

SUMMARY
Necroptosis mediated by Z-nucleic-acid-binding protein (ZBP)1 (also called DAI or DLM1) contributes to innate host defense against viruses by triggering cell death to eliminate infected cells. During infection, vaccinia virus (VACV) protein E3 prevents death signaling by competing for Z-form RNA through an N-terminal Zα domain. In the absence of this E3 domain, Z-form RNA accumulates during the early phase of VACV infection, triggering ZBP1 to recruit receptor interacting protein kinase (RIPK)3 and execute necroptosis. The C-terminal E3 double-strand RNA-binding domain must be retained to observe accumulation of Z-form RNA and induction of necroptosis. Substitutions of Zα from either ZBP1 or the RNA-editing enzyme double-stranded RNA adenosine deaminase (ADAR)1 yields fully functional E3 capable of suppressing virus-induced necroptosis. Overall, our evidence reveals the importance of Z-form RNA generated during VACV infection as a pathogen-associated molecular pattern (PAMP) unleashing ZBP1/RIPK3/MLKL-dependent necroptosis unless suppressed by viral E3.
Graphical abstract
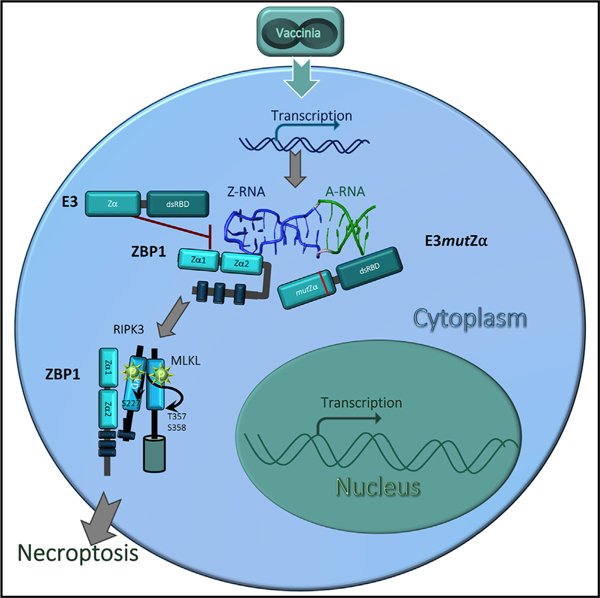
In brief
ZBP1-activated necroptosis is a host-defense mechanism against vaccinia virus. Koehler and colleagues delineate how the vaccinia-encoded protein E3, a virulence factor composed of both dsRNA and Z-nucleic-acid-binding domains, suppresses ZBP1-activated necroptosis. Disruption of E3 Z-RNA binding triggers necroptosis by exposing Z-RNA in the cytoplasm early after infection.
INTRODUCTION
Necroptosis is an important host-defense mechanism against viruses (Guo et al., 2018; Kaiser et al., 2013; Koehler et al., 2017; Mocarski et al., 2015; Nogusa et al., 2016; Shubina et al., 2020; Upton and Chan, 2014; Upton et al., 2010, 2012) that is mediated by receptor interacting protein (RIP) kinase (RIPK)3 and mixed-lineage kinase-domain-like (MLKL), a pseudokinase that drives plasma membrane leakage (Sun et al., 2012). RIPK3 contains an N-terminal kinase domain and a C-terminal RIP homotypic interaction motif (RHIM). To trigger necroptosis, RIPK3 must first be recruited through RHIM-dependent interactions with RIPK1, TIR-domain-containing adapter-inducing interferon (IFN)β (TRIF) or Z-nucleic acid (NA)-binding protein 1 (ZBP1). TNF-induced RIPK1/RIPK3-dependent necroptosis (Cho et al., 2009) and virus-induced ZBP1/RIPK3-dependent necroptosis (Koehler et al, 2017) act in concert against VACV as this virus replicates in the cytoplasm of infected cells.
The ZBP1/RIPK3 pathway of necroptosis kills cells during infection with E3 N-terminal deletion mutant VACV, E3Δ83N (Koehler et al, 2017). This mutant completely lacks a Zα-motif-containing domain known to bind left-handed Z-form double-stranded (ds)NA (Z-NA) (Kim et al., 2004) but retains a C-terminal dsRNA-binding domain (dsRBD) known to interact with conventional right-handed A-form dsRNA (Figure 1). The dsRBD is best known for preventing protein kinase R (PKR) activation (Chang et al., 1992). E3 must retain the ability to contact Z-NA to preserve virulence and immunomodulation of interferon (IFN) signaling (Brandt et al., 2005; Kim et al., 2003; White and Jacobs, 2012). Notably, alanine substitutions replacing conserved amino acids, either P63 (E3P63A) or Y48 (E3Y48A), yield attenuated viruses that replicate poorly in mice similar to viruses with complete deletion of the N-terminal Zα-motif-containing region. Given LD50 of Zα mutants are multiple orders of magnitude higher than wild-type (WT) VACV, E3 Z-NA-binding contribution to VACV pathogenesis has been attributed to suppression of virus-induced necroptosis (Koehler et al, 2017). The precise mechanisms through which E3 Zα promotes replication and Zα-deficient VACV triggers ZBP1-dependent necroptosis have not been explored.
Figure 1. Amino acids in the N terminus of E3 that impart Z-NA binding are required to restrict VACV-induced necroptosis.
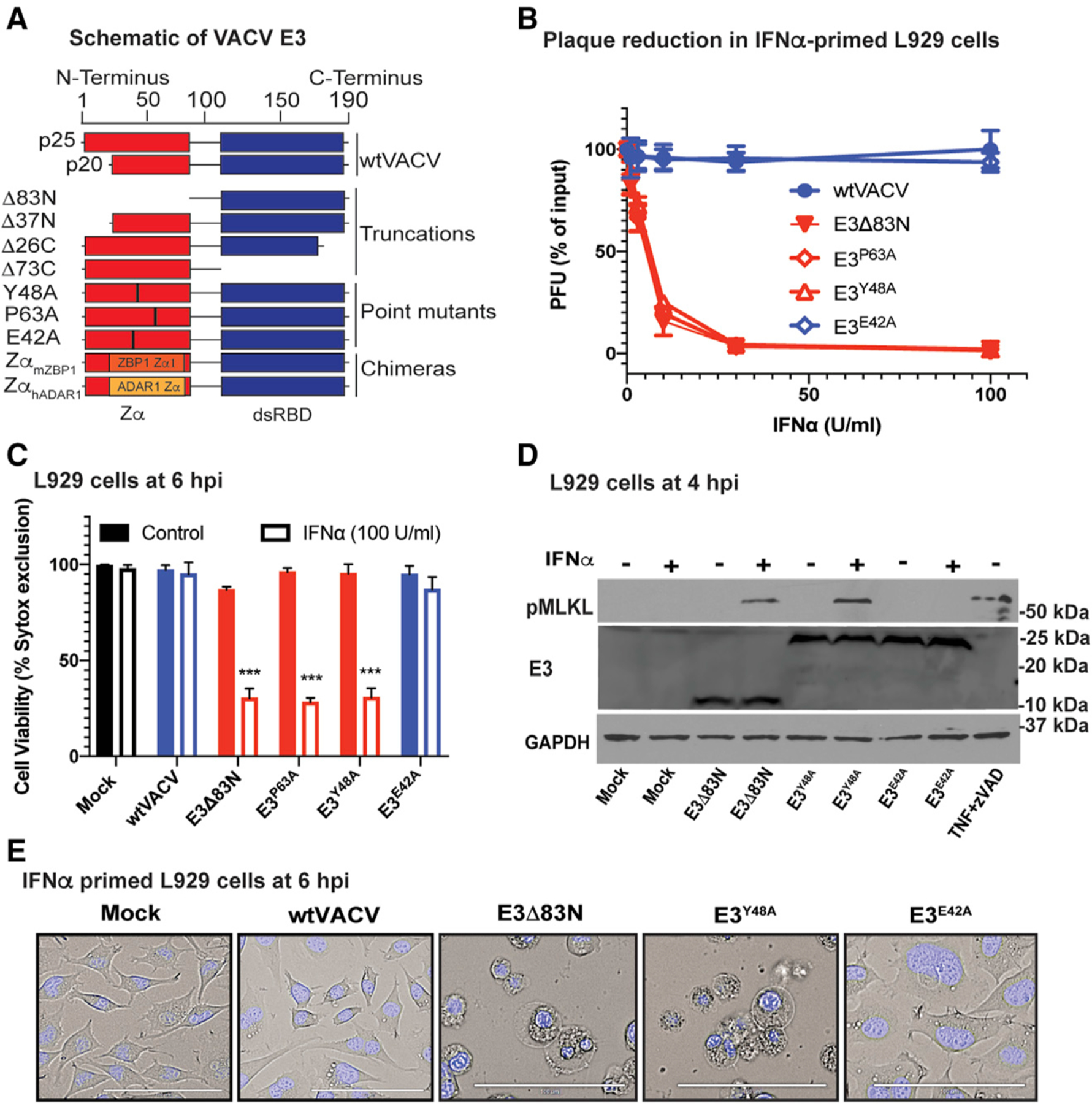
(A) A schematic diagram of VACV E3 and E3 derivatives made previously (Kim et al., 2003) and used in this study, including truncation mutants (E3Δ83N, E3Δ37N, E3Δ26C, and E3Δ73C), single amino acid substitution mutants (E3Y48A, E3P63A, and E3E42A) and chimeric E3 with Zα substitutions (ZαhADAR1 and ZαmZBP1).
(B) Plaque reduction assay performed by pretreating L929 cells with increasing concentrations of mouse IFN-α for 18 h prior to infection. Percent reduction of PFU was calculated as a proportion of input PFU for each virus evaluated. E3Y48A and E3P63A point mutations disrupt Z-NA binding; whereas, E3E42A mutation is at a non-essential location of the Zα so this protein retains Z-NA binding and is included as a control (Kim et al., 2003).
(C) L929 cells either left untreated or treated with mouse IFN-α (100 U/mL) for 18 h and subsequently infected at a MOI of 5 with each of the viruses shown. Cell viability was determined by SYTOX dye exclusion by counting an average of 10 fields microscopically at 6 hpi in three individual experiments.
(D)Immunoblot (IB) of L929 cell lysates either left untreated (−) or pretreated (+) with IFN-α and prepared at 4 hpi (MOI of 5 with indicated viruses) for phospho-MLKL, total E3 levels and GAPDH (as a loading control) following separation by SDS-PAGE. Mock infected cells and cells induced into necroptosis by TNF at 25 ng/mL in the presence of 50 µM zVAD-fmk from 1 h prior to TNF (Koehler et al., 2017) are included as a negative and positive controls, respectively.
(E) Phase contrast micrographs showing the morphology of L929 cells pretreated with IFN-α and left uninfected (mock) or infected at a MOI of 5 with the indicated viruses before being stained at 6 hpi with cell-permeant Hoechst 33342 and visualized. Bar indicates 100 µM.
Error bars represent the standard deviation (SD). Figures are representative of three independent replicates, except (B) and (C), which compiles the results of the replicates. Statistical significance was determined by a nonparametric Mann-Whitney test using GraphPad Prism9 with p ≤ 0.05 considered significant (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
ZBP1 has long been recognized as an interferon-inducible protein (called DLM-1) later implicated as a DNA-dependent activator of IFN regulatory factors (DAI) (Takaoka et al., 2007) as well as an inducer of NFκB-dependent transcription through RHIM interactions with RIPK1 and RIPK3 (Kaiser et al., 2008). Although ZBP1 contributes to IFN induction during infection with herpes simplex virus (HSV)1 (Pham et al., 2013) and human cytomegalovirus (HCMV) (DeFilippis et al., 2010), this specialized pathogen sensor is best known for triggering virus-induced death pathways (Mocarski et al., 2015; Upton and Chan, 2014). The characterization of murine cytomegalovirus (MCMV) mutants lacking the viral inhibitor of RIP activation (vIRA) brought to light RHIM signaling suppression to prevent recruitment and activation of RIPK3 during infection (Upton et al., 2010, 2012). In this setting, the viral inhibitor of caspase-8 activation (vICA) sensitizes cells to necroptosis (Kaiser et al., 2011; Mocarski et al., 2011) depending on the cell type that is infected (Daley-Bauer et al., 2017). HSV1 relies on a homologous RHIM suppressor to prevent activation of ZBP1 by newly synthesized RNA in human cells (Guo et al., 2018). During infection with viruses such as influenza A virus (IAV) that fail to suppress caspase-8, ZBP1 senses RNA and triggers combined apoptotic and necroptotic cell death pathways (Kuriakose et al., 2016; Nogusa et al., 2016; Thapa et al., 2016; Zhang et al., 2020). Combined death pathways are observed during infection with cell death suppressor mutants of either HSV1 (Guo et al., 2015) or MCMV (Daley-Bauer et al., 2017). In some settings, markers of inflammasome activation and pyroptosis are also observed, prompting the term PANoptosis (Samir et al., 2020). The combined activation of apoptosis and necroptosis by RIPK3 initially emerged from studies of kinase mutants and inhibitors (Mandal et al., 2014). The Zα domain characteristic of the ZBP1/ADAR1 family has long been known to recognize either Z-form DNA or RNA (Placido et al., 2007; Schwartz et al., 2001). Studies with IAV (Nogusa et al., 2016; Thapa et al., 2016; Zhang et al., 2020), MCMV (Maelfait et al., 2017; Sridharan et al., 2017), and HSV1 (Guo et al., 2018) have implicated ZBP1 as a natural sensor of newly transcribed RNA rather than DNA, extending and correcting earlier interpretations that had not considered the potential role for RNA (Pham et al., 2013; Takaoka et al., 2007). In each of these settings, dsRNA accumulates within infected cells as a pathogen-associated molecular pattern (PAMP) that is recognized by ZBP1 dependent on the presence of the Zα1 and/or Zα2. Most recently, restriction of VACV replication and pathogenesis by Zα-deficient E3Δ83N mutant virus (Figure 1A) was revealed to be dependent on ZBP1/RIPK3-dependent necroptosis (Koehler et al., 2017). Although E3 Zα appears to compete for Z-NA species produced in the course of virus infection, evidence of Z-NA accumulation in VACV-infected cells as well as the means through which ZBP1 becomes activated remain unresolved.
Here, we show that Z-form RNA accumulates during the early phase of Zα-deficient VACV infection, triggering ZBP1 to initiate necroptosis. Z-RNA serves as the ZBP1-activating PAMP; however, distinct from observations with IAV defective interfering particles in the nucleus of infected cells (Zhang et al., 2020), this DNA virus promotes the accumulation of Z-RNA in the cytoplasm dependent on the viral E3 C-terminal dsRNA-binding domain (dsRBD). Thus, expression of mutant E3 that retains dsRNA-binding capacity but lacks Z-NA-binding capacity leaves VACV susceptible to ZBP1-mediated sensing through the exposure of Z-RNA in the cytoplasm of infected cells. This scenario implicates the E3 dsRBD in promoting PAMP formation and the Zα domain in suppressing ZBP1 activation during WT VACV infection.
RESULTS
Amino acids in the N terminus of E3 that impart Z-NA binding are required to restrict VACV-induced necroptosis
As a virulence factor, E3 is known to confer type I IFN resistance on VACV (Brandt and Jacobs, 2001; Chang et al., 1992) through the action of its N-terminal Zα domain together with its C-terminal dsRBD (Figure 1A). Both domains contribute to type I IFN resistance as well as to viral pathogenesis in mice (Brandt et al., 2005; Kim et al., 2003; White and Jacobs, 2012). The N-terminal domain suppresses virus-induced necroptosis (Koehler et al., 2017). Type I IFN priming boosts ZBP1 levels in L929 cells rendering these cells susceptible to necroptosis following infection with Zα-deficient E3Δ83N. To determine more precisely whether E3 suppression of virus-induced necroptosis involves interaction with Z-NA, Zα loss-of-function point mutants (Kim et al., 2003) generated by alanine substitution (E3Y48A or E3P63A) were evaluated in IFNα-primed L929 cells along with WT VACV as well as control E3E42A (Figure 1A). When infected with either loss-of-function point mutant, L929 cells showed a pattern of compromised replication (Figure 1B), accompanied by loss of membrane integrity (Figure 1C), activated MLKL (Figure 1D), and necrotic morphology (Figure 1E). In contrast, E3E42A, like WT VACV, failed to trigger appreciable death of infected cells. These data demonstrate that the suppression of VACV-induced necroptosis depends on an E3 domain characterized for its ability to recognize Z-NA (Ha et al., 2004; Kim et al., 2004).
ZBP1 Zα2 domain senses VACV and triggers necroptosis
Necroptosis-sensitive L929 and SVEC4–10 cells were employed to investigate the contribution of type I IFN priming to cell death following E3 Zα-deficient VACV infection. L929 cells required priming with IFN-β to exhibit robust MLKL phosphorylation (Figure 2A, left) and loss of viability (Figure 2B) indicative of necroptosis. In contrast, E3 mutant VACV triggered phosphorylation of MLKL (Figure 2A, left) and necroptosis (Figure 2B) in SVEC4–10 cells independent of priming, consistent with constitutive high ZBP1 levels independent of pretreatment (Figure 1A). SVEC4–10-derived ZBP1-null empty vector (EV) control cells (Sridharan et al., 2017) resisted death unless stably reconstituted with FLAG epitope-tagged full-length WT ZBP1 (Figure 2A, middle, and 2B). Likewise, siRNA-mediated inhibition of ZBP1 expression in IFN-β-primed L929 cells prevented VACV-induced necroptosis (Figure 2A, right, and 2B). Together, these data establish that type I IFN sensitizes cells to VACV-induced necroptosis by boosting ZBP1 levels as noted for other settings (Ingram et al., 2019; Kuriakose et al., 2016; Thapa et al., 2016; Yang et al., 2020). SVEC4–10 cells express constitutive levels of ZBP1 that support ZBP1-dependent necroptosis without a requirement for IFN priming consistent with previous observations (Upton et al., 2010, 2012).
Figure 2. ZBP1 requires Zα2 to sense VACV and trigger necroptosis.
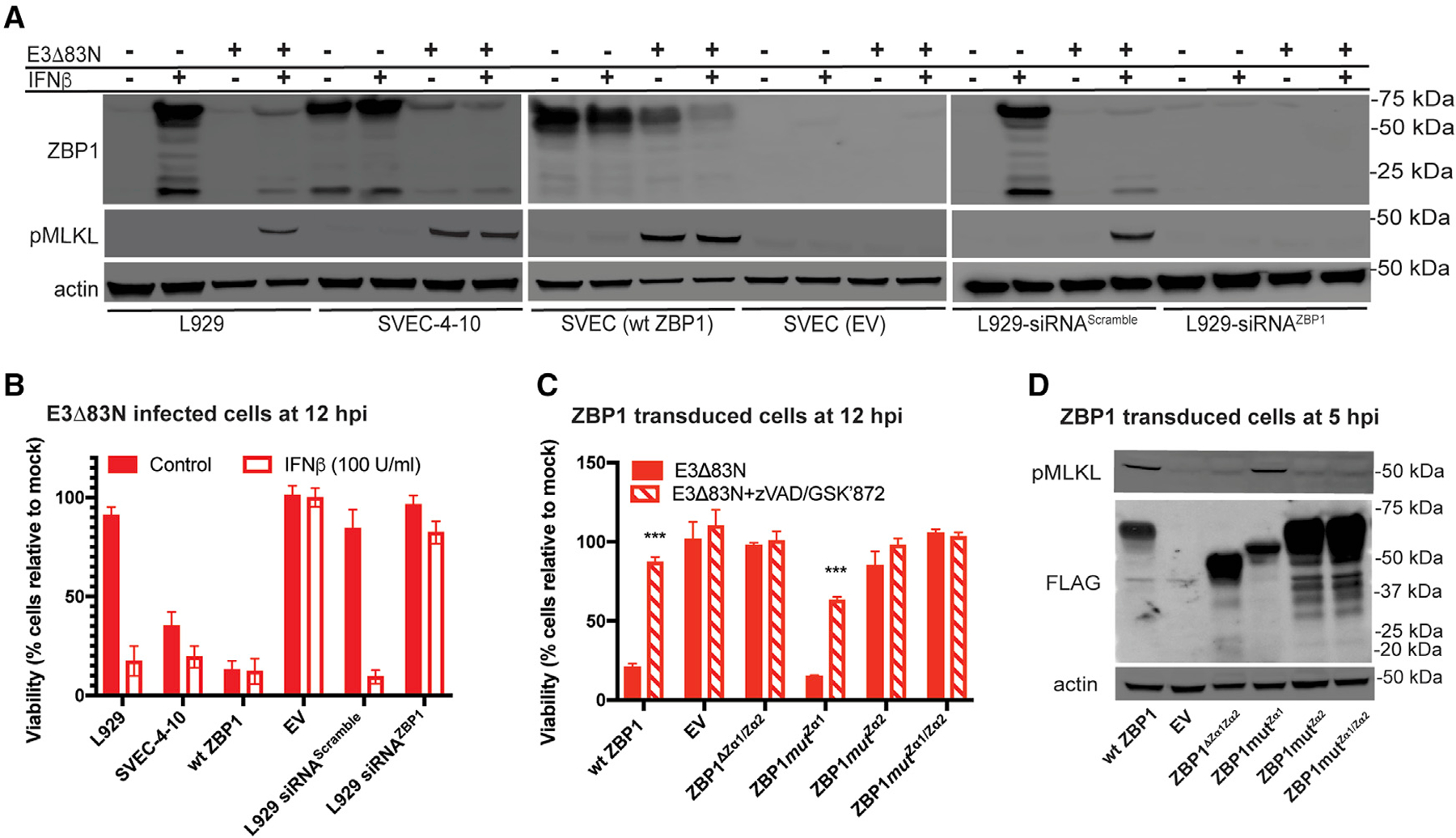
(A) IB of L929, SVEC4–10, FLAG-ZBP1-reconstituted SVEC (WT ZBP1) and ZBP1-null SVEC (EV) (Sridharan et al., 2017) cells either left untreated (−) or pretreated with 100 U/mL of mouse IFN-β for 18 h (+) and either left uninfected (−) or infected at an MOI of 5 with E3Δ83N (+). L929 cells in rightmost panel were transfected with either a scrambled siRNA pool or an siRNA pool targeting ZBP1 for 36 h prior to IFN-β treatment. Lysates were harvested at 4 hpi, and, following SDS-PAGE, evaluated for phospho-MLKL and ZBP1.
(B) Viability of L929, SVEC4–10, WT ZBP1 or EV cells either untreated or pretreated with IFN-β and then infected at an MOI of 5 with E3Δ83N for 12 h determined by measuring intracellular ATP levels with a Cell Titer-Glo luminescent cell viability assay kit. Rightmost bars show results from cells transfected with either a scrambled siRNA pool or an siRNA pool targeting ZBP1 as described in (A). CellTiter-Glo assay (Promega).
(C) Viability of ZBP1-null SVEC29–11 cells reconstituted with indicated ZBP1 constructs infected with E3Δ83N either alone or in combination with zVAD-fmk plus GSKʹ872 assessed at 12 hpi by CellTiter-Glo assay as described in (B).
(D) IB of ZBP1-reconstituted SVEC29–11 cells infected at a MOI of 5. Lysates were prepared as described in (A) and evaluated for phospho-MLKL and FLAG-ZBP1.
Error bars represent the SD. Each set of data is representative of two replicates except for (B) and (C), which compiles the results of the replicates. Statistical significance was determined as described in Figure 1.
In order to interrogate the contribution of ZBP1 Z-NA-binding domains, cells expressing epitope-tagged WT ZBP1 as well as Zα1 and Zα2 loss-of-function mutants (Sridharan et al., 2017) known to disrupt binding to Z-NA (Brown et al., 2000; Placido et al., 2007) were evaluated for susceptibility to VACV-induced necroptosis. SVEC4–10-derived ZBP1-null cells stably transduced with WT ZBP1 exhibited sensitivity to necroptosis compared with EV control (Figure 2C). Cells transduced with ZBP1 deletion mutant of Zα1 together with Zα2 (ZBP1ΔZα1/Zα2) or combined loss-of-function point mutants in Zα1 (N46D/Y50A) together with Zα2 (N122D/Y126A) (ZBP1mutZα1Zα2) resisted cell death. Mutation of Zα2 (ZBP1mutZα2) alone conferred resistance as well, whereas mutation of Zα1 (ZBP1mutZα1) alone did not. Necroptosis-sensitive cells treated with a death suppressor cocktail composed of RIPK3-inhibitor GSKʹ872 and caspase inhibitor zVAD-fmk did not succumb to E3Δ83N-induced death. These combined inhibitors preserved cell viability in all settings as expected from prior characterization of prominent RIPK3-dependent cell death pathways (Mandal et al., 2014). The dependence of VACV-induced necroptosis on ZBP1 Zα2 was confirmed by immunoblots showing the absence of phosphorylated MLKL in ZBP1ΔZα1/Zα2, ZBP1mutZα2, and ZBP1mutZα1/Zα2 as well as EV control (Figure 2D). In contrast, cells transduced with either WT ZBP1 or ZBP1mutZα1 showed the expected activation of MLKL consistent with triggering necroptosis. Together, these results implicate ZBP1 Zα2 in sensing VACV to initiate necroptosis. These results are reminiscent of a requirement for Zα2 in other characterized virus-induced death settings where ZBP1 has been implicated as the pathogen sensor (Guo et al., 2018; Kesavardhana et al., 2020; Sridharan et al., 2017; Thapa et al., 2016) as well as settings where RIPK1-deficient cells have shown sensitivity to IFN-induced, ZBP1-dependent necroptosis (Ingram et al., 2019; Yang et al., 2020).
VACV E3 Zα and dsRNA-binding domains collaborate to regulate necroptosis
To further characterize the requirements for VACV-induced necroptosis, we evaluated E3-domain-specific mutants affecting the N-terminal Z-NA-binding domain and/or C-terminal A-form-specific dsRBD (Chang and Jacobs, 1993). Infection with N-terminal deletion mutants E3Δ37N unleashed necroptosis with a pattern identical to E3Δ83N (Koehler et al., 2017), including loss of membrane integrity by 6 hpi (Figure 3A) subsequent to activation of MLKL (Figure 3B). Two different dsRBD mutants (E3Δ26C or E3Δ73C) resisted death, suggesting that A-RNA binding did not contribute to cell death suppression. Surprisingly, E3-deficient VACV (ΔE3L) resisted necroptosis. Thus, the E3 dsRBD must be retained in order for E3 Zα-deficient virus to unleash necroptosis. To evaluate whether E3 dsRBD and Zα domain function when expressed as separate proteins, IFN-primed L929 cells were simultaneously infected with VACV carrying the C- and N-terminal mutations such that both domains were expressed on separate E3 derivatives. As expected, WT VACV or E3Δ73C infections alone did not cause the death of cells and E3Δ37N-induced death (Figure 3C) associated with activated MLKL (Figure 3D). There was no change in this pattern of necroptosis during coinfections of E3Δ37N, capable of binding dsRNA, together with E3Δ73C, capable of recognizing Z-NA. Thus, the E3 Zα domain and the dsRBD must be retained on the same molecule to observe cell death suppression. These studies identify the E3 dsRNA binding as necessary to either sensitize cells to ZBP1-dependent death or to prohibit other pathways on which necroptosis depends.
Figure 3. VACV E3 Zα domain and dsRBD collaborate to regulate cell death.
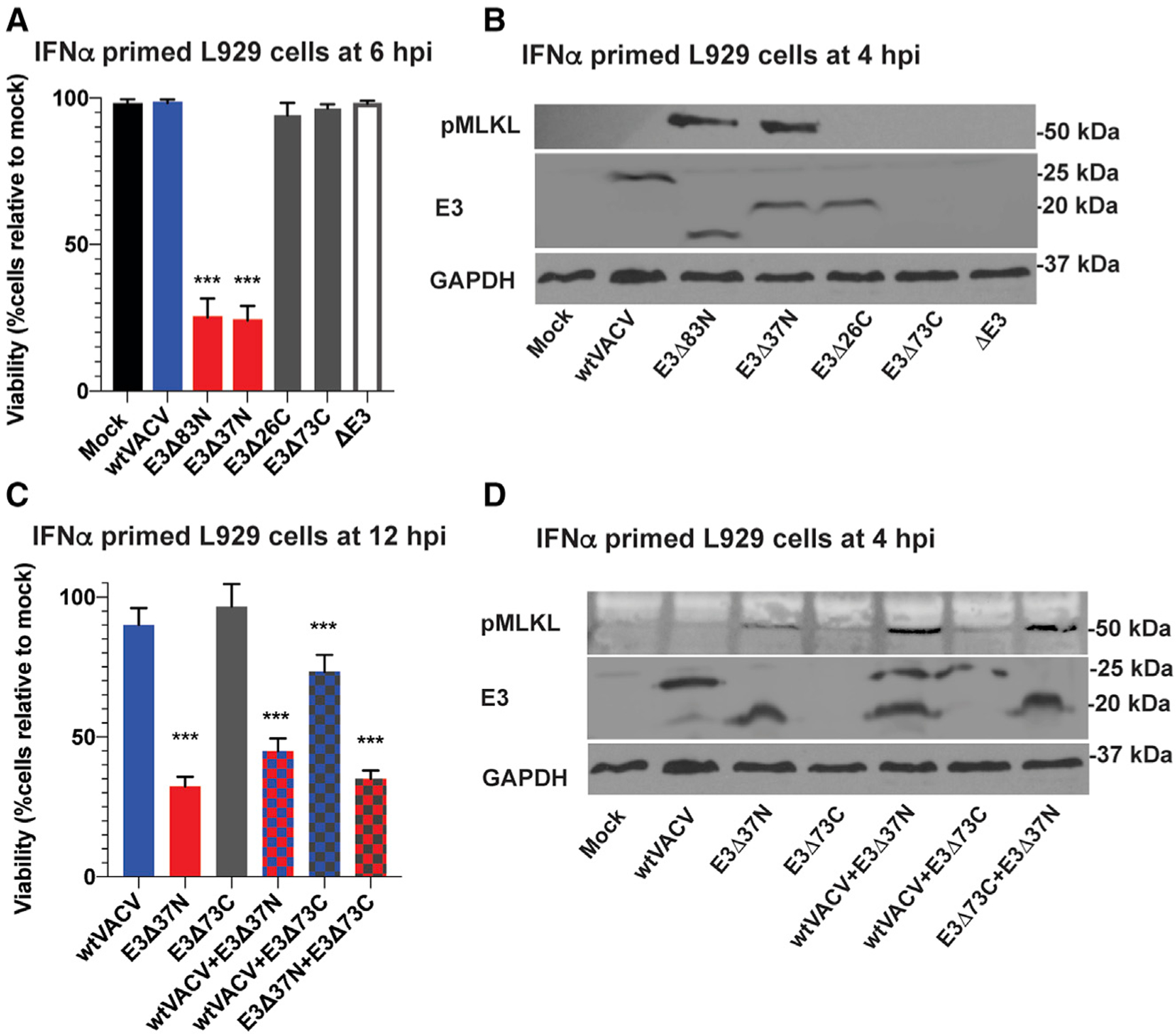
(A) Viability of L929 cells that were IFN-α-pretreated as described in Figure 1 and subsequently infected at an MOI of 5 with the indicated viruses. Cell viability was determined at 6 hpi by Sytox dye exclusion as described in Figure 1.
(B) IB of L929 cells that were IFN-α-pretreated as described in Figure 1 prior to infection at an MOI of 5 with the indicated viruses. Lysates were harvested at 4 hpi and, following SDS-PAGE, evaluated for phospho-MLKL.
(C) Viability of IFN-β-pretreated L929 cells were infected with a single virus at an MOI of 10 or coinfected with two viruses each at an MOI of 5. Cell viability was determined at 12 hpi by CellTiter-Glo assay as described in Figure 2.
(D) IB of IFN-β-primed L929 cells infected with single virus or coinfected with two viruses as described in (C). Lysates were harvested at 4 hpi and, following SDS-PAGE, evaluated for phospho-MLKL. Error bars represent the SD. Each set of data is representative of three replicates except for (A) and (C) which compiles the results of the replicates. Statistical significance was determined as described in Figure 1.
VACV E3 Zα suppresses transcription-dependent necroptosis independent of PKR
To determine the steps in viral replication necessary to trigger necroptosis, we utilized a series of chemical inhibitors known to block stages of VACV replication. We first showed that virus entry was necessary, given that UV-inactivated E3Δ83N particles failed to activate MLKL (Figure 4A). E3Δ83N-infected cells treated with inhibitors of protein synthesis (cytosine arabinoside or cycloheximide) or DNA synthesis (hydroxyurea) supported activation of MLKL, whereas a general inhibitor of RNA transcription (actinomycin D) or specific inhibitor of VACV transcription (Sarrsina) (Arndt et al., 2012) blocked MLKL phosphorylation (Figure 4B). These data reveal a role for early transcription prior to viral DNA replication in the induction of necroptosis reminiscent of the requirement for transcription in other settings where virus-induced ZBP1 activation initiates a cell death cascade (Guo et al., 2018; Kesavardhana et al., 2020; Sridharan et al., 2017; Thapa et al., 2016).
Figure 4. VACV E3 Zα regulates transcription-dependent induction of necroptosis without any contribution of PKR.
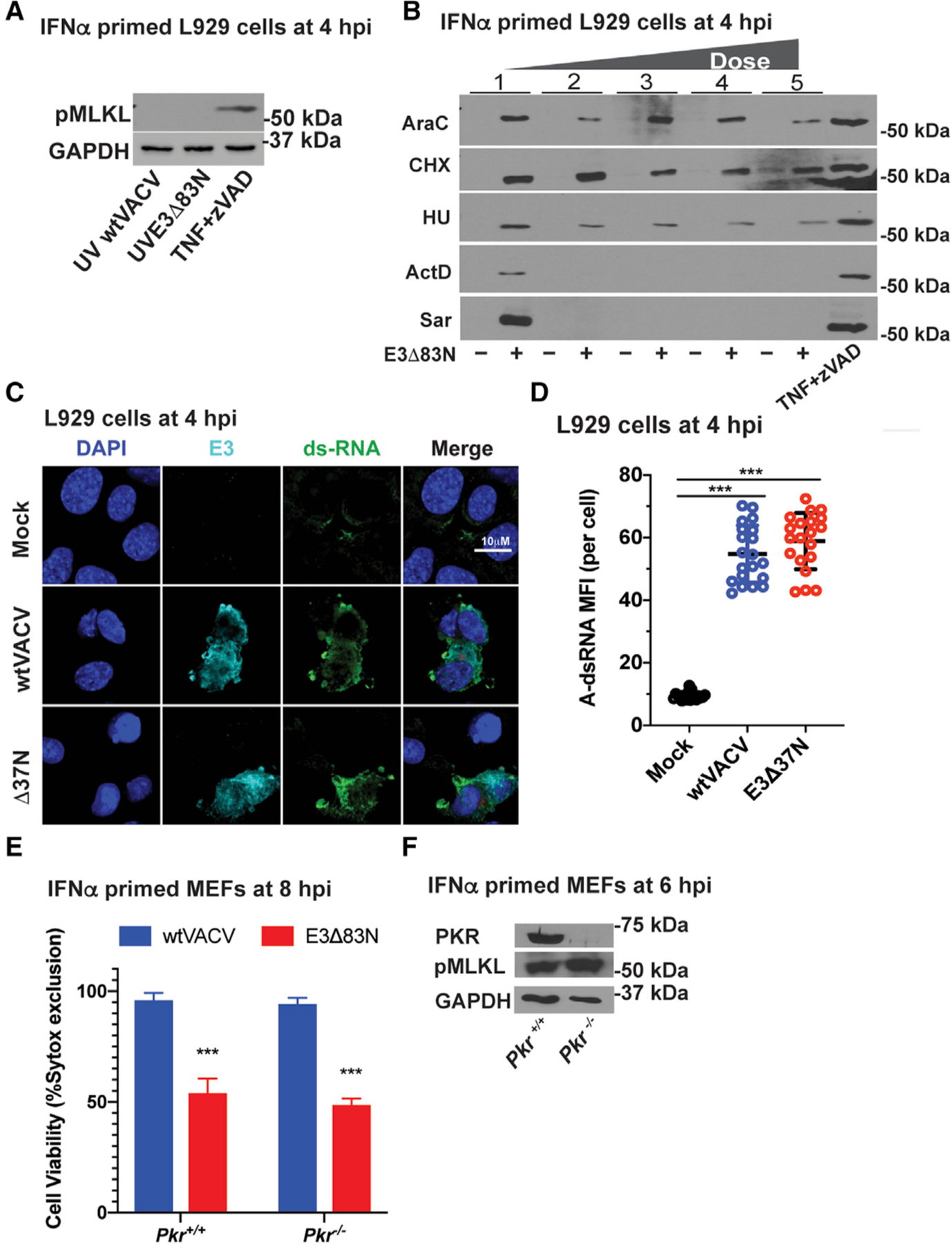
(A) IB of L929 cells that were IFN-α-pretreated as described in Figure 1 and then infected with an equivalent MOI of 5 using UV-inactivated viral particles. TNF plus zVAD-fmk is shown as a positive control as described in Figure 1. Lysates were harvested at 4 hpi and, following SDS-PAGE, evaluated for phospho-MLKL.
(B) IB of L929 cells that were IFN-α-pretreated as described in Figure 1 and then treated with increasing doses of the following inhibitors: cytosine arabinoside (AraC), at 200, 400, 800, and 1,600 mg/mL; cycloheximide (CHX) at 20, 40, 80, and120 mg/mL; hydroxyurea (HU) at 5, 10, 20, and 40 mM: actinomycin D (ActD) at 1, 2, 4, and 8 mg/mL; and, Sarracenia extract (Sar) at 10, 30, 60, and 120 µL/mL. Treatment with AraC, HU, ActD, and Sar started 1 h prior to infection and continued throughout infection. Treatment with CHX started 30-min post-adsorption and continued throughout infection. Positive control TNF plus zVAD-fmk is described in Figure 1. Lysates were harvested at 4 hpi and, following SDS-PAGE, evaluated for phospho-MLKL.
(C) Confocal immunofluorescent micrographs of L929 cells either uninfected (mock) or virus-infected at a MOI of 5 with WT VACV or E3Δ37N, fixed and permeabilized then subsequently stained with J2 anti-A-RNA (green), anti-E3 (cyan), and DAPI nuclear dye (blue). Bar represents 10 μM.
(D) Quantification of the median fluorescence intensity of A-form dsRNA-specific staining by J2 antibody analyzed with Leica LAS X software to generate mean fluorescence intensity of 20 individual cells with mean indicated.
(E) Viability of IFN-primed Pkr+/+ and Pkr−/− MEFs infected at a MOI of 5 with WT VACV or E3Δ83N. Cell viability was determined at 8 hpi by Sytox dye exclusion as described in Figure 1.
(F) IB of Pkr+/+ and Pkr −/− MEFs primed with IFN for 18 h and then infected with E3Δ83N as in (E). Lysates were harvested at 6 hpi and, following SDS-PAGE, evaluated for PKR, phospho-MLKL, or GAPDH.
Error bars represent SD. Statistical significance was determined as described in Figure 1. Each set of data is representative of two replicates except for (F), which compiles the results of the replicates. Statistical significance was determined as described in Figure 1.
Given the importance of the E3 dsRBD in PKR activation as well as the early activation of PKR following infection with ΔE3L virus (Chang et al., 1992), we sought to characterize the accumulation of A-RNA in infected cells. A-RNA accumulated in L929 cells within the first 4 hpi based on immunofluorescent imaging with conventional dsRNA-specific J2 antibody, as observed previously (Schönborn et al., 1991; Weber et al., 2006). A-RNA accumulated to equivalent levels in WT and E3 Zα-deficient VACV-infected cells by this time (Figures 4C and 4D). Thus, dsRNA accumulates in VACV-infected cells independently of the E3 Zα domain. In order to determine whether IFN-α-responsive A-RNA sensor PKR contributes to virus-induced necroptosis, we compared the susceptibility of PKR-deficient (Pkr−/−) and WT MEFs. When MEFs were primed with IFN-α and subsequently infected with E3Δ83N, necroptosis remained robust in Pkr−/− cells (Figures 4E and 4F) indicating that PKR does not contribute to activation or suppression of VACV-induced, ZBP1-mediated necroptosis.
VACV terminates early transcription at precise sites but is imprecise in the termination of intermediate and late transcripts, resulting in the generation of overlapping transcripts that form dsRNA species, particularly at late times of infection (Duesberg and Colby, 1969). In order to evaluate the impact of accumulating A-RNA levels on the induction of necroptosis, we utilized isatin β-thiosemicarbazone (IBT), an agent known to enhance processivity of the VACV intermediate/late RNA polymerase and increase the abundance of viral A-RNA (Condit and Niles, 2002). IBT-resistant (IBTR) mutants (E3Δ37NIBTR3 and E3Δ37NIBTR7) produced less A-RNA than parental virus (Figure S1A) and failed to activate PKR (Figure S1B) similar to viruses previously characterized to be IBTR, such as monkeypox virus (MPXV) or VACV-mutant A24R-1R carrying a mutation in the viral RNA polymerase (Arndt et al., 2016). Nevertheless, IBTR E3Δ37N derivatives showed patterns of growth sensitivity (Figure S1C), cell death (Figure S1D), and activated MLKL (Figure S1E) similar to the IBT-sensitive parental virus. Thus, increased dsRNA character of intermediate and late transcripts did not influence the induction of necroptosis.
Z-RNA accumulates during VACV infection
Given that accumulating A-RNA did not appear to dictate necroptosis outcomes, we next asked whether Z-NA accumulated during VACV infection. To this end, infected cells were fixed and stained at 4 hpi with monoclonal antibody (clone Z22) raised against Z-DNA (Staiano-Coico et al., 1985) and recently employed to characterize Z-RNA (Zhang et al., 2020). Z-NA was present in the cytoplasm of either E3Δ37N or WT VACV-infected cells but was absent from mock-infected cells (Figures 5A and S2A). The mean fluorescence intensity appeared to be significantly greater in the absence the E3 Zα domain (Figure 5B). Importantly, this signal resisted DNase I treatment but was sensitive to RNase A treatment (Figures 5C and S2B) suggesting accumulation of cytoplasmic Z-RNA within infected cells. The Z-RNA-specific signal was significantly more intense in cells infected with the Zα-deficient virus (E3Δ83N) compared with either WT VACV or E3-null virus (ΔE3), indicating that the accumulation of Z-RNA depended on E3 dsRBD function as well as the absence of the E3 Zα domain (Figure S2C). Furthermore, proteinase K treatment reduced the Z-NA-specific signal intensity in E3Δ37N-infected cells (Figures 5A, 5B, and S2A), consistent with a contribution of protein to the Z-RNA signal within infected cells. This result contrasts the Z-NA signal attributed to defective viral dsRNA in the nucleus of IAV-infected cells (Zhang et al., 2020). In VACV-infected cells, the most robust cytoplasmic Z-RNA signal occurs in the presence of Zα-deficient E3 that has retained the ability to bind A-RNA. Once induced to form, this Z-RNA may recruit ZBP1, resulting in RIPK3-mediated activation of MLKL and execution of necroptosis.
Figure 5. E3 sequesters Z-RNA that accumulates during VACV infection.
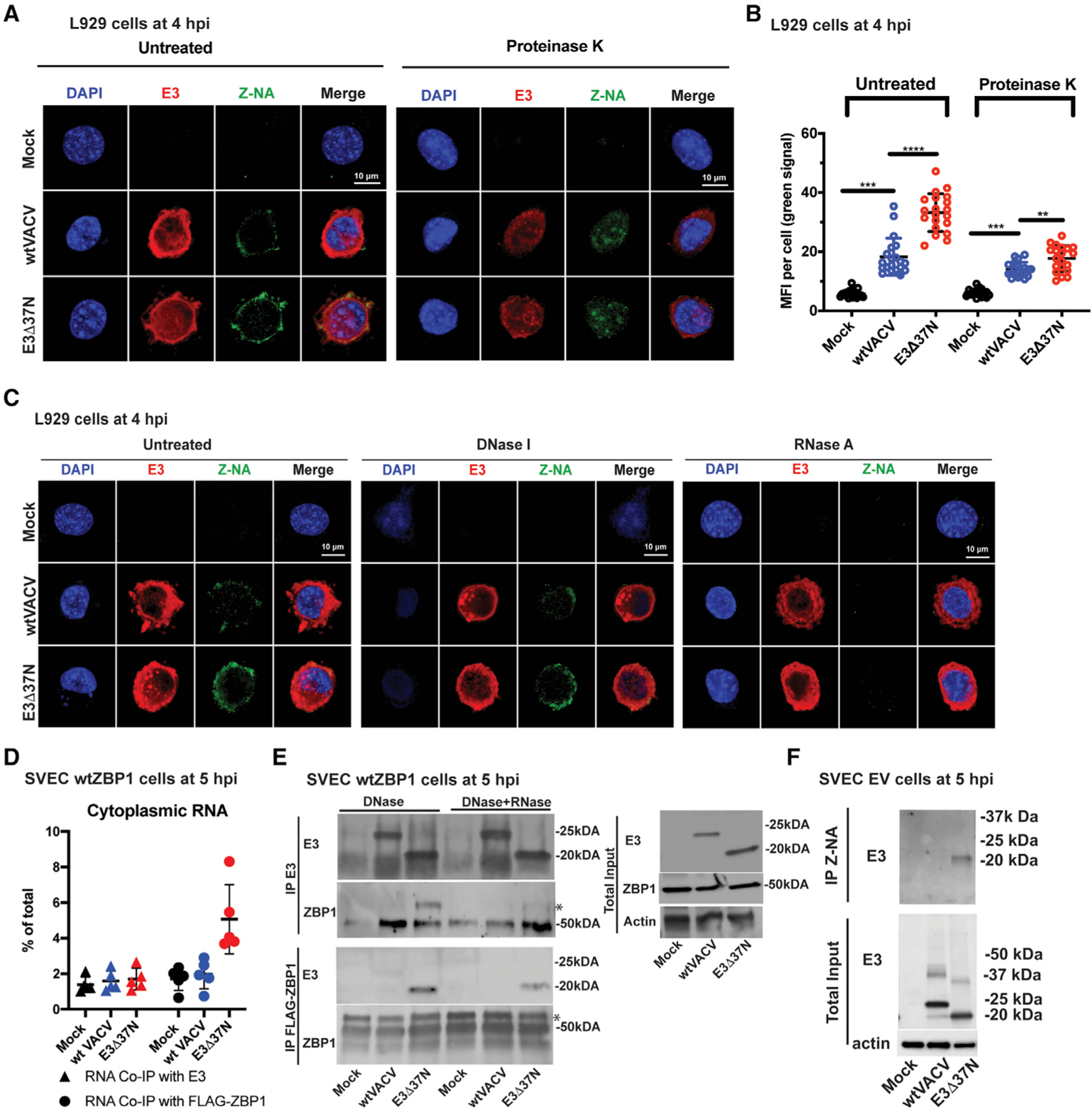
(A) Confocal immunofluorescent micrographs of single L929 cells at 4 hpi. Cells were either uninfected (mock) or infected with the indicated viruses at an MOI of 5, fixed and permeabilized prior to staining with anti-Z-NA antibody (green), anti-E3 antibody (red) and DAPI nuclear dye (blue). Bar indicates 10 µM.
(B) Quantification of the median florescence intensity (MFI) of Z-NA-specific monoclonal antibody staining in individual L929 cells using Leica LAS X software. Each point represents an individual cell and mean represents the average intensity from 20 cells.
(C) Confocal immunofluorescent micrographs of single L929 cells at 4 hpi. Cells were either uninfected (mock) or infected with the indicated viruses at an MOI of 5 and fixed and permeabilized then subsequently left untreated or digested with DNase I or RNase A. Cells were stained as described in (A).
(D) Coimmunoprecipitation (coIP) of RNA bound to E3 or ZBP1. SVEC-derived WT ZBP1 cells were left uninfected (mock) or infected with indicated viruses at an MOI of 5. At 5-hpi cells were UV crosslinked, lysed and either ZBP1- or E3-associated RNA was isolated by coimmunoprecipitation with E3-specific antibody (triangles) or FLAG-specific antibody. RNA associates with either of these immunoprecipitates were isolated by Trizol, quantified by nanodrop, and the proportion recovered was compared with total cytoplasmic RNA.
(E) Coimmunoprecipitation of E3 and ZBP1. SVEC-derived WT ZBP1 cells were either left uninfected (mock) or infected at an MOI of 5 with WT VACV (expresses p20 and p25) or E3Δ37N (expresses only p20). Cells were UV crosslinked at 5 hpi, lysed, and cell lysates were subjected to immunoprecipitation with either mouse anti-E3-specific antibody (BEI) or rabbit anti-FLAG antibody (cell signaling) specific for ZBP1 here). Immunoprecipitates were treated with DNase I alone or in combination with RNase T1 and subsequently washed to remove unbound proteins. Total lysates (right panel) and immunoprecipitates were subjected to SDS-PAGE and evaluated for ZBP1 with a mouse anti-ZBP1 antibody (Adipogen) or for E3 with rabbit polyclonal anti-E3 antibody. * indicates specific bands.
(F) CoIP of E3 with anti-Z-NA antibody. SVEC EV cells were either left uninfected (mock) or infected at an MOI of 5 with WT VACV or E3Δ37N. Cells were UV crosslinked at 6 hpi and treated with DNase I. Z-NA was immunoprecipitated from whole cell lysates and total lysates, and immunoprecipitated samples were subjected to SDS-PAGE and IB evaluation with rabbit polyclonal anti-E3 antibody.
Error bars represent the SD. Each set of data is representative of three replicates. Statistical significance was determined as described in Figure 1
E3 dsRBD promotes formation of Z-RNA and recruitment of ZBP1
The E3L gene naturally encodes a full-length 190-amino-acid protein, p25, and a shorter protein, p20, arising from translation initiation at M38 (Chang et al., 1992). E3Δ37N encodes only the Zα-deficient shorter protein (Figure 1A) that retains the ability to bind A-RNA via its C-terminal dsRBD with an affinity similar to full-length p25 (Chang and Jacobs, 1993). To determine how these E3 isoforms impacted the association of cytoplasmic RNA with ZBP1, we infected FLAG-ZBP1-reconstituted SVEC cells with either WT VACV or E3Δ37N, and, at 5 hpi, subjected cells to UV crosslinking and lysis. After E3- or FLAG-ZBP1-associated RNA was isolated by immunoprecipitation with anti-E3 or anti-FLAG antibody, respectively, RNA was subsequently recovered, purified and quantified by nanodrop. Overall, similar proportions of E3-associated RNA were detected in WT VACV- and E3Δ37N-infected cells (Figures 5D and S2H). However, more RNA was associated with FLAG-ZBP1 in E3Δ37N-infected cells compared with WT VACV-infected cells. To determine whether Zα-deficient E3 p20 promoted bridging to ZBP1 via RNA, FLAG-ZBP1 reconstituted cells were infected and subjected to UV crosslinking, lysis and incubation with either anti-E3 or anti-FLAG antibody immobilized on beads. Following incubation, antibody:bead complexes were washed and then either left untreated or RNase treated for 15 min. After additional washing, bound proteins were recovered, separated, and subjected to immunoblot evaluation. Whereas full-length E3 p25 did not associate with ZBP1, E3Δ37N-encoded p20 and ZBP1 clearly associated in reciprocal immunoprecipitation assays (Figure 5E). This interaction was substantially reduced when anti-body:bead complexes were treated with RNase T1, consistent with an RNA bridge between Zα-deficient E3 and ZBP1. Finally, the failure to detect any bridging between E3 p25 and ZBP1 during WT VACV infection appeared to reflect efficient Zα-mediated competition by E3 for Z-RNA thereby preventing recognition by ZBP1.
We next sought to evaluate whether the RNA bound to Zα-deficient E3 exhibited Z-form character by employing Z-NA-specific antibody for immunoprecipitation on DNase-treated cytoplasmic lysates. E3Δ37N-encoded p20 was expressed at levels similar to p25 by WT VACV (Figure 5F). Z-NA-specific antibody immunoprecipitated E3Δ37N-encoded p20 but not WT VACV-encoded p25 or the smaller amounts of p20 made during WT VACV infection. Although Zα-deficient E3 consistently bound Z-RNA, full-length E3 either failed to bind or was bound to Z-RNA in a way that prevented access by Z-NA-specific antibody. To distinguish between these possibilities, we utilized flow cytometry to detect free Z-RNA in permeabilized DNase-treated SVEC4–10 cells that were either deficient in ZBP1 (EV), or reconstituted with WT ZBP1 or ZBP1mut Zα1/Zα2 (Sridharan et al., 2017). These experiments revealed an increase in fluorescent signal following infection with E3-Zα-deficient mutant virus, but only in ZBP1mutZα1/Zα2 cells (Figures S2E–S2G). Both frequency of Z-RNA-positive cells and median fluorescence intensity were reduced in ZBP1-expressing cells, suggesting that WT VACV-encoded E3 or cellular ZBP1 occlude access of anti-Z-NA-specific antibody depending on Zα function. Most importantly, these data strongly reinforce a mechanism where Zα-deficient E3 binds to dsRNA, exposing Z-RNA that promotes recruitment of ZBP1 to trigger virus-induced necroptosis.
Equivalence of Z-NA binding family Zα substitutions in suppressing necroptosis
A conserved structure is characteristic of the Zα family consisting of a helix-turn-helix motif with an additional β-sheet (Rich and Zhang, 2003) even though minimal sequence identity is retained outside of amino acids that make contact with Z-NA (Figure 6A). Given evidence that E3 Zα sequestered Z-RNA from ZBP1, we sought to determine the capacity of Zα from other family members to substitute for E3 Zα. Prior studies revealed substitution of E3 Zα with either Zα1 from murine ZBP1 or Zα from human ADAR1 fully complemented pathogenesis (Brandt et al., 2005; Kim et al., 2003; White and Jacobs, 2012). Virulence correlated with the ability of E3 to bind Z-NA (Kim et al., 2003). Here, substitution of mouse ZBP1 Zα1 (E3ZαmZBP1) or human ADAR1 Zα (E3ZαhADAR1) restored type I IFN resistance (Figure 6B), restricted loss of viability (Figure 6C) and blocked MLKL activation (Figure 6D). These data further implicate the function of Zα within E3 as a competitor and critical suppressor of VACV-induced necroptosis.
Figure 6. Equivalence of Z-NA binding family Zα substitutions in suppressing necroptosis.
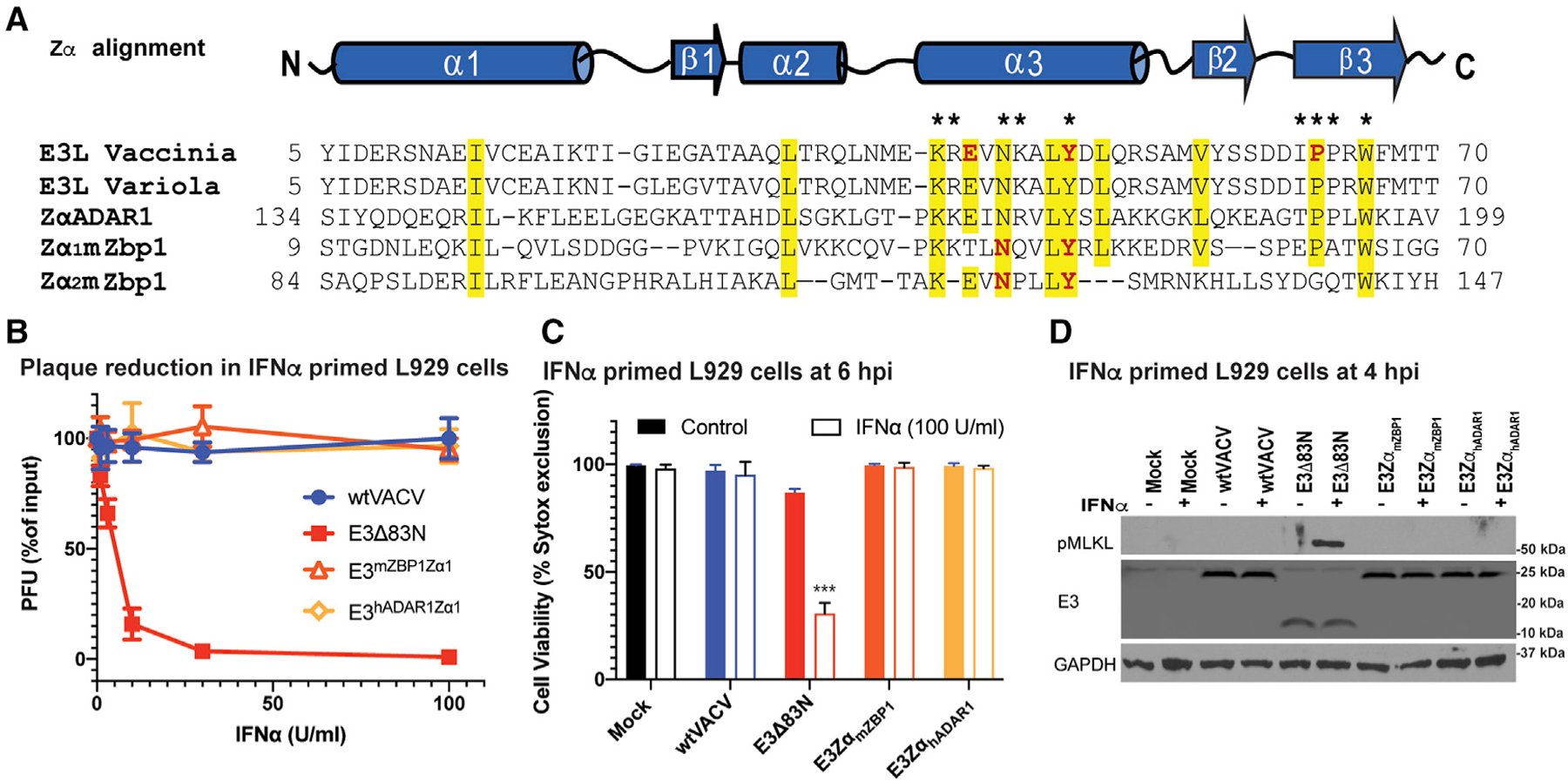
(A) Sequence alignment of the Za of VACV E3, variola E3, hADAR1 and mouse Zα1 and Zα2. Yellow signifies >70% sequence identity, amino acids with an asterisk above are known to be required for ZNA binding, and those in red have been targeted by point mutations (Kim et al., 2004, 2003). The GenBank accession numbers for the various sequences are as follows: GenBank: AAA02759 (vaccinia virus); GenBank: NP_042088 (variola virus); ADAR 1 (Homo sapiens): GenBank: AAB06697; ZBP1 (Mus musculus): GenBank: NP_067369.
(B) Plaque reduction assays using Zα chimeric viruses were performed on IFNα-primed L929 cells as described in Figure 1.
(C) L929 cells left untreated or treated with IFNα for 18 h and subsequently infected at a MOI of 5 with the indicated viruses. Cell viability was determined at 6 hpi using by Sytox dye exclusion as described in Figure 1.
(D) IB of L929 cell lysates from cells either left untreated or IFN-α-pretreated as described in Figure 1 and then infected with a MOI of 5 with the indicated viruses. Lysates were harvested at 4 hpi, subjected to SDS-PAGE and evaluated for phospho-MLKL.
Error bars represent the SD. Statistical significance was determined as described in Figure 1. Each set of data is representative of three replicates except for (B) and(C), which compiles the results of the replicates. Statistical significance was determined as described in Figure 1.
DISCUSSION
ZBP1-dependent necroptosis is a critical antiviral pathway that plays out against DNA viruses as well as the dsRNA virus IAV. Suppression of virus-induced necroptosis provides a survival advantage for replicating VACV and supports pathogenesis in mice. The importance of blocking cell death pathways during poxvirus infection first emerged from studies showing the caspase inhibitor B13R sensitizes to TNF-dependent necroptosis (Chan et al., 2003; Cho et al., 2009; Li and Beg, 2000) as well as an earlier report suggesting that dsRNA triggers apoptosis during infections with E3-deficient VACV (Kibler et al., 1997). Upon detailed study, virus lacking the E3 Z-NA-binding domain (Koehler et al., 2017) revealed the importance of virus-induced necroptosis (ZBP1→RIPK3→MLKL) during VACV infection. Here, we have shown this pathway relies on ZBP1 sensing of Z-RNA in infected cells and requires the E3 dsRBD. Both the N-terminal Z-NA- and C-terminal A-RNA-binding domains are retained and clearly important to the biology of a wide range of orthopoxviruses, including the human smallpox virus, variola, in addition to vaccinia strains. Suppression of PKR is also important in all of these poxviruses; however, suppression of necroptosis proceeds independently of PKR. VACV E3 suppresses virus-induced necroptosis by competing with ZBP1 sensing of virus-induced Z-RNA. E3 has no impact on TNF-induced necroptosis (TNF→TNFR1→RIPK1→RIPK3→MLKL). As with other virus-induced death settings (Guo et al., 2018; Kaiser et al., 2013; Koehler et al., 2017; Nogusa et al., 2016; Shubina et al., 2020; Upton and Chan, 2014; Upton et al., 2010, 2012), ZBP1-dependent necroptosis does not require either TNF signaling or RIPK1 (Koehler et al., 2017).
VACV E3 has long been recognized as a suppressor of PKR and other dsRNA-dependent antiviral mediators via its C-terminal dsRBD (Jacobs et al., 2009; Rahman and McFadden, 2020). Here, E3 emerges as a promiscuous RNA-binding protein, relying on its A-RNA interaction dictated by the C-terminal region as well as an N-terminal Zα domain to interact with Z-RNA and prevent activation of ZBP1. Induction of necroptosis in the absence of E3 Zα appears to hinge upon recruitment of the host-encoded sensor ZBP1 via Zα2 sensing of newly transcribed Z-RNA that accumulates shortly after infection. The process depends on the interaction of the E3 dsRBD with A-form RNA in a way that promotes Z-RNA character that is recognized as a PAMP by ZBP1. While a complete understanding will require structural studies, this scenario is supported by our demonstration of an RNA bridge between Zα-deficient E3 and ZBP1 prior to induction of necroptosis as well as the reduction of cytosolic Z-RNA signal following protease treatment of fixed infected cells. The presence of E3 Zα-deficient p20:Z-RNA complexes in E3 Zα mutant-infected cells, but not in WT VACV-infected cells, supports a model where the A-RNA:protein complex promotes formation of the appropriate Z-NA-character PAMP. Full-length E3 seems to rely on its Z-NA-binding domain to prevent ZBP1 access. In this way, competitive binding via Zα motifs in E3 and ZBP1 apparently dictate Z-RNA accessibility and VACV-associated death or survival outcomes. A-RNA-binding proteins, several of which inhibit PKR like E3, are expressed by many DNA viruses as well as IAV and may be involved in the induction of Z-RNA that triggers this pathway in other systems.
The mechanism described here, as previously (Koehler et al., 2017), relied on necroptosis-sensitive cells as well as infections of mice. Many common cell lines that have been employed for VACV lack necroptosis machinery present in mice as well as in cell lines used in our studies. While the possible consequences of E3:ZBP1 competition has been evident in mouse pathogenesis studies (Brandt et al., 2005; Brandt and Jacobs, 2001; White and Jacobs, 2012), a focus on modulation of PKR function prevailed because this activity predominates in necroptosis-insensitive cells (Jacobs et al., 2009; Rahman and McFadden, 2020). The N-terminal, Zα-function long recognized as biologically significant from in vivo studies of virulence in mice can now be attributed to suppression of necroptosis. When cells are competent, ZBP1-dependent cell death predominates as an early event in the virus replication cycle, proceeding in a PKR-independent manner. Some cell types, like SVEC4–10 endothelial cells, express constitutive ZBP1; whereas, other cell types, such as the L929 fibroblasts, require IFN priming to induce sufficient ZBP1 to support the necroptotic pathway. It is likely that both are important in the physiological response to virus infection against viruses but as likely to vary between viruses. For example, the type I IFN response does not impact the restriction of MCMV replication through unleashed necroptosis (Upton et al., 2010, 2012); whereas, IFN has been central to the observations with IAV (Samir et al., 2020). Our work emphasizes both the importance of the Z-RNA sensing pathway and the crucial contribution made by a viral competitor of Z-RNA PAMP recognition. The importance of this PAMP in a growing number of DNA and RNA viruses combined with the various mechanisms that DNA viruses have adopted to suppress sensing, signaling, and execution of cell death pathways reinforce the importance of this innate immune pathway in host defense.
E3 suppression of necroptosis is critical to VACV infection. ZBP1 represents a potent host-defense pathway poised to cut short infection by driving the death of cells early in infection. Z-RNA-triggered, ZBP1-mediated cell death described here for E3 mutant VACV has been observed in other viruses, including herpesviruses and IAV (Guo et al., 2018; Kesavardhana et al., 2020; Maelfait et al., 2017; Sridharan et al., 2017; Zhang et al., 2020). ZBP1 has also been implicated as a trigger of broad cell death outcomes in nonviral IFN-stimulated settings such as in RIPK1-deficient cells (Ingram et al., 2019; Yang et al., 2020). Orthopoxvirus E3 suppresses virus-induced necroptosis through a competition for the PAMP rather than by RHIM signaling suppression characterized in herpesviruses (Guo et al., 2018; Upton et al., 2010). Recently the viral inducer of RIPK3 degradation (vIRD) was identified in cowpox and was shown to regulate virus-induced inflammation and pathogenesis (Liu et al., 2021). Other poxviruses that typically lack E3 institute a block to this same pathway at the downstream execution stage of MLKL activation (Petrie et al., 2019). Thus, poxviruses have evolved several distinct mechanisms to escape the consequences of necroptosis and other programed death pathways.
It has long been known from genetic studies that VACV E3 Zα confers type I IFN resistance as well as virulence in mice (Brandt et al., 2005; Brandt and Jacobs, 2001; White and Jacobs, 2012). Similarly, this capacity has been tied to Z-NA binding (Kim et al., 2003). We now recognize that Z-NA binding by E3 is crucial for prevention of necroptosis, a mechanism that involves competition with type-I-IFN-regulated ZBP1. Unquestionably, our data demonstrate that the induction of necroptosis by Zα-deficient mutants of VACV relies on a mechanism where the dsRBD associates with newly transcribed RNA, inducing Z-NA character that acts as a PAMP to recruit and bridge to ZBP1. In the course of this recruitment, ZBP1 triggers RIPK3 to mediate the activation of MLKL followed by execution of cell death.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Edward Mocarski (mocarski@emory.edu).
Materials availability
This study did not generate new unique reagents. All viruses and cell lines can be obtained from the corresponding authors of original publications. Contact Bertram Jacobs (bjacobs@asu.edu) for poxviruses and Jason Upton (jwu0003@auburn.edu) for ZBP1-transduced SVEC29–11 cells.
Data and code availability
This study did not generate any datasets/code. Protein sequences were analyzed from the GenBank accession numbers for the various sequences are as follows: ADAR1 AAB06697.1 (amino acids 134–199) (Homo sapiens); ZBP1 (Mus musculus) NP_067369 (amino acids 9–70 and 84–147); the E3L proteins, AAA02759 (amino acids 5–70) (vaccinia virus); AAC08018 (amino acids 5–70) (variola virus).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells
SVEC4–10 (CRL—2181) and SVEC-derived ZBP1-deficient clone29–11 cells and various ZBP1-transduced cells were previously generated (Sridharan et al., 2017), Cells were cultured in DMEM containing 4.5 g/ml glucose, 10% FBS (Peak Serum), 2 mM L-glutamine, 100 U penicillin/ml, and 100 U streptomycin/ml (Invitrogen).
BHK21 (ATCC-CCL-10) and L929 (ATCC-CRL-6364) were cultured in MEM (GIBCO) supplemented with 5% FBS, 0.1 mM nonessential amino acid solution (GIBCO) and 50 μg gentamycin/ml.
HeLa cells (kind gift of George Pavlakis, NCI) were maintained in DMEM; (GIBCO) supplemented with 5% FBS (HyClone).
Pkr−/− and wt MEFs (129/SV background) were generously provided by Lynda Morrison, Saint Louis University, and were maintained in DMEM with 10% FBS and 50 μg gentamicin/ml.
RK-E3L (RK13) cells (ATCC CCL-37) have been stably transfected with a plasmid expressing the E3L gene under regulatory control of the Tet-Off system (Clontech) maintained in MEM (Cellgro) supplemented with 5% FBS.
All cells were incubated at 37°C in the presence of 5% CO2.
VIRUS
The Western Reserve (WR) strain was employed as parental wt VACV to generate E3 truncation, substitution or loss of function alanine substitution point mutant viruses by DNA transfection-virus infection in RK cells using lacZ as an indicator marker as previously described (Brandt and Jacobs, 2001; Chang et al., 1995; Kim et al., 2003; Shors et al., 1997). Zα chimeric substitution viruses were previously generated by replacing nucleotides 61–261 (amino acids 1–67) of the vaccinia virus E3L gene with nucleotides 554–742 (Zα) from human ADAR1 or nucleotides 116–316 (Zα) from murine ZBP1 (Kim et al., 2003). We confirmed the identity of all recombinant viruses by sequencing a PCR product using E3L flanking primers. All experiments with these viruses were performed using an MOI of 5 unless specified otherwise.
Virus stock preparation
BHK21 cells were used to propagate wt and recombinant viruses except for ΔE3L which was propagated on RK-E3L cells. Cell culture medium was removed from the monolayers and virus infections were performed in MEM (GIBCO) supplemented with 2% FBS, 50 µg gentamycin/ml, and 0.1 mM nonessential amino acid solution (GIBCO). Cells were infected at a MOI of 0.01 at 37°C with rocking every 10 min. After 1 hr adsorption, the inoculum was removed and cell culture medium was replaced. At 100% CPE, virus was released from cells by three rounds of freeze/thaw (−80°C/37°C) cycles followed by 30s sonication with a probe type sonicator on ice. Cell debris was removed by centrifugation (700 × g for 10 min at 4°C) and supernatant was stored in aliquots at −80°C. The virus titer in the supernatant was determined by plaque assay.
Spontaneous isatin-β-thiosemicarbazone (IBT)-resistant mutants were previously generated from parental E3Δ37N as described (Cresawn et al., 2007). After 1-h incubation, inoculum was aspirated and replaced with fresh medium containing 45 μM IBT. 24 hr later, virus was collected as describe above. IBT-resistant progeny were isolated by plaque purification in the presence of 45 μM IBT.
METHOD DETAILS
Plaque reduction assays
L929 cell monolayers were treated with 0 to 300 U/ml of mouse IFNα (Calbiochem) and then infected with 1 × 102 PFU of wt VACV or E3 mutant viruses (Koehler et al., 2017). Plaques were counted after fixation with 50% methanol and staining with crystal violet at 3 to 4 dpi. Percent plaque reduction was calculated by comparing the input PFU to the number of plaques formed in treated monolayers.
Cell viability
Viability was assessed with a SYTOX nuclear stain exclusion assay or by a CellTiter-Glo Luminescent Cell Viability Assay (Promega). For SYTOX nuclear stain, Hoechst 33342 (2’-[4-ethoxyphenyl]-5-[4-methyl-1-piperazinyl]-2,5’-bi-1H-benzimidazole) and SYTOX® were applied for 15 min prior to infection and throughout the time following infection. Cells were incubated at 37°C in the presence of 5% CO2 on an EVOS™ FL Auto Imaging System (Invitrogen) with onstage incubator system. An image was collected every 2 min for 5 h. Sytox+ cells were considered dead, so the percentage of cells excluding this dye were determined in 10 fields at 100× magnification for each independent experiment (N=3). CellTiter-Glo® luminescent Cell Viability Assay was performed at 12 hpi according to manufacturer’s recommendations (Mandal et al., 2014).
Cell extracts and immunoblot
50% confluent cell monolayers in 60 mm dishes infected with viruses at a MOI of 5 for various times were washed twice with PBS, scraped, pelleted and lysed in 100 µl of 100 mM Tris-Cl (pH 6.8) with 2% SDS and 100 mM 2-mercaptoethanol and 1X Halt Protease and Phosphatase Inhibitor Cocktail (Pierce Thermo Scientific). Cell lysates were further processed utilizing QIAshredder columns (Qiagen) according to manufacturer’s recommendations. Boiled samples were subjected to denaturing SDS-PAGE using 10% gels. Membranes were blocked in 3% skim milk in Tris-buffered saline with 0.1% Tween20 Detergent (TBST) for 1 hr and then incubated in primary antibody diluted 1:1000 in PBS overnight at 4°C. Membranes were then washed and incubated in secondary antibody (diluted 1:5000) for 2 hr at room temperature. Immunoreactive bands were visualized by chemiluminescence with horse radish peroxidase substrate.
Immune adsorption
ZBP1-null 29–11 cell derivatives (Sridharan et al., 2017) were plated on 10 cm dishes at 50% confluency. At 24 hr post-seeding, cells were infected, incubated until 4 hpi, scraped and resuspended in ice cold Ca+2- and Mg+2-free 1X Hanks balanced salt solution (HBSS*) supplemented with 10 mM HEPES, pH 7.3 (Gibco), and irradiated on ice at 400 mJ/cm2 in a Spectrolinker XL-1500 to crosslink nucleic acids and proteins. Following crosslinking cells were scraped, sedimented and lysed in 0.5 ml of ice-cold Ca+2- and Mg+2- free 1X phosphate-buffered saline (PBS) supplemented with 0.1% SDS, 0.5% deoxycholate, 0.5% Nonidet (N)P-40 (PXL buffer) with SUPERase•In RNase Inhibitor (Invitrogen) and 1X Halt Protease and Phosphatase Inhibitor Cocktail (Pierce Thermo Scientific) for 15 min and subjected to brief sedimentation at 10,000xg for 10 min to remove insoluble cell debris. 10% of lysates were reserved to assess total protein or RNA. Mouse monoclonal anti-Z-NA antibody Clone Z22 or mouse monoclonal E3 NR-4547 was incubated with anti-mouse IgG Dynabeads (Thermo). After washing unbound antibody, Dynabead-bound anti-Z-NA antibody was then added to lysates and incubated while rotating for 4 hr at 4°C, washed 3X in a magnetic stand in 1 ml of 1X PXL buffer followed by two washes with 50mM Tris-Cl, pH7.4, 10mM MgCl2 and 0.5% NP-40 (PNK+ buffer). For RNA bridging studies, beads were treated in between the two wash steps with DNase I with or without RNase T1 at 10 U/ml in 1 mM EDTA and 10 mM Tris-Cl, pH 7.8 (TE buffer). For RNA quantification beads were incubated in Trizol for 10 min then RNA was isolated according to manufacturer’s instructions and quantified on a Nanodrop instrument. For protein association, beads were then incubated in 1X SDS lysis, heated at 95°C for 10 min and placed back on magnetic stand to remove the supernatant for SDS-PAGE using 4–20% gradient gels and immunoblotting after transfer to polyvinylidene difluoride (PVDF) membranes.
Live cell imaging
L929 cells were seeded in 12-well CytoOne tissue culture treated plates (USA Scientific) and pre-treated with 100 U/ml of mouse IFNα (Calbiochem) for 18 hr prior to infection. Live cell nuclear stain Hoechst 33342 was applied 15 min prior to infection. Cells were infected and incubated for an additional 5 hr prior to evaluation of alterations in morphology with an EVOS™ FL Auto Imaging System (Invitrogen). For time-lapse imaging, cells were overlaid with MEM supplemented with 1 µM SYTOX Green NA stain (Thermo Fisher Scientific) and 5 µg/ml Hoechst 33342 (Thermo Fisher Scientific). Cells were incubated at 37°C supplemented with 5% CO2 on the EVOS™ System with onstage incubator system (Koehler et al., 2017). An image was taken every 2 min for 5 hr.
Cell treatments
Cell death suppressor cocktail including GSK inhibitor composed of N-(6-(Isopropylsulfonyl)quinolin-4-yl)benzo[d]thiazol-5-amine (GSK’872, GlaxoSmithKline) at 3 µM and zVAD-fmk (Enzo Life Sciences) at 50 µM was applied to cells 1 hr prior to and during infection (Mandal et al., 2014).
IFN treatment:
Mouse cells were treated with IFNa (Calbiochem) or mouse IFNβ (PBL assay).
Necroptosis positive control:
To induce TNF-dependent necroptosis, cells were treated with 50 µM zVAD-fmk (Enzo life sciences) for 1 hr then stimulated with 25 ng TNF/ml (PeproTech) and lysates were harvested 6 hr after addition of TNF (Koehler et al., 2017).
Viral replication inhibitors:
Cells were then treated with increasing concentration of inhibitors throughout infection.
Cytosine arabinoside (AraC) was used at 200 µg/ml, 400 µg/ml, 800 µg/ml and 1600 µg/ml starting at 1 hr prior to infection
Cycloheximide (CHX) was used at 20 µg/ml, 40 µg/ml, 80 µg/ml, and 120 µg/ml starting at 30 min after infection.
Hydroxyurea (HU) was used at 5 µM, 10 µM, 20 µM and 40 µM starting 1 hr prior to infection
Actinomycin D (ActD) treated throughout infection at 1 µg/ml, 2 µg/ml, 4 µg/ml, 8 µg/ml starting 1 hr prior to infection
Sarracenia extract (Sar) made from an ethanol extract (Arndt et al., 2012) was used at 10 µl/ml, 30 µl/ml, 60 µl/ml, 120 µl/ml starting at 1 hr prior to infection.
siRNA transfection:
L929 cells were plated in 6-well dishes at 30% confluency, allowed to adhere for 24 hr and transfected with ON-TARGETplus siRNA (Dharmacon) or control siRNA using INTERFERin transfection reagent (Polyplus) according to manufacturer’s recommendations. Following transfection, cells were incubated for 36 hr, either mock- or IFNα-treated for 18 hr and subjected to infection for times indicated.
Immunofluorescence imaging
Cells were plated on 8-well glass slides (EMD Millipore), allowed to adhere for at least 24 hr and infected at a MOI of 5. Following infection, cells were fixed in 4% (w/v) paraformaldehyde, permeabilized in 0.2% (v/v) Triton X-100, blocked with MAX-block Blocking Medium (Active Motif), and incubated overnight in a humidified chamber with primary antibodies at 4°C. After three washes in PBS, slides were incubated with fluorophore-conjugated secondary antibodies for 1 hr at room temperature. Cells were then washed 3X in PBS, slides were mounted in ProLong Gold Antifade Reagent (Thermo Fisher Scientific) and imaged by confocal microscopy on a Leica SP8 instrument. Fluorescence intensity was quantified and subjected to statistical calculations using Leica LAS X software. In some experiments, cells were subjected to additional treatments post fixation. Proteinase K treatment was performed with (0.008 U/ml) for 20 to 40 min at 37°C. RNase A (1 µg/ml) or DNase I (25 U/ml) was used for 1 hr at 37°C. Primary antibodies were used at the following dilutions Z-NA (Z22 clone Ab, 1:200), A-RNA (J2 clone, 1:50), E3 (rabbit polyclonal, 1:500).
Flow cytometry
Z-RNA
Infected SVEC4–10-derived EV and wt ZBP1 29–11 cells were UV crosslinked at 4 hpi, stained with fixable viability 700 (BD Horizon) and permeabilized with CytoPerm/Fix kit (BD) for 20 min at 4°C. Following permeabilization and DNase I treatment, cells were blocked in FACS buffer (2% BSA, 0.9% sodium azide in PBS) with 10% normal rat serum and then probed with mouse monoclonal anti-Z-NA antibody clone Z22 at 1:100 in Permwash buffer (BD) for 30 min at 4°C then washed 3X with Permwash buffer followed by secondary anti-mouse-IgG 488 antibody (Invitrogen). Cells were evaluated by flow cytometry using a BD LSR Fortessa Cell Analyzer and gated on FlowJo9 software to exclude doublets and dead cells to determine the frequency and median fluorescence intensity of Z-NA+ cells.
dsRNA (A-RNA)
HeLa cells were mock- or virus-infected with either wt VACV, ΔE3, E3Δ37N, E3Δ37NIBTR−3, E3Δ37NIBTR-7, VACV-A24R-R1, or MPXV. At 6 hpi, cells were trypsinized and fixed with CytoPerm/Fix kit (BD). Cells were stained with rabbit anti-VACV (Abcam) and mouse anti-dsRNA (J2 clone, Scicons) overnight at 4°C. Cells were washed and stained with anti-rabbit-IgG Pacific Blue (BD) and anti-mouse-IgG 488 (Invirtogen) for 4 °C. Cells were washed, resuspended in FACS buffer (2% BSA, 0.9% sodium azide in PBS) and were analyzed by flow cytometry using a BD FACSymphony™ A3 Cell Analyzer and cells were gated to exclude doublet and non-viable cells.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all comparisons between mean values from mock, infected, control or treated samples, significance was determined by a nonparametric Mann-Whitney test using Graphpad Prism9 with p ≤ 0.05 considered significant (*,p ≤ 0.05,**, p ≤ 0.01 ***, p ≤ 0.001). Error bars represent SD with an N≥3 as indicated in the figure legends. All statistical analysis was performed using Prism 8 (Graphpad).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Anti-ZBP1, mAb (Zippy-1) | Adipogen | Cat# AG-20B-0010-C100; RRID:AB_2892147 |
| DYKDDDDK Tag (D6W5B) Rabbit mAb | Cell Signaling | Cat# 14793; RRID:AB_2572291 |
| Monoclonal Anti-Vaccinia Virus E3L, Clone TW2.3 (produced in vitro), NR-4547 | BEI Resources, NIAID, NIH | Cat# NR-4547; RRID:AB_2892148 |
| E3L Rabbit polyclonal Ab | Generated in Jacobs Lab | 10.1016/0042-6822(91)90768-7; RRID:AB_2892150 |
| Anti-β-Actin (Clone AC-74 mAb) | Sigma | Cat# A5316; RRID:AB_476743 |
| Anti-GAPDH (Clone G-9 mAb) | Santa Cruz Biotechnology | Cat# sc-365062; RRID:AB_10847862 |
| Anti-MLKL (phospho S345) [EPR9515(2) mAb] | Abcam | Cat# ab196436; RRID:AB_2687465 |
| Anti-double-stranded RNA (Clone J2) | SCICONS | Cat# 10010200; RRID:AB_2651015 |
| Anti-PKR (phospho T446) antibody (Clone E120) | Abcam | Cat# ab32036; RRID:AB_777310 |
| Anti-Z-DNA (Clone Z22 mAb) | Absolute antibody | Cat# Ab00783–3.0; RRID:AB_2820286 |
| Anti-Vaccinia Virus polyclonal antibody | Abcam | Cat# ab35219; RRID:AB_778768 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Halt™ Protease and Phosphatase Inhibitor Cocktail | Thermo Scientific | Cat# 78446 |
| SUPERase•In™ RNase Inhibitor | Invitrogen | Cat# AM2694 |
| TRIzol™ Reagent | Invitrogen | Cat# 15596026 |
| zVAD-fmk | SM Biochemicals | Cat# SMFMK001 |
| GSK’872 | Sigma-Calbiochem | Cat# 5303890001 |
| Istatin-b-thiosemicarbazone (IBT) | Pfaltz & Baur | Cat# I04639 lot 14888 |
| Cycloheximide | Sigma-Aldrich | Cat# 01810 |
| Hydroxyurea | Sigma-Aldrich | Cat# H8627 |
| Actinomycin D | Sigma-Aldrich | Cat# A9415 |
| Cytosine β-D-arabinofuranoside (AraC), | Sigma-Aldrich | Cat# C1768 |
| Alpha-interferon, mouse, recombinant, E. coli. | Millipore Sigma | Cat# 407293 |
| Recombinant Murine TNF-α | Peprotech | Cat# 315–01A |
| Fixation/Permeabilization Solution Kit | BD Bioscience | Cat# 554714 |
| ON-TARGETplus Mouse Zbp1 siRNA | Horizon | Cat# L-048021–00-0020 |
| ON-TARGETplus Non-targeting Control Pool | Horizon | Cat# D-001810–10-05 |
| Proteinase K | New England BioLabs | Cat# P8107 |
| RNase A | Thermofisher | Cat# EN0531 |
| RNase T1 | Thermofisher | Cat# EN0541 |
| Fixable Viability Stain 700 | BD Horizon | Cat# 564997 |
| DNase I | Thermofisher | Cat# 90083 |
| SYTOX™ Green Nucleic Acid Stain | Invitrogen | Cat# S7020 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) | Invitrogen | Cat# D1306 |
| INTERFERin transfection reagent | Polyplus | Cat# 409–10 |
| MAXblock Blocking Medium | Active Motif | Cat# 15252 |
| ProLong Gold Antifade Reagent | Thermo Fisher | Cat# P36930 |
| Mouse Interferon Beta | PBL Assay Science | Cat# 12401–1 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# G7570 |
| BS3 (bis(sulfosuccinimidyl)suberate) | Thermo Fisher | Cat# 21580 |
| Dynabeads™ M-280 Sheep Anti-Mouse IgG | Thermo Fisher | Cat# 11202D |
|
| ||
| Cells and Virus | ||
|
| ||
| L929 (NCTC clone 929) | ATCC | ATCC® CCL-1 |
| HeLa cells | Gift from George Pavlakis, NCI | Bert Jacobs |
| SVEC4–10 derived EV and FLAG-ZBP1 | https://doi.org/10.15252/embr.201743947 | Jason Upton |
| Vaccinia virus (VACV) |
https://doi.org/10.1073/pnas.0431131100
https://doi.org/10.1128/JVI.69.10.6605-6608 https://doi.org/10.1128/JVI.75.2.850-856 https://doi.org/10.1006/viro.1996 https://doi.org/10.1016/j.virol.2005.01.006 |
Bert Jacobs |
| Monkey poxvirus | BEI Resources | (MPXV) 7–61 (WRAIR) |
|
| ||
| Software and Algorithms | ||
|
| ||
| Prism9 | https://www.graphpad.com/ | Version 9.1.0 (216) |
| FlowJo | Becton Dickinson & Company (BD) | Version FlowJo 10.7.1 |
| Clustal Omega | EMBL-EBI | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| Leica LAS X software | Leica Microsystems | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
|
| ||
| Other | ||
|
| ||
| BD FACSymphony™ A3 Flow cytometer | BD | A3 |
| Leica SP8 instrument confocal microscope | Leica Microsystems | SP8 |
Highlights.
Vaccinia virus infection results in production of cytoplasmic Z-RNA
Zα domains of vaccinia E3 and host ZBP1 compete for Z-RNA
ZBP1 senses Z-RNA and drives necroptosis unless prevented by vaccinia E3 Zα
E3 complexed with dsRNA promotes Z-RNA formation
ACKNOWLEDGMENTS
We thank Hongyan Guo, Ph.D. and Liliana Hernandez-Villalobos for assistance as well as other laboratory members for helpful discussions. PHS grants from NIH supported this work (R01 AI095394 to B.L.J., R01 AI135025 to S.B., R01 AI020211 to E.S.M., 2R56DK074731–08A1 and R01DK074731 to D.K.). This research was also supported in part by the Emory University School of Medicine Flow Cytometry Core in addition to the NIH Cancer Center Support grant P30CA006927 awarded to Fox Chase Cancer Center.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.chom.2021.05.009.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arndt W, Mitnik C, Denzler KL, White S, Waters R, Jacobs BL, Rochon Y, Olson VA, Damon IK, and Langland JO (2012). In vitro characterization of a nineteenth-century therapy for smallpox. PLoS One 7, e32610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt WD, White SD, Johnson BP, Huynh T, Liao J, Harrington H, Cotsmire S, Kibler KV, Langland J, and Jacobs BL (2016). Monkeypox virus induces the synthesis of less dsRNA than vaccinia virus, and is more resistant to the anti-poxvirus drug, IBT, than vaccinia virus. Virology 497, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt T, Heck MC, Vijaysri S, Jentarra GM, Cameron JM, and Jacobs BL (2005). The N-terminal domain of the vaccinia virus E3L-protein is required for neurovirulence, but not induction of a protective immune response. Virology 333, 263–270. [DOI] [PubMed] [Google Scholar]
- Brandt TA, and Jacobs BL (2001). Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J. Virol 75, 850–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BA 2nd, Lowenhaupt K, Wilbert CM, Hanlon EB, and Rich A (2000). The Zalpha domain of the editing enzyme dsRNA adenosine deaminase binds left-handed Z-RNA as well as Z-DNA. Proc. Natl. Acad. Sci. USA 97, 13532–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, and Lenardo MJ (2003). A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J. Biol. Chem 278, 51613–51621. [DOI] [PubMed] [Google Scholar]
- Chang HW, and Jacobs BL (1993). Identification of a conserved motif that is necessary for binding of the vaccinia virus E3L gene products to double-stranded RNA. Virology 194, 537–547. [DOI] [PubMed] [Google Scholar]
- Chang HW, Uribe LH, and Jacobs BL (1995). Rescue of vaccinia virus lacking the E3L gene by mutants of E3L. J. Virol 69, 6605–6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HW, Watson JC, and Jacobs BL (1992). The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA 89, 4825–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, and Chan FK (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condit RC, and Niles EG (2002). Regulation of viral transcription elongation and termination during vaccinia virus infection. Biochim. Biophys. Acta 1577, 325–336. [DOI] [PubMed] [Google Scholar]
- Cresawn SG, Prins C, Latner DR, and Condit RC (2007). Mapping and phenotypic analysis of spontaneous isatin-beta-thiosemicarbazone resistant mutants of vaccinia virus. Virology 363, 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley-Bauer LP, Roback L, Crosby LN, McCormick AL, Feng Y, Kaiser WJ, and Mocarski ES (2017). Mouse cytomegalovirus M36 and M45 death suppressors cooperate to prevent inflammation resulting from antiviral programmed cell death pathways. Proc. Natl. Acad. Sci. USA 114, E2786–E2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFilippis VR, Alvarado D, Sali T, Rothenburg S, and Früh K (2010). Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J. Virol 84, 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duesberg PH, and Colby C (1969). On the biosynthesis and structure of double-stranded RNA in vaccinia virus-infected cells. Proc. Natl. Acad. Sci. USA 64, 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Gilley RP, Fisher A, Lane R, Landsteiner VJ, Ragan KB, Dovey CM, Carette JE, Upton JW, Mocarski ES, and Kaiser WJ (2018). Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis 9, 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, and Mocarski ES (2015). Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 17, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha SC, Lokanath NK, Van Quyen D, Wu CA, Lowenhaupt K, Rich A, Kim YG, and Kim KK (2004). A poxvirus protein forms a complex with left-handed Z-DNA: crystal structure of a Yatapoxvirus Zalpha bound to DNA. Proc. Natl. Acad. Sci. USA 101, 14367–14372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram JP, Thapa RJ, Fisher A, Tummers B, Zhang T, Yin C, Rodriguez DA, Guo H, Lane R, Williams R, et al. (2019). ZBP1/DAI drives RIPK3-mediated cell death induced by IFNs in the absence of RIPK1. J. Immunol 203, 1348–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs BL, Langland JO, Kibler KV, Denzler KL, White SD, Holechek SA, Wong S, Huynh T, and Baskin CR (2009). Vaccinia virus vaccines: past, present and future. Antiviral Res 84, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, and Mocarski ES (2011). RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, and Mocarski ES (2008). Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J. Immunol 181, 6427–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, and Mocarski ES (2013). Viral modulation of programmed necrosis. Curr. Opin. Virol 3, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesavardhana S, Malireddi RKS, Burton AR, Porter SN, Vogel P, Pruett-Miller SM, and Kanneganti TD (2020). The Zalpha2 domain of ZBP1 is a molecular switch regulating influenza-induced panoptosis and perinatal lethality during development. J. Biol. Chem 295, 8325–8330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibler KV, Shors T, Perkins KB, Zeman CC, Banaszak MP, Biesterfeldt J, Langland JO, and Jacobs BL (1997). Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J. Virol 71, 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YG, Lowenhaupt K, Oh DB, Kim KK, and Rich A (2004). Evidence that vaccinia virulence factor E3L binds to Z-DNA in vivo: implications for development of a therapy for poxvirus infection. Proc. Natl. Acad. Sci. USA 101, 1514–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YG, Muralinath M, Brandt T, Pearcy M, Hauns K, Lowenhaupt K, Jacobs BL, and Rich A (2003). A role for Z-DNA binding in vaccinia virus pathogenesis. Proc. Natl. Acad. Sci. USA 100, 6974–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler H, Cotsmire S, Langland J, Kibler KV, Kalman D, Upton JW, Mocarski ES, and Jacobs BL (2017). Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc. Natl. Acad. Sci. USA 114, 11506–11511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, and Kanneganti TD (2016). ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol 1, aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, and Beg AA (2000). Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J. Virol 74, 7470–7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Nailwal H, Rector J, Rahman MM, Sam R, McFadden G, and Chan FK (2021). A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation. Immunity 54, 247–258, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, and Rehwinkel J (2017). Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J 36, 2529–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, et al. (2014). RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 56, 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocarski ES, Guo H, and Kaiser WJ (2015). Necroptosis: the Trojan horse in cell autonomous antiviral host defense. Virology 479–480, 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocarski ES, Upton JW, and Kaiser WJ (2011). Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat. Rev. Immunol 12, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH 3rd, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm O, et al. (2016). RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie EJ, Sandow JJ, Lehmann WIL, Liang LY, Coursier D, Young SN, Kersten WJA, Fitzgibbon C, Samson AL, Jacobsen AV, et al. (2019). Viral MLKL homologs subvert necroptotic cell death by sequestering cellular RIPK3. Cell Rep 28, 3309–3319.e5. [DOI] [PubMed] [Google Scholar]
- Pham TH, Kwon KM, Kim YE, Kim KK, and Ahn JH (2013). DNA sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J. Virol 87, 3076–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placido D, Brown BA 2nd, Lowenhaupt K, Rich A, and Athanasiadis A (2007). A left-handed RNA double helix bound by the Zalpha domain of the RNA-editing enzyme ADAR1. Structure 15, 395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MM, and McFadden G (2020). Myxoma virus-encoded host range protein M029: a multifunctional antagonist targeting multiple host antiviral and innate immune pathways. Vaccines (Basel) 8 (8), 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich A, and Zhang S (2003). Timeline: Z-DNA: the long road to biological function. Nat. Rev. Genet 4, 566–572. [DOI] [PubMed] [Google Scholar]
- Samir P, Malireddi RKS, and Kanneganti TD (2020). The PANoptosome: a deadly protein complex driving pyroptosis, apoptosis, and necroptosis (panoptosis). Front. Cell. Infect. Microbiol 10, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonborn J, Oberstrass J, Breyel E, Tittgen J, Schumacher J, and Lukacs N (1991). Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res 19, 2993–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz T, Behlke J, Lowenhaupt K, Heinemann U, and Rich A (2001). Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat. Struct. Biol 8, 761–765. [DOI] [PubMed] [Google Scholar]
- Shors T, Kibler KV, Perkins KB, Seidler-Wulff R, Banaszak MP, and Jacobs BL (1997). Complementation of vaccinia virus deleted of the E3L gene by mutants of E3L. Virology 239, 269–276. [DOI] [PubMed] [Google Scholar]
- Shubina M, Tummers B, Boyd DF, Zhang T, Yin C, Gautam A, Guo XJ, Rodriguez DA, Kaiser WJ, Vogel P, et al. (2020). Necroptosis restricts influenza A virus as a stand-alone cell death mechanism. J. Exp. Med 217, e20191259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan H, Ragan KB, Guo H, Gilley RP, Landsteiner VJ, Kaiser WJ, and Upton JW (2017). Murine cytomegalovirus IE3-dependent transcription is required for DAI/ZBP1-mediated necroptosis. EMBO Rep 18, 1429–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staiano-Coico L, Stollar BD, Darzynkiewicz Z, Dutkowski R, and Weksler ME (1985). Binding of anti-Z-DNA antibodies in quiescent and activated lymphocytes: relationship to cell cycle progression and chromatin changes. Mol. Cell. Biol 5, 3270–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, and Wang X (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, et al. (2007). Dai (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505. [DOI] [PubMed] [Google Scholar]
- Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, Sridharan H, Kosoff R, Shubina M, Landsteiner VJ, et al. (2016). DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, and Chan FK (2014). Staying alive: cell death in antiviral immunity. Mol. Cell 54, 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, and Mocarski ES (2010). Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7, 302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, and Mocarski ES (2012). Dai/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F, Wagner V, Rasmussen SB, Hartmann R, and Paludan SR (2006). Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol 80, 5059–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SD, and Jacobs BL (2012). The amino terminus of the vaccinia virus E3 protein is necessary to inhibit the interferon response. J. Virol 86, 5895–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Liang Y, Zhao S, Ding Y, Zhuang Q, Shi Q, Ai T, Wu SQ, and Han J (2020). ZBP1 mediates interferon-induced necroptosis. Cell. Mol. Immunol 17, 356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, Ragan KB, Ishizuka T, Crawford JC, Tummers B, et al. (2020). Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell 180, 1115–1129.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any datasets/code. Protein sequences were analyzed from the GenBank accession numbers for the various sequences are as follows: ADAR1 AAB06697.1 (amino acids 134–199) (Homo sapiens); ZBP1 (Mus musculus) NP_067369 (amino acids 9–70 and 84–147); the E3L proteins, AAA02759 (amino acids 5–70) (vaccinia virus); AAC08018 (amino acids 5–70) (variola virus).