

ABSTRACT
Gout is the most common form of inflammatory arthritis in adults. Elevation serum uric acid (SUA) concentration is known to be the key to gout pathogenesis. Since the first genome-wide association study (GWAS) for SUA was performed in 2007, the number of gene loci known to be associated with hyperuricemia and gout has grown rapidly. GWASs and Mendelian randomization studies have also reported numerous novel results regarding the genetics of hyperuricemia and gout since 2018. We concisely review recent advances in scholarship on the effects of genetics on hyperuricemia and gout risk. We also review data from genetic association studies in Taiwan and perform GWASs of SUA levels among Taiwan Biobank participants.
KEYWORDS: Genetics, Genome-wide association study, Gout, Hyperuricemia, Mendelian randomization study
INTRODUCTION
The incidence of hyperuricemia has increased over the last century in many populations [1,2]. Hyperuricemia is central to the pathogenesis of gout [3,4,5,6]. Serum uric acid (SUA) is the end product of purine metabolism, and elevated SUA concentration resulting from hepatic overproduction or renal or intestinal underexcretion [7] may lead to the saturation of urate levels and the formation of monosodium urate (MSU) crystals. These MSU crystals are deposited in the synovial fluid of joints. Cascading inflammatory responses follow, and gout develops. Gout is currently the most common form of inflammatory arthritis in adults, and its prevalence is increasing [1,2,8]. The progression from hyperuricemia to gout can develop in four stages: hyperuricemia only, MSU crystal deposition without symptomatic gout, acute gout flares and tophi, and chronic and advanced gouty arthritis [3,6,9]. Each stage from asymptomatic hyperuricemia (AHUA) toward gout has different etiological factors that have different causal relationships with comorbidities. Cardiometabolic comorbidities that are common in patients with gout include obesity, hypertension, type 2 diabetes mellitus, chronic kidney disease, dyslipidemia, and coronary artery disease [10,11,12,13,14]. However, the causal relationships between hyperuricemia, gout, and various lifestyle effects and cardiometabolic disorders remain to be determined [15,16,17,18,19].
Since 2007, Li et al. [20] first reported the genome-wide association study (GWAS) for SUA levels. Major et al. [4] reported an updated review of genetics of hyperuricemia and gout for both GWASs and Mendelian randomization (MR) studies in 2018. Thereafter, a lot of publications from East Asian population and from transethnic studies with meta-analysis have been reported. Herein, we will briefly summarize the data of GWAS and MR studies for hyperuricemia and gout, especially focused on publications after 2018.
HERITABILITY OF HYPERURICEMIA AND GOUT
Heritability is defined as the percentage variance in phenotype that is explained by genetic variations between individuals in a population. Heritability can be estimated by studying phenotypic correlations between related individuals—typically twins. A genome-wide association study (GWAS) can also reveal candidate gene loci that may partly explain the genetic variance of a phenotype. Twin studies have demonstrated that the heritability of serum urate is 45%–73% [21,22,23], whereas the heritability of the renal clearance of urate was estimated to be 46%–96% [24,25,26]. GWASs have identified gene loci by using a prevalence of >1% for each allele. The estimated heritability of SUA levels by GWAS was 45%–68% in Europeans [27]. However, index single-nucleotide polymorphisms (SNPs) explained only 5.3% of the variance in SUA concentrations in populations with European ancestry that had 28 loci identified [27]. Larger studies of up to 457,690 individuals identifying 183 loci were only estimated to explain 7.7% of SUA variance [28], which is significantly less than what is predicted to be heritable. In addition, in a general population-based pedigree study, the 183 index SNPs explained 17% of SUA heritability, in which 5% was attributed to three index SNPs on ABCG2, SLC2A9, and SLC22A12 [28]. The missing heritability may be due to copy number variants, rare or population-specific genetic variants, mitochondrial variants, epigenetic effects, and gene–gene and gene–environment interactions. The prevalence of gout increases with age and plateaus at age 70. The incidence is 2–6 times higher in men than in women [8,29,30]. Some ethnic groups, such as aboriginal Taiwanese, Pacific Islanders living in New Zealand, and Maoli (native Hawaiian) people (specifically, men), have a higher prevalence of gout than other ethnic groups [8,29]. Gout is heritable and tends to cluster in families. By using segregation and linkage analysis of familial gout in aboriginal Taiwanese, an autosomal–arbitrary major gene model was found to best fit the data, indicating a genetic basis for familial gout [30]. Using data from the National Health Insurance Research Database in Taiwan, Kuo et al. [31] conducted a nationwide study enrolling more than 1,000,000 individuals with physician-diagnosed gout to estimate the degree of familial aggregation for gout. The results revealed that the relative contribution of heritability was sexually dimorphic at 35.1% in men and 17.0% in women.
GENOME-WIDE ASSOCIATION STUDIES FOR SERUM URIC ACID LEVEL AND GOUT BEFORE 2018
New scholarly advances on the genetic basis of hyperuricemia have improved our understanding of the pathogenesis of hyperuricemia and gout [4,32,33,34]. By using a genome-wide scan, Li et al. 20 first reported that GLUT9 gene variants are associated with SUA levels. Vitart et al. [35] further revealed that SLC2A9, encoded by the GLUT9 gene, is a newly identified urate transporter influencing SUA concentrations, urate excretion, and gout. Analyzing a total of 26,714 participants from the Framingham Heart Study (FHS), Rotterdam study, and the Atherosclerosis Risk in Communities (ARIC) study, Dehghan et al. [36] revealed that three gene loci, ABCG2, SLC2A9I, and SLC17A3, are associated with uric acid concentrations and gout risk. A meta-analysis of 28,141 individuals discovered five new loci – SLC22A11, SLC16A9, GCKR, LRRC16A, and PDZK1 – that influence uric acid levels [37]. Two more loci, RBEB1 and INHBC, were also reported to be associated with gout by Yang et al. [38]. In 2013, by combining data from >140,000 individuals of European ancestry, 18 new gene regions for SUA concentrations were later identified and replicated in or near TRIM46, INHBB, SFMBT1, TMEM171, VEGFA, BAZ1B, PRKAG2, STC1, HNF4G, A1CF, ATXN2, UBE2Q2, IGF1R, NFAT5, MAF, HLF, ACVR1B-ACVRL1, and B3GNT4 gene loci [27]. In an updated review of genetics of hyperuricemia and gout in 2018 [4], the authors summarized earlier GWAS findings and reported that 28 GWAS loci for serum urate levels were detected in European populations, whereas only four loci were detected in East Asian populations and three in African-American populations, suggesting that the molecular genetics of hyperuricemia and gout had been underestimated in Asian and African populations in earlier studies.
RECENT GENOME-WIDE ASSOCIATION STUDIES FOR HYPERURICEMIA AND GOUT
Since 2018, many new GWASs have used more Asians participants and have recruited transethnic populations or used meta-analyses to discover more genome-wide loci that are related to hyperuricemia and gout. Kanai et al. [39] conducted a GWAS investigating 58 quantitative traits in 162,255 individuals from the Biobank Japan Project and identified 27 gene loci for SUA levels in Japanese people. A total of 10 loci were novel including MAFTRR, AC022166.1, AC092130.1, AC009159.1, LRP2, and several other gene loci (NXRN2, SLC22A11-SLC22A12, and others) were located in chromosome 11. By performing a transethnic GWAS of serum urate for 457,690 individuals, Tin et al. [28] identified 183 loci (with 147 novel loci) for serum urate levels that improve the prediction of gout in an independent cohort of 334,880 individuals. By conducting a GWAS investigating SUA levels among 6881 Korean individuals, Cho et al. [40] identified two low-frequency and six common independent variants including LC22A12 p.W258X and an East Asian-specific missense variant (rs671) in ALDH2 associated with SUA. The NHGRI-EBI GWAS Catalog is a publicly available resource for GWAS, and it contains useful visualizations of variant-trait associations. All variants are mapped onto chromosomal positions on the human genome (https://www.ebi.ac.uk/gwas/). When the keywords of “gout” and “uric acid measurement” were searched, a total of 193 associations from 23 studies regarding gout and 380 associations from 38 studies regarding uric acid were identified in the GWAS Catalog. The identified gene loci were compared; in an analysis of only protein-coding genes, a total of 41 gene loci were found to overlap the categories of “gout” and “uric acid measurement” [Table 1].
Table 1.
Gene loci associated with uric acid measurement and/ or gout: Data derived from genome-wide association study catalog
| Phenotype/disease | Protein-coding gene loci |
|---|---|
| Uric acid measurement only | A1CF, ALDH3B1, ANKRD33-FIGNL2-DT, ARHGAP26, B4GALT1, BANF1-CST6, BAZ1B, BCAS1, BICC1, BRAP, C11orf80, C1QTNF9B-ANKRD20A19P, CARMIL1, CCDC18, CNTN4, COMMD4-ANP32BP1, CPS1, CYB5B-NFAT5, DACH1, DCDC2, DIP2C-AL157709.1, DNAH3, DPEP1, EIF1AD, ETV5, F5, FAT4-ANKRD50, FLRT1-MACROD1, FRAS1, FRMD8, GAD1, GAREM1, GNAS, GPLD1, HECTD4, HLA-DQA1-HLA-DQB1, HLF, HNF4A, HNF4G, IGF1R, INHBC, INSR, IPO11-ISCA1P1, KCNE1, KIAA0319, KLHL5, RNU1-130P, LMAN2-MXD3, LRP2, MAFTRR, MAJIN, MALAT1, MAP4K2, MAPKAPK5-ADAM1A, MARK4-EXOC3L2, METTL6, MPPED2-AS1, MTCYBP37, MUC1, MYO9A, MYOF, NDUFAF6, NFATC2, NFKB1, NR5A2, NRAD1, NRG4, NTRK1-INSRR, ORC4, OVOL1, PACS1, PCNX3, PIGN, PKD2, PLAAT2-PLAAT4, PLAAT5, PNPLA3, POLA2, PPP2R5B, PSORS1C1-PSORS1C2, PYGM, QRICH2, RAI14, RBM14-RBM4, RGS20, RN7SKP182, RNASEH2C-KRT8P26, RNF115, RNU1-148P-STC1, RPH 3A, RPL21P41, RPL32P31, RPL7P45, RPS6KA4, RREB1-AL139390.1, RSPO3, RTN4RL1-RPA1, SBF2, SELENOO, SESN2, SESTD1, SHF, SHROOM3, SIPA1L3, SLC10A2, SLC14A2, SLC17A3, SLC17A4, SLC22A10, SLC22A24, SLC25A26-LRIG1, SLC27A5, SLC29A2, STK32B, SYN3, TENM2, TM4SF4, TMC6, TMEM171, TMEM18, TSGA10IP-DRAP1, TSHR, TTLL1, UMOD, UNCX, USP2, USP34, VEGFA, VPS37D-MLXIPL, VPS45, VPS51, WDR1, WDR72, WNT7A, ZBTB4, ZNF318, ZNF584 |
| Gout only | AADACL2-AS1, ABCA1, ABCC8, ABCC9, ABCG1, AC002463.1, RBFOX1, ZNF724, RN7SKP93, CLDN24, ACSM2B, H4C5, AL157886.1, OR13C8, ALDH1A2, ALX4, LINC02724, ARID5B, ASB10, ATP1A4, BDKRB2, CFTR, CNIH2, CNPY4, CNTN5, CYB5AP4, CYP2C8, CYP2E1, DST, EIF3IP1, EVA1A, FAM53A, FGFR2, FSTL4, FTH1P22, GABPA, GLUD1, GRID1, HGF, JAZF1, LHFPL3, LMCD1-AS1, MALRD1, MRPS36P2, MSC-AS1, NIPAL1, NSUN3-ARMC10P1, NTNG2-MED27, PDE1C, PIBF1, POLD3, PPARD, PPARGC1A-DHX15, PRKAG2, PRKCA, PTPRD, PXDNL, RARB, RFX3-AS1, RUNX2-CLIC5, SLC13A3, SLC22A1, SLC22A18AS, SLC38A1, SMARCC1, SMYD3, SV2B, TPST1, VDR, WNT5B |
| Both uric acid measurement and gout | ABCG2, BCAS3, RN7SL496P, CACNA2D3, SFMBT1, ZMYM6, ACAD10, THEM4, TRIM46, ALDH2, ANO2, BAIAP2, CCDC75P1, CDC42BPG-MEN1, CUX2, FAAHP1, PRDM8-FGF5, GCKR-C2orf16, GREM2, KCNQ1, MYL2, MAP3K11, MAPK6, MLXIP, MRPS33P3-RPF2P2, MYH11, NAA25, NALCN, NRXN2, OR4D5-OR8D4, OR6M1-OR6M2P, CD160-PDZK1, PKNOX2, RAB27B, SHLD2, SLC16A9, SLC17A1, SLC22A11-SLC22A12, SLC2A9, SNX18P26-CYP4F34P, WWOX |
Urate excretion is regulated by the kidney and gut, and urate excretion in the kidneys is controlled by a suite of urate transporters. Variations in SLC2A9, ABCG2, and SLC22A12 gene loci are most strongly associated with both SUA levels and gout in multiple populations. Dysfunction in ABCG2 also mediates the underexcretion of urate in the gut and causes an overload of renal urate [4]. Four distinct subtypes of gout have been classified according to patient clinical parameters. These subtypes are classified according to the causes of gout and are renal underexcretion (RUE), renal overload, combined, and normal [41]. GWAS meta-analyses of clinically defined gout were performed to identify subtype-specific susceptibility loci using 3053 clinically defined gout cases and 4554 controls from Japanese men. Both ABCG2 and ALDH2 were associated with all gout subtypes; SLC2A9, SLC22A11-SLC2A12, and SHLD2 were associated with the RUE and combined gout subtypes; BCAS3 was associated with the combined gout subtype, and CD2–PTGFRN and SLC28A3-NTRK2 were associated with normal gout cases [41,42]. These findings, including two novel genes CD2–PTGFRN and SLC28A3-NTRK2, can elucidate the molecular pathophysiology of different gout subtypes and facilitate the development of precision medicines for gout and hyperuricemia prevention and therapy. Elevated SUA concentrations are known to be central to the pathogenesis of gout, however, most individuals with AHUA do not develop gout. By analyzing a total of 2860 gout cases and 3149 AHUA controls in Japanese men, Kawamura et al. [43] revealed that six gene loci (SLC2A9, ABCG2, CNTN5, ALDH2, MIR302F, and ZNF724) were associated with crystal-induced inflammation (i.e., the final stage of gout development). Sandoval-Plata et al. [44], in the analysis of 4934 gout cases and 56,948 AHUA controls as the discovery cohort and 2115 gout cases and 24,406 AHUA controls as the replication cohorts from the UK Biobank, further revealed 13 independent SNPs in ABCG2, SLC2A9, SLC22A11, GCKR, MEPE, PPM1K-DT, LOC105377323, and ADH1B gene loci reached genome-wide significance and replicated as predictors of AHUA to gout transition. These results aid our understanding of the molecular mechanisms of gout development and the prevention of gout attacks in high-risk asymptomatic individuals with hyperuricemia [43] [Table 2].
Table 2.
Gene loci associated with transition from asymptomatic hyperuricemia to gout (data derived from references [44,45])
| Phenotype/disease | Gene loci |
|---|---|
| GWAS catalog available | |
| Uric acid measurement only | Nil |
| Gout only | ZNF724, CNTN5 |
| Both uric acid measurement and gout | ALDH2, GCKR-C2orf16, SHLD2, SLC22A11-SLC22A12, SLC2A9 |
| GWAS catalog not available | CD2–PTGFRN, SLC28A3-NTRK2, MIR302F, MEPE, PPM1K-DT, LOC105377323, ADH1B |
GWAS: Genome-wide association study
ETHNIC HETEROGENEITY FOR GENETICS OF HYPERURICEMIA AND GOUT
Several studies have revealed ethnic heterogeneity in the genetics of hyperuricemia and gout. In a GWAS of SUA levels, ABCG2 variants were reported to have a prominent effect (≈1%) on SUA levels in East Asian and European populations, whereas the ABCG2 lead SNP (rs22331142, Gln141Lys) is less common in the African-American population but with a similar effect size [36]. ALDH2 rs671 (Glu504Lys) is another variant prevalent in East Asian populations associated with hyperuricemia and gout [45,46]. The Lys allele causes substantially reduced ALDH2 activity, and by reducing plasma and urinary hypoxanthine levels, is associated with reduced SUA after alcohol ingestion; it, therefore, reduces the risk of gout. However, ALDH2 rs671 is rare in other populations and Glu allele-positive individuals have elevated hypoxanthine and urate levels, which increase the risk of gout. Liu et al. [47] reported interactive effects between body mass index and alcohol intake on the association between rs671 genotypes and risk of gout. Sulem et al. [48] identified a low-frequency missense variant (c.1580C>G) in ALDH16A1 (rs150414818; Pro476Arg) associated with gout and SUA levels in the Icelandic population, which was not reported in other populations. However, multiple gene loci identified by GWAS as controlling urate are common across multiple populations, indicating the critical role of urate excretion in the pathogenesis of hyperuricemia and gout [4].
STUDIES OF GENETIC BASIS OF HYPERURICEMIA AND GOUT FROM TAIWAN
We have analyzed 459 participants from cardiovascular health examination and found the association between ABCG2 rs2231142 genotypes and SUA level occurred predominantly in male or obese Taiwanese individuals [49]. SNPs in the alpha-protein kinase 1 (ALPK1) gene were found to be associated with gout in aboriginal and in Han Chinese Taiwanese, and the ALPK1 gene variant was identified as being able to effectively interfere with microRNA target recognition and to modulate mRNA expression [50]. Tu et al. [51,52] reported that a combination of variants of ALPK1 with ABCG2, SLC2A9, and SLC22A12 increased the positive predictive value for gout and that the synergistic effect of high-risk alleles influencing composite genetic urate scores combined with alcohol use increased the risk of gout and the occurrence of tophi in the Taiwanese population. By conducting a GWAS of 747 patients with gout, 747 patients with hyperuricemia, and 2071 age-matched controls, Chen et al. [53] demonstrated that ABCG2 variants contributed to not only hyperuricemia but also to gout and were involved in inflammation dysregulation through augmented IL-8 release in endothelial cells. Chang et al. [54] enrolled 448 patients with gout and 943 population controls of Han Chinese descent and reported a significant association with gout in variants of peroxisome PPARGC1B with a missense mutation rs45520937 augmenting NLPR3 (NOD-, LRR- and pyrin domain-containing protein 3) and interleukin-1β expression. Using whole-genome sequencing and promoter-wide methylation, Tseng et al. [55] identified the seven CpG loci of CNTN5, PGGT1B, INSIG1, ANGPTL2, JNK1, UBAP1, and RAPTOR that are involved in interleukin-1β production and associated with an increased risk of gout. Recently, we performed an MR study with participants from Taiwan Biobank (TWB) [56]. By using the same study population, we analyzed the genetic determinants of SUA levels in 23,067 TWB participants. As shown in Figure 1, the candidate gene loci for SUA levels were TRIM46, GCKR, LRP2, SLC2A9, ABCG2, SLC17A3, NRG1, BICC1, SHLD2, and SLC22A12. The Taiwan Biobank (TWB) conducted a large-scale population-based cohort study by recruiting volunteers aged between 30 and 70 years with no history of cancer [57,58]. Although the number of the candidate gene loci in Figure 1 is relatively limited, the target sample size of TWB is aimed at 200,000 participants. The future expansion of the available study population for TWB participants will help to elucidate more of the molecular basis of hyperuricemia and gout in Taiwan.
Figure 1.
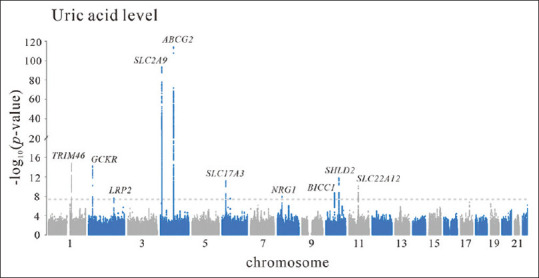
Manhattan plot of a genome-wide association study of serum uric acid levels. Data derived from 27,720 Taiwan Biobank participants with no history of cancer, stroke, coronary artery disease, or systemic disease. A total of 4653 participants were excluded from the analysis, according to the following criteria: fasting for <6 h (622), quality control for the genome-wide association study (2737), and history of gout (1294). The final study population was 23,067 participants. Ten candidate gene loci with genome-wide significant associations were detected
NEW PERSPECTIVES FOCUSED ON NONCODING RNA AND ITS IMPACT ON FUTURE THERAPEUTIC STRATEGY
Noncoding RNA is a type of RNA, such as microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) that does not encode protein. Although initially considered redundancy, many studies have revealed that noncoding RNA plays a critical role in many diseases and has the potential to be a new target for therapy 59. Multiple miRNAs and lncRNAs are involved in various stages of gout development [60]. GWASs also identified several noncoding RNA as candidate gene loci [39,43,44]. Ko et al. [50] showed that the candidate ALPK1 gene variant for gout was identified as being able to effectively interfere with microRNA target recognition and to modulate mRNA expression. Medications for hyperuricemia and gout, such as colchicine, allopurinol, and benzbromarone, may upregulate different miRNAs [60]. Noncoding RNAs can directly regulate the transcriptional level of coding genes and display faster regulation of the occurrence and development of diseases. Therefore, exploration of miRNAs and lncRNAs may be considered as a target for future gout therapy.
RISK OR COMORBIDITY ASSOCIATED WITH HYPERURICEMIA AND GOUT: EVALUATED BY MENDELIAN RANDOMIZATION STUDY
Although observational studies are commonly used to discover correlations between quantitative traits and disease status, the causality of the associations is uncertain in these studies due to their inability to control for confounding factors and biases from reverse causation. An MR study is performed by using either genetic variants or genetic risk scores as an instrumental variable for modifiable exposure. MR is widely used in GWASs to investigate causality [61,62]. In an umbrella review of evidence from observational studies, randomized controlled trials, and MR studies, Li et al. [63] comprehensively analyzed 107 MR studies identified from 36 publications (with a median number of 7158 participants and 2225 cases) from before 2017 with SUA level as an exposure for multiple health outcomes. Instrumental variables explained 2%–6% of the variance of SUA. The authors found a significant association at P < 0.01 for the four outcomes of diabetic macrovascular disease, arterial stiffness, renal events, and gout. However, only the findings for the outcome of gout were robust because they were obtained from 71,501 participants at P = 3.55 × 10−40 and power >99%. By contrast, negative results were obtained for common cardiac metabolic disorders, such as type 2 diabetes, hypertension, chronic kidney disease, ischemic heart disease, and congestive heart failure.
Since 2018, MR studies have used SUA level or gout as an exposure or outcome [Table 3]. Larsson et al. [66] and Larsson et al. [65], analyzing data from the Global Urate Genetics Consortium, reported that coffee intake may lower the risk of gout and that obesity, as determined by body mass index, causes increased SUA levels and an increased likelihood of gout attacks. By contrast, a genetically higher waist–hip ratio (adjusted for body mass index) was not associated with SUA levels or gout risk [66]. This result is similar to that reported by Lyngdoh et al. [77], which suggested that elevated SUA levels are a consequence rather than a cause of adiposity. Yuan et al. [69] reported genetically high iron status, such as high serum iron, ferritin, and transferrin saturation, and low transferrin levels (causally) increase the odds of gout but lower the odds of rheumatoid arthritis in 48,972 individuals of European descent. By contrast, Lee [67] reported that smoking is not causally associated with gout in a study involving data from 85,997 individuals of European descent as the exposure and data from 2115 patients with gout and 67,259 controls as the outcome. By analyzing data regarding 106,147 individuals from the Copenhagen General Population Study, Kobylecki et al. [64] discovered that genetically high plasma Vitamin C is not causally related to SUA levels. In addition, with 333,214 participants from UK Biobank, Nicolopoulos et al. [68] demonstrated no causal association between coffee intake and gout. Topless et al. 70 used data from the ARIC study, the FHS, the Coronary Artery Risk Development in Young Adults (CARDIA) study, the Cardiovascular Health Study (CHS), and the UK Biobank (419,060 individuals in total) and found only weak causal effects of four dietary habits – preferentially drinking skim milk, consuming tub margarine, preferentially drinking milk with higher fat content, and consuming dried fruit – on SUA levels. These effects were mediated by body mass index. These results indicate that diet has a relatively minor effect on SUA levels, whereas urate-lowering therapy is the largest factor for hyperuricemia leading to gout. When SUA was used as an exposure in MR studies, gout and inflammatory polyarthropathies were the most consistent outcomes for hyperuricemia [71,72]. Chronic kidney disease, reduced bone mineral density, and urolithiasis were also not demonstrated to be outcomes of hyperuricemia [72,74,76], and neither bone mineral density nor Alzheimer's disease [74,75] was demonstrated to be outcomes of gout. Biradar et al. [15] analyzed 10,000 TWB participants and found that elevated SUA levels may increase blood pressure and triglyceride levels and decrease high-density lipoprotein cholesterol levels, resulting in a higher risk of metabolic syndrome, whereas high waist circumference may be causative for all the components of metabolic syndrome including hyperuricemia. By analyzing the same study population, Chiang et al. [16] also found that high SUA levels are a precursor to the development of cardiometabolic diseases. However, by using a phenome-wide association study together with a Bayesian analysis of a tree-structure phenotypic model, Li et al. [73] identified 13 distinct phecodes associated with genetically determined SUA levels after using multiple testing correction. These results, and the conclusions of Kanai et al. [39], either suggest that a range of interrelated disease outcomes is associated or that the genetic correlations are, in fact, due to the existence of common upstream pathological elements that influence both SUA levels and metabolic traits but may not themselves be causal.
Table 3.
Risk or comorbidity associated with hyperuricemia and gout: Evaluated by recent mendelian randomization studies
| Author/year/reference number | Dominant ancestry | Number of study participants | Exposure | Outcome | Conclusions |
|---|---|---|---|---|---|
|
| |||||
| Hyperuricemia or gout as an outcome | |||||
| Kobylecki et al., 2018 [64] | EA | 106,147 from CGPS (24.099 with HUA) | Plasma Vitamin C | SUA | No causal relationships detected |
| Larsson and Carlström, 2018 [65] | EA | 110,347 from GUGC | Coffee | SUA | OR=−0.15 mg/dl (95% CI−0.22-−0.09, P=7.9×10−6) for SUA, for 1 cup/day coffee consumption |
| Larsson and Carlström, 2018 [65] | EA | 2115 gout and 110,347 controls, from GUGC | Coffee | Gout | OR=0.56 (95% CI 0.38-0.84; P=0.005) for gout, for 1 cup/day coffee consumption |
| Larsson et al., 2018 [66] | EA | 110,347 from GUGC | Adiposity | SUA | OR=0.30 mg/dl (95% CI 0.25-0.35, P=1.6×10−36) for SUA, for each SD increase of BMI |
| Larsson et al., 2018 [66] | EA | 2115 gout and 67,259 controls from GUGC | Adiposity | Gout | OR=2.24 (95% CI 1.70-2.95; P=8.4×10−9) for gout, for each SD increase of BMI |
| Lee, 2018 [67] | EA | Exposure dataset: 85,997 Outcome dataset: 2115 gout and 67,259 controls |
Smoking | Gout | No causal relationships detected |
| Nicolopoulos et al., 2020 [68] | EA | 333,214 from UKB | Coffee | MR-PheWAS for 1117 phecodes | Association with gout was due to pleiotropy. Causal association for increased odds of osteoarthrosis, other arthropathies, and overweight, and lower odds of postmenopausal bleeding |
| Yuan and Larsson, 2020 [69] | EA | 140,000 (2115 gout) from GUGC | Iron status* | Gout, rheumatoid arthritis and inflammatory bowel disease | Genetically high iron status was positively associated with gout and inversely associated with rheumatoid arthritis |
| Topless et al., 2021 [70] | EA | 419,060 (ARIC, FHS, CARDIA, CHS) and UKB | Diet | Hyperuricemia | Weak causal effect of four dietary habits, such as preferentially drinking skim milk, consuming tub margarine, preferentially drinking milk with a higher fat content and dried fruit, on SUA levels, which were mediated by BMI |
|
| |||||
| Hyperuricemia or gout as an exposure | |||||
|
| |||||
| Li et al., 2018 [71] | EA | 120,091 from UKB | SUA | MR-PheWAS for 568 phecodes | OR=4.58 (95% CI 2.72-7.72) for gout OR=1.15 (95% CI 1.01-1.31) for inflammatory polyarthropathies |
| Jordan et al., 2019 [72] | EA | 110,347–335,212 (from GUGC, EMR-linked UKB, ARIC, FHS, CARDIA, CHS) | SUA | CKD, eGFR, Gout | OR=3.41-6.04 for gout, per 1 mg/dl increase in SUA, all P<0.001 |
| Biradar et al., 2020 [15] | Taiwan | 10,000 TWB participants | SUA | MetS components | SUA increment may augment the risk of MetS through increase blood pressure and triglyceride levels and decrease high-density lipoprotein cholesterol levels |
| Chiang et al., 2019 [16] | Taiwan | 10,000 TWB participants | SUA | CVD | OR=1.62 (95% CI 1.17-2.23), P=0.003 for high versus. low SUA WGRS group |
| Li et al., 2019 [73] | EA | 339,256 from UKB | SUA | MR-PheWAS for 1431 disease outcomes | Associations with circulatory and metabolic disorders were due to pleiotropy. For the association with inflammatory polyarthropathies, only gout had a significant association in PheWAS analysis |
| Lee and Song, 2019 [74] | EA | Exposure dataset: 28,141 Outcome dataset: 4807 gout, 332,352 controls |
SUA, gout | Bone mineral density | No causal relationships detected |
|
| |||||
| Hyperuricemia or gout as an exposure | |||||
|
| |||||
| Lee, 2019 [75] | EA | 17,008 Alzheimer’s disease and 37,154 controls | Gout | Alzheimer’s disease | No causal relationships detected |
| Narang et al., 2021 [76] | EA | 359,827 (6398 urolithiasis) from UKB | SUA | Urolithiasis | No causal relationships detected |
*Including serum iron, ferritin, transferrin saturation, and transferrin levels. ARIC: Atherosclerosis risk in communities, CARDIA: Coronary artery risk development in young adults, CGPS: Copenhagen general population study, CHS: Cardiovascular health study, CI: Confidence interval, CKD: Chronic kidney disease, CVD: Cardiovascular disease, EA: European ancestry, eGFR: Estimated glomerular filtration rate, EMR: Electronic medical record, FHS: Framingham heart study, GUGC: Global urate genetics consortium, HUA: Hyperuricemia, MetS: Metabolic syndrome, OR: Odds ratio, PheWAS: Phenome-wide association study, SD: Standard deviation, SUA: Serum uric acid, TWB: Taiwan Biobank, UKB: UK Biobank, WGRS: Weighted genetic risk score, MR: Mendelian randomization, BMI: Body mass index
CONCLUSION
Since 2018, the rapid expansion of GWAS and MR studies has increased our understanding for the genetics of hyperuricemia and gout. With Biobank studies and transethnic analysis, gene loci of East Asian as well as other populations have been greatly expanded. The understanding of the molecular basis of gout has progressed from initially urate transporter genes to many gene loci for different stages of gout development, from AHUA to gout and from different etiology of hyperuricemia to gout. The elucidation of noncoding RNA as candidate loci for hyperuricemia and gout also provides a hope for future target drug therapy. Till now, gout as an outcome of elevated uric acid levels was confirmed in most MR studies. When uric acid level or gout was used as an exposure or as an outcome, the causal relationships with other lifestyle factors, phenotypes, or diseases have made much progress in recent large population studies, however, we still need more replication studies to reach final conclusions.
Studies of the genetics of hyperuricemia and gout are ongoing. By comparing 35 different blood and urine laboratory measurements of individuals in the UK Biobank (n = 363,228 individuals, mostly British or non-British individuals of European descent), Sinnott-Armstrong et al. [17] found 363 independent loci associated with SUA levels. In these 363 gene variants, 3 were protein-truncating variants, 40 were protein-altering variants, and 320 were noncoding variants. These results further expand our understanding of novel gene loci for hyperuricemia and for gout in transethnic studies. We believe that efforts to include different ethnic populations, especially in Biobank studies, and performing meta-analyses will facilitate the discovery of gene loci for hyperuricemia and gout and also in understanding more of the causal inference between SUA level/gout and various candidate exposures and outcomes in the future.
Financial support and sponsorship
This study was supported by grants from Buddhist Tzu Chi Medical Foundation Academic Advancement (TCMF-EP 108-05) to Y. L. Ko.
Conflicts of interest
Dr. Yu-Lin Ko, an editorial board member at Tzu Chi Medical Journal, had no role in the peer review process of or decision to publish this article.
REFERENCES
- 1.Butler F, Alghubayshi A, Roman Y. The epidemiology and genetics of hyperuricemia and gout across major racial groups: A literature review and population genetics secondary database analysis. J Pers Med. 2021;11:231. doi: 10.3390/jpm11030231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roman YM. The Daniel K. Inouye College of Pharmacy Scripts: Perspectives on the epidemiology of gout and hyperuricemia. Hawaii J Med Public Health. 2019;78:71–6. [PMC free article] [PubMed] [Google Scholar]
- 3.Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet. 2016;388:2039–52. doi: 10.1016/S0140-6736(16)00346-9. [DOI] [PubMed] [Google Scholar]
- 4.Major TJ, Dalbeth N, Stahl EA, Merriman TR. An update on the genetics of hyperuricaemia and gout. Nat Rev Rheumatol. 2018;14:341–53. doi: 10.1038/s41584-018-0004-x. [DOI] [PubMed] [Google Scholar]
- 5.Riches PL, Wright AF, Ralston SH. Recent insights into the pathogenesis of hyperuricaemia and gout. Hum Mol Genet. 2009;18:R177–84. doi: 10.1093/hmg/ddp369. [DOI] [PubMed] [Google Scholar]
- 6.Dalbeth N, Gosling AL, Gaffo A, Abhishek A. Gout. Lancet. 2021;397:1843–55. doi: 10.1016/S0140-6736(21)00569-9. [DOI] [PubMed] [Google Scholar]
- 7.Ichida K, Matsuo H, Takada T, Nakayama A, Murakami K, Shimizu T, et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat Commun. 2012;3:764. doi: 10.1038/ncomms1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuo CF, Grainge MJ, Zhang W, Doherty M. Global epidemiology of gout: Prevalence, incidence and risk factors. Nat Rev Rheumatol. 2015;11:649–62. doi: 10.1038/nrrheum.2015.91. [DOI] [PubMed] [Google Scholar]
- 9.Dalbeth N, Stamp L. Hyperuricaemia and gout: Time for a new staging system? Ann Rheum Dis. 2014;73:1598–600. doi: 10.1136/annrheumdis-2014-205304. [DOI] [PubMed] [Google Scholar]
- 10.Bardin T, Richette P. Impact of comorbidities on gout and hyperuricaemia: An update on prevalence and treatment options. BMC Med. 2017;15:123. doi: 10.1186/s12916-017-0890-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borghi C, Agabiti-Rosei E, Johnson RJ, Kielstein JT, Lurbe E, Mancia G, et al. Hyperuricaemia and gout in cardiovascular, metabolic and kidney disease. Eur J Intern Med. 2020;80:1–11. doi: 10.1016/j.ejim.2020.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Lee SJ, Oh BK, Sung KC. Uric acid and cardiometabolic diseases. Clin Hypertens. 2020;26:13. doi: 10.1186/s40885-020-00146-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richette P, Clerson P, Périssin L, Flipo RM, Bardin T. Revisiting comorbidities in gout: A cluster analysis. Ann Rheum Dis. 2015;74:142–7. doi: 10.1136/annrheumdis-2013-203779. [DOI] [PubMed] [Google Scholar]
- 14.Sumpter NA, Saag KG, Reynolds RJ, Merriman TR. Comorbidities in gout and hyperuricemia: Causality or epiphenomena? Curr Opin Rheumatol. 2020;32:126–33. doi: 10.1097/BOR.0000000000000691. [DOI] [PubMed] [Google Scholar]
- 15.Biradar MI, Chiang KM, Yang HC, Huang YT, Pan WH. The causal role of elevated uric acid and waist circumference on the risk of metabolic syndrome components. Int J Obes (Lond) 2020;44:865–74. doi: 10.1038/s41366-019-0487-9. [DOI] [PubMed] [Google Scholar]
- 16.Chiang KM, Tsay YC, Vincent Ng TC, Yang HC, Huang YT, Chen CH, et al. Is hyperuricemia, an early-onset metabolic disorder, causally associated with cardiovascular disease events in Han Chinese? J Clin Med. 2019;8:1202. doi: 10.3390/jcm8081202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinnott-Armstrong N, Tanigawa Y, Amar D, Mars N, Benner C, Aguirre M, et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat Genet. 2021;53:185–94. doi: 10.1038/s41588-020-00757-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teumer A, Li Y, Ghasemi S, Prins BP, Wuttke M, Hermle T, et al. Genome-wide association meta-analyses and fine-mapping elucidate pathways influencing albuminuria. Nat Commun. 2019;10:4130. doi: 10.1038/s41467-019-11576-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keenan T, Zhao W, Rasheed A, Ho WK, Malik R, Felix JF, et al. Causal assessment of serum urate levels in cardiometabolic diseases through a mendelian randomization study. J Am Coll Cardiol. 2016;67:407–16. doi: 10.1016/j.jacc.2015.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li S, Sanna S, Maschio A, Busonero F, Usala G, Mulas A, et al. The GLUT9 gene is associated with serum uric acid levels in Sardinia and Chianti cohorts. PLoS Genet. 2007;3:e194. doi: 10.1371/journal.pgen.0030194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalousdian S, Fabsitz R, Havlik R, Christian J, Rosenman R. Heritability of clinical chemistries in an older twin cohort: The NHLBI Twin Study. Genet Epidemiol. 1987;4:1–11. doi: 10.1002/gepi.1370040102. [DOI] [PubMed] [Google Scholar]
- 22.Krishnan E, Lessov-Schlaggar CN, Krasnow RE, Swan GE. Nature versus nurture in gout: A twin study. Am J Med. 2012;125:499–504. doi: 10.1016/j.amjmed.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Whitfield JB, Martin NG. Inheritance and alcohol as factors influencing plasma uric acid levels. Acta Genet Med Gemellol (Roma) 1983;32:117–26. doi: 10.1017/s0001566000006401. [DOI] [PubMed] [Google Scholar]
- 24.Emmerson BT, Nagel SL, Duffy DL, Martin NG. Genetic control of the renal clearance of urate: A study of twins. Ann Rheum Dis. 1992;51:375–7. doi: 10.1136/ard.51.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monga M, Macias B, Groppo E, Hargens A. Genetic heritability of urinary stone risk in identical twins. J Urol. 2006;175:2125–8. doi: 10.1016/S0022-5347(06)00272-2. [DOI] [PubMed] [Google Scholar]
- 26.Nilsson SE, Read S, Berg S, Johansson B. Heritabilities for fifteen routine biochemical values: Findings in 215 Swedish twin pairs 82 years of age or older. Scand J Clin Lab Invest. 2009;69:562–9. doi: 10.1080/00365510902814646. [DOI] [PubMed] [Google Scholar]
- 27.Köttgen A, Albrecht E, Teumer A, Vitart V, Krumsiek J, Hundertmark C, et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet. 2013;45:145–54. doi: 10.1038/ng.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tin A, Marten J, Halperin Kuhns VL, Li Y, Wuttke M, Kirsten H, et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat Genet. 2019;51:1459–74. doi: 10.1038/s41588-019-0504-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winnard D, Wright C, Taylor WJ, Jackson G, Te Karu L, Gow PJ, et al. National prevalence of gout derived from administrative health data in Aotearoa New Zealand. Rheumatology (Oxford) 2012;51:901–9. doi: 10.1093/rheumatology/ker361. [DOI] [PubMed] [Google Scholar]
- 30.Wang WH, Chang SJ, Wang TN, Cheng LS, Feng YP, Chen CJ, et al. Complex segregation and linkage analysis of familial gout in Taiwanese aborigines. Arthritis Rheum. 2004;50:242–6. doi: 10.1002/art.11441. [DOI] [PubMed] [Google Scholar]
- 31.Kuo CF, Grainge MJ, See LC, Yu KH, Luo SF, Valdes AM, et al. Familial aggregation of gout and relative genetic and environmental contributions: A nationwide population study in Taiwan. Ann Rheum Dis. 2015;74:369–74. doi: 10.1136/annrheumdis-2013-204067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dalbeth N, Stamp LK, Merriman TR. The genetics of gout: Towards personalised medicine? BMC Med. 2017;15:108. doi: 10.1186/s12916-017-0878-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merriman TR. Population heterogeneity in the genetic control of serum urate. Semin Nephrol. 2011;31:420–5. doi: 10.1016/j.semnephrol.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 34.Tai V, Merriman TR, Dalbeth N. Genetic advances in gout: Potential applications in clinical practice. Curr Opin Rheumatol. 2019;31:144–51. doi: 10.1097/BOR.0000000000000571. [DOI] [PubMed] [Google Scholar]
- 35.Vitart V, Rudan I, Hayward C, Gray NK, Floyd J, Palmer CN, et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008;40:437–42. doi: 10.1038/ng.106. [DOI] [PubMed] [Google Scholar]
- 36.Dehghan A, Köttgen A, Yang Q, Hwang SJ, Kao WL, Rivadeneira F, et al. Association of three genetic loci with uric acid concentration and risk of gout: A genome-wide association study. Lancet. 2008;372:1953–61. doi: 10.1016/S0140-6736(08)61343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kolz M, Johnson T, Sanna S, Teumer A, Vitart V, Perola M, et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009;5:e1000504. doi: 10.1371/journal.pgen.1000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Q, Köttgen A, Dehghan A, Smith AV, Glazer NL, Chen MH, et al. Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ Cardiovasc Genet. 2010;3:523–30. doi: 10.1161/CIRCGENETICS.109.934455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanai M, Akiyama M, Takahashi A, Matoba N, Momozawa Y, Ikeda M, et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat Genet. 2018;50:390–400. doi: 10.1038/s41588-018-0047-6. [DOI] [PubMed] [Google Scholar]
- 40.Cho SK, Kim B, Myung W, Chang Y, Ryu S, Kim HN, et al. Polygenic analysis of the effect of common and low-frequency genetic variants on serum uric acid levels in Korean individuals. Sci Rep. 2020;10:9179. doi: 10.1038/s41598-020-66064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakayama A, Nakaoka H, Yamamoto K, Sakiyama M, Shaukat A, Toyoda Y, et al. GWAS of clinically defined gout and subtypes identifies multiple susceptibility loci that include urate transporter genes. Ann Rheum Dis. 2017;76:869–77. doi: 10.1136/annrheumdis-2016-209632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakayama A, Nakatochi M, Kawamura Y, Yamamoto K, Nakaoka H, Shimizu S, et al. Subtype-specific gout susceptibility loci and enrichment of selection pressure on ABCG2 and ALDH2 identified by subtype genome-wide meta-analyses of clinically defined gout patients. Ann Rheum Dis. 2020;79:657–65. doi: 10.1136/annrheumdis-2019-216644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawamura Y, Nakaoka H, Nakayama A, Okada Y, Yamamoto K, Higashino T, et al. Genome-wide association study revealed novel loci which aggravate asymptomatic hyperuricaemia into gout. Ann Rheum Dis. 2019;78:1430–7. doi: 10.1136/annrheumdis-2019-215521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandoval-Plata G, Morgan K, Abhishek A. Variants in urate transporters, ADH1B, GCKR and MEPE genes associate with transition from asymptomatic hyperuricaemia to gout: results of the first gout versus asymptomatic hyperuricaemia GWAS in Caucasians using data from the UK Biobank. Ann Rheum Dis. 2021;80:1220–6. doi: 10.1136/annrheumdis-2020-219796. [DOI] [PubMed] [Google Scholar]
- 45.Sakiyama M, Matsuo H, Nakaoka H, Yamamoto K, Nakayama A, Nakamura T, et al. Identification of rs671, a common variant of ALDH2, as a gout susceptibility locus. Sci Rep. 2016;6:25360. doi: 10.1038/srep25360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang D, Yang M, Zhou D, Li Z, Cai L, Bao Y, et al. The polymorphism rs671 at ALDH2 associated with serum uric acid levels in Chinese Han males: A genome-wide association study. Gene. 2018;651:62–9. doi: 10.1016/j.gene.2018.01.064. [DOI] [PubMed] [Google Scholar]
- 47.Liu YR, Tantoh DM, Lin CC, Hsiao CH, Liaw YP. Risk of gout among Taiwanese adults with ALDH-2 rs671 polymorphism according to BMI and alcohol intake. Arthritis Res Ther. 2021;23:115. doi: 10.1186/s13075-021-02497-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sulem P, Gudbjartsson DF, Walters GB, Helgadottir HT, Helgason A, Gudjonsson SA, et al. Identification of low-frequency variants associated with gout and serum uric acid levels. Nat Genet. 2011;43:1127–30. doi: 10.1038/ng.972. [DOI] [PubMed] [Google Scholar]
- 49.Cheng ST, Wu S, Su CW, Teng MS, Hsu LA, Ko YL. Association of ABCG2 rs2231142 – A allele and serum uric acid levels in male and obese individuals in a Han Taiwanese population. J Formos Med Assoc. 2017;116:18–23. doi: 10.1016/j.jfma.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 50.Ko AM, Tu HP, Liu TT, Chang JG, Yuo CY, Chiang SL, et al. ALPK1 genetic regulation and risk in relation to gout. Int J Epidemiol. 2013;42:466–74. doi: 10.1093/ije/dyt028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tu HP, Chung CM, Min-Shan Ko A, Lee SS, Lai HM, Lee CH, et al. Additive composite ABCG2, SLC2A9 and SLC22A12 scores of high-risk alleles with alcohol use modulate gout risk. J Hum Genet. 2016;61:803–10. doi: 10.1038/jhg.2016.57. [DOI] [PubMed] [Google Scholar]
- 52.Tu HP, Min-Shan Ko A, Lee SS, Lee CP, Kuo TM, Huang CM, et al. Variants of ALPK1 with ABCG2, SLC2A9, and SLC22A12 increased the positive predictive value for gout. J Hum Genet. 2018;63:63–70. doi: 10.1038/s10038-017-0368-9. [DOI] [PubMed] [Google Scholar]
- 53.Chen CJ, Tseng CC, Yen JH, Chang JG, Chou WC, Chu HW, et al. ABCG2 contributes to the development of gout and hyperuricemia in a genome-wide association study. Sci Rep. 2018;8:3137. doi: 10.1038/s41598-018-21425-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang WC, Jan Wu YJ, Chung WH, Lee YS, Chin SW, Chen TJ, et al. Genetic variants of PPAR-gamma coactivator 1B augment NLRP3-mediated inflammation in gouty arthritis. Rheumatology (Oxford) 2017;56:457–66. doi: 10.1093/rheumatology/kew337. [DOI] [PubMed] [Google Scholar]
- 55.Tseng CC, Wong MC, Liao WT, Chen CJ, Lee SC, Yen JH, et al. Systemic investigation of promoter-wide methylome and genome variations in gout. Int J Mol Sci. 2020;21:4702. doi: 10.3390/ijms21134702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hsu LA, Chou HH, Teng MS, Wu S, Ko YL. Circulating chemerin levels are determined through circulating platelet counts in nondiabetic Taiwanese people: A bidirectional Mendelian randomization study. Atherosclerosis. 2021;320:61–9. doi: 10.1016/j.atherosclerosis.2021.01.014. [DOI] [PubMed] [Google Scholar]
- 57.Chen CH, Yang JH, Chiang CWK, Hsiung CN, Wu PE, Chang LC, et al. Population structure of Han Chinese in the modern Taiwanese population based on 10,000 participants in the Taiwan Biobank project. Hum Mol Genet. 2016;25:5321–31. doi: 10.1093/hmg/ddw346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fan CT, Lin JC, Lee CH. Taiwan Biobank: A project aiming to aid Taiwan's transition into a biomedical island. Pharmacogenomics. 2008;9:235–46. doi: 10.2217/14622416.9.2.235. [DOI] [PubMed] [Google Scholar]
- 59.Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. 2017;16:167–79. doi: 10.1038/nrd.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu YT, Leng YR, Liu MM, Dong RF, Bian J, Yuan LL, et al. MicroRNA and long noncoding RNA involvement in gout and prospects for treatment. Int Immunopharmacol. 2020;87:106842. doi: 10.1016/j.intimp.2020.106842. [DOI] [PubMed] [Google Scholar]
- 61.Benn M, Nordestgaard BG. From genome-wide association studies to Mendelian randomization: Novel opportunities for understanding cardiovascular disease causality, pathogenesis, prevention, and treatment. Cardiovasc Res. 2018;114:1192–208. doi: 10.1093/cvr/cvy045. [DOI] [PubMed] [Google Scholar]
- 62.Davey Smith G, Hemani G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:R89–98. doi: 10.1093/hmg/ddu328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li X, Meng X, Timofeeva M, Tzoulaki I, Tsilidis KK, Ioannidis JP, et al. Serum uric acid levels and multiple health outcomes: Umbrella review of evidence from observational studies, randomised controlled trials, and Mendelian randomisation studies. BMJ. 2017;357:j2376. doi: 10.1136/bmj.j2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kobylecki CJ, Afzal S, Nordestgaard BG. Genetically high plasma vitamin C and urate: A Mendelian randomization study in 106 147 individuals from the general population. Rheumatology (Oxford) 2018;57:1769–76. doi: 10.1093/rheumatology/key171. [DOI] [PubMed] [Google Scholar]
- 65.Larsson SC, Carlström M. Coffee consumption and gout: A Mendelian randomisation study. Ann Rheum Dis. 2018;77:1544–6. doi: 10.1136/annrheumdis-2018-213055. [DOI] [PubMed] [Google Scholar]
- 66.Larsson SC, Burgess S, Michaëlsson K. Genetic association between adiposity and gout: A Mendelian randomization study. Rheumatology (Oxford) 2018;57:2145–8. doi: 10.1093/rheumatology/key229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee YH. Assessing the causal association between smoking behavior and risk of gout using a Mendelian randomization study. Clin Rheumatol. 2018;37:3099–105. doi: 10.1007/s10067-018-4210-3. [DOI] [PubMed] [Google Scholar]
- 68.Nicolopoulos K, Mulugeta A, Zhou A, Hyppönen E. Association between habitual coffee consumption and multiple disease outcomes: A Mendelian randomisation phenome-wide association study in the UK Biobank. Clin Nutr. 2020;39:3467–76. doi: 10.1016/j.clnu.2020.03.009. [DOI] [PubMed] [Google Scholar]
- 69.Yuan S, Larsson S. Causal associations of iron status with gout and rheumatoid arthritis, but not with inflammatory bowel disease. Clin Nutr. 2020;39:3119–24. doi: 10.1016/j.clnu.2020.01.019. [DOI] [PubMed] [Google Scholar]
- 70.Topless RK, Major TJ, Florez JC, Hirschhorn JN, Cadzow M, Dalbeth N, et al. The comparative effect of exposure to various risk factors on the risk of hyperuricaemia: Diet has a weak causal effect. Arthritis Res Ther. 2021;23:75. doi: 10.1186/s13075-021-02444-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li X, Meng X, Spiliopoulou A, Timofeeva M, Wei WQ, Gifford A, et al. MR-PheWAS: Exploring the causal effect of SUA level on multiple disease outcomes by using genetic instruments in UK Biobank. Ann Rheum Dis. 2018;77:1039–47. doi: 10.1136/annrheumdis-2017-212534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jordan DM, Choi HK, Verbanck M, Topless R, Won HH, Nadkarni G, et al. No causal effects of serum urate levels on the risk of chronic kidney disease: A Mendelian randomization study. PLoS Med. 2019;16:e1002725. doi: 10.1371/journal.pmed.1002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li X, Meng X, He Y, Spiliopoulou A, Timofeeva M, Wei WQ, et al. Genetically determined serum urate levels and cardiovascular and other diseases in UK Biobank cohort: A phenome-wide mendelian randomization study. PLoS Med. 2019;16:e1002937. doi: 10.1371/journal.pmed.1002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee YH, Song GG. Uric acid level, gout and bone mineral density: A Mendelian randomization study. Eur J Clin Invest. 2019;49:e13156. doi: 10.1111/eci.13156. [DOI] [PubMed] [Google Scholar]
- 75.Lee YH. Gout and the risk of Alzheimer's disease: A Mendelian randomization study. Int J Rheum Dis. 2019;22:1046–51. doi: 10.1111/1756-185X.13548. [DOI] [PubMed] [Google Scholar]
- 76.Narang RK, Gamble GG, Topless R, Cadzow M, Stamp LK, Merriman TR, et al. Assessing the Relationship Between Serum Urate and Urolithiasis Using Mendelian Randomization: An Analysis of the UK Biobank. Am J Kidney Dis. 2021;78:210–8. doi: 10.1053/j.ajkd.2020.11.018. [DOI] [PubMed] [Google Scholar]
- 77.Lyngdoh T, Vuistiner P, Marques-Vidal P, Rousson V, Waeber G, Vollenweider P, et al. Serum uric acid and adiposity: Deciphering causality using a bidirectional Mendelian randomization approach. PLoS One. 2012;7:e39321. doi: 10.1371/journal.pone.0039321. [DOI] [PMC free article] [PubMed] [Google Scholar]