

Abstract
Background
Sex‐specific differences in vasodilation are mediated in part by differences in cytosolic Ca2+ handling, but how variations in mitochondrial Ca2+ contributes to this effect remains unknown. Here, we investigated the extent to which mitochondrial Ca2+ entry via the MCU (mitochondrial Ca2+ uniporter) drives sex differences in vasoreactivity in resistance arteries.
Methods and Results
Enhanced vasodilation of mesenteric resistance arteries to acetylcholine (ACh) was reduced to larger extent in female compared with male mice in 2 genetic models of endothelial MCU ablation. Ex vivo Ca2+ imaging of mesenteric arteries with Fura‐2AM confirmed higher cytosolic Ca2+ transients triggered by ACh in arteries from female mice versus male mice. MCU inhibition both strongly reduced cytosolic Ca2+ transients and blocked mitochondrial Ca2+ entry. In cultured human aortic endothelial cells, treatment with physiological concentrations of estradiol enhanced cytosolic Ca2+ transients, Ca2+ buffering capacity, and mitochondrial Ca2+ entry in response to ATP or repeat Ca2+ boluses. Further experiments to establish the mechanisms underlying these effects did not reveal significant differences in the expression of MCU subunits, at either the mRNA or protein level. However, estradiol treatment was associated with an increase in mitochondrial mass, mitochondrial fusion, and the mitochondrial membrane potential and reduced mitochondrial superoxide production.
Conclusions
Our data confirm that mitochondrial function in endothelial cells differs by sex, with female mice having enhanced Ca2+ uptake capacity, and that these differences are attributable to the presence of more mitochondria and a higher mitochondrial membrane potential in female mice rather than differences in composition of the MCU complex.
Keywords: calcium, mitochondrial Ca2+ uniporter, sex differences, vasoreactivity
Subject Categories: Endothelium/Vascular Type/Nitric Oxide, Oxidant Stress, Physiology, Vascular Biology, Women
Nonstandard Abbreviations and Acronyms
- eNOS
endothelial nitric oxide synthase
- HAECs
human aortic endothelial cells
- MCU
mitochondrial Ca2+ uniporter
Clinical Perspective.
What Is New?
Recent evidence points toward sex differences in mitochondrial function, including metabolic activity and Ca2+ uptake and retention capacity.
Here, we demonstrate that mitochondrial Ca2+ uptake via the MCU (mitochondrial Ca2+ uniporter) promotes NO‐dependent endothelial function by affecting cytosolic Ca2+ levels.
Sex differences in Ca2+ handling and vasodilation correlated with mitochondrial superoxide production, copy numbers, mitochondrial length, and membrane potential.
What Are the Clinical Implications?
Sex differences in mitochondrial function may be leveraged for the development of therapies for vascular disease targeted to men or women.
Reports on sex differences in vasodilation and vasoconstriction in different vascular beds have provided evidence for regulation by the sex hormones estradiol and testosterone. 1 Overall, estradiol is regarded as a vasodilator whose effects are mediated in part by enhanced NO bioavailability. Estradiol reduces endothelial oxidative stress and asymmetric dimethylarginines, 2 , 3 thus leading to an increase in NO bioavailability. It also augments the intracellular free Ca2+ concentration ([Ca2+]i) that promotes the activity of endothelial nitric oxide synthase (eNOS), 4 through either Ca2+‐dependent phosphorylation of eNOS or its direct binding to Ca2+/calmodulin. 5 , 6 , 7 In small resistance vessels, dilation also depends on other endothelium‐dependent hyperpolarization factors, which is enhanced by activation of endothelial estradiol receptors. 8 These studies have provided valuable mechanistic insights but did not consider a potential role of sex differences in mitochondrial function that have been reported in other organs in the past decade. 9 , 10
Men and women differ significantly with respect to various aspects of mitochondrial function, 9 , 10 including in reactive oxygen species (ROS) production and Ca2+ uptake capacity. 11 , 12 One potential way by which sex differences in mitochondria may affect endothelial function is via mitochondrial Ca2+ entry or uptake capacity that affects Ca2+ levels in other compartments, such as the cytosol and endoplasmic reticulum of endothelial cells. 13 , 14
That mitochondrial Ca2+ handling in endothelium may be different between men and women can be speculated based on work in cardiac myocytes. 11 , 12 One study in rat cardiomyocytes reported a lower Ca2+ uptake capacity and higher opening thresholds in cells from female mice compared with cells from male mice. 11 Of note, this study was performed years before the molecular identity of the MCU (mitochondrial Ca2+ uniporter) complex, through which Ca2+ enters the mitochondrial matrix, was identified. 15 , 16
The MCU complex consists of the pore‐forming subunit mitochondrial calcium uniporter a (MCUa), its negative regulator mitochondrial calcium uniporter b (MCUb), and the additional regulatory subunits essential MCU regulator (EMRE), mitochondrial calcium uptake 1 (MICU1),2,3, and MCUR. 17 , 18 , 19 Given emerging evidence that the composition of the MCU complex is variable in different tissues and cell types leading to differences in the thresholds for Ca2+uptake and uniporter activation, 20 it is tempting to speculate that sex differences in mitochondrial Ca2+ handling may be driven by differential composition of the MCU complex. Although insights into the transcriptional and posttranscriptional regulation of MCU subunits have emerged, 20 , 21 , 22 , 23 no data on differences in MCU subunit expression between men and women are currently available, including for endothelial cells.
The aim of this study was to determine whether sex differences in mitochondrial function contribute to differences in endothelial function and vascular reactivity in mesenteric resistance arteries and, if so, to identify the underlying mechanisms. Here, we deployed 2 mouse models in which specifically endothelial cells were depleted of MCU activity and performed Ca2+ imaging and vasodilation studies in second‐order mesenteric resistance arteries. We also investigated the molecular mechanisms that underlie these differences in cultured human aortic endothelial cells (HAECs), including in the MCU complex, and other parameters that could potentially influence mitochondrial Ca2+ uptake, such as mitochondrial abundance, shape, and membrane potential. Collectively, our data provide evidence that sex‐specific differences in mitochondrial Ca2+ handling contribute to previously observed sex‐specific differences in vasoreactivity.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Reagents
The following antibodies and reagents were used in this study: ATP (Research Product International; A300030), mitoTracker Green FM (Life Technology; M7514), and tetramethylrhodamine methyl ester (Invitrogen; I34361), anti‐MCUa (Prestige Antibodies; HPA016480), anti‐EMRE (Sigma; HPA060340), anti‐MICU1 (Sigma; HPA037480), and anti‐MCUb (Abgent; C109B, AP12355b). Anti‐CoxIV (4850S) and anti‐GAPDH (2178), anti‐phosho‐eNOS (p‐eNOS Ser1177 and Thr495; 9571 and 9574), anti‐eNOS antibodies (9586), anti‐phospho pDRP‐1 (Ser616, D9A1; 4494), anti‐DRP1 (D6C7, 8570), and anti‐MFN2 (D2D10, 9482) were purchased from Cell Signaling and anti‐OPA1 (612806) from BD Biosciences. 17β‐Estradiol (E8875) and testosterone (T1500) were obtained from Sigma, and stock solutions in absolute ethanol were stored at −20 °C.
Mice
All experimental procedures were approved by the University of Iowa and the Iowa City Veterans Affairs Health Care System Institutional Animal Care and Use Committees and in accordance with institutional guidelines and with the standards for the care and use of laboratory animals of the Institute of Laboratory Animal Resource, National Academy of Science. To study the effects of deletion of MCU in endothelial cells, we used mice in which exons 5 and 6 are flanked by loxP sites (Jackson Laboratories, 029817). In a second transgenic model, the mitochondria‐targeted inhibitor peptide of the Ca2+/calmodulin‐dependent kinase II (CaMKII), mitochondrially targeted peptide inhibitor of CaMKII (mtCaMKIIN), was expressed in a transgenic model (e‐mtCaMKIIN). Of note, the inhibitor peptide CaMKIIN is the most potent (IC50 ≈10 nmol/L) and specific inhibitor (no measurable activity against Ca2+/calmodulin‐dependent protein kinase IV or protein kinase C) and blocks all isoforms of CaMKII. 24 , 25 These mice were generated by cloning cDNA for the hemagglutinin‐tagged CaMKII inhibitor peptide CaMKIIN 25 fused with the mitochondria targeting Cox8‐palmitoylation sequence into a construct containing the CX‐1 promoter and a floxed enhanced green fluorescent protein sequence. These mice express enhanced green fluorescent protein in all tissues before cre recombination. Mice double‐transgenic for floxed MCU and cre or for mtCaMKIIN and cre were generated by crossing to Tie2‐CreERT2 mice, 26 followed by injection of tamoxifen (2 mg per mouse) on day 1 to 5 and day 15 to 19 to produce MCU deletion (e‐MCU−/−). mtCaMKIIN expression by cre recombination was achieved as described above for e‐MCU−/− mice. Littermate control mice carried the cre transgene and underwent tamoxifen treatment. Mice were housed at a controlled temperature (23 °C) and a dark/light cycle of 12 hours. All mice had free access to water and standard rodent chow. Experiments were performed no earlier than 4 weeks after cre recombination was initiated. The samples from all animals underwent the same protocols, and no data were excluded.
Vasoreactivity
Vascular responses in second‐order mesenteric arteries were measured as described previously. 27 Briefly, mesenteric arteries were isolated and cleaned of adherent fat. They were cannulated with micropipettes in an organ bath filled with Krebs buffer and pressurized to 40 mm Hg. Lumen diameter was recorded using a video camera and an electronic dimension analyzer. After a 30‐minute equilibration period, arteries were submaximally preconstricted using phenylephrine (1.0–3.0 μmol/L). After constrictions stabilized, cumulative concentration curves for the responses to ACh or sodium nitroprusside were generated. Relaxation responses were expressed as the percentage decrease in tension from preconstriction values. NO‐dependent vasoreactivity were estimated using mesenteric segments that were preincubated with the nitric oxide synthase (NOS) inhibitor Nω‐Nitro‐l‐arginine (100 μmol/L) for 30 minutes before being stimulated with ACh or sodium nitroprusside.
Ca2+ Imaging in En Face Preparations of Mesenteric Arteries
The imaging procedure was adapted from a previous report by Wilson and colleagues. 28 , 29 Following euthanasia, the mesenteric vessel bed was removed and placed in PSS (145 mmol/L NaCl, 4.7 mmol/L KCl, 2.0 mmol/L MOPS, 1.2 mmol/L NaH2PO4, 5.0 mmol/L glucose, 2.0 mmol/L pyruvate, 0.02 mmol/L EDTA, 1.17 mmol/L MgCl2, 2.0 mmol/L CaCl2, adjusted to pH 7.4 with NaOH). Second‐order mesenteric arteries were cleaned of connective tissue and fat, removed from the mesenteric bed, opened using microscissors, and pinned with the endothelial side‐up on glass bottom dishes coated with Sylgard. The endothelial‐cell layer was then preferentially loaded with the Ca2+ indicators Fura‐2 AM and Rhod‐2 (5 and 10 μmol/L, respectively with 0.04% Pluronic F127) at 37 °C for 40 minutes. Following this incubation, the preparations were rinsed in PSS twice for 5 minutes. Following a 30‐minute equilibration period, real‐time shifts in Fura‐2 AM and Rhod‐2 fluorescence were recorded at room temperature every 5 seconds for periods of 5 minutes, in the luminal‐most cells after ACh was added (10−6 mol/L) with a Nikon Eclipse Ti2 inverted light microscope. Approximately 50 endothelial cells were visualized in the resulting image field. Ca2+ recordings were obtained from 3 separate vessels per mouse, in samples from 3 mice. Three or 4 arteries per mouse from 4 mice per condition were used.
Cell Culture
HAECs were cultured in endothelial cell medium‐phenol red free (ScienceCell; 1001‐prf) supplemented with 25 mL (5%) fetal bovine serum (ScienceCell; 0025), 5 mL endothelial cell growth supplement (ScienceCell; 1052), and 5 mL penicillin/streptomycin solution (ScienceCell; 0503) at 37 °C in a humidified 95% air and 5% CO2 incubator. Subconfluent cells at passages 4 to 7 were used. These cells were initially cultured for 3 passages in medium without sex hormones. Thereafter, sex hormones were added at physiological concentrations for 72 hours. Stock solutions were added to a final concentration of 10 nmol/L estradiol or 20 nmol/L testosterone. Nontreated cells (with addition of diluent without sex hormones) were used as controls.
Mitochondrial Ca2+ Imaging in HAECs
HAECs were seeded at equal density onto 35‐mm glass‐bottom microwell dishes (MatTek) and infected with adenovirus expressing the mitochondria‐targeted ratiometric Ca2+ indicator protein Pericam and then treated with sex hormones for 72 hours. Pericam fluorescence was determined in Tyrode solution (140 mmol/L NaCl, 10 mmol/L glucose, 5.4 mmol/L KCl, 1.8 mmol/L CaCl2, 2.0 mmol/L MgCl2, 1.2 mmol/L KH2PO4, 5 mmol/L HEPES, pH 7.4 with NaOH). Imaging was performed at room temperature, using a customized Nikon Eclipse Ti2 Inverted microscope. Pericam was excited at 405 and 480 nm, and its emission was recorded at 535 nm. ATP (100 μmol/L) was added by micropipet (10 μL) to trigger mitochondrial Ca2+ uptake. Recordings were performed every 5 seconds for at least 10 minutes. Pericam signals were quantified using ImageJ. The rise in amplitude above baseline and the area under the curve (AUC) after ATP application were calculated. Peak amplitude (R) was calculated using Rpeak−Rbaseline. The AUC and the peak amplitude were determined by subtracting out the baseline fluorescence signal before addition of the agonist.
Cytosolic Ca2+ Imaging in HAECs
HAECs were cultured at equal density on 35‐mm glass‐bottom microwell dishes in the presence of sex hormones and incubated with the ratiometric indicator dye Fura‐2 AM (1 μmol/L; Invitrogen) in endothelial cell medium‐phenol red free for 10 minutes at 37 °C. For recording of cytosolic Ca2+ transients, images were acquired continuously for 90 seconds before and at least 240 seconds after carbonilcyanide p‐triflouromethoxyphenylhydrazone (FCCP) treatment. Additionally, images were acquired continuously for 90 seconds before ATP treatment, 10 minutes after ATP treatment, and 5 minutes after FCCP treatment. The cells were excited alternately at 340 and 380 nm. Fluorescence signal intensity was acquired at 510 nm. Cells with no response, with multiple peaks after addition of an agonist, or whose amplitudes did not show return back to baseline were excluded. Data are presented as peak amplitude or AUC. For the analysis, the baseline fluorescence before addition of the agonist (ATP or FCCP) was subtracted.
Ca2+ Retention Assay
Calcium Green‐5N was used to monitor extramitochondrial Ca2+ in hormone‐treated, digitonin‐permeabilized HAECs (2×106 cells). Cells were trypsinized, resuspended in respiration buffer, counted using a Z1 Coulter Particle counter, and 2×106 cells were used for each treatment group. Mitochondrial Ca2+ uptake was recorded for cells in a 96‐well plate. Each well contained 100 μL respiration buffer (125 mmol/L KCl, 2 mmol/L K2HPO4, 1 mmol/L MgCl2, 20 mmol/L HEPES, 5 mmol/L glutamate, and 5 mmol/L malate, pH 7.4) supplemented with 1% digitonin and 1 μmol/L Calcium Green‐5N (Invitrogen) at 30 °C. Calcium Green‐5N fluorescence was monitored at 500 nm excitation and 532 nm emission, after the addition of CaCl2 (10 μmol/L free Ca2+).
Tetramethylrhodamine Methyl Ester Imaging in HAECs
The mitochondrial membrane potential was measured using tetramethylrhodamine methyl ester. Hormone‐treated HAECs cultured on 35‐mm glass‐bottom microwell (matTek) dishes were incubated in PBS with 20 nmol/L tetramethylrhodamine methyl ester and 200 nmol/L mitoTrackerTM Green FM (Invitrogen) for 20 minutes. Cells were rinsed once with endothelial cell medium‐phenol red free media. Images were acquired using a customized Nikon Eclipse Ti2 inverted microscope fitted with a ×40 objective. For each treatment group, 8 to 10 separate microscopic fields were imaged. Imaging was performed with the same parameters in each experiment. The mitochondria per each cell were selected as the area of interest and analyzed. The fluorescence signal was quantified using the ImageJ software and expressed as arbitrary units.
Quantitative Real‐Time Polymerase Chain Reaction
For quantification of mitochondrial copy number, total DNA was isolated using the DNeasy Kit (Qiagen; 69506). To determine mRNA levels of MCU subunits, total RNA was isolated from HAECs using the Micro RNeasy Kit (Qiagen, 74104) and reverse transcribed using RTIII (Invitrogen). cDNA was amplified by quantitative polymerase chain reaction using 1‐μg RNA and SuperScript VILO MasterMix in PowerSYBR Green and an ABI real‐time polymerase chain reaction machine. The following primers, purchased from Integrated DNA Technologies were used: MCUa (forward: 5′‐gca gaa ttt ggg agc tgt tt‐3′ reverse: 5′‐gtc aat tcc ccg atc ctc tt‐3′), MCUb (forward: 5′‐ggt ctt ccc ttg gta aca ct‐3′ reverse: 5′‐gtt gcc atc tgc tgt gaa ga‐3′), MICU1 (forward: 5′‐gag gca gct caa gaa gca ct‐3′ reverse: 5′‐caa aca cca cat cac aca cg‐3′), MICU2 (forward: 5′‐ggc agt ttt aca gtc tcc gc‐3′ reverse: 5′‐aag agg aag tct cgt ggt gtc‐3′), EMRE (forward: 5′‐tgt cgg gac act cat tag ca‐3′ reverse: 5′‐gct gat agg gaa ggc aga ga‐3′), and β‐actin (forward: 5′‐ata gca cag cct gga tag caa cgt ac‐3′ reverse: 5′‐cac ctt cta caa tga gct gcg tgt g‐3′), mitochondrial tRNA‐Leu (forward: 5′‐cac cca aga aca ggg ttt gt‐3′ reverse: 5′‐tgg cca tgg gta tgt tgt ta‐3′), and NADH‐ubiquinone oxidoreductase chain 1 (forward: 5′‐ata ccc atg gcc aac ctc ct‐3′ reverse: 5′‐ggg cct ttg cgt act tgt at‐3′).
Cell Lysis and Fractionation
For whole‐cell lysates, cells were lysed in RIPA buffer (20 mmol/L Tris, 150 mmol/L NaCl, 5 mmol/L EDTA, 5 mmol/L EGTA, 1% Triton X‐100, 0.5% deoxycholate, 0.1% SDS, pH 7.4) supplemented with protease (Mini cOmplete; Roche) and phosphatase inhibitors (PhosSTOP; Roche). Lysates were sonicated and debris pelleted by centrifugation at 10 000g for 10 minutes at 4 °C. For the preparation of mitochondrial fractions, cells were washed in PBS and in MSEM buffer (5 mmol/L MOPS, 70 mmol/L sucrose, 2 mmol/L EGTA, 220 mmol/L Mannitol, pH 7.5 with protease inhibitors) before homogenization in cold MSEM buffer using ≈50 strokes in a Potter‐Elvehjem homogenizer. Nuclei and cell debris were pelleted by centrifugation at 600g for 5 minutes at 4 °C. Mitochondria were separated from the cytosolic fraction by centrifugation at 8000g for 10 minutes at 4 °C. Protein concentrations were determined by Pierce BCA protein assay (Thermo Scientific).
Immunoblotting
Equivalent amounts of protein (50–60 μg) from the mitochondrial fraction were separated on NuPAGE 4% to 12% Bis‐Tris gels (Life Technologies) and transferred to polyvinyl difluoride membranes (BioRad). Membranes were blocked in 5% BSA for 60 minutes and incubated overnight at 4 °C with primary antibodies. Blots were washed 3 times for 10 minutes with 0.05% Tween‐20 in TBS and incubated for 60 minutes at room temperature with secondary antibodies at a dilution of 1:5000. Blots were developed with ECL chemiluminescent substrate, scanned, and analyzed using ImageJ software.
Detection of Mitochondrial ROS
Mitochondrial ROS were measured in live cells using the dihydroethidium derivative, mitoSOX red (5 μmol/L). HAECs had been transduced with control or with mtCaMKIIN adenovirus (MOI 25) for 72 hours together with hormone treatment. Cells were loaded with mitoSOX for 20 minutes at 37 °C and then washed with HBSS and imaged via Nikon Eclipse Ti2 under ×40 objective. Analysis was performed by measuring the mitoSox fluorescence per visual field with NS Elements software (Nikon). Data are presented as relative fluorescent units.
Immunofluorescent Labeling in Mouse Aortas
Mouse thoracic aorta was fixed by immersion in 4% paraformaldehyde/PBS. After 48 hours, the aorta was cryopreserved at −80 °C in Tissue Freezing Medium. Ten‐micrometer frozen sections were cut, postfixed on Superfrost Plus glass slides, and washed in PBS. They were blocked in 5% nonfat dry milk. After washing with PBS for 10 minutes, the sections were incubated overnight with primary antibody for hemagglutinin‐tagged mtCaMKIIN (anti‐hemagglutinin). After rinsing in PBS, sections were incubated for 2 hours with biotinylated secondary antibodies diluted 1:250 in PBS. Sections were then washed with PBS for 15 minutes and incubated with Alexa 568‐conjugated secondary. After rinsing in PBS, the sections were counterstained with ToPro‐3 iodide to visualize nuclei and mounted in Vectashield. Negative controls without primary antibody were performed in every experiment. Images were captured with Zeiss LSM 710 m microscope.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism (version 9.0.0; GraphPad Software). Data are expressed as mean±SEM. Normal data distribution was assessed using the D‐Agostino‐Pearson omnibus normality test. When the distribution was normal, the data were analyzed using a 2‐tailed Student t test or ANOVA followed by the appropriate correction. Otherwise, the Mann‐Whitney test or the Kruskal‐Wallis test was used. For data sets with samples sizes of <7, a Kruskal‐Wallis test was used. When data of experiments were normalized to a control condition, a Friedman test was used for the analysis of the data set. Dunn multiple comparison tests or Tukey test were used for post hoc pairwise comparisons after Kruskal‐Wallis and Friedman or ANOVA test, respectively. A 2‐way repeated measure ANOVA was performed to assess concentration curves and to evaluate the effect of MCU reduction on vasodilation or constriction in an arterial segment. A 3‐way repeated measure ANOVA was performed to determine the interactions between sex and genotype. Two‐way ANOVA was used for other analyses. A probability value of <0.05 was considered significant.
Results
In Mesenteric Resistance Arteries, Sex Differences in Vasodilation Depend on Mitochondrial Ca2+ Uptake
We studied vasodilation and vasoconstriction of second‐order mesenteric arteries in 2 different in vivo models of reduced mitochondrial Ca2+ entry in the endothelium. First, we tested mice in which MCU had been deleted selectively in the endothelium (MCU reduction model). Consistent with previous reports, in wild‐type mice, maximal vasodilation in response to ACh was stronger in arteries from female versus male mice, with no change in the half maximal effective concentration of Ach (Figure 1A through 1C, Table S1). The deletion of MCU in e‐MCU−/− mice reduced the maximal dilation in mesenteric arteries of female mice only. This finding suggests that sex differences in mitochondrial Ca2+ entry are present and modulate endothelium‐dependent vasodilation. In contrast, the response to sodium nitroprusside as well as vasoconstriction in response to KCl were not significantly impacted by MCU deletion (Figure 1D and 1E). We also ascertained that, whereas addition of the NOS inhibitor Nω‐Nitro‐L‐arginine strongly reduced ACh‐triggered vasodilation in wild‐type mice (Figure S1A), its effect in e‐MCU−/− mice was significantly smaller. As a second in vivo model, we used mice in which activity of the MCU activator CaMKII (Ca2+/calmodulin‐dependent kinase II), which is expressed in the mitochondrial matrix, was blocked specifically in the endothelium by expressing a mitochondria‐targeted inhibitor peptide (CaMKIIN) in this tissue (MCU inhibition model, e‐mtCaMKIIN). 30 , 31 , 32 Vasoreactivity studies in this model largely recapitulated our findings from direct deletion of MCU (Figure 2A through 2E, Figure S1B and Table S1): expression of mtCaMKIIN reduced endothelium‐dependent vasodilation, and it did so to a greater extent in female versus male mice (Figure 2A and 2C). These findings demonstrate that mitochondrial Ca2+ entry via MCU drives sex differences in vasodilation and that endothelial loss of MCU leads to impaired release of NO.
Figure 1. In mesenteric resistance arteries from female mice, MCU (mitochondrial Ca2+ uniporter) deletion reduces vasodilation to a greater extent than in male mice.
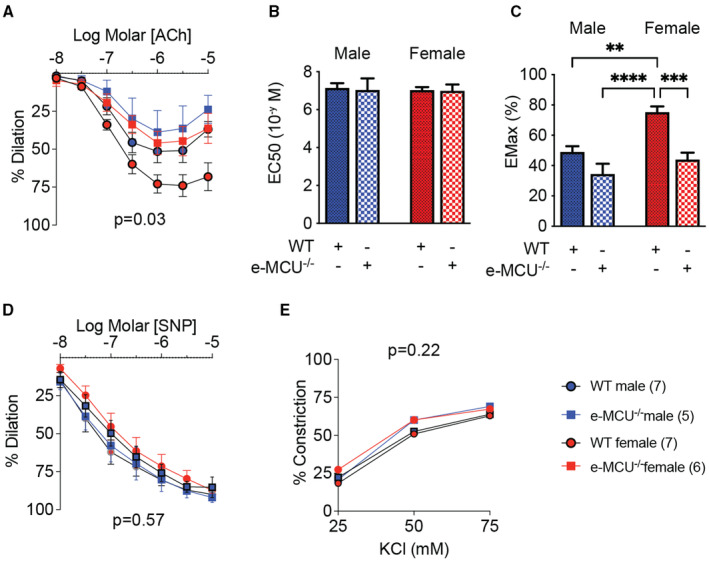
A, Acetylcholine (Ach)‐induced vasodilation of second‐order mesenteric arteries from male and female mice with endothelial MCU deletion (e‐MCU−/−), and in littermate control (wild‐type [WT]) mice. B, EC50 of vasodilation to Ach as in (A) and (C) and EMax of vasodilation to Ach as in (A). (D) Endothelium‐independent vasodilation to sodium nitroprusside (SNP). E, Vasoconstriction to KCl. Analysis by repeated‐measure 2‐way ANOVA (A, D, and E) and 2‐way ANOVA (B and C). No significant interactions between sex and genotype were seen for (B and C). P values for source of variations in treatment groups as shown. **P<0.01, ***P<0.005, ****P<0.001. EC50 indicates half maximal effective concentration; EMax, maximal effect; KCl, potassium chloride.
Figure 2. In mesenteric resistance arteries from female mice, inhibition of MCU (mitochondrial Ca2+ uniporter) by treatment with mtCaMKIIN reduces vasodilation to a larger extent than in male mice.
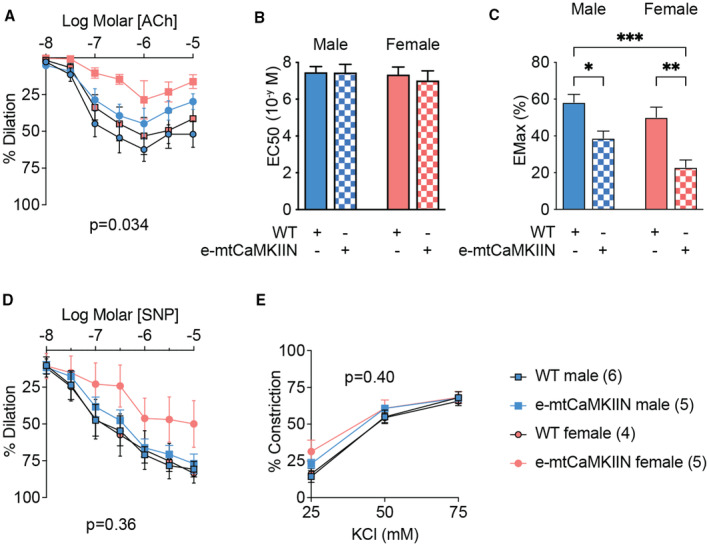
A, Acetylcholine (ACh)‐induced vasodilation of second‐order mesenteric arteries from male and female mice with endothelium‐specific expression of mitochondria‐targeted CaMKIIN (e‐mtCaMKIIN) and in littermate control (wild‐type [WT]) mice. B, EC50 of vasodilation to Ach as in (A). C, EMax of vasodilation to ACh as in (A). D, Endothelium‐independent vasodilation to sodium nitroprusside (SNP). E, Vasoconstriction to KCl. Analysis by repeated‐measure 2‐way ANOVA (A, D, and E) and 2‐way ANOVA (B and C). No significant interactions between sex and genotype were seen for Figure 1B and 1C. P values for source of variations in treatment groups as shown. *P<0.05, **P<0.01, ***P<0.005. EC50 indicates half maximal effective concentration; EMax, maximal effect; mtCaMKIIN, mitochondrially targeted peptide inhibitor of CaMKII; KCl, potassium chloride.
To confirm that mtCaMKIIN effectively blocks mitochondrial Ca2+ entry in the endothelium, we performed Ca2+ imaging with Rhod‐2 in en‐face preparations of mesenteric arteries. As anticipated, mitochondrial Ca2+ uptake by Rhod‐2 imaging following the addition of ACh was strongly reduced in arteries from e‐mtCaMKIIN mice (Figure S2). Next, we tested the effects of MCU inhibition on cytosolic Ca2+ transients by Fura‐2 imaging. In arteries from wild‐type mice, the endothelial Ca2+ transients were larger than in samples from female versus male mice (Figure 3A). The inhibition of MCU by mtCaMKIIN significantly reduced the cytosolic Ca2+ transients with greater differences in female mice. The peak amplitude and AUC were reduced in male mice and female mice in which mtCaMKIIN was expressed. In particular, the peak amplitude was lowered to a greater extent in arteries from female mice (Figure 3B and 3C). These data demonstrate that sex differences in vasodilation correlate with cytosolic Ca2+ levels and imply that inhibition of vasodilation caused by a reduction of MCU activity is driven by reduced cytosolic Ca2+.
Figure 3. In mesenteric resistance arteries, MCU (mitochondrial Ca2+ uniporter) inhibition reduces cytosolic Ca2+ transients to a greater extent in female than in male mice.
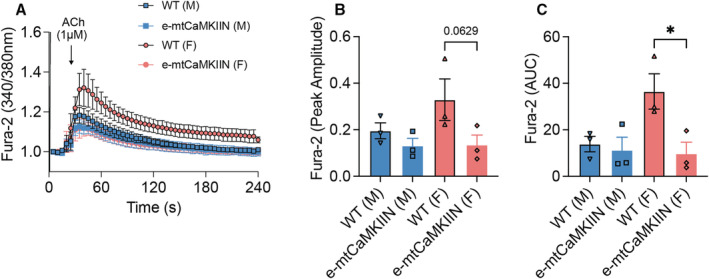
En‐face endothelial Fura‐2 imaging of second‐order mesenteric arteries. A, Representative cytosolic Ca2+ transients measured as Fura‐2 traces after addition of 1 μmol/L acetylcholine (Ach) to samples from wild‐type (WT) and e‐mtCaMKIIN mice. B and C, Quantification of tracings as in (A), as (B) peak amplitude and (C) area under the curve (AUC). *P<0.05, by Kruskal‐Wallis test with group‐wise comparison as indicated. N=3 mice per sex/genotype. F indicates female; M, male; mtCaMKIIN, mitochondrially targeted peptide inhibitor of CaMKII.
In Human Endothelial Cells, Mitochondrial Ca2+ Entry Is Enhanced by Estradiol Treatment
The above‐described ex vivo studies were complemented by mechanistic studies in cultured HAECs. This cell type was selected to ascertain that our observations in mice can be extended to human physiology.
First, we tested the capacity for mitochondrial Ca2+ uptake using Calcium Green 5N. Repeated addition of Ca2+ boluses (10 μmol/L) revealed higher Ca2+ uptake capacity in HAECs (ie, cytosolic [Ca2+]) following treatment with estradiol versus testosterone (Figure 4A). Next, we evaluated the effect of sex hormones on cytosolic Ca2+ transients by Fura‐2 recording. Consistent with this finding, Ca2+ release from mitochondria following their depolarization by FCCP was significantly elevated in estradiol‐treated HAECs (Figure 4B and 4C). Because ACh elicited only minimal Ca2+ transients in cultured HAECs, we recorded the more robust responses to ATP. This treatment triggered Ca2+ peaks of similar amplitude in estradiol‐ and testosterone‐treated cells (Figure 4D and 4E). Mitochondrial Ca2+ release was then induced by depolarization with FCCP; this experiment confirmed that the capacity for mitochondrial Ca2+ retention was higher in samples treated with estradiol (Figure 4D and 4F).
Figure 4. In human aortic endothelial cells (HAECs), mitochondrial Ca2+ uptake capacity is increased by treatment with sex hormones.
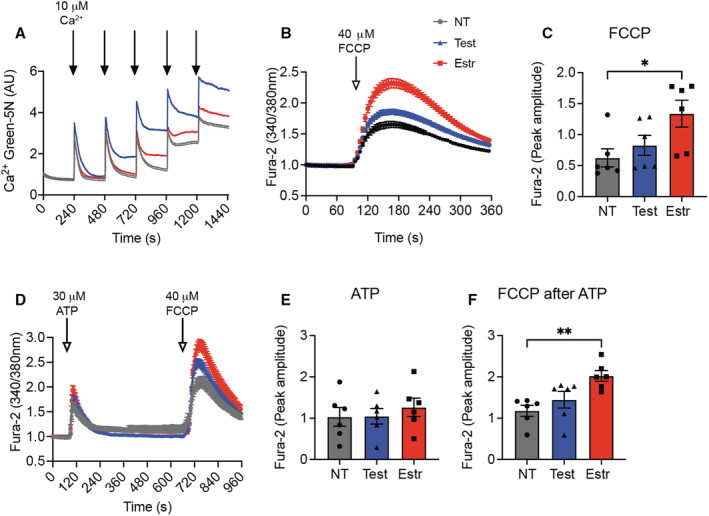
A, Mitochondrial Ca2+ uptake, as assessed by Ca2+ Green 5N assay, in HAECs treated with testosterone, estradiol, or diluent (nontreated [NT]) for 72 hours. Treatment with digitonin (0.001%), Ca2+ (1 μmol/L). n=6 independent experiments. B, FCCP‐evoked cytosolic Ca2+ transients, measured using Fura‐2, in HAECs treated with testosterone, estradiol, or diluent (NT) for 72 hours. C, Peak amplitude for (B). n=6 independent experiments. D, Cytosolic Ca2+ transients, measured by Fura‐2 imaging, in HAECs treated with testosterone, estradiol, or diluent (NT) for 72 hours, in response to ATP and subsequently to FCCP. E, Peak amplitude for the response to ATP in (D). F, Peak amplitude for the response to FCCP in (D). n=6 independent experiments. *P<0.05, **P<0.01 by Kruskal‐Wallis test. Estr indicates estradiol; FCCP, carbonilcyanide p‐triflouromethoxyphenylhydrazone; Test, testosterone.
We also directly tested mitochondrial Ca2+ entry by imaging with mitochondria‐targeted Pericam (Figure 5A). Comparison of both the peak amplitude and AUC revealed that mitochondrial Ca2+ entry in response to ATP was higher in cells pretreated with estradiol versus testosterone, as demonstrated by comparison of both AUC and peak amplitude (Figure 5B and 5C).
Figure 5. In human aortic endothelial cells (HAECs), mitochondrial Ca2+ entry is enhanced by treatment with estradiol.
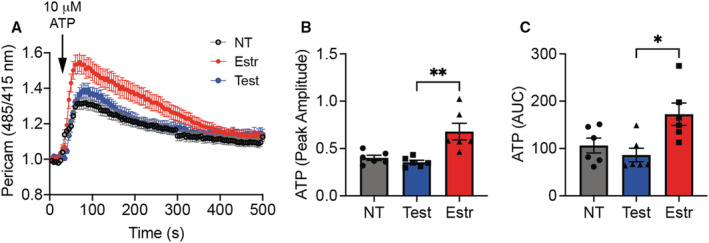
A, Mitochondrial Ca2+ transients, as assessed by Pericam imaging, in response to ATP (10 μmol/L), in HAECs treated with testosterone, estradiol, or diluent (nontreated [NT]) for 72 hours. B, Peak amplitude for the response to ATP in (A). C, Area under the curve (AUC) after addition of ATP as in (A). *P<0.05, **P<0.01 by Kruskal‐Wallis test. n=6 independent experiments. Estr indicates estradiol; Test, testosterone.
Given that increases in cytosolic Ca2+ levels can affect eNOS activation, we immunoblotted for activated eNOS (p‐Ser1177). Specifically, HAECs were pretreated with estradiol or testosterone and then stimulated with ATP. Following ATP administration, eNOS phosphorylation was higher in cells pretreated with estradiol when compared with untreated than in those treated with testosterone (Figure S3A and S3B). These data provide additional evidence that estradiol has a greater effect on the induction of NO release than does testosterone, consistent with our in vivo data indicating larger maximal endothelium‐dependent vasodilation in female mice (Figures 1 and 2).
Sex Hormones Do Not Significantly Affect the MCU Subunit Composition
Next, we tested whether treatment with sex hormones affects the expression of mRNAs encoding MCU subunits MCUa, MCUb, EMRE, MICU1. We found that HAECs treated with estradiol or testosterone for 72 hours did not differ with regard to transcript levels of MCUa, MCUb, MICU1, MICU2, or EMRE (Figure S4A through S4E). Also, no differences were seen at earlier time points (data not shown), and immunoblots did not reveal significant differences in levels of the MCU subunits at the protein level (Figure 6A through 6E). These data imply that the sex differences in Ca2+ uptake are not because of differences in the composition of the MCU complex.
Figure 6. In human aortic endothelial cells (HAECs), levels of MCU (mitochondrial Ca2+ uniporter) subunit proteins are not altered by sex hormones.
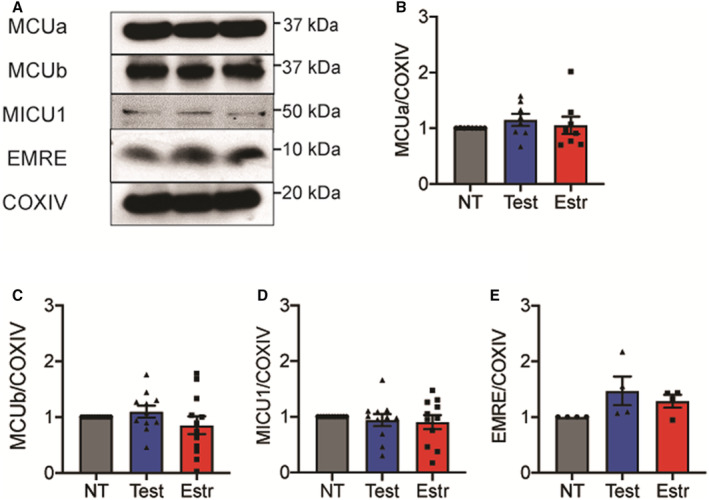
A, Representative immunoblots for MCU subunits MCUa, MCUb, EMRE, and MICU1, as well as the housekeeping protein COXIV (control), in mitochondrial fractions of HAECs treated with testosterone, estradiol, or diluent (nontrearted [NT]) for 72 hours. B through E, Quantification of levels of the (B) MCUa, (C) MCUb, (D) MICU1, and (E) EMRE proteins, normalized to COXIV. N=8 (B), 12 (C), 11 (D), 4 (E) biological replicates. Analysis by Friedman test. COXIV indicates cytochrome c oxidase subunit IV; EMRE, essential MCU regulator; Estr, estradiol; MCUa, mitochondrial calcium uniporter a; MCUb, mitochondrial calcium uniporter b; MICU1, mitochondrial calcium uptake 1; NT, nontrearted; Test, testosterone.
Mitochondrial Membrane Potential and Mitochondrial DNA Copy Number Are Increased by Treatment With Estradiol
Apart from the abundance and composition of MCU, several other factors influence mitochondrial Ca2+ entry and retention. Previous studies revealed that in pancreatic β‐cells, Ca2+ uptake is promoted by a high mitochondrial membrane potential, high mitochondrial DNA copy number, and the presence of elongated mitochondrial networks. 33 , 34 We analyzed mitochondrial mass in HAECs pretreated with sex hormones by both imaging with mitoTracker (Figure 7A) and quantitative polymerase chain reaction for the mitochondria‐encoded genes tRNA and NADH‐ubiquinone oxidoreductase chain 1 (Figure 7D and 7E, Figure S5A and S5B). The mitochondrial mass was significantly greater in HAECs pretreated with estradiol than in counterparts treated with testosterone and in nontreated cells. These data were further supported by analysis of the mitochondrial membrane potential (Figure 7A and 7B). Moreover, mitochondria of HAECs treated with estradiol were longer than those in the control samples (form factor analysis; Figure 7F). Immunoblots for dynamin‐related protein 1, optic atrophy type 1, and mitofusin 2 (MFN2) that modulate mitochondrial fission and fusion, respectively, provided additional insight (Figure 7G through 7I). Increased optic atrophy type 1 levels and a similar trend for MFN2 were seen with estradiol treatment. Lastly, we assessed mitochondrial ROS levels, which were increased in HAECs incubated with testosterone compared with estradiol as well as control conditions (Figure 7J). Of note, adenoviral expression of mtCaMKIIN in HAECs concomitantly with sex hormone treatment reduced mitoSox levels compared to those seen in control cells. These data support the notion that the sex‐hormone‐mediated modulation of mitochondrial Ca2+ entry into endothelial cells is driven by mitochondrial mass, form factor, and membrane potential. They also suggest that the modulation of mitochondrial Ca2+ entry by sex hormones impacts on ROS production, which may further modulate vasoreactivity.
Figure 7. In human aortic endothelial cells (HAECs), mitochondrial DNA copy number, shape, and membrane potential are modulated by sex hormones.
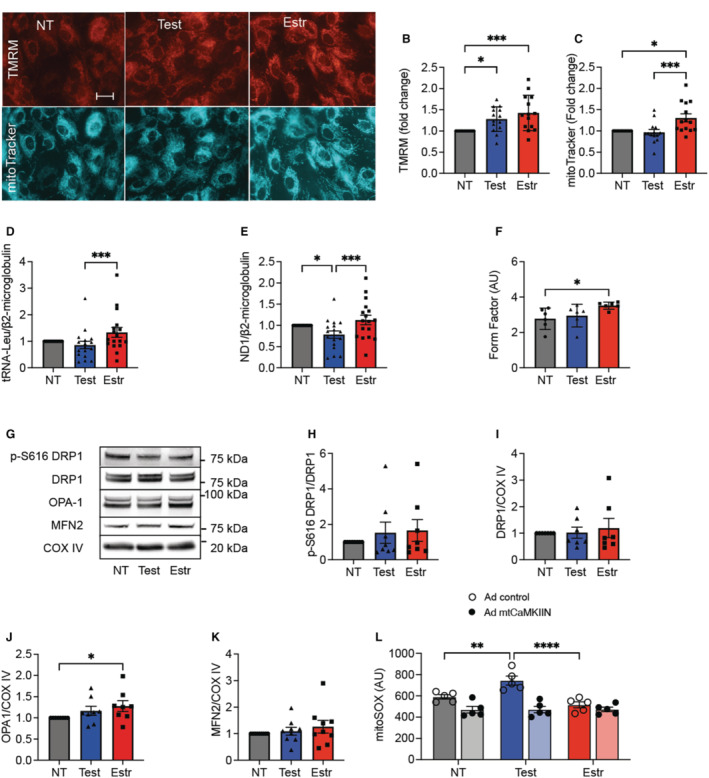
A, Representative images of tetramethylrhodamine methyl ester (TMRM) (mitochondrial membrane potential) and mitoTracker signal in HAECs treated with sex hormones or diluent (nontreated [NT]) for 72 hours. Scale bar=50 μm, ×20. B, Quantification of the TMRM fluorescence. C, Quantification of mitoTracker fluorescence. B and C, Each data point indicates 1 independent experiment (n=14). D and E, Quantitative polymerase chain reaction for genes encoded by mitochondrial DNA normalized to β2‐microglobulin: (D) tRNA‐Leu and (E) for NADH‐ubiquinone oxidoreductase chain 1. n=16 (D), n=17 (E) biological replicates. F, Quantification of mitochondrial form factor. Cells labeled with mitoTracker were analyzed. G through K, Representative immunoblot and quantification for active phosphorylated (p‐)DRP1, DRP1, OPA1, and MFN2 in mitochondrial fractions treated with sex hormones or diluent (NT) for 72 hours. COXIV as loading control. n=8 (H through J) and 9 (K) independent immunoblots. (L) Mitochondrial superoxide levels by mitoSOX fluorescence in HAECs transduced with adenovirus expressing mtCaMKIIN or control and treated with sex hormones or diluent (NT) for 72 hours. n=5 independent experiments. *P<0.05, **P<0.01, ***P<0.005, ****P<0.001 by Kruskal‐Wallis test (F), Friedman test (B through E and H through K), 2‐way ANOVA (L). COXIV indicates cytochrome c oxidase subunit IV; DRP1, dynamin‐related protein 1; Estr, estradiol; MFN2, mitofusin 2; mtCaMKIIN, mitochondrially targeted peptide inhibitor of CaMKII; OPA1, optic atrophy type 1; Test, testosterone.
Discussion
In this study of mitochondria‐regulated sex differences in endothelial function, we present 4 major findings on the role of mitochondrial Ca2+ uptake. First, in second‐order mesenteric resistance arteries, inhibition of MCU was sufficient to decrease ACh‐triggered cytosolic Ca2+ transients, and these effects were more pronounced in arteries from female versus male mice. Second, in these arteries, MCU depletion abolished differences in vasodilation in female versus male mice. Third, in HAECs, treatment with estradiol (at physiologic concentrations) led to enhanced mitochondrial Ca2+ entry and Ca2+ storage capacity. Fourth, levels of MCU subunits were unaffected by physiological levels of estradiol, yet mitochondrial DNA content, mitochondrial mass, and mitochondrial fusion were enhanced. We posit that these latter findings provide an explanation for the observed sex differences in endothelial Ca2+ handling in endothelium. Our studies also reveal increased Ca2+‐dependent mitochondrial production of superoxide after testosterone pretreatment.
In recent years, several studies reported on sex‐related divergences in mitochondria 10 and revealed tissue‐ and cell type–dependent diversity in mitochondrial structure, density, and function. Most studies of sex differences in mitochondrial function were performed in organs in which energy production strongly depends on oxidative phosphorylation (eg, liver, 12 , 35 skeletal muscle,36 and heart 37 , 38 , 39 ). Therefore, they may not be applicable to endothelial cells, which depend mainly on glycolysis for energy production. 40
Although previous studies of sex differences involved detailed phenotyping, the mechanisms underlying the sex differences were dissected only rarely. 36 , 38 , 39 , 41 , 42 , 43 In the studies that tested the effects of select proteins, most targets were downstream effectors of pathways 37 rather than upstream regulators with broad effects on mitochondrial function like Ca2+ handling. Few studies focused on pathways regulating sex differences in the endothelium; in brain microvascular endothelial cells, estradiol increased mitochondrial cytochrome c transcription via its action on the estradiol receptor α. 44 Consistent with such a mechanism, Tcherepanova and colleagues reported that in cerebral blood vessels, estradiol receptor α is coactivated by peroxisome proliferator‐activated receptor‐gamma coactivator 1α, the master regulator of mitochondrial biogenesis. 45 This interaction could potentially be responsible for the higher mitochondrial mass in samples from female mice but was not directly tested in this study.
Mitochondrial Ca2+ uptake via the MCU complex is fundamental to the regulation of mitochondrial function. It controls the activities of the tricarboxylic acid cycle 45 , 46 and the electron transport chain, as well as opening of the transition pore and the production of ROS. 21 , 46 In contrast to cardiac myocytes, in which 30% of the cytoplasmic volume is occupied by mitochondria, endothelial cells have a low mitochondrial content (≈5%). Nonetheless, mitochondria account for 25% of the Ca2+ storage capacity of these cells. Further evidence for a role of mitochondrial Ca2+ handling in endothelial cell function was provided by a report that MCU knockdown reduced (Ca2+) transients and oscillations induced by fluid shear stress and regulated the temporal profile of shear‐induced endoplasmic reticulum Ca2+ release. 47 However, to date, sex differences in mitochondrial Ca2+ handling have not been tested in endothelial cells, in contrast to cardiac and skeletal myocytes. 11 , 15 , 16 In mitochondria isolated from cardiomyocytes of female rats, Ca2+ uptake following the application of Ca2+ boluses was lower than in their male counterparts, 11 and this difference was abolished by applying a pharmacologic inhibitor of MCU. In our hands, ATP induced strong increases in cytosolic Ca2+ and greater mitochondrial Ca2+ transients in cultured HAECs treated with estradiol. These differences may be attributable to differences in methodologies (eg, the measurements in isolated mitochondria versus in intact cells). The greater mitochondrial mass in HAECs treated with estradiol would be expected to lead to mitochondrial Ca2+ transients.
In the absence of differences in MCU composition after treatment with sex hormones, we attribute the observed differences in HAECs to the increased mitochondrial mass, membrane potential, and fusion after estradiol. This interpretation is consistent with data from a myoblast cell line. In this study, forcing mitochondrial fission reduced mitochondrial Ca2+ uptake. 33
Several alternative explanations for the observed sex differences should be considered for further investigation, for example, sex differences in proteins (MFN2, voltage‐dependent anion‐selective channel 1) that are part of the mitochondrial endoplasmic reticulum contact sites, MERCS. These are crucial for Ca2+ transition from endoplasmic reticulum to mitochondria, and thus, such changes could account for differential mitochondrial Ca2+ entry. Our analysis did not reveal significant differences in MFN2 expression (Figure 7K). It is also possible that additional proteins known to modulate MCU function are expressed or regulated differently in male and female mice. One example is the mitochondrial Rho GTPase 1, which promotes MCU function by associating with its N‐terminus. Small sex‐specific differences in mitochondrial Rho GTPase 1 protein levels by mass spectroscopy were reported in brain endothelial cells. 42 Lastly, in yeast, MCU stability and functionality are modulated by lipids in the mitochondrial inner membrane, specifically by cardiolipin. 48 This may be relevant given recent evidence that estradiol induces cardiolipin synthesis. 49
The results from the 2 systems used in this study, mesenteric arteries isolated from male and female mice and cultured HAECs treated with estradiol and testosterone, gave largely consistent results. However, some disparities were seen. For example, the differences in peak ACh‐triggered Ca2+ transients were larger in arteries than the ATP‐induced peak (Ca2+) in cultured HAECs after pretreatment with estradiol or testosterone (Figures 3 and 4). This discrepancy could be because of differences in Ca2+ mobilization that develop in cultured cells. Our vasodilation studies imply that with MCU deletion, endothelium‐dependent vasodilation is stronger in female versus male mice and driven with higher NOS activity (Figure 1 and Figure S1). Moreover, the increased levels of mitochondrial ROS may further modulate vasoreactivity. Both vasodilation and constriction with hydrogen peroxide have been described. 50 We also found that cytosolic Ca2+ transients triggered by ACh are more pronounced in arteries from female than male mice and is strongly reduced in mice with MCU inhibition. This finding implies reduced Ca2+‐dependent NOS activity when MCU is blocked, in accordance with a previous study that described that increased mitochondrial Ca2+ uptake facilitated Ca2+‐triggered NO formation. 14 The higher cytosolic Ca2+ transients in arteries from female mice would predict greater NOS‐dependent dilation. Our finding that eNOS activation is higher in estradiol‐treated HAECs compared with baseline than in testosterone‐treated cells is also consistent with data from human umbilical vein endothelial cells isolated from female and male donors. 1 Together, our findings suggest that sex differences in mitochondrial Ca2+ handling enhance cytosolic Ca2+ transients that drive increases in eNOS activity and NO production. Further studies are needed to establish whether novel agents capable of modulating MCU activity 51 can be leveraged to selectively promote endothelial function in men and women.
Sources of Funding
This project was supported in part by grants from the US Department of Veterans Affairs (I01 BX000163) and the National Institutes of Health (R01 HL108932) to Dr Grumbach and from the American Heart Association (22PRE902649) to B. T. Endoni.
Disclosures
Dr Grumbach reports grants from the National Institutes of Health, Department of Veterans Affairs, and the American Heart Association. B. T. Endoni declares a grant from the American Heart Association. The remaining authors have no disclosures to report.
Supporting information
Table S1
Figures S1–S5
Acknowledgments
The authors thank Dr Blaumueller of the Scientific Editing and Research Communication Core at the University of Iowa for critical reading of the article.
This article was sent to Hossein Ardehali, Guest Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.023912
For Sources of Funding and Disclosures, see page 13.
Contributor Information
Olha M. Koval, Email: olha-koval@uiowa.edu.
Isabella M. Grumbach, Email: isabella-grumbach@uiowa.edu.
References
- 1. Cattaneo MG, Vanetti C, Decimo I, Di Chio M, Martano G, Garrone G, Bifari F, Vicentini LM. Sex‐specific enos activity and function in human endothelial cells. Sci Rep. 2017;7:9612. doi: 10.1038/s41598-017-10139-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wassmann S, Baumer AT, Strehlow K, van Eickels M, Grohe C, Ahlbory K, Rosen R, Bohm M, Nickenig G. Endothelial dysfunction and oxidative stress during estrogen deficiency in spontaneously hypertensive rats. Circulation. 2001;103:435–441. doi: 10.1161/01.CIR.103.3.435 [DOI] [PubMed] [Google Scholar]
- 3. Monsalve E, Oviedo PJ, Garcia‐Perez MA, Tarin JJ, Cano A, Hermenegildo C. Estradiol counteracts oxidized ldl‐induced asymmetric dimethylarginine production by cultured human endothelial cells. Cardiovasc Res. 2007;73:66–72. doi: 10.1016/j.cardiores.2006.09.020 [DOI] [PubMed] [Google Scholar]
- 4. Rubio‐Gayosso I, Sierra‐Ramirez A, Garcia‐Vazquez A, Martinez‐Martinez A, Munoz‐Garcia O, Morato T, Ceballos‐Reyes G. 17Beta‐estradiol increases intracellular calcium concentration through a short‐term and nongenomic mechanism in rat vascular endothelium in culture. J Cardiovasc Pharmacol. 2000;36:196–202. doi: 10.1097/00005344-200008000-00009 [DOI] [PubMed] [Google Scholar]
- 5. Murthy S, Koval OM, Ramiro Diaz JM, Kumar S, Nuno D, Scott JA, Allamargot C, Zhu LJ, Broadhurst K, Santhana V, et al. Endothelial CaMKII as a regulator of eNOS activity and NO‐mediated vasoreactivity. PLoS One. 2017;12:e0186311. doi: 10.1371/journal.pone.0186311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Busse R, Mülsch A. Calcium‐dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett. 1990;265:133–136. doi: 10.1016/0014-5793(90)80902-U [DOI] [PubMed] [Google Scholar]
- 7. Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin‐dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:E68–E75. doi: 10.1161/hh1101.092677 [DOI] [PubMed] [Google Scholar]
- 8. Sobrino A, Oviedo PJ, Novella S, Laguna‐Fernandez A, Bueno C, Garcia‐Perez MA, Tarin JJ, Cano A, Hermenegildo C. Estradiol selectively stimulates endothelial prostacyclin production through estrogen receptor‐{alpha}. J Mol Endocrinol. 2010;44:237–246. doi: 10.1677/JME-09-0112 [DOI] [PubMed] [Google Scholar]
- 9. Klinge CM. Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem. 2008;105:1342–1351. doi: 10.1002/jcb.21936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ventura‐Clapier R, Moulin M, Piquereau J, Lemaire C, Mericskay M, Veksler V, Garnier A. Mitochondria: a central target for sex differences in pathologies. Clin Sci (Lond). 2017;131:803–822. doi: 10.1042/CS20160485 [DOI] [PubMed] [Google Scholar]
- 11. Arieli Y, Gursahani H, Eaton MM, Hernandez LA, Schaefer S. Gender modulation of Ca(2+) uptake in cardiac mitochondria. J Mol Cell Cardiol. 2004;37:507–513. doi: 10.1016/j.yjmcc.2004.04.023 [DOI] [PubMed] [Google Scholar]
- 12. Chweih H, Castilho RF, Figueira TR. Tissue and sex specificities in Ca2+ handling by isolated mitochondria in conditions avoiding the permeability transition. Exp Physiol. 2015;100:1073–1092. doi: 10.1113/EP085248 [DOI] [PubMed] [Google Scholar]
- 13. Olson ML, Chalmers S, McCarron JG. Mitochondrial Ca2+ uptake increases Ca2+ release from inositol 1,4,5‐trisphosphate receptor clusters in smooth muscle cells. J Biol Chem. 2010;285:2040–2050. doi: 10.1074/jbc.M109.027094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Charoensin S, Eroglu E, Opelt M, Bischof H, Madreiter‐Sokolowski CT, Kirsch A, Depaoli MR, Frank S, Schrammel A, Mayer B, et al. Intact mitochondrial Ca(2+) uniport is essential for agonist‐induced activation of endothelial nitric oxide synthase (eNOS). Free Radic Biol Med. 2017;102:248–259. doi: 10.1016/j.freeradbiomed.2016.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher‐Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty‐kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mammucari C, Gherardi G, Zamparo I, Raffaello A, Boncompagni S, Chemello F, Cagnin S, Braga A, Zanin S, Pallafacchina G, et al. The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep. 2015;10:1269–1279. doi: 10.1016/j.celrep.2015.01.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Finkel T, Menazza S, Holmstrom KM, Parks RJ, Liu J, Sun J, Liu J, Pan X, Murphy E. The ins and outs of mitochondrial calcium. Circ Res. 2015;116:1810–1819. doi: 10.1161/CIRCRESAHA.116.305484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu JC, Parks RJ, Liu J, Stares J, Rovira II, Murphy E, Finkel T. The in vivo biology of the mitochondrial calcium uniporter. Adv Exp Med Biol. 2017;982:49–63. doi: 10.1007/978-3-319-55330-6_3 [DOI] [PubMed] [Google Scholar]
- 20. Paillard M, Csordás G, Szanda G, Golenár T, Debattisti V, Bartok A, Wang N, Moffat C, Seifert EL, Spät A, et al. Tissue‐specific mitochondrial decoding of cytoplasmic Ca2+ signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep. 2017;18:2291–2300. doi: 10.1016/j.celrep.2017.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koval OM, Nguyen EK, Santhana V, Fidler TP, Sebag SC, Rasmussen TP, Mittauer DJ, Strack S, Goswami PC, Abel ED, et al. Loss of MCU prevents mitochondrial fusion in G1‐S phase and blocks cell cycle progression and proliferation. Sci Signal. 2019;12:eaav1439. doi: 10.1126/scisignal.aav1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shanmughapriya S, Rajan S, Hoffman NE, Zhang X, Guo S, Kolesar JE, Hines KJ, Ragheb J, Jog NR, Caricchio R, et al. Ca2+ signals regulate mitochondrial metabolism by stimulating creb‐mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci Signal. 2015;8:ra23. doi: 10.1126/scisignal.2005673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu C, Wang Y, Peng J, Shen Q, Chen M, Tang W, Li X, Cai C, Wang B, Cai S, et al. Mitochondrial calcium uniporter as a target of microRNA‐340 and promoter of metastasis via enhancing the Warburg effect. Oncotarget. 2017;8:83831–83844. doi: 10.18632/oncotarget.19747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol. 2014;5:21. doi: 10.3389/fphar.2014.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang BH, Mukherji S, Soderling TR. Characterization of a calmodulin kinase II inhibitor protein in brain. Proc Natl Acad Sci USA. 1998;95:10890–10895. doi: 10.1073/pnas.95.18.10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Forde A, Constien R, Gröne HJ, Hämmerling G, Arnold B. Temporal Cre‐mediated recombination exclusively in endothelial cells using Tie2 regulatory elements. Genesis. 2002;33:191–197. doi: 10.1002/gene.10117 [DOI] [PubMed] [Google Scholar]
- 27. Prasad AM, Morgan DA, Nuno DW, Ketsawatsomkron P, Bair TB, Venema AN, Dibbern ME, Kutschke WJ, Weiss RM, Lamping KG, et al. Calcium/calmodulin‐dependent kinase II inhibition in smooth muscle reduces angiotensin II‐induced hypertension by controlling aortic remodeling and baroreceptor function. J Am Heart Assoc. 2015;4:e001949. doi: 10.1161/JAHA.115.001949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilson C, Zhang X, Buckley C, Heathcote HR, Lee MD, McCarron JG. Increased vascular contractility in hypertension results from impaired endothelial calcium signaling. Hypertension. 2019;74:1200–1214. doi: 10.1161/HYPERTENSIONAHA.119.13791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wilson C, Lee MD, Heathcote HR, Zhang X, Buckley C, Girkin JM, Saunter CD, McCarron JG. Mitochondrial ATP production provides long‐range control of endothelial inositol trisphosphate‐evoked calcium signaling. J Biol Chem. 2019;294:737–758. doi: 10.1074/jbc.RA118.005913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nguyen EK, Koval OM, Noble P, Broadhurst K, Allamargot C, Wu M, Strack S, Thiel WH, Grumbach IM. CaMKII (Ca2+/calmodulin‐dependent kinase II) in mitochondria of smooth muscle cells controls mitochondrial mobility, migration, and neointima formation. Arterioscler Thromb Vasc Biol. 2018;38:1333–1345. doi: 10.1161/ATVBAHA.118.310951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sebag SC, Koval OM, Paschke JD, Winters CJ, Comellas AP, Grumbach IM. Inhibition of the mitochondrial calcium uniporter prevents IL‐13 and allergen‐mediated airway epithelial apoptosis and loss of barrier function. Exp Cell Res. 2018;362:400–411. doi: 10.1016/j.yexcr.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, et al. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491:269–273. doi: 10.1038/nature11444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kowaltowski AJ, Menezes‐Filho SL, Assali EA, Gonçalves IG, Cabral‐Costa JV, Abreu P, Miller N, Nolasco P, Laurindo FRM, Bruni‐Cardoso A, et al. Mitochondrial morphology regulates organellar Ca. FASEB J. 2019;33:13176–13188. doi: 10.1096/fj.201901136R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Montemurro C, Vadrevu S, Gurlo T, Butler AE, Vongbunyong KE, Petcherski A, Shirihai OS, Satin LS, Braas D, Butler PC, et al. Cell cycle‐related metabolism and mitochondrial dynamics in a replication‐competent pancreatic beta‐cell line. Cell Cycle. 2017;16:2086–2099. doi: 10.1080/15384101.2017.1361069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Valle A, Guevara R, García‐Palmer FJ, Roca P, Oliver J. Sexual dimorphism in liver mitochondrial oxidative capacity is conserved under caloric restriction conditions. Am J Physiol Cell Physiol. 2007;293:C1302–C1308. doi: 10.1152/ajpcell.00203.2007 [DOI] [PubMed] [Google Scholar]
- 36. Colom B, Alcolea MP, Valle A, Oliver J, Roca P, García‐Palmer FJ. Skeletal muscle of female rats exhibit higher mitochondrial mass and oxidative‐phosphorylative capacities compared to males. Cell Physiol Biochem. 2007;19:205–212. doi: 10.1159/000099208 [DOI] [PubMed] [Google Scholar]
- 37. Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res. 2010;106:1681–1691. doi: 10.1161/CIRCRESAHA.109.213645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Colom B, Oliver J, Roca P, Garcia‐Palmer FJ. Caloric restriction and gender modulate cardiac muscle mitochondrial H2O2 production and oxidative damage. Cardiovasc Res. 2007;74:456–465. doi: 10.1016/j.cardiores.2007.02.001 [DOI] [PubMed] [Google Scholar]
- 39. Ribeiro RF, Ronconi KS, Morra EA, Do Val Lima PR, Porto ML, Vassallo DV, Figueiredo SG, Stefanon I. Sex differences in the regulation of spatially distinct cardiac mitochondrial subpopulations. Mol Cell Biochem. 2016;419:41–51. doi: 10.1007/s11010-016-2748-4 [DOI] [PubMed] [Google Scholar]
- 40. Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015;116:1231–1244. doi: 10.1161/CIRCRESAHA.116.302855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Straface E, Vona R, Campesi I, Franconi F. Mitochondria can orchestrate sex differences in cell fate of vascular smooth muscle cells from rats. Biol Sex Differ. 2015;6:34. doi: 10.1186/s13293-015-0051-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rutkai I, Dutta S, Katakam PV, Busija DW. Dynamics of enhanced mitochondrial respiration in female compared with male rat cerebral arteries. Am J Physiol Heart Circ Physiol. 2015;309:H1490–H1500. doi: 10.1152/ajpheart.00231.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sanz A, Hiona A, Kujoth GC, Seo AY, Hofer T, Kouwenhoven E, Kalani R, Prolla TA, Barja G, Leeuwenburgh C. Evaluation of sex differences on mitochondrial bioenergetics and apoptosis in mice. Exp Gerontol. 2007;42:173–182. doi: 10.1016/j.exger.2006.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Razmara A, Sunday L, Stirone C, Wang XB, Krause DN, Duckles SP, Procaccio V. Mitochondrial effects of estrogen are mediated by estrogen receptor alpha in brain endothelial cells. J Pharmacol Exp Ther. 2008;325:782–790. doi: 10.1124/jpet.107.134072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tcherepanova I, Puigserver P, Norris JD, Spiegelman BM, McDonnell DP. Modulation of estrogen receptor‐alpha transcriptional activity by the coactivator PGC‐1. J Biol Chem. 2000;275:16302–16308. doi: 10.1074/jbc.M001364200 [DOI] [PubMed] [Google Scholar]
- 46. Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci USA. 2015;112:9129–9134. doi: 10.1073/pnas.1504705112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Scheitlin CG, Julian JA, Shanmughapriya S, Madesh M, Tsoukias NM, Alevriadou BR. Endothelial mitochondria regulate the intracellular Ca2+ response to fluid shear stress. Am J Physiol Cell Physiol. 2016;310:C479–C490. doi: 10.1152/ajpcell.00171.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ghosh S, Basu Ball W, Madaris TR, Srikantan S, Madesh M, Mootha VK, Gohil VM. An essential role for cardiolipin in the stability and function of the mitochondrial calcium uniporter. Proc Natl Acad Sci USA. 2020;117:16383–16390. doi: 10.1073/pnas.2000640117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Acaz‐Fonseca E, Ortiz‐Rodriguez A, Lopez‐Rodriguez AB, Garcia‐Segura LM, Astiz M. Developmental sex differences in the metabolism of cardiolipin in mouse cerebral cortex mitochondria. Sci Rep. 2017;7:43878. doi: 10.1038/srep43878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lucchesi PA, Belmadani S, Matrougui K. Hydrogen peroxide acts as both vasodilator and vasoconstrictor in the control of perfused mouse mesenteric resistance arteries. J Hypertens. 2005;23:571–579. doi: 10.1097/01.hjh.0000160214.40855.79 [DOI] [PubMed] [Google Scholar]
- 51. Woods JJ, Nemani N, Shanmughapriya S, Kumar A, Zhang M, Nathan SR, Thomas M, Carvalho E, Ramachandran K, Srikantan S, et al. A selective and cell‐permeable mitochondrial calcium uniporter (MCU) inhibitor preserves mitochondrial bioenergetics after hypoxia/reoxygenation injury. ACS Cent Sci. 2019;5:153–166. doi: 10.1021/acscentsci.8b00773 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Figures S1–S5