

ABSTRACT
Coronavirus disease 2019 (COVID-19) pandemic is currently the most serious public health threat faced by mankind. Thus, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes COVID-19, is being intensively investigated. Several vaccines are now available for clinical use. However, owing to the highly mutated nature of RNA viruses, the SARS-CoV-2 is changing at a rapid speed. Breakthrough infections by SARS-CoV-2 variants have been seen in vaccinated individuals. As a result, effective therapeutics for treating COVID-19 patients is urgently required. With the advance of computer technology, computational methods have become increasingly powerful in the biomedical research and pharmaceutical drug discovery. The applications of these techniques have largely reduced the costs and simplified processes of pharmaceutical drug developments. Intensive and extensive studies on SARS-CoV-2 proteins have been carried out and three-dimensional structures of the major SARS-CoV-2 proteins have been resolved and deposited in the Protein Data Bank. These structures provide the foundations for drug discovery and design using the structure-based computations, such as molecular docking and molecular dynamics simulations. In this review, introduction to the applications of computational methods in the discovery and design of novel drugs and repurposing of existing drugs for the treatments of COVID-19 is given. The examples of computer-aided investigations and screening of COVID-19 effective therapeutic compounds, functional peptides, as well as effective molecules from the herb medicines are discussed.
KEYWORDS: Bioinformatics, Coronavirus disease 2019, Molecular docking, Molecular dynamics simulations, Severe acute respiratory syndrome coronavirus 2
INTRODUCTION
Coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is currently the most serious public health threat to mankind leading to a worldwide humanity disaster. SARS-CoV-2 is one of the seven coronaviruses (CoVs) known to infect human. Among the seven, four of them, human CoVs OC43, HKU1, 229E, and NL63, cause common cold [1]. Infection with SARS-CoV-1 or Middle East Respiratory Syndrome (MERS)-CoV, on the other hand, results in serious symptoms leading to high fatality rates of approximately 10% and 47% in infected individuals, respectively [2]. However, the seriousness and high mortality rates of the infections are considered disadvantages for virus transmission, as the infected individuals are less mobile, thus reducing the virus transmission efficiency and are easily identified, as a result quarantined. The SARS-CoV-2 seems to be a “smarter” virus. Infected individuals with severe symptoms account for only <20%, and the fatality rate is indicated to be approximately 1%–6% in confirmed cases, depending on the regions where the cases are reported [3,4,5,6,7]. The large proportion of the mild symptomatic and asymptomatic infections increases the difficulty in the identification of infected individuals, and a large number of infected individuals are still able to travel actively, thus speed up the disease spreading. There have been several vaccines approved by authorities worldwide for clinical use [8], and dozens of vaccine candidates are under investigations and clinical trials [9]. Although some of the vaccines were tested to be effective in preventing symptomatic infections, none of these vaccines is able to 100% terminate the virus infection. In addition, owing to the nature of single-stranded RNA viruses, the mutation rate of SARS-CoV-2 is high. The evolution of the virus might lead to decreased or lost protective efficacy of the vaccines in a very near future [9,10]. Therefore, it is always necessary to have feasible therapeutic strategies in hands for treating COVID-19 patients. With the advancement of computer technology, bioinformatic tools are nowadays very powerful in the development of pharmaceutics and have been applied in the design of therapeutics for COVID-19. This article aims to review recent studies of computer-assisted design, analysis, and developments of therapeutics against SARS-CoV-2 infections and to give examples to applications of the computational approaches related to these issues.
CURRENT UNDERSTANDING OF SEVERE ACUTE RESPIRATORY SYNDROME CORONAVIRUS 2 INFECTION AND PATHOGENESIS
It is believed that human-to-human transmission of SARS-CoV-2 is mainly through respiratory droplets and indirect contact through contaminated surfaces. SARS-CoV-2 is currently suggested to have a zoonotic origin. Sequence analysis has indicated a >75% sequence identity of the virus spike glycoprotein between SARS-CoV-2 and several bat CoVs [11], among which, bat CoV RaTG13 has the highest spike protein sequence identity of 97.56% [11]. SARS-CoV-2 and pangolin CoV share 92.3% amino acid identity in their spike proteins [12,13,14]. Despite these, there are still arguments about the roles played by bat and pangolin CoVs in SARS-CoV-2 evolution [15,16]. SARS-CoV-2, approximately 125 nm in diameter [17], contains four structural proteins, the spike, envelope, membrane (M), and nucleocapsid proteins. The virus infection to host cells is initiated when the receptor-binding domain (RBD) of spike protein engages to the host cell receptor angiotensin-converting enzyme 2 (ACE2) [18]. The spike protein is then primed by the cellular transmembrane serine protease 2 (TMPRSS2) [19] allowing the release of a fusion peptide to facilitate the SARS-CoV-2 entry into the host cells. In the host cells, the released positive-stranded virus genomic RNA is directly translated into a polyprotein by exploiting cellular machinery. The polyprotein is then processed to produce nonstructural proteins (NSPs) which form replicase-transcriptase complex. Following these processes, negative-sense RNA templates are generated for genomic RNA replication. Sub-genomic RNA then encodes structural proteins, spike, M, and envelope, which are inserted in the endoplasmic reticulum (ER), and are transported to ER-Golgi intermediate compartment (ERGIC). In cytosol, the newly synthesized viral genomic RNA is encapsulated by nucleocapsids formed by nucleocapsid protein, and the RNA containing nucleocapsids are condensed with the envelope components in ERGIC. The assembled viruses are released from the host cell through exocytosis and spread to other cells and organs [17]. The sensing of the SARS-CoV-2 infection by the host should follow the similar pathway as those for other CoVs, in which Toll-Like receptors, cytosolic retinoic acid-inducible gene I, and melanoma differentiation-associated protein are involved [14]. The sensing of the virus triggers signaling cascades resulting in the activation of immune cells, including dendritic cells, macrophages, and polymorphonuclear neutrophils, and elevated productions of complex combinations of pro-inflammatory and anti-inflammatory cytokines [14,17], including interleukin (IL)-1β, IL-1RA, IL-2RA, IL-6, IL-7, IL-8, IL-9, IL-10, basic FGF, G-CSF, GM-CSF, HGF, interferon gamma, MCP-1, MIP-1a, MIP-1b, PDGF, tumor necrosis factor-alpha (TNF-α), IP-10, and MCP-1 are measured in mild and moderate cases [17,20]. In severe cases, patients encounter a deadly acute respiratory distress syndrome (ARDS), which is thought to be caused by a cytokine storm, most likely induced by the stimulation and activation of the IL-6-STAT3 pathway or NF-κB signaling [21]. A variety of therapeutic strategies targeting the virus lifecycle or reducing effects caused by the virus are currently under investigations. As computer technology is nowadays a powerful tool for solving problems human encountered, it has currently been applied for drug design. Intensive and extensive researches have yielded structures of the host receptor ACE2, as well as most of the SARS-CoV-2 proteins with X-ray crystallography, cryo-electron microscopy (cryo-EM), or NMR experiments. These structures have been deposited in the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) (https://www.rcsb.org/), allowing computational investigation and design of possible/potential therapeutics, based on the analysis of interactions between SARS-CoV-2 and host proteins. Among many in silico approaches in used for drug discovery and modifications, molecular docking and molecular dynamics (MD) simulations are the most applied techniques and will be discussed in the next few paragraphs.
MOLECULAR DOCKING AND MOLECULAR DYNAMICS SIMULATIONS
Molecular docking is currently one of the most important computational techniques in drug discovery which allows effective investigation on interactions between two or more molecules and prediction of how these molecules fit together. The molecules studied can be proteins and their ligands, which can be small molecules, nucleic acids, or other proteins. Molecular docking is structure based. It describes the binding between ligand and protein, including orientations and poses. As a result, to perform this technique, structures of the molecules of interest are required. The protein structures can be determined by X-ray crystallography, NMR spectroscopy, cryo-EM, or computational homology modeling. In general, molecular docking includes rigid-body docking and induced-fit docking. In rigid-body docking, the ligand and protein are set rigid, thus the docking is fast and requires lower computing cost. However, it is relatively less accurate. On the other hand, in induced-fit docking, the ligand and protein are set flexible, thus the docking requires higher computing cost but is able to provide a higher accuracy. Molecular docking programs generate all possible poses, which are potential orientations and conformations of the protein interacted with its ligand(s). To find out the best fits, scoring functions are introduced. Most scoring functions are physical chemistry-based molecular mechanics force fields which calculate the free energy of the poses. The lowest calculated free energy indicates the most possible binding orientation. In general terms, the binding free energy of the protein with its ligand in solvent can be expressed as [22,23]:
△Gbind = Gcomplex - (Gprotein+Gligand) ≅ △EMM - T△S+Gpolar + Gnon-polar (1)
Where, Gcomplex is the total free energy of the protein-ligand complex, and Gprotein and Gligand are total free energies of the isolated protein and ligand in solvent, respectively; T is temperature and S denotes the entropy; EMM is the vacuum potential energy calculated based on the molecular mechanics (MM) force-field parameters. The free energy for each individual entity (Gx) can be given by [22]:
Gx =⟨EMM⟩ - TS + ⟨Gsolvation⟩ (2)
where ⟨Gsolvation⟩ is free energy of solvation.
EMM is expressed as:
EMM = Ebonded + Enon-bonded = Ebonded + (Eelec + Evdw) (3)
Where, Ebonded is the energy of bonded interactions consisting of bond, angle, dihedral, and improper interactions. The energy of nonbonded interactions (Enon-bonded) includes both electrostatic (Eelec) and van der Waals (Evdw) interactions [22]. The energy of nonbonded interactions also includes hydrogen bond energy. In a broad sense, hydrogen bond and van der Waals interactions are both dipole-dipole interactions, and in energy point of view, there is no clear “energy border” which distinguishes hydrogen bond and van der Waals interactions [24]. As a result, in many of the estimations, the hydrogen bond energy is included in the term Evdw. In EMM calculations, Eelec is modeled with the Coulomb potential function, and Evdw is modeled using the Lennard-Jones potential function, which also includes hydrogen bond energy in general force fields. In a single trajectory, the conformation of protein and ligand in the bound and unbound forms is assumed to be the same. As a result, ΔEbonded is set as zero. The solvation free energy in equation 2 can then be expressed as Gpolar and Gnon-polar (in equation 1), which are the electrostatic and nonelectrostatic contributions to the solvation free energy. ΔGpolar is electrostatic solvation energy and ΔGnon-polar is the nonelectrostatic solvation energy and is considered proportional to the solvent accessible surface area (SASA) [22,23]:
ΔGnon-polar =γ × SASA +β (4)
Molecular docking relies on approximations, and in most of the time, the receptor flexibility is not included in the docking process. As a result, in some cases, MD simulations, which calculate more detailed interaction energies, are required for providing complementary to molecular docking and more reliable results [25]. MD simulations can be used independently for investigations of conformational changes of specific molecules over a period of time [26,27]. These simulations can also be applied to optimize the structures of final complexes from molecular docking and provide insights of the ligand binding mechanism [23,25,28].
Molecular docking and MD simulations can be used to study interactions between drugs and their receptors, for providing clues for drug designs and for virtual screening of new compounds for specific protein biomolecular targets. In the following sections, the examples of applications of the computational tools in drug discovery for the treatments of COVID-19 are discussed.
REPURPOSED THERAPEUTIC COMPOUNDS
Since the first cases reported, the SARS-CoV-2 infection has spread around the world at an extremely high speed, causing serious medical and humanity crisis in almost all the regions on this planet. Finding effective therapeutic strategies for treating COVID-19 patients has thus become a crucial and urgent task. Some existing drugs, such as hydroxychloroquine (HCQ), chloroquine, remdesivir, and lopinavir/ritonavir, were repurposed and evaluated for their antiviral activities against SARS-CoV-2 [29,30,31,32]. Remdesivir [Figure 1a], a nucleoside analog clinically trialed for the treatment of Ebola virus infection previously, was designed to inhibit RNA-dependent RNA polymerases (RdRp) of viruses and also showed possible inhibitory effects on MERS and SARS. Therefore, it was considered for COVID-19 treatments. The treatment using remdesivir was found to improve the clinical condition of some COVID-19 patients [33] and was one of the only few authorized means for COVID-19 treatments. However, the actual therapeutic improvements by this drug to COVID-19 patients were found to be only slightly better than that of the patients receiving placebo [34]. A report from NIH clinical trials indicated that the treatment of remdesivir can only speed up the disease progression and shorten the recovery time in infected patients [35]. The mortality rate for the group receiving remdesivir versus that for the placebo group was not statistically different [35]. Antimalarial drugs chloroquine and [HCQ, Figure 1b] have also been proposed to be potential drugs for treating COVID-19 [32]. However, the actual antiviral mechanism of chloroquine and HCQ in the human body is still not clear, although an in vitro study suggested that they might inhibit the acidification of endosome important for virus infection and replication [36]. The effects of HCQ in clinical applications remain controversial, despite the positive results obtained in the in vitro tests. While an open-label nonrandomized clinical trial suggested that HCQ treatment was significantly associated with viral load reduction or disappearance in COVID-19 patients [37], randomized controlled open-label clinical trials indicated that the patients received HCQ did not have a lower incidence of death or an improved clinical status as compared to those who only received usual care [38,39]. In addition to RdRp, interactions between nucleoside analog drugs and the equilibrative nucleoside transporters (ENTs), which function in nucleoside and nucleobase uptake, were analyzed with computational methods. Molnupiravir [Figure 1c, EIDD-2801], a synthetic nucleoside analog originally developed to treat influenza, is currently in clinical trials for COVID-19 treatments in many countries [40,41,42] and has been approved for medical use in the United Kingdom in October 2021 [43]. Molnupiravir is able to inhibit the replication of certain RNA viruses and is suggested to have a potent ability to inhibit RdRp of SARS-CoV-2. On the other hand, Miller et al. investigated ENT-drug interactions on nucleoside analogs remdesivir, molnupiravir, and molnupiravir's metabolite β-D-N4-Hydroxycytidine (EIDD-1931) by using Bayesian machine learning models, which constructed statistical models based on Bayes’ Theorem, to identify potential interactions with the transporters [44]. Together with in vitro experiments, the authors found that remdesivir and EIDD-1931 are substrates of ENTs 1 and 2 and are potent inhibitors of ENT-mediated uridine cellular uptake [44]. SARS-CoV-2 main protease (Mpro, or 3C-like protease), which cleaves the virus polyprotein at 11 conserved sites, is a crucial enzyme for the productions of mature virus proteins. A molecular docking and MD simulation study by Mishra et al. found that HCQ/remdesivir/tetrahydrocannabinol might interact and inhibit the SARS-CoV-2 Mpro [45]. Based on the computational study, they also modified the structures of the original compounds, leading to the designs of 18 derivatives. Among these derivatives, two of them showed great affinity to the Mpro, and as a result, were suggested to have the potential to be developed into SARS-CoV-2 inhibitory drugs [45]. The outbreak of SARS in 2003 had triggered intensive research into the treatments of SARS-CoV-1 infection. A homology model of SARS-CoV-1 Mpro constructed based on the crystal structures for human coronavirus 229E Mpro was published [46], and a Mpro inhibitor rupintrivir, originally developed for the treatment of human rhinovirus, was investigated for its potential for inhibiting SARS-CoV-1 Mpro [47]. A rupintrivir derivative, PF-00835231 [Figure 1d], was designed and selected as a development candidate for SARS-CoV-1 treatments. However, the project was ended with the ending of 2003 pandemic. Following the COVID-19 outbreak, PF-00835231 was again considered as a promising drug and has been tested for its inhibitory activity against SARS CoV-2 Mpro. A cocrystal structure (PDB: 6XHL) with PF-00835231 bound in the Mpro active site has been solved [47], and the preclinical characterization of PF-00835231 and its prodrug PF-07304814 [lufotrelvir, Figure 1d] has been published [48]. This drug is currently under clinical trials [49]. PF-07321332 [Figure 1e] is developed as an orally administered SARS-CoV-2 inhibitor by Pfizer, Inc. and is currently under clinical trials [50]. The binding mechanism of PF-07321332 onto Mpro has been investigated with MD and binding-free energy simulations [51] and its affinity toward the Mpro were found to be greater than those of α-ketoamide, lopinavir, and ritonavir [51]. These analyses might be helpful for future development and optimization of specific compounds targeting COVID.
Figure 1.

Structures of representative potential antiviral compounds against severe acute respiratory syndrome coronavirus 2. (a) remdesivir. (b) hydroxychloroquine. (c) molnupiravir. (d) PF-00835231 (R1: hydroxyl group)/PF-07304814 (R1: phosphate). (e) PF-07321332
The development of novel therapeutic drugs is a long and complex process and is sometimes too slow to deal with emerging health threats. In addition, as the drug development costs are high and most of the drug development failed in between the drug discovery and being put on market, the pharmaceutical industries normally invest into new drug developments with great cautiousness and are sometimes reluctant to do so. Repurposing existing drugs for new applications is therefore considered as a practical and fast-track approach for combating newly emerging medical situations [52], because of the fact that these drugs have already been tested for their safety and fulfilled many of the requirements set by the authorities. In 2021, a study carried out by Jang et al. [53] virtually screened 6218 approved and clinical trial drugs against Mpro and RdRp of SARS-CoV-2 using molecular docking and MD simulations. They also introduced a filtering strategy to reduce false-positive results. This mentioned study identified 15 and 23 potential drug candidates targeting the Mpro structure [Figure 2a, PDB: 6Y2F [54]] and the RdRp structure [Figure 2b, PDB: 6M71 [55]], respectively. Cellular experiments showed that 7 of these drugs were able to inhibit SARS-CoV-2 replication in Vero cells, and 3 of them, emodin, omipalisib, and tipifarnib, showed inhibitory effects on SARS-CoV-2 in human lung cell line Cali-3 [53]. It was also found that the anti-SARS-CoV-2 activity of omipalisib [an anti-cancer PI3K/mTOR inhibitor also known as GSK2126458, Figure 3a] is much greater than that of remdesivir in Calu-3 cells [53]. As mentioned previously, the deadly ARDS in severe COVID-19 patients is caused by cytokine storm. As a result, COVID-19 associated cytokines and their receptors are also considered as targets for drug developments. An in silico study indicated that the FDA approved drugs rifampicin [Figure 3b] and letermovir [Figure 3c] have the potential to be repurposed for COVID-19 treatments [56]. In addition to targeting Mpro, these two drugs were found to show excellent affinity to TNF-α, IL-6, and IL-1β [56]. Computational methods were also applied for large-scale screening of potential lead drugs. A method of deep representation learning on heterogeneous drug networks has been established for the discovery of anti-inflammatory agents for COVID-19 patients [57]. With this method, 22 anti-inflammatory drugs for COVID-19 were identified, in which 9 of them were suggested to be involved in TNF-α related mechanism, 12 of them interact with mechanisms related to IL-6, and a drug acarbose binds to both TNF-α and IL-6 [57]. The TMPRSS2 enzyme of SARS-CoV-2 is also considered a target for the suppression of virus infection. Elbadwi et al. applied structure-based virtual screening to search for drugs with the potential to target SARS-CoV-2 TMPRSS2, and identified 5 commercially available drugs amikacin, isepamicin, butikacin, lividomycin, and paromomycin, possibly having inhibitory abilities against the TMPRSS2 enzyme [58]. Hamdy et al. applied an iterated virtual screening method to re-screened Mpro effective compounds against a TMPRSS2 structure (PDB: 2OQ5) using molecular docking [59]. After MD simulations, five compounds were identified to possess dual-binding affinity to Mpro and TMPRSS2, and one of them were tested to exhibit an improved in vitro antiviral activity and safety [59].
Figure 2.
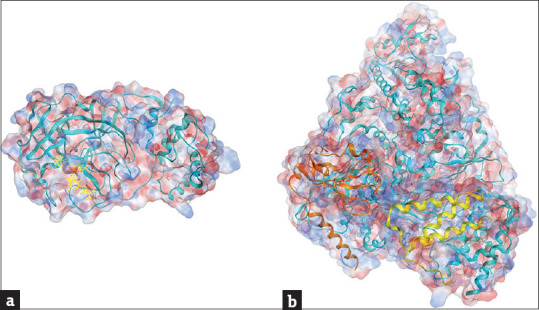
Resolved structures of severe acute respiratory syndrome coronavirus 2 Mpro and RdRp. (a) A crystal structure of Mpro (PDB: 6Y2F [54]). The structure in cyan color is the Mpro; the molecule in yellow color is a α-ketoamide inhibitor binding to the protein. (b) A cryo-EM structure (PDB: 6M71 [55]) of RdRp (cyan) in complex with co-factors non-structural protein 7 (yellow) and non-structural protein 8 (brown). Blue and red colors on the sphere presentation of the protein structures indicate the positive and negative charged force fields, respectively
Figure 3.

Examples of approved drugs with potential for repurposing to treat severe acute respiratory syndrome coronavirus 2 infection. (a) omipalisib. (b) rifampicin. (c) letermovir
Because of the nature of RNA viruses, the SARS-CoV-2 is a virus with a high mutation rate. The changes in its genomic RNA sequence cause the changes in the structures of target viral proteins, leading to the possible losses of efficacies of vaccines and therapeutics in use or under clinical trials. Efficient approaches for creating the structures of mutant proteins will provide great help in the future development of vaccines and therapeutics for emerging and mutated viruses. Alphafold by DeepMind, now part of Google's parent firm, is a sequence-based artificial intelligence (AI) algorithmic prediction tool for constructing tertiary structures of proteins with outstanding accuracy [60,61]. Robertson et al. created the structure models of SARS-CoV-2 Mpro with the Alphafold2 (Alphafold version 2) program and evaluated the concordance of the X-ray and AlphaFold models of Mpro with the results from residual dipolar couplings measured in solution [62]. The results showed that although the structures from X-ray crystallography and Alphafold predictions were similar, as compared to the best crystal structures, AlphaFold Mpro models agreed more closely with the experimental results of solution residual dipolar couplings [62], suggesting that the AI tools can provide new opportunities for structure-based analysis and simulations for drug discovery and design. SARS-CoV-2 uses its NSP6 to interact with the host cell sigma receptors involved in lipid remodeling and ER stress response. Pandey et al. utilized an Alphafold created NSP6 structure to study the binding mechanism of dextromethorphan, a cough suppressant, and haloperidol, an antipsychotic drug, unto the NSP6 with molecular docking and MD simulations [63]. It was found that the binding of dextromethorphan, identified previously to have pro-viral activity [64], destabilized the structure of drug-NSP6 complex and led to an increase in conformational dynamics and energetic frustrations [63]. On the other hand, the strong binding of the haloperidol, found to be antiviral, caused minimal structural and dynamical perturbations to NSP6 [63]. As a result, haloperidol was concluded in this mentioned study to be a potential candidate drug for COVID-19. The M protein of SARS-CoV-2 is crucial for virus assembly, and is also considered as a drug target. Peele et al. [65] applied an Alphafold created SARS-CoV-2 M protein structure to screen approved drugs in SuperDRUG2 database for drug repurposing. A total of 3639 SuperDRUG2 database drugs and 14 potential SARS-CoV-2 drugs were selected for examinations. After molecular docking screening, nine drugs were found to bind to the M protein active site. MD simulation analyses and binding free energy calculations suggested that 4 of the 9 bound to M protein with desired binding stability. Among these four, colchicine, normally used to treat gout flares and Familial Mediterranean fever, was found to be the top most binder to the M protein [65]. Because of this, the authors searched colchicine-like substructures in PubChem database (https://pubchem.ncbi.nlm.nih.gov/) for the identification of effective compounds with less toxicities. Among 683 compounds retrieved, 10 were found to have better binding affinity to the M protein than colchicine, as revealed by docking analyses. The pharmacokinetic properties of these compounds were further calculated with an online software SwissADME [66], and the calculations indicated that 4 of the compounds display comparable pharmacokinetic properties with that of colchicine. The compound with PubChem ID 6711380 (IUPAC: N-[(7S)-1,2,10-trimethoxy-9-oxo-3-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl] oxy-6,7-dihydro-5H-benzo[a]heptalen-7-yl]acetamide) was calculated to be the best among all selected derivatives [65].
For the screening and study of repurposed drugs against COVID-19, computational tools have proved themselves to be powerful aids in understanding drug-target interactions, revealing unknown mechanisms of known effective drugs, finding new functions of existing drugs, as well as screening of possible drug candidates from drug libraries.
THERAPEUTIC PEPTIDES
Although there are currently effective monoclonal antibody therapies developed for direct targeting the virus spike protein [67], these therapies are extremely expensive. As a result, they may not be the solutions for the worldwide crisis. Relatively inexpensive therapeutics is required. Peptides are biomaterials assembled with amino acids. As they can be easily chemically synthesized with different amino acid sequences, functional peptides are emerging as a popular group of agents for therapeutic purposes. There have been attempts in designing peptides for interrupting the interactions between host ACE2 and SARS-CoV-2 spike protein by using computational approaches. Han and Král [68] analyzed the interactions between host ACE2 and virus spike protein RBD interface using a resolved complex structure [Figure 4a], PDB: 6M17 [69]]. They identified that in total 15 residues of ACE2 interact with the virus spike protein RBD. These residues include Q24, T27, D30, K31, H34, E35, E37, D38, Y41, and Q42 of α1 helix, M82 of α2 helix, K353, G354, D355, and R357 from the linker between β3 and β4. They designed four inhibitory peptides [inhibitor 1–4 in Table 1]. By using classical MD simulations, they found that the inhibitors formed by two sequential self-supporting α-helices (α1 and α2) derived from the protease domain of ACE2 bind to the SARS-CoV-2 spike protein RBD, and the α-helical peptides maintain their secondary structure and provide a highly specific and stable binding [68]. On the other hand, Cao et al. applied two strategies for the design of mini-proteins to neutralize the SARS-CoV-2 spike protein RBD [70]. They firstly designed mini-protein incorporated with a derived helix of ACE2 (residues from 23 to 46) responsible for the interactions with the virus RBD by using the Rosetta blueprint builder [73]. They also de novo designed RBD-binding proteins by using rotamer interaction field docking [74] with large in silico mini-protein libraries [75], followed by the design to generate binders to the distinct regions of the RBD surface [70], the sequences of peptides designed are shown in Table 1 (AHB1-2, LCB1-8). The neutralization activity of the designed mini-proteins was experimentally measured with a focus reduction neutralization test on cell monolayers. Effective concentration (EC50) values of less than 50 nM were achieved [70]. Karoyan et al. designed human ACE2 peptide-mimics composed of 27 residues based on the computational analysis of a crystal structure [Figure 4b, PDB: 6M0J] of SARS-CoV-2 spike protein RBD bound with ACE2. They identified that amino acid sequence from S19 to L45 of the hACE2 H1 helix interacts with SARS-CoV-2 spike protein, and 12 residuals in the sequence are important for the interaction. From these finding, they designed/optimized 12 peptide mimics for in vitro and cellular experimental tests. 3 peptide-mimics [P8-10 in Table 1] were found to be able to block SARS-CoV-2 pulmonary cell infection with an inhibitory concentration (IC50) of within nanomolar range [71]. For screening of peptides, Chitsike et al. designed several candidate peptides [72] from motifs in ACE2 and spike protein RBD by analyzing a crystal complex structure (PDB: 6LZG). Peptides with and without modifications (indicated with # in Table 1) to the native sequences were screened for their inhibitory potential to ACE2-RBD binding with a proximity-based AlphaScreen™ assay [72]. The sequence between the 21th amino acid to the 45th amino acid of ACE2 is commonly found in the results from different research groups to interact with SARS-CoV-2 spike protein RBD and should be an important consideration for future peptide drug design. As mentioned, Mpro is also an important target for the development of anti-SARS-CoV-2 drugs. Peptide inhibitors have been investigated using bioinformatic approaches for their applications against CoV Mpro [76]. For SARS-CoV-2 specific inhibitors, the previously identified CoV inhibitor M3 peptide [76] was analyzed for its interactions with SARS-CoV-2, and the interactions were further proved by X-ray crystallography [Figure 4c, PDB: 6LU7] [77]. In addition, analysis of large biological data sets with computational approaches to extract meaningful information has been applied for the identification of virus inhibitory peptides. Several machine learning approaches have been developed for predicting antiviral peptides [78,79,80,81,82,83,84,85,86,87,88]. Among these, the recently published few methods have been specifically developed for the prediction of peptides with anti-coronavirus activities [84,85,87]. A neural network-based method developed by Timmons and Hewage, with an external test accuracy of 93.9%, was found to outperform other methods [87].
Figure 4.

Molecular docking of severe acute respiratory syndrome coronavirus 2 proteins with receptors/ligands. (a) A cryo-EM structure of severe acute respiratory syndrome coronavirus 2 spike protein RBD (green) in complex with human ACE2 (cyan) (PDB: 6M17 [69]). (b) A crystal structure of severe acute respiratory syndrome coronavirus 2 spike protein RBD (cyan) bound with ACE2 (pink) (PDB: 6M0J [18]). (c) A crystal structure of Mpro (green) in complex with a peptide-like inhibitor N3 (yellow) (PDB: 6 LU7 [77]). Blue and red colors on the sphere presentation of the protein structures indicate the positive and negative charged force fields, respectively
Table 1.
Examples of inhibitory peptides targeting spike protein - angiotensin-converting enzyme 2 interactions
| Sequences (names) | Sequence source |
|---|---|
| 21IEEQAKTFLDKFNHEAEDLFYQSSLASWNYNTNIT55 (inhibitor 1) | ACE2 [68] |
| 21IEEQAKTFLDNFNHEAEDLFYQSSLASWNYNTNITEENVQNMNNAGDKWSAFLKEQSTLAQMYPLQEI88 | ACE2 [68] |
| 349WDLGKGDFR357(inhibitor 2) | |
| 21IEEQAKTFLDNFNHEAEDLFYQSSLASWNYNTNITEENVQNMNNAGDKWSAFLKEQSTLAQMYPLQEIQALTVKLQLQALQQNGS105 | ACE2 [68] |
| 323MTQGFWENSMLTDPGNVQKAVCHPTAWDLGKGDFRILMCT362 (inhibitor 3) | |
| 21IEEQAKTFLDNFNHEAEDLFYQSSLASWNYNTNITEENVQNMNNAGDKWSAFLKEQSTLAQMYPLQEIQALTVKL95 | ACE2 [68] |
| 335DPGNVQKAVCHPTAWDLGKGDFRILMCTKVTMDDFLTAHHEMGHIQYDMAYAAQPFLLRNGANEGF400 (inhibitor 4) | |
| DEDLEELERLYRKAEEVAKEAKDASRRGDDERAKEQMERAMRLFDQVFELAQELQEKQTDGNRQKATHLDKAVKEAADELYQRVR (AHB1) | de Novo [70] |
| ELEEQVMHVLDQVSELAHELLHKLTGEELERAAYFNWWATEMMLELIKSDDEREIREIEEEARRILEHLEELARK (AHB2) | de Novo [70] |
| DKEWILQKIYEIMRLLDELGHAEASMRVSDLIYEFMKKGDERLLEEAERLLEEVER (LCB1) | de Novo [70] |
| SDDEDSVRYLLYMAELRYEQGNPEKAKKILEMAEFIAKRNNNEELERLVREVKKRL (LCB2) | de Novo [70] |
| NDDELHMLMTDLVYEALHFAKDEEIKKRVFQLFELADKAYKNNDRQKLEKVVEELKELLERLLS (LCB3) | de Novo [70] |
| QREKRLKQLEMLLEYAIERNDPYLMFDVAVEMLRLAEENNDERIIERAKRILEEYE (LCB4) | de Novo [70] |
| SLEELKEQVKELKKELSPEMRRLIEEALRFLEEGNPAMAMMVLSDLVYQLGDPRVIDLYMLVTKT (LCB5) | de Novo [70] |
| DREQRLVRFLVRLASKFNLSPEQILQLFEVLEELLERGVSEEEIRKQLEEVAKELG (LCB6) | de Novo [70] |
| DDDIRYLIYMAKLRLEQGNPEEAEKVLEMARFLAERLGMEELLKEVRELLRKIEELR (LCB7) | de Novo [70] |
| PIIELLREAKEKNDEFAISDALYLVNELLQRTGDPRLEEVLYLIWRALKEKDPRLLDRAIELFER (LCB8) | de Novo [70] |
| SALEEQLKTFLDKFMHELEDLLYQLAL (P8) | Derived* [71] |
| SALEEQYKTFLDKFM HELEDLLYQLSL (P9) | Derived* [71] |
| SALEEQYKTFLDKFMHELEDLLYQLAL (P10) | Derived* [71] |
| 19STIEEQAKTFLDKFNHEAEDLFYQSSL45 | ACE2 WT [72] |
| 24QAKTFLDKFNHEAEDLFYQSS44GLGKGDFR | ACE2 WT [72] |
| QVKYFLDKFNHEAEDRDYQSSL | ACE2 MT [72] |
| PFLEKLLHEAEDLLYQLELA | ACE2 MT [72] |
| PFLEKLLHEcdEDCLYQLELA | ACE2 MT [72] |
| 483VEGFNCYFPLQSYGFQPTNGVGY505 | RBD WT [72] |
*Peptides derived from ACE2 sequence“19STIEEQAKTFLDKFNHEAEDLFYQSSL45”. WT: Wild type, MT: Mutant, cd: D-cysteine, ACE2: Angiotensin-converting enzyme 2, RBD: Receptor-binding domain
PHYTOCHEMICALS
Herbal medicines and their active phytochemicals have long been important sources for the developments of therapeutic drugs. Large-scale screenings of SARS-CoV-2 inhibitory compounds have been performed in several research groups. Yang et al. applied computational molecular docking to screen 1800 natural compounds for the identification of SARS-CoV-2 spike protein inhibitors [89]. A compound corilagin derived from an annual perennial herbal species Phyllanthus urinaria was identified to have a strong binding affinity to both the SARS-CoV-2 spike protein RBD and human ACE2. The binding was further confirmed by experimental methods such as biolayer interferometry (BLI), ELISA and immunocytochemistry assay [89]. Zhang et al. screened a library of 1871 natural compounds by using molecular docking combined with BLI measurements, and identified 4 compounds, epigallocatechin gallate, isobavachalcone, salvianolic acid A, and isoliensinine, to have effective inhibitory effects on the SARS-CoV-2 entry. The effects were further proven by plaque formation assay in Vero E6 cells [90]. Perrella et al. identified two natural polyphenols, polydatin and resveratrol, to possess activities to interact with the spike protein of SARS-CoV-2 [91]. Molecular docking simulations revealed that both polyphenols can bind to the spike protein, ACE2 and the ACE2: spike protein complex [91]. SARS-CoV-2 Mpro has been mentioned previously to be a popular target for inhibitory compound screening. Kumar et al. applied molecular docking and MD simulations to screen effective compounds from purple nutsedge (Cyperus rotundus) against Mpro of SARS-CoV-2 [92]. Two compounds β-amyrin and stigmasta-5,22-dien-3-ol were identified to exhibit excellent binding abilities to the virus Mpro and were suggested by the authors to be possible inhibitors for SARS-CoV-2 [92]. Giofrè et al. screened 14 natural compounds from limonoids and terpenoids for their ability to inhibit the key target proteins of SARS-CoV-2 by using molecular docking and MD simulations, and identified two limonoids, deacetylnomilin and ichangin, able to directly interact with the catalytic dyad of Mpro [93]. Silva et al. investigated the pharmacokinetic and toxicological properties of molecules in a natural products database of Brazilian semiarid region and performed site prediction and druggability analysis on the SARS-CoV-2 Mpro [94]. After molecular docking and MD simulation, among 10 molecules selected, two of them were suggested to have better potential to interact with the Mpro and to be worth further studying [94]. Gupta et al. screened more than 53,500 bioactive natural molecules from six different natural product databases for the identification of effective molecules against Mpro [95]. The top three screened molecules were further validated by MD simulations, and one of the three was found to possess highest binding affinity as indicated by relative binding energy analysis [95]. The effects of chromenes, flavonoids, and hydroxamic acid compounds on SARS-CoV-2 Mpro have been investigated [96], and compounds in two herbal methanolic extracts, from Averrhoa carambola (star fruit) leaves and Ageratum conyzoides aerial part, were found to demonstrate significant inhibition on SARS-CoV-2 Mpro. In this study, the in vitro experiment results were supported by in silico molecular docking analysis [96]. Li et al. applied ensemble and cooperative docking, as well as molecular simulations, to investigate potential interactions of more than 600 compounds from an herbal medicine with eight SARS-CoV-2 proteins including spike protein, nucleocapsid protein, Mpro, Papain-like protease, RdRp, NSP3, and cat/human ACE2 [97]. This study identified more than nine compounds which may effectively bind to SARS-CoV-2 proteins [97]. In addition, it was found that some of these compounds simultaneously bind to the same target sites. Thus, these compounds might serve as cooperative inhibitors for SARS-CoV-2 proteins [97]. Altogether it has been demonstrated that computational methodologies not only provide useful tools for systematically assess potential antiviral activities of molecules but also indicate new avenues for the search of cooperative compounds to target SARS-CoV-2-related proteins.
CONCLUSION
With the advanced capability of computer technology, computational methods have become powerful tools for biomedical investigations. The computational approaches also provide meaningful, rapid, and cost-effective ways in drug design and screening. They speed up the process of understanding how structurally complicated molecules interact with one another. Owing to the great efforts of research scientists around the world, structures of the major proteins of SARS-CoV-2 have been resolved with biophysical techniques such as X-ray crystallography and NMR, providing the foundation for applying structure-based computational methods, such as molecular docking and MD simulations, to study virus-host protein interactions for the design of therapeutic drugs against COVID-19. These structure-based methods can also be applied to identify effective molecules from compound/drug banks as well as from traditional medicines. Among all the SARS-CoV-2 proteins, the most frequently used targets are the spike protein RBD and Mpro. Owing to the nature of RNA viruses, the SARS-CoV-2 proteins are mutating at a fast speed, thus changing their structures. These might result in their escape from the targeting of specific therapeutics. To combat these situations, powerful AI tools, such as Alphafold, will play increasing important roles in the future for the generations of viral protein structures for investigations. In addition to structure-based analytical tools, machine learn algorithms have been applied in drug discovery for the effective treatments of COVID-19. Efforts by research scientists have been focused on the discovery and design of novel drug candidates, or repurposing and modifying existing approved drugs. Projects on theses purposes have been either exclusively computational or computational-experimental combined studies. It is expected that the computer-aided methods will continue to play central and crucial roles in the battle against COVID-19.
Financial support and sponsorship
This work is supported by Buddhist Compassion Relief Tzu Chi Foundation, Grant number: TCIRP106001-01; Collaborative Grant by Tzu Chi Medical Foundation, Tzu Chi University, and Academia Sinica, Grand number: TCAS-111-03 (Je-Wen Liou); and Ministry of Science and Technology, Taiwan, Grand number: MOST 108-2221-E-320-002.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
The authors would like to thank Professor Ji-Hshiung Chen for valuable suggestions to this manuscript. The chemical structures of compounds in the manuscript were sketched with the software ChemSketch. The PDB structures were prepared with the software Molecular Operating Environment (MOE).
REFERENCES
- 1.Corman VM, Muth D, Niemeyer D, Drosten C. Hosts and sources of endemic human coronaviruses. Adv Virus Res. 2018;100:163–88. doi: 10.1016/bs.aivir.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu L, Zhong W, Bian Z, Li Z, Zhang K, Liang B, et al. A comparison of mortality-related risk factors of COVID-19, SARS, and MERS: A systematic review and meta-analysis. J Infect. 2020;81:e18–25. doi: 10.1016/j.jinf.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang C, Horby PW, Hayden FG, Gao GF. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470–3. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo G, Zhang X, Zheng H, He D. Infection fatality ratio and case fatality ratio of COVID-19. Int J Infect Dis. 2021;113:43–6. doi: 10.1016/j.ijid.2021.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korean Society of Infectious Diseases and Korea Centers for Disease Control and Prevention. Analysis on 54 mortality cases of coronavirus disease 2019 in the republic of Korea from January 19 to March 10, 2020. J Korean Med Sci. 2020;35:e132. doi: 10.3346/jkms.2020.35.e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mortazavi H. Managing older adults’ fear of coronavirus disease: A new role for social work practice. Qual Soc Work. 2021;20:507–12. doi: 10.1177/1473325020973295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ikoona EN, Kitara DL. A proposed framework to limit post-lockdown community transmission of COVID-19 in Africa. Pan Afr Med J. 2021;38:303. doi: 10.11604/pamj.2021.38.303.24008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fortner A, Schumacher D. First COVID-19 vaccines receiving the US FDA and EMA emergency use authorization. Discoveries (Craiova) 2021;9:e122. doi: 10.15190/d.2021.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gómez CE, Perdiguero B, Esteban M. Emerging SARS-CoV-2 Variants and impact in global vaccination programs against SARS-CoV-2/COVID-19. Vaccines (Basel) 2021;9:243. doi: 10.3390/vaccines9030243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.To KK, Hung IF, Ip JD, Chu AW, Chan WM, Tam AR, et al. Coronavirus disease 2019 (COVID-19) re-infection by a phylogenetically distinct severe acute respiratory syndrome coronavirus 2 strain confirmed by whole genome sequencing. Clin Infect Dis. 2021;73:e2946–51. doi: 10.1093/cid/ciaa1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fraguas Bringas C, Booth D. Identification of a SARS-like bat coronavirus that shares structural features with the spike glycoprotein receptor-binding domain of SARS-CoV-2. Access Microbiol. 2020;2:acmi000166. doi: 10.1099/acmi.0.000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang T, Wu Q, Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol. 2020;30:1346–51.e2. doi: 10.1016/j.cub.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopes LR, de Mattos Cardillo G, Paiva PB. Molecular evolution and phylogenetic analysis of SARS-CoV-2 and hosts ACE2 protein suggest Malayan pangolin as intermediary host. Braz J Microbiol. 2020;51:1593–9. doi: 10.1007/s42770-020-00321-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol. 2020;38:1–9. doi: 10.12932/AP-200220-0772. [DOI] [PubMed] [Google Scholar]
- 15.Malaiyan J, Arumugam S, Mohan K, Gomathi Radhakrishnan G. An update on the origin of SARS-CoV-2: Despite closest identity, bat (RaTG13) and pangolin derived coronaviruses varied in the critical binding site and O-linked glycan residues. J Med Virol. 2021;93:499–505. doi: 10.1002/jmv.26261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu P, Jiang JZ, Wan XF, Hua Y, Li L, Zhou J, et al. Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)? PLoS Pathog. 2020;16:e1008421. doi: 10.1371/journal.ppat.1008421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lebeau G, Vagner D, Frumence É, Ah-Pine F, Guillot X, Nobécourt E, et al. Deciphering SARS-CoV-2 virologic and immunologic features. Int J Mol Sci. 2020;21:E5932. doi: 10.3390/ijms21165932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–20. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 19.Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–80.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The COVID-19 cytokine storm; what we know so far. Front Immunol. 2020;11:1446. doi: 10.3389/fimmu.2020.01446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hojyo S, Uchida M, Tanaka K, Hasebe R, Tanaka Y, Murakami M, et al. How COVID-19 induces cytokine storm with high mortality. Inflamm Regen. 2020;40:37. doi: 10.1186/s41232-020-00146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumari R, Kumar R. Open Source Drug Discovery Consortium; Lynn A. g_mmpbsa – A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model. 2014;54:1951–62. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 23.Chang CC, Hsu HJ, Yen JH, Lo SY, Liou JW. A Sequence in the loop domain of hepatitis C virus E2 protein identified in silico as crucial for the selective binding to human CD81. PLoS One. 2017;12:e0177383. doi: 10.1371/journal.pone.0177383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tantardini C. When does a hydrogen bond become a van der Waals interaction? A topological answer. J Comput Chem. 2019;40:937–43. doi: 10.1002/jcc.25774. [DOI] [PubMed] [Google Scholar]
- 25.Santos LH, Ferreira RS, Caffarena ER. Integrating molecular docking and molecular dynamics simulations. Methods Mol Biol. 2019;2053:13–34. doi: 10.1007/978-1-4939-9752-7_2. [DOI] [PubMed] [Google Scholar]
- 26.Chang CC, Liou JW, Dass KT, Li YT, Jiang SJ, Pan SF, et al. Internal water channel formation in CXCR4 is crucial for G i-protein coupling upon activation by CXCL12. Commun Chem. 2020;3:133. doi: 10.1038/s42004-020-00383-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen MC, Hsiao YC, Chang CC, Pan SF, Peng CW, Li YT, et al. Valine-279 deletion-mutation on arginine vasopressin receptor 2 causes obstruction in G-Protein binding site: A clinical nephrogenic diabetes insipidus case and its sub-molecular pathogenic analysis. Biomedicines. 2021;9:301. doi: 10.3390/biomedicines9030301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liou JW, Chang FT, Chung Y, Chen WY, Fischer WB, Hsu HJ. In silico analysis reveals sequential interactions and protein conformational changes during the binding of chemokine CXCL-8 to its receptor CXCR1. PLoS One. 2014;9:e94178. doi: 10.1371/journal.pone.0094178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fantini J, Di Scala C, Chahinian H, Yahi N. Structural and molecular modelling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS-CoV-2 infection. Int J Antimicrob Agents. 2020;55:105960. doi: 10.1016/j.ijantimicag.2020.105960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, et al. A trial of lopinavir-ritonavir in adults hospitalized with severe COVID-19. N Engl J Med. 2020;382:1787–99. doi: 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–71. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P, et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Clin Infect Dis. 2020;71:732–9. doi: 10.1093/cid/ciaa237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez MA. Compounds with therapeutic potential against novel respiratory 2019 coronavirus. Antimicrob Agents Chemother. 2020;64:e00399–20. doi: 10.1128/AAC.00399-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of COVID-19 – Final report. N Engl J Med. 2020;383:1813–26. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Bari MA. Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol Res Perspect. 2017;5:e00293. doi: 10.1002/prp2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gautret P, Lagier JC, Parola P, Hoang VT, Meddeb L, Mailhe M, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. 2020;56:105949. doi: 10.1016/j.ijantimicag.2020.105949. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Group RC, Horby P, Mafham M, Linsell L, Bell JL, Staplin N, et al. Effect of hydroxychloroquine in hospitalized patients with COVID-19. N Engl J Med. 2020;383:2030–40. doi: 10.1056/NEJMoa2022926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Self WH, Semler MW, Leither LM, Casey JD, Angus DC, Brower RG, et al. Effect of hydroxychloroquine on clinical status at 14 days in hospitalized patients with COVID-19: A randomized clinical trial. JAMA. 2020;324:2165–76. doi: 10.1001/jama.2020.22240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wahl A, Gralinski LE, Johnson CE, Yao W, Kovarova M, Dinnon KH, 3rd, et al. SARS-CoV-2 infection is effectively treated and prevented by EIDD-2801. Nature. 2021;591:451–7. doi: 10.1038/s41586-021-03312-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holman W, Holman W, McIntosh S, Painter W, Painter G, Bush J, et al. Accelerated first-in-human clinical trial of EIDD-2801/MK-4482 (molnupiravir), a ribonucleoside analog with potent antiviral activity against SARS-CoV-2. Trials. 2021;22:561. doi: 10.1186/s13063-021-05538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khoo SH, Fitzgerald R, Fletcher T, Ewings S, Jaki T, Lyon R, et al. Optimal dose and safety of molnupiravir in patients with early SARS-CoV-2: A Phase I, open-label, dose-escalating, randomized controlled study. J Antimicrob Chemother. 2021;76:3286–95. doi: 10.1093/jac/dkab318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.First Oral Antiviral for COVID-19, Lagevrio (Molnupiravir), Approved by MHRA (Press Release). Medicines and Healthcare Products Regulatory Agency, U. K. [Last accessed on 2021 Nov 05]. Available from: https://www.gov.uk/government/news/firstoral-antiviral-for-covid-19-lagevrio-molnupiravir-approved-by-mhra .
- 44.Miller SR, McGrath ME, Zorn KM, Ekins S, Wright SH, Cherrington NJ. Remdesivir and EIDD-1931 interact with human equilibrative nucleoside transporters 1 and 2: Implications for reaching SARS-CoV-2 viral sanctuary sites. Mol Pharmacol. 2021;100:548–57. doi: 10.1124/molpharm.121.000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mishra D, Maurya RR, Kumar K, Munjal NS, Bahadur V, Sharma S, et al. Structurally modified compounds of hydroxychloroquine, remdesivir and tetrahydrocannabinol against main protease of SARS-CoV-2, a possible hope for COVID-19: Docking and molecular dynamics simulation studies. J Mol Liq. 2021;335:116185. doi: 10.1016/j.molliq.2021.116185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science. 2003;300:1763–7. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 47.Hoffman RL, Kania RS, Brothers MA, Davies JF, Ferre RA, Gajiwala KS, et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J Med Chem. 2020;63:12725–47. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boras B, Jones RM, Anson BJ, Arenson D, Aschenbrenner L, Bakowski MA, et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat Commun. 2021;12:6055. doi: 10.1038/s41467-021-26239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Vries M, Mohamed AS, Prescott RA, Valero-Jimenez AM, Desvignes L, O’Connor R, et al. A comparative analysis of SARS-CoV-2 antivirals characterizes 3CL (pro) inhibitor PF-00835231 as a potential new treatment for COVID-19. J Virol. 2021;95:e01819–20. doi: 10.1128/JVI.01819-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vandyck K, Deval J. Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection. Curr Opin Virol. 2021;49:36–40. doi: 10.1016/j.coviro.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahmad B, Batool M, Ain QU, Kim MS, Choi S. Exploring the binding mechanism of PF-07321332 SARS-CoV-2 protease inhibitor through molecular dynamics and binding free energy simulations. Int J Mol Sci. 2021;22:9124. doi: 10.3390/ijms22179124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, et al. Drug repurposing: Progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- 53.Jang WD, Jeon S, Kim S, Lee SY. Drugs repurposed for COVID-19 by virtual screening of 6,218 drugs and cell-based assay. Proc Natl Acad Sci U S A. 2021;118:e2024302118. doi: 10.1073/pnas.2024302118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–12. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368:779–82. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pathak Y, Mishra A, Choudhir G, Kumar A, Tripathi V. Rifampicin and Letermovir as potential repurposed drug candidate for COVID-19 treatment: Insights from an in-silico study. Pharmacol Rep. 2021;73:926–38. doi: 10.1007/s43440-021-00228-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Xin B, Tan W, Xu Z, Li K, Li F, et al. DeepR2cov: Deep representation learning on heterogeneous drug networks to discover anti-inflammatory agents for COVID-19. Brief Bioinform. 2021;22:bbab226. doi: 10.1093/bib/bbab226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elbadwi FA, Khairy EA, Alsamani FO, Mahadi MA, Abdalrahman SE, Ahmed ZA, et al. Identification of novel transmembrane protease serine type 2 drug candidates for COVID-19 using computational studies. Inform Med Unlocked. 2021;26:100725. doi: 10.1016/j.imu.2021.100725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hamdy R, Fayed B, Mostafa A, Shama NM, Mahmoud SH, Mehta CH, et al. Iterated virtual screening-assisted antiviral and enzyme inhibition assays reveal the discovery of novel promising anti-SARS-CoV-2 with dual activity. Int J Mol Sci. 2021;22:9057. doi: 10.3390/ijms22169057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thornton JM, Laskowski RA, Borkakoti N. AlphaFold heralds a data-driven revolution in biology and medicine. Nat Med. 2021;27:1666–9. doi: 10.1038/s41591-021-01533-0. [DOI] [PubMed] [Google Scholar]
- 61.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robertson AJ, Courtney JM, Shen Y, Ying J, Bax A. Concordance of X-ray and AlphaFold2 models of SARS-CoV-2 main protease with residual dipolar couplings measured in solution. J Am Chem Soc. 2021;143:19306–10. doi: 10.1021/jacs.1c10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pandey P, Prasad K, Prakash A, Kumar V. Insights into the biased activity of dextromethorphan and haloperidol towards SARS-CoV-2 NSP6: In silico binding mechanistic analysis. J Mol Med (Berl) 2020;98:1659–73. doi: 10.1007/s00109-020-01980-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–68. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peele KA, Kumar V, Parate S, Srirama K, Lee KW, Venkateswarulu TC. Insilico drug repurposing using FDA approved drugs against membrane protein of SARS-CoV-2. J Pharm Sci. 2021;110:2346–54. doi: 10.1016/j.xphs.2021.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Daina A, Michielin O, Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cohen MS. Monoclonal antibodies to disrupt progression of early COVID-19 infection. N Engl J Med. 2021;384:289–91. doi: 10.1056/NEJMe2034495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Han Y, Král P. Computational design of ACE2-based peptide inhibitors of SARS-CoV-2. ACS Nano. 2020;14:5143–7. doi: 10.1021/acsnano.0c02857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–8. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cao L, Goreshnik I, Coventry B, Case JB, Miller L, Kozodoy L, et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science. 2020;370:426–31. doi: 10.1126/science.abd9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Karoyan P, Vieillard V, Gómez-Morales L, Odile E, Guihot A, Luyt CE, et al. Human ACE2 peptide-mimics block SARS-CoV-2 pulmonary cells infection. Commun Biol. 2021;4:197. doi: 10.1038/s42003-021-01736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chitsike L, Krstenansky J, Duerksen-Hughes PJ. ACE2:S1 RBD interaction-targeted peptides and small molecules as potential COVID-19 therapeutics. Adv Pharmacol Pharm Sci. 2021;2021:1828792. doi: 10.1155/2021/1828792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.An L, Lee GR. De novo protein design using the blueprint builder in Rosetta. Curr Protoc Protein Sci. 2020;102:e116. doi: 10.1002/cpps.116. [DOI] [PubMed] [Google Scholar]
- 74.Dou J, Vorobieva AA, Sheffler W, Doyle LA, Park H, Bick MJ, et al. De novo design of a fluorescence-activating β-barrel. Nature. 2018;561:485–91. doi: 10.1038/s41586-018-0509-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chevalier A, Silva DA, Rocklin GJ, Hicks DR, Vergara R, Murapa P, et al. Massively parallel de novo protein design for targeted therapeutics. Nature. 2017;550:74–9. doi: 10.1038/nature23912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang H, Xie W, Xue X, Yang K, Ma J, Liang W, et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3:e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, et al. Structure of M (pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–93. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 78.Thakur N, Qureshi A, Kumar M. AVPpred: Collection and prediction of highly effective antiviral peptides. Nucleic Acids Res. 2012;40:W199–204. doi: 10.1093/nar/gks450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Beltrán Lissabet JF, Belén LH, Farias JG. AntiVPP 1.0: A portable tool for prediction of antiviral peptides. Comput Biol Med. 2019;107:127–30. doi: 10.1016/j.compbiomed.2019.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chowdhury AS, Reehl SM, Kehn-Hall K, Bishop B, Webb-Robertson BM. Better understanding and prediction of antiviral peptides through primary and secondary structure feature importance. Sci Rep. 2020;10:19260. doi: 10.1038/s41598-020-76161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chung CR, Kuo TR, Wu LC, Lee TY, Horng JT. Characterization and identification of antimicrobial peptides with different functional activities. Brief Bioinform. 2019;21:1098–114. doi: 10.1093/bib/bbz043. [DOI] [PubMed] [Google Scholar]
- 82.Gull S, Shamim N, Minhas F. AMAP: Hierarchical multi-label prediction of biologically active and antimicrobial peptides. Comput Biol Med. 2019;107:172–81. doi: 10.1016/j.compbiomed.2019.02.018. [DOI] [PubMed] [Google Scholar]
- 83.Meher PK, Sahu TK, Saini V, Rao AR. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou's general PseAAC. Sci Rep. 2017;7:42362. doi: 10.1038/srep42362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pang Y, Wang Z, Jhong JH, Lee TY. Identifying anti-coronavirus peptides by incorporating different negative datasets and imbalanced learning strategies. Brief Bioinform. 2021;22:1085–95. doi: 10.1093/bib/bbaa423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pang Y, Yao L, Jhong JH, Wang Z, Lee TY. AVPIden: A new scheme for identification and functional prediction of antiviral peptides based on machine learning approaches. Brief Bioinform. 2021;22:bbab263. doi: 10.1093/bib/bbab263. [DOI] [PubMed] [Google Scholar]
- 86.Schaduangrat N, Nantasenamat C, Prachayasittikul V, Shoombuatong W. Meta-iAVP: A sequence-based meta-predictor for improving the prediction of antiviral peptides using effective feature representation. Int J Mol Sci. 2019;20:E5743. doi: 10.3390/ijms20225743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Timmons PB, Hewage CM. ENNAVIA is a novel method which employs neural networks for antiviral and anti-coronavirus activity prediction for therapeutic peptides. Brief Bioinform. 2021;22:bbab258. doi: 10.1093/bib/bbab258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wei L, Zhou C, Su R, Zou Q. PEPred-Suite: Improved and robust prediction of therapeutic peptides using adaptive feature representation learning. Bioinformatics. 2019;35:4272–80. doi: 10.1093/bioinformatics/btz246. [DOI] [PubMed] [Google Scholar]
- 89.Yang LJ, Chen RH, Hamdoun S, Coghi P, Ng JP, Zhang DW, et al. Corilagin prevents SARS-CoV-2 infection by targeting RBD-ACE2 binding. Phytomedicine. 2021;87:153591. doi: 10.1016/j.phymed.2021.153591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang D, Hamdoun S, Chen R, Yang L, Ip CK, Qu Y, et al. Identification of natural compounds as SARS-CoV-2 entry inhibitors by molecular docking-based virtual screening with bio-layer interferometry. Pharmacol Res. 2021;172:105820. doi: 10.1016/j.phrs.2021.105820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perrella F, Coppola F, Petrone A, Platella C, Montesarchio D, Stringaro A, et al. Interference of polydatin/resveratrol in the ACE2: Spike recognition during COVID-19 infection. A focus on their potential mechanism of action through computational and biochemical assays. Biomolecules. 2021;11:1048. doi: 10.3390/biom11071048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kumar SB, Krishna S, Pradeep S, Mathews DE, Pattabiraman R, Murahari M, et al. Screening of natural compounds from Cyperus rotundus Linn against SARS-CoV-2 main protease (M (pro)): An integrated computational approach. Comput Biol Med. 2021;134:104524. doi: 10.1016/j.compbiomed.2021.104524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Giofrè SV, Napoli E, Iraci N, Speciale A, Cimino F, Muscarà C, et al. Interaction of selected terpenoids with two SARS-CoV-2 key therapeutic targets: An in silico study through molecular docking and dynamics simulations. Comput Biol Med. 2021;134:104538. doi: 10.1016/j.compbiomed.2021.104538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Silva RC, Freitas HF, Campos JM, Kimani NM, Silva CH, Borges RS, et al. Natural products-based drug design against SARS-CoV-2 Mpro 3CLpro. Int J Mol Sci. 2021;22:11739. doi: 10.3390/ijms222111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gupta SS, Kumar A, Shankar R, Sharma U. In silico approach for identifying natural lead molecules against SARS-COV-2. J Mol Graph Model. 2021;106:107916. doi: 10.1016/j.jmgm.2021.107916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hariono M, Hariyono P, Dwiastuti R, Setyani W, Yusuf M, Salin N, et al. Potential SARS-CoV-2 3CLpro inhibitors from chromene, flavonoid and hydroxamic acid compound based on FRET assay, docking and pharmacophore studies. Results Chem. 2021;3:100195. doi: 10.1016/j.rechem.2021.100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li J, McKay KT, Remington JM, Schneebeli ST. A computational study of cooperative binding to multiple SARS-CoV-2 proteins. Sci Rep. 2021;11:16307. doi: 10.1038/s41598-021-95826-6. [DOI] [PMC free article] [PubMed] [Google Scholar]