

Abstract

The oxidation state of the redox noninnocent tetra-amido macrocyclic ligand (TAML) scaffold was recently shown to affect the formation of nitrene radical species on cobalt(III) upon reaction with PhI=NNs [van Leest N. P.; et al. J. Am. Chem. Soc. 2020, 142, 552−563]. For the neutral [CoIII(TAMLsq)] complex, this leads to the doublet (S = 1/2) mono-nitrene radical species [CoIII(TAMLq)(N•Ns)(Y)] (bearing an unidentified sixth ligand Y in at least the frozen state), while a triplet (S = 1) bis-nitrene radical species [CoIII(TAMLq)(N•Ns)2]– is generated from the anionic [CoIII(TAMLred)]– complex. The one-electron-reduced Fischer-type nitrene radicals (N•Ns–) are formed through single (mono-nitrene) or double (bis-nitrene) ligand-to-substrate single-electron transfer (SET). In this work, we describe the reactivity and mechanisms of these nitrene radical complexes in catalytic aziridination. We report that [CoIII(TAMLsq)] and [CoIII(TAMLred)]– are both effective catalysts for chemoselective (C=C versus C–H bonds) and diastereoselective aziridination of styrene derivatives, cyclohexane, and 1-hexene under mild and even aerobic (for [CoIII(TAMLred)]–) conditions. Experimental (Hammett plots; [CoIII(TAML)]-nitrene radical formation and quantification under catalytic conditions; single-turnover experiments; and tests regarding catalyst decomposition, radical inhibition, and radical trapping) in combination with computational (density functional theory (DFT), N-electron valence state perturbation theory corrected complete active space self-consistent field (NEVPT2-CASSCF)) studies reveal that [CoIII(TAMLq)(N•Ns)(Y)], [CoIII(TAMLq)(N•Ns)2]–, and [CoIII(TAMLsq)(N•Ns)]– are key electrophilic intermediates in aziridination reactions. Surprisingly, the electrophilic one-electron-reduced Fischer-type nitrene radicals do not react as would be expected for nitrene radicals (i.e., via radical addition and radical rebound). Instead, nitrene transfer proceeds through unusual electronically asynchronous transition states, in which the (partial) styrene substrate to TAML ligand (single-) electron transfer precedes C–N coupling. The actual C–N bond formation processes are best described as involving a nucleophilic attack of the nitrene (radical) lone pair at the thus (partially) formed styrene radical cation. These processes are coupled to TAML-to-cobalt and cobalt-to-nitrene single-electron transfer, effectively leading to the formation of an amido-γ-benzyl radical (NsN––CH2–•CH–Ph) bound to an intermediate spin (S = 1) cobalt(III) center. Hence, the TAML moiety can be regarded to act as a transient electron acceptor, the cobalt center behaves as a spin shuttle, and the nitrene radical acts as a nucleophile. Such a mechanism was hitherto unknown for cobalt-catalyzed hypovalent group transfer and the more general transition-metal-catalyzed nitrene transfer to alkenes but is now shown to complement the known concerted and stepwise mechanisms for N-group transfer.
Keywords: aziridination, ligand-to-substrate single-electron transfer, substrate-to-ligand single-electron transfer, electronically asynchronous transition states, nitrene radical, spin shuttle
Introduction
The aziridine fragment (three-membered N–C–C ring) is a frequently encountered motif in antibiotic and antitumor drugs such as azicemicins, azinomycins, mitomycins, miraziridine, etc.1 In addition, the aziridine ring is a versatile intermediate to afford 1,2-functionalized products via selective ring-opening reactions.2 The prevalence of the aziridine motif and its utility as a synthetic building block and a functional group are reflected in a wide variety of methods to assemble this small N-heterocyclic ring. Carbene and ylide addition to imines and cyclization of 1,2-amino alcohols, 1,2-aminohalides, and 1,2-azido-alcohols are well-established.1b,3 However, these methods rely on the pre-functionalization of the substrate. Direct N-group transfer to (unactivated) alkene bonds using transition-metal catalysis has therefore gained momentum in the last few decades.3 Mechanistic studies have revealed that these reactions commonly proceed through concerted (Figure 1A) or stepwise (i.e., sequential radical addition and radical rebound, Figure 1B) N-group transfer. The former is more common for electrophilic nitrenoids on late transition metals, while a stepwise mechanism is often observed for nitrogen-centered radical intermediates.4 Notably, in the stepwise mechanism, the (α) spin density is effectively transferred from the nitrene radical intermediate to the alkene, giving rise to the formation of an amido-γ-benzyl radical that bears the same (α) spin density (see Figure 1B).
Figure 1.

Schematic representation of concerted (A) and stepwise (B) nitrene transfer to an alkene. In (B), the formation of an amido-γ-benzyl (R = Ph) intermediate is depicted bearing similar (α) spin as the nitrene radical.
Recently, a copper(II) complex bearing two redox noninnocent ortho-aminophenol ligands was shown to form aziridines from C=C double bonds and N-tosyliminoiodinane (PhINTs) as the nitrene precursor.5 Mechanistic studies revealed the importance of the redox-active ligands in the stepwise mechanism, as copper retains its +II oxidation state and nitrene formation is accomplished via ligand-to-substrate single-electron transfer (SET). Similar ligand-centered redox reactions6 have been reported in nitrene transfer reactions on e.g., Rh7 and Pd.8
Examples of cobalt complexes bearing redox-active ligands that catalyze reactions, wherein cobalt retains its initial oxidation state throughout the catalytic cycle and only ligand-centered oxidation state changes occur, are currently limited to C–C bond-forming reactions.9 To the best of our knowledge, there is no example of a cobalt-catalyzed hypovalent carbon-, oxygen-, or nitrogen-group transfer reaction, wherein all redox reactions are ligand-centered.
The use of cobalt(II)-porphyrin ([CoII(por)]) catalysts as N-group transfer catalysts has been studied thoroughly.4a−4d It was found that hypovalent N-group transfer reactions (e.g., aziridination and C–H amination) proceed via [CoIII(por2–)(N•–R)] or [CoIII(por•–)(N•–R)2] intermediates, depending on the type of nitrene precursor employed.10 These nitrene radicals on cobalt are the result of metal-to-substrate SET and subsequent ligand-to-substrate SET for the bis-nitrene species (Figure 2). During catalytic N-group transfer, cobalt is therefore oxidized from the +II to the +III oxidation state and consequently acts as the primary redox-active center, whereas the ligand acts as the secondary redox-active center.
Figure 2.

Formation of mono- and bis-nitrene radial complexes at [CoII(por)] involving metal-to-substrate and ligand-to-substrate SET, respectively.10a
The tetra-amido macrocyclic ligand (TAML)11 scaffold has been used in manganese and iron-mediated imido/nitrene transfer.12 In the latter case, the TAML backbone acts as a redox-active ligand on iron, and the three possible ligand oxidation states of the TAML scaffold (red, sq, and q) are depicted in Figure 3A. Addition of PhINTs to [FeIII(TAMLred)]− was shown to afford [FeV(TAMLred)(NTs)]−, which is active in stoichiometric C–H amination of substrates with weak C–H bonds and nitrene transfer to functionalized thioanisoles.12a Ligand-centered single-electron oxidation afforded the neutral [FeV(NTs)(TAMLsq)] complex, with iron retaining its +V oxidation state.12b This neutral complex is 2.5 times more reactive in C–H amination of substrates featuring weak C–H bonds and 17 000 times more reactive in nitrene transfer to thioanisoles as compared to the anionic complex. A similar trend was observed for [MnV(TAMLred)(NMes)]− (Mes = mesityl), which is only active for hydrogen atom transfer from C–H bonds or nitrene transfer to thioanisole after one-electron oxidation to [MnVI(TAMLred)(NMes)].12c In these cases, oxidation of the TAML complexes affords (more) reactive nitrene transfer complexes.
Figure 3.
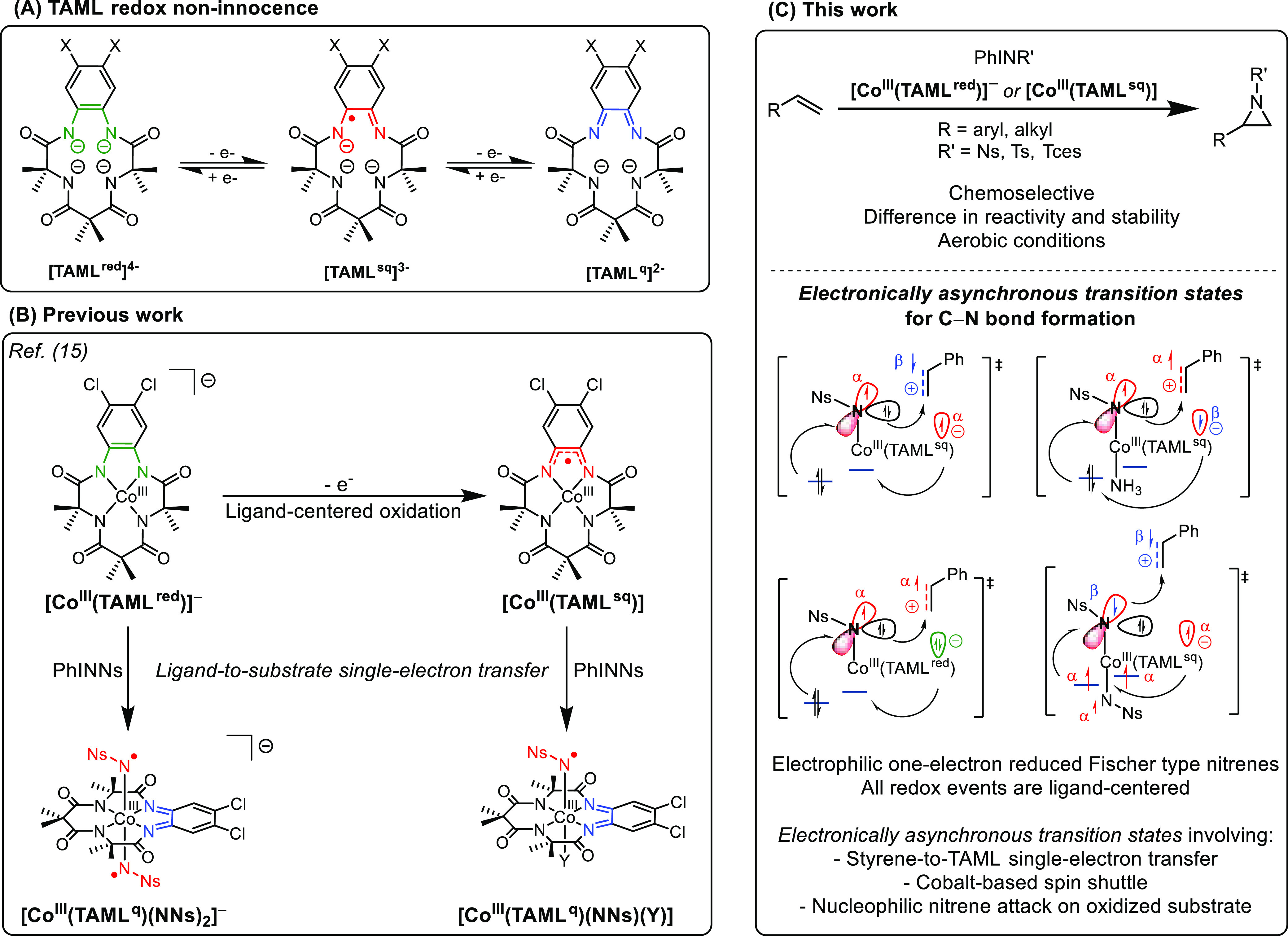
(A) Redox noninnocence of the TAML scaffold. (B) Previous work on the formation of [CoIII(TAML)-nitrene] complexes. (C) The first use of [CoIII(TAML)-nitrene] species in catalytic aziridination reactions via electronically asynchronous transition states.16
We recently disclosed that oxidation of PPh4[CoIII(TAMLred)](13) (cobalt-centered triplet spin state) with thianthrenium tetrafluoroborate ((Thi)BF4) afforded [CoIII(TAMLsq)] via ligand-centered oxidation.14,15 Addition of excess PhINNs (Ns = nosyl) to solutions of [CoIII(TAMLsq)] or PPh4[CoIII(TAMLred)] selectively afforded the neutral mono-nitrene adduct [CoIII(TAMLq)(N•Ns)(Y)] and the anionic bis-nitrene adduct PPh4[CoIII(TAMLq)(N•Ns)2], respectively (Figure 3B). The presence of an unidentified sixth ligand Y in the mono-nitrene adduct was inferred in the frozen state on the basis of Co K-edge X-ray absorption near-edge spectroscopy (XANES) analysis and is confirmed in this work by extended X-ray absorption fine structure (EXAFS) studies. The nitrenes are best described as one-electron reduced Fischer-type nitrene radicals that are formed through single (mono-nitrene) or double (bis-nitrene) ligand-to-substrate single-electron transfer, whereby cobalt retains its +III oxidation state. The cobalt center does undergo a spin flip from an intermediate spin (S = 1) in [CoIII(TAMLsq)] and PPh4[CoIII(TAMLred)] to low spin (S = 0) in the nitrene radical complexes.
In continuation of this work, we herein explore the potential of the nitrene radical species [CoIII(TAMLq)(NNs)(Y)] and PPh4[CoIII(TAMLq)(NNs)2] in catalytic nitrene transfer reactions (Figure 3C). Moreover, as the nitrene complexes are generated through ligand-based oxidation reactions, while cobalt retains its +III oxidation state, we investigate whether the cobalt-TAML platform can be used to perform nitrene transfer catalysis via a hitherto unknown mechanism, potentially involving the redox noninnocence of the TAML moiety. More specifically, using a combined experimental and computational approach, we address the following research questions in this contribution:
-
(1)
Are the neutral ([CoIII(TAMLq)(NNs)(Y)]) and anionic (PPh4[CoIII(TAMLq)(NNs)2]) nitrene species catalytically active in the aziridination of alkenes?
-
(2)
Is there a difference in reactivity, stability, and/or selectivity between the neutral ([CoIII(TAMLsq)]) and anionic ([CoIII(TAMLred)]–) aziridination catalysts?
-
(3)
Do the catalytically active intermediates react as nitrene radicals or rather as electrophilic nitrenes, and what is the mechanism of C–N bond formation?
-
(4)
What is the role of the redox-active ligand and the CoIII center in the mechanism of nitrene transfer, and can the ligand be used as the redox-active site with cobalt retaining its +III oxidation state?
Results and Discussion
Catalytic Reactivity of [CoIII(TAMLred)]− and [CoIII(TAMLsq)] in Alkene Aziridination
We suspected that the anionic bis-nitrene-radical species [CoIII(TAMLq)(NNs)2]– and the neutral mono-nitrene-radical species [CoIII(TAMLq)(NNs)(Y)] (Y being an unidentified sixth ligand) could have a pronouncedly different activity in catalytic nitrene transfer reactions, similar to previously disclosed examples with Mn- and Fe-TAML complexes.12 To adequately compare the performance of both [CoIII(TAML)] complexes, we first explored the temperature influence on the aziridination of styrene with PhINNs in toluene as a benchmark reaction. Catalytic tests were performed using 5.0 mol % PPh4[CoIII(TAMLred)] or [CoIII(TAMLsq)] as a catalyst (Figure 4). Using PPh4[CoIII(TAMLred)] as a catalyst generally led to a higher yield for aziridine product 1, with a maximum yield of 50% at 45 °C. At higher temperatures, lower yields were obtained, which we attribute to the instability of the bis-nitrene-radical intermediate at elevated temperatures. Interestingly, [CoIII(TAMLsq)] performed best at 60 °C to yield 21% of 1. However, [CoIII(TAMLsq)] is less soluble in toluene than PPh4[CoIII(TAMLred)], which might explain the reduced yield. The previously characterized15 nitrene complexes PPh4[CoIII(TAMLq)(NNs)2] and [CoIII(TAMLq)(NNs)(Y)] were demonstrated to form under similar conditions (albeit in the absence of styrene) and are therefore plausible intermediates in the aziridination reactions (vide infra). However, the two [CoIII(TAML)] complexes clearly have a different activity and/or stability.
Figure 4.
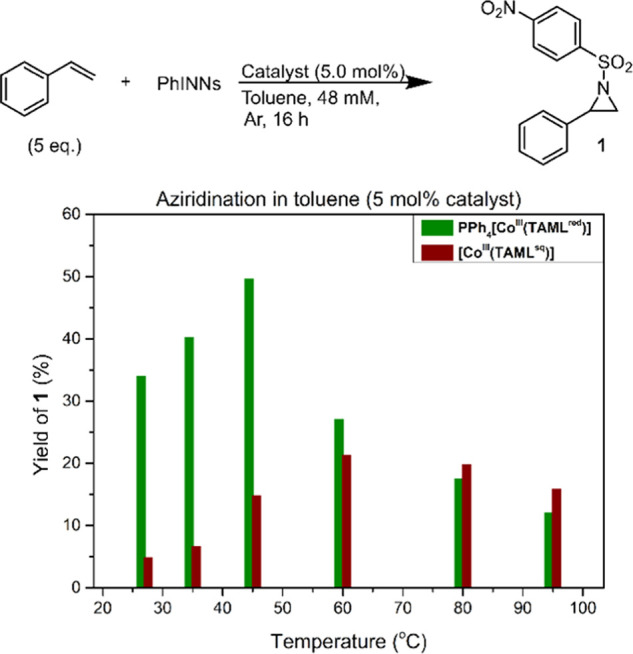
Production of 1 at various temperatures in toluene, using PPh4[CoIII(TAMLred)] (green) or [CoIII(TAMLsq)] (red). Reaction conditions: PhINNs (1.0 equiv, 48 mM), styrene (5.0 equiv), catalyst (5.0 mol %), argon, 16 h.
To further optimize the reaction conditions for the formation of 1 with 5.0 mol % PPh4[CoIII(TAMLred)] or [CoIII(TAMLsq)], we screened benzene, MeCN, and CH2Cl2 as solvents at 35 °C (entries 1–4 and 11–12 in Table 1).17 In CH2Cl2, both complexes afforded 1 in equal yield (58 and 57% for PPh4[CoIII(TAMLred)] and [CoIII(TAMLsq)], respectively). An equimolar amount of styrene relative to PhINNs proved detrimental to the yield (entry 5), whereas reducing the PhINNs concentration by a factor of 2 led to an increase in yield (entry 6). Interestingly, the maximum yield for PPh4[CoIII(TAMLred)] and [CoIII(TAMLsq)] was achieved within 2 h, affording 1 in 76 and 74% yield, respectively (entry 7 and 13). We therefore used the same reaction time (2 h) in most subsequent studies.
Table 1. Optimization of the Reaction Conditions for the Formation of 1, Catalyzed by PPh4[CoIII(TAMLred)] or [CoIII(TAMLsq)]e.

| entry | catalyst loading (mol %) | solvent | concentration PhINNs (mM) (time, h) | yield (%) |
|---|---|---|---|---|
| PPh4[CoIII(TAMLred)] | ||||
| 1 | 5.0 | toluene | 48 (16) | 40 |
| 2 | 5.0 | benzene | 48 (16) | 41 |
| 3 | 5.0 | MeCN | 48 (16) | 18 |
| 4 | 5.0 | CH2Cl2 | 48 (16) | 58 |
| 5a | 5.0 | CH2Cl2 | 48 (16) | 44 |
| 6 | 5.0 | CH2Cl2 | 24 (16) | 77 |
| 7 | 5.0 | CH2Cl2 | 24 (2) | 76 |
| 8 | 2.5 | CH2Cl2 | 24 (2) | 64 |
| 9b | 2.5 | CH2Cl2 | 24 (2) | 67 |
| 10c | – | CH2Cl2 | 24 (2) | 0 |
| [CoIII(TAMLsq)] | ||||
| 11 | 5.0 | toluene | 48 (16) | 7 |
| 12 | 5.0 | CH2Cl2 | 48 (16) | 57 |
| 13 | 5.0 | CH2Cl2 | 24 (2) | 74 |
| 14 | 2.5 | CH2Cl2 | 24 (2) | 35 |
| 15b | 2.5 | CH2Cl2 | 24 (2) | –d |
1.0 equiv styrene was used.
Aerobic conditions.
No catalyst added.
[CoIII(TAMLsq)] is not stable under aerobic conditions.
Yields based on 1H NMR integration using 1,3,5-trimethoxybenzene as an internal standard..
Reducing the catalyst loading to 2.5 mol % led to marginal reduction in the yield of 1 for PPh4[CoIII(TAMLred)] (64%, entry 8) but to a large drop for [CoIII(TAMLsq)] (35%, entry 14), indicating that [CoIII(TAMLsq)] is less robust under the applied conditions. Moreover, we observed 13% conversion of aziridine 1 to unknown product(s) in 1H NMR spectroscopy upon prolonged standing (16 h, see Figure S19 in the Supporting Information (SI)) under the conditions as defined in entry 13, Table 1.
Furthermore, [CoIII(TAMLsq)] is not stable under aerobic conditions due to reaction with atmospheric H2O, resulting in one-electron reduction of the complex.13,15 In contrast, PPh4[CoIII(TAMLred)] is stable in solution in the presence of styrene, PhI, NsNH2, H2O, and aziridine 1.18 Interestingly, a reaction with 2.5 mol % PPh4[CoIII(TAMLred)] under aerobic conditions afforded 1 in 67% yield (entry 9), equal to the reaction outcome using an argon atmosphere. Under the optimal reaction conditions, PPh4[CoIII(TAMLred)] already reached the maximum yield of 1 after 20 min, whereas this process takes 2 h for [CoIII(TAMLsq)] (Figure 5). A control reaction without a cobalt catalyst, to test for possible background reactions with PhINNs,19 did not yield any aziridine 1 (Table 1, entry 10).
Figure 5.
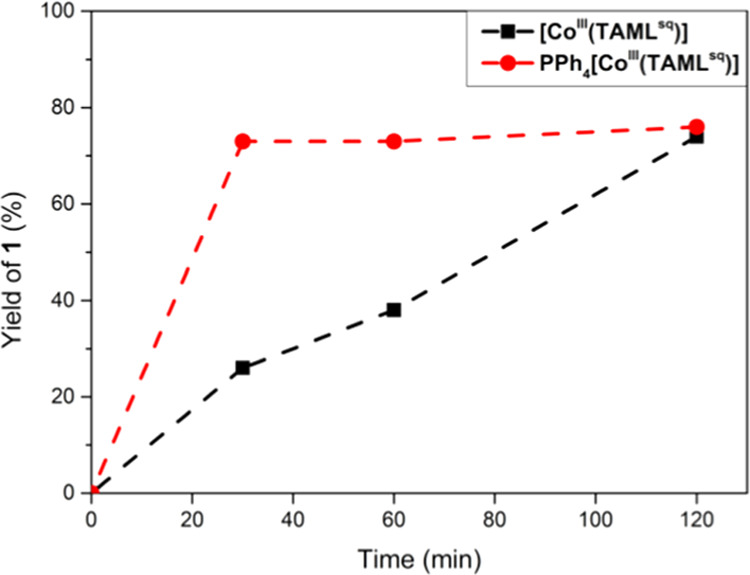
Kinetic profiles for formation of 1 in CH2Cl2, catalyzed by PPh4[CoIII(TAMLred)] (red) and [CoIII(TAMLsq)] (black). Reaction conditions: PhINNs (1.0 equiv, 24 mM), styrene (5.0 equiv), catalyst (5.0 mol %), 35 °C, argon. Yields based on 1H NMR integration using 1,3,5-trimethoxybenzene as an internal standard.
Having established the optimal reaction conditions, we investigated the general suitability of both catalysts for the aziridination of alkenes (Table 2). To probe whether PPh4[CoIII(TAMLred)] and [CoIII(TAMLsq)] would perform differently, all reactions were screened with both 5.0 and 2.5 mol % catalyst loading. Moreover, to demonstrate the general applicability of PPh4[CoIII(TAMLred)], we also performed the reactions under aerobic conditions with 2.5 mol % catalyst loading.
Table 2. Substrate Scope for the Aziridination Reaction, Catalyzed by PPh4[CoIII(TAMLred)] or [CoIII(TAMLsq)]a.

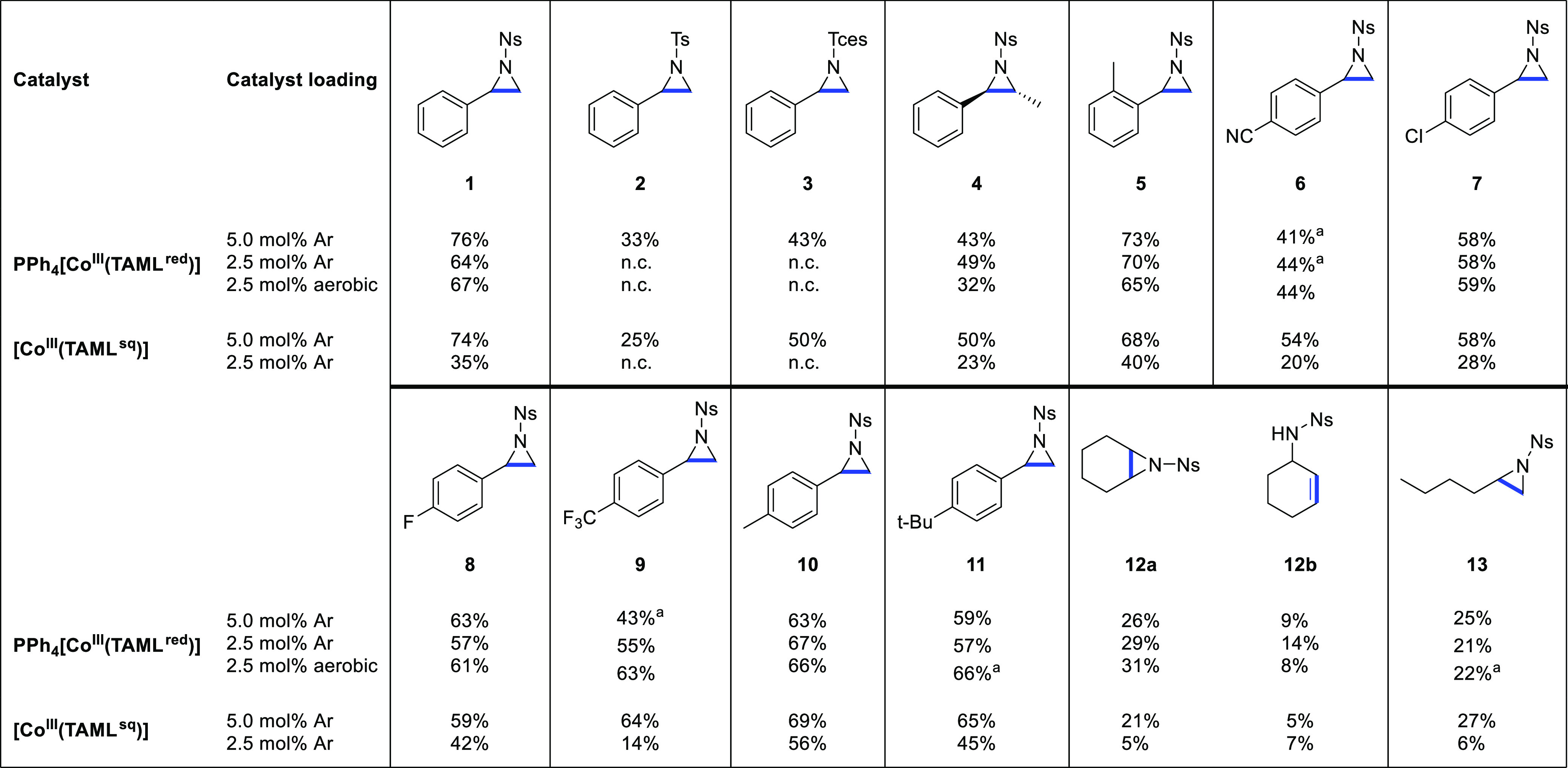
Yields based on 1H NMR integration using 1,3,5-trimethoxybenzene as an internal standard. n.c: not conducted. Reaction performed in duplicate, the average of two yields.
First, the influence of the nitrene precursor was investigated. The reactions proceeded best with more electron-withdrawing nitrene precursors, as the yields of 1, 2, and 3 reflect the following trend: PhINNs > PhINTces > PhINTs (Ns = nosyl, Tces = 2,2,2-trichloroethoxysulfonamide, and Ts = tosyl).20 Next, styrene derivatives were investigated as substrates for catalytic aziridination with PhINNs as the nitrene precursor. β-trans-Methylstyrene afforded diastereoselective formation of trans-aziridine 4 in all cases, as indicated by 1H NMR analysis. This is indicative of either concerted nitrene transfer or a very rapid (i.e., faster than C–C bond rotation) radical rebound step in a stepwise mechanism (vide infra).21ortho-Methylstyrene afforded 5 in 73% (PPh4[CoIII(TAMLred)]) or 68% ([CoIII(TAMLsq)]) yield. Functionalization of the arene ring at the para-position using electron-withdrawing (−CN, −Cl, −F, −CF3) or -donating (−Me, −tBu) groups afforded products 6–11 in moderate yields.
Next, the aziridination protocol was tested for aliphatic substrates. Cyclohexene as a substrate afforded a mixture of the aziridination (12a) and allylic C(sp3)–H amination (12b) product but with the predominant formation of the aziridine product, albeit in poor yields. Interestingly, 1-hexene was converted to aziridine 13 in comparatively low yield but no C(sp3)–H aminated product was observed. Cyclohexene features four allylic C(sp3)–H bonds, whereas only two are present in 1-hexene. The observed C–H amination in cyclohexene is therefore likely a result of increased probability for competition between C–H amination and aziridination. Moreover, the low yields obtained for aliphatic alkenes indicate that these cobalt systems are more suitable for aziridination of aryl-substituted alkenes.
In general, PPh4[CoIII(TAMLred)] performs better in the aziridination of styrene derivatives than [CoIII(TAMLsq)], giving higher conversions at lower catalyst loadings and affording the desired products 1–11 in moderate yields, even under aerobic conditions. In some cases, using a lower (2.5 mol % instead of 5 mol %) loading of the anionic catalyst gives slightly higher yields.22 The inferior performance of [CoIII(TAMLsq)], in comparison to PPh4[CoIII(TAMLred)], is attributed to its relative instability and reactivity with the formed aziridine (vide supra). Interestingly, no C(sp3)–H amination was observed for any substrates with relatively weak (i.e., benzylic or allylic) C–H bonds, except for cyclohexene. However, when using excess PhINNs, prolonged reaction times in toluene, or catalysis in the presence of 1,3,5-trimethoxybenzene, we did observe reaction with benzylic and −OCH3 C(sp3)–H bonds.
Competence of Detected [CoIII(TAMLq)(NNs)2]− and [CoIII(TAML)(NNs)(Y)] Species in Catalytic Aziridination
Intrigued by the catalytic activity of the cobalt-TAML complexes in catalytic aziridination reactions, we first decided to confirm that the previously characterized15 nitrene radical complexes PPh4[CoIII(TAMLq)(NNs)2] and [CoIII(TAMLq)(NNs)(Y)] (which are readily generated from PPh4[CoIII(TAMLred)] and [CoIII(TAMLsq)] upon reaction with PhINNs) are indeed catalytically competent species.
We therefore re-examined the formation of the nitrene radical species and investigated their reactivity with styrene. Previously reported XANES analysis of the nitrene radical complexes showed that the coordination number around cobalt had increased with respect to the four-coordinate starting compounds [CoIII(TAMLsq)] and PPh4[CoIII(TAMLred)], consistent with the formation of six-coordinate Co-(nitrene) bonds.15 EXAFS studies23 indeed indicated an octahedral (six-coordinated) geometry around cobalt in [CoIII(TAMLq)(NNs)(Y)] (see Table S3 and Figure S16 in the SI), very similar to a previously described cobalt-porphyrin mono-nitrene radical complex.10a Although the exact nature of the ligand Y remains elusive, we expect that NsNH2, which might be formed in the reaction, coordinates to the mono-nitrene radical complex.18 The formation of a Co–nitrene bond in [CoIII(TAMLq)(NNs)(Y)] was also indicated by electron paramagnetic resonance (EPR) spectroscopy, as the obtained EPR spectrum revealed hyperfine coupling interactions with both cobalt and the nitrene-nitrogen atom.15
Electrospray ionization high-resolution mass spectrometry (ESI-HRMS–) of [CoIII(TAMLq)(NNs)2]– and [CoIII(TAMLq)(NNs)(Y)], which were generated at 25 °C, is exceptionally clean and only shows the desired nitrene radical complexes and the starting cobalt complexes. Notably, the mono-nitrene complex is detected as [CoIII(TAMLq)(NNs)], consistent with our hypothesis that the sixth coordinating ligand might be a weakly bound NsNH2 moiety. To quantify the conversion to [CoIII(TAMLq)(NNs)(Y)], we performed an EPR spin counting experiment (double integration of the nitrene radical signal with respect to that of the starting material), which revealed its quantitative formation from [CoIII(TAMLsq)] and PhINNs (see Table S6 in the SI).24 The formation of the bis-nitrene species was achieved via reaction of PPh4[CoIII(TAMLred)] with the soluble iminoiodinane OMePhINTs, which enabled a UV–vis titration study. Sequential addition of 0.5–10 equiv OMePhINTs resulted in disappearance of the PPh4[CoIII(TAMLred)] starting material (λmax = 510 nm) with concomitant formation of the bis-tosyl-nitrene complex [CoIII(TAMLq)(NTs)2]–. The clear isosbestic points at 406 and 685 nm (see Figure S17 in the SI) confirm the clean conversion of the starting complex to the bis-nitrene species.25 Gratifyingly, also under catalytically relevant conditions (CH2Cl2, 35 °C), the formation of the nitrene radical complexes [CoIII(TAMLq)(NNs)2]– and [CoIII(TAMLq)(NNs)(Y)] (again observed as [CoIII(TAMLq)(NNs)]) was detected with ESI-HRMS– (see Figures S20 and S21 in the SI). It should, however, be noted that the stability of the nitrene radical complexes at 35 °C is lower than that at room temperature. To investigate whether the nitrene radical complexes are indeed intermediates in the aziridination reactions, we exposed freshly prepared solutions of [CoIII(TAMLq)(NNs)2]– and [CoIII(TAMLq)(NNs)(Y)] to excess styrene at 35 °C (see Schemes S3 and S4 in the SI), which afforded aziridine 1 in yields of 40 and 59%, respectively. These combined spectroscopic and single-turnover reactivity studies point toward the quantitative formation of [CoIII(TAMLq)(NNs)2]– and [CoIII(TAMLq)(NNs)(Y)] under catalytic conditions and confirm their competence in the aziridination of styrene. As such, it is reasonable to propose that these nitrene radical complexes are key reactive intermediates in the above-described catalytic nitrene transfer reactions.
Experimental Mechanistic Studies
In an attempt to understand the modes of action for PPh4[CoIII(TAMLred)] and [CoIII(TAMLsq)] in catalytic aziridination of alkenes with iminoiodinanes, we set out to study the underlying reaction mechanism(s) for both systems. First, the possible deactivation of PPh4[CoIII(TAMLred)] under (synthetically useful) aerobic conditions was investigated. Using the conditions defined in Table 1, entry 9, one additional equivalent of PhINNs was added to the catalytic reaction mixture after 30 and 60 min. The three 30-min periods provided 28, 7, and 3 turnovers in the selective formation of 1, respectively (see Figure S29 in the SI). Subsequent addition of additional 2.5 mol % of PPh4[CoIII(TAMLred)] after 90 min provided 25 additional turnovers. Both experiments point to significant catalyst deactivation after 28 turnovers under the applied reaction conditions.
Next, we investigated the possible involvement of radical intermediates in the catalysis by performing the reactions in the presence of the well-known radical traps 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO), N-tert-butyl-α-phenylnitrone (PBN), and 5,5-dimethyl-1-pyrroline N-oxide (DMPO). Addition of 5 equiv TEMPO or DMPO with respect to PhINNs to the reaction mixtures with either PPh4[CoIII(TAMLred)] or [CoIII(TAMLsq)] under anaerobic conditions (see entries 8 and 13 in Table 1, respectively) completely inhibited aziridine product formation. Usage of PBN under identical conditions led to reduction in the formation of aziridine and afforded 1 in only 28% (PPh4[CoIII(TAMLred)]) or 13% ([CoIII(TAMLsq)]) yield. Given the known reactivity of TEMPO, DMPO, and PBN as radical scavengers, this indicates that radical-type intermediates are generated for both catalysts and trapped by the spin trapping reagents. Indeed, HRMS analysis of the trapping studies with DMPO afforded signals at m/z values corresponding to [DMPO + styrene + H]+, [DMPO + NNs-H]−, and [DMPO + NNs]+.26 X-band EPR analysis of the DMPO-trapped radical intermediates afforded a mixture of paramagnetic DMPO-adducts, again indicating that multiple radical-type intermediates are involved in the catalytic reactions (see Figure S28 in the SI).
The outcome of a Hammett analysis using a range of para-functionalized styrenes in aziridination with PhINNs is depicted in Figure 6. We used the optimized aerobic conditions for PPh4[CoIII(TAMLred)] (entry 9, Table 1) and the optimized conditions for [CoIII(TAMLsq)] (entry 13, Table 1). Apart from the classic electronic Hammett constants (σ+), also the σJJ• radical spin-delocalization substituent constants27 were included to account for the involvement of radical-type intermediates (vide supra).28 Plotting log(kx/kH) versus ρ•σJJ• + ρ+σ+ and applying multiple coefficient linear regression, we found linear correlations for PPh4[CoIII(TAMLred)] (R2 = 0.99, ρJJ• = 0.14, ρ+ = −0.80, slope ρ = 1.00) and [CoIII(TAMLsq)] (R2 = 0.96, ρ• = 0.14, ρ+ = −1.21, slope ρ = 1.00).29 The large |ρ+/ρ•| ratios measured for PPh4[CoIII(TAMLred)] (5.71) and [CoIII(TAMLsq)] (8.64) show that electronic effects dominate the reaction rate.27 The large negative ρ+ values for both catalysts reveal a dominant buildup of positive charge at the styrene moiety in the rate-limiting transition state of both reactions. Besides these dominant electronic effects, the significant positive ρ• values of +0.14 reveal non-negligible radical stabilization effects by delocalization of spin density over the styrene moiety.27
Figure 6.
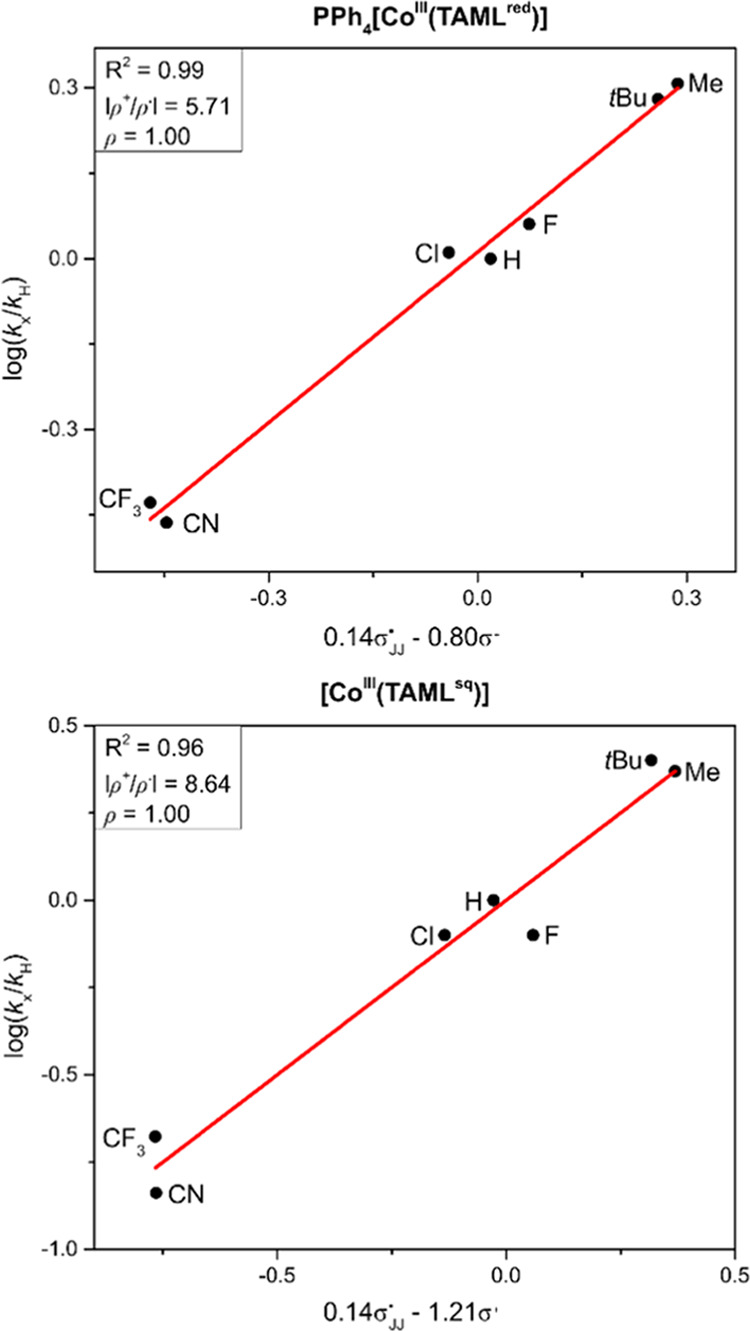
Hammett plots for the PPh4[CoIII(TAMLred)]- and [CoIII(TAMLsq)]-catalyzed aziridination of styrene derivatives.30
Electronic effects dominate when the |ρ/ρ•| ratio is close to or larger than unity (ρ = ρ+ or ρmb).27 The |ρ/ρ•| ratio provides valuable information and has been used in mechanistic studies of various active styrene aziridination catalysts. For example, the |ρ/ρ•| ratio found for [CuI(TMG3trphen)(NR)]+ is dependent on the N-group substituent (0.50 for R = Ts, 0.98 for R = Ns),31 while various [Cu(Tpx)(NTs)]32,33 and [Ag(Tpx)(NTs)]33 complexes have ratios between 0.82–1.282 and 1.141–1.625, respectively. Electronic effects dominate in the transition state for aziridination reactions catalyzed by [RuIV(por)(NTs)2] (|ρ/ρ•| = 2.02).34
Radical-type pathways were proposed for anionic MnII, FeII, and CoII complexes of a triphenylamido-amine ligand, with |ρ/ρ•| ratios of 0.75 (Mn), 1.17 (Fe), and 1.00 (Co).35 Low |ρ/ρ•| ratios were reported for styrene aziridination with [FeII(dipyrrinato)(Cl)(NAd)] (0.04)36 and toluene C(sp3)–H amination with [CoII(por)(NAr)] (0.008),37 which are both dominated by spin-delocalization effects and are meanwhile known to react via radical-type mechanisms involving nitrene radicals.10
The nitrene transfer reactions mediated by PPh4[CoIII(TAMLred)] and [CoIII(TAMLsq)] are associated with unusually large |ρ+/ρ•| ratios combined with significantly positive ρ• values. These values are at odds with the Hammett parameters37 found for the abovementioned [CoII(por)(NR)] nitrene radical species.10 Hence, despite the fact that the [CoIII(TAMLq)(N•Ns)(Y)] and [CoIII(TAMLq)(N•Ns)2]– intermediates were shown to have clear nitrene radical character,15 the Hammett data (Figure 6) provide strong evidence that the reactions do not proceed via direct (nitrene) radical addition to styrene (as observed for [CoII(por)(NR)] species), as |ρ/ρ•| values much smaller than 1 are then expected.27
Large |ρ/ρ•| values (8.45–47.5) are reported for [CuI(Tpx)] catalyzed cyclopropanation of styrene derivatives, which proceeds via an asynchronous concerted carbene addition to the C=C double bond (i.e., nonsimultaneous formation of the two C–C bonds), leading to a substantial buildup of carbocation character at the benzylic position in the transition states.33 However, these reactions show much smaller ρ• values (<0.05), suggesting that the mechanisms for nitrene transfer from [CoIII(TAMLq)(N•Ns)2]– and [CoIII(TAMLq)(N•Ns)(Y)] are unique. Combined with their unusual electronic structures and nitrene radical character (N•Ns–), the measured Hammett parameters are suggestive of (partial) single-electron transfer from styrene to the electrophilic nitrene intermediate, leading to (partial) styrene radical cation formation in the rate-limiting transition states (see the Computational Mechanistic Studies below).
Computational Mechanistic Studies
To obtain more insight in the aziridine formation with [CoIII(TAMLred)]– and [CoIII(TAMLsq)] as the catalysts, density functional theory (DFT) calculations were performed to evaluate the free energy reaction profile (ΔG298K° in kcal mol–1). We used the solvent (CH2Cl2) adducts of the starting complexes and nitrene species (A, C, D, H, HNH3, and J, Schemes 1 and 2) and exchanged solvent for the incoming substrate during all our calculations (see the SI for additional details).38 Previously reported multiconfigurational N-electron valence state perturbation theory corrected complete active space self-consistent field (NEVPT2-CASSCF) calculations showed that both [CoIII(TAMLsq)] and [CoIII(TAMLq)(NNs)(Y)] (wherein Y is a vacant site) have a net-doublet (S = 1/2) electronic ground state, which is why we evaluated the nitrene transfer mechanism at the doublet spin surface.15 Analogously, the triplet spin surface was used to calculate the [CoIII(TAMLred)]– catalyzed reaction, based on the triplet (S = 1) electronic ground state of [CoIII(TAMLred)]– and [CoIII(TAMLq)(NNs)2]– derived from NEVPT2-CASSCF calculations. Moreover, we envisioned that under catalytic conditions with [CoIII(TAMLred)]–, an anionic mono-nitrene species ([CoIII(TAMLsq)(NNs)]–) might also be active in aziridination. NEVPT2-CASSCF(14,13) calculations revealed that also [CoIII(TAMLsq)(NNs)]– has a triplet electronic ground state, arising from ferromagnetic coupling between a TAML ligand-centered radical and a nitrene nitrogen-centered radical bound to a low spin cobalt(III) center (see C in Schemes 1 and S7). In line with our previous observations, this species arises from TAML ligand-to-substrate single-electron transfer upon reaction with PhINNs, resulting in the formation of a one-electron reduced Fischer-type nitrene-radical complex.
Scheme 1. Proposed Mechanism for the [CoIII(TAMLred)]− Catalyzed Aziridination of Styrene to Afford 1 via a Mono-nitrene (Right) and Bis-nitrene (Left) Pathway.
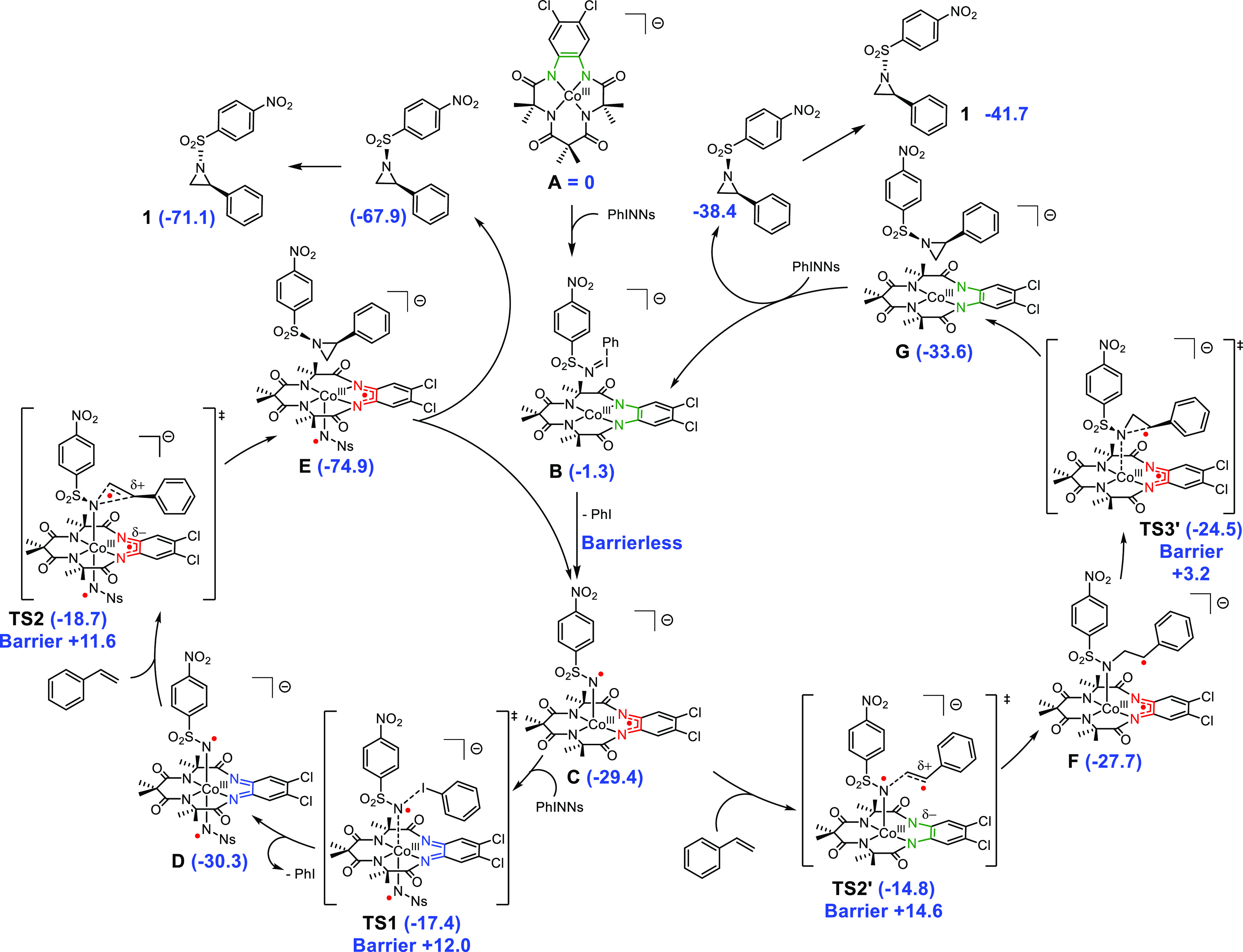
Free energies (ΔG298K° in kcal mol–1) calculated with DFT at the BP86/def2-TZVP/disp3 (m4-grid) level of theory at the triplet (S = 1) spin surface.
Scheme 2. Proposed Mechanism for the [CoIII(TAMLsq)] Catalyzed Aziridination of Styrene to Afford Aziridine 1.

Free energies (ΔG298K° in kcal mol–1) calculated with DFT at the BP86/def2-TZVP/disp3 (m4-grid) level of theory at the doublet (S = 1/2) spin surface. The superscript describes the nature of Y: vacant site or NH3.
The computed mechanism for the aziridination reaction catalyzed by the anionic [CoIII(TAMLred)]– complex is depicted in Scheme 1. Exchange of CH2Cl2 (solvent) with PhINNs on [CoIII(TAMLred)]– (A, reference point) affords adduct B with ΔG° = −1.3 kcal mol–1. The anionic mono-nitrene [CoIII(TAMLsq)(NNs)]– (C, ΔG° = −29.4 kcal mol–1) is formed through barrierless ligand-to-substrate single-electron transfer. Formation of the bis-nitrene [CoIII(TAMLq)(NNs)2]– (D, ΔG° = −30.3 kcal mol–1) is slightly exergonic (ΔΔG° = −0.9 kcal mol–1) and proceeds via ligand-to-substrate single-electron transfer in TS1 (ΔG° = −17.4 kcal mol–1) with a relative free energy barrier of + 12.0 kcal mol–1. Nitrene transfer to styrene proceeds via an electronically asynchronous TS2 (ΔG° = −18.7 kcal mol–1, ΔΔG‡ = +11.6 kcal mol–1, vide infra), immediately followed by barrierless cis-aziridine formation to form E in a highly exergonic reaction (ΔG° = −74.9 kcal mol–1) and concomitant one-electron reduction of the ligand. The cis isomer of the product is released in an endergonic manner (ΔΔG° = +7.0 kcal mol–1) to regenerate mono-nitrene C. The free cis-aziridine then isomerizes in solution to the more stable trans-aziridine (1, ΔG° = −71.1 kcal mol–1) via N-pyramidal inversion.4d The overall Gibbs free reaction energy for aziridine 1 formation is thus −41.7 kcal mol–1 as intermediate C is found at ΔG° = −29.4 kcal mol–1. C can re-enter the bis-nitrene mechanism via TS1 or directly react with styrene via the electronically asynchronous TS2′ (ΔG° = −14.8 kcal mol–1, ΔΔG‡ = +14.6 kcal mol–1, vide infra) to afford benzylic radical F (ΔG° = −27.7 kcal mol–1) in an exergonic fashion. The radical rebound via TS3′ (ΔG° = −24.5 kcal mol–1, ΔΔG‡ = +3.2 kcal mol–1) affords the coordinated cis-aziridine in G (ΔG° = −33.6 kcal mol–1), concomitant with one-electron reduction of the ligand and regeneration of B after product dissociation (ΔG° = −38.4 kcal mol–1). The trans-aziridine (1, ΔG° = −41.7 kcal mol–1) is then obtained after N-pyramidal inversion. The calculated activation energies for both anionic mechanistic cycles are relatively low and of comparable magnitude. Both [CoIII(TAMLsq)(NNs)]– (C) and [CoIII(TAMLq)(NNs)2]– (D) could therefore be active in the aziridination reaction under the optimized mild conditions.
Scheme 2 describes the proposed mechanism for aziridination catalyzed by the neutral [CoIII(TAMLsq)] complex. We computed the full mechanistic cycle for a five-coordinate mono-nitrene complex [CoIII(TAMLq)(NNs)(Y)] (Y = vacant site) and a six-coordinate mono-nitrene complex, wherein Y = NH3 as an archetypical sixth ligand, as indicated by a superscript NH3 in the compound labeling. Exchange of CH2Cl2 (solvent) with PhINNs on [CoIII(TAMLsq)(Y)] (H and HNH3) affords adduct I (ΔG° = −1.6 kcal mol–1) and INH3 (ΔG° = −8.2 kcal mol–1) and mono-nitrene [CoIII(TAMLq)(NNs)(Y)] (J, ΔG° = −21.2 kcal mol–1 and JNH3, ΔG° = −14.2 kcal mol–1) through barrierless ligand-to-substrate single-electron transfer. Nitrene addition onto styrene occurs through the electronically asynchronous TS4 (ΔG° = −14.8 kcal mol–1, ΔΔG‡ = +6.4 kcal mol–1, vide infra) and TS4NH3 (ΔG° = −6.1 kcal mol–1, ΔΔG‡ = +8.1 kcal mol–1, vide infra). For Y = NH3, this transition state is immediately followed by barrierless cis-aziridine formation to afford LNH3 in an exergonic manner (ΔG° = −35.0 kcal mol–1). For Y = vacant site, the radical rebound on the formed benzylic radical K (ΔG° = −26.8 kcal mol–1) occurs via TS5 (ΔG° = −25.3 kcal mol–1, ΔΔG‡ = +1.5 kcal mol–1) to yield the coordinated cis-aziridine on the complex and one-electron reduction of the ligand (L, ΔG° = −36.3 kcal mol–1). Product dissociation from L or LNH3 then regenerates H or HNH3, respectively, and the trans-aziridine after N-pyramidal inversion (ΔG° = −41.7 kcal mol–1).
The mechanistic cycles depicted in Schemes 1 and 2 are consistent with the diastereoselective formation of 4 from β-trans-methylstyrene and PhINNs, as the radical rebound from the benzylic radicals is (nearly) barrierless, and therefore outcompetes rotation around the N–C–C–Ph bond. Moreover, all transition states have low activation energies (ΔΔG‡ ≤ +14.6 kcal mol–1), and short reaction times are therefore expected. Experimentally, the relatively long reaction times required (20 min to 2 h, Table 2 and Figure 5) are likely due to the low solubility of PhINNs and relative instability of [CoIII(TAMLsq)] (vide supra). In addition, endergonic product dissociation from intermediate E might hamper the liberation of the free anionic cobalt catalyst.
Asynchronous Electron Transfer in the Rate-Limiting C–N Bond-Forming Transition States
A close inspection of the spin densities and charge distributions (Figure 7 and Table S14 in the SI) in TS2, TS2′, TS4NH3, and TS4 revealed interesting asynchronous electron transfer16 processes in the first step of the C–N bond-forming transition states of the stepwise nitrene transfer mechanism (Scheme 3). Pure (two-electron) electrophilic addition of the nitrenes to styrene could explain the large Hammett |ρ+/ρ•| ratios but would not result in the observed large spin densities on styrene in TS2, TS2′, TS4NH3, and TS4 (Figure 7 and Scheme 3) or even in the formation of benzyl radicals F and K, and hence, this pathway can be discarded. On the other hand, stepwise radical addition of the (α-spin) nitrene radicals to styrene does not fit with the Hammett analysis, as the |ρ/ρ•| ratios are too large for that (vide supra). Furthermore, such a stepwise radical addition mechanism would result in (near-complete) spin transfer of the nitrene radical to the benzylic position of styrene with a straightforward relocation of α-spin density from the nitrene moiety to the γ-position of the developing Co–N(Ns)–CH2CH2•Ph benzyl radical moiety (Figure 1B).
Figure 7.

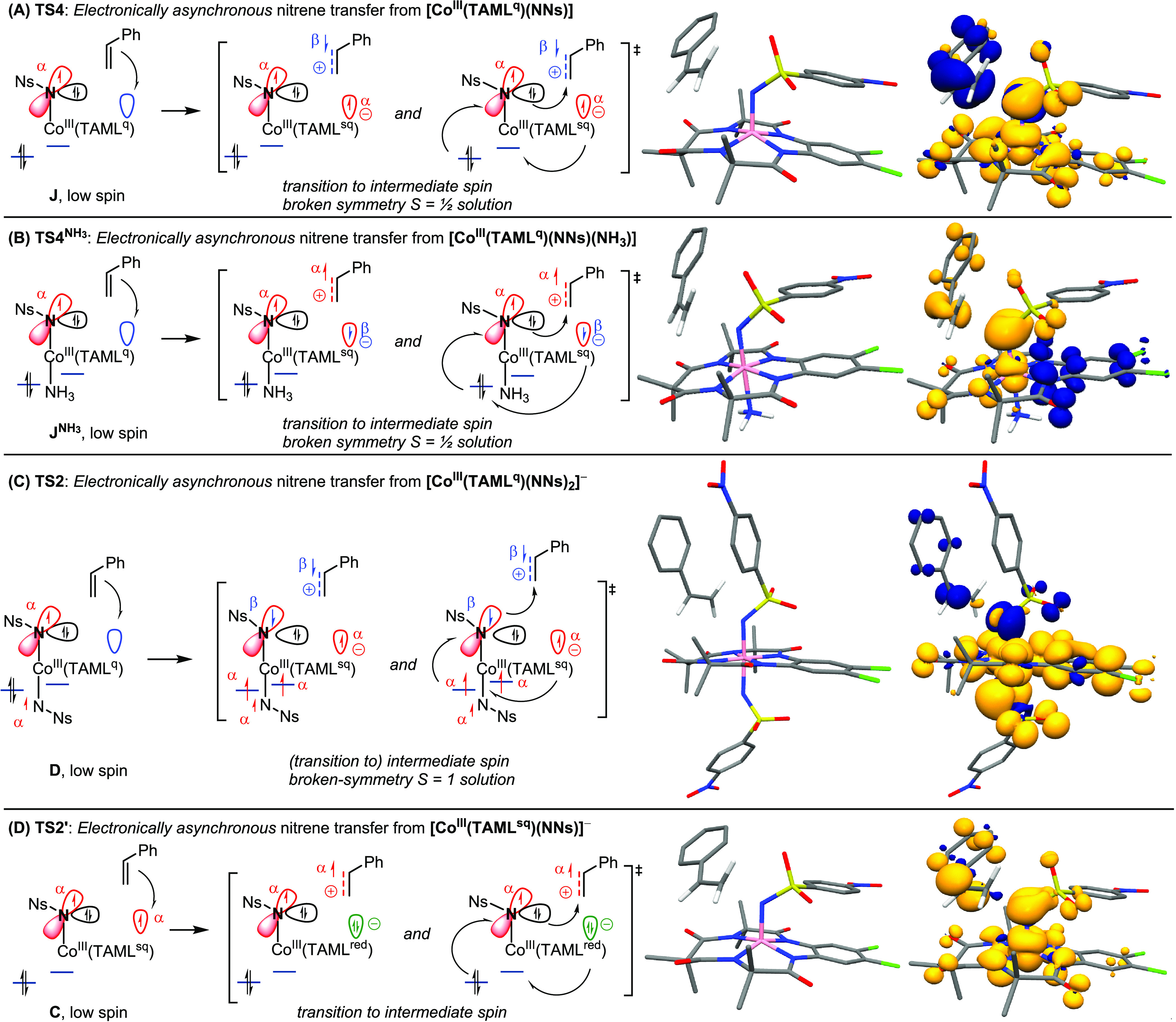
Development of natural population analysis (NPA) charges (black, italics) and spin densities (positive α: red; negative β: blue) in TS4 (A), TS4NH3 (B), TS2 (C), and TS2′ (D). See Table S14 in the SI for assignment of TAML oxidation states.
Scheme 3. Unusual Spin Density Flow Observed in the DFT-Computed Electronically Asynchronous Transition States for C–N Bond Formation, Graphical Representations and Spin Densities (α–β) for TS4 (A), TS4NH3 (B), TS2 (C), and TS2′ (D).
Blue bars describe cobalt-centered orbitals. Blue, red, and green lobes represent empty, 1e– filled and 2e– filled TAML-, and nitrene-based orbitals. Yellow lobes in the graphical representations indicate α-spin density, and blue lobes indicate β-spin density. H atoms (except for vinylic protons) are omitted for clarity.
This is not what is observed in TS4 and TS2, wherein styrene attack results in unusual transformation in electron spin from α-spin density on the nitrene moiety to β-spin density at the developing Co–N(Ns)–CH2CH2•Ph benzyl radical moiety (see Figure 7 and Scheme 3). Moreover, it is clear from Scheme 3A that in TS4 the styrene in fact is attacked by the nucleophilic, non-spin-bearing sp2-hybridized lone pair of the nitrene moiety instead of the expected radical addition of styrene to the α-spin-containing p-orbital of the nitrene radical moiety (which would be the anticipated reactivity of nitrene radicals, see Figure 1B). Furthermore, analysis of the natural population analysis (NPA) atomic charges in TS4, TS4NH3, TS2, and TS2′ reveals a clear and substantial decrease in charge density (i.e., the buildup of net positive charge) on the styrene moiety (largest effect in TS4, in good agreement with the Hammett data). Another striking observation is the increase of (positive) α-spin density in the redox-active orbital of the TAML ligand in TS4 and TS2, and the increase in β-spin density on the same orbital of the TAML ligand in TS4NH3. On the other hand, the α-spin density in this orbital vanishes in TS2′ (Scheme 3 and Table S14 in the SI). In all cases, an increase in (positive) spin density at cobalt is observed, indicating a transition from a low spin (S = 0) to an intermediate spin (S = 1) cobalt(III) center. The positive (α) spin density at the reacting nitrene fragment decreases in each of the transition states but (as explained above) is not straightforwardly transferred to the styrene moiety. The combined data are indicative for (partial) styrene radical cation formation in all three C–N bond-forming transition states, with the redox-active orbital of the TAML ligand being the initial acceptor orbital in the redox process and cobalt undergoing a transition from low spin to intermediate spin (see Scheme 3).
In the neutral five-coordinate mono-nitrene transition state TS4, (partial) electron transfer from styrene to the empty redox-active TAML orbital leads to buildup of α-spin at the TAML ligand and β-spin at the styrene moiety (Scheme 3A). The developing charge separation (reduced complex and oxidized styrene substrate) collapses in the same transition state by simultaneous two-electron nucleophilic attack of the lone pair on the nitrene moiety to the (developing) styrene radical cation to generate a benzylic radical K. The C–N bond formation step is accompanied by single-electron transfer from the mono-reduced TAML backbone to cobalt and from cobalt to the nitrene moiety, thus producing an intermediate spin cobalt(III) center. During this electronically asynchronous transition state, cobalt accommodates the spin-changing events by acting as a spin shuttle while the TAML scaffold acts as a transient electron acceptor. The resulting spin directionalities (β-spin on the benzyl radical fragment and two α-spins on cobalt) in K are consistent with the formation of an intermediate spin cobalt(III) center and an overall S = 1/2 spin state.
A similar multistep electron-flow process explains the development of spin densities and NPA atomic charges in the six-coordinate neutral mono-nitrene transition state TS4NH3 (Scheme 3B) and the anionic bis-nitrene transition state TS2 (Scheme 3C). While in TS4 initial substrate-to-ligand single-electron transfer involves an α-spin electron, the electron transfer in TS4NH3 involves a β-spin electron as evident from the buildup of β-spin density on the redox-active orbital on the TAML ligand. The axial NH3 donor leads to a more advanced S = 1 state for cobalt on the Co–Ns fragment in the transition state, which adopts a BS(2,1) (broken-symmetry) DFT solution with β-spin density on the TAML fragment. Likewise, a subtle difference between TS2 and TS4 is that the (partial) electron transfer from styrene to the empty redox-active TAML orbital results in an S = 1 state with a more advanced development of the intermediate spin state on the cobalt(III) site, which consequently has to adopt a BS(3,1) DFT solution with a β-spin at the nitrene radical moiety.
Another subtle difference between TS2 and TS4 or TS4NH3 is that in TS2 the styrene moiety approaches the β-spin-bearing p-orbital, while in TS4 and TS4NH3 it approaches the sp2-hybridized lone pair. However, this (β-spin-containing) p-orbital still acts as a two-electron nucleophile in the C–N bond formation step, in a process involving simultaneous (α-spin) electron transfer from the mono-reduced TAML ligand to cobalt and from cobalt to the (β-spin-bearing) p-orbital (see Scheme 3C). While TS4 leads to a discrete benzyl radical intermediate (K), TS2 and TS4NH3 do not lead to a stable intermediate, as the benzyl radical formed in these processes collapses in a barrierless manner to an aziridine adduct E or LNH3, respectively. Nonetheless, despite these subtle differences, the C–N bond formation steps in TS2, TS4, and TS4NH3 are in essence very similar and clearly electronically asynchronous.
The development of spin densities and NPA charges in the anionic mono-nitrene transition state TS2′ can also be explained by a similar electron-flow process (Scheme 3D). For this transition state, we observed reduction of α-spin density on the TAML moiety and a substantial development of α-spin density on the styrene fragment, which is an opposite spin density development than in TS4 and TS2. This is, however, to be expected, as the redox-active TAML orbital is already half-filled in C (in contrast to J and D, for which this orbital is empty), so that SET from styrene to this α-spin-containing orbital must involve a β-spin electron, thus reducing the α-spin population at TAML and leaving α-spin density on the developing styrene radical cation in this case. As observed in TS4 and TS4NH3, styrene radical cation formation and C–N bond formation again occur simultaneously, involving two-electron nucleophilic attack of the sp2-hybridized nitrene lone pair at the (developing) styrene radical cation to generate benzylic radical intermediate F. The C–N bond-forming step is again accompanied by a cobalt-facilitated spin shuttle event (i.e., SET from the reduced TAML backbone to cobalt and from cobalt to the nitrene moiety) to produce an intermediate spin cobalt(III) center.
The unusual electron-flow processes in the electronically asynchronous transition states TS4, TS4NH3, TS2, and TS2′, as detailed in Scheme 3, correlate well with both the positive ρ• values and the large |ρ+/ρ•| ratios obtained from the Hammett plots in Figure 6 and the spin trapping studies. To the best of our knowledge, such a mechanism is unprecedented for cobalt-catalyzed hypovalent group transfer and transition-metal-catalyzed nitrene transfer to alkenes in general. Somewhat related substrate-to-catalyst single-electron transfer coupled to σ-bond formation has been observed in some other instances though. Iron-tosylimido complexes acting as (iron-centered) one-electron oxidants toward thioanisole and aromatic amines have recently been disclosed, producing sulfonimides/sulfonamides and oxidized amines, respectively.39,40 Recently, electron transfer from p-methoxythioanisole to [FeV(TAMLsq)(NTs)] was observed prior to S–N bond formation.12d Moreover, single-electron transfer from styrene to an Fe(IV)-nitride was proposed to occur during C–N bond formation and was found to be coupled to a spin state change from the singlet to triplet surface.41 Charge separation was also observed in the transition state for C–O bond formation with electron-rich styrene derivatives and Fe(IV)-porphyrin-oxo radical cations, which was interpreted as electron transfer from the substrate to the complex during the transition state.42 Furthermore, rate-limiting electron transfer to form a substrate radical cation was described for a dicationic Mn(IV)-oxo complex.43 Herein, the initial electron transfer precedes C/S/H–O bond formation in epoxidation, sulfoxidation, and hydroxylation reactions. Finally, charge transfer in the transition state was observed in Fe(II)(porphyrin)-carbene catalyzed cyclopropanation of styrene.44
To the best of our knowledge, transition states involving substrate-to-ligand single-electron transfer have not been described for transition-metal-mediated nitrene transfer to styrenes or olefins. Moreover, the electronically asynchronous transition states described in this work are accompanied by a cobalt-facilitated spin shuttle and nucleophilic (two-electron) C–N bond formation on a nitrene radical, which are hitherto unreported, to the best of our knowledge. Current investigations in our group are focussed on the applicability of these electronically asynchronous transition states in related nitrene transfer reactions to other substrates, which will be reported in due time.
Conclusions
We have presented a mechanistic study for olefin aziridination with [CoIII(TAML)] catalysts in two ligand oxidation states, [CoIII(TAMLred)]– and [CoIII(TAMLsq)]. A variety of styrene derivatives, cyclohexene, and 1-hexene could be converted to the corresponding aziridines in a chemoselective (i.e., nearly always without C–H amination) and diastereoselective fashion (for the formation of 4) under mild conditions, at short reaction times, and with relatively low catalyst loadings. Especially, [CoIII(TAMLred)]– proved to be a practically useful catalyst, as the reaction can be performed under aerobic conditions. In contrast to previous reports on nitrene/imido transfer with iron- or manganese-TAML complexes, the reduced complex ([CoIII(TAMLred)]–) was shown to be more stable and reactive toward nitrene transfer than the oxidized ([CoIII(TAMLsq)]) complex.
HRMS studies were used to demonstrate that both [CoIII(TAMLq)(NNs)2]– and [CoIII(TAMLq)(NNs)(Y)] are formed under catalytically relevant conditions (CH2Cl2, 35 °C). Moreover, the formation of [CoIII(TAMLq)(NNs)(Y)] from [CoIII(TAMLred)] was found to be quantitative based on EPR spin counting experiments and a UV–vis titration showed clean conversion of [CoIII(TAMLred)]– to the bis-tosyl-nitrene complex on the basis of clear isosbestic points. Single-turnover experiments confirmed that [CoIII(TAMLq)(NNs)2]– and [CoIII(TAMLq)(NNs)(Y)] are active intermediates in the aziridination of styrene.
Experimental (Hammett plots, radical trapping) and computational mechanistic studies have shown that the C–N bond formation proceeds via unusual electronically asynchronous transition states. In these reactions, a (partial) styrene substrate to a TAML ligand (single) electron transfer precedes C–N coupling. The actual C–N bond formation is best described as a nucleophilic attack of the nitrene (radical) lone pair at the thus (partially) formed styrene radical cation, coupled to TAML-to-cobalt and cobalt-to-nitrene SET, leading to the formation of an amido-γ-benzyl radical bound to an intermediate spin (S = 1) cobalt(III) center. These complex multiple (intra- and intermolecular) electron-transfer/reorganization processes are coupled to the C–N bond formation in a single electronically asynchronous transition state, wherein the TAML ligand acts as a transient electron acceptor, while the cobalt center acts as a spin shuttle and facilitates an unexpected change in electron spin at the amido-γ-benzyl radical moiety via its transition from a low spin (S = 0) to an intermediate spin (S = 1) configuration. To the best of our knowledge, such a mechanism for cobalt-catalyzed hypovalent group transfer or transition-metal-catalyzed nitrene transfer to styrenes or alkenes has not been reported before and complements the often proposed concerted and stepwise mechanisms for N-group transfer.
Interestingly, the proposed mechanisms proceed exclusively via ligand-centered redox reactions whereby cobalt retains the +III oxidation state in each intermediate of the reaction. This was hitherto unknown for cobalt-catalyzed nitrene transfer reactions. Future work in our group focusses on the use of the herein described electronically asynchronous transition state to steer product formation. More specifically, we are exploring the use of [CoIII(TAMLred)]– as a chemoselective catalyst in nitrene transfer reactions with substrates that have low oxidation potentials.
Acknowledgments
Financial support from The Netherlands Organization for Scientific Research (NWO TOP-Grant 716.015.001 to B.d.B. and NWO VIDI grant 723.014.010 to M.T.) and the research priority area Sustainable Chemistry of the University of Amsterdam (RPA SusChem, UvA) is gratefully acknowledged. Ed Zuidinga is thanked for HRMS measurements. The authors thank the staff of beamline B18, Diamond Light Source (proposal number SP22432) in Didcot, U.K., for support and access to their facilities.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c01343.
Experimental details, synthetic procedures, NMR and EPR spectra, HRMS data, geometries (xyz coordinates) and energies of stationary points and transition states (DFT), and the description of the CASSCF calculation (PDF)
Author Present Address
§ Institute of Chemistry, Carl von Ossietzky University Oldenburg, Carl-von-Ossietzky-Strasse 9-11, 26129 Oldenburg, Germany.
Author Present Address
∥ Georg-August-Universität Göttingen, Tammanstrasse 4, 37077 Göttingen, Germany.
Author Present Address
⊥ Institut für Chemie, Sekr. PC14, Technische Universität Berlin, Strasse des 17. Juni 135, 10623 Berlin, Germany.
Author Present Address
# Zernike Institute for Advanced Materials, University of Groningen, 9747 AG Groningen, The Netherlands.
The authors declare no competing financial interest.
Scheme 3 was incomplete in the version published on June 24, 2020 and was correctly restored on June 26, 2020.
Supplementary Material
References
- a Sweeney J. B. Aziridines: epoxides’ ugly cousins?. Chem. Soc. Rev. 2002, 31, 247–258. 10.1039/B006015L. [DOI] [PubMed] [Google Scholar]; b Watson I. D. G.; Yu L.; Yudin A. K. Advances in Nitrogen Transfer Reactions Involving Aziridines. Acc. Chem. Res. 2006, 39, 194–206. 10.1021/ar050038m. [DOI] [PubMed] [Google Scholar]
- Hu X. E. Nucleophilic ring opening of aziridines. Tetrahedron 2004, 60, 2701–2743. 10.1016/j.tet.2004.01.042. [DOI] [Google Scholar]
- a Pellissier H. Recent developments in asymmetric aziridination. Tetrahedron 2010, 66, 1509–1555. 10.1016/j.tet.2009.11.089. [DOI] [Google Scholar]; b Jenkins D. M. Atom-Economical C2 + N1 Aziridination: Progress towards Catalytic Intermolecular Reactions Using Alkenes and Aryl Azides. Synlett 2012, 23, 1267–1270. 10.1055/s-0031-1290977. [DOI] [Google Scholar]; c Uchida T.; Katsuki T. Asymmetric Nitrene Transfer Reactions: Sulfimidation, Aziridination and C–H Amination Using Azide Compounds as Nitrene Precursors. Chem. Rec. 2014, 14, 117–127. 10.1002/tcr.201300027. [DOI] [PubMed] [Google Scholar]
- a van Leest N. P.; Epping P. F. J.; van Vliet K. M.; Lankelma M.; van den Heuvel E. J.; Heijtbrink N.; Broersen R.; de Bruin B.. Single-Electron Elementary Steps in Homogeneous Organometallic Catalysis. Advances in Organometallic Chemistry; Elsevier, 2018; Vol. 70, pp 71–180. [Google Scholar]; b Kuijpers P. F.; van der Vlugt J. I.; Schneider S.; de Bruin B. Nitrene Radical Intermediates in Catalytic Synthesis. Chem. – Eur. J. 2017, 23, 13819–13829. 10.1002/chem.201702537. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Caselli A.; Gallo E.; Fantauzzi S.; Morlacchi S.; Ragaini F.; Cenini S. Allylic Amination and Aziridination of Olefins by Aryl Azides Catalyzed by CoII(tpp): A Synthetic and Mechanistic Study. Eur. J. Inorg. Chem. 2008, 3009–3019. 10.1002/ejic.200800156. [DOI] [Google Scholar]; d Olivos Suarez A. I.; Jiang H.; Zhang X. P.; de Bruin B. The radical mechanism of cobalt(II) porphyrin-catalyzed olefin aziridination and the importance of cooperative H-bonding. Dalton Trans. 2011, 40, 5697–5705. 10.1039/c1dt10027k. [DOI] [PubMed] [Google Scholar]; e Chang J. W. W.; Ton T. M. U.; Chan P. W. H. Transition-metal-catalyzed aminations and aziridinations of C–H and C–C bonds with iminoiodinanes. Chem. Rec. 2011, 11, 331–357. 10.1002/tcr.201100018. [DOI] [PubMed] [Google Scholar]; f Varela-Álvarez A.; Yang T.; Jennings H.; Kornecki K. P.; Macmillan S. N.; Lancaster K. M.; Mack J. B. C.; Du Bois J.; Berry J. F.; Musaev D. G. Rh2(II,III) Catalysts with Chelating Carboxylate and Carboxamidate Supports: Electronic Structure and Nitrene Transfer Reactivity. J. Am. Chem. Soc. 2016, 138, 2327–2341. 10.1021/jacs.5b12790. [DOI] [PubMed] [Google Scholar]; g Kornecki K. P.; Berry J. F. Evidence for a One-Electron Mechanistic Regime in Dirhodium-Catalyzed Intermolecular C–H Amination. Chem. – Eur. J. 2011, 17, 5827–5832. 10.1002/chem.201100708. [DOI] [PubMed] [Google Scholar]; h Park Y.; Kim Y.; Chang S. Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]
- Ren Y.; Cheaib K.; Jacquet J.; Vezin H.; Fensterbank L.; Orio M.; Blanchard S.; Desage-El Murr M. Copper-Catalyzed Aziridination with Redox-Active Ligands: Molecular Spin Catalysis. Chem. – Eur. J. 2018, 24, 5086–5090. 10.1002/chem.201705649. [DOI] [PubMed] [Google Scholar]
- van der Vlugt J. I. Radical-Type Reactivity and Catalysis by Single-Electron Transfer to or from Redox-Active Ligands. Chem. – Eur. J. 2019, 25, 2651–2662. 10.1002/chem.201802606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fujita D.; Sugimoto H.; Shiota Y.; Morimoto Y.; Yoshizawa K.; Itoh S. Catalytic C–H amination driven by intramolecular ligand-to-nitrene one-electron transfer through a rhodium(III) centre. Chem. Commun. 2017, 53, 4849–4852. 10.1039/C7CC01840A. [DOI] [PubMed] [Google Scholar]; b Fujita D.; Sugimoto H.; Morimoto Y.; Itoh S. Noninnocent Ligand in Rhodium(III)-Complex-Catalyzed C–H Bond Amination with Tosyl Azide. Inorg. Chem. 2018, 57, 9738–9747. 10.1021/acs.inorgchem.8b00289. [DOI] [PubMed] [Google Scholar]
- a Broere D. L. J.; de Bruin B.; Reek J. N. H.; Lutz M.; Dechert S.; van der Vlugt J. I. Intramolecular Redox-Active Ligand-to-Substrate Single-Electron Transfer: Radical Reactivity with a Palladium(II) Complex. J. Am. Chem.Soc. 2014, 136, 11574–11577. 10.1021/ja502164f. [DOI] [PubMed] [Google Scholar]; b Broere D. L. J.; van Leest N. P.; de Bruin B.; Siegler M. A.; van der Vlugt J. I. Reversible Redox Chemistry and Catalytic C(sp3)–H Amination Reactivity of a Paramagnetic Pd Complex Bearing a Redox-Active o-Aminophenol-Derived NNO Pincer Ligand. Inorg. Chem. 2016, 55, 8603–8611. 10.1021/acs.inorgchem.6b01192. [DOI] [PubMed] [Google Scholar]
- a Smith A. L.; Hardcastle K. I.; Soper J. D. Redox-Active Ligand-Mediated Oxidative Addition and Reductive Elimination at Square Planar Cobalt(III): Multielectron Reactions for Cross-Coupling. J. Am. Chem. Soc. 2010, 132, 14358–14360. 10.1021/ja106212w. [DOI] [PubMed] [Google Scholar]; b Dzik W. I.; van der Vlugt J. I.; Reek J. N. H.; de Bruin B. Ligands that Store and Release Electrons during Catalysis. Angew. Chem., Int. Ed. 2011, 50, 3356–3358. 10.1002/anie.201006778. [DOI] [PubMed] [Google Scholar]; c van der Meer M.; Rechkemmer Y.; Peremykin I.; Hohloch S.; van Slageren J.; Sarkar B. (Electro)catalytic C–C bond formation reaction with a redox-active cobalt complex. Chem. Commun. 2014, 50, 11104–11106. 10.1039/C4CC03309D. [DOI] [PubMed] [Google Scholar]
- a Goswami M.; Lyaskovskyy V.; Domingos S. R.; Buma W. J.; Woutersen S.; Troeppner O.; Ivanović-Burmazović I.; Lu H.; Cui X.; Zhang X. P.; Reijerse E. J.; DeBeer S.; van Schooneveld M. M.; Pfaff F. F.; Ray K.; de Bruin B. Characterization of Porphyrin-Co(III)-“Nitrene Radical” Species Relevant in Catalytic Nitrene Transfer Reactions. J. Am. Chem. Soc. 2015, 137, 5468–5479. 10.1021/jacs.5b01197. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lyaskovskyy V.; Olivos Suárez A. I.; Lu H.; Jiang H.; Zhang X. P.; de Bruin B. Mechanism of Cobalt(II) Porphyrin-Catalyzed C–H Amination with Organic Azides: Radical Nature and H-Atom Abstraction Ability of the Key Cobalt(III)–Nitrene Intermediates. J. Am. Chem. Soc. 2011, 133, 12264–12273. 10.1021/ja204800a. [DOI] [PubMed] [Google Scholar]; c Kuijpers P. F.; Tiekink M. J.; Breukelaar W. B.; Broere D. L. J.; van Leest N. P.; van der Vlugt J. I.; Reek J. N. H.; de Bruin B. Cobalt-Porphyrin-Catalysed Intramolecular Ring-Closing C–H Amination of Aliphatic Azides: A Nitrene-Radical Approach to Saturated Heterocycles. Chem. – Eur. J. 2017, 23, 7945–7952. 10.1002/chem.201700358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins T. J.; Ryabov A. D. Targeting of High-Valent Iron-TAML Activators at Hydrocarbons and Beyond. Chem. Rev. 2017, 117, 9140–9162. 10.1021/acs.chemrev.7b00034. [DOI] [PubMed] [Google Scholar]
- a Hong S.; Sutherlin K. D.; Vardhaman A. K.; Yan J. J.; Park S.; Lee Y.-M.; Jang S.; Lu X.; Ohta T.; Ogura T.; Solomon E. I.; Nam W. A. Mononuclear Nonheme Iron(V)-Imido Complex. J. Am. Chem. Soc. 2017, 139, 8800–8803. 10.1021/jacs.7b04695. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hong S.; Lu H.; Lee Y.-M.; Seo M. S.; Ohta T.; Ogura T.; Clémancey M.; Maldivi P.; Latour J.-M.; Sarangi R.; Nam W. Achieving One-Electron Oxidation of a Mononuclear Nonheme Iron(V)-Imido Complex. J. Am. Chem. Soc. 2017, 139, 14372–14375. 10.1021/jacs.7b08161. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shi H.; Xie J.; Lam W. W. Y.; Mab W.-L.; Mak C.-K.; Yiu S.-M.; Lee H. K.; Lau T.-C. Generation and Reactivity of a One-Electron-Oxidized Manganese(V) Imido Complex with a Tetraamido Macrocyclic Ligand. Chem. – Eur. J. 2019, 25, 12895–12899. 10.1002/chem.201902405. [DOI] [PubMed] [Google Scholar]; d Lu X.; Li X.-X.; Lee Y.-M.; Jang Y.; Seo M. S.; Hong S.; Cho K.-B.; Fukuzumi S.; Nam W. Electron-Transfer and Redox Reactivity of High-Valent Iron Imido and Oxo Complexes with the Formal Oxidation States of Five and Six. J. Am. Chem. Soc. 2020, 142, 3891–3904. 10.1021/jacs.9b11682. [DOI] [PubMed] [Google Scholar]
- Collins T. J.; Powell R. D.; Slebodnick C.; Uffelman E. S. Stable highly oxidizing cobalt complexes of macrocyclic ligands. J. Am. Chem. Soc. 1991, 113, 8419–8425. 10.1021/ja00022a033. [DOI] [Google Scholar]
- The electronic ground state of [CoIII(TAMLsq)] was investigated by electron paramagnetic resonance (EPR), UV–vis, X-ray absorption near edge structure (XANES) and multiconfigurational complete active space self-consistent field (CASSCF) calculations, and was best described as an S = 1 cobalt center antiferromagnetically coupled to a ligand-centered radical (S = 1/2). See ref 15.
- van Leest N. P.; Tepaske M. A.; Oudsen J.-P. H.; Venderbosch B.; Rietdijk N. R.; Siegler M. A.; Tromp M.; van der Vlugt J. I.; de Bruin B. Ligand Redox Non-Innocence in [CoIII(TAML)]0/– Complexes Affects Nitrene Formation. J. Am. Chem. Soc. 2020, 142, 552–563. 10.1021/jacs.9b11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- We define an electronically asynchronous transition state as a process where (C–N) bond formation is preceded by an electron transfer event from the substrate to the complex, in a single step without forming discrete redox intermediates.
- We chose 35 °C as the reaction temperature to perform the reactions under atmospheric pressure without the need for a reflux condenser (boiling point CH2Cl2 = 39.6 °C). Moreover, the low solubility of PhINNs and removal of styrene during workup prevented calculation of the conversion. However, the crude reaction mixtures for reactions in CH2Cl2 demonstrated clean formation of the aziridine product (see Figures S1–S13 in the SI).
- NsNH2 can be formed through hydrolysis of PhINNs or via (degradative) side reactions with the other substrates and/or catalyst. See ref 10. In addition, formation of NsNH2 is detrimental to the yield of the desired aziridine, as PhINNs is the limiting reagent.
- van Leest N. P.; Grooten L.; van der Vlugt J. I.; de Bruin B. Uncatalyzed Oxidative C–H Amination of 9,10-Dihydro-9-Heteroanthracenes: A Mechanistic Study. Chem. – Eur. J. 2019, 25, 5987–5993. 10.1002/chem.201900377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This trend correlates well with the proposed electronically asynchronous transition states (see the “Asynchronous electron transfer in the rate-limiting C–N bond forming transition states” section), i.e. a more electrophilic (oxidizing) CoIII(TAML)–nitrene complex favours the initiating substrate-to-complex single-electron transfer.
- Nebra N.; Lescot C.; Dauban P.; Mallet-Ladeira S.; Martin-Vaca B.; Bourissou D. Intermolecular Alkene Aziridination: An Original and Efficient CuI···CuI Dinuclear Catalyst Deriving from a Phospha-Amidinate Ligand. Eur. J. Org. Chem. 2013, 984–990. 10.1002/ejoc.201201478. [DOI] [Google Scholar]
- Degradation of [CoIII(TAMLq)(NNs)2]– is faster at higher concentrations, which might contribute to the lower yields obtained in some instances at 5.0 mol % instead of 2.5 mol % PPh4[CoIII(TAMLred)] catalyst loading. This concentration-dependent trend was also observed in the optimization of the reaction conditions; a lower concentration afforded higher yields of the aziridine. This behavior points to intermolecular reactions between reactive catalytic intermediates with the catalysts species themselves.
- EXAFS spectra and fits (see Tables S1 and S2 in the SI) of PPh4[CoIII(TAMLred)] and [CoIII(TAMLsq)] are consistent with the bond distances and coordination geometry determined by XRD and DFT, respectively. Unfortunately, the low solubility of PPh4[Co(TAMLq)(NNs)2] in toluene hampered the data quality to the extent that no reliable fit of the EXAFS spectrum could be obtained.
- [Co(TAMLq)(NNs)2]– is EPR and 1H NMR silent, and its formation from [CoIII(TAMLred)]– cannot be quantified by EPR spin counting.
- Formation of the tosyl-bis-nitrene species was followed by UV–vis using OMePhINTs as the nitrene precursor (which has sufficient solubility in CH2Cl2, whereas PhINNs and PhINTs are poorly soluble). The UV–vis spectra of [Co(TAMLq)(NNs)2]– and [Co(TAMLq)(NTs)2]– are (as expected) similar. See ref 15 and Figure S17 in the SI.
- [DMPO+styrene+H]+; ESI-HRMS+ calcd 218.1545, found 218.1560 and 218.1597 for [CoIII(TAMLsq)] and PPh4[CoIII(TAMLred)], respectively. [DMPO+NNs-H]–; ESI-HRMS– calcd 312.0654, found 312.0728 and 312.0600 for [CoIII(TAMLsq)] and PPh4[CoIII(TAMLred)], respectively. [DMPO+NNs]+; field desorption-HRMS+ calcd 313.0732, found 313.0719 for [CoIII(TAMLsq)] as the catalyst.
- Jiang X.-K. Establishment and Successful Application of the σJJ• Scale of Spin-Delocalization Substituent Constants. Acc. Chem. Res. 1997, 30, 283–289. 10.1021/ar950121h. [DOI] [Google Scholar]
- Hammett plots using only the classical electronic Hammett constants (σ+) are included in Figures S32 and S33 in the SI, and give lower R 2 values. Furthermore, as the involvement of radical intermediates was also shown by spin trapping studies, we considered including σJJ• in the Hammett analysis to be relevant. It is clear, however, that the relative rates are primarily determined by the electronic Hammett constants (σ+).
- See the Supporting Information for a detailed description on the construction of these Hammett plots, including error bars and a comparison with the pure electronic Hammett plots.
- A constant (C) to describe the linear trend line and error-bars per data point are reported in Figures S30 and S31 in the SI.
- Bagchi V.; Paraskevopoulou P.; Das P.; Chi L.; Wang Q.; Choudhury A.; Mathieson J. S.; Cronin L.; Pardue D. B.; Cundari T. R.; Mitrikas G.; Sanakis Y.; Stravropoulos P. A Versatile Tripodal Cu(I) Reagent for C–N Bond Construction via Nitrene-Transfer Chemistry: Catalytic Perspectives and Mechanistic Insights on C–H Aminations/Amidinations and Olefin Aziridinations. J. Am. Chem. Soc. 2014, 136, 11362–11381. 10.1021/ja503869j. [DOI] [PubMed] [Google Scholar]
- Díaz-Requejo M. M.; Pérez P. J.; Bookhart M.; Templeton J. L. Substituent Effects on the Reaction Rates of Copper-Catalyzed Cyclopropanation and Aziridination of para-Substituted Styrenes. Organometallics 1997, 16, 4399–4402. 10.1021/om970382q. [DOI] [Google Scholar]
- Maestre L.; Sameera W. M. C.; Díaz-Raquejo M. M.; Maseras F.; Pérez P. J. A General Mechanism for the Copper- and Silver-Catalyzed Olefin Aziridination Reactions: Concomitant Involvement of the Singlet and Triplet Pathways. J. Am. Chem. Soc. 2013, 135, 1338–1348. 10.1021/ja307229e. [DOI] [PubMed] [Google Scholar]
- Au S.-M.; Huang J.-S.; Yu W.-Y.; Fung W.-H.; Che C.-M. Aziridination of Alkenes and Amidation of Alkanes by Bis(tosylimido)ruthenium(VI) Porphyrins. A Mechanistic Study. J. Am. Chem. Soc. 1999, 121, 9120–9132. 10.1021/ja9913481. [DOI] [Google Scholar]
- Bagchi V.; Kalra A.; Das P.; Paraskevopoulou P.; Gorla S.; Ai L.; Wang Q.; Mohapatra S.; Choudhury A.; Sun Z.; Cundari T. R.; Stavroloulos P. Comparative Nitrene-Transfer Chemistry to Olefinic Substrates Mediated by a Library of Anionic Mn(II) Triphenylamido-Amine Reagents and M(II) Congeners (M = Fe, Co, Ni) Favoring Aromatic over Aliphatic Alkenes. ACS Catal. 2018, 8, 9183–9206. 10.1021/acscatal.8b01941. [DOI] [Google Scholar]
- Hennessy E. T.; Liu R. Y.; Iovan D. A.; Duncan R. A.; Betley T. A. Iron-mediated intermolecular N-group transfer chemistry with olefinic substrates. Chem. Sci. 2014, 5, 1526–1532. 10.1039/C3SC52533C. [DOI] [Google Scholar]
- Ragaini F.; Penomi A.; Gallo E.; Tollari S.; Gotti C. L.; Lapadula M.; Mangiono E.; Cenini S. Amination of Benzylic C–H Bonds by Arylazides Catalyzed by CoII-Porphyrin Complexes: A Synthetic and Mechanistic Study. Chem. – Eur. J. 2003, 9, 249–259. 10.1002/chem.200390018. [DOI] [PubMed] [Google Scholar]
- In the DFT calculations presented in ref 15 regarding theformation of the neutral and anionic nitrene complexes we used the non-solvated [CoIII(TAMLred)]− and [CoIII(TAMLsq)] complexes asreference points. The relative Gibbs free energies for the formation of the nitrene adducts reported therein are therefore slightly less negative (ΔΔG298K° [Co(TAMLq)(NNs)] = 0.9 kcal mol–1, ΔΔG298K° [Co(TAMLq)(NNs)2]− = 0.4 kcal mol–1, ΔΔG298K° [Co(TAMLsq)(NNs)]− = 1.5 kcal mol–1). These small differences haveno impact on the interpretation of the presented data in this manuscriptor in ref 15.
- Sabenya G.; Gamba I.; Gómez J.; Clémancy M.; Frisch J. R.; Klinker E. J.; Blondin G.; Torelli S.; Que L. Jr; Mar-tin-Diaconescu V.; Latour J.-M.; Lloret-Fillol J.; Costat M. Ocatedral iron(IV)-tosylimido complexes exhibiting single-electron-oxidation reactivity. Chem. Sci. 2019, 10, 9513–9529. 10.1039/C9SC02526J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardhaman A. K.; Lee Y.-M.; Jung J.; Ohkubo K.; Nam W.; Fukuzumi S. Enhanced Electron Transfer Reactivity of a Nonheme Iron(IV)–Imido Complex as Compared to the Iron(IV)-Oxo Analogue. Angew. Chem., Int. Ed. 2016, 55, 3709–3713. 10.1002/anie.201600287. [DOI] [PubMed] [Google Scholar]
- Crandell D. W.; Muñoz S. B. III; Smith J. M.; Baik M.-H. Mechanistic study of styrene aziridination by iron(IV) nitrides. Chem. Sci. 2018, 9, 8542–8552. 10.1039/C8SC03677B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D.; Latifi R.; Kumar S.; Rybak-Akimova E. V.; Sainna M. A.; de Visser S. P. Rationalization of the Barrier Height for p-Z-styrene Epoxidation by Iron(IV)-Oxo Porphyrin Cation Radicals with Variable Axial Ligands. Inorg. Chem. 2013, 52, 7968–7979. 10.1021/ic4005104. [DOI] [PubMed] [Google Scholar]
- Lee Y.-M.; Kim S.; Ohkubo K.; Kom K.-H.; Nam W.; Fukuzumi S. Unified Mechanism of Oxygen Atom Transfer and Hydrogen Atom Transfer Reactions with a Triflic Acid-Bound Nonheme Manganese(IV)–Oxo Complex via Outer-Sphere Electron Transfer. J. Am. Chem. Soc. 2019, 141, 2614–2622. 10.1021/jacs.8b12935. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Tinoco A.; Steck V.; Fasan R.; Zhang Y. Cyclopropanations via Heme Carbenes: Basic Mechanism and Effects of Carbene Substituent, Protein Axial Ligand, and Porphyrin Substitution. J. Am. Chem. Soc. 2018, 140, 1649–1662. 10.1021/jacs.7b09171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.