

Abstract
Differing from the mouse Foxp3 gene that encodes only one protein product, human FOXP3 encodes two major isoforms through alternative splicing – a longer isoform (FOXP3 FL) containing all the coding exons, and a shorter isoform lacking the amino acids encoded by exon 2 (FOXP3 ΔE2). The two isoforms are naturally expressed in humans, yet their differences in controlling regulatory T cell phenotype and functionality remains unclear. In this study, we show that patients expressing only the shorter isoform fail to maintain self-tolerance and develop Immunodeficiency, Polyendocrinopathy, and Enteropathy X-Linked (IPEX) syndrome. Mice with Foxp3 exon 2 deletion have excessive follicular helper T (TFH) and geminal center B (GC B) cell responses, and develop systemic autoimmune disease with anti-dsDNA and anti-nuclear autoantibody production, as well as immune-complex glomerulonephritis. Despite having normal suppressive function in in vitro assays, regulatory T cells expressing FOXP3 ΔE2 are unstable and sufficient to induce autoimmunity when transferred into Tcrb-deficient mice. Mechanistically, the FOXP3 ΔE2 isoform allows increased expression of selected cytokines, but decreased expression of a set of Foxp3 positive regulators without altered binding to these gene loci. These findings uncover indispensable functions of the FOXP3 exon 2 region, highlighting a role in regulating a transcriptional program that maintains Treg stability and immune homeostasis.
One Sentence Summary:
Tregs expressing a FOXP3 isoform lacking exon 2 are unstable and sufficient to induce a systemic autoimmune disease.
INTRODUCTION
Regulatory T cells (Tregs) expressing the X-chromosome-encoded transcription factor forkhead box P3 (FOXP3) are a specialized lineage of CD4+ T cells that provide a critical mechanism to suppress T cell responses to self, the commensal microbiota, and dietary and environmental antigens (1–4). Decreased or absent functional FOXP3 leads to widespread autoimmunity and atopy caused by the absence of Treg cells.
Human and mouse FOXP3 genes are highly conserved in both amino acid sequence (with 87% overall identity) and gene structure. While the mouse Foxp3 gene only encodes a single protein, the human FOXP3 gene encodes two major isoforms – a “full length” isoform including all the coding exons as seen in mice, and an isoform lacking the sequence encoded by exon 2 (referred to as FOXP3 FL and FOXP3 ΔE2 respectively hereafter) due to alternative RNA splicing. The FL and ΔE2 isoforms of FOXP3 were regarded as equally functional in mediating Treg differentiation and function, as ectopic expression of each isoform separately or in combination causes CD4+ T cells to acquire Treg phenotypes and lose their cytokine-secreting capacity (5–7). Since in vitro studies using ectopic expression of FOXP3 ΔE2 or FOXP3 FL isoforms in CD4+ T cells (5–7) may result in supra-physiological expression levels and are not likely to represent the function of Tregs in vivo, previous studies are still open to interpretation.
The in vivo study of isoform-specific regulation of Treg development and function is hindered by the fact that the two alternatively spliced isoforms are co-expressed in human Tregs. In patients with antineutrophil cytoplasmic antibody-associated vasculitis (8), Hashimoto’s thyroiditis or Graves’ disease (9), giant cell arteritis (10), and coeliac disease (11), FOXP3 ΔE2 isoform expression is upregulated to become the dominant isoform, suggesting a possible correlation of this isoform with autoimmunity. Conversely, in patients with rheumatoid arthritis, the ratio of FOXP3 ΔE2 to FOXP3 FL mRNA is either decreased (12, 13) or unchanged (14) compared to healthy controls. Similarly, FOXP3 FL but not FOXP3 ΔE2 mRNA expression is significantly increased in peripheral blood mononuclear cells from coronary artery disease patients (15). Thus, an association of increased FOXP3 FL isoform with autoimmune and inflammatory diseases is suggested (16). With the divergent conclusions of these patient studies, how the two major isoforms FOXP3 FL and FOXP3 ΔE2 differ in regulating functionality and biology of Tregs remains unclear.
In vitro studies have shown that the FOXP3 exon 2 region is associated with lineage-defining transcription factors ROR-α and ROR-γt to suppress Th17 differentiation (17, 18). The LXXLL motif of FOXP3 exon 2 has been shown to interact with ROR-α, and the expression of FOXP3 FL was necessary to block ROR-γt-mediated Th17 cell differentiation in retroviral transduction experiments (19). However, increased Th17 differentiation was only seen in morpholino antisense oligonucleotide-mediated deletion of both exons 2 and 7, but not exon 2 alone, from endogenous FOXP3 pre-mRNA in human CD4+ T cells (20). FOXP3 ΔE2ΔE7 is a minor isoform incapable of mediating functional Treg development (21), likely due to the fact that the exon 7 encoded leucine zipper domain is required for FOXP3 dimerization and Treg function (22, 23).
Treg therapy and immunotherapy have been regarded as promising approaches to treat autoimmune disease and chronic allergic inflammation (24–26). While quite effective in mouse studies (e.g. (3, 27)), clinical studies on human diseases often give mixed, or even limited, benefits (e.g. (28–30)). Delineating the impact of the FOXP3 ΔE2 isoform on the stability and function of Tregs will not only shed light on the pathophysiology of autoimmunity but is also important for advancing understanding of Treg therapies.
In this manuscript, we show that patients who only express the FOXP3 ΔE2 isoform developed immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Deletion of Foxp3 exon 2 in mice did not impact thymocyte development, but resulted in excessive TFH and GC B cell responses, with systemic autoimmune disease featuring anti-dsDNA and anti-nuclear autoantibody production, as well as immune-complex glomerulonephritis. In vitro, FOXP3 ΔE2 Tregs has comparable suppressive ability to FOXP3 FL Tregs, whilst FOXP3 ΔE2 Tregs in vivo exhibited intrinsic defects in expression of phenotypic molecules including CD25, FOXP3, and CTLA-4. When transferred into Tcrb-deficient mice, purified FOXP3 ΔE2 Tregs lost FOXP3 expression and were sufficient to induce systemic autoimmunity, similar to that seen in Foxp3 ΔE2 mice. The FOXP3 ΔE2 isoform up-regulated selected inflammatory cytokines but downregulated a set of Foxp3 positive regulators in Tregs, in the absence of changes in binding to gene loci. Our studies establish a crucial role of FOXP3 exon 2 region in mediating Treg cell stability and homeostasis of the immune system.
RESULTS
Patients expressing only the FOXP3 ΔE2 isoform develop IPEX syndrome
IPEX syndrome is a rare disease linked to mutations of FOXP3, a key transcription factor required for the differentiation and function of Tregs. The dysfunction of Tregs is the main pathogenic event leading to multi-organ autoimmunity and eczema. We identified four IPEX patients carrying deletion mutations in exon 2 of the FOXP3 gene (Table S1). While these mutations result in a frame shift in exon 2-containing mRNA and thus altered and truncated the amino acid sequence, the alternatively spliced FOXP3 ΔE2 isoform – excluding the mutated exon 2 – is predicted to be intact. When peripheral blood mononuclear cells (PBMCs) from patient #3 were stained with two anti-FOXP3 antibodies (clone 259D recognizing a common epitope C-terminal of exon 2, and clone 150D specific for exon 2), a population of CD4+ T cells (2.9%) stained positive for 259D but negative for 150D, indicating expression of the FOXP3 ΔE2 isoform only (Fig. 1A). Compared to IPEX patients, 4.4% CD4+ T cells in the healthy donor stained positive for 150D indicating expression of the FOXP3 FL isoform (Fig. 1A). The population of CD4+ T cells expressing only the FOXP3 ΔE2 isoform in IPEX patients was more prominent when PBMCs were activated with anti-CD3/anti-CD28 coated beads for 24 hr that upregulated FOXP3 and CD25 expression (Fig. 1B). Patient CD4+FOXP3+ cells at rest expressed lower FOXP3 and CD25 than cells from the healthy donors but upregulated to similar levels after activation (Fig. 1C).
Figure 1. Expression of FOXP3 ΔE2 isoform in IPEX patients with deletion mutations within exon 2 of FOXP3 gene.
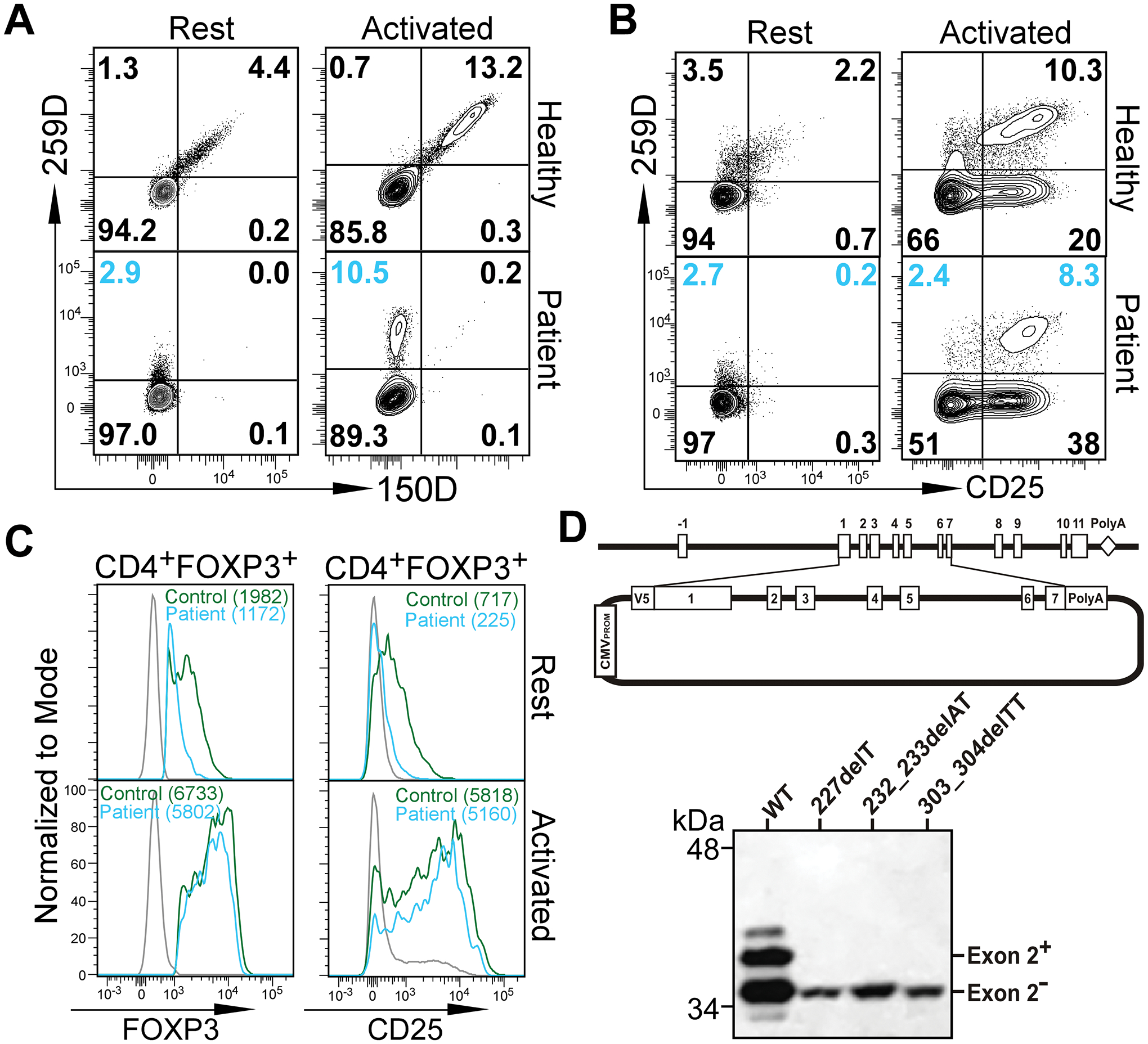
(A) Flow cytometric analysis of FOXP3 ΔE2 isoform expression in IPEX patient #3. PBMCs from the patient and a healthy donor were stained with two anti-FOXP3 antibodies. Clone 150D is exon 2-specific while clone 259D recognizes an epitope after exon 2 common for both isoforms. Cells were analyzed either ex vivo (Rest) or stimulated with anti-CD3/anti-CD28 coated beads for 24 hours (Activated). Gated on CD4+ T cells. (B) Flow cytometric analysis of CD25 expression on CD4+ T cells before and after activation. (C) Expression of FOXP3 and CD25 by Tregs (gated on CD4+259D+) from healthy control (green line) and patient #3 (blue line) before or after activation. Gray lines were 259D− cells. Numbers in the parenthesis represent mean fluorescence intensity (MFI). Similar results as in A-C were obtained with PBMCs from patient #2. (D) Deletion mutations in FOXP3 exon 2 identified in IPEX patients #1, #2 and #4 lead to expression of only the FOXP3 ΔE2 isoform. Genomic DNA fragment containing exons 1–7 of the FOXP3 gene from a healthy control and patients #1, #2, and #4 were cloned into the pcDNA3-NV5 vector in-frame with an N-terminal V5 epitope tag (upper panel). Jurkat T cells transfected with the expression constructs were examined by Western blot with an anti-V5 antibody.
To further confirm the ability of the mutated FOXP3 alleles in these patients to express the FOXP3 ΔE2 isoform, we cloned the genomic DNA fragment spanning exon 1 to exon 7 into the pcDNA3 vector with an N-terminal V5 epitope tag. Jurkat T cells were transfected with the expression constructs and Western blots were performed with the cell lysates with an anti-V5 antibody. While the genomic DNA fragment from the healthy donor encoded two FOXP3 isoforms (exon 2+ and exon 2−), genomic fragments from three of the IPEX patients (#1, 2 and 4) only produce the FOXP3 isoform lacking the exon 2 (Fig. 1D). Therefore, in contrast with prior in vitro analyses suggesting equal function of the isoforms (5–7), the FOXP3 ΔE2 isoform is insufficient to maintain self-tolerance leading to autoimmune responses seen in these IPEX patients.
Foxp3 exon 2 deletion results in altered immune homeostasis.
To study the functional difference of the two FOXP3 isoforms, we deleted exon 2 of the mouse Foxp3 gene (Foxp3 ΔE2), while leaving intact the intronic regulatory elements (Fig. S1A). Foxp3 ΔE2 mice were viable and morphologically normal with unaffected thymocyte development (Fig. S1B,C). The percentage of FOXP3+ CD4+ cells and the expression level of FOXP3 (mean fluorescent intensity, MFI) in the thymus of Foxp3 ΔE2 mice was comparable to WT mice (Fig. S1D). Sequencing of the PCR amplified Foxp3 transcripts shows correct splicing/joining of exon 1 to exon 3 and Western blotting detected exon 2-deleted FOXP3 with the expected size in iTregs generated from naïve CD4+ T cells isolated from Foxp3 ΔE2 mice (Fig. S1E–G).
In the periphery, however, Foxp3 ΔE2 mice had increased lymph node size and cellularity (Fig. 2A), as well as expansion of B and T cells in peripheral lymph nodes, but not in spleens, at 8 weeks of age (Fig. 2B). Foxp3 ΔE2 mice had an increased frequency of activated CD62LloCD44hi cells, as well as elevated numbers of IFN-γ and IL-4 expressing CD4+ T cells (Fig. 2C&D). In contrast to previous studies implicating sequences within the FOXP3 exon 2 region in the inhibition of IL-17 expression (18), IL-17-expressing CD4+ T cells were unchanged in Foxp3 ΔE2 mice as compared to wild type mice (Fig. 2E). To further study the effects of FOXP3 ΔE2 on Th17 differentiation, we isolated naïve CD4+ T cells from WT or Foxp3 ΔE2 mice and cultured under Th17 differentiation conditions, with varying concentration of IL-6 for three days. While increasing IL-6 concentration increased the percentage of Th17 cells, there were no differences between WT and Foxp3 ΔE2 CD4+ T cells (Fig. S2A). Consistent with previous studies showing that human FOXP3 exon 7, rather than exon 2, suppressed IL-17A production (20), we also demonstrated that a morpholino oligo was able to significantly shift endogenous FOXP3 expression to the FOXP3 ΔE2 isoform (Fig. S2B). While increasing IL-6 from 10 ng/ml to 20 ng/ml in the culture significantly increased RORC expression, shifting FOXP3 isoform expression with the morpholino had no effects on RORC and IL17A expression (Fig. S2B). These data demonstrate that Foxp3 ΔE2 mice have disrupted immune homeostasis with increased CD4+ T cell activation and inflammatory cytokine IFN-γ and IL-4 production.
Figure 2. Altered immune homeostasis in Foxp3 ΔE2 mice at 8 weeks of age.
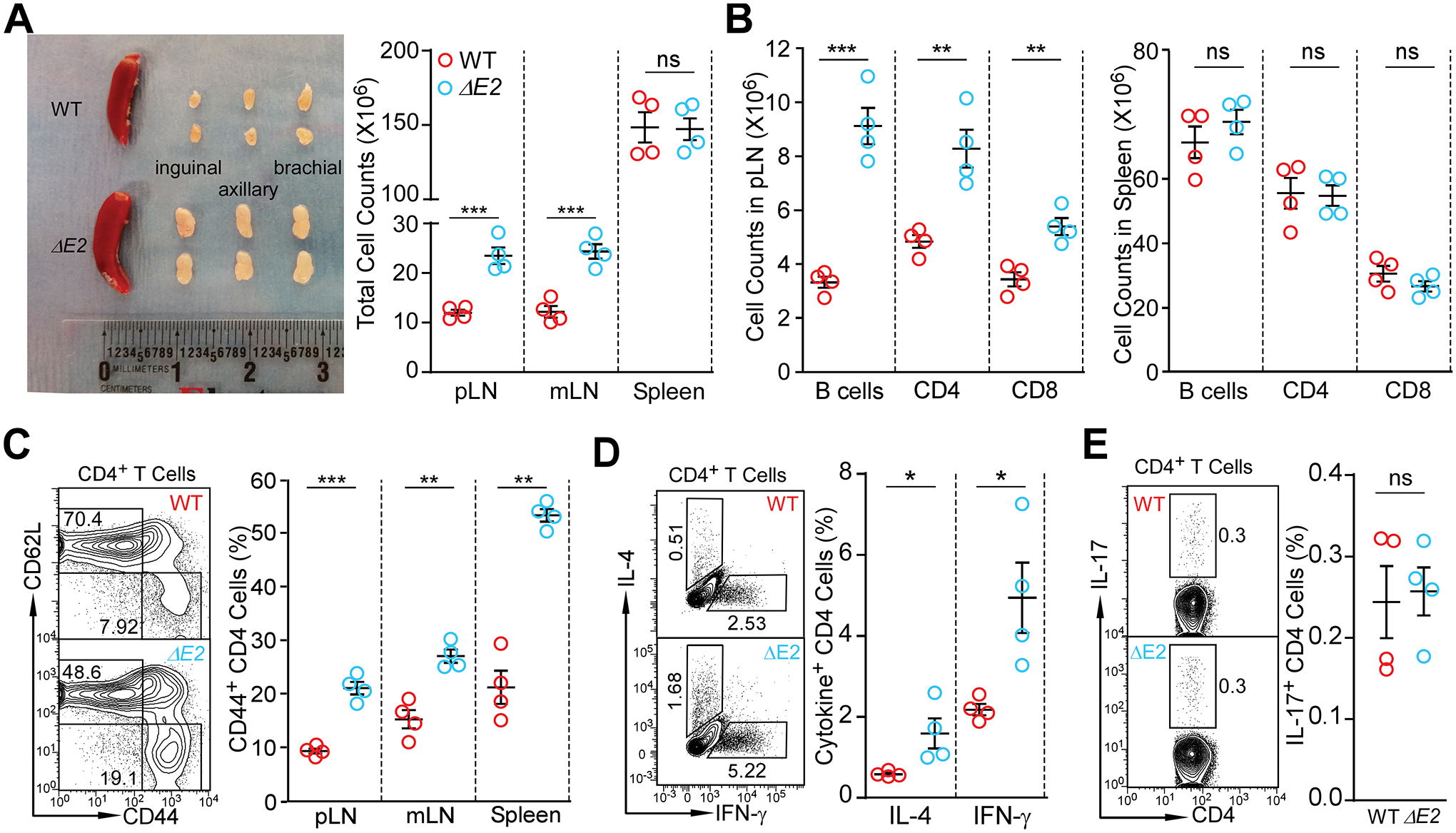
(A) Size and cellularity of lymph nodes and spleen in WT and Foxp3 ΔE2 mice. (B) Flow cytometric analysis of B220+ B cells, CD4+ and CD8+ T cells in peripheral (pLN) and spleen of WT and Foxp3 ΔE2 mice. (C) Expression of CD62L and CD44 in CD4+ T cells of WT and Foxp3 ΔE2 mice. (D) Expression of IL-4 and IFN-γ in CD4+ T cells ex vivo from pLN of WT and Foxp3 ΔE2 mice. (E) Expression of IL-17 in CD4+ T cells ex vivo from pLN of WT and Foxp3 ΔE2 mice. Data represent mean ± SEM from one of ≥ 2 experiments. ns: not significant; *: p < 0.05; **: p < 0.01; ***: p < 0.001 by multiple t-tests (A - D) or two-tailed t-test (E).
Foxp3 ΔE2 mice develop systemic autoimmune disease
Systemic autoimmune diseases are often characterized by the existence of autoantibodies accompanied by increased follicular helper T cells (TFH) and germinal center B cells (GC B). Relative to WT mice, Foxp3 ΔE2 mice at 8 weeks of age showed a marked increase in TFH (CD4+CXCR5+PD-1+) cells (Fig. 3A). Consistent with the expanded TFH population, Foxp3 ΔE2 mice had significantly increased GC B cells (CD95+GL7+Bcl6+) (Fig. 3B), enlarged spontaneous germinal centers (Fig. 3C), and elevated serum anti-dsDNA IgG concentration (Fig. 3D) that could be detected in the serum starting around 40 days of age (Fig. 3E). The elevated anti-dsDNA autoantibodies in Foxp3 ΔE2 mice were mainly IgG1 and IgM isotypes (Fig. S3A) with IgA comparable to WT mice (Fig. S3B). Similar to hemizygous Foxp3 ΔE2 male mice, homozygous Foxp3 exon 2 deletion (ΔE2Hom) female mice also exhibited increased total cellularity and percentages of Tregs in peripheral lymph nodes (Fig. S4A) and serum anti-dsDNA autoantibodies (Fig. S4B). However, heterozygous Foxp3 exon 2 deletion (ΔE2Het) female mice resembled WT mice with respect to LN cellularity and percentage of Tregs (Fig. S4A), levels of serum autoantibodies (Fig. S4B). Foxp3 ΔE2Het female mice also generated similar frequency of TFH and GC B cells to WT mice 7 days after sheep red blood cell immunization (Fig. S4 C&D).
Figure 3. Foxp3 exon 2 deletion results in a systemic autoimmune disease.
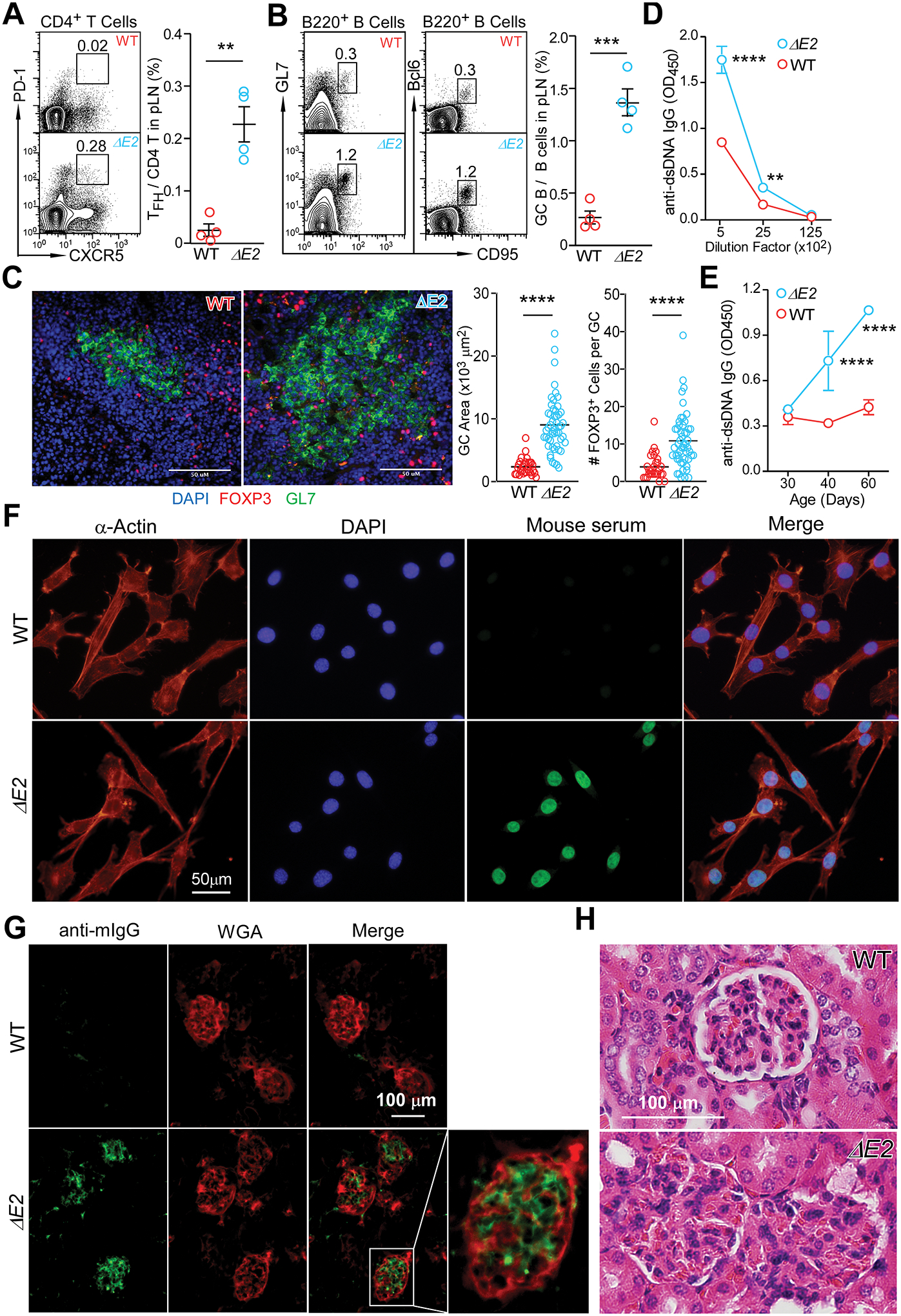
(A) Flow cytometric analysis of CD4+PD-1+CXCR5+ follicular helper T cells (TFH) in peripheral lymph nodes of WT and Foxp3 ΔE2 mice. (B) Flow cytometric analysis of B220+GL-1+CD95+Bcl6+ germinal center B (GC B) cells in peripheral lymph nodes of WT and Foxp3 ΔE2 mice. (C) Spontaneous germinal centers in WT and Foxp3 ΔE2 mice were examined by immunohistochemistry. Representative images (left) and quantification of GCs (right). (D) ELISA quantification of anti-dsDNA IgG in the serum of WT and Foxp3 ΔE2 mice (n = 6–7). (E) Time course of anti-dsDNA IgG in the serum of Foxp3 ΔE2 mice. Sera from WT and Foxp3 ΔE2 mice at indicated ages were diluted at 1:500 (n = 10 mice). (F) Representative images of serum anti-ANA IgG (at 1:160 dilution of serum) detected with fixed mouse 3T3 fibroblast cells. Mouse serum: sera collected from WT or Foxp3 ΔE2 mice were used as primary antibody and FITC anti-mouse IgG was used as secondary antibody to detect the existence of anti-ANA IgG in sera. (G) Immunofluorescence of kidney glomerulus sections showing IgG deposits in Foxp3 ΔE2 mice. WGA: wheat germ agglutinin. (H) Kidney glomerulus sections stained with hematoxylin and eosin (H&E). Data represent mean ± SEM (A-B) or mean ± SD (D&E) from one of ≥ 2 independent experiments. **: p < 0.01; ***: p < 0.001; ****: p < 0.0001 by two-tailed t-test (A-C) or two-way ANOVA with Bonferroni post-hoc test (D&E).
The presence of antinuclear antibodies and renal deposition of immune complexes are hallmarks of systemic autoimmune diseases. Consistent with this, sera from two-month-old Foxp3 ΔE2 mice strongly stained the nuclei of mouse 3T3 fibroblast cells (Fig. 3F). Immunohistochemistry with frozen sections of kidney showed prominent IgG deposition in the glomeruli (Fig. 3G) and hematoxylin and eosin staining indicated reduced Bowman’s space of the glomeruli (Fig. 3H) in Foxp3 ΔE2 mice. The autoimmune disease in Foxp3 ΔE2 mice progressed with age. By 10 months of age, Foxp3 ΔE2 mice developed splenomegaly in addition to further enlarged lymph nodes (Fig. S5A). About 50% of Foxp3 ΔE2 mice at this age had skin lesions on the neck and face (Fig. S5B). Histological analysis of the skin from Foxp3 ΔE2 mice revealed a skin pathology seen in cutaneous lupus erythematosus, including epidermal hypertrophy, hyperkeratosis, and a dermal mononuclear cell infiltrate (Fig. S5C). Consistent with increased mast cell counts in cutaneous lupus erythematosus (31), number of mast cells in the skin of Foxp3 ΔE2 mice was also significantly increased (Fig. S5D). Inflammatory infiltrates were also detected in the liver (Fig. S5E). Taken together, our data demonstrate that Foxp3 ΔE2 mice developed a spontaneous systemic autoimmune disease that resembles systemic lupus erythematosus.
FOXP3 ΔE2 Tregs have reduced expression of phenotypic genes but normal suppressive function
Despite having relatively normal thymic Treg development (Fig. S1D), Foxp3 ΔE2 mice had significantly increased Treg frequency and proliferation in secondary lymphoid organs (Fig. 4A&B). A significantly higher proportion of FOXP3 ΔE2 Tregs had dim staining of FOXP3 compared to Tregs from WT mice, while mean fluorescent intensity (MFI) in FOXP3 expression was similar, likely due to the presence of more activated Tregs in Foxp3 ΔE2 mice (Fig. 4C). Tregs in Foxp3 ΔE2 mice (both hemizygous males and homozygous females) showed significantly lower CD25, but comparable CTLA-4 and GITR, expression compared to FOXP3 FL Tregs in WT mice (Fig. 4D). Similar to conventional CD4+ T cells (Fig. 2C), Tregs from Foxp3 ΔE2 mice were also more activated, with a higher proportion of CD44hiCD62Llo cells than in WT mice (Fig. 4E). Surprisingly, Tregs purified from either Foxp3 ΔE2 or WT mice similarly suppressed proliferation of activated naïve CD4+ T cells in vitro (Fig. 4F). Consistent with having normal suppressive function, purified FOXP3 ΔE2 Tregs showed similar demethylation patterns in the Treg-specific demethylation region (TSDR) to FOXP3 FL Tregs (Fig. S6A&B). These data suggest that the FOXP3 ΔE2 isoform are capable of mediating Treg differentiation and suppressive function, despite the systemic autoimmunity observed in Foxp3 ΔE2 mice.
Figure 4. FOXP3 ΔE2 Tregs display more activated phenotypes, reduced phenotypic markers, but similar suppressive function.
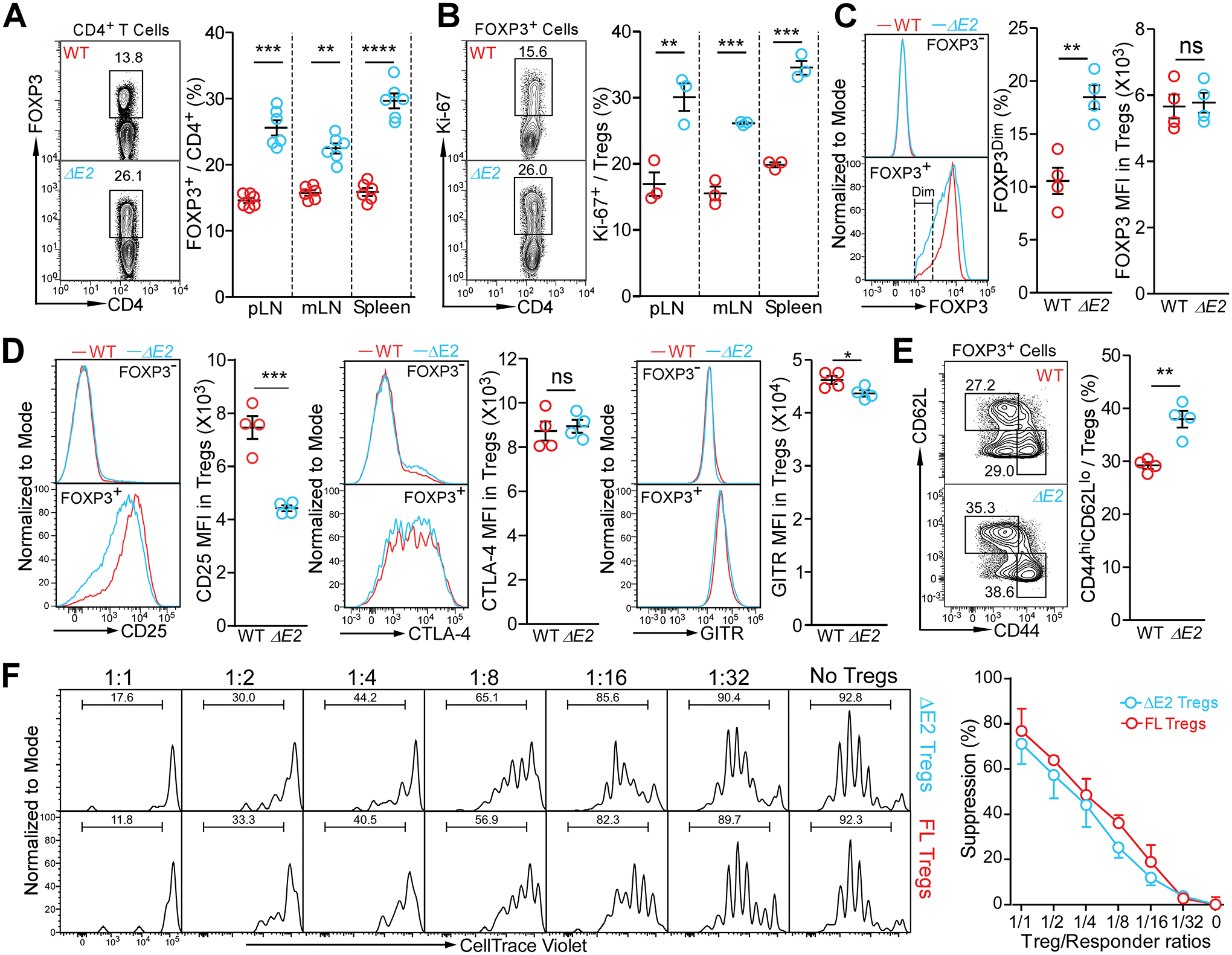
(A) Flow cytometric analysis of Treg frequencies in secondary lymphoid organs of WT and Foxp3 ΔE2 mice. (B) Proliferation of Treg cells in WT and Foxp3 ΔE2 mice as marked with Ki-67 expression. (C) Mean fluorescent intensity (MFI) of FOXP3 and percentage of FOXP3Dim in Treg cells of WT and Foxp3 ΔE2 mice. (D) Mean fluorescent intensity (MFI) of CD25, CTLA-4 and GITR in Treg cells of WT and Foxp3 ΔE2 mice. (E) Flow cytometric analysis of CD44 and CD62L in Treg cells of WT and Foxp3 ΔE2 mice. Data in (A - E) represent mean ± SEM from one of ≥ 2 independent experiments. **: p < 0.01; ***: p < 0.001; ****: p < 0.0001 by multiple t-tests (A & B) or two-tailed t-test (C – E). (F) Suppressive function of Foxp3 ΔE2 Tregs vs Foxp3 FL Tregs in vitro. Tregs were FACS purified from Foxp3 ΔE2;BAC-Foxp3eGFP and Foxp3 FL;BAC-Foxp3eGFP mice. Data represent mean ± SEM (n = 3 mice) from one of 2 independent experiments. Means were compared by two-way ANOVA with Bonferroni post-hoc test. No difference in suppression by FOXP3 ΔE2 and FOXP3 FL Tregs.
Since Foxp3 ΔE2 mice displayed systemic autoimmunity, the observed Tregs phenotypes could be attributed to an inflammatory environment. To determine the cell intrinsic effects of the FOXP3 ΔE2 isoform on Treg phenotype in the absence of systemic inflammation, we examined Tregs in heterozygous Foxp3 exon 2 deletion (Foxp3 ΔE2Het) female mice. Due to random X chromosome inactivation, these mice have two populations of Tregs, expressing either FOXP3 ΔE2 or FOXP3 FL, but not both (Fig. 5A). Consistent with the expected 1:1 ratio of FOXP3 FL Tregs to FOXP3 ΔE2 Tregs due to random X inactivation, there were roughly equal numbers of the two Treg populations in the thymus of Foxp3 ΔE2Het female mice (Fig. 5A). However, FOXP3 ΔE2 Tregs only accounted for 20–25% of total Tregs in secondary lymphoid organs (Fig. 5A). The reduced percentage of the FOXP3 ΔE2 Treg population was also seen in mucosal tissues such as lung and gut (Fig. 5A). To eliminate the possible impact of biased X chromosome inactivation on the homeostasis of FOXP3 ΔE2 Tregs, we generated mixed bone marrow chimeras, where sub-lethally irradiated male Rag2-deficient recipients were adoptively transferred with 1:1 mixed bone marrow from WT and Foxp3 ΔE2 male mice. Despite similar numbers of FOXP3 FL and FOXP3 ΔE2 Tregs being generated in the thymus (Fig S7A), FOXP3 ΔE2 Tregs in the peripheral lymphoid organs only accounted for 20–25% of total Tregs (Fig. S7B). A reduced frequency of FOXP3 ΔE2 Tregs in the periphery of heterozygous Foxp3 ΔE2 female mice was also observed in a heterozygous healthy women carrying one allele with c.305delT mutation in the FOXP3 exon 2 (32). Notably, similar to the frequency of FOXP3 ΔE2 Tregs in Foxp3 ΔE2Het female mice, the frequency of Tregs expressing only the FOXP3 ΔE2 isoform was ~17% of total Tregs in circulation from this heterozygous c.305delT carrier (32). These data further suggest that our Foxp3 ΔE2 mice are a valid model to study the biology of FOXP3 ΔE2 isoform in Treg development and function.
Figure 5. Intrinsic defects of FOXP3 ΔE2 Tregs in heterozygous Foxp3 exon 2 deletion female mice.
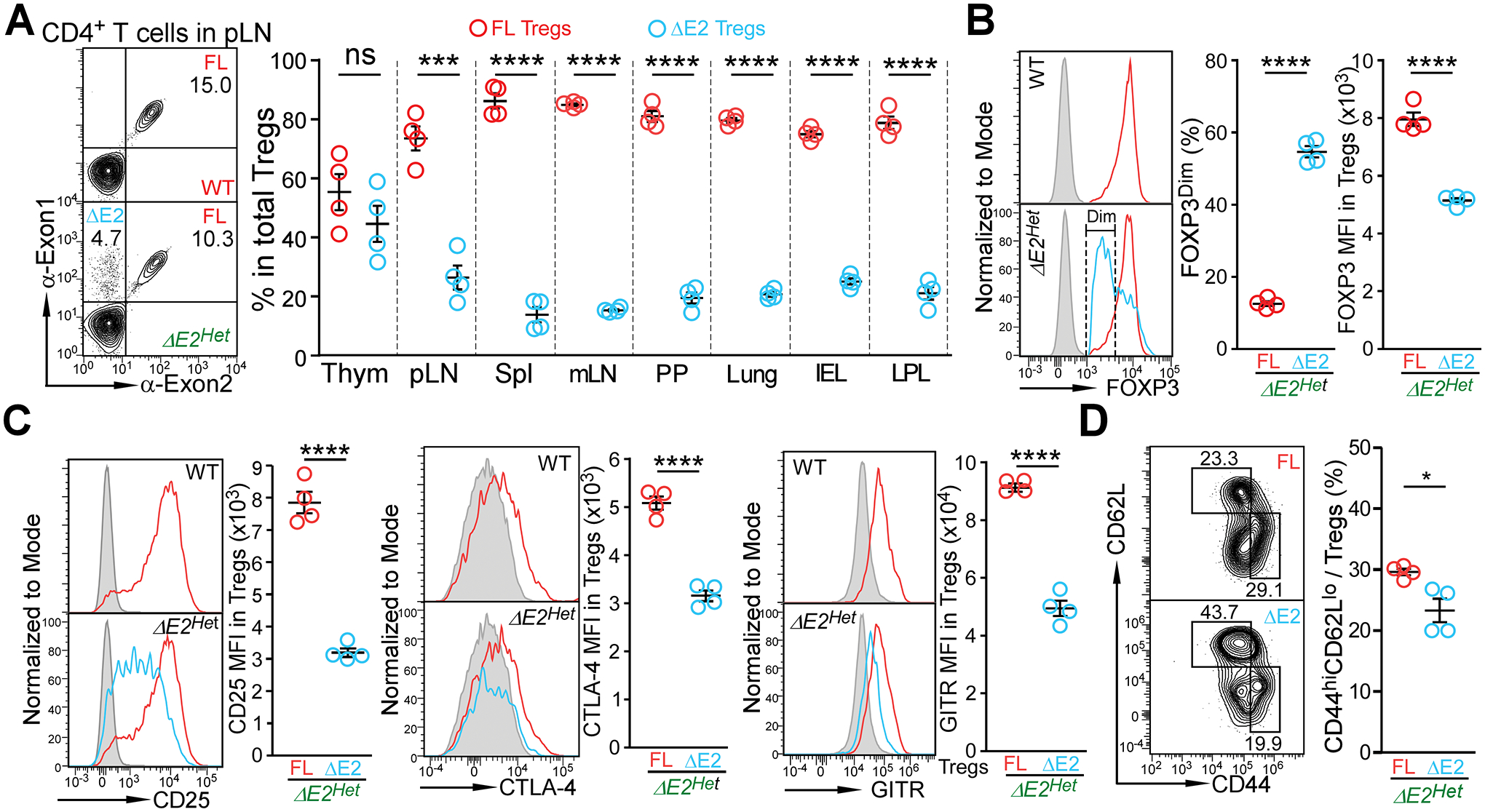
(A) FOXP3 FL and FOXP3 ΔE2 Treg populations in heterozygous Foxp3 exon 2 deletion (ΔE2Het) female mice at the age of 2 months. Thym: thymus; pLN: peripheral lymph nodes; Spl: spleen; mLN: mesenteric lymph nodes; PP: Peyer’s Patches; IEL: intraepithelial lymphocytes of the gut; LPL: lamina propria lymphocytes of the gut. (B) Expression of FOXP3 in FOXP3 FL and FOXP3 ΔE2 Tregs in ΔE2Het female mice. (C) Mean fluorescent intensity (MFI) of phenotypic Treg markers CD25, CTLA-4, and GITR in FOXP3 FL and FOXP3 ΔE2 Tregs in ΔE2Het female mice. Grey line: FOXP3− cells; Red line: FOXP3 FL Tregs; Blue line: FOXP3 ΔE2 Tregs. (D) Flow cytometric analysis of CD44 and CD62L in Treg cells expressing either the FOXP3 FL (red) or FOXP3 ΔE2 (blue) isoform in pLN of ΔE2Het female mice. Data represent mean ± SEM from one of > 2 independent experiments.*: p < 0.05 **: p < 0.01; ***: p < 0.001; ****: p < 0.001 by paired two-tailed t-test.
Unlike Tregs from Foxp3 ΔE2 mice (Fig. 4C), FOXP3 ΔE2 Tregs from Foxp3 ΔE2Het female mice showed a significant reduction in FOXP3 expression, with over 50% staining dim for FOXP3, with significantly lower MFI compared to FOXP3 FL Tregs in the same mice (Fig. 5B). In addition to even lower CD25 expression, CTLA-4 and GITR expression were also significantly reduced compared to FOXP3 FL Tregs from the same Foxp3 ΔE2Het female mice (Fig. 5C). They also displayed a less activated phenotype with a lower proportion of CD44hiCD62Llo cells than FOXP3 FL Tregs (Fig. 5D) in contrast to more activated FOXP3 ΔE2 Tregs in Foxp3 ΔE2 mice (Fig. 4E). These data indicate that deletion of Foxp3 exon 2 results in intrinsic defects in expression of Treg phenotypic genes.
FOXP3 exon 2 region is required for Treg stability
Despite purified CD25hi FOXP3 ΔE2 Tregs having normal suppressor function in vitro (Fig. 4F), as well as largely normal demethylated TSDR (Fig. S6A&B) compared to FOXP3 FL Tregs, our observation that FOXP3 ΔE2 Tregs had reduced expression of CD25 and FOXP3 (Fig. 4&5) and frequency in the periphery of Foxp3 ΔE2Het female mice may suggest that FOXP3 ΔE2 Tregs have reduced stability. To test our hypothesis that FOXP3 ΔE2 Tregs had a reduced ability to maintain FOXP3 expression and lineage identity, we FACS sorted FOXP3 ΔE2 and FOXP3 FL Tregs based upon eGFP and Thy1.1/Thy1.2 expression from mixed bone marrow chimera mice, and transferred purified Tregs into Tcrb deficient mice at a 1:1 ratio (Fig. 6A). The expression of eGFP (encoded by a transgenic bacterial artificial chromosome transgene (33)) strongly mirrored endogenous FOXP3 expression with similar Nrp1+ percentages for both FOXP3 FL and FOXP3 ΔE2 Tregs (Fig. S8A–C). After transfer, the identity of FOXP3 ΔE2 and FOXP3 FL Tregs in recipient mice can be tracked based upon Thy1.1/Thy1.2 and FOXP3 expression. Total Thy1.1+ (FOXP3 ΔE2 Treg-derived) and Thy1.1− (FOXP3 FL Treg-derived) cells within the CD4+TCRβ+ donor population maintained a 1:1 ratio over the 4-week period (Fig. 6B), suggesting that the overall survival rate of FOXP3 ΔE2 Treg-derived cells was not decreased compared with FOXP3 FL Treg-derived cells. However, the percentage of FOXP3+ cells in the Thy1.1+ cell population quickly decreased to less than 20%, while over 55% of Thy1.1− FOXP3 FL Treg derived cells retained FOXP3 expression (Fig. 6C). At the end of the 4-week period, ~75% FOXP3+ cells were FOXP3 FL Treg derived (Fig. 6D upper panels) whereas ~75% FOXP3− cells are FOXP3 ΔE2 Treg-derived (Fig. 6D lower panels). The instability of FOXP3 ΔE2 Tregs was not rescued when naïve CD4+ T cells were co-transferred with Tregs (Fig. S9), and the percentage of FOXP3 ΔE2 Tregs (3G3+150D−) was reduced from 57% before transfer to ~ 20% 4 weeks after cell transfer.
Figure 6. Instability of FOXP3 ΔE2 Tregs.
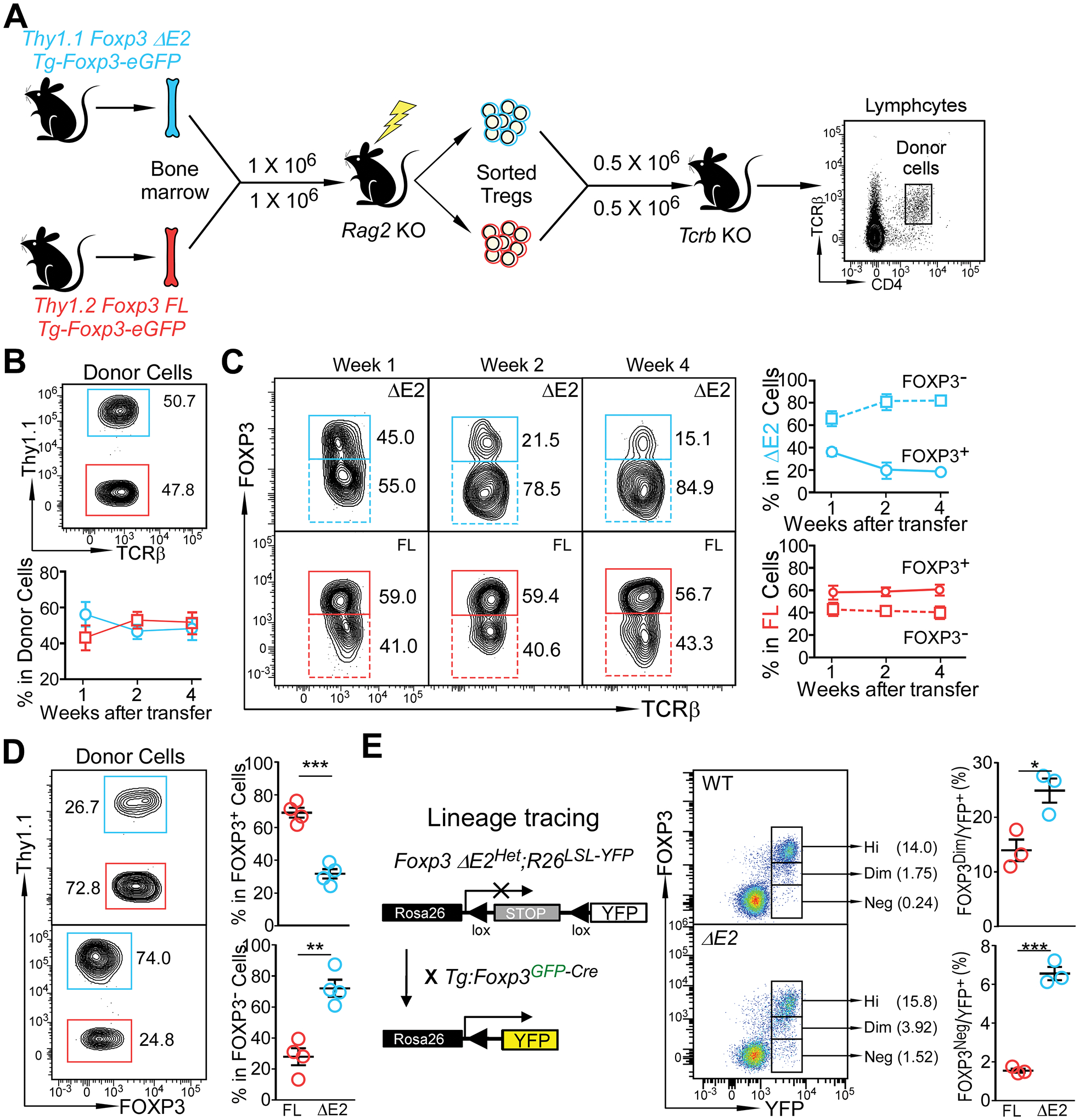
(A) Experimental design to examine stability of FOXP3 FL and FOXP3 ΔE2 Tregs. FOXP3 ΔE2 Tregs (Thy1.1+) and FOXP3 FL Tregs (Thy1.2+) were sort purified from mixed bone marrow chimeras. The Tregs were mixed at a 1:1 ratio, adoptively transferred (i.v.) into Tcrb deficient mice, and their identity was tracked by flow cytometry. (B) Percentage of Thy1.1+ (FOXP3 ΔE2 Treg derived) and Thy1.1− (FOXP3 FL Treg derived) donor cells in the blood over the 4 week period. (C) Percentage of Thy1.1+ (FOXP3 ΔE2 Treg derived) and Thy1.1− (FOXP3 FL Treg derived) donor cells still remaining FOXP3+ over time. (D) Flow cytometric analysis of FOXP3 expression in FOXP3 ΔE2 and FOXP3 FL Treg derived donor cells four weeks after transfer. (E) Tracking Treg stability using lineage tracing mice. Data represent mean ± SEM (n = 4 mice for B-D; and n = 3 mice for E) from one of 2 independent experiments. **: p < 0.01; ***: p < 0.001 by two-tailed t-test.
To further determine the stability of FOXP3 ΔE2 Tregs in a more physiological environment, we crossed Foxp3 ΔE2Het;R26LSL-YFP female mice (C57BL/6 background) with Tg:Foxp3GFP-Cre male mice (129S background) to obtain F1 male mice carrying either Foxp3 ΔE2 allele of Foxp3 FL (WT) alleles (Fig. 6E). The Foxp3-Cre–mediated excision of the floxed STOP cassette upstream of YFP at the Rosa26 locus results in constitutive, heritable expression of YFP, even for Tregs that have lost Foxp3 expression (Ex-Tregs, YFP+FOXP3−). Foxp3 ΔE2 mice had significantly more YFP+ cells that were dim or negative for FOXP3 than Foxp3 FL (WT) mice, suggesting the preferential loss of FOXP3 expression in FOXP3 ΔE2 Tregs (Fig. 6E). Taken together, these data indicate that FOXP3 ΔE2 Tregs are unstable and easily lose FOXP3 expression, and thus Treg lineage identity.
Sufficiency of FOXP3 ΔE2 Tregs to induce systemic autoimmunity
Komatsu et al. demonstrated that ex-FOXP3 cells undergo transdifferentiation into pathogenic TH17 cells leading to more severe autoimmune arthritis after immunization (34). The fact that thymus-derived Tregs express self-reactive TCRs (35, 36) suggested the possibility that FOXP3 ΔE2 Tregs could become autoreactive effectors upon loss of FOXP3 expression, leading to autoimmunity. To test this hypothesis, we transferred FACS sorted FOXP3 ΔE2 Tregs into Tcrb-deficient recipient mice. Control recipient mice received purified FOXP3 FL Tregs or FOXP3 FL + FOXP3 ΔE2 Tregs at 1:1 ratio as depicted in Fig. 7A. Three months after cell transfer, recipient mice that received FOXP3 ΔE2 Tregs developed anti-dsDNA IgG autoantibodies, as well as anti-nuclear antibodies (Fig. 7B&C). Consistent with the report showing that Tregs lost FOXP3 expression after transferring into T cell deficient mice (37), we also observed that approximately 60% of CD4+TCRβ+ donor cells became Foxp3− in the recipient mice that received either FOXP3 FL Tregs alone or mixed with FOXP3 ΔE2 Tregs (Fig. 7D). The FOXP3 ΔE2 Treg-transferred group, however, had expanded CD4+TCRβ+FOXP3+ and FOXP3− cells (Fig. 7D), a phenomenon similar to what was seen in hemizygous male and homozygous female Foxp3 ΔE2 mice (Fig. 4A and Fig. S4A). Significantly higher percentages of TFH and TFR cells (Fig. 7E), as well as GC B cells (Fig. 7F), were also observed in FOXP3 ΔE2 Treg-transferred recipient mice.
Figure 7. FOXP3 ΔE2 Tregs are sufficient to induce autoantibody and enhanced TFH and GC B cell response.
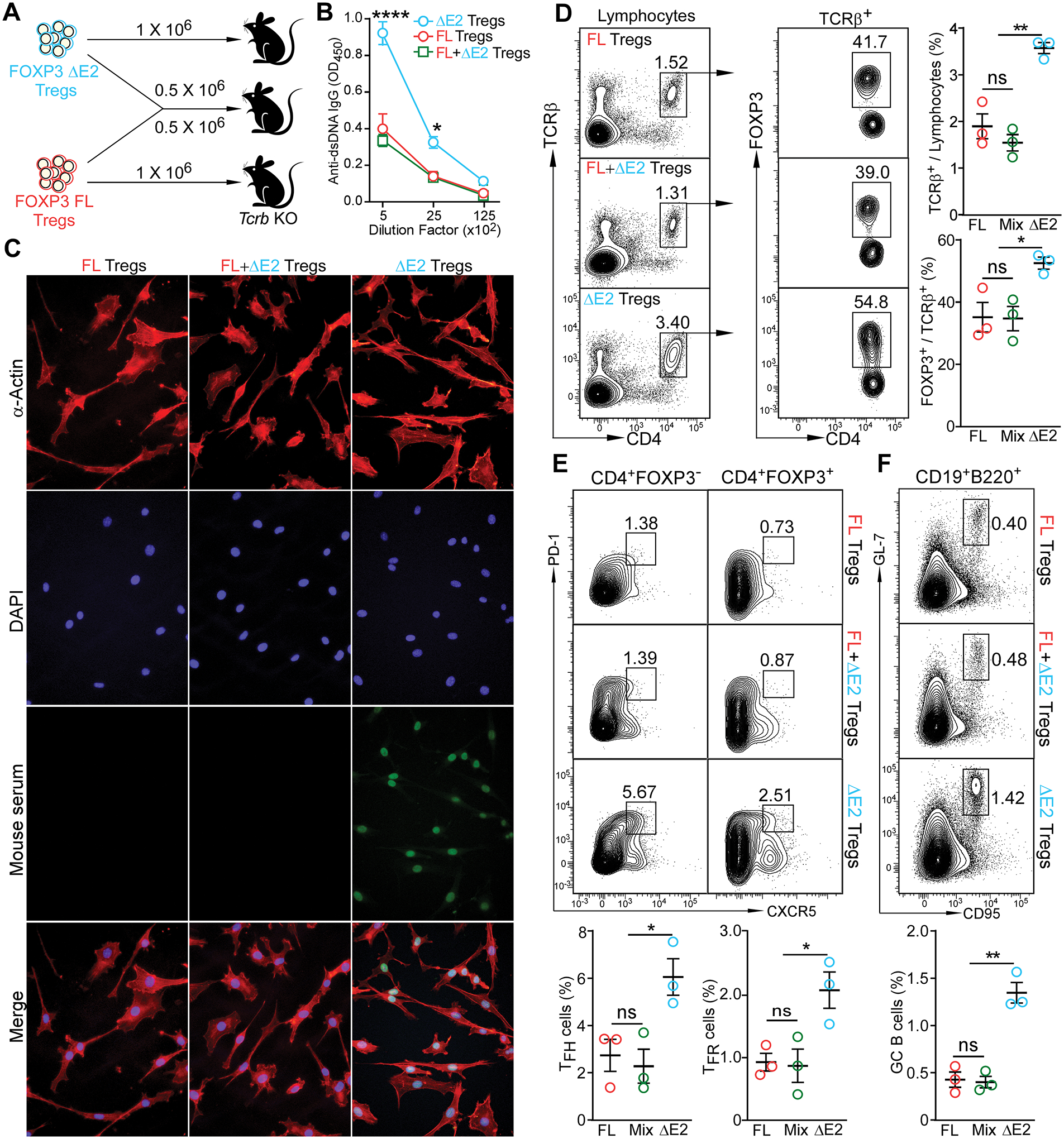
(A) Experimental design. FOXP3 FL and FOXP3 ΔE2 Tregs were purified by FACS sorting. 1 × 106 purified Tregs were adoptively transferred into Tcrb deficient recipients by tail vail injection. Recipient mice were analyzed 3 months after cell transfer. (B) ELISA quantification of anti-dsDNA IgG in the serum of the recipient mice. (C) Representative images of serum anti-ANA IgG (at 1:80 dilution of serum) in the recipient mice detected with fixed mouse 3T3 fibroblast cells. Mouse serum: sera collected from recipient mice receiving indicated Tregs were used as primary antibody and FITC anti-mouse IgG was used as secondary antibody to detect the existence of anti-ANA IgG in sera. (D) Percentage of donor cells and percentage of donor cells still remaining FOXP3+ in the spleen of recipient mice. (E) TFH (left panels) and TFR (right panels) in the spleen of recipient mice. (F) Germinal center B cells in the spleen of recipient mice. Data represent mean ± SEM (n = 3 mice) from one of 2 independent experiments. ns: not significant; *: p < 0.05; **: p < 0.01; ****: p < 0.0001 by two-way ANOVA (B) or one-way ANOVA (D – F) with Bonferroni post-hoc test.
Tcrb-deficiency represents a harsh environment for transferred Treg cells due to an absence of IL2 and other factors from conventional CD4+ T cells important for maintaining the Treg niche. To exclude the possibility that the induction of autoantibody in FOXP3 ΔE2 Treg-transferred Tcrb-deficient mice was due to a lack of conventional CD4+ T cells, we co-transferred 2 × 106 FOXP3 ΔE2 or FOXP3 FL Tregs with 8 × 106 naïve CD4+ T cells into Tcrb-deficient recipient mice (Fig. S10A) and tracked anti-dsDNA autoantibody in these mice. Four weeks after the cell transfer, sera from mice that received FOXP3 ΔE2 Tregs and naïve CD4+ T cells had significantly higher reactivity to dsDNA than that from mice received FOXP3 FL Tregs and naïve CD4+ T cells (Fig. S10B&C). Four months after the cell transfer, the anti-dsDNA IgG titer in mice received naïve CD4+ T cells with FOXP3 ΔE2 Tregs reached the level seen in 4 month-old Foxp3 ΔE2 mice (Fig. S11A). Histological studies revealed that these mice had reduced Bowman’s space (Fig. S11B) with significant “wire loop” lesions of the glomeruli (Fig. S11C). Interestingly, both FOXP3 ΔE2 and FOXP3 FL Tregs were able to prevent the development of naïve CD4+ T cell-induced colitis. The mice received naïve CD4+ T cells plus FOXP3 ΔE2 Tregs had similar body weight, colon length and microstructure as the mice that received naïve CD4+ T cells plus FOXP3 FL Tregs (Fig. S11D–F). Thus, transferring FOXP3 ΔE2 Tregs into a T cell-deficient host recapitulated the phenotype seen in Foxp3 ΔE2 mice.
FOXP3 ΔE2 regulates inflammatory cytokines and Foxp3 positive regulators without changes in genomic binding
To explore the molecular mechanisms of FOXP3 ΔE2 Treg instability, we used RNA-seq to compare the transcriptional profile of FOXP3 ΔE2 and WT Tregs. 342 genes were up-regulated and 275 genes down-regulated with greater than 1.5-fold change (FDR < 0.05) in FOXP3 ΔE2 Tregs compared with WT Tregs (Fig. 8A and Supplementary Data File 1). Genes up-regulated in FOXP3 ΔE2 Tregs were enriched in inflammatory response and allograft rejection pathways (Fig. 8B). Notably, expression of a set of inflammatory cytokines such as Ifng, Il4 and Il9 was increased in FOXP3 ΔE2 Tregs (Fig. 8C). Using CRISPR-based screening, a recent study identified a set of 25 FOXP3 positive regulators and Treg-specific deletion of the positive regulator Usp22 destabilized FOXP3 expression leading to spontaneous autoimmunity (38). The majority of these FOXP3 positive regulators were found to be down-regulated in FOXP3 ΔE2 Tregs (Fig. 8D).
Figure 8. FOXP3 isoforms regulate target gene expression mostly independent of DNA binding.
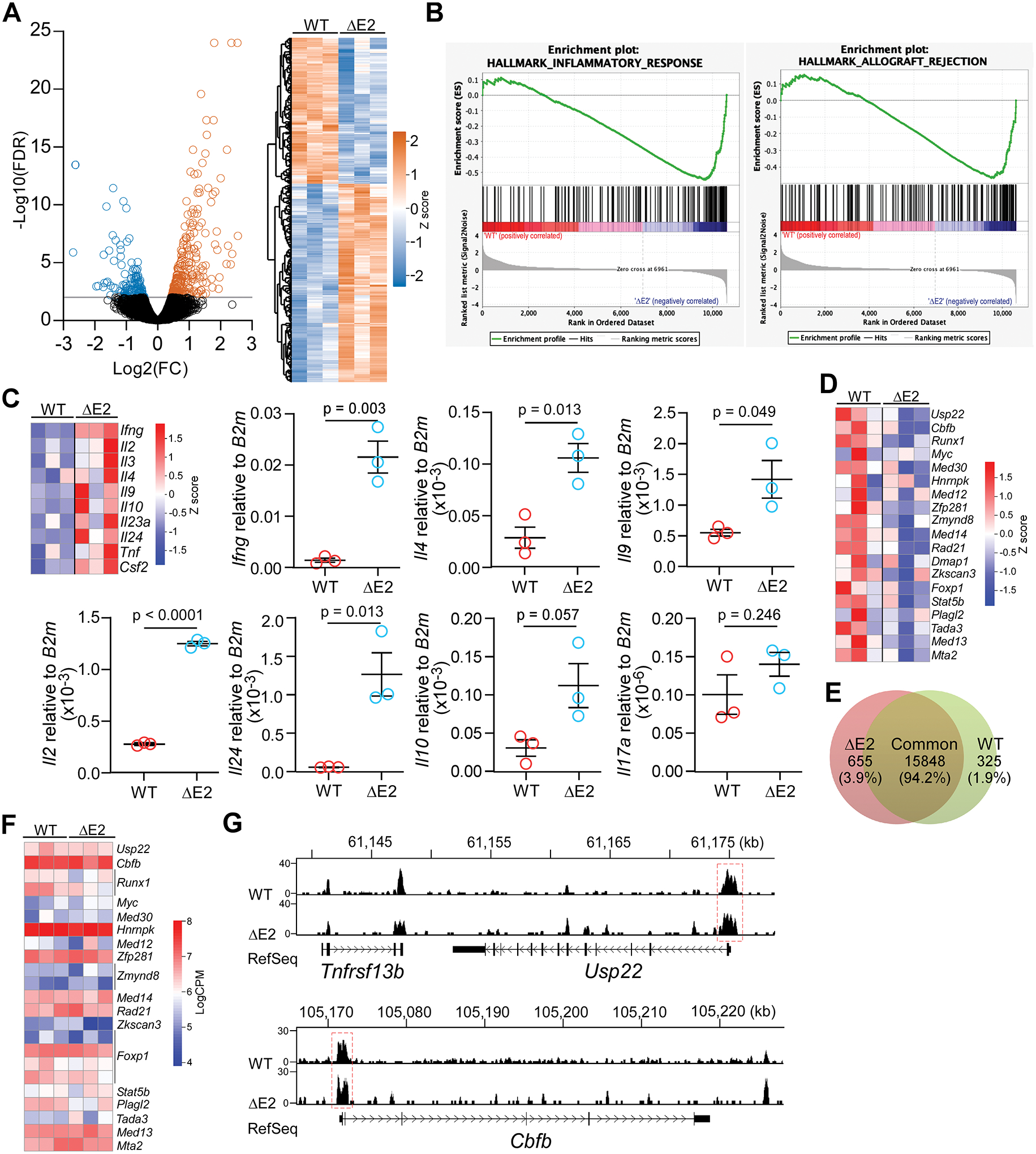
(A) Volcano plot and hierarchical clustering heatmap showing differential gene expression in FOXP3 ΔE2 Tregs. (B) Up-regulated genes in FOXP3 ΔE2 Tregs are enriched in inflammatory response and allograft rejection gene sets. (C) Heatmap (RNA-seq) and RT-qPCR showing up-regulation of cytokines in FOXP3 ΔE2 Tregs. (D) Heatmap of Foxp3 positive regulators in FOXP3 ΔE2 and FOXP3 FLTregs. (E) FOXP3 binding peaks enriched in FOXP3 ΔE2 vs WT (FOXP3 FL) Tregs (change ≥ 1.5 folds, p < 0.05). (F) Heatmap of FOXP3 ΔE2 and FOXP3 FL binding (LogCPM) to the loci of Foxp3 positive regulators in (D). (G) Representative plots of FOXP3 ChIP-seq reads from FOXP3 ΔE2 and FOXP3 FL (WT) Tregs at the Usp22 and Cbfb gene loci. n = 3 mice/group for both RNA-seq and ChIP-seq analysis. Data in (C) represent mean ± SEM (n = 3 mice) with p by two tailed t-test.
Next, we performed FOXP3 ChIP-seq to determine whether the FOXP3 ΔE2 isoform had changed DNA binding specificity. Out of total 16,828 binding peaks, 655 peaks (3.89%) were enriched in FOXP3 ΔE2 Tregs and 325 peaks (1.93%) were enriched in WT (FOXP3 FL) Tregs (p < 0.05, Fig. 8E and Supplementary Data File 2). Despite increased expression of cytokine genes in FOXP3 ΔE2 Tregs (Fig. 8C), neither FOXP3 ΔE2 isoform nor FOXP3 FL isoform bound to these gene loci. On the contrary, FOXP3 had binding peak(s) at all the Foxp3 positive regulators (Fig. 8F) but Dmap1 locus, mostly at the promoter of the genes (Supplementary Data File 3). No significant differences of FOXP3 ΔE2 and FOXP3 FL bindings to these loci were identified (Fig. 8F and Supplementary Data File 3) as shown at the promoter regions of Usp22 and Cbfb (Fig. 8G). Our results suggest that FOXP3 ΔE2 and FOXP3 FL isoforms regulate their target gene expression independent of DNA binding.
DISCUSSION
Self-tolerance is maintained by thymic-derived FOXP3+CD4+ T cells (2, 39). In this study, we demonstrated that patients expressing only the FOXP3 ΔE2 isoform in CD4+ T cells due to deletion mutations in exon 2 of the FOXP3 gene developed IPEX syndrome. Consistent with this observation, mice with Foxp3 exon 2 deletion had increased TFH and GC B responses and developed systemic autoimmune disease with increased anti-dsDNA autoantibodies, glomerular immune complex deposition, and nephritis. Although capable of suppressing CD4+ T cell proliferation in vitro, FOXP3 ΔE2 Tregs have intrinsic defects in FOXP3 and CD25 expression, and when transferred into Tcrb-deficient recipient mice lost FOXP3 expression and induced systemic immunity as seen in Foxp3 ΔE2 mice. While FOXP3 ΔE2 is sufficient to generate suppressive Tregs, it is less efficient at either inducing or maintaining expression of several genes that reinforce FOXP3 expression. We speculate that this decreased function leads to decreased Treg stability and an increased inflammatory environment that may further potentiates the switch to a less stable T regulatory cell phenotype. Our findings emphasize a critical requirement for the exon 2 encoded region of FOXP3 in maintaining the lineage stability of Tregs and immune homeostasis in vivo.
The majority of the FOXP3 mutations that cause IPEX syndrome alter the DNA-binding Forkhead (FKH) domain followed by the N-terminal proline-rich (ProR) domain and the leucine-zipper (LZ) domain (40, 41). Affected males typically die within the first or second year of life if not treated with immunosuppressive agents or by hematopoietic stem cell transplantation. The deletion mutations in the FOXP3 exon 2 region were reported previously (32, 40, 42–47). Most patients with these mutations showed typical IPEX syndrome days to weeks after birth. The ability of these mutant alleles to produce the FOXP3 ΔE2 isoform in early reports was not examined, but one report showed that the patient with c.227delT mutation had no detectable CD4+CD25+FOXP3+ Tregs (44). We and a recent case report (32) demonstrated that patients with deletion mutations in the FOXP3 exon 2 region are able to generate Tregs expressing only the FOXP3 ΔE2 isoform. Similar to what we saw in Foxp3 ΔE2 mice, the circulating FOXP3+CD127lo population in the P2 patient with c.305delT mutation was expanded 2 times compared with the healthy donor (32). This individual was largely healthy until 40 years of age when he developed psoriatic arthritis and late-onset diabetes. The wide range of FOXP3+ Treg frequencies seen in the patients with deletion mutation in FOXP3 exon 2 suggests a yet to be uncovered mechanism that regulates the expression/stability of the FOXP3 ΔE2 isoform in the absence of the FOXP3 FL isoform and likely severity of the disease.
FOXP3 binds to thousands of genomic sites (48, 49) and interacts with hundreds of partners (50) to activate or repress gene expression in Treg cells. Several recent studies demonstrated that missense point mutations in the DNA binding FKH domain changed the mutated FOXP3 binding to Batf locus (51) and Th2 cytokine locus (52), leading to impaired Treg function and autoimmunity. Despite increased cytokine expression and decreased Foxp3 positive regulatory genes, we did not identify altered binding specificity of FOXP3 ΔE2 at these gene loci. It has been shown that FOXP3 may regulate gene expression independent of direct DNA binding and the N-terminal proline-rich domain (ProR, exon 1 – exon 3) is important in FOXP3-mediated transcriptional regulation (53). The interaction of ProR with class I HDACs is critical for FOXP3-mediated Il2 suppression (53). However, co-immunoprecipitation showed that FOXP3 ΔE2 isoform is equally capable of interacting with HDAC1 and HDAC3 as FOXP3 FL isoform (Fig. S12), suggesting that FOXP3 exon 2 region regulates Tregs transcriptome without direct interaction with HDACs.
Our study demonstrates that lack of FOXP3 exon 2 results in reduced FOXP3 and CD25 expression in human FOXP3 ΔE2 Tregs but are restored upon activation (Fig. 1). Likewise, reduction of FOXP3 and CD25 expression on FOXP3 ΔE2 Tregs is alleviated in the autoimmune Foxp3 ΔE2 mice (Fig. 4) comparing to that in heterozygous mice that do not develop autoimmunity (Fig. 5). The hypomethylation of the Treg-sepcfic demethylated region (TSDR) (Fig. S6), suppressive function (Fig. 4F), and protein and mRNA stability (Fig. S13) are also comparable between FOXP3 FL and FOXP3 ΔE2 Tregs. These data suggest that FOXP3 ΔE2 can support Treg development and function. However, RNA-seq analysis showed that majority of FOXP3 positive regulators identified by CRISPR screen (38) are down-regulated (Fig. 8D). Although the moderate reduction in these positive regulators does not result in functional deficiency of FOXP3 ΔE2 Tregs as severe as USP22 (38) and RUNX1/CBFB (54) deficient Tregs, we speculate that the reduction in all these genes would overtime reduce FOXP3 ΔE2 expression and destabilize Tregs. FOXP3 ΔE2 Tregs also have increased expression of inflammatory cytokines and the increased inflammatory environment that may further potentiates the switch to a less stable T regulatory cell phenotype.
Human FOXP3 isoforms arise from alternative RNA splicing, and Tregs that express predominately the FOXP3 ΔE2 isoform exist even in healthy donors. The frequency of this population is increased in the PBMC of patients with anti-neutrophil cytoplasmic antibody-associated vasculitis (AAV) (8) and giant cell arteritis (10), accounting in many cases for over 80% of the Treg population (8). The ratio of FOXP3 ΔE2 mRNA to total FOXP3 mRNA increases from ~30% in the PBMC of healthy donors to ~65% in that from patients with Graves’ disease and Hashimoto’s thyroiditis (9). The imbalanced expression of FOXP3 ΔE2 to FOXP3 FL isoform is likely more prominent in the diseased tissue. FOXP3 ΔE2 mRNA is nearly 17 times more than FOXP3 FL mRNA in CD4+ T cells isolated from small intestinal biopsies of patients with active coeliac disease while only ~ 3 times in those from healthy controls (11). Taken together, these data indicate that increased FOXP3 ΔE2 isoform expression is strongly associated with autoimmune diseases.
The interaction between FOXP3 exon 2 region and RORα and RORγt was shown to be important for FOXP3 to suppress IL-17 expression and Th17 differentiation (17, 18). Yet our results demonstrate significantly increased IFN-γ and IL-4, but not IL-17, expression in CD4+ T cells in Foxp3 ΔE2 mice at 8 weeks of age. Naïve CD4 T cells isolated from Foxp3 ΔE2 mice does not show enhanced Th17 differentiation over a wide range of IL-6 concentration in vitro (Fig. S2A). This discrepancy was not because the FOXP3 ΔE2 isoform does not exist in mice. Using a morpholino antisense oligo to force splicing of FOXP3 to ΔE2 isoform in human cells had no impact on Th17 differentiation (Fig. S2B and (20)), while forced ΔE7 isoform in human cells strongly favored Th17 differentiation in vitro and increased ΔE7, but not ΔE2 level, correlating with IL-17A expression in vivo (20). These results suggest that suppression of IL-17 expression and/or Th17 differentiation is not a major function for FOXP3 exon 2, but rather the instability of FOXP3 ΔE2 Tregs leads to interruption of immune homeostasis.
There are limitations in our study as our research focuses on mouse models to study the function of the FOXP3 ΔE2 isoform. Comparing to the IPEX patients carrying deletion mutations in the FOXP3 exon 2 region, our Foxp3 ΔE2 mice have much milder autoimmune disease. One important difference between these patients and Foxp3 ΔE2 mice is that these deletion mutation alleles produce both FOXP3 ΔE2 mRNA and FOXP3 FL mRNA containing the mutated exon 2, while our mice with germline exon 2 deletion only produce Foxp3 ΔE2 mRNA. In patients, the mutated FOXP3 FL mRNA might interfere with the translation of the FOXP3 ΔE2 mRNA, resulting in reduced production of FOXP3 ΔE2 protein. Furthermore, the mutated FOXP3 FL mRNA could be translated into peptides containing exon 1, partial exon 2 and frame shifted amino acids after the deletion point. The partial FOXP3 + frame shifted peptides would likely interact with the FOXP3 ΔE2 isoform and interfere with their function. How the frame shifted/truncated peptide (depending on the position of the deletion, and how far the shifted reading frame reaching the stop codon) impacts number and function of the Tregs needs to be further defined. Another limitation is that our Foxp3 ΔE2 mice, despite being an indispensable tool to study FOXP3 ΔE2 function in vivo, would not provide information on how FOXP3 isoforms are regulated. Are there any benefits of the expression and balance of the two isoforms as seen in humans? How are the expression and balance of the two isoforms regulated in health and disease? Glycolysis may control the expression of the FOXP3 FL isoform, possibly through the binding of Elonase-1 to FOXP3 regulatory elements (57), although that would not explain the fact that the two isoforms are generated by pre-mRNA alternative splicing. It is currently unclear whether the increased FOXP3 ΔE2:FOXP3 FL ratio seen in several autoimmune diseases (8–11) is determined by genetic disposition, thus leading to the development of autoimmunity, or whether the inflammatory environment of autoimmunity shifts the expression of FOXP3 isoforms. The fact that IFN-γ plus butyrate treatment tends to increase FOXP3 ΔE2:FOXP3 FL protein ratio in PBMCs from patients with coeliac disease (but not from healthy donors) suggests the balance of the two FOXP3 isoforms might be determined by both genetics and environment.
Treg therapy is a promising approach to treat autoimmune diseases (24–26). While relatively effective in mouse studies to prevent or even reverse type 1 diabetes (27, 55, 56), clinical studies in the context of human disease often give mixed, or even limited, benefits (28–30). The possible shift of FOXP3 isoforms from FL to ΔE2 due to the inflammatory environment in the patients and thus results in instability of the Tregs would certainly affect the efficacy of Treg therapy. The transdifferentiation of ex-Tregs into self-reactive effector T cells might make the transferred Tregs detrimental rather than beneficial. The instability/transdifferentiation of Tregs expressing predominant the FOXP3 ΔE2 isoform should be an important factor to be considered in future studies of Treg therapy. We speculate that manipulating FOXP3 alternative splicing to enhance Treg stability and/or function could be exploited as a therapeutic approach for immune-mediated diseases and cancer immunotherapy.
MATERIALS AND METHODS
Study design
The aim of this study was to reveal the functional differences between the two major splicing isoforms of FOXP3 – FOXP3 FL and FOXP3 ΔE2 – that are normally expressed in human Tregs. We analyzed FOXP3 expression in several IPEX patients with nucleotide(s) deletion in FOXP3 exon 2 region by FACS and Western blot to determine the FOXP3 ΔE2 isoform expression. We generated a mouse line with Foxp3 exon 2 deletion to determine the function of FOXP3 ΔE2 Tregs. In vitro Treg suppression assay and FACS analysis were used to compare the differences between FOXP3 ΔE2 and FOXP3 FL Tregs. Enzyme-linked immunosorbent assay (ELISA) and fluorescent immunohistochemistry were performed to determine anti-dsDNA IgG and antinuclear antibodies (ANA) in serum. Adoptive cell transfer and lineage tracing were used to show that FOXP3 ΔE2 Tregs were unstable and sufficient to induce autoimmunity when transferred into TCRβ-deficient mice. RNA-seq and ChIP-seq analyses showed that mouse FOXP3 ΔE2 Tregs had changed transcriptomic program that is largely independent of its DNA binding capability. The number of mice and statistics used in the studies are included in the figure legend.
Cell culture media
T cells were cultured in RPMI 1640 (Gibco Life Technologies, 11875–093) supplemented with 10% FBS (Atlanta Biologicals, S11150), 100 unit/ml penicillin (Hyclone, SV30010), 100 μg/ml streptomycin (Hyclone, SV30010), 50 μM β-mercaptoethanol, 10mM HEPES buffer (gibco/life technologies, 15630–080), 1mM sodium pyruvate (Gibco Life Technologies, 11360–070), 1X MEM non-essential amino acids (Gibco Life Technologies, 11140–050).
NIH3T3 cells were cultured in DMEM (Gibco Life Technologies, 11965–092) supplemented with 10% FBS (Atlanta Biologicals, S11150), 100 unit/ml penicillin (Hyclone, SV30010), and 100 μg/ml streptomycin (Hyclone, SV30010).
Antibodies for FACS analysis
FITC anti-mouse Ki-67 (clone SolA15, 11–5698-82) and PE anti-mouse Foxp3 (clone NRRF30, 12–4771) are from eBioscience. APC anti-mouse CD4 (clone GK1.5, 100412), PerCP/Cy5.5 anti-mouse CD4 (clone GK1.5, 100434), APC/Cy7 anti-mouse CD4 (clone GK1.5, 100414), PE anti-mouse CD4 (clone GK1.5, 100408), FITC anti-mouse CD4 (clone GK1.5, 100406), PerCP/Cy5.5 anti-mouse B220 (clone RA3–6B2, 103236), FITC anti-mouse CD8 (clone 5.3–6.7, 100706), APC anti-mouse CD8 (clone 5.3–6.7, 100712), APC/Cy7 anti-mouse CD38 (clone 90, 102727), biotin anti-mouse CXCR5 (clone L138D7, 145510), PerCP/Cy5.5 anti-mouse CD44 (clone IM7, 103032), FITC anti-mouse CD44 (clone IM7, 103006), APC anti-mouse CD62L (clone MEL-14, 104412), PE anti-mouse CD62L (clone MEL-14, 104408), APC anti-mouse CD73 (clone TY/11.8, 127209), PE anti-mouse CD95 (clone SA367H8, 152607), APC anti-mouse Nrp-1 (clone 3E12, 145206), PerCP/Cy5.5 anti-mouse PD-1 (clone 29F.1A12, 135208), PE anti-mouse CD90.1 (Thy1.1, clone OX-7, 202524), FITC anti-mouse GITR (clone YGITR 765, 120205), APC anti-mouse IL-4 (clone 11B11, 504106), PE anti-mouse IL-4 (clone 11B11, 504104), APC anti-mouse IFN-γ (clone XMG1.2, 505810), Alexa Fluor® 647 anti-mouse IL-17a (clone TC11–18H10.1, 506912), APC/Cy7 anti-mouse TCRβ (clone H57–597, 109220), APC anti-mouse TIGIT (clone 1G9, 142105), FITC anti-mouse GL7 (clone GL7, 144604), Alexa Fluor® 488 anti-mouse/rat/human FOXP3 (clone 150D, 320012), APC-Cy7-straptavidin (405208), PE-straptavidin (405204), FITC-straptavidin (405202) are from BioLegend. APC anti-mouse FOXP3 (clone 3G3, 20–5773-U100) are from TONBO biosciences.
Mice and genotyping
Breeding and maintenance of the mouse colony, and all experiments involving animals were approved by the IACUC of Indiana University School of Medicine. Both female and male mice were used between the age of 8 to 16 weeks except for experiments in Fig. 6 (only heterozygous females used as males have only one X chromosome) and Fig. 6E (F1 males for lineage tracing, see below for details). For adoptive cell transfer experiments, sex and age matched donor and recipient mice were used. All the mice were maintained in SPF animal facilities (ambient temperature 70–72°F, humidity 50%, light/dark cycle 12/12hr).
Foxp3 exon2 deletion mice were generated with CRISPR-Cas9 system. The upstream guide RNA matches 5’CCATTGTTGCTACCGTGTGAGAC3’ of intron 1. The downstream guide RNA matches 5’GGTATGGAATCGGAGCAGGCTGG3’ of intron 2. Ninety nine bp of intron 1 and 16 bp of intron 2 that are adjacent to exon 2 were deleted along with exon 2. Four founder lines were obtained and showed the same phenotypes. Foxp3 exon2 deletion mice were backcrossed to wild-type C57BL/6 for 9 generations. Genotyping PCR was carried out with primer set of Foxp3-ScF1 (5’CTCCCAATCCTCATCCCGATAG 3’) and Foxp3-ScR1 (5’TGGACGCACTTGGAGCACAG3’). PCR amplified one 552bp band for the wildtype Foxp3 allele and one 331bp band for the Foxp3 exon 2 deletion allele.
Tcrbtm1Mom mice that are deficient in beta T-cell receptor were purchased from the Jackson Laboratory (Stock No: 002118). Rag2 knockout (Rag2tm1.1Cgn) mice were obtained from the Jackson Laboratory (Stock No: 008449).
Foxp3-DTR/eGFP transgenic mice (also named DEREG, stock No: 32050) and Foxp3-eGFP/iCre transgenic mice (stock No: 023161) were obtained from the Jackson Laboratory. Foxp3-DTR/eGFP mice were used to sort Tregs based upon eGFP expression. Male Foxp3-eGFP/iCre mice (129S background) were crossed to Foxp3 ΔE2Het; R26-stop-eYFP female mice to generate F1 generation. The resulted F1 male mice were Foxp3-eGFP/iCre;R26-stop-eYFP with either Foxp3 ΔE2 or Foxp3 FL under same genetic background (half C57BL/6 and half 129S) and thus can be used in lineage tracing of the stability of FOXP3 ΔE2 Tregs and FOXP3 FL Tregs.
Naïve CD4 T cell isolation and iTreg induction
Mouse naïve CD4 T cells (CD4+CD25−CD62L+) were purified with CD4+CD62L+ T Cell Isolation Kit II (Miltenyi Biotec, 130–093-227). For iTregs induction, 24-well plate was coated with 400 μl of 1 μg/ml anti-mouse CD3ε in phosphate-buffered saline solution (PBS) per well at 37°C for 4 h, followed by rinse with PBS. Naïve CD4 T cells were diluted at 0.3 × 106 /ml in complete RPMI 1640 supplemented with 1 μg/ml of anti-CD28, 5ng/ml of recombinant human TGFβ, and 20ng/ml of recombinant human IL-2 and were cultured for 3 to 5 days.
Peripheral Treg sorting
CD4+ T cells were first enriched from lymphocytes of lymph nodes and spleens from Foxp3 FL (WT);DEREG or Foxp3 ΔE2;DEREG mice with MoJoSort mouse CD4 T cell isolation kit (BioLegend, 480006). Enriched CD4 T cells were then stained with anti-mouse CD4 and Tregs were sorted based upon eGFP expression and CD4 staining.
Treg in vitro suppressive assay
FOXP3 FL and FOXP3 ΔE2 Tregs (CD45.2) were sorted as described above from three of each WT and Foxp3 ΔE2 mice. CD4+CD19−CD8−CD44−CD25− cells were sorted out from CD45.1 mice as responder cells and labeled with CellTrace™ Violet (ThermoFisher, C34557). Tregs were mixed with 1.0 × 105 responder cells at ratios from 1:1 to 1:128 with Tregs only and responders only as controls. Mixed cells were cultured in 96 well round bottom pate in the presence of 2.5 × 105 irradiated APCs and 2.5μg/ml soluble anti-CD3 for 3 days. On day 3, cells were stained with anti-CD4- BV605, anti-CD45.1-PECy7 (responder cells), anti-CD45.2-PE (Tregs) and LIVE/DEAD™ Fixable Near-IR dye (ThermoFisher, L10119). Dilution of CellTrace Violet in the responder cells were examined with flow cytometry. Percentage of repression = (T0-T[i])/T0, where T0 is percentage of proliferated cells without Tregs (responders only); T[i] is percentage of proliferated cells at a given Treg:responder ratio.
Immunofluorescent detection of anti-nuclear antigen IgG
Poly-L-lysine coated cover slip (Neuvitro, GG-18-PLL) was placed in each well of a 12-well plate, and NIH-3T3 cells were then seeded and cultured overnight. Cells on the cover slip were fixed with 1ml of 4% formaldehyde diluted in PBS at room temperature for 10 min, and washed with ice-cold PBS. Cells were permeabilized with 1ml of 0.1% TritonX-100 in PBS for 10min followed by wash with PBS. After blocked with blocking buffer (2% BSA, 0.1% Tween-20, and 10% normal RAT serum in PBS) at room temperature for 1 h, cells were incubated successively with serum diluted in dilution buffer (1% BSA , 0.1% Tween-20 in PBS) at 4°C overnight, FITC-Rat-anti-mouse IgG (BioLegend, 406001) at room temperature for 1h, and 100 nM Rhodamine Phalloidin (Cytoskeleton, PHDR1) at room temperature for 30 min. Cells were washed for 3 times with PBS after each incubation and mounted on glass slide with Vectashield Mounting Medium with DAPI and sealed with nail polish.
Immunohistochemistry
To detect immune complex deposition in kidney, mouse kidneys were embedded in OCT compound (TED PELLA, 27050) and frozen on dry ice followed by cryosection at 12 μm thickness. Sections were fixed and permeabilized with ice-cold acetone for 5 min and air dried at room temperature. Sections were then washed 2X with PBS and 2X with PBST (0.1% Tween-20 in PBS) followed by blocking with block buffer (2% BSA, 0.1% Tween-20, and 10% normal RAT serum in PBS) at room temperature for 1 h. After two time of wash with PBST, sections were incubated with 5 μg/ml Texas red-Wheat Germ Agglutinin (Life technologies, W21405) and 2 μg/ml FITC-RAT anti-mouse IgG (BioLegend, 406001) at 4°C overnight. Sections were then washed for three times with PBS and mounted with Vectashield Mounting Medium with DAPI.
Histological analysis
Kidney, colon, and liver were fixed in 4% formaldehyde diluted in PBS. Fixed samples were embedded in paraffin and the sections were stained with hematoxylin and eosin (H&E) and Periodic acid–Schiff (PAS).
Flow cytometry analysis
For cytokine staining, lymphocytes were stimulated with 50 ng/ml PMA and 500 ng/ml ionomycin for 5 h. Monensin (Biolegend, 420701) was added 2 h after stimulation. Cells were then stained with antibodies for the surface molecules followed by fixation with 4% formaldehyde diluted in PBS at room temperature for 10 min. Fixed cells were permeabilized and stained with fluorescence labeled antibodies. For FOXP3 staining, surface stained lymphocytes were fixed at 4°C overnight with 1X Fixation/Permeabilization buffer (eBioscience 00–5123) diluted in Fixation/Permeabilization Diluent (eBioscience 00–5223). Cells were then stained with FOXP3 antibody in Permeabilization Buffer (eBioscience 00–5523).
Enzyme-linked immunosorbent assay for anti-dsDNA antibodies
Coating buffer (5 μg/ml of sheared salmon sperm DNA and 1mM EDTA in PBS) and dilution buffer (1% BSA, 1mM EDTA, 0.05% Tween20 in PBS) were made fresh before use. Flat-bottom 96-well plates were coated with coating buffer at 4°C overnight. Plates were blocked at room temperature for 30 min and then incubated successively with 50 μl per well of diluted serum at 4°C overnight, 1μg/ml biotin-goat anti-mouse IgG at room temperature for 1 h, diluted Avidin-HRP at room temperature for 30 min with shaking. Plates were washed 3X with wash buffer (1mM EDTA and 0.05% of Tween20 in PBS) after each incubation. Color was developed with 1-step Ultra TMB-ELISA (Thermo Scientific, 34028) and stopped with stop solution (2N H2SO4). Absorbance at 450 nm was read within 30 min.
Bulk RNA-seq
Differentiated Tregs were snap frozen in liquid nitrogen and shipped on dry ice to Otogenetics Corporation. Illumina sequencing library were prepared from polyA mRNA by the company with Q.C. 100bp paired-end sequencing of the library were performed on HiSeq2500 (Illumina, Inc.). The bcl2fastq2 Conversion software were used to convert base call (bcl) files to FASTQ files, and trim adapter sequence at the same time. The reads were mapped to the mouse genome using STAR (v2.5) RNA-seq aligner with the following parameter: “--outSAMmapqUnique 60” and uniquely mapped sequencing reads were assigned to mm10 genes using featureCounts (from subread v1.5.1) with the following parameters: “-s 0 –p –Q 10”. The data was filtered using read count per million (CPM) > 0.5 in more than 2 of the samples, normalized using TMM (trimmed mean of M values) method and subjected to differential expression analysis using likelihood ratio test method in edgeR (v3.20.8).
ChIP-seq
Differentiated Tregs were crosslinked and chromatin was isolated by using the truChIP Chromatin Shearing Kit with Formaldehyde (Covaris). Briefly, 5 million cells were fixed with 1% paraformaldehyde for 10 mins, the reaction was quenched by using the quenching buffer. After washing with PBS twice, the cells were lysed for 10 mins with rotation at 4°C. Nuclei were collected by centrifugation at 17000g for 5 mins at 4°C. The pellet was washed twice with wash buffer. The washed nuclei were resuspended in shearing buffer and the chromatin was sheared with an AFA Focus-ultrasonicator for 30 mins. Chromatin immunoprecipitation against 5 μg polyclonal rabbit anti-FOXP3 antibody was performed with Magna ChIP A/G Chromatin Immunoprecipitation Kit (Milipore Sigma) according to manufacturer’s protocol. The precipitated DNA and input DNA samples were processed and sequenced by BGI Americas Corporation. 50bp single-end sequencing was performed on Illumina NextSeq 2000. The reads were mapped to the mm10 mouse genome using Bowtie2 (v2.4.4) and duplicated reads were removed using Picard (v.2.18.26). BigWig files were generated using bedtools (v2.2.9) and bedGraphToBigWig (v2.8) which were normalized by total number of reads per 10 million reads.
Statistics analysis
Statistical was analyzed by using GraphPad Prism (GraphPad Software) and presented as means ± SEM. Unpaired Student t tests, multiple t test, and one-way or two-way ANOVA analysis with Bonferroni post-hoc test were used in data analysis. A p value <0.05 was considered statistically significant. ns: p>0.05, *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001.
Supplementary Material
Supplementary Data File 1. RNA-seq analysis of differential gene expression in FOXP3 ΔE2 and WT Tregs.
Supplementary Data File 3. FOXP3 binding to Foxp3 positive regulators in FOXP3 ΔE2 and WT Tregs.
Supplementary Data File 2. ChIP-seq analysis of FOXP3 whole genome binding in FOXP3 ΔE2 and WT Tregs
Supplementary Data File 4. Raw data excel file
Figure S1. Generation and characterization of Foxp3 exon 2 deletion (Fox3 ΔE2) mice.
Figure S2. FOXP3 exon 2 deletion does not impact mouse and human Th17 differentiation in vitro.
Figure S3. Isotypes of anti-dsDNA antibodies and total IgE in the serum of Foxp3 ΔE2 mice at the age of two months.
Figure S4. Homozygous, but not heterozygous, Foxp3 exon 2 deletion females develop a similar systemic autoimmune disease.
Figure S5. More severe disease in aged (10 month old) Foxp3 ΔE2 mice.
Figure S6. Bisulfite sequencing of the Treg-sepcfic demethylated region (TSDR), or conserved non-coding sequence 2 (CNS2), of Foxp3 gene locus.
Figure S7. Reduced FOXP3 ΔE2 Treg population in peripheral lymphoid organs in mixed bone marrow chimera males.
Figure S8. Sorting of FOXP3 FL and FOXP3 ΔE2 Treg cells from mixed bone marrow chimera mice.
Figure S9. Co-transfer of naive CD4+ T cells did not rescue the instability of FOXP3 ΔE2 Tregs.
Figure S10. Induction of auto-antibody by FOXP3 ΔE2 Tregs was not ameliorated by co-transferred naive CD4+ T cells.
Figure S11. FOXP3 ΔE2 Tregs induced SLE-like disease but not colitis in the presence of naive CD4+ T cells.
Figure S12. FOXP3 ΔE2 isoform interacts with HDACs similar to FOXP3 FL isoform.
Figure S13 Protein and mRNA stability of FOXP3 isoforms.
Table S1. Clinical characteristics of IPEX patients carrying deletion mutations within exon 2 of FOXP3 gene.
Acknowledgements:
The authors thank Dr. George Sandusky, Professor of Pathology and Laboratory Medicine IU School of Medicine, for analyzing kidney pathology presented in Figure S11, and Dr. Ye Zheng, Associate Professor Salk Institute, for providing the antibody used in FOXP3 ChIP-seq.
Funding:
The work in the study was supported by NIH grants R01 AI085046, R21 AI110773, R21 AI159804 and Wells Center for Pediatric Research translational fund to B.Z.; R01 AI112323 and R01 AI136475 to S.F.Z.; The Mark Foundation for Cancer Research ASPIRE Award to X.L. and B.Z.. Flow Cytometry Resource Facility usage was also supported by IU Simon Cancer Center Support Grant P30 CA082709 and U54 DK106846. Support provided by the Herman B Wells Center was in part from the Riley Children’s Foundation.
Footnotes
Competing interests: Authors declare no conflict of interests.
Data and materials availability:
RNA-seq and ChIP-seq data sets are deposited to GEO and available under accession number: GSE199494. All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References
- 1.Sakaguchi S, Miyara M, Costantino CM, Hafler DA, FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10, 490–500 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Sakaguchi S, Yamaguchi T, Nomura T, Ono M, Regulatory T cells and immune tolerance. Cell 133, 775–787 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Curotto de Lafaille MA, Kutchukhidze N, Shen S, Ding Y, Yee H, Lafaille JJ, Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity 29, 114–126 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, Umetsu DT, Rudensky AY, Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 482, 395–399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allan SE, Passerini L, Bacchetta R, Crellin N, Dai M, Orban PC, Ziegler SF, Roncarolo MG, Levings MK, The role of 2 FOXP3 isoforms in the generation of human CD4+ Tregs. J Clin Invest 115, 3276–3284 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aarts-Riemens T, Emmelot ME, Verdonck LF, Mutis T, Forced overexpression of either of the two common human Foxp3 isoforms can induce regulatory T cells from CD4(+)CD25(−) cells. Eur J Immunol 38, 1381–1390 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Smith EL, Finney HM, Nesbitt AM, Ramsdell F, Robinson MK, Splice variants of human FOXP3 are functional inhibitors of human CD4+ T-cell activation. Immunology 119, 203–211 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Free ME, Bunch DO, McGregor JA, Jones BE, Berg EA, Hogan SL, Hu Y, Preston GA, Jennette JC, Falk RJ, Su MA, Patients with antineutrophil cytoplasmic antibody-associated vasculitis have defective Treg cell function exacerbated by the presence of a suppression-resistant effector cell population. Arthritis Rheum 65, 1922–1933 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kristensen B, Hegedus L, Madsen HO, Smith TJ, Nielsen CH, Altered balance between self-reactive T helper (Th)17 cells and Th10 cells and between full-length forkhead box protein 3 (FoxP3) and FoxP3 splice variants in Hashimoto’s thyroiditis. Clin Exp Immunol 180, 58–69 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyabe C, Miyabe Y, Strle K, Kim ND, Stone JH, Luster AD, Unizony S, An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Annals of the rheumatic diseases 76, 898–905 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serena G, Yan S, Camhi S, Patel S, Lima RS, Sapone A, Leonard MM, Mukherjee R, Nath BJ, Lammers KM, Fasano A, Proinflammatory cytokine interferon-gamma and microbiome-derived metabolites dictate epigenetic switch between forkhead box protein 3 isoforms in coeliac disease. Clin Exp Immunol 187, 490–506 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryder LR, Woetmann A, Madsen HO, Odum N, Ryder LP, Bliddal H, Danneskiold-Samsoe B, Ribel-Madsen S, Bartels EM, Expression of full-length and splice forms of FoxP3 in rheumatoid arthritis. Scandinavian journal of rheumatology 39, 279–286 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Ryder LR, Bartels EM, Woetmann A, Madsen HO, Odum N, Bliddal H, Danneskiold-Samsoe B, Ribel-Madsen S, Ryder LP, FoxP3 mRNA splice forms in synovial CD4+ T cells in rheumatoid arthritis and psoriatic arthritis. Apmis 120, 387–396 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Suzuki K, Setoyama Y, Yoshimoto K, Tsuzaka K, Abe T, Takeuchi T, Decreased mRNA expression of two FOXP3 isoforms in peripheral blood mononuclear cells from patients with rheumatoid arthritis and systemic lupus erythematosus. International journal of immunopathology and pharmacology 24, 7–14 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Lundberg AK, Jonasson L, Hansson GK, Mailer RKW, Activation-induced FOXP3 isoform profile in peripheral CD4+ T cells is associated with coronary artery disease. Atherosclerosis 267, 27–33 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Mailer RKW, Alternative Splicing of FOXP3-Virtue and Vice. Frontiers in immunology 9, 530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du J, Huang C, Zhou B, Ziegler SF, Isoform-specific inhibition of ROR alpha-mediated transcriptional activation by human FOXP3. J Immunol 180, 4785–4792 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR, TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453, 236–240 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, Feng X-H, Jetten AM, Dong C, Molecular Antagonism and Plasticity of Regulatory and Inflammatory T Cell Programs. Immunity 29, 44–56 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mailer RK, Joly AL, Liu S, Elias S, Tegner J, Andersson J, IL-1beta promotes Th17 differentiation by inducing alternative splicing of FOXP3. Scientific reports 5, 14674 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joly AL, Liu S, Dahlberg CI, Mailer RK, Westerberg LS, Andersson J, Foxp3 lacking exons 2 and 7 is unable to confer suppressive ability to regulatory T cells in vivo. Journal of autoimmunity 63, 23–30 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Chae WJ, Henegariu O, Lee SK, Bothwell AL, The mutant leucine-zipper domain impairs both dimerization and suppressive function of Foxp3 in T cells. Proc Natl Acad Sci U S A 103, 9631–9636 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song X, Li B, Xiao Y, Chen C, Wang Q, Liu Y, Berezov A, Xu C, Gao Y, Li Z, Wu SL, Cai Z, Zhang H, Karger BL, Hancock WW, Wells AD, Zhou Z, Greene MI, Structural and biological features of FOXP3 dimerization relevant to regulatory T cell function. Cell reports 1, 665–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akdis CA, Therapies for allergic inflammation: refining strategies to induce tolerance. Nat Med 18, 736–749 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Riley JL, June CH, Blazar BR, Human T regulatory cell therapy: take a billion or so and call me in the morning. Immunity 30, 656–665 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Boehmer H, Daniel C, Therapeutic opportunities for manipulating TReg cells in autoimmunity and cancer. Nat Rev Drug Discov 12, 51–63 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Mahne AE, Klementowicz JE, Chou A, Nguyen V, Tang Q, Therapeutic regulatory T cells subvert effector T cell function in inflamed islets to halt autoimmune diabetes. J Immunol 194, 3147–3155 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zolkipli Z, Roberts G, Cornelius V, Clayton B, Pearson S, Michaelis L, Djukanovic R, Kurukulaaratchy R, Arshad SH, Randomized controlled trial of primary prevention of atopy using house dust mite allergen oral immunotherapy in early childhood. J Allergy Clin Immunol 136, 1541–1547 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, Wujtewicz MA, Witkowski P, Mlynarski W, Balcerska A, Mysliwska J, Trzonkowski P, Administration of CD4+CD25highCD127- regulatory T cells preserves beta-cell function in type 1 diabetes in children. Diabetes care 35, 1817–1820 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, Liu W, Long SA, Masiello LM, Nguyen V, Putnam AL, Rieck M, Sayre PH, Tang Q, Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Science translational medicine 7, 315ra189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaczmarczyk-Sekuła K, Dyduch G, Kostański M, Wielowieyska-Szybińska D, Szpor J, Białas M, Okoń K, Mast cells in systemic and cutaneous lupus erythematosus. Polish journal of pathology : official journal of the Polish Society of Pathologists 66, 397–402 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Frith K, Joly AL, Ma CS, Tangye SG, Lohse Z, Seitz C, Verge CF, Andersson J, Gray P, The FOXP3Delta2 isoform supports Treg cell development and protects against severe IPEX syndrome. J Allergy Clin Immunol 144 317–320.e318 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T, Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med 204, 57–63 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H, Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 20, 62–68 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY, Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity 21, 267–277 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ, Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol 2, 301–306 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, Hori S, Fagarasan S, Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science 323, 1488–1492 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Cortez JT, Montauti E, Shifrut E, Gatchalian J, Zhang Y, Shaked O, Xu Y, Roth TL, Simeonov DR, Zhang Y, Chen S, Li Z, Woo JM, Ho J, Vogel IA, Prator GY, Zhang B, Lee Y, Sun Z, Ifergan I, Van Gool F, Hargreaves DC, Bluestone JA, Marson A, Fang D, CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature 582, 416–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan X, Cheng G, Malek TR, The importance of regulatory T-cell heterogeneity in maintaining self-tolerance. Immunol Rev 259, 103–114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, Segundo GRS, Allenspach EJ, Favre C, Ochs HD, Torgerson TR, Clinical, Immunological, and Molecular Heterogeneity of 173 Patients With the Phenotype of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome. Frontiers in immunology 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barzaghi F, Passerini L, Bacchetta R, Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Frontiers in immunology 3, 211 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobayashi I, Shiari R, Yamada M, Kawamura N, Okano M, Yara A, Iguchi A, Ishikawa N, Ariga T, Sakiyama Y, Ochs HD, Kobayashi K, Novel mutations of FOXP3 in two Japanese patients with immune dysregulation, polyendocrinopathy, enteropathy, X linked syndrome (IPEX). Journal of medical genetics 38, 874–876 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fuchizawa T, Adachi Y, Ito Y, Higashiyama H, Kanegane H, Futatani T, Kobayashi I, Kamachi Y, Sakamoto T, Tsuge I, Tanaka H, Banham AH, Ochs HD, Miyawaki T, Developmental changes of FOXP3-expressing CD4+CD25+ regulatory T cells and their impairment in patients with FOXP3 gene mutations. Clin Immunol 125, 237–246 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Otsubo K, Kanegane H, Kamachi Y, Kobayashi I, Tsuge I, Imaizumi M, Sasahara Y, Hayakawa A, Nozu K, Iijima K, Ito S, Horikawa R, Nagai Y, Takatsu K, Mori H, Ochs HD, Miyawaki T, Identification of FOXP3-negative regulatory T-like (CD4(+)CD25(+)CD127(low)) cells in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Clin Immunol 141, 111–120 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi I, Kubota M, Yamada M, Tanaka H, Itoh S, Sasahara Y, Whitesell L, Ariga T, Autoantibodies to villin occur frequently in IPEX, a severe immune dysregulation, syndrome caused by mutation of FOXP3. Clin Immunol 141, 83–89 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Moudgil A, Perriello P, Loechelt B, Przygodzki R, Fitzerald W, Kamani N, Immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: an unusual cause of proteinuria in infancy. Pediatric nephrology (Berlin, Germany) 22, 1799–1802 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Owen CJ, Jennings CE, Imrie H, Lachaux A, Bridges NA, Cheetham TD, Pearce SH, Mutational analysis of the FOXP3 gene and evidence for genetic heterogeneity in the immunodysregulation, polyendocrinopathy, enteropathy syndrome. The Journal of clinical endocrinology and metabolism 88, 6034–6039 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, Li MO, Leslie C, Stamatoyannopoulos JA, Rudensky AY, Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151, 153–166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY, Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 445, 936–940 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, Leslie C, Shaffer SA, Goodlett DR, Rudensky AY, Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol 13, 1010–1019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hayatsu N, Miyao T, Tachibana M, Murakami R, Kimura A, Kato T, Kawakami E, Endo TA, Setoguchi R, Watarai H, Nishikawa T, Yasuda T, Yoshida H, Hori S, Analyses of a Mutant Foxp3 Allele Reveal BATF as a Critical Transcription Factor in the Differentiation and Accumulation of Tissue Regulatory T Cells. Immunity 47, 268–283.e269 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Van Gool F, Nguyen MLT, Mumbach MR, Satpathy AT, Rosenthal WL, Giacometti S, Le DT, Liu W, Brusko TM, Anderson MS, Rudensky AY, Marson A, Chang HY, Bluestone JA, A Mutation in the Transcription Factor Foxp3 Drives T Helper 2 Effector Function in Regulatory T Cells. Immunity 50, 362–377.e366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie X, Stubbington MJ, Nissen JK, Andersen KG, Hebenstreit D, Teichmann SA, Betz AG, The Regulatory T Cell Lineage Factor Foxp3 Regulates Gene Expression through Several Distinct Mechanisms Mostly Independent of Direct DNA Binding. PLoS genetics 11, e1005251 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kitoh A, Ono M, Naoe Y, Ohkura N, Yamaguchi T, Yaguchi H, Kitabayashi I, Tsukada T, Nomura T, Miyachi Y, Taniuchi I, Sakaguchi S, Indispensable Role of the Runx1-Cbfβ Transcription Complex for In Vivo-Suppressive Function of FoxP3+ Regulatory T Cells. Immunity 31, 609–620 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA, In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med 199, 1455–1465 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM, CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med 199, 1467–1477 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, Romano A, Simone S, Procaccini C, Rocca C, Carrieri PB, Maniscalco GT, Salvetti M, Buscarinu MC, Franzese A, Mozzillo E, Cava A, Matarese G, Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data File 1. RNA-seq analysis of differential gene expression in FOXP3 ΔE2 and WT Tregs.
Supplementary Data File 3. FOXP3 binding to Foxp3 positive regulators in FOXP3 ΔE2 and WT Tregs.
Supplementary Data File 2. ChIP-seq analysis of FOXP3 whole genome binding in FOXP3 ΔE2 and WT Tregs
Supplementary Data File 4. Raw data excel file
Figure S1. Generation and characterization of Foxp3 exon 2 deletion (Fox3 ΔE2) mice.
Figure S2. FOXP3 exon 2 deletion does not impact mouse and human Th17 differentiation in vitro.
Figure S3. Isotypes of anti-dsDNA antibodies and total IgE in the serum of Foxp3 ΔE2 mice at the age of two months.
Figure S4. Homozygous, but not heterozygous, Foxp3 exon 2 deletion females develop a similar systemic autoimmune disease.
Figure S5. More severe disease in aged (10 month old) Foxp3 ΔE2 mice.
Figure S6. Bisulfite sequencing of the Treg-sepcfic demethylated region (TSDR), or conserved non-coding sequence 2 (CNS2), of Foxp3 gene locus.
Figure S7. Reduced FOXP3 ΔE2 Treg population in peripheral lymphoid organs in mixed bone marrow chimera males.
Figure S8. Sorting of FOXP3 FL and FOXP3 ΔE2 Treg cells from mixed bone marrow chimera mice.
Figure S9. Co-transfer of naive CD4+ T cells did not rescue the instability of FOXP3 ΔE2 Tregs.
Figure S10. Induction of auto-antibody by FOXP3 ΔE2 Tregs was not ameliorated by co-transferred naive CD4+ T cells.
Figure S11. FOXP3 ΔE2 Tregs induced SLE-like disease but not colitis in the presence of naive CD4+ T cells.
Figure S12. FOXP3 ΔE2 isoform interacts with HDACs similar to FOXP3 FL isoform.
Figure S13 Protein and mRNA stability of FOXP3 isoforms.
Table S1. Clinical characteristics of IPEX patients carrying deletion mutations within exon 2 of FOXP3 gene.
Data Availability Statement
RNA-seq and ChIP-seq data sets are deposited to GEO and available under accession number: GSE199494. All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.