fluctuations until the end of the simulation run. The latter dynamic behavior highlighted a significant conformation shift for the GC376 ligand but with the retainment of the ligand at the MPRO-active site.
The above differential ligand-MPRO complex stabilities were confirmed through conformational analysis of the simulated complexes at the initial and final timelines of the simulated runs (Figure 6). Extracted and 0.00001 kcal/mol.Å2 gradient-minimized frames at 0 ns and 100 ns only showed ligand-pocket confinement for PAD-, X77-, and GC376-bound MPRO complexes. The latter came in great concordance with the above-obtained RMSD trajectories for the latter three top-stable complexes. Limited conformation alterations were depicted for X77, which corresponded to the lowest RMSD values across all simulated models. Furthermore, a significant conformational shift was illustrated for the scaffold of the GC376, where at the end of the simulation run, this functional group was shifted away from the pocket contact towards the solvent side. This could be the reason why there was a slight elevation at the RMSDs tones beyond the 60 ns timeframes. Regarding the other three simulated complexes, the bound ligands drifted away towards the solvent side for both THY and GDA at the end of their respective simulation runs. However, GNG rested at a surface cleft ~25 Å far from the canonical MPRO binding site when it reached the final simulation frame. The latter could describe the steady RMSD tones depicted for GNG across 90–100 ns timeframes (Figure 5).
Figure 6.


Conformational analysis of the simulated ligand-MPRO complexes at the start and end of the 100 ns explicit molecular dynamics simulation runs. (A) PAD; (B) GNG; (C) GDA; (D) THY; (E) X77; (F) GC376. Overlaid snapshots at 0 ns and 100 ns are presented in green and red colors, respectively; the target proteins (cartoon) and ligands (sticks) are colored corresponding to the extracted frame.
Exploring the nature of ligand-MPRO binding as well as the individual ligand contributions was finally completed by calculating the free binding energies for each simulated model using the Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) calculation [72]. While a greater negative binding energy correlates to a higher ligand–target affinity, the MM/PBSA binding energy calculation accounts for more accurate ligand–target affinity compared to static or even most sophisticated flexible molecular docking techniques. The MM/PBSA approach is considered of comparable accuracy to Free-Energy Perturbation approaches, but at much less computational expenditure [41]. To our delight, significant free binding energies were depicted for the top-stable isolated compound, PAD, in relation to the reference potent MPRO inhibitors (Table 5). Slightly high negative free binding energy was assigned to the covalent ligand over the non-covalent, which was higher than that of PAD. Moderate and comparable free binding energies were assigned to GDA and GNG, correlating to their weak affinity towards the MPRO pocket. A poor energy value was depicted for THY, which came in great agreement with the preliminary docking scores as well as the above-described RMSD and conformational analyses.
Table 5.
Binding-free energies and dissected contribution energy terms of the ligand-MPRO complexes.
| Energy (kJ/mol ± SE) |
Ligand-MPRO Complexes | |||||
|---|---|---|---|---|---|---|
| PAD | GNG | GDA | THY | X77 | GC376 | |
| van der Waal | −200.525 ± 14.859 | −208.826 ± 36.878 | −297.374 ± 20.396 | −81.297 ± 80.752 | −290.680 ± 14.694 | −338.201 ± 19.508 |
| Electrostatic | −55.142 ± 6.607 | −88.152 ± 21.814 | −96.964 ± 5.328 | −32.173 ± 31.394 | −127.985 ± 6.912 | −130.781 ± 3.844 |
| Solvation; Polar | 145.170 ± 45.751 | 228.655 ± 30.934 | 318.944 ± 16.085 | 108.316 ± 92.295 | 274.061 ± 9.408 | 340.822 ± 20.096 |
| Solvation; Apolar-SASA | −29.505 ± 2.352 | −33.672 ± 1.436 | −36.409 ± 2.272 | −9.327 ± 12.288 | −20.797 ± 1.356 | −40.481 ± 0.628 |
| Total binding energy | −140.002 ± 24.642 | −101.995 ± 30.019 | −111.803 ± 13.409 | −14.481 ± 32.140 | −165.401 ± 13.960 | −168.641 ± 16.706 |
Dissecting the total free binding energy in terms of its constituting energy terms illustrated dominant energy contributions of the van der Waal potentials over the Coulomb’s electrostatic attraction forces. This came with the cited reports that the MPRO pocket is considered a large surface area that is mainly hydrophobic in nature [3,30,57,59,62]. Due to differential energy contributions of the closely related molecules, PAD and GNG, it was noticed that higher electrostatic energy was assigned for GNG, which could be correlated with the existence of an extra free polar hydroxyl group at the GNG linker. However, this could be double-bladed since GNG was assigned a much higher polar solvation energy (almost two-fold) compared to that of PAD, which might contribute to the earlier compromised stability and solvent drift. This is highly plausible since ligand–target binding is considered a solvent-displacement process. Similar findings were also depicted for the second unstable ligand, GDA, where the presence of a high number of polar oxygen functionalities on the flavonoid scaffold contributed to both high electrostatic and unfavored polar solvation energies, with the latter compromising the ligand affinity. Despite the high polar-solvation energies of the two control inhibitors, as they harbor several hydrogen bond donors/acceptors, both ligands managed to compensate for these repulsive forces due to their aromatic and hydrophobic functionalities. This was obvious by their furnishing of the highest high van der Waal energy contributions.
The depicted double-bladed influence of polar functionalities on SARS-CoV-2 Mpro ligand binding came in agreement with reported studies. Investigated scalarane sesterterpene metabolites isolated from Hyrtios erectus marine sponge were of limited solvation penalty towards Mpro binding compared to the more hydrophilic reference control, lopinavir [63]. The authors suggested that possessing the hydrophobic cage-like sesterterpene skeleton was beneficial for limiting the solvation penalty against their preferential binding by furnishing balanced compensatory van der Waal binding potentials. Similarly, Hassan and his research group reported higher solvation energies for investigated marine natural polyketides through molecular-docking-coupled dynamics investigations [60]. The hydrophilic functionalities decorating the ligands terminal tail were suggested to impose high solvation entropy against their Mpro binding. The authors suggested future optimization of the isolated compounds through the introduction of ionizable groups with higher hydrophobic characteristics (i.e., a tetrazole scaffold). This would have been beneficial for minimizing the solvation entropy and extending ligand/Mpro binding. In this regard, the ability of the ligand to exhibit balanced hydrophobic and hydrophilic potentialities compensating for the unfavored polar solvation repulsion forces would guarantee high affinity towards the MPRO-active site.
4. Conclusions
The emergence of SARS-CoV-2 caused more than 6 million deaths, and it also had major economic consequences on various countries of the world. Further, SARS-CoV-2 virus mutation increased the virus’s capacity to spread and infect. Despite the emergence of many vaccines, the lack of an effective treatment so far may exacerbate the problem. In this scenario, broad-spectrum and safe anti-SARS-Cov-2 treatments are urgently required. Researchers in various countries of the world are struggling to find a treatment for this virus and are exploring different methods. Molecular docking is used to accelerate the discovery of antiviral hits against SARS-CoV-2 by studying the effect of known phyto-constituents as well as known antiviral drugs on different targets of the virus that play an important role in its life. In this regard, medicinal plants could be a valuable source of simple bioactive compounds. In this work, thirty-three plants belonging to seventeen different families were tested against SARS-CoV-2 MPRO using a FRET assay and GC376 as a positive control. P. punctulata, A. melegueta, and N. sativa extracts showed a high percentage of inhibition. Their isolated major constituents are gardenins A and B (from P. punctulata), 6-gingerol and 6-paradol (from A. melegueta), and thymoquinone (from N. sativa). Among them, only THY, GDA, GNG, and PAD showed SARS-CoV-2 MPRO inhibition potential. It is worth noting that these compounds were tested for the first time in vitro on SARS-CoV-2 MPRO. Moreover, the tested compounds displayed moderate-to-low antiviral activity against SARS-CoV-2 ((hCoV-19/Egypt/NRC-03/2020). PDA showed the highest potency against the enzyme, but it demonstrated a cytotoxic effect on the tested VERO cells, which indicated the unsuitability of this method to detect its activity. THY showed relatively high cytotoxicity and strong anti-SARS-CoV-2 activity with a low selectivity index. Future studies on this compound via the preparation of different derivatives may help in increasing the selectivity index. Meanwhile, GNG had moderate in vitro activity at non-cytotoxic concentrations, with a significant selectivity index. Moreover, GDA possessed weak activity against the SARS-CoV-2 virus at non-cytotoxic concentrations in vitro.
An in silico study replicated the depicted in vitro findings and showed the stability of several isolated metabolites, particularly PAD, towards the SARS-CoV-2 MPRO main enzyme. These findings introduced PAD as a promising clinical candidate for further drug optimization and development processes.
Acknowledgments
This project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, under grant no. (RG-4-130-42). The authors, therefore, acknowledge DSR’s technical and financial support with thanks. The simulations in this work were performed at King Abdulaziz University’s High Performance Computing Center (Aziz Supercomputer) (http://hpc.kau.edu.sa, accessed on 13 December 2021). The authors, therefore, acknowledge the center with thanks for the technical support.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/plants11151914/s1, Figure S1. 1HNMR spectra of paradol, Figure S2. 13CNMR spectra of paradol, Figure S3. 1HNMR spectra of gingerol, Figure S4. 13CNMR spectra of gingerol, Figure S5. 1HNMR spectra of gardenin B, Figure S6. 13CNMR spectra of gardenin B, Figure S7. 1HNMR spectra of gardenin A, Figure S8. 13CNMR spectra gardenin A, Figure S9. 1HNMR spectra of thymoquinone, Figure S10. 13CNMR spectra of thymoquinone, Figure S11. Superimposing the co-crystallized (blue sticks) and redocked (green sticks) ligands. (A) Non-covalent X77 (PDB: 6W63); (B) covalent GC376 (PDB: 7CBT) forming hemi-thioacetal adduct with Cys145 (lines) to validate the adopted docking protocols, Figure S12. Thymoquinone HPLC chromatogram, Figure S13. 6-Gingerol HPLC chromatogram, Figure S14. 6-Paradol HPLC chromatogram, Figure S15. Gardenin A (A) and gardenin B (B) HPLC chromatogram.
Author Contributions
Conceptualization, H.M.A. and A.M.E.-H.; methodology, H.M.A., A.M.E.-H., M.A.E., K.M.D., A.E.K., S.A.F. and S.S.E.; validation, A.M.E.-H., M.A.E., S.S.E., M.M.A., A.A.A. and A.E.K.; formal analysis, H.M.A.; resources, M.M.A.; writing—original draft preparation, H.M.A., M.A.E., S.S.E. and A.M.E.-H.; writing—review and editing, H.M.A., G.A.M. and S.R.M.I.; project administration, A.B.A.-N.; funding acquisition, M.M.A. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data included in the main text.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, under grant no. (RG-4-130-42). Therefore, the authors acknowledge the DSR with thanks for the technical and financial support.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zu Z.Y., Jiang M.D., Xu P.P., Chen W., Ni Q.Q., Lu G.M., Zhang L.J. Coronavirus disease 2019 (COVID-19): A perspective from China. Radiology. 2020;296:E15–E25. doi: 10.1148/radiol.2020200490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A., Schulz L., Widera M., Mehdipour A.R., Tascher G. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahlquist P. RNA-dependent RNA polymerases, viruses, and RNA silencing. Science. 2002;296:1270–1273. doi: 10.1126/science.1069132. [DOI] [PubMed] [Google Scholar]
- 5.Kim Y., Jedrzejczak R., Maltseva N.I., Wilamowski M., Endres M., Godzik A., Michalska K., Joachimiak A. Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020;29:1596–1605. doi: 10.1002/pro.3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016;3:237–261. doi: 10.1146/annurev-virology-110615-042301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shang J., Wan Y., Liu C., Yount B., Gully K., Yang Y., Auerbach A., Peng G., Baric R., Li F. Structure of mouse coronavirus spike protein complexed with receptor reveals mechanism for viral entry. PLoS Path. 2020;16:e1008392. doi: 10.1371/journal.ppat.1008392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jo S., Kim S., Shin D.H., Kim M.-S. Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzym. Inhib. Med. Chem. 2020;35:145–151. doi: 10.1080/14756366.2019.1690480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khaerunnisa S., Kurniawan H., Awaluddin R., Suhartati S., Soetjipto S. Potential inhibitor of COVID-19 main protease (Mpro) from several medicinal plant compounds by molecular docking study. Preprint. 2020:2020030226. doi: 10.20944/preprints202003.0226.v1. [DOI] [Google Scholar]
- 10.Denaro M., Smeriglio A., Barreca D., De Francesco C., Occhiuto C., Milano G., Trombetta D. Antiviral activity of plants and their isolated bioactive compounds: An update. Phytother. Res. 2020;34:742–768. doi: 10.1002/ptr.6575. [DOI] [PubMed] [Google Scholar]
- 11.Siddiqui A.J., Danciu C., Ashraf S.A., Moin A., Singh R., Alreshidi M., Patel M., Jahan S., Kumar S., Alkhinjar M.I. Plants-derived biomolecules as potent antiviral phytomedicines: New insights on ethnobotanical evidences against coronaviruses. Plants. 2020;9:1244. doi: 10.3390/plants9091244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sayed A.M., Khattab A.R., AboulMagd A.M., Hassan H.M., Rateb M.E., Zaid H., Abdelmohsen U.R. Nature as a treasure trove of potential anti-SARS-CoV drug leads: A structural/mechanistic rationale. RSC Adv. 2020;10:19790–19802. doi: 10.1039/D0RA04199H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ali S., Alam M., Khatoon F., Fatima U., Elasbali A.M., Adnan M., Islam A., Hassan M.I., Snoussi M., De Feo V. Natural products can be used in therapeutic management of COVID-19: Probable mechanistic insights. Biomed. Pharmacother. 2022;147:112658. doi: 10.1016/j.biopha.2022.112658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ullah S., Munir B., Al-Sehemi A.G., Muhammad S., Haq I.-U., Aziz A., Ahmed B., Ghaffar A. Identification of phytochemical inhibitors of SARS-CoV-2 protease 3CL-Pro from selected medicinal plants as per molecular docking, bond energies and amino acid binding energies. Saudi J. Biol. Sci. 2022;29:103274. doi: 10.1016/j.sjbs.2022.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shree P., Mishra P., Selvaraj C., Singh S.K., Chaube R., Garg N., Tripathi Y.B. Targeting COVID-19 (SARS-CoV-2) main protease through active phytochemicals of ayurvedic medicinal plants–Withania somnifera (Ashwagandha), Tinospora cordifolia (Giloy) and Ocimum sanctum (Tulsi)—A molecular docking study. J. Biomol. Struct. Dyn. 2022;40:190–203. doi: 10.1080/07391102.2020.1810778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alhadrami H.A., Burgio G., Thissera B., Orfali R., Jiffri S.E., Yaseen M., Sayed A.M., Rateb M.E. Neoechinulin A as a promising SARS-CoV-2 Mpro inhibitor: In vitro and in silico study showing the ability of simulations in discerning active from inactive enzyme inhibitors. Mar. Drugs. 2022;20:163. doi: 10.3390/md20030163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdallah H.M., El-Halawany A.M., Sirwi A., El-Araby A.M., Mohamed G.A., Ibrahim S.R., Koshak A.E., Asfour H.Z., Awan Z.A., Elfaky M.A. Repurposing of some natural product isolates as SARS-CoV-2 main protease inhibitors via in vitro cell free and cell-based antiviral assessments and molecular modeling approaches. Pharmaceuticals. 2021;14:213. doi: 10.3390/ph14030213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alhadrami H.A., Sayed A.M., Hassan H.M., Youssif K.A., Gaber Y., Moatasim Y., Kutkat O., Mostafa A., Ali M.A., Rateb M.E. Cnicin as an anti-SARS-CoV-2: An integrated in silico and in vitro approach for the rapid identification of potential COVID-19 therapeutics. Antibiotics. 2021;10:542. doi: 10.3390/antibiotics10050542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akindele A.J., Sowemimo A., Agunbiade F.O., Sofidiya M.O., Awodele O., Ade-Ademilua O., Orabueze I., Ishola I.O., Ayolabi C.I., Salu O.B. Bioprospecting for Anti-COVID-19 Interventions From African Medicinal Plants: A Review. Nat. Prod. Commun. 2022;17:1934578X221096968. doi: 10.1177/1934578X221096968. [DOI] [Google Scholar]
- 20.Abdallah H.M., Zakaria E.M., El-Halawany A.M., Mohamed G.A., Safo M.K., El-Bassossy H.M. Psiadia punctulata major flavonoids alleviate exaggerated vasoconstriction produced by advanced glycation end products. PLoS ONE. 2019;14:e0222101. doi: 10.1371/journal.pone.0222101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El Halawany A.M., Sayed N.S.E., Abdallah H.M., El Dine R.S. Protective effects of gingerol on streptozotocin-induced sporadic Alzheimer’s disease: Emphasis on inhibition of β-amyloid, COX-2, alpha-, beta-secretases and APH1a. Sci. Rep. 2017;7:2902. doi: 10.1038/s41598-017-02961-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Rafeeq M., Murad H.A.S., Abdallah H.M., El-Halawany A.M. Protective effect of 6-paradol in acetic acid-induced ulcerative colitis in rats. BMC Complement. Med. Ther. 2021;21:28. doi: 10.1186/s12906-021-03203-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu L., George S., Schmidt M.F., Al-Gharabli S.I., Rademann J., Hilgenfeld R. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antivir. Res. 2011;92:204–212. doi: 10.1016/j.antiviral.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 25.Mostafa A., Kandeil A., AMM Elshaier Y., Kutkat O., Moatasim Y., Rashad A.A., Shehata M., Gomaa M.R., Mahrous N., Mahmoud S.H. FDA-approved drugs with potent in vitro antiviral activity against severe acute respiratory syndrome coronavirus 2. Pharmaceuticals. 2020;13:443. doi: 10.3390/ph13120443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Rabia M.W., Alhakamy N.A., Ahmed O.A.A., Eljaaly K., Alaofi A.L., Mostafa A., Asfour H.Z., Aldarmahi A.A., Darwish K.M., Ibrahim T.S., et al. Repurposing of Sitagliptin- Melittin Optimized Nanoformula against SARS-CoV-2; Antiviral Screening and Molecular Docking Studies. Pharmaceutics. 2021;13:307. doi: 10.3390/pharmaceutics13030307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mesecar A.D. A Taxonomically-Driven Approach to Development of Potent, Broad-Spectrum Inhibitors of Coronavirus Main Protease Including SARS-CoV-2 (COVID-19) Center for Structural Genomics of Infectious Diseases (CSGID); Chicago, IL, USA: 2020. RcSB Protein Data Bank. [Google Scholar]
- 28.Sharun K., Tiwari R., Dhama K. Protease inhibitor GC376 for COVID-19: Lessons learned from feline infectious peritonitis. Ann. Med. Surg. 2021;61:122–125. doi: 10.1016/j.amsu.2020.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Douangamath A., Fearon D., Gehrtz P., Krojer T., Lukacik P., Owen C.D., Resnick E., Strain-Damerell C., Aimon A., Ábrányi-Balogh P., et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020;11:5047. doi: 10.1038/s41467-020-18709-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gimeno A., Mestres-Truyol J., Ojeda-Montes M.J., Macip G., Saldivar-Espinoza B., Cereto-Massagué A., Pujadas G., Garcia-Vallvé S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. Int. J. Mol. Sci. 2020;21:3793. doi: 10.3390/ijms21113793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El Raey M.A., El-Hagrassi A.M., Osman A.F., Darwish K.M., Emam M. Acalypha wilkesiana flowers: Phenolic profiling, cytotoxic activity of their biosynthesized silver nanoparticles and molecular docking study for its constituents as Topoisomerase-I inhibitors. Biocatal. Agric. Biotechnol. 2019;20:101243. doi: 10.1016/j.bcab.2019.101243. [DOI] [Google Scholar]
- 32.Almalki A.J., Ibrahim T.S., Elhady S.S., Hegazy W.A.H., Darwish K.M. Computational and Biological Evaluation of β-Adrenoreceptor Blockers as Promising Bacterial Anti-Virulence Agents. Pharmaceuticals. 2022;15:110. doi: 10.3390/ph15020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The PyMOL Molecular Graphics System, 2.0.6. Schrödinger, LLC; New York, NY, USA: 2021. [Google Scholar]
- 34.Ross G.A., Rustenburg A.S., Grinaway P.B., Fass J., Chodera J.D. Biomolecular Simulations under Realistic Macroscopic Salt Conditions. J. Phys. Chem. B. 2018;122:5466–5486. doi: 10.1021/acs.jpcb.7b11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaki A.A., Ashour A., Elhady S.S., Darwish K.M., Al-Karmalawy A.A. Calendulaglycoside A Showing Potential Activity Against SARS-CoV-2 Main Protease: Molecular Docking, Molecular Dynamics, and SAR Studies. J. Tradit. Complement. Med. 2021;12:16–34. doi: 10.1016/j.jtcme.2021.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saleh A.H., Abdelwaly A., Darwish K.M., Eissa A., Chittiboyina A., Helal M.A. Deciphering the molecular basis of the kappa opioid receptor selectivity: A Molecular Dynamics study. J. Mol. Graph. Modell. 2021;106:107940. doi: 10.1016/j.jmgm.2021.107940. [DOI] [PubMed] [Google Scholar]
- 37.Tuble S.C., Anwar J., Gale J.D. An Approach to Developing a Force Field for Molecular Simulation of Martensitic Phase Transitions between Phases with Subtle Differences in Energy and Structure. J. Am. Chem. Soc. 2004;126:396–405. doi: 10.1021/ja0356131. [DOI] [PubMed] [Google Scholar]
- 38.Darden T., York D., Pedersen L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 39.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 40.Páll S., Hess B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013;184:2641–2650. doi: 10.1016/j.cpc.2013.06.003. [DOI] [Google Scholar]
- 41.Kumari R., Kumar R., Lynn A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 42.Rizvi S.M.D., Hussain T., Moin A., Dixit S.R., Mandal S.P., Adnan M., Jamal Q.M.S., Sharma D.C., Alanazi A.S., Unissa R. Identifying the most potent dual-targeting compound (s) against 3CLprotease and NSP15exonuclease of SARS-CoV-2 from Nigella sativa: Virtual screening via physicochemical properties, docking and dynamic simulation analysis. Processes. 2021;9:1814. doi: 10.3390/pr9101814. [DOI] [Google Scholar]
- 43.Esharkawy E.R., Almalki F., Hadda T.B. In vitro Potential Antiviral SARS-CoV-19-Activity of Natural Product Thymohydroquinone and Dithymoquinone from Nigela sativia. Bioorg. Chem. 2022;120:105587. doi: 10.1016/j.bioorg.2021.105587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khazdair M.R., Ghafari S., Sadeghi M. Possible therapeutic effects of Nigella sativa and its thymoquinone on COVID-19. Pharm. Biol. 2021;59:696–703. doi: 10.1080/13880209.2021.1931353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kadil Y., Mouhcine M., Filali H. In silico investigation of the SARS CoV2 protease with thymoquinone, the Major Constituent of Nigella Sativa. Curr. Drug Disc. Technol. 2021;18:570–573. doi: 10.2174/1570163817666200712164406. [DOI] [PubMed] [Google Scholar]
- 46.Xu H., Liu B., Xiao Z., Zhou M., Ge L., Jia F., Liu Y., Jin H., Zhu X., Gao J. Computational and experimental studies reveal that thymoquinone blocks the entry of coronaviruses into in vitro cells. Infect. Dis. Ther. 2021;10:483–494. doi: 10.1007/s40121-021-00400-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Al Amri A.M., Bamosa A.O. Phase I safety and clinical activity study of thymoquinone in patients with advanced refractory malignant disease. Shiraz E. Med. J. 2009;10:107–111. [Google Scholar]
- 48.Alkharfy K.M., Ahmad A., Khan R., Al-Shagha W.M. Pharmacokinetic plasma behaviors of intravenous and oral bioavailability of thymoquinone in a rabbit model. Eur. J. Drug Metab. Pharmacokinet. 2015;40:319–323. doi: 10.1007/s13318-014-0207-8. [DOI] [PubMed] [Google Scholar]
- 49.Oso B.J., Adeoye A.O., Olaoye I.F. Pharmacoinformatics and hypothetical studies on allicin, curcumin, and gingerol as potential candidates against COVID-19-associated proteases. J. Biomol. Struct. Dyn. 2022;40:389–400. doi: 10.1080/07391102.2020.1813630. [DOI] [PubMed] [Google Scholar]
- 50.Kanjanasirirat P., Suksatu A., Manopwisedjaroen S., Munyoo B., Tuchinda P., Jearawuttanakul K., Seemakhan S., Charoensutthivarakul S., Wongtrakoongate P., Rangkasenee N. High-content screening of Thai medicinal plants reveals Boesenbergia rotunda extract and its component Panduratin A as anti-SARS-CoV-2 agents. Sci. Rep. 2020;10:19963. doi: 10.1038/s41598-020-77003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zick S.M., Djuric Z., Ruffin M.T., Litzinger A.J., Normolle D.P., Alrawi S., Feng M.R., Brenner D.E. Pharmacokinetics of 6-gingerol, 8-gingerol, 10-gingerol, and 6-shogaol and conjugate metabolites in healthy human subjects. Cancer Epidemiol. Prev. Biomark. 2008;17:1930–1936. doi: 10.1158/1055-9965.EPI-07-2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim Y., Lovell S., Tiew K.C., Mandadapu S.R., Alliston K.R., Battaile K.P., Groutas W.C., Chang K.O. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 2012;86:11754–11762. doi: 10.1128/JVI.01348-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vuong W., Khan M.B., Fischer C., Arutyunova E., Lamer T., Shields J., Saffran H.A., McKay R.T., van Belkum M.J., Joyce M.A., et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020;11:4282. doi: 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perera K.D., Galasiti Kankanamalage A.C., Rathnayake A.D., Honeyfield A., Groutas W., Chang K.O., Kim Y. Protease inhibitors broadly effective against feline, ferret and mink coronaviruses. Antivir. Res. 2018;160:79–86. doi: 10.1016/j.antiviral.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi Y., Shuai L., Wen Z., Wang C., Yan Y., Jiao Z., Guo F., Fu Z.F., Chen H., Bu Z., et al. The preclinical inhibitor GS441524 in combination with GC376 efficaciously inhibited the proliferation of SARS-CoV-2 in the mouse respiratory tract. Emerg. Microbes Infect. 2021;10:481–492. doi: 10.1080/22221751.2021.1899770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang B., He F., Liu D., Fang M., Wu Z., Xu D. AI-aided design of novel targeted covalent inhibitors against SARS-CoV-2. Biomolecules. 2022;12:746. doi: 10.3390/biom12060746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Owis A.I., El-Hawary M.S., El Amir D., Aly O.M., Abdelmohsen U.R., Kamel M.S. Molecular docking reveals the potential of Salvadora persica flavonoids to inhibit COVID-19 virus main protease. RSC Adv. 2020;10:19570–19575. doi: 10.1039/D0RA03582C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kontoyianni M., McClellan L.M., Sokol G.S. Evaluation of Docking Performance: Comparative Data on Docking Algorithms. J. Med. Chem. 2004;47:558–565. doi: 10.1021/jm0302997. [DOI] [PubMed] [Google Scholar]
- 59.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 60.El-Demerdash A., Al-Karmalawy A.A., Abdel-Aziz T.M., Elhady S.S., Darwish K.M., Hassan A.H.E. Investigating the structure-activity relationship of marine natural polyketides as promising SARS-CoV-2 main protease inhibitors. RSC Adv. 2021;11:31339–31363. doi: 10.1039/D1RA05817G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Al-Karmalawy A.A., Farid M.M., Mostafa A., Ragheb A.Y., Mahmoud S.H., Shehata M., Shama N.M.A., GabAllah M., Mostafa-Hedeab G., Marzouk M.M. Naturally Available Flavonoid Aglycones as Potential Antiviral Drug Candidates against SARS-CoV-2. Molecules. 2021;26:6559. doi: 10.3390/molecules26216559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen Y.W., Yiu C.B., Wong K.Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure. F1000Research. 2020;9:129. doi: 10.12688/f1000research.22457.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elhady S.S., Abdelhameed R.F.A., Malatani R.T., Alahdal A.M., Bogari H.A., Almalki A.J., Mohammad K.A., Ahmed S.A., Khedr A.I.M., Darwish K.M. Molecular Docking and Dynamics Simulation Study of Hyrtios erectus Isolated Scalarane Sesterterpenes as Potential SARS-CoV-2 Dual Target Inhibitors. Biology. 2021;10:389. doi: 10.3390/biology10050389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guterres H., Im W. Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J. Chem. Inf. Modeling. 2020;60:2189–2198. doi: 10.1021/acs.jcim.0c00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arnittali M., Rissanou A.N., Harmandaris V. Structure of Biomolecules through Molecular Dynamics Simulations. Procedia Comput. Sci. 2019;156:69–78. doi: 10.1016/j.procs.2019.08.181. [DOI] [Google Scholar]
- 66.Liu K., Watanabe E., Kokubo H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput. Aided Mol. Des. 2017;31:201–211. doi: 10.1007/s10822-016-0005-2. [DOI] [PubMed] [Google Scholar]
- 67.Soltan M.A., Eldeen M.A., Elbassiouny N., Kamel H.L., Abdelraheem K.M., El-Gayyed H.A., Gouda A.M., Sheha M.F., Fayad E., Ali O.A.A., et al. In Silico Designing of a Multitope Vaccine against Rhizopus microsporus with Potential Activity against Other Mucormycosis Causing Fungi. Cells. 2021;10:3014. doi: 10.3390/cells10113014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hamed M.I.A., Darwish K.M., Soltane R., Chrouda A., Mostafa A., Abo Shama N.M., Elhady S.S., Abulkhair H.S., Khodir A.E., Elmaaty A.A., et al. β-Blockers bearing hydroxyethylamine and hydroxyethylene as potential SARS-CoV-2 Mpro inhibitors: Rational based design, in silico, in vitro, and SAR studies for lead optimization. RSC Adv. 2021;11:35536–35558. doi: 10.1039/D1RA04820A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alnajjar R., Mostafa A., Kandeil A., Al-Karmalawy A.A. Molecular docking, molecular dynamics, and in vitro studies reveal the potential of angiotensin II receptor blockers to inhibit the COVID-19 main protease. Heliyon. 2020;6:e05641. doi: 10.1016/j.heliyon.2020.e05641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abo Elmaaty A., Hamed M.I.A., Ismail M.I., Elkaeed E.B., Abulkhair H.S., Khattab M., Al-Karmalawy A.A. Computational Insights on the Potential of Some NSAIDs for Treating COVID-19: Priority Set and Lead Optimization. Molecules. 2021;26:3772. doi: 10.3390/molecules26123772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Elmaaty A.A., Alnajjar R., Hamed M.I.A., Khattab M., Khalifa M.M., Al-Karmalawy A.A. Revisiting activity of some glucocorticoids as a potential inhibitor of SARS-CoV-2 main protease: Theoretical study. RSC Adv. 2021;11:10027–10042. doi: 10.1039/D0RA10674G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cavasotto C.N. Binding Free Energy Calculation Using Quantum Mechanics Aimed for Drug Lead Optimization. Methods Mol. Biol. 2020;2114:257–268. doi: 10.1007/978-1-0716-0282-9_16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data included in the main text.