

Abstract
The genetic determinants of fasting glucose (FG) and fasting insulin (FI) have been studied mostly through genome arrays, resulting in over 100 associated variants. We extended this work with high-coverage whole genome sequencing analyses from fifteen cohorts in NHLBI’s Trans-Omics for Precision Medicine (TOPMed) program. Over 23,000 non-diabetic individuals from five race-ethnicities/populations (African, Asian, European, Hispanic and Samoan) were included. Eight variants were significantly associated with FG or FI across previously identified regions MTNR1B, G6PC2, GCK, GCKR and FOXA2. We additionally characterize suggestive associations with FG or FI near previously identified SLC30A8, TCF7L2, and ADCY5 regions as well as APOB, PTPRT, and ROBO1. Functional annotation resources including the Diabetes Epigenome Atlas were compiled for each signal (chromatin states, annotation principal components, and others) to elucidate variant-to-function hypotheses. We provide a catalog of nucleotide-resolution genomic variation spanning intergenic and intronic regions creating a foundation for future sequencing-based investigations of glycemic traits.
Subject terms: Sequencing, Genetics research, Diabetes, Quantitative trait
This study of 23,000 non-diabetic individuals highlights loci associated with fasting glucose and fasting insulin in diverse cohorts with whole genome sequence data.
Introduction
Type 2 diabetes (T2D) and insulin resistance are complex genetic conditions often characterized by disruptions of normal levels of fasting glucose (FG) and fasting insulin (FI)1. These traits are influenced by a spectrum of common to rare genetic variation2–7 with most evidence coming from genome-wide association studies (GWAS)8,9, exome arrays2,3,6, whole-exome sequencing2, and small samples of low-pass whole-genome sequencing (WGS)4,10. These efforts have found over 100, mostly common (minor allele frequency (MAF) > 0.05), variants associated with FG or FI, including those in non-coding and intergenic spaces2–4,6,8,9. We expand on these previous studies by assessing common, low frequency (MAF < 0.05), and rare (MAF < 0.01) variants through comprehensive WGS association analysis in diverse population cohorts in NHLBI’s Trans-Omics for Precision Medicine (TOPMed) program. The current study aims to better understand the variants at GWAS loci through multiple approaches including discovery and fine-mapping in both coding and non-coding regions as well as aggregate rare variant testing using both protein-coding variants and intergenic variants. In addition, we explore ancestry-specific results through our four included race/ethnicities and one population group: African, Asian, European, Hispanic, and Samoan, respectively. Finally, we characterize all reported regions with annotations including chromatin states, annotation principal components (PCs), expression quantitative trait loci (eQTL), and others from recent annotation accumulation efforts including the Diabetes Genome Atlas (DGA).
Results
Phenotypes and genotypes in the NHLBI TOPMed program
We identified and characterized common and rare variants associated with FG and (natural log-transformed) FI through association tests using WGS data from fifteen cohorts in TOPMed (Supplementary Table 1). As in prior quantitative trait analyses, we excluded individuals with diabetes (by glycemia or medication), resulting in a total sample size of 26,807 individuals with FG and 23,211 individuals with FI. This represents a diverse sample of four self-reported race/ethnicities and one population group including African, Asian, European, Hispanic, and Samoan, respectively, and our total sample was >40% non-European (Supplementary Tables 2–3). In addition to use of genetic ancestry adjustments in our models (see the “Methods” section), we used participant’s self-reported race/ethnicity to assign individuals to one of five groups for stratified analyses or inclusion as a covariate. Individuals were given a single label to infer their ancestry, but each group represents a diverse cross-section of race, culture, or admixture. Trait measures were harmonized across cohorts and assays and adjusted for self-reported race/ethnicity, study age, sex, and body mass index (BMI; Supplementary Tables 2–3, “Methods”). We assessed 60 M variants from the TOPMed Freeze 5b WGS data freeze for each trait using single-variant testing (minor allele count, MAC > 20) in pooled and race/ethnicity-specific approaches. We used a significance threshold of P < 1.0 × 10−9 as has been established for WGS studies including African ancestry11. We also separately report signals identified with P < 5.0 × 10−8 as suggestively associated with either trait. These suggestively associated signals are reported to characterize potential regions of interest with our available annotations for use in future higher-powered studies. We further performed rare variant testing (MAF < 0.01) using aggregate burden and SKAT tests in both gene centric and genetic region approaches. We computed 95% credible sets for each distinct common variant signal conditioned on any other identified signal at the locus (“Methods”, Supplementary Data 1). 99% credible sets are also reported for signals identified through the pooled analysis (Supplementary Data 2), with a median size of 12 variants. This is 20% smaller than a recent multi-ethnic GWAS of glycemic traits12 which reported a median 99% credible set size of 15.
Whole-genome sequence significant associations with fasting glucose and insulin
We identified eight distinct variants significantly associated with FG or FI across five gene regions in the pooled race/ethnicity analysis (P < 1.0 × 10−9, Table 1). These include previously identified MTNR1B, G6PC2, GCK, GCKR, and FOXA2 gene regions (Supplementary Data 3–4). MTNR1B had a distinct secondary signal after conditioning on the lead variant. G6PC2 had three distinct association signals, one of which was rare (MAF < 0.01), as determined by sequential conditional analysis. These distinct secondary and tertiary signals are also reported in Table 1 (locus-wide significance threshold of 1.0 × 10−5, “Methods”) and further described in the following sections. Manhattan and QQ plots for single variant analyses of FG and FI are shown in Supplementary Fig. 1.
Table 1.
Distinct signals at loci significantly associated with glycemic traits FG and FI in TOPMed, P < 1 × 10−9.
| Trait | Population | Nearest gene | MarkerIDa | EA | rsID | Annotation | EAF | P-value | Beta | SE | Conditioned on |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fasting glucose | Pooled | MTNR1B | 11:92975544:C:G | G | rs10830963 | Intronic | 0.24 | 9.1 × 10−46 | 0.07 | 0.01 | |
| 11:92884161:G:Ab | G | rs73560545 | intronic | 0.83 | 5.7 × 10−6 | 0.03 | 0.01 | rs10830963 | |||
| G6PC2 | 2:168906638:T:C | C | rs560887 | Intronic | 0.82 | 6.8 × 10−37 | 0.07 | 0.01 | |||
| 2:168900420:A:Gb | A | rs540524 | 2KB upstream | 0.66 | 9.9 × 10−14 | 0.04 | 0.01 | rs560887 | |||
| 2:168907981:T:C c | T | rs2232326 | Missense | 0.99 | 5.0 × 10−6 | 0.15 | 0.03 | rs560887, rs540524 | |||
| GCK | 7:44189469:C:T | T | rs1799884 | 2KB upstream | 0.18 | 3.9 × 10−28 | 0.06 | 0.01 | |||
| GCKR | 2:27508073:T:C | C | rs1260326 | Missense | 0.65 | 6.1 × 10−21 | 0.04 | 0.01 | |||
| FOXA2 | 20:22581688:A:AG | A | rs3833331 | 3′ UTR | 0.85 | 5.4 × 10−10 | 0.04 | 0.01 | |||
| Fasting insulin | Pooled | GCKR | 2:27508073:T:C | C | rs1260326 | Missense | 0.67 | 7.2 × 10−13 | 0.03 | 0.01 |
EA effect allele, EAF effect allele frequency, EU European, HS/L Hispanic/Latinx.
aChromosome, position(Hg38), reference allele, alternative allele of the genetic variant with the lowest P-value and highest posterior probability representing a distinct association at a locus.
bIndicates secondary signal.
cIndicates tertiary signal for association at significance level P < 1 × 10−5 and MAC > 20 after conditional analysis.
Our significantly associated variants replicate previous GWAS findings, which are summarized in Supplementary Data 3–4. We further characterize these variants in the context of sequencing and related available resources as summarized in Fig. 1. We used the Diabetes Epigenome Atlas (DGA) and TOPMed resources to provide functional annotations including chromatin states, annotation principal components (aPCs)11 that each provide a summary of related functional annotations via PCA (“Methods”), and expression quantitative trait loci (eQTL) from adipose, pancreas, liver and skeletal muscle. In addition to variant descriptions in Fig. 1, regional locus plots with tissue-specific annotations for reported loci in Supplementary Fig. 2, and associations of reported loci with related traits in Supplementary Fig. 3 and Supplementary Data 5. Selected regions are further described based on the data below.
Fig. 1. Characterization of significant and suggestive single-variant signals associated with fasting glucose and fasting insulin in TOPMed.

TOPMed features of distinct, significant and suggestive signals associated with fasting glucose and fasting insulin (log-transformed) in pooled analysis. P-values (unconditional −log10-transformed) for glycemic and related traits (HbAa1c and type 2 diabetes) and effect allele frequency (with respect to the pooled analysis effect allele) across race/ethnicities in TOPMed are reported. Chromatin states at relevant tissues were drawn from two sets of experiments from DGA46,47; annotation PCs provide summaries of multi-faceted variant function; variants that are significant eQTLs in relevant tissues are denoted. EAF, effect allele frequency for TOPMed sample; EnhA1, Active Enhancer 1; EnhA2, Active Enhancer 2; Het, Heterochromatin; Quies, Quiescent/Low; ReprPC, Repressed PolyComb; ReprPCWk, Weak Repressed PolyComb; TssA, Active TSS; TssFlnk, Flanking TSS; TxWk, Weak Transcription; ZNF/Rpts, ZNF genes & repeats.
MTNR1B intronic variant rs10830963 (P = 9.1 × 10−46) has been characterized as a strong signal for insulin secretion13; this variant is in a weak transcription chromatin state in islets, is a metabolite QTL for glucose14,15, and is a pancreatic-islet-specific eQTL associated with the expression of MTNR1B16. Identified after conditioning on the primary variant rs10830963 in the MTNR1B region, intronic variant rs73560545 occurs upstream of the primary signal and had a lowering effect on FG, in contrast to the primary signal which had a glucose-raising effect. While this is a well-known region in the context of these traits, this secondary variant at rs73560545 has not been previously identified in the reviewed literature (Supplementary Data 3–4).
The GCKR locus had a significant association with both FG and FI at rs1260326 (P = 6.1 × 10−21, 7.2 × 10−13, respectively) with functional activity suggested by its relatively high aPC-Epigenetics and aPC-Transcription-Factor values. This variant is also an eQTL and pQTL associated with many genes and proteins, most relevantly with insulin17. GCKR and rs1260326 have been previously described in previous studies for both traits (Supplementary Data 3–4).
The FOXA2 locus has also been previously found to be associated with glycemic traits, regulating gene expression for glucose sensing in pancreatic beta cells18. We observe one FG-associated signal at rs3833331 (P = 5.4 × 10–10), a variant moderately linked to previously identified FOXA2 lead variants rs6048205 and rs6048216, and based on conditional analysis is likely the same signal. The variant rs3833331 is in the 3′ UTR of the gene and classified as a CAGE promoter, GeneHancer, and SuperEnhancer. It is in an active TSS chromatin state for both pancreas and islets. Our identified variant rs3833331 is frequent in African individuals, while it has relatively low frequency in both European and Hispanic race/ethnicities.
Rare variant aggregate testing performed using both gene-centric and genetic region approaches identified one significantly associated region with FG at the known G6PC2 locus, described in the next section (Supplementary Data 6–7). No rare variant aggregate signals were found to be associated with FI that were not composed mostly of singletons (Supplementary Data 8–9). Manhattan plots for region-based rare variant aggregate analysis in Supplementary Fig. 4.
Refinement of the multi-allelic associations at the known G6PC2 locus
At the known FG and T2D-associated G6PC2 locus2,3, we observed several previously identified variant associations with FG (Fig. 2). In single variant analyses, we identified three distinct association signals, the third of which was a previously identified association after conditioning on two previously reported common GWAS variants, rs560887 (primary signal, P = 6.8 × 10−37) and rs540524 (secondary signal, P = 9.9 × 10−14). The rare missense variant rs2232326 (tertiary signal, P = 5.0 × 10−6) is annotated19 by the aPCs as disruptive and likely damaging, with a score falling in the top 7% distribution of the aPC representing “protein function” (aPC-Protein Function = 31.5, top 7% genome-wide). In addition, rs2232326 appears to be highly conserved, with a score falling in the top 0.13% of the distribution of an aPC representing “conservation” (aPC-Conservation = 28.8, top 0.13% genome-wide). The genomic region surrounding rs2232326 is annotated to be in a weakly transcribed chromatin state, relative to the genome, in islets and this variant is near the transcription end site (Fig. 2). The frequency of the C allele at rs2232326 was <0.01 in all race/ethnicity groups except for the Asian group where the frequency was 0.03 (gnomAD: East Asian AF = 0.05, Overall AF = 0.01). In aggregate gene-centric testing of all 75 rare missense variants in G6PC2, this previously identified rare (MAF = 0.01) variant rs2232326, along with variant rs2232323 (MAF = 0.01), contributed the most to the significant association test statistic (PBurden,1,1 = 1.4 × 10−10, Supplementary Data 6).
Fig. 2. Regional investigation of three conditionally significant signals associated with fasting glucose in the G6PC2 locus in TOPMed.
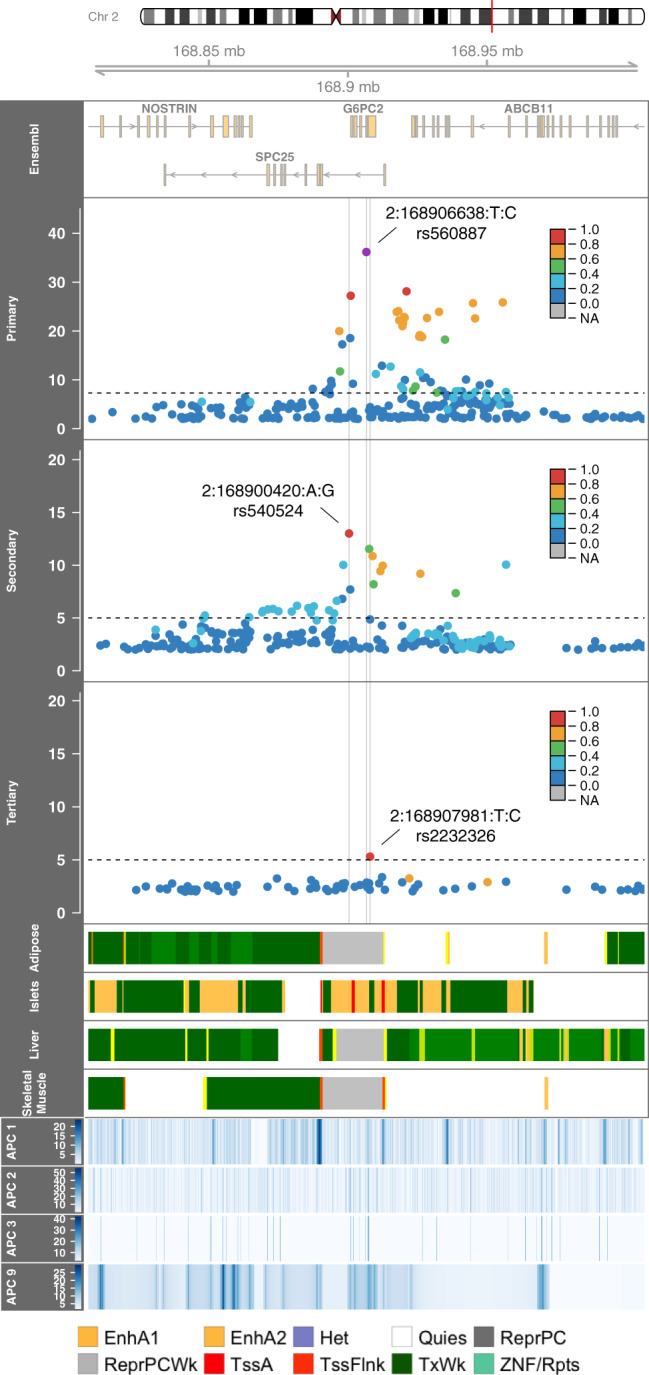
Regional association plot of −log10 P values by genomic position for sequential conditional single-variant analyses. The linkage disequilibrium (r2) between the primary signal (rs560887, 2:168906638:T:C), as defined by the highest posterior probability, and variants in the region for each panel as calculated in TOPMed is indicated in the colors of the points. The chromatin states at four relevant tissues47 and annotation PCs are provided across the region. APC1, APC Epigenetics, APC2, APC Conservation, APC3, APC Protein, APC9, APC Distance to TSS/TSE; EnhA1, Active Enhancer 1; EnhA2, Active Enhancer 2; Het, Heterochromatin; Quies, Quiescent/Low; ReprPC, Repressed PolyComb; ReprPCWk, Weak Repressed PolyComb; TssA, Active TSS; TssFlnk, Flanking TSS; TxWk, Weak Transcription; ZNF/Rpts, ZNF genes & repeat.
Given the multiple distinct signals at G6PC2, we performed a haplotype analysis to evaluate the contribution of rare variants and identify allele-specific effects. We extended the haplotype analysis of Mahajan et al.3 (rs560887, rs138726309, rs2232323, rs492594) to include our secondary (rs540524) and tertiary (rs2232326) signals. Our secondary signal is in moderate linkage (r2 = 0.58) to the previously haplotyped rs492594 and the effect allele A has a glucose-raising effect in both marginal and conditional analyses (Supplementary Table 4 and Table 1). We observed consistent direction of effects as the previous haplotype analysis, demonstrating the reliability of associations identified in the present TOPMed sample. Both haplotypes containing the C allele of the tertiary signal at rs2232326, the variant with the largest effect size included in the analysis, had glucose-lowering effects. The largest glucose-lowering effects at G6PC2 were observed at the two haplotypes each carrying a rare allele: rs2232326 (rs560887-C, rs138726309-C, rs2232323-A, rs492594-C, rs540524-G, rs2232326-C, Beta = −0.15 +/− 0.00002) and rs2232323 (rs560887-T, rs138726309-C, rs2232323-C, rs492594-G, rs540524-A, rs2232326-T, Beta = −0.11+/−0.00008, Supplementary Table 4).
Additional suggestive associations with fasting glucose and insulin
We further report twelve distinct variants suggestively associated with FG or FI across ten gene regions in the pooled race/ethnicity analysis and ancestry-specific analyses (P < 5.0 × 10−8; Table 2). These include previously identified SLC30A8, TCF7L2, and ADCY5 gene regions (Supplementary Data 3–4). Other regions not previously identified include APOB, PTPRT, ROBO1 and those described in the ancestry-specific section below. SLC30A8 and PTPRT have distinct secondary signals identified through conditional analysis, which are also reported in Table 2 (locus-wide significance threshold P < 1.0 × 10−5). We outline these suggestive signals and the corresponding gene regions below to provide annotation and description and to provide context for investigation of these signals in future, larger studies.
Table 2.
Distinct signals at loci suggestively associated with glycemic traits FG and FI in TOPMed, P < 5 × 10−8.
| Trait | Population | Nearest gene | MarkerIDa | EA | rsID | Annotation | EAF | P-value | Beta | SE | Conditioned on |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fasting glucose | Pooled | SLC30A8 | 8:117179236:C:T | C | rs35859536 | 2.5KB downstream | 0.74 | 1.0 × 10−9 | 0.03 | 0.01 | |
| 8:117258547:C:Tb | T | rs542965166 | Intronic | 0.001 | 1.9 × 10−6 | 0.50 | 0.11 | rs35859536 | |||
| APOB | 2:21074277:A:G | A | rs478588 | 30KB upstream | 0.29 | 2.9 × 10−9 | 0.03 | 0.01 | |||
| TCF7L2 | 10:112998590:C:T | T | rs7903146 | Intronic | 0.25 | 2.0 × 10−8 | 0.03 | 0.01 | |||
| ADCY5 | 3:123335923:A:C | A | rs72964564 | Intronic | 0.80 | 2.8 × 10−8 | 0.03 | 0.01 | |||
| HS/L | HS6ST3 | 13:96407609:A:G | G | rs1328056 | Intronic | 0.02 | 3.6 × 10−8 | 0.33 | 0.06 | ||
| CTD-2199C04.4 | 5:10169711:T:C | C | rs13361160 | Intergenic | 0.45 | 3.1 × 10−8 | 0.10 | 0.02 | |||
| Fasting insulin | Pooled | PTPRT | 20:42752773:G:A | A | rs185250851 | Intronic | 0.002 | 2.1 × 10−8 | 0.30 | 0.05 | |
| 20:43230137:C:Tb | T | rs78618809 | Intergenic | 0.02 | 5.9 × 10−6 | 0.08 | 0.02 | rs185250851 | |||
| ROBO1 | 3:79812347:C:A | C | rs539973028 | 44KB upstream | 0.99 | 4.7 × 10−8 | 0.51 | 0.09 | |||
| EU | LINC00704, LINC00705 | 10:4656482:GAAAAT:G | G | rs775018107 | ncRNA intronic | 0.002 | 4.5 × 10−8 | 0.33 | 0.06 | ||
| Samoan | RP11/IGSF11 | 3:118656074:T:Gc | G | rs117592405 | ncRNA intronic | 0.01 | 3.3 × 10−8 | 0.80 | 0.14 |
EA effect allele, EAF effect allele frequency, EU European, HS/L Hispanic/Latinx.
aChromosome, position(Hg38), reference allele, alternative allele of the genetic variant with the lowest P-value and highest posterior probability representing a distinct association at a locus.
bIndicates secondary signal for association at significance level P < 1 × 10−5 and MAC > 20 after conditional analysis.
cThis signal was not replicated (Supplementary Table 7).
In the ADCY5 region, variant rs72964564 (P = 2.8 × 10−8) showed suggestive association with FG and is highly linked (r2 = 0.86 in the present study sample) with the known FG-associated variant rs11708067. Both ADCY5 variants are designated GeneHancer and SuperEnhancer variants, and rs72964564 is in an active enhancer state for adipose tissue and is an eQTL associated with ADCY5 expression in pancreatic islets16. ADCY5 and rs72964564 have been previously identified in studies of FG (Supplementary Data 3–4).
Near the APOB region a suggestively associated variant at rs478588 (P = 2.9 × 10−9) has not previously identified (Supplementary Data 4). Variant rs478588 has robust associations with lipid traits20 and significant parent-of-origin effects on metabolic traits21. Lipid traits have been studied for pleiotropy with glycemic traits but have been inconclusive with respect to APOB. Replication was attempted in UK-BioBank (UKBB) with consistent direction of effect and P = 0.01, but it should be noted UKBB sample used was not based on WGS data (Supplementary Table 5).
We identified a pair of suggestively FG-associated signals in islet-specific active enhancer regions at the known SLC30A8 locus. The primary signal is at variant rs35859536 (P = 1.0 × 10−9), which is an intergenic variant 2.5KB downstream of SLC30A8. This variant is highly linked (r2 > 0.95) to previously identified lead variants rs11558471 and rs3802177 at SLC30A8, both of which are in the 3′ UTR. This is a known T2D susceptibility locus and has been identified as associated with triglyceride levels22. Our lead variant is also significantly associated with T2D in TOPMed (Supplementary Data 5)23. To evaluate potential causal variants (“Methods”) we performed credible set analyses and found rs35859536 has a posterior probability (PP) of 0.48; other variants in the 95% credible set with PP of at least 0.05 were either missense or in the 3′ UTR, are highly linked with this lead variant (r2 > 0.97), and were significantly associated with FG in previous studies2,8,24. Our lead variant, along with other previous lead variants, is in an active enhancer 2 region for islets; rs35859536 is also mapped as an accessible chromatin site in islet of Langerhans given inflammatory-inducing cytokine exposure25. Replication of these signals was attempted in the METSIM cohorts, and we observe nominal significance of the primary signal with a consistent direction of effect, while the secondary signal was too low frequency in this cohort to estimate an effect (Supplementary Table 6).
The secondary suggestively FG-associated signal at the SLC30A8 locus is at variant rs542965166 (P = 1.9 × 10−6). This intergenic variant is only observed in individuals in the Asian population (Asian EAF = 0.007); this race/ethnicity-specificity is replicated in gnomAD26 where the allele is only observed in East Asians at a rare frequency. This secondary, race/ethnicity-specific signal is not highly linked to other variants in the region, which may indicate that this is a distinct, secondary signal and requires further follow-up in an Asian population.
Upstream of the ROBO1 locus we identified a suggestively novel (to the best of our knowledge) FI-associated rare variant, rs539973028 (P = 4.7 × 10−8). This locus has previously been studied for SLIT-ROBO signaling and expression in T2D complication diabetic retinopathy27. ROBO1 has been associated with the glycemia-related traits of BMI and waist-to-hip ratio28–30 and is commonly expressed in muscle and skin31. This variant is only observed in the African population of TOPMed and gnomAD26. It is intergenic and in a weakly transcribed region in islets.
We identified a pair of distinct, suggestively novel (to the best of our knowledge) rare variant signals associated with FI near the PTPRT gene (Table 2). The primary signal, rs185250851 (P = 2.1 × 10−8), is an intronic variant. It is rare in all tested population groups and not observed in Asian individuals, as validated in gnomAD26. The secondary signal at variant rs78618809 (P = 5.9 × 10−6) is in an intergenic region. This variant is within the top 5% of variants with respect to an aPC representing “Distance to TSS/TSE,” a composite measure of individual annotations indicating low variant distance to the endpoints of the intergenic region. This variant is rare overall, but observed frequently (EAF = 0.07) in the African ancestry population. This gene has previously been associated with BMI, which has moderate genetic correlation (previously estimated as ρg = 0.48) with FI20,21. The expression of this gene is most commonly associated with variants as eQTLs in pancreas in GTEx31. The multi-ethnic TOPMed sample permits the identification of this signal, which requires a sufficiently diverse sample.
Race/ethnicity-specific analyses associated with fasting glucose and insulin
In race/ethnicity-specific analyses, we observed two not previously identified race/ethnicity-specific rare variant suggestive associations with FG in individuals of the Hispanic/Latinx population (Table 2). The first signal, rs1328056 (P = 3.6 × 10−8), is an intronic variant in the HS6ST2 gene, which has been associated with obesity and impaired glucose metabolism in mouse studies13. The second signal is an intergenic variant near the ATPSCKMT gene, rs13361160 (P = 3.1 × 10−8) which is associated with eosinophil counts, a measure that has been negatively correlated with FG14. We would require further data from individuals from the Hispanic/Latinx population in order to replicate these suggestive signals.
We identified two suggestively novel (to the best of our knowledge) race/ethnicity-specific rare alleles associated with FI. In the European population, rs775018107 (P = 4.5 × 10−8) at the LINC00704/LINC00705 locus was suggestively associated with FI (Table 2). We also identified a suggestive FI association in the Samoan population cohort at rs117592405 (P = 3.3 × 10−8); this intronic variant was not replicated in an independent Samoan cohort using imputed genotypes (N = 1401, Supplementary Table 7).
Enrichment of trait-associated variants in chromatin states
We assessed whether our trait-associated variants were found more often than expected in a particular chromatin state using the tool GREGOR (Genomic Regulatory Elements and Gwas Overlap algorithm)15 (“Methods”). We observe that fasting glucose-associated variants are found more often in “Active Enhancers”, “Weak Transcription”, and “Genic Enhancer” chromatin states in Islets (P < 0.05, Supplementary Table 8). This complements findings from Chen et al.12 showing similar enrichment of glycemic trait-associated signals in islet enhancers.
Discussion
In this paper, we leveraged high-coverage WGS data in large multi-ethnic population-based cohorts to assemble a comprehensive catalog of nucleotide-resolution genomic variation associated with the key diabetes-related quantitative traits FG and FI. Our analysis covered intergenic and intronic regions to a MAC of 20 in single variant analysis and combines base pair variation with tissue-specific epigenomic annotation to illuminate variant-to-function hypotheses in diabetes pathobiology.
A strength of the present analysis is the inclusion of individuals from 15 cohorts, comprised of four major race/ethnicity groups and one population group(African, Asian, European, Hispanic/Latinx, and Samoan, respectively). Some of our reported regions were either mostly or exclusively present in a single race/ethnic group. These include the secondary SLC30A8 variant rs542965166 only observed in the Asian group, ROBO1’s rs539973028 only present in African group, and others. Previous genetic studies of glycemic traits have included samples primarily from individuals of European ancestry, but increasingly a larger degree of African ancestry. The most recent meta-analysis by the MAGIC consortium included ~30% non-European ancestry individuals, demonstrating that a number of trait-associated loci that would have been undetected in samples exclusively of European ancestry18. While extending the genetic ancestries studied beyond European populations, the MAGIC results were subject to the limitation of imputation by the 1000 Genomes Project reference panel, so most rare and ancestry-specific variation was still not assessed. In addition, we observe a 20% decrease in average 99% credible set size from the MAGIC results suggesting value of WGS in fine-mapping.
This analysis benefits from the availability of whole-genome sequencing data provided by the TOPMed Program of the NHLBI’s Precision Medicine Initiative10,32. Previous studies have been limited by reliance on imputation or minimal sample sizes for data with sequencing paired with glycemic phenotypes. The GoT2D study has performed WGS in a limited sample, contributing to the larger DIAMANTE meta-analysis of summary statistics but relying on imputation for complete genotyping of most samples33. The UKBB study includes a large set of primarily European individuals with whole-exome sequencing; however, the sample size with measured fasting glycemic traits is limited as described in the validation study. One of the most expansive efforts, a MAGIC collaboration8,34, has performed extensive analyses for glycemic traits, but results rely primary on Exome Chip data and thus have limited coverage of intergenic and intronic regions6. Our significant findings replicate previous GWAS findings in terms of gene regions, but we are able to characterize these regions in great detail and report on specific variants which may not previously described in these known regions, such as the secondary MTNR1B-associated variant.
In addition to reporting significant and suggestive associations, we provide detailed characterization of each locus in terms of functional annotations, chromatin states, quantitative trait loci, related trait associations and more. The G6PC2 in particular was described in terms of allelic effects and provided functional characterization of low-frequency signal, demonstrating the glucose-lowering effects of rare alleles and islet-specificity of this locus’s associations. Many of our reported regions lie in enhancer or transcription start site chromatin states, and we particularly see significant enrichment in enhancer states in islets. This agrees with findings of previous GWAS and the expected relevant tissues for glycemic traits. We provide this data and the visualizations for use in future investigation of these loci.
A limitation of this study is our smaller sample size compared to the most recent GWAS. Our significant single variant results are all found near previously identified gene regions. Also, many of our suggestively novel results lack substantiating replication, particularly those which are race/ethnicity specific. We analyzed independent studies with genetic data to investigate associations significant in TOPMed; we were unable to replicate potentially novel signals in these external cohorts. This may be attributable to limitations in the available replication studies’ samples with respect to size, race/ethnicity and imputed versus WGS genotypes. To support the understanding of these signals, we consider a set of tissue-specific chromatin states, an effort that would benefit from further tissue-specific characterization across functional measures. This could also help inform the underlying biological mechanisms of glycemic regulation and its role in diabetes.
This multi-ethnic WGS study provides the foundation for future sequencing-based investigation of glycemic traits. Our results from common and rare variant analysis comprised multiple suggestive hits, including those with exceedingly rare variants that require further investigation, indicating the potential for the identification of novel signals given larger sequencing studies and external validation studies. The value of diverse studies like TOPMed is further evidenced by the specificity of such signals to certain populations and cohorts. This value is also demonstrated by the intronic and intergenic location of many such suggested signals. These signals, in both single variant and rare variant set-based testing, indicated that many associations lie outside gene boundaries and it is important to perform genome-wide single variant testing but also complement gene-centric RV testing with region-based RV testing to fully capture signal. Future TOPMed study phases will permit the continued investigation of these signals empowered by increased sample sizes, with future directions including detailed fine-mapping of signal regions and assessment of glycemic trait heritability. To support future research, all results from this analysis have been made available to the research community through the Type 2 Diabetes Knowledge Portal (Genetic Association Data will be released in January 2021)17.
Methods
Whole-genome sequencing
Whole-genome sequencing of blood samples for all participants included deep coverage (>30x on average) sequencing from blood samples provided by the NHLBI TOPMed program. Sequencing was performed across six centers (Broad Institute of MIT and Harvard, Northwest Genomics Center, New York Genome Center, Illumina Genomic Services, Macrogen, and Baylor College of Medicine Human Genome Sequencing Center) as previously described35. The TOPMed Informatics Research Center at the University of Michigan performed data harmonization and joint variant discovery and genotype calling, requiring DNA sample contamination below 3% and at least 95% of the genome with at least 10x coverage. Freeze 5b was aligned to GRCh38 reads from the 1000 Genomes Project reference sequences36. The samples were further processed by a centralized pipeline by the TOPMed Data Coordinating Center at the University of Washington, where further quality control and sample-identity resolution were performed, including sex and relatedness concordance and selection of variants with missingness <5% and QUAL > 127. Variants were also checked via an excess heterozygosity filter (EXHET), which removed the variant if the Hardy-Weinberg disequilibrium p-value was <1 × 10−6, after accounting for population structure. After processing, Freeze 5b contained 54,508 samples with 438 million single nucleotide variants (SNVs) and 33 million short insertion-deletion variants.
Population structure principal components were calculated across all Freeze 5b TOPMed participants using PC-AiR; a genetic relatedness matrix was calculated across all Freeze 5b TOPMed participants using PC-Relate accounting for population structure. Race/ethnicity was determined by self-report from each study. Self-reported race/ethnicity was used in conjunction with principal component and/or genetic relatedness matrix adjustment to control for both genetic and individually identified ancestry37.
Phenotype harmonization
Phenotype harmonization proceeded following a protocol defined by the TOPMed Diabetes Working Group for participating TOPMed studies. Duplicated individuals were excluded following the TOPMed Diabetes Working Group protocol. Within a study, monozygotic twins were retained and the duplicate to be kept was chosen based on verification of cohort characteristics, including proper cohort sequencing center designation, and then by highest call rate. Across studies, duplicates were selected by removing missing trait data, prioritizing population-based cohorts, and retaining individual records with the longest follow-up period. All study participants provided informed consent and each study was approved by their respective institutional review boards.
Glycemic traits (fasting glucose (FG) and fasting insulin (FI)) were analyzed for individuals who did not have diabetes at the time of glycemic trait measurement. This subset was defined as those not taking anti-diabetes medication, with fasting glucose <7 mmol/l and/or HbA1c < 6.5%. For individuals with multiple blood draws, the earliest exam or most complete exam was used. Age, sex, and BMI covariates were reported at the time of glycemic trait measurement. Fasting was defined to be at least 8 h without food or drink; measurements from blood were converted to plasma values using a 1.13 correction factor38. The units for glucose are mmol/l; units for insulin are pmol/l. Fasting insulin was natural log-transformed prior to analysis in order to address non-normality.
Study sample and power
The present analysis included 23,211 (FI) and 26,807 (FG) individuals from the NHLBI TOPMed program. The cohorts included consist of participants of self-reported African American (FI n = 6803; FG n = 7174), East Asian (FI n = 572; FG n = 2217), European (FI n = 13,281; FG n = 14,513), Hispanic/Latinx (FI n = 1641; FG n = 1989), and Samoan (FI n = 914; FG n = 914) race/ethnicity. Our analysis of fasting insulin included 14 cohorts and fasting glucose included 15 cohorts. The sample is predominantly of European race/ethnicity (FI 57.2%; FG 54.1%) and female (FI 66.5%; FG 65.2%); full cohort descriptions are given in Supplementary Tables 2 and 3.
We performed a post hoc power calculation to evaluate the power to detect genetic signal at the genome-wide threshold for statistical significance of 5 × 10−8. Given the study sample size, this analysis was powered to detect 0.54–4.21% and 0.57–4.21% percent variation in glycemic trait explained by a genotype in race/ethnicity-specific analyses for FG and FI, respectively. The pooled study including all samples was powered to detect 0.16% and 0.17% percent variation in glycemic trait explained by a genotype for FG and FI, respectively.
Single-variant analysis
We performed single variant analysis in Freeze 5b of TOPMed using race/ethnicity-specific and pooled approaches. We tested 64,675,008 variants for associations with FG and 58,759,883 with FI in both pooled and race/ethnicity-specific analyses, and restricted analysis to variants with minor allele count >= 20. We used linear mixed effects models and adjusted for age, age squared, sex, body mass index, study-race/ethnicity, with heterogeneous variance permitted across study-race/ethnicity groups and empirical kinship for relatedness and population structure. Models were fit using GENetic Estimation and Inference in Structured samples (GENESIS)39 in the Analysis Commons cloud-computing platform40. P-Values reported are for a two-sided Wald test from the mixed model. Fasting glucose and natural log-transformed fasting insulin were used as outcomes in separate models. We define the standard genome-wide threshold for statistical significance as 1 × 10−9. We also report variants with P < 5 × 10−8 as suggestively associated to provide context for regions of interest for future, higher-powered studies.
Stepwise conditional analysis was performed at each identified locus, defined to be a 500 kb region centered on the most significant variant, in order to identify distinct signals. This analysis proceeded by first including the most significant variant as a covariate, and repeating until no variants were associated with the phenotype with p-value <1 × 10−5. For each distinct signal, a final model was run conditioning on the set of other distinct signals; we report these potentially distinct signals.
Towards fine-mapping the identified loci, we generated 95% credible sets to investigate likely causal variants (LocalZoom). For each locus, we calculated Bayes factors for all variants from their single variant p-value; p-values were taken from conditional analyses on all other identified variants at the locus where multiple distinct signals were identified in the stepwise conditional analysis. We calculated posterior probability of association (PP) of each variant as the proportion contributed to the summation of all BFs in the region. The variants were sorted by descending PP, indicating decreasing probability that the variant is truly associated with the glycemic trait. The 95% credible set was constructed by including variants, starting with the highest PPA, until their cumulative PPA exceeded 0.95. 99% credible sets were similarly constructed for association signals from the pooled analysis only.
Rare variant analysis
We performed gene-based and genetic region aggregate testing to identify sets of rare variants associated with fasting glucose and log-transformed fasting insulin. We first fit a heteroscedastic linear mixed model for fasting glucose and log-transformed fasting insulin. Both traits were adjusted for age, age2, sex, body mass index (BMI), study-race/ethnicity group indicators, and ten population structure principal components. A variance component was included for the empirically derived sparse kinship matrix and residual variances were permitted to be different for study-race/ethnicity groups to account for family relatedness, population structure, and study-race/ethnicity differences.
The heteroscedastic linear mixed model was used to perform variant set analyses for rare variants with MAF < 1%. Sets were defined by genetic regions and gene-centric categories. Genetic regions allowed the complete analysis of the genome, particularly non-coding regions that have not been previously captured by arrays. The regional analysis used 2 kb sliding windows to scan the genome with 1 kb skip length. The gene-centric analysis examined all protein-coding genes in Ensembl using functionally determined masks to aggregate variants together by coding and non-coding annotations. Coding annotations were used to define three SNV filters categorized by GENCODE based on consequence: (a) putative loss of function (stop gain, stop loss, splicing), (b) missense, and (c) synonymous variants. Leveraging the whole-genome sequencing, we used non-coding annotations to test sets of variants that are not protein coding. We constructed masks (d) characterized as promoters given they were within +/− 3 kb of a transcription start site with CAGE signal overlay, or (e) characterized as enhancers given they were identified by GeneHancer with CAGE signal overlay.
The burden test and SKAT were used for testing the association of the rare variant sets and FG and FI. In these approaches, a weight based on the MAF can be used to upweight rarer variants. We considered two common weighting schemes based on , where and or and .
Statistical significance was defined for each glycemic trait, separately for gene-centric and genetic region analysis. For gene-centric analysis, a threshold was defined by a Bonferroni-corrected significance threshold of , correcting for all five masks and all protein-coding genes when considering the minimum p-value across the burden and SKAT tests (Supplementary Table 9). The threshold for the genetic region analysis was determined given the total number of 2 kb sliding windows tested, yielding a Bonferroni-corrected threshold of . We report sets that include variant(s) with effective minor allele count greater than five and that are not exclusively composed of singletons; complete results based on the significance threshold are provided in Supplementary Data 6–9.
Haplotype analysis
We performed haplotype analysis for variants associated with fasting glucose. This analysis considered a set of 18,071 unrelated individuals, identified by PC-AiR41 by the TOPMed Program with a threshold of third-degree relatives. We performed regression of fasting glucose on haplotype using a two-step EM algorithm on the unphased genotypes, as implemented in the haplo.stats R package42. The posterior probabilities of haplotypes were computed for variants in the G6PC2 gene; the variants were included based on the variants included in a previous G6PC2 haplotype analysis, variants driving the G6PC2 missense set signal, and distinct G6PC2 signals from the single variant analysis. The association was adjusted for age, age2, sex, body mass index, study-race/ethnicity, and ten population structure principal components.
Annotation
In order to characterize the functional impact of associated variants, we assembled functional annotations from multiple publicly available databases. We considered annotations from the Diabetes Epigenome Atlas, FAVOR, InsPIRE, and GTEx projects. From the Diabetes Epigenome Atlas, we obtained chromatin states from four tissues relevant to glycemic traits: adipose, islet, liver, and muscle. These were available from two experiments, Parker lab ChromHMM 13-state model under accession TSTSR679993 & AMP-T2D ChromHMM 18-state model under accession TSTSR043890. We also report annotation PCs from the FAVOR database43, which are summaries calculated as the first principal component of individual functional annotations across functional categories including conservation, epigenetics, local nucleotide diversity, mutation density, protein function, proximity to TSS/TSE, proximity to coding, and transcription factor binding. The individual annotations contributing to the aPCs are previously described19. Annotation PCs are calculated at the variant level and reported as PHRED-scaled scores derived from the first PC from the category’s PCA, providing the interpretation that variants with scores >10 are in the top 10% of category across all TOPMed variants. We obtained pancreatic islet-specific signals from the InsPIRE consortium and tissue-specific signals from the GTEx project (Version 8) to assess colocalization with gene expression at signal variants and those highly linked to signal variants via look-up. We reported eQTLs in the following tissues, reported for their importance in glycemic phenotypes: adipose subcutaneous, adipose visceral, muscle skeletal, and pancreas.
Replication
We sought to replicate our findings in the METSIM study44, using data from 10,058 individuals with fasting glucose, fasting insulin, and TOPMed-imputed genotypes. EMMAX was used to test for associations with fasting glucose or log-transformed fasting insulin at the variants reported in Table 1 with age, age2, and BMI as covariates and kinship; sex was not included as a covariate as the study is all males.
We additionally performed replication analysis in a sample from the UK Biobank. A sample of 12,854 European ancestry individuals from the UK Biobank with glucose was selected from all individuals with glucose measurement, excluding individuals with diabetes (Data-field 2443), on diabetes medication (Data-field 6177/6153), or with fasting time <8 h (Data-field 74). Glucose values were taken from variable 30740. The model included age (Data-field 21022), age2, sex (Data-field 31), BMI (Data-field 21001), and ten population structure PCs. Association models were run using Scalable and Accurate Implementation of GEneralized mixed model (SAIGE)45 to analyze UKBB phenotype data and the imputed chip genetic data.
This research has been conducted using the UK Biobank Resource under Application Number 42614.
We also performed replication analysis of the Samoan-specific association of rs117592405 with fasting insulin in a cohort of 1401 Samoans without WGS from the Samoan Study. rs117592405 was imputed using a Samoan-specific reference panel that was developed from the WGS of 1284 Samoans as part of TOPMed. R version 3.6.0 was used to replicate the association with fasting insulin in individuals without a previous diabetes diagnosis or diabetes medication use. Age, age2, BMI, and sex were included in the model.
Enrichment
The tool GREGOR was used to assess if our trait-associated variants in Table 1 were significantly enriched in a particular chromatin state annotation. Using computed LD from the 1000-genomes reference panel and the 18-state chromatin model described in the text and shown in Fig. 1, we obtained an expected number of variants to lie within each chromatin state. This was compared to the observed number of variants in each chromatin state to generate a P-value. Any P-values <0.05 are reported in the text and Supplementary Table 8.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
The Analysis Commons was funded by R01HL131136. Diabetes Genome Atlas (DGA) Processed data files were downloaded from the DGA website. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 04/05/20. Alisa K. Manning was supported by NIH K01 DK107836. Bertha Hidalgo was supported by K01 HL130609 01. Brian E. Cade is supported by K01-HL135405, American Thoracic Society Foundation Unrestricted Grant (Sleep). Ching-Ti Liu was supported by U01DK078616. Chloé Sarnowski was supported by U01DK078616. Daniel DiCorpo was supported by U01DK078616. Daniel E. Weeks was supported by R01 HL093093, R01 HL133040. Heather M was funded by NHLBI training grant T32 HL007055, T32 HL129982-03, ADA Grant #1-19-PDF-045, R01HL142825, R56HG010297, R01HG010297, U01HG007416, U01HG004803, R56HG010297, R01HG010297, U01HG007416, and U01HG004803. Heming Wang Sleep Research Society Foundation Career Development Award 018-JP-18, R35-HL135818, R01-HL113338. James B Meigs was supported by U01DK078616, NIDDK T2D AMP Opportunity Pool OP6. Jiang He was supported by U01HL072507, R01HL087263, and R01HL090682. Jose Florez was supported by NIH/NIDDK U01 DK105554 (J. Florez, PI), NIH/NIDDK U01 DK078616 (J. Meigs, PI) and NIH/NIDDK K24 DK110550 (J. Florez, PI). Josée Dupuis was supported by U01DK078616. Sheila Gaynor was supported by NIH/NHLBI 1OT3HL147154-01. Natalie Hasbani was supported by American Heart Association grant number 18CDA34110116. Paul de Vries was supported by American Heart Association grant number 18CDA34110116. Peitao Wu is supported by U01DK078616. Robert Sladek was supported by NIH/NIDDK U01 DK078616 (J. Meigs, PI). Ryan L. Minster was supported by R01 HL093093, R01 HL133040. Samantha Lent was supported by NIH/NIDDK (U01DK078616) and NIGMS (grant T32GM074905). Simin Liu was supported by AHA/18UNPG33750001; R01DK125403; R01ES029082. Xihong Lin was supported by NIH/NCI R35 CA197449, NIH/NHGRI U01HG009088. Stephen T McGarvey was supported by R01 HL093093, R01 HL133040. Laura M. Raffield was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant KL2TR002490 (LMR). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. LMR was also funded by T32 HL129982. Susan Redline was supported by R35-HL135818, R01-HL113338. Lu Chen Weng is supported by the American Heart Association (18SFRN34110082). Jennifer Wessel is supported by American Heart Association 15SDG22210003, and Indiana University Grand Challenge Precision Health Initiative-Diabetes. Huichun Xu was supported by the AHA grant (19CDA34760258). Amish Study: We gratefully thank our Amish community and research volunteers for their long-standing partnership in research, and acknowledge the dedication of our Amish liaisons, field workers, and the Amish Research Clinic staff, without which these studies would not have been possible. The Amish studies are supported by grants and contracts from the NIH, including R01 AG18728, R01 HL088119, U01 GM074518, U01 HL072515, U01 HL84756, U01 HL137181, R01 DK54261, the University of Maryland General Clinical Research Center, grant M01 RR 16500, and the Mid-Atlantic Nutrition Obesity Research Center grant P30 DK72488, the Baltimore Diabetes Research and Training Center grant P60DK79637. Atherosclerosis Risk in Communities Study: The Atherosclerosis Risk in Communities study has been funded in whole or in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services (contract numbers HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I, and HHSN268201700005I). The authors thank the staff and participants of the ARIC study for their important contributions. CFS Study: The Cleveland Family Study has been supported by National Institutes of Health grants (R01-HL046380, KL2-RR024990, R35-HL135818, and R01-HL113338). CHS Study: This research was supported by contracts 75N92021D00006, HHSN268201200036C, HHSN268200800007C, HHSN268201800001C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, and grants R01HL120393, U01HL080295 and U01HL130114 from the National Heart, Lung, and Blood Institute (NHLBI), with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629 from the National Institute on Aging (NIA). A full list of principal CHS investigators and institutions can be found at CHS-NHLBI.org. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Framingham Heart Study: WGS for “NHLBI TOPMed: Whole Genome Sequencing and Related Phenotypes in the Framingham Heart Study (phs000974.v1.p1) was performed at the Broad Institute of MIT and Harvard (3R01HL092577-06S1 (AFGen)). This research was conducted in part using data and resources from the Framingham Heart Study of the National Heart Lung and Blood Institute of the National Institutes of Health and Boston University School of Medicine. The Framingham Heart Study (FHS) acknowledges the support of contracts NO1-HC-25195, HHSN268201500001I, and 75N92019D00031 from the National Heart, Lung and Blood Institute and grant supplement R01 HL092577-06S1 for this research. We also acknowledge the dedication of the FHS study participants without whom this research would not be possible. GeneSTAR Study: GeneSTAR was supported by grants from the National Institutes of Health/National Heart, Lung, and Blood Institute (U01 HL72518, HL087698, HL49762, HL59684, HL58625, HL071025, HL092165, HL099747, K23HL105897, HL112064), by grants from the National Institutes of Health/National Institute of Nursing Research (NR0224103, NR008153), by a grant from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NS062059), by a grant from the National Institutes of Health/National Center for Research Resources (M01-RR000052) to the Johns Hopkins General Clinical Research Center, and by a grant from the National Center for Advancing Translational Sciences, National Institutes of Health (UL1TR001079) to the Johns Hopkins Institute for Clinical & Translational Research. GENOA Study: Support for GENOA was provided by the National Heart, Lung and Blood Institute (HL054457, HL054464, HL054481, HL119443, HL085571, and HL087660) of the National Institutes of Health. We would like to thank the Mayo Clinic Genotyping Core, the DNA Sequencing and Gene Analysis Center at the University of Washington, and the Broad Institute for their genotyping and sequencing services. We would also like to thank the GENOA participants. Gen Salt Study: Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number P20GM109036 and by research grants U01HL072507, R01HL087263, and R01HL090682 from the National Heart Lung and Blood Institute of the National Institutes of Health, Bethesda, Maryland, USA. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. GOLDN Study: GOLDN biospecimens, baseline phenotype data, and intervention phenotype data were collected with funding from National Heart, Lung and Blood Institute (NHLBI) grant U01 HL072524. Whole-genome sequencing in GOLDN was funded by NHLBI grant R01 HL104135-04S1. We thank GOLDN participants and investigators for their significant contributions. HyperGEN Study: The HyperGEN Study is part of the National Heart, Lung, and Blood Institute (NHLBI) Family Blood Pressure Program; collection of the data represented here was supported by grants U01 HL054472 (MN Lab), U01 HL054473 (DCC), U01 HL054495 (AL FC), and U01 HL054509 (NC FC). The HyperGEN: Genetics of Left Ventricular Hypertrophy Study was supported by NHLBI grant R01 HL055673 with whole-genome sequencing made possible by supplement—18S1. Jackson Heart Study: The Jackson Heart Study is supported and conducted in collaboration with Jackson State University (HHSN268201800013I), Tougaloo College (HHSN268201800014I), the Mississippi State Department of Health (HHSN268201800015I) and the University of Mississippi Medical Center (HHSN268201800010I, HHSN268201800011I, and HHSN268201800012I) contracts from the National Heart, Lung, and Blood Institute (NHLBI) and the National Institute on Minority Health and Health Disparities (NIMHD). The authors thank the participants and data collection staff of the Jackson Heart Study. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services. MESA & MESA Family Study: Whole-genome sequencing (WGS) for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). WGS for “NHLBI TOPMed: Multi-Ethnic Study of Atherosclerosis (MESA)” (phs001416.v1.p1) was performed at the Broad Institute of MIT and Harvard (3U54HG003067-13S1). Centralized read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1, contract HHSN268201800002I). Phenotype harmonization, data management, sample-identity QC, and general study coordination, were provided by the TOPMed Data Coordinating Center (3R01HL-120393; U01HL-120393; contract HHSN268180001I). MESA and the MESA SHARe projects are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420. MESA Family is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support is provided by grants and contracts R01HL071051, R01HL071205, R01HL071250, R01HL071251, R01HL071258, R01HL071259, by the National Center for Research Resources, Grant UL1RR033176. Also supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. San Antonio Family Study: Collection of the San Antonio Family Study (SAFS) data was supported in part by the following grants from the National Institutes of Health: P01 HL045522; R01 MH078111; R01 MH078143; R01 MH083824; R01 DK042273; R01 DK047482; and R01 DK053889. Whole-genome sequencing of the SAFS subjects was supported by U01 DK085524 and R01 HL113323. We are very grateful to the participants of the San Antonio Family Study for their continued involvement in our research programs. Samoan Study: Samoan Study - Data collection was funded by NIH grant R01-HL093093 and data analysis was funded by NIH grant R01-HL133040. We thank the Samoan participants of the study and local village authorities. We acknowledge the support of the Samoan Ministry of Health and the Samoa Bureau of Statistics for their support of this research. WHI Study: The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201600018C, HHSN268201600001C, HHSN268201600002C, HHSN268201600003C, and HHSN268201600004C.
Author contributions
D.D. and S.M.G. contributed equally to this work. D.D., S.M.G., E.M.R., K.E.W., L.M.R., X.L, J.D., J.B.M., J.W., and A.K.M. contributed to writing. D.D., S.M.G., T.D.M., and A.K.M. analyzed data. D.D., E.M.R., and A.J. provided replication results. D.D., T.D.M., P.W., C.S., H.M.H., J.W., and A.K.M. contributed to trait harmonization efforts. S.M.G., E.M.R., K.E.W., R.D’O.A., and A.V. provided annotation information. H.M.H., J.I.R., X.L., J.D., J.B.M., J.W., and A.K.M. provided leadership. D.D, S.M.G, E.M.R., K.E.W., L.M.R., T.D.M., P.W., C.S., H.M.H., A.J, N.R.H., P.S.V., J.A.B., B.H., X.G., J.A.P., J.R.O., S.Len., M.E.M., B.E.C., D.J., H.W., R.D.A., A.V., L.R., L.L., N.D.P., M.A., J.M.P., S.A., A.S.B., A.G.B., L.F.B., C.S.C., Y.D.I.C., W.J.C., M.O.G., J.S.F., M.R.I., R.R.K., T.N.K, S.Lee., C.T.L., D.L., J.E.M., R.L.M., T.N., J.S.P., L.J.R.T., A.P.R., M.S.R., E.S., J.A.S., D.E.W., H.X., J.Y., W.Z., S.P., A.A., D.K.A., J.B., E.B., A.C., L.A.C., J.E.C., R.D., J.H., S.R.H., S.L.R.K., R.W.K., C.K., S.Li., R.A.M., S.T.M., B.D.M., A.C.M., P.A.P., B.M.P., S.R., A.R.S., K.D.T., R.S.V., K.A.V.M., J.C.F., J.G.W., R.S., S.S.R., J.I.R., X.L., J.D., J.B.M., J.W, and A.K.M. contributed to manuscript review.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Chiea Chuen Khor and Luke R. Grinham.
Data availability
The summary results generated during this study are available at the AMP-T2D Portal, http://t2d.hugeamp.org/. Fasting Insulin: https://t2d.hugeamp.org/dinspector.html?dataset=TOPMed_frz5b_pooled_FI_WGS. Fasting Glucose: https://t2d.hugeamp.org/dinspector.html?dataset=TOPMed_frz5b_pooled_FG_WGS. Accession codes for genotype and phenotype files by cohort may be found in Supplementary Table 1.
Code availability
This study did not rely on custom code or mathematical algorithms.
Competing interests
Jose Florez has received a consulting honorarium from Goldfinch Bio. and speaker fees from Novo Nordisk. James S. Floyd has consulted for Shionogi Inc. Bruce M. Psaty serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. Stella Aslibekyan holds equity in 23andMe, Inc.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Daniel DiCorpo, Sheila M. Gaynor.
Contributor Information
Jennifer Wessel, Email: wesselj@iu.edu.
Alisa K. Manning, Email: amanning@broadinstitute.org
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-022-03702-4.
References
- 1.Saeedi P, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pr. 2019;157:107843. doi: 10.1016/j.diabres.2019.107843. [DOI] [PubMed] [Google Scholar]
- 2.Wessel J, et al. Low-frequency and rare exome chip variants associate with fasting glucose and type 2 diabetes susceptibility. Nat. Commun. 2015;6:5897. doi: 10.1038/ncomms6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahajan A, et al. Identification and functional characterization of G6PC2 coding variants influencing glycemic traits define an effector transcript at the G6PC2-ABCB11 locus. PLoS Genet. 2015;11:e1004876. doi: 10.1371/journal.pgen.1004876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jun G, et al. Evaluating the contribution of rare variants to type 2 diabetes and related traits using pedigrees. Proc. Natl Acad. Sci. USA. 2018;115:379–384. doi: 10.1073/pnas.1705859115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manning A, et al. A low-frequency inactivating AKT2 variant enriched in the finnish population is associated with fasting insulin levels and type 2. Diabetes Risk. Diabetes. 2017;66:2019–2032. doi: 10.2337/db16-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ng, N. H. J. et al. Tissue-specific alteration of metabolic pathways influences glycemic regulation. https://www.biorxiv.org/content/10.1101/790618v1 (2020).
- 7.Sarnowski C, et al. Impact of rare and common genetic variants on diabetes diagnosis by hemoglobin A1c in multi-ancestry cohorts: the trans-omics for precision medicine program. Am. J. Hum. Genet. 2019;105:706–718. doi: 10.1016/j.ajhg.2019.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manning AK, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat. Genet. 2012;44:659–669. doi: 10.1038/ng.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dupuis J, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuchsberger C, et al. The genetic architecture of type 2 diabetes. Nature. 2016;536:41–47. doi: 10.1038/nature18642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pulit SL, de With SA, de Bakker PI. Resetting the bar: statistical significance in whole-genome sequencing-based association studies of global populations. Genet. Epidemiol. 2017;41:145–151. doi: 10.1002/gepi.22032. [DOI] [PubMed] [Google Scholar]
- 12.Chen J, et al. The trans-ancestral genomic architecture of glycemic traits. Nat. Genet. 2021;53:840–860. doi: 10.1038/s41588-021-00852-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pessentheiner AR, Ducasa GM, Gordts P. Proteoglycans in obesity-associated metabolic dysfunction and meta-inflammation. Front. Immunol. 2020;11:769. doi: 10.3389/fimmu.2020.00769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu L, et al. Eosinophil inversely associates with type 2 diabetes and insulin resistance in Chinese adults. PLoS ONE. 2013;8:e67613. doi: 10.1371/journal.pone.0067613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt EM, et al. GREGOR: evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics. 2015;31:2601–2606. doi: 10.1093/bioinformatics/btv201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viñuela A, et al. Genetic variant effects on gene expression in human pancreatic islets and their implications for T2D. Nat. Commun. 2020;11:4912. doi: 10.1038/s41467-020-18581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Type 2 Diabetes Knowledge Portal. Genetic Association Data Sets. type2diabetesgenetics.org. https://t2d.hugeamp.org/datasets.html (2020).
- 18.Chen, J., Spracklen, C. N., Marenne, G. & Varshney, A. The trans-ancestral genomic architecture of glycaemic traits. https://www.biorxiv.org/content/10.1101/2020.07.23.217646v1 (2020). [DOI] [PMC free article] [PubMed]
- 19.Li X, et al. Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nat. Genet. 2020;52:969–983. doi: 10.1038/s41588-020-0676-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pena GG, Dutra MS, Gazzinelli A, Correa-Oliveira R, Velasquez-Melendez G. Heritability of phenotypes associated with glucose homeostasis and adiposity in a rural area of Brazil. Ann. Hum. Genet. 2014;78:40–49. doi: 10.1111/ahg.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gervais O, et al. Genomic heritabilities and correlations of 17 traits related to obesity and associated conditions in the Japanese population. G3 (Bethesda) 2020;10:2221–2228. doi: 10.1534/g3.120.401242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richardson TG, et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: a multivariable Mendelian randomisation analysis. PLoS Med. 2020;17:e1003062. doi: 10.1371/journal.pmed.1003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wessel, J. et al. Rare non-coding variation identified by large scale whole genome sequencing reveals unexplained heritability of type 2 diabetes. Preprint at medRxiv10.1101/2020.11.13.20221812 (2020).
- 24.Nagy R, et al. Exploration of haplotype research consortium imputation for genome-wide association studies in 20,032 Generation Scotland participants. Genome Med. 2017;9:23. doi: 10.1186/s13073-017-0414-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramos-Rodríguez M, et al. The impact of proinflammatory cytokines on the β-cell regulatory landscape provides insights into the genetics of type 1 diabetes. Nat. Genet. 2019;51:1588–1595. doi: 10.1038/s41588-019-0524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karczewski KJ, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou W, et al. The expression of the Slit-Robo signal in the retina of diabetic rats and the vitreous or fibrovascular retinal membranes of patients with proliferative diabetic retinopathy. PLoS ONE. 2017;12:e0185795. doi: 10.1371/journal.pone.0185795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pulit SL, et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 2019;28:166–174. doi: 10.1093/hmg/ddy327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kichaev G, et al. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 2019;104:65–75. doi: 10.1016/j.ajhg.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winkler TW, et al. The influence of age and sex on genetic associations with adult body size and shape: a large-scale genome-wide interaction study. PLoS Genet. 2015;11:e1005378. doi: 10.1371/journal.pgen.1005378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Consortium GT. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flannick J, et al. Sequence data and association statistics from 12,940 type 2 diabetes cases and controls. Sci. Data. 2017;4:170179. doi: 10.1038/sdata.2017.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahajan A, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018;50:1505–1513. doi: 10.1038/s41588-018-0241-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scott RA, et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet. 2012;44:991–1005. doi: 10.1038/ng.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taliun D, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–299. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Regier AA, et al. Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat. Commun. 2018;9:4038. doi: 10.1038/s41467-018-06159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khan, A. T. et al. Recommendations on the use and reporting of race, ethnicity, and ancestry in genetic research: experiences from the NHLBI Trans-Omics for Precision Medicine (TOPMed) program. Preprint at https://arxiv.org/abs/2108.07858 (2021).
- 38.Haeckel R, et al. Comparability of blood glucose concentrations measured in different sample systems for detecting glucose intolerance. Clin. Chem. 2002;48:936–939. doi: 10.1093/clinchem/48.6.936. [DOI] [PubMed] [Google Scholar]
- 39.Gogarten SM, et al. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics. 2019;35:5346–5348. doi: 10.1093/bioinformatics/btz567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brody JA, et al. Analysis commons, a team approach to discovery in a big-data environment for genetic epidemiology. Nat. Genet. 2017;49:1560–1563. doi: 10.1038/ng.3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conomos MP, Miller MB, Thornton TA. Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genet. Epidemiol. 2015;39:276–293. doi: 10.1002/gepi.21896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lake SL, et al. Estimation and tests of haplotype-environment interaction when linkage phase is ambiguous. Hum. Hered. 2003;55:56–65. doi: 10.1159/000071811. [DOI] [PubMed] [Google Scholar]
- 43.The NHGRI Genome Sequencing Program (GSP). Functional Annotation of Variants - Online Resource (FAVOR) Server. http://favor.genohub.org (2020).
- 44.Laakso M, et al. The Metabolic Syndrome in Men study: a resource for studies of metabolic and cardiovascular diseases. J. Lipid Res. 2017;58:481–493. doi: 10.1194/jlr.O072629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou W, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 2018;50:1335–1341. doi: 10.1038/s41588-018-0184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaulton KJ, et al. A map of open chromatin in human pancreatic islets. Nat. Genet. 2010;42:255–259. doi: 10.1038/ng.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Varshney A, et al. Genetic regulatory signatures underlying islet gene expression and type 2 diabetes. Proc. Natl Acad. Sci. USA. 2017;114:2301–2306. doi: 10.1073/pnas.1621192114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The summary results generated during this study are available at the AMP-T2D Portal, http://t2d.hugeamp.org/. Fasting Insulin: https://t2d.hugeamp.org/dinspector.html?dataset=TOPMed_frz5b_pooled_FI_WGS. Fasting Glucose: https://t2d.hugeamp.org/dinspector.html?dataset=TOPMed_frz5b_pooled_FG_WGS. Accession codes for genotype and phenotype files by cohort may be found in Supplementary Table 1.
This study did not rely on custom code or mathematical algorithms.