

Abstract
Background
Mice with cardiomyocyte‐specific deletion of Bmal1, a core clock gene, had spontaneous abnormal cardiac metabolism, dilated cardiomyopathy, and shortened lifespan. However, the role of cardiomyocyte Bmal1 in pressure overload induced cardiac remodeling is unknown. Here we aimed to understand the contribution of cardiomyocyte Bmal1 to cardiac remodeling in response to pressure overload induced by transverse aortic constriction or chronic angiotensin Ⅱ (AngⅡ) infusion.
Methods and Results
By generating a tamoxifen‐inducible cardiomyocyte‐specific Bmal1 knockout mouse line (cKO) and challenging the mice with transverse aortic constriction or AngⅡ, we found that compared to littermate controls, the cKO mice displayed remarkably increased cardiac hypertrophy and augmented fibrosis both after transverse aortic constriction and AngⅡ induction, as assessed by echocardiographic, gravimetric, histologic, and molecular analyses. Mechanistically, RNA‐sequencing analysis of the heart after transverse aortic constriction exposure revealed that the PI3K/AKT signaling pathway was significantly activated in the cKOs. Consistent with the in vivo findings, in vitro study showed that knockdown of Bmal1 in cardiomyocytes significantly promoted phenylephrine‐induced cardiomyocyte hypertrophy and triggered fibroblast‐to‐myofibroblast differentiation, while inhibition of AKT remarkedly reversed the pro‐hypertrophy and pro‐fibrosis effects of Bmal1 knocking down.
Conclusions
These results suggest that postnatal deletion of Bmal1 in cardiomyocytes may promote pressure overload‐induced cardiac remodeling. Moreover, we identified PI3K/AKT signaling pathway as the potential mechanistic ties between Bmal1 and cardiac remodeling.
Keywords: Bmal1, cardiac remodeling, circadian clock, fibrosis, hypertrophy
Subject Categories: Cardiomyopathy, Hypertrophy, Remodeling
Nonstandard Abbreviations and Acronyms
- AngⅡ
angiotensin Ⅱ
- NRCFs
neonatal rat cardiac fibroblasts
- NRVMs
neonatal rat ventricular myocytes
- TAC
transverse aortic constriction.
Clinical Perspective
What Is New?
Postnatal deletion of Bmal1 appears to be a good strategy to study the cardiac function of Bmal1 and the circadian machinery of the heart.
Conditional deletion of Bmal1 in adult cardiomyocytes promotes pressure overload‐induced cardiac remodeling.
The PI3K/AKT signaling pathway acts as a mechanistic tie between this circadian clock gene and cardiac remodeling.
What Are the Clinical Implications?
Identification of Bmal1‐PI3K/AKT signaling pathway in promoting pressure overload‐induced cardiac remodeling implies potential implications for circadian regulation in the remodeling of cardiovascular tissue.
Genetic manipulation of the cardiac molecular clock may represent a therapeutic target for the treatment of pressure overload induced cardiac remodeling and heart failure.
Emerging evidence indicates that most cardiovascular physiology exhibits time‐of‐day‐dependent oscillations, for example the daily variations of blood pressure, heart rate, and cardiac output. 1 , 2 , 3 The onset of many adverse cardiovascular events (e.g., myocardial infarction, sudden cardiac death) is time‐of‐day‐dependent in humans. 4 , 5 , 6 Disturbed diurnal rhythms are associated with increased risk of adverse cardiovascular events and poorer prognosis. 7 Moreover, genetic deficiency of the core clock components in mice, such as BMAL1 and CLOCK, has been reported to be related with altered circadian oscillations of blood pressure and heart rate and accompanied with abnormalities in cardiac function. 8 , 9
Cardiac remodeling, typically cardiac hypertrophy and fibrosis, is initially an adaptive response of the heart to pathophysiologic stimuli in an attempt to counterbalance ventricular wall stress and preserve cardiac function. However, sustained hypertrophy and fibrosis in response to cardiac insults, such as hypertension or myocardial infarction, can eventually lead to arrhythmias, dilated cardiomyopathy, and heart failure, leading causes of cardiac morbidity and mortality. 10 By using a murine model of TAC and subjecting the mice to a rhythm‐disruptive 20‐hour versus rhythm‐normal 24‐hour environment, Martino et al proved that environmental circadian desynchrony may adversely affect pressure overload induced cardiac remodeling, reflected by less cardiac hypertrophy and reduced left ventricular contractile strength. 11 Alibhai et al demonstrated that a short‐term (5 days) disruption of the diurnal rhythm may even exacerbate maladaptive cardiac remodeling and adversely affect long‐term cardiac healing after myocardial infarction. 12 Previous studies have reported that mice with genetic disrupted cardiac circadian mechanism, where Bmal1 or Clock was prenatally deleted only in cardiomyocytes, had abnormal cardiac metabolism, signaling, and contractile function, eventually gave rise to age‐associated dilated cardiomyopathy and shortened lifespan. 13 , 14 However, the effect of genetic circadian disruption on pressure overload induced cardiac remodeling had not been explored.
In the present study, by generating a mouse line with tamoxifen inducible, cardiac‐restricted, ablation of Bmal1, and challenging the mice with TAC or chronic AngⅡ (angiotensin Ⅱ) infusion to simulate pressure overload induced cardiac remodeling, we found that postnatal depletion of Bmal1 in cardiomyocytes may promote pressure overload induced cardiac hypertrophy and fibrosis without worsening cardiac dysfunction. Mechanically, we proved that over activation of the PI3K/AKT signaling might be the major contributor to the pro‐remodeling effect of cardiac Bmal1 absence.
Methods
Animal
For generation of the postnatally cKO mice, Bmal1flox/flox mice (from C. Bradfield, University of Wisconsin, Madison, WI, USA) were crossed onto the α‐MHC‐MerCreMer mice; gene recombination was induced by intraperitoneal administration of tamoxifen at 40 mg/kg per day for 5 continuous days at 8 weeks of age. 15 , 16 Bmal1flox/flox littermates with tamoxifen administration were served as Ctrl. All experiments were performed 6 weeks after the last tamoxifen administration. All mice were C57BL/6 genetic background and fed on a standard diet and maintained on a 12 hour light/12 hour dark schedule under a temperature‐controlled room (22–25◦C) with 45%–65% humidity. The room lights were turned on at 8:00 am local time, this was defined as Zeitgeber time 0 (ZT0); and lights were turned off at 8:00 pm (ZT12). All animal care and experimental procedures complied with the guidelines of National Institutes of Health and were approved by the Institute for Animal Care and Use Committee of Dalian Medical University. The data will be made available upon request to the corresponding author.
Transverse Aortic Constriction and AngⅡ Infusion
TAC and chronic AngⅡ infusion were performed to establish two mouse models of pressure overload‐induced cardiac remodeling, in accordance with previously described methods. 17 , 18 For TAC surgery, mice were anaesthetized with 1% isoflurane and subjected to thoracotomy, the exposed aortic arch was ligated using a 4‐0 silk suture between the brachiocephalic and left common carotid arteries with an overlaying 27‐gauge needle for either 2 weeks (TAC 2W) or 4 weeks (TAC 4W). Operated mice without constriction served as controls (Sham). For AngⅡ infusion, mice were anesthetized with 1% isoflurane. AngⅡ (1000 ng/kg per minute) dissolved in sterile saline was infused using an osmotic minipump (Alzet model 2004; Alza Corp) inserted subcutaneously for 28 days. All surgeries were performed between ZT1 and ZT4, and the operations were performed alternately between the Ctrl and cKO mice. Systolic blood pressure was measured using a computerized noninvasive tail‐cuff device as we previously described and was recorded as the mean value of three measurements at both light (1 hour after the light on) and dark (1 hour after the light off) phases for 3 consecutive days. 19 At the end of the experiments, all mice were euthanized with overdose of CO2.
Echocardiography
M‐mode echocardiography was performed using high‐frequency ultrasound with a VEVO 3100 echography device (VisualSonics, Toronto, Canada), and data were analyzed using VEVO 3100 software (version 1.5.0). Temperature and heart rate of mice were monitored and stabilized at ranges of 37.0 ± 0.5 °C and 500 ± 30 beats/min, respectively. The mouse was placed in supine position on adjustable rail to allow coordination of the ultrasound transducer and anesthetized using 1% isoflurane. M‐mode images and recordings were acquired from the short‐axis view of the left ventricle at the level of the papillary muscles. The left ventricular internal diameter (LVID); left ventricular anterior wall thickness (LVAW); left ventricular posterior wall thickness (LVPW); ejection fraction (EF) and fractional shortening (FS) at diastole and systole were measured from the M‐mode recordings.
Behavioral and Metabolic Monitoring
Mice for behavioral activity recording were individually housed in running wheel‐equipped cages contained within a ventilated and light‐tight isolation chamber with a computer‐controlled lighting system. The locomotor activities were continuously recorded at regular light/dark cycles (12 hour light:12 hour dark, LD) for 4 days and then at the constant dark cycles (DD) for another for 4 days. Data were analyzed using ClockLab software (Actimetrics). For metabolic parameter collection, continuous three‐day monitoring was performed using the CLAMS (Columbus Instruments Inc., Columbus, OH), and the food intake, water intake, energy expenditure was measured in an automated fashion.
Cell Culture
H9c2 cells (CRL 1446; American Type Culture Collection [ATCC], Rockville, MD) were grown in the DMEM medium supplemented with 10% FBS and 1% penicillin‐streptomycin at 37 ℃ and 5% CO2. Neonatal rat ventricular myocytes (NRVMs) and neonatal rat cardiac fibroblasts (NRCFs) were obtained from 1 to 3 days old neonatal Sprague‐Dawley rat pups as we previously described. 20 Briefly, heart tissues were minced into pieces and digested with trypsin and collagenase II. Digested cells were pelleted and plated on 10‐cm dishes and cultured in DMEM supplemented with 10% FBS plus 1% penicillin and streptomycin. A 2–3 hour pre‐plating period was used to separate early attached NRCFs and unattached NRVMs. Cells at passage 0 to 1 were used for the experiment and analysis. Both NRVMs and NRCFs were changed to serum‐free DMEM for 12 hour before phenylephrine (PE) stimulation or other treatment. For PE and MK2206 (a highly selective allosteric pan‐AKT inhibitor) treatment, cells were treated with PE at 200 µmol/L for 6 hours and MK2206 at 10 nmol/L for 1 hours. Notably, for the phosphorylated AKT detection, PE was treated for 30 minutes. For the conditional medium collection, primary cultured NRVMs were treated with or without phenylephrine, MK2206 and Bmal1 siRNA as indicated, and the supernatant were collected as conditional medium.
RNA Interference
NRVMs were transfected with 100 pmol of small interfering RNA (siRNA) specific for Bmal1 or negative control with Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) for 24 hours. The sequence of siRNA‐Bmal1 is (5’‐3’) GCAACAGGCCUUCAGUAAA. The sequence of negative control is (5’‐3’) UUCUCCGAACGUGUCACGU.
Western Blot
Cardiac tissues and cells were lysed with RIPA lysis buffer containing the protease inhibitor. Protein concentrations were determined by the Pierce BCA kit (Thermo scientific). Equal amounts of protein were loaded and separated by 10% SDS‐PAGE, and then transferred onto nitrocellulose filter membranes for blotting. Membranes were briefly washed and blocked in 5% Bovine Serum Albumin (BioFroxx) for 1 hour and then incubated with primary antibody solution overnight at 4 ℃. The following antibodies were used: GAPDH (Proteintech, 10494‐1‐AP) with 1:5000 dilution, Bmal1 (Abcam, ab93806) with 1:2000 dilution, α‐SMA (Abcam, ab124964) with 1:10000 dilution, Fibronectin (abcam, ab2413) with 1:5000 dilution, collagen I (Proteintech, 14695‐1‐AP) with 1:5000 dilution, AKT (T‐AKT) (CST, 4691) with 1:2000 dilution, phosphorylated AKT (P‐AKT) (CST, 4060) with 1:2000 dilution, ANP (atrial natriuretic peptide; abcam, ab225844) with 1:2000 dilution. Membranes were incubated with either goat‐rabbit or anti‐mouse horse radish peroxidase‐conjugated secondary antibody (Proteintech) for 1 hour. Finally, the membranes were transferred to the Super Lumia ECL Plus HRP Substrate Reagent (advansta), and images were collected using Chemiluminescent Imaging System (Tanon, China). Quantification analysis was performed using Image J software.
Quantitative RT‐PCR
Total RNA was isolated from tissues and cells with Trizol reagent (Takara) and the reverse transcription was performed using a reagent kit (Vazyme). Real‐time PCR was performed using LightCycler96 (Roche, Switzerland). For quantitative RT‐PCR (qRT‐PCR), specific Taqman assays were designed for each gene from mouse sequences available in GenBank. The following primer sequences were used: Bmal1 (R: GCCCCACCGACCTACTCT; F: CTTTGTCTGTGTCCATACTTTCTTG), Fibronectin (R: AAGGTTCGGGAAGAGGTTGT; F: GAGCTTAAAGCCAGCGTCAG), collagen I (R: GAGTACTGGATCGACCCTAACCA; F: GACGGCTGAGTAGGGAACACA), collagen Ⅲ (R: TGCCACCCCGAACTCAAG; F: AGATCAGGCAGGGCCATAGCT), ANP (R: TGCTTCCTCAGTCTGCTCACTC; F: TACAGTGCGGTGTCCAACACAG), BNP (R: GGTCCTTCAAGAGCTGTCTCTG; F: TCCTAGCCAGTCTCCAGAGCA), and β‐MHC (myosin heavy chain beta; R: AGGCCTTCACCTTCAGCTGC; F: CTGAAGGGCATGAGGAAGAGT). All mRNA measurements were calculated by the 2‐△△ct method and normalized to 18S or beta‐actin mRNA levels.
RNA‐seq
Total RNA was extracted from heart tissues using TRIzol regent. RNA quantification and qualification were performed using RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). The mRNA was purified from total RNA using poly‐T oligo‐attached magnetic beads and fragmentated using divalent cations. cDNA was synthesized and the fragments were purified with AMPure XP system (Beckman Coulter, Beverly, USA). The quality was assessed by Agilent Bioanalyzer 2100 system. Sequencing was performed on an Illumina Novaseq platform (Novogene, Beijing, China). FeatureCounts v1.5.0‐p3 was used to count the reads numbers mapped to each gene. And then FPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene. Differential expression analysis of two groups was performed using the DESeq2 package (1.20.0). Gene Ontology (GO) enrichment analysis of differentially expressed genes was performed by the clusterProfiler software (v3.4.4).
Histology
Heart tissues were harvested and fixed in 4% paraformaldehyde, embedded in paraffin wax, and cut into 5 µm sections. Serial heart sections were stained with hematoxylin and eosin (H&E) or wheat germ agglutinin (WGA). Masson trichrome staining and Picrosirius red staining were performed according to the standard protocol. 21 , 22 The percentage of fibrotic area was quantified by Image J.
Cell Immunofluorescence
The H9c2 cells were fixed in 4% paraformaldehyde for 20 minutes and then permeabilized with 0.1% TritonX‐100. The cells were stained with phalloidin using Phalloidin–Tetramethylrhodamine B isothiocyanate (sigma) following the manufacturer’s instructions. The stained cells were visualized by confocal microscopy (Leica, Germany).
Statistical Analysis
All data are presented as the mean±standard error of the mean (SEM). Comparisons were performed by using student t test between two groups or ANOVA among multiple groups. P<0.05 was considered statistically significant.
Results
Generation and Characterization of the cKO Mice
To generate an inducible cardiomyocyte‐specific Bmal1 knock‐out mouse line, we crossed the Bmal1flox/flox mice and the α‐MHC‐MerCreMer mice to yield Bmal1flox/flox‐αMHC‐Cre+ mice (cKO). The mice were injected intraperitoneally with tamoxifen for 5 consecutive days at an age of 8 weeks to induce deletion of exons 4 of the Bmal1 gene. Bmal1flox/flox‐αMHC‐Cre‐ mice treated with tamoxifen were used as controls (Ctrl) (Figure S1A and S1B). Real‐time PCR and western blots were used for validating the deletion of Bmal1 in cardiomyocytes. As shown, the Bmal1 mRNA and protein were significantly suppressed in the cKO hearts, the remaining expression might reflect the existence of non‐cardiomyocytes in the heart (Figure S1C and S1D). Next, we examined if the postnatal cardiomyocyte‐specific Bmal1 deletion has effect on whole‐body behavior and metabolism, wheel running data revealed that the locomotor activity of the cKO mice was undisturbed at both regular light cycles and constant darkness conditions (Figure S1E). Consistently, the food and water intake, oxygen consumption, CO2 excretion, heat production, and RER showed no difference between cKO and Ctrl mice (Figure S1F through S1K). Given that mice expressing α‐MHC‐MerCreMer recombinase may be subjected to a transient loss of cardiac function when activated by tamoxifen, 23 we examined the cardiac performance of the cKO mice 6 weeks after tamoxifen injection as in order to be sure that pressure overload is imposed in mice with normal cardiac function. As shown in Table S1, no significant distinctions were observed between cKO and Ctrl mice and we didn’t detect any markers of cardiac dysfunction (reflective by equal ejection fraction and fractional shortening), cardiac hypertrophy (reflective by comparable left ventricular wall thickness, LVAW and LVPW), and dilated cardiomyopathy (reflective by indistinguishable left ventricular diameter LVID and LV volume) for the cKO mice.
Deficiency of Bmal1 in Cardiomyocytes Promotes TAC‐Induced Myocardial Hypertrophy
To explore the functional contribution of cardiac Bmal1 in pressure overload induced cardiac dysfunction, we performed TAC or sham surgery and evaluated cardiac function and morphology at 2 and 4 weeks after surgery. As shown, although no comparable differences were observed in the 2‐week sham surgery groups (Figure 1A through 1C), a slightly increased gravimetric heart weight was seen in the cKO mice after 4‐week sham surgery (Figure 1D through 1F). However, since we did not observe any significant differences of the LV mass and systolic function between the 4‐week sham groups (Figure 1H, 1M and 1N), we assume the increase of heart weight might reflect a transit induction of cardiac hypertrophy by Cre‐positive mice receiving tamoxifen injections. Nevertheless, after TAC surgery, cardiac hypertrophy was observed in both groups, while compared with the Ctrl mice, the cKO mice exhibited even greater hypertrophic effect, as reflected by further increased ratios of heart weight to body weight (HW/BW) and heart weight to tibia length (HW/TL) (Figure 1A through 1F). Similar effects were observed in echocardiography‐determined left ventricular (LV) mass (Figure 1G and 1H). The aggravated hypertrophic response in cKO mice was further evidenced by greater cardiomyocyte size by histological analyses of ventricular sections stained with WGA (Figure 1I and 1J). Cardiac function was also measured by echocardiography (Table S2 and S3). The parameters of EF% and FS% indicated that short‐term TAC surgery (2 weeks) did not worsen cardiac function while long‐term TAC (4 weeks) did induce heart failure in the Ctrl mice (Figure 1K through 1N). However, compared with the Ctrl mice, the cKO mice displayed preserved heart function after 4 weeks of TAC treatment (Figure 1M and 1N), suggesting that Bmal1 deficiency in cardiomyocytes may protect cardiac function in response to pressure overload, and the augmented cardiac hypertrophy might be an adaptive compensation response. At the molecular level, we also observed greater induction of hypertrophic‐related genes, ANP, BNP, and myosin heavy chain beta (β‐MHC) in cKO mice compared with Ctrl mice following 2 weeks TAC surgery (Figure S2A through S2C). While after 4 weeks TAC, equally increased mRNA expression of ANP, BNP and β‐MHC was observed in the cKO and Ctrl mice (Figure S2D through S2F), which might be explained by a ceiling effect or saturation effect at high expression levels. Taken together, these findings implicated that cardiomyocyte‐specific Bmal1 deficiency may promote TAC‐induced cardiac hypertrophy and prevent cardiac dysfunction.
Figure 1. Deficiency of Bmal1 in cardiomyocytes promotes TAC‐induced myocardial hypertrophy.
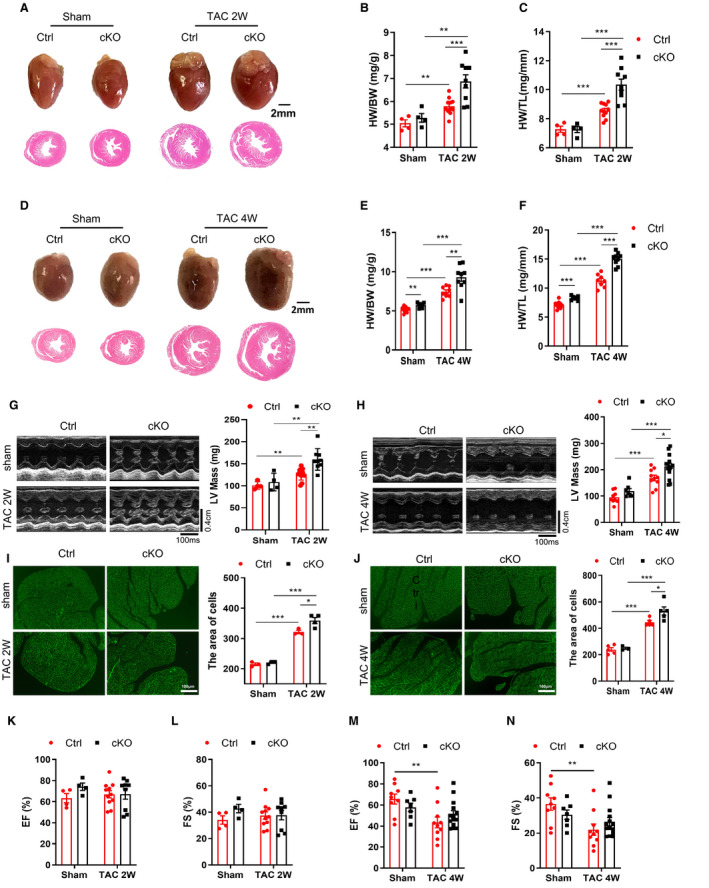
A, Representative photographs (top) and H&E‐stained sections (bottom) of hearts after 2 weeks TAC surgery. Scale bar: 2 mm. B and C, The ratios of heart weight (HW) to body weight (BW) and to tibia length (TL) after 2 weeks TAC surgery. n=4 to 11 mice per group. D, Representative photographs (top) and H&E‐stained sections (bottom) of hearts after 4 weeks TAC surgery. Scale bar: 2 mm. E and F, The ratios of HW/BW and HW/TL after 4 weeks TAC surgery. n=7 to 9 mice per group. G and H, Representative echocardiographic images and assessment of left ventricle mass (LV Mass) in mice subjected to 2 weeks (G) and 4 weeks (H) TAC surgery. Transverse scale bar: 100 milliseconds. Vertical scale bar: 0.4 cm. n=4 to 14 mice per group. I and J, WGA‐staining and quantification of myocyte cross‐sectional area in mice subjected to 2 weeks (I) and 4 weeks (J) TAC surgery. Scale bar: 100 μm. n=3 to 5 mice per group. K and L, Echocardiographic assessment of ejection fraction (EF%) (K) and fractional shortening (FS%) (L) in mice following 2 weeks TAC surgery. n=4 to 11 mice per group. M and N, Echocardiographic assessment of ejection fraction (EF%) (M) and fractional shortening (FS%) (N) in mice following 4 weeks TAC surgery. n=7 to 14 mice per group. cKO indicates cardiomyocyte‐specific Bmal1 knockout mice; Ctrl, control mice; and TAC 2/4W, transverse aortic constriction 2/4 weeks. Data were presented as mean±SEM, *P<0.05, **P<0.01, ***P<0.00.
Deficiency of Bmal1 in Cardiomyocytes Accelerates TAC‐Induced Cardiac Fibrosis
Because fibrosis, characterized by accumulation of collagen and perivascular fibrosis in the heart, is a classical feature of pathological cardiac remodeling, we further evaluated the effects of cardiomyocyte Bmal1 deletion on cardiac fibrosis in TAC challenged hearts. Masson trichrome and Sirius red staining showed that, compared with the Ctrl mice, the cKO mice exhibited significantly augmented collagen deposition after both 2‐ and 4‐weeks TAC treatment (Figure 2A through 2D). Correspondingly, substantially enhanced upregulation of fibrotic genes including Col1α1 (collagen, type I, α1), Col3α1 (collagen, type III, α1), and FN (fibronectin) (Figure 2E through 2J) was displayed in the cKO groups. Thus, deletion of Bmal1 in cardiomyocytes may accelerate the development of TAC‐induced cardiac fibrosis.
Figure 2. Deficiency of Bmal1 in cardiomyocytes accelerates TAC‐induced cardiac fibrosis.
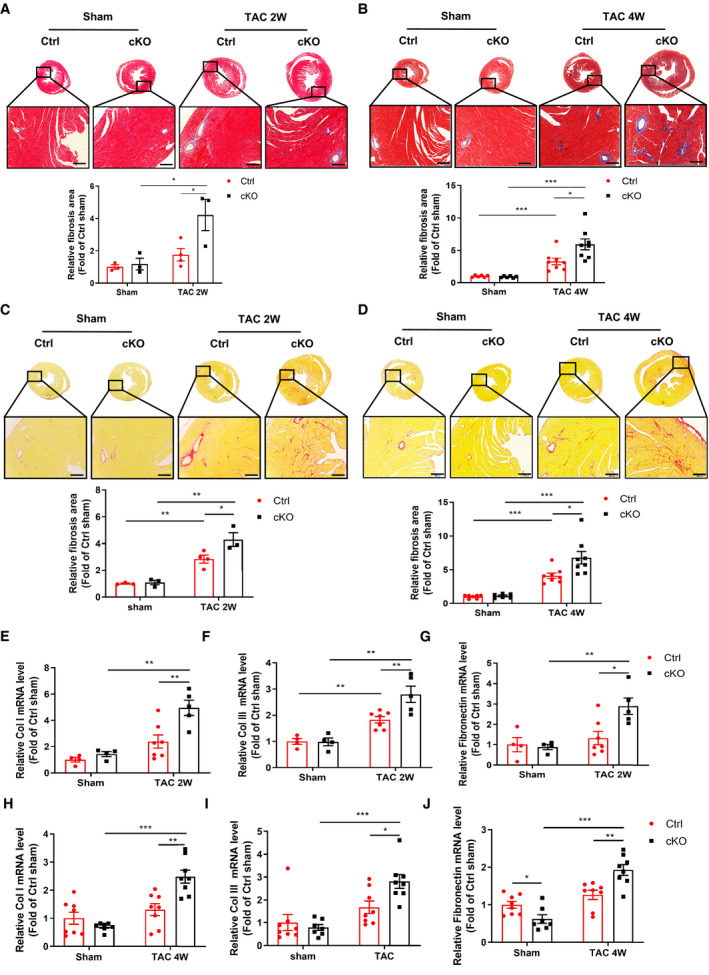
A and B, Representative images of Masson’s trichrome staining and corresponding quantitative analysis of fibrotic (blue) areas in hearts after 2 weeks (A) and 4 weeks (B) of TAC surgery. Relative fibrosis area was analyzed by image J software. Scale bar: 200 μm. n=3 to 8 mice per group. C and D, Representative images of Sirius red staining and corresponding quantitative analysis of fibrotic (red) areas in hearts after 2 weeks (C) and 4 weeks (D) of TAC surgery. Relative fibrosis area was analyzed by image J software. Scale bar: 200 μm. n=3 to 8 mice per group. E through J, qRT‐PCR analysis of the mRNA levels of Col1α1, Col3α1, and fibronectin in the hearts of Ctrl and cKO mice in response to 2 weeks (E through G) and 4 weeks (H through J) TAC surgery. n=4 to 8 mice per group. cKO indicates cardiomyocyte‐specific Bmal1 knockout mice; Ctrl, control mice; and TAC 2/4W, transverse aortic constriction 2/4 weeks. Data were presented as mean±SEM, *P<0.05, **P<0.01, ***P<0.001.
Deficiency of Bmal1 in Cardiomyocytes Promotes AngⅡ‐Induced Cardiac Remodeling
To determine whether cardiomyocyte Bmal1 deficiency had similar effects on other experimental cardiac remodeling induced by pressure overload, we exposed the cKO and Ctrl mice to continuous AngⅡ infusion for 4 weeks. As expected, AngⅡ stimulation induced pathological hypertrophy was further exacerbated in the cKO mice. Heart morphology in gross appearance, HW/BW, HW/TL and echocardiography‐detected LV mass were all significantly increased in the cKO mice (Figure 3A through 3E, Table S4), which is in accordance with increased cardiomyocyte size (Figure 3F). Consistently, AngⅡ induced cardiac fibrosis was also dramatically augmented by cardiomyocyte Bmal1 depletion (Figure 3G and 3H). Regarding cardiac function, although AngⅡ failed to induce statistically impaired LV function in the Ctrl mice, the cKO mice did display preserved LV function after AngⅡ exposure, whereas the EF% and FS% were significantly higher than the Ctrl group (Figure 3I and 3J). These data suggest that like the TAC model, cardiomyocyte‐specific Bmal1 deficiency may promote AngⅡ‐induced cardiac hypertrophy and slow the deterioration of cardiac function. To test the possible involvement of blood pressure in the phenotypes of the cKO mice, systolic blood pressure (SBP) was measured before and after AngⅡ treatment, the results showed that chronic AngⅡ infusion did significantly raise SBP, however, no obvious difference was observed between the 2 genotypes. The day‐night variations of the SBP were not affected in the cKO mice either (Figure 3K), indicating that the promoted cardiac remodeling in the cKO mice was not associated with blood pressure alteration.
Figure 3. Deficiency of Bmal1 in cardiomyocytes promotes AngⅡ‐induced cardiac remodeling.
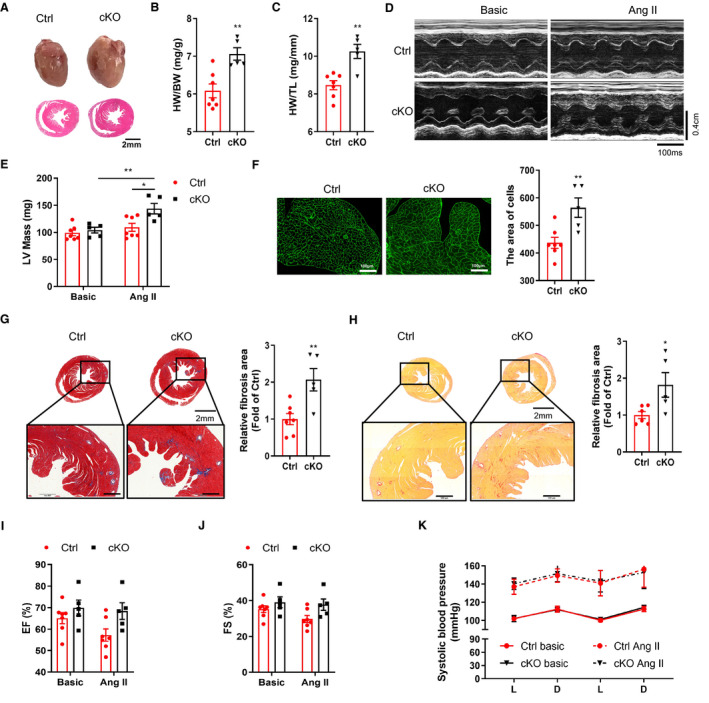
A, Representative photographs (top) and H&E‐stained section (bottom) of hearts after 4 weeks AngⅡ treatment. Scale bar: 2 mm. B and C, The ratios of HW/BW (B) and HW/TL (C) in Ctrl and cKO mice after 4 weeks AngⅡ treatment. n=5 to 7 mice per group. D and E, Representative echocardiographic images (D) and assessment of left ventricle mass (LV Mass) (E) in Ctrl and cKO mice after 4 weeks AngⅡ treatment. Transverse scale bar: 100 milliseconds. Vertical scale bar: 0.4 cm. n=5 to 7 mice per group. F, WGA‐staining and quantification of myocyte cross‐sectional area in Ctrl and cKO mice after 4 weeks AngⅡ treatment. Scale bar: 100 μm. n=5 to 7 mice per group. G, Representative images of Masson’s trichrome staining and the corresponding quantitative analysis of fibrotic (blue) areas in hearts after AngⅡ infusion. Relative fibrosis area was analyzed by image J software. Scale bar: 500 μm. n=5 to 7 mice per group. H, Representative images of Sirius red staining and the corresponding quantitative analysis of fibrotic (red) areas in hearts after AngⅡ infusion. Relative fibrosis area was analyzed by image J software. Scale bar: 500 μm. n=5 to 6 mice per group. I and J, Echocardiographic assessment of ejection fraction (EF%) (I) and fractional shortening (FS%) (J) in mice before and after AngⅡ treatment. n=5 to 7 mice per group. K, Diurnal rhythms of blood pressures in Ctrl and cKO mice before and after AngⅡ treatment. n=5 to 7 mice per group. AngⅡ indicates angiotensin Ⅱ; cKO, .cardiomyocyte‐specific Bmal1 knockout mice; Ctrl, control mice; HW/BW, heart weight/body weight; and HW/TL, heart weight/tibial length. Data were presented as mean±SEM, *P<0.05, **P<0.01.
Knockdown of Bmal1 in Cardiomyocytes Augments Hypertrophy and Fibrogenesis in vitro
To further define the function of Bmal1 in the regulation of cardiomyocyte hypertrophy, we employed an in vitro model of cardiomyocyte hypertrophy induced by phenylephrine (PE) in H9c2 cells. We silenced endogenous Bmal1 expression via specific siRNA targeting rat Bmal1. qRT‐PCR and western blot analysis showed that mRNA and protein levels of Bmal1 were markedly reduced in Bmal1 siRNA transfected cells (Figure 4A through 4C). The cells were treated with PE and immunostained with phalloidin to determine cell size or collected for the analysis of ANP expression. As shown, Bmal1 silencing remarkably promoted PE‐induced increase in cell size and enhanced the expression of ANP (Figure 4D through 4F), illustrating a direct role of Bmal1 in regulating cardiomyocyte hypertrophy. Cardiac fibroblasts play a critical role in the pathological process of cardiac fibrosis. To identify the mechanism by which cardiomyocyte Bmal1 deletion accelerating cardiac fibrosis, we applied the conditional medium from Bmal1 siRNA and PE treated NRVMs to NRCFs (Figure 4G) and detected the expression levels of protein that associated with fibrosis, including Col1α1,α‐SMA and FN. As displayed in Figure 4H through 4K, the induced protein expression of Col1α1, α‐SMA and FN were all further augmented by silencing Bmal1 in NRVMs, suggesting that deletion Bmal1 in cardiomyocytes triggers fibroblast‐to‐myofibroblast differentiation and contributes to cardiac fibrosis. These findings are supportive to previous reports that cardiomyocytes directly respond to pathological stimuli and secrete paracrine factors such as connective tissue growth factor (CTGF) and vascular endothelial growth factor (VEGF) and then activate fibroblasts to augment cardiac fibrosis. 24 Indeed, knock down of Bmal1 did significantly enhance the VEGF and CTGF gene expression in PE‐treated NRVMs (Figure S3). In summary, these in vitro results demonstrated that lack of Bmal1 in cardiomyocytes may not only directly promote PE‐induced cardiomyocyte hypertrophy but also trigger fibrogenesis in fibroblasts indirectly.
Figure 4. Silencing Bmal1 in cardiomyocytes promotes cardiomyocyte hypertrophy and triggers fibroblast‐to‐myofibroblast differentiation in vitro.
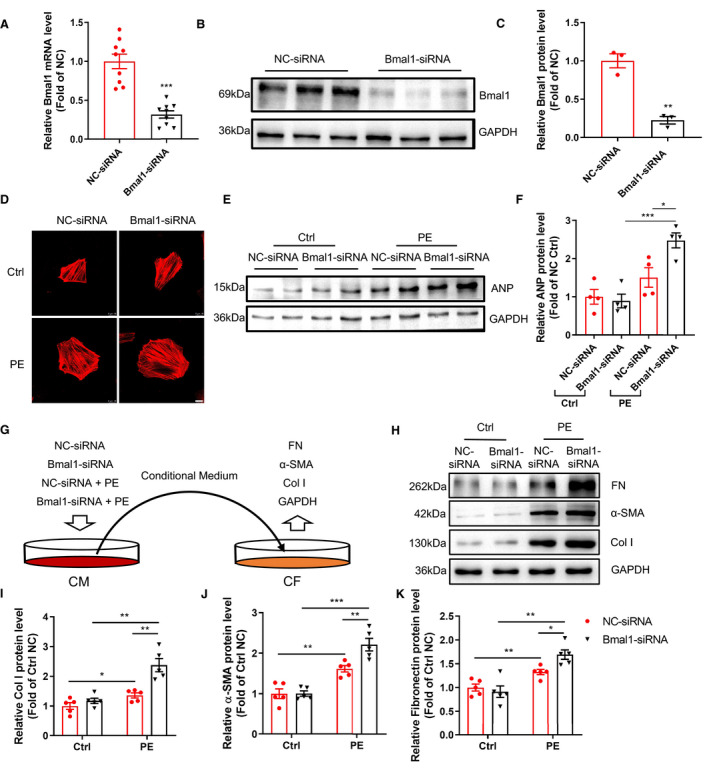
A through C, H9c2 cells were transfected with negative control (NC) or Bmal1 siRNA for 24 hour. Bmal1 expression was determined by qRT‐PCR (A) and western blot (B), the quantification was performed by image J (C). n=9 per group for mRNA, n=3 per group for western blot. D through F, H9c2 cells transfected with NC or Bmal1 siRNA were further treated with PE (200 µmol/L for 6 hour). Cells were stained with Phalloidin (red) to reflect cell size. Scale bar: 10 μm. Hypertrophic marker protein ANP was determined by western blot (E) and the quantification was performed by image J (F). n=4 per group. G, A schematic showing of an in‐direct co‐culture model: NRVMs were transfected with NC or Bma1 siRNA for 24 hour and treated with PE for another 6 hour. After PE stimulation, the supernatant was collected as conditional medium and added to NRCFs and further incubated for 24 hour. H through K, Western blot analysis and quantification of Col1α1, α‐SMA, and FN protein levels in NRCFs. n=5 per group. α‐SMA indicates alpha‐smooth muscle actin. CM indicates cardiac myocytes; CF, cardiac fibroblast; Col I, collagen 1a1; Ctrl, control group; and FN, fibronectin; PE, phenylephrinr group. Data were presented as mean±SEM, *P<0.05, **P<0.01, ***P<0.001.
Cardiomyocyte‐Specific Bmal1 Knockout Promotes Cardiac Remodeling via the PI3K/AKT Signaling Pathway
To further elucidate the underlying mechanisms by which cardiomyocyte Bmal1 deletion promotes cardiac remodeling, we performed RNA‐Seq using heart tissues from Ctrl and cKO mice in response to TAC for 4 weeks. According to volcano plot analysis, there were 846 differentially expressed genes between the 2 genotypes, the number of gene upregulated and downregulated in the cKO mice were 314 and 532, respectively (Figure 5A). Gene ontology (GO) analysis indicated that in the cKO mice, pressure overload provoked the different gene profile predominantly clustered into extracellular matrix compared with Ctrl mice (Figure 5B), which is consistent with in vivo phenotype. Kyoto Encyclopedia of Genes and Genomes (KEGG)pathway enrichment analysis identified that PI3K/AKT signaling pathway was the most prominent mediator of cardiac remodeling under Bmal1 deficiency in cardiomyocytes (Figure 5C). By further analysis of the enrichment map of genes related to PI3K/AKT signaling pathway, we observed that 13 genes down‐regulated and 14 genes up‐regulated in the cKO mice (Figure 5D). Consistent with the findings by RNA‐seq, the significant upregulation of cyclin D1 (Ccnd1), collagen type Ⅳ, α2 (Col4α2), secreted phosphoprotein 1 (Spp1), lysophosphatidic acid receptor 1(Lpar1), thrombospondin 4 (Thbs4) and as well as downregulation of fibroblast growth factor 23 (Fgf23), fibroblast growth factor 10 (Fgf10), interleukin 2 receptor and beta chain (IL2rb) were independently validated in cKO hearts upon TAC induction, using qRT‐PCR (Figure 5E through 5L). These findings indicated that the pro‐remodeling effect of cardiomyocyte Bmal1depletion might be largely associated with the activation of PI3K/AKT signaling pathway.
Figure 5. RNA‐sequence analysis of heart tissues from Ctrl and cKO mice after TAC surgery for 4 weeks.
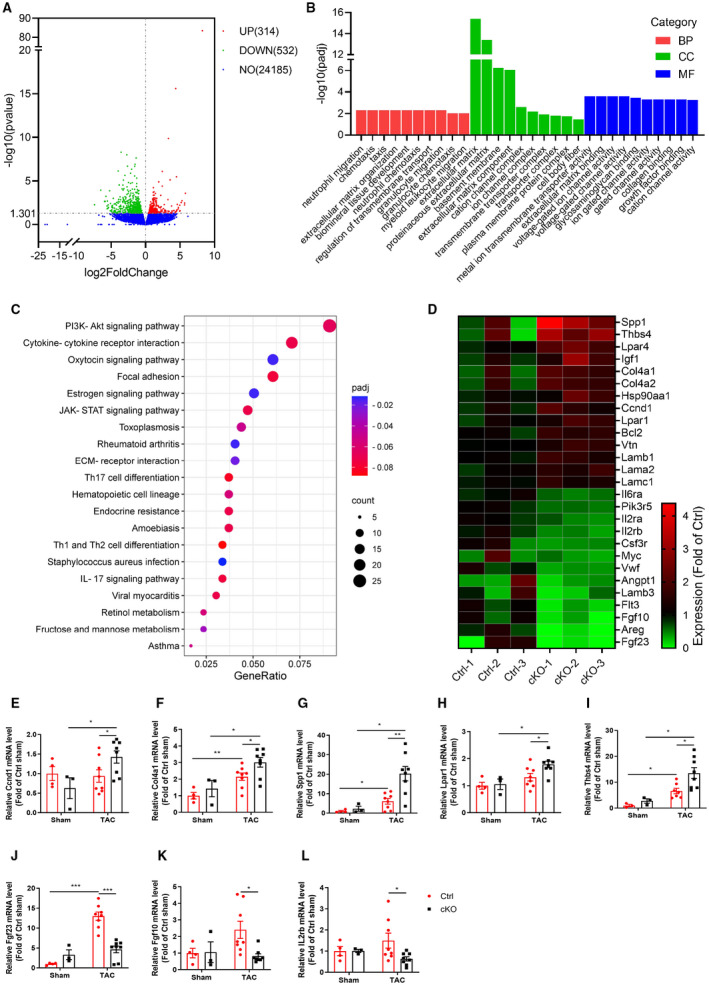
A, Volcano plot of the differentially expressed genes (DEGs). The red dots represent genes upregulated in cKO mice, the green dots represent genes downregulated in cKO mice, and the blue dots represent non‐differentially expressed genes. B, Gene ontology (GO) analysis of the DEGs indicated that, compared with the Ctrl mice, the different gene profile predominantly clustered into extracellular matrix in the cKO mice. C, KEGG pathway enrichment analysis revealed that pathways associated with PI3K/AKT signaling pathway were the most enriched by DEGs. D, Heatmap of the DEGs related to PI3K/AKT signaling pathway. The map is color coded with red corresponding to upregulation and blue to downregulation. E through L, Independent qRT‐PCR validation of Ccnd1 (E), Col4α2 (F), Spp1 (G), Lpar1 (H), Thbs4 (I) and Fgf23 (J), Fgf10 (K), IL2rb (L) expression in heart tissues from Ctrl and cKO mice. n=3 to 8 mice per group. BP, biological process; CC, cell component; cKO, cardiomyocyte‐specific Bmal1 knockout mice; Ctrl, control mice; MF, molecular function; NO, no difference; and TAC, transverse aortic constriction. Data were presented as mean±SEM, *P<0.05, **P<0.01, ***P<0.001.
Inhibition of PI3K/AKT Signaling Reverses the Pro‐Hypertrophic and Fibrogenic Effects of Bmal1 Silence in vitro
In vitro experiments were further performed to prove that the activation of PI3K/AKT pathway is essential for Bmal1 deficiency‐mediated cardiac hypertrophy and fibrogenesis. We found that PE treatment could significantly induce AKT phosphorylation in H9c2 cells, while this induction was dramatically augmented by Bma1 silence (Figure 6A). More importantly, in Bmal1 siRNA and PE treated hypertrophic cardiomyocytes, pre‐treatment with a pan‐AKT inhibitor MK2206 observably reversed the cardiomyocyte size and the expression of ANP (Figure 6B and 6C), implying that the pro‐hypertrophic effect of Bmal1 silence was largely dependent on excessive activation of PI3K/AKT signaling. Consistently, adding MK2206 to Bmal1 siRNA and PE treated NRVMs also remarkedly rescued the conditional medium evoked fibroblast‐to‐myofibroblast differentiation in NRCFs (Figure 6D through 6H). Collectively, the foregoing data verified that the pro‐hypertrophic and fibrogenic effects of Bmal1 deletion in cardiomyocytes predominantly depends on the activation of PI3K/AKT signaling.
Figure 6. Inhibition of PI3K/AKT reversed Bmal1 knockdown‐mediated cardiomyocyte hypertrophy and fibroblost‐to‐myofibroblast differentiation.
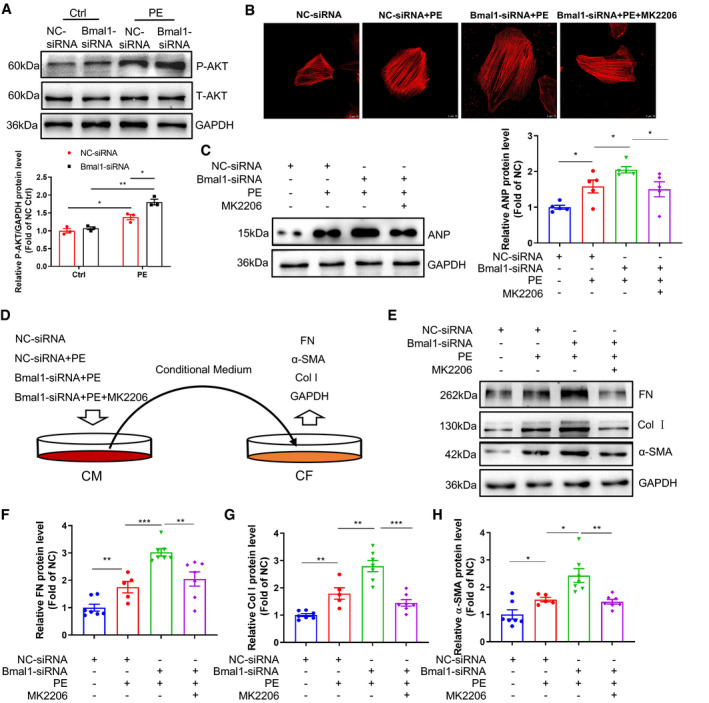
A, H9c2 cells transfected with negative control (NC) or Bmal1 siRNA for 24 hour, and then treated with or without PE (200 µmol/L) for 30 minutes. The levels of phosphorylated‐AKT (P‐AKT) and total‐AKT (T‐AKT) were detected by western blot. n=3 per group. B and C, H9c2 cells were transfected with NC or Bmal1 siRNA for 24 hour, and then treated with or without PE (200 µmol/L, 6 hour) and MK2206 (10 nmol/L, 1 hour). Cells were stained with Phalloidin (red) to reflect cell size (B). Scale bar: 10 μm. Hypertrophic marker protein ANP was determined by western blot (C). n=5 per group. D, A schematic showing of an in‐direct co‐culture model: NRVMs were pre‐transfected with NC or Bma1 siRNA for 24 hour and then exposed with or without PE (200 µmol/L, 6 hour) and MK2206 (10 nmol/L, 1 hour). After PE and MK2206 stimulation, the supernatant was collected as conditional medium and added to NRCFs and further incubated for 24 hour. E through H. Western blot analysis and quantification of FN, Col1α1 and α‐SMA protein levels in NRCFs. n=5 to 7 per group. Ctrl, control group; PE, phenylephrine; CM, cardiac myocytes; CF, cardiac fibroblasts; NRVMs, neonatal rat ventricular myocytes; NRCFs, neonatal cardiac fibroblasts. FN: fibronectin; Col I: collagen 1a1; a‐SMA: alpha smooth muscle actin. Data were presented as mean±SEM, *P<0.05, **P<0.01.
Discussion
Circadian rhythms are daily variations of physiological functions that are found in every living organism. At the molecular level, several core clock genes that consists of the rhythmic transcription‐translation feedback loop, including Bmal1, Clock, Per ,and Cry, are essential and expressed in all mammalian cells, including cardiomyocytes. 25 , 26 Among them, Bmal1 is the only one whose sole deletion results in complete loss of behavioral circadian rhythms. In the cardiovascular system, it has been shown that the conventional systemic Bmal1 knockout mice lack the diurnal variation in heart rate and blood pressure 8 and may eventually develop to premature cardiomyopathy. 27 Moreover, a recently published study demonstrated that mice with conventional cardiomyocyte specific Bmal1 deletion (CBK) also displayed an age‐associated dilated cardiomyopathy, characterized by thinning of the myocardial walls, dilation of the left ventricle, and decreased cardiac performance. 14 , 28 However, what cannot be ignored is that germline Bmal1 global deficiency may result in an early aging phenotype and the CBK mice showed similarly reduced life span, which might comprise the etiopathogenesis of their premature cardiomyopathy. 29 , 30 In addition, the CBK mouse who exhibits abnormalities in glucose utilization as well as abnormal metabolic responses also complicates its cardiac phenotype analysis. 14 Another confounding factor is that Bmal1 may play a crucial role in regulating the timing of cardiac development and growth. Upon analysis of cardiomyocyte size, Mellani et al. found that germline Bmal1 knockout hearts at 4 weeks of age had enlarged heart with reduced average cardiomyocyte size, 27 suggesting the possible existence of hyperplasia of cardiomyocyte. Since cardiomyocyte hyperplasia occurs mainly during embryonic development, 31 thus it’s possible that Bmal1 plays critical roles during embryonic cardiac development.
In this study, we aimed to clarify the role of Bmal1 in the cKO mice to possibly bypass the adverse effect of Bmal1 deficiency during early development and exclude the premature aging impact of the traditional Bmal1 knockout on cardiac function. Like the CBK mice, the diurnal variations of the whole‐body behavior (locomotor activity and food/water intake) and energy metabolism (respiratory exchange ratio, heat production) were not changed by the adulthood cardiomyocyte specific Bmal1 depletion, suggesting that the cardiomyocyte specific Bmal1 ablation did not impact the central circadian function. However, different from the CBK mice which exhibited age‐onset cardiomyopathy starting from 20 weeks of age and early death with mean survival age for 33 weeks, 14 we failed to observe obvious difference of the cardiac function between the cKO mice and littermate controls at 28 weeks of age (20 weeks post tamoxifen injection) (data not shown), implying that the postnatal cardiomyocyte specific Bmal1 depletion might be a better strategy to study the cardiac function of Bmal1 and the circadian machinery of the heart.
Next, by subjecting the cKO mice to TAC surgery and chronic AngⅡ infusion to mimic aortic valve stenosis or systemic hypertension and induce overload of the myocardium, we found that the postnatal cardiomyocyte Bmal1 deficiency significantly promoted pressure‐overload induced cardiac remodeling, reflective by increased heart weight, left ventricular mass, cardiomyocyte size, and augmented cardiac fibrosis. Regarding the hypertrophy‐specific gene expression in the heart, although we failed to detect difference of ANP, BNP and β‐MHC expression after 4 weeks TAC or AngⅡ treatment, whereas after 2 weeks TAC challenge, these genes did show significant upregulation in the cKO mice, implying a ceiling effect in the long‐term treatment groups. As known, under persistent pressure‐overload condition, cardiac remodeling evolved progressively from the initial compensatory hypertrophy and fibrosis to decompensation which will lead to diastolic dysfunction and ultimately heart failure and sudden death. 32 , 33 Intriguingly, based on the echocardiographic analysis of the cardiac function, we found that the left ventricular ejection fraction and left ventricular fractional shortening was significantly declined in the control mice after TAC surgery, however the contractile function remained unimpaired in the cKO groups. The heart function of the cKO mice was even enhanced after chronic AngⅡ infusion. This suggests that the promoted cardiac hypertrophy and fibrosis of the cKO mice is likely an adaptive and compensatory response to the pressure overload stimuli and depletion of Bmal1 in the cardiomyocytes may retard the decompensated heart failure and appears to be cardioprotective at this stage. However, if the pathological remodeling would be deleterious and lead to decompensation in the cKO mice with prolonged stimuli, such as TAC for 8 weeks, may need further investigation. Nevertheless, our current study did suggest that genetic manipulation of the cardiac molecular clock may significantly affect disease progressing and represent a therapeutic target for the treatment of pressure overload induced cardiac remodeling and heart failure. However, extrapolating the current data from mice to humans may require in‐depth investigation. Supportive to our current findings, a most recent study by He et al. demonstrated that pressure‐overload induced hypertrophy in rats was associated with a marked decrease in cardiac Bmal1 expression after 12 weeks of ascending aortic stenosis and the compound 'choline' which restored Bmal1 expression was associated with an attenuation of cardiac hypertrophy and fibrosis. 34 Indeed, several lines of evidence had previously demonstrated that normal day‐night rhythms are critical to the compensatory remodeling of cardiovascular tissue, and environmental circadian disruption (disrupted light/dark cycle) may augment myocardial maladaptation and exacerbate disease pathophysiology. 11 , 12 The importance of circadian rhythms and the molecular clock system in cardiovascular diseases merits in depth consideration and evaluation. Moreover, what requires to be clarified in our current study is that the promoted cardiac remodeling in the cKO mice does not appear to be due to the tamoxifen exposure to the MerCreMer mice, 23 , 35 as the hearts of the cKO mice in the sham groups (exposed to the same tamoxifen injection but without TAC surgery) exhibit negligible fibrosis and similar cardiac function relative to littermate controls. More importantly, similar effects were observed in in vitro experiments which are independent to Cre recombination and tamoxifen injection. Nevertheless, lack of the tamoxifen treated MerCreMer littermate controls is still a limitation of the present study. Another limitation of the current study is that the TAC surgery was performed only in male mice, but not in female mice. Considering the potential sex‐dependent differences in early cardiac response to pressure overload in mice, 36 , 37 future experiments on female mice are necessarily required.
The pathogenesis of cardiac remodeling is complicated. Studies have indicated that several signaling pathways could affect the progression of cardiac remodeling, such as MEK/JNK, Calcineurin/NFATc3 PI3K/AKT, and AMPK signal pathways. 38 With the aim for a more detailed view on the molecular changes driving the promoted hypertrophy and fibrosis in the cKO mice, we performed a RNAseq analysis of the TAC hearts with or without Bmal1 expression. Gene Ontology enrichment analysis of differentially expressed genes (DEGs) were mainly enriched in the extracellular matrix cluster which is consistent with the promoted remodeling in the cKO mice in vivo. KEGG pathway analysis further revealed the association of the DEGs in extracellular matrix and PI3K/AKT signaling, implying that activation of the PI3K/AKT pathway might be related to the promoted hypertrophy and fibrosis in the cKO mice. Indeed, the regulation of circadian clock on AKT phosphorylation had been reported in both cardiac and non‐cardiac tissues previously. 39 Martin E. Young and his colleges had also proved the direct regulation of Pik3r1 (the regulatory subunit of PI3K) by the cardiomyocyte Bmal1, 14 and both baseline and insulin‐mediated AKT activation was augmented in clock disrupted hearts. 40 Here we uncovered that under pathological conditions, typically pressure overloading, lack of Bmal1 expression in cardiomyocytes would similarly active the PI3K/AKT signaling pathway. Moreover, we confirmed that the activation of PI3K/AKT was associated with a functional impact on the pro‐hypertrophy and pro‐fibrosis effect of Bmal1 deficiency. By conducting the in vitro experiments of cultured cardiomyocytes with Bmal1 gene silencing, we demonstrated that identical to the in vivo findings, Bmal1 silencing significantly enlarged the cell surface area and augmented the AKT phosphorylation after phenylephrine stimuli, whereas when the cells were treated with AKT inhibitor, the pro‐hypertrophic effect of Bmal1 silencing was remarkedly reversed. Furthermore, cardiac fibroblasts interact with cardiomyocytes are essential for the progression of cardiac remodeling. 41 When we applied the conditional medium from Bmal1 inhibited cardiomyocytes to the primary cultured cardiac fibroblasts, the expression of phenylephrine evoked fibrosis‐related genes was further augmented, while this augmentation was reversed by AKT inhibitor pretreatment as well. This supports an idea that cardiomyocytes may directly response to the pro‐hypertrophic stimuli and secrete paracrine factors such as TGF‐β and fibroblast growth factor, and then activate fibroblasts to induce cardiac fibrosis. 24 Lack of Bmal1 in cardiomyocytes would augment this crosstalk and facilitate cardiac remodeling via promoting the AKT phosphorylation. However, the contribution of Bmal1 in fibroblasts to cardiac fibrosis cannot be excluded and more evidence is required.
In summary, the present study demonstrated that Bmal1 plays a prominent role in pressure overload induced cardiac remodeling in mice. Moreover, we identified PI3K/AKT as a significant pathway that may contribute to the adverse effect of Bmal1 in cardiac remodeling. These observations provide a new potential insight into the understanding of the relationship between circadian clock and pathological cardiac remodeling.
Sources of Funding
This work was supported by grants from National Key R&D Program of China (No. 2019YFA0802400) and National Natural Science Foundation of China (Nos. 32071157, 31871190, and 81900267).
Disclosures
None.
Supporting information
Tables S1–S4
Figures S1–S3
Supplemental Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.025021
For Sources of Funding and Disclosures, see page 15.
References
- 1. Smolensky MH, Hermida RC, Portaluppi F. Circadian mechanisms of 24‐hour blood pressure regulation and patterning. Sleep Med Rev. 2017;33:4–16. doi: 10.1016/j.smrv.2016.02.003 [DOI] [PubMed] [Google Scholar]
- 2. Douma LG, Gumz ML. Circadian clock‐mediated regulation of blood pressure. Free Radic Biol Med. 2018;119:108–114. doi: 10.1016/j.freeradbiomed.2017.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Crnko S, Du Pre BC, Sluijter JPG, Van Laake LW. Circadian rhythms and the molecular clock in cardiovascular biology and disease. Nat Rev Cardiol. 2019. doi: 10.1038/s41569-019-0167-4 [DOI] [PubMed] [Google Scholar]
- 4. Takeda N, Maemura K. Circadian clock and the onset of cardiovascular events. Hypertens Res. 2016;39:383–390. doi: 10.1038/hr.2016.9 [DOI] [PubMed] [Google Scholar]
- 5. López Messa JB, Garmendia Leiza JR, Aguilar García MD, Andrés de Llano JM, Alberola López C, Fernández JA, Grupo de Estudio A. Cardiovascular risk factors in the circadian rhythm of acute myocardial infarction. Rev Esp Cardiol. 2004;57:850–858. doi: 10.1016/S1885-5857(06)60650-6 [DOI] [PubMed] [Google Scholar]
- 6. Lopez‐Messa JB, Alonso‐Fernandez JI, Andres‐de Llano JM, Garmendia‐Leiza JR, Ardura‐Fernandez J, de Castro‐Rodriguez F, Gil‐Gonzalez JM. Circadian rhythm and time variations in out‐hospital sudden cardiac arrest. Med Intensiva. 2012;36:402–409. doi: 10.1016/j.medin.2011.11.007 [DOI] [PubMed] [Google Scholar]
- 7. Morris CJ, Purvis TE, Hu K, Scheer FA. Circadian misalignment increases cardiovascular disease risk factors in humans. Proc Natl Acad Sci USA. 2016;113:E1402–E1411. doi: 10.1073/pnas.1516953113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Curtis AM, Cheng Y, Kapoor S, Reilly D, Price TS, Fitzgerald GA. Circadian variation of blood pressure and the vascular response to asynchronous stress. Proc Natl Acad Sci USA. 2007;104:3450–3455. doi: 10.1073/pnas.0611680104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alibhai FJ, LaMarre J, Reitz CJ, Tsimakouridze EV, Kroetsch JT, Bolz S‐S, Shulman A, Steinberg S, Burris TP, Oudit GY, et al. Disrupting the key circadian regulator clock leads to age‐dependent cardiovascular disease. J Mol Cell Cardiol. 2017;105:24–37. doi: 10.1016/j.yjmcc.2017.01.008 [DOI] [PubMed] [Google Scholar]
- 10. Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139 [DOI] [PubMed] [Google Scholar]
- 11. Martino TA, Tata N, Belsham DD, Chalmers J, Straume M, Lee P, Pribiag H, Khaper N, Liu PP, Dawood F, et al. Disturbed diurnal rhythm alters gene expression and exacerbates cardiovascular disease with rescue by resynchronization. Hypertension. 2007;49:1104–1113. doi: 10.1161/HYPERTENSIONAHA.106.083568 [DOI] [PubMed] [Google Scholar]
- 12. Alibhai FJ, Tsimakouridze EV, Chinnappareddy N, Wright DC, Billia F, O'Sullivan ML, Pyle WG, Sole MJ, Martino TA. Short‐term disruption of diurnal rhythms after murine myocardial infarction adversely affects long‐term myocardial structure and function. Circ Res. 2014;114:1713–1722. doi: 10.1161/CIRCRESAHA.114.302995 [DOI] [PubMed] [Google Scholar]
- 13. Bray MS, Shaw CA, Moore MWS, Garcia RAP, Zanquetta MM, Durgan DJ, Jeong WJ, Tsai J‐Y, Bugger H, Zhang D, et al. Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function, metabolism, and gene expression. Am J Physiol Heart Circ Physiol. 2008;294:H1036–H1047. doi: 10.1152/ajpheart.01291.2007 [DOI] [PubMed] [Google Scholar]
- 14. Young ME, Brewer RA, Peliciari‐Garcia RA, Collins HE, He L, Birky TL, Peden BW, Thompson EG, Ammons B‐J, Bray MS, et al. Cardiomyocyte‐specific BMAL1 plays critical roles in metabolism, signaling, and maintenance of contractile function of the heart. J Biol Rhythms. 2014;29:257–276. doi: 10.1177/0748730414543141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Volland C, Schott P, Didié M, Männer J, Unsöld B, Toischer K, Schmidt C, Urlaub H, Nickels K, Knöll R, et al. Control of p21Cip by BRCA1‐associated protein is critical for cardiomyocyte cell cycle progression and survival. Cardiovasc Res. 2020;116:592–604. doi: 10.1093/cvr/cvz177 [DOI] [PubMed] [Google Scholar]
- 16. Zhang J, Sheng J, Dong L, Xu Y, Yu L, Liu Y, Huang X, Wan S, Lan HY, Wang H. Cardiomyocyte‐specific loss of RNA polymerase II subunit 5‐mediating protein causes myocardial dysfunction and heart failure. Cardiovasc Res. 2019;115:1617–1628. doi: 10.1093/cvr/cvy307 [DOI] [PubMed] [Google Scholar]
- 17. Liu S, Sun WC, Zhang YL, Lin QY, Liao JW, Song GR, Ma XL, Li HH, Zhang B. Socs3 negatively regulates cardiac hypertrophy via targeting GRP78‐mediated ER stress during pressure overload. Front Cell Dev Biol. 2021;9:629932. doi: 10.3389/fcell.2021.629932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yan W, Dong ZC, Wang JJ, Zhang YL, Wang HX, Zhang B, Li HH. Deficiency of the immunoproteasome LMP10 subunit attenuates angiotensin II‐induced cardiac hypertrophic remodeling via autophagic degradation of gp130 and IGF1R. Front Physiol. 2020;11:625. doi: 10.3389/fphys.2020.00625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu HU, Fang B, Du S, Wang S, Li Q, Jia X, Bao C, Ye L, Sui X, Qian L, et al. Endothelial cell prostaglandin E2 receptor EP4 is essential for blood pressure homeostasis. JCI Insight. 2020;5. doi: 10.1172/jci.insight.138505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ji S, Guo R, Wang J, Qian L, Liu M, Xu H, Zhang J, Guan Y, Yang G, Chen L. Microsomal prostaglandin E2 synthase‐1 deletion attenuates isoproterenol‐induced myocardial fibrosis in mice. J Pharmacol Exp Ther. 2020;375:40–48. doi: 10.1124/jpet.120.000023 [DOI] [PubMed] [Google Scholar]
- 21. Lorenzen JM, Schauerte C, Hübner A, Kölling M, Martino F, Scherf K, Batkai S, Zimmer K, Foinquinos A, Kaucsar T, et al. Osteopontin is indispensible for AP1‐mediated angiotensin ii‐related MIR‐21 transcription during cardiac fibrosis. Eur Heart J. 2015;36:2184–2196. doi: 10.1093/eurheartj/ehv109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non‐myocyte proliferation and requires TGF‐beta. J Clin Investig. 2010;120:3520–3529. doi: 10.1172/JCI42028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hall ME, Smith G, Hall JE, Stec DE. Systolic dysfunction in cardiac‐specific ligand‐inducible MerCreMer transgenic mice. Am J Physiol Heart Circ Physiol. 2011;301:H253–H260. doi: 10.1152/ajpheart.00786.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nuamnaichati N, Sato VH, Moongkarndi P, Parichatikanond W, Mangmool S. Sustained beta‐ar stimulation induces synthesis and secretion of growth factors in cardiac myocytes that affect on cardiac fibroblast activation. Life Sci. 2018;193:257–269. doi: 10.1016/j.lfs.2017.10.034 [DOI] [PubMed] [Google Scholar]
- 25. Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–320. doi: 10.1016/S0092-8674(02)00722-5 [DOI] [PubMed] [Google Scholar]
- 26. Zhang J, Sun R, Jiang T, Yang G, Chen L. Circadian blood pressure rhythm in cardiovascular and renal health and disease. Biomolecules. 2021;11:868. doi: 10.3390/biom11060868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lefta M, Campbell KS, Feng HZ, Jin JP, Esser KA. Development of dilated cardiomyopathy in bmal1 deficient mice. Am J Physiol. 2012;303:H475–H485. doi: 10.1152/ajpheart.00238.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ingle KA, Kain V, Goel M, Prabhu SD, Young ME, Halade GV. Cardiomyocyte‐specific BMAL1 deletion in mice triggers diastolic dysfunction, extracellular matrix response, and impaired resolution of inflammation. Am J Physiol Heart and Circ Physiol. 2015;309:H1827–H1836. doi: 10.1152/ajpheart.00608.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang G, Chen L, Grant GR, Paschos G, Song WL, Musiek ES, Lee V, McLoughlin SC, Grosser T, Cotsarelis G, et al. Timing of expression of the core clock gene BMAL1 influences its effects on aging and survival. Sci Transl Med. 2016;8:324ra316. doi: 10.1126/scitranslmed.aad3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age‐related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 2006;20:1868–1873. doi: 10.1101/gad.1432206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kamo T, Akazawa H, Komuro I. Cardiac nonmyocytes in the hub of cardiac hypertrophy. Circ Res. 2015;117:89–98. doi: 10.1161/CIRCRESAHA.117.305349 [DOI] [PubMed] [Google Scholar]
- 32. Liu X, Shi GP, Guo J. Innate immune cells in pressure overload‐induced cardiac hypertrophy and remodeling. Front Cell Dev Biol. 2021;9:659666. doi: 10.3389/fcell.2021.659666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu Y, Chen X, Zhang HG. Editorial: Cardiac Hypertrophy: From Compensation to Decompensation and Pharmacological Interventions. Front Pharmacol. 2021;12:665936. doi: 10.3389/fphar.2021.665936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. He X, Yang S, Deng J, Wu Q, Zang WJ. Amelioration of circadian disruption and calcium‐handling protein defects by choline alleviates cardiac remodeling in abdominal aorta coarctation rats. Lab Invest. 2021;101:878–896. doi: 10.1038/s41374-021-00578-6 [DOI] [PubMed] [Google Scholar]
- 35. Lexow J, Poggioli T, Sarathchandra P, Santini MP, Rosenthal N. Cardiac fibrosis in mice expressing an inducible myocardial‐specific Cre driver. Dis Models Mech. 2013;6:1470–1476. doi: 10.1242/dmm.010470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Witt H, Schubert C, Jaekel J, Fliegner D, Penkalla A, Tiemann K, Stypmann J, Roepcke S, Brokat S, Mahmoodzadeh S, et al. Sex‐specific pathways in early cardiac response to pressure overload in mice. J Mol Med. 2008;86:1013–1024. doi: 10.1007/s00109-008-0385-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ritterhoff J, McMillen TS, Villet O, Young S, Kolwicz SC Jr, Senn T, Caudal A, Tian R. Increasing fatty acid oxidation elicits a sex‐dependent response in failing mouse hearts. J Mol Cell Cardiol. 2021;158:1–10. doi: 10.1016/j.yjmcc.2021.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schirone L, Forte M, Palmerio S, Yee D, Nocella C, Angelini F, Pagano F, Schiavon S, Bordin A, Carrizzo A, et al. A review of the molecular mechanisms underlying the development and progression of cardiac remodeling. Oxid Med Cell Longev. 2017;2017:3920195. doi: 10.1155/2017/3920195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ko ML, Shi L, Tsai JY, Young ME, Neuendorff N, Earnest DJ, Ko GY. Cardiac‐specific mutation of clock alters the quantitative measurements of physical activities without changing behavioral circadian rhythms. J Biol Rhythms. 2011;26:412–422. doi: 10.1177/0748730411414170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McGinnis GR, Tang Y, Brewer RA, Brahma MK, Stanley HL, Shanmugam G, Rajasekaran NS, Rowe GC, Frank SJ, Wende AR, et al. Genetic disruption of the cardiomyocyte circadian clock differentially influences insulin‐mediated processes in the heart. J Mol Cell Cardiol. 2017;110:80–95. doi: 10.1016/j.yjmcc.2017.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. 2002;91:1103–1113. doi: 10.1161/01.RES.0000046452.67724.B8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S4
Figures S1–S3