

Abstract
A nucleic acid sequence-based amplification (NASBA) technique for the detection of hepatitis A virus (HAV) in foods was developed and compared to the traditional reverse transcription (RT)-PCR technique. Oligonucleotide primers targeting the VP1 and VP2 genes encoding the major HAV capsid proteins were used for the amplification of viral RNA in an isothermal process resulting in the accumulation of RNA amplicons. Amplicons were detected by hybridization with a digoxigenin-labeled oligonucleotide probe in a dot blot assay format. Using the NASBA, as little as 0.4 ng of target RNA/ml was detected per comparison to 4 ng/ml for RT-PCR. When crude HAV viral lysate was used, a detection limit of 2 PFU (4 × 102 PFU/ml) was obtained with NASBA, compared to 50 PFU (1 × 104 PFU/ml) obtained with RT-PCR. No interference was encountered in the amplification of HAV RNA in the presence of excess nontarget RNA or DNA. The NASBA system successfully detected HAV recovered from experimentally inoculated samples of waste water, lettuce, and blueberries. Compared to RT-PCR and other amplification techniques, the NASBA system offers several advantages in terms of sensitivity, rapidity, and simplicity. This technique should be readily adaptable for detection of other RNA viruses in both foods and clinical samples.
Hepatitis A virus (HAV) is a foodborne pathogen frequently implicated in cases of acute gastroenteritis around the world. The virus is transmitted among humans via the fecal-oral route, and infection by HAV represents the most serious form of viral illness acquired from foods. The most common food vehicles for the transmission of HAV to humans are shellfish (21), fruits, and vegetables, (8, 9, 20). These foods may be contaminated by irrigation water tainted with untreated sewage or by contact with infected human food handlers (9, 18, 22).
HAV and other viruses implicated in foodborne illness are often found in very low concentrations in contaminated foods since they are not able to multiply in vitro. The ability to detect traces of HAV contamination in foods and related samples is essential in developing tools for the investigation and possible prevention of viral disease outbreaks. Traditional methods for the detection of enteric viruses in foods involve their extraction from the sample matrix and concentration, followed by viral multiplication in sensitive cell cultures in which characteristic cytopathic effects can be visualized (24). These assays are cumbersome, costly, and time consuming, with the cytopathic effect often requiring more than 1 week to observe. Furthermore, there is no single cell culture system available which allows the propagation of all human enteric viruses. Moreover, some human enteric viruses replicate in cell culture without any apparent cytopathic effect (19). For many other epidemiologically significant enteric viruses, propagation in cell culture is not possible.
Molecular biology techniques have been proposed for the sensitive and specific detection of some enteric viruses. Nucleic acid hybridization techniques have been used to detect viruses in foods. Hybridization of DNA or RNA probes to target viral RNA has been reported to detect from 104 to 105 PFU (13, 28). These hybridization techniques lack sufficient sensitivity for the direct detection of low numbers of viral particles in foods. More recently, nucleic acid amplification techniques have been developed for the detection of low numbers of viral particles. To date, the reverse transcription-PCR (RT-PCR) technique is the only published method offering the possibility of direct detection of enteric viruses in foods (6, 10). In RT-PCR the viral target nucleic acid (RNA) is first converted to complementary double-stranded DNA (cDNA) in a reverse-transcription step, followed by PCR amplification of the target cDNA sequences to a level detectable by a DNA probe or gel electrophoresis. Lopez-Sabater et al. (15) have shown that levels of HAV ranging from 10 to 105 PFU were successfully detected in artificially contaminated samples of shucked American oysters by use of magnetic immunoseparation followed by RT-PCR. Jothikumar et al. (11) have detected 0.04 PFU of cell culture-adapted HAV added to water and sewage samples by RT-PCR after immunomagnetic concentration. However, RT-PCR procedures have the disadvantages of requiring a two-step amplification process and relying on the use of expensive thermal cycling equipment, which add to the complexity and cost of their implementation for routine testing programs.
An alternative to RT-PCR that is potentially applicable to the detection of enteric viruses is the nucleic acid sequence-based amplification (NASBA) technique. NASBA is a homogeneous, isothermal RNA amplification process involving the action of three enzymes, reverse transcriptase, T7 RNA polymerase, and RNase-H, as well as two target sequence-specific oligonucleotide primers (one of which bears a bacteriophage T7 promoter sequence appended to its 5′ end), acting in concert to amplify target sequences more than 108-fold (5). Its advantage over the traditional RT-PCR technique is its suitability for the direct amplification of RNA targets, obviating the need for thermal cycling equipment. NASBA has been successfully applied to the detection of pathogenic bacteria, such as Listeria monocytogenes (2, 3), Campylobacter (26), and Mycobacterium (27). NASBA has also been used for the amplification of RNA from the human immunodeficiency virus in infected cells, blood, and plasma (12). However, it has never been used for the detection of enteric viruses in either food or clinical samples. A major interest of our research team is developing alternative nucleic acid amplification techniques such as NASBA for the ultrasensitive detection of various enteric viruses in foods. In this paper, we focus on the development of a NASBA technique for the detection of HAV in both experimentally inoculated waste water and food samples.
MATERIALS AND METHODS
Preparation of viral and bacterial stocks.
The cytopathic HAV strain HM-175 (provided by S. Bidawid, Bureau of Microbial Hazards, Health Canada) was chosen as the viral model for this work. The HM-175 strain has the advantage of being readily quantifiable by plaque assay in cell culture (7). The virus was propagated in FRhK-4 cells as described by Mbithi et al. (16, 17). The viral stock solution was concentrated by precipitation with 8% (wt/vol) PEG 6000 followed by centrifugation at 10,000 × g for 20 min. The pellet was resuspended in RNase-free water at a final concentration of 109 PFU/ml (14), and the concentrated stock solution was stored in 1-ml fractions at −80°C until use. Rotavirus (Institut Armand-Frappier, Montréal, Canada) and various bacterial strains, including L. monocytogenes (ATCC-43256) and Escherichia coli O157:H7 (ATCC-11775), were used to evaluate whether or not the presence of nonhomologous microorganisms interferes with the amplification reaction by NASBA. Bacterial strains were grown in tryptic soy broth (Difco Laboratories, Detroit, Mich.), and bacterial counts were determined by plating on tryptic soy agar (Difco).
Plaque assay.
The HAV concentration in the stock suspension was determined by plaque assay on a FRhK-4 cell monolayer (16, 17). Wells of six cluster plates (Falcon, Becton Dickinson, Franklin Lakes, N.J.) were inoculated with 0.1 ml of sequential dilutions of HAV in Eagle minimum essential medium (Life Technologies, Burlington, Canada) with 10% fetal bovine serum (HyClone, Logan, Utah), 2 mM glutamine (Life Technologies), 0.1 mM nonessential amino acids (Life Technologies), 0.015 M HEPES (Life Technologies), and 0.113% sodium bicarbonate (Life Technologies). The virus was allowed to adsorb for 90 min at 37°C in the presence of 5% CO2. Each well was overlaid with 2 ml of a semisolid agarose-containing overlay medium. The plates were incubated in a humid atmosphere at 37°C with 5% CO2 for 8 days. The monolayers were fixed and stained as described by Sattar et al. (23) and examined for the characteristic plaque lysis resulting from the cytopathic effect.
Primers and oligonucleotide probes.
A variety of oligonucleotides were selected from published HAV nucleic acid sequences, synthesized, and gel purified before use (Table 1). NASBA primers bearing the bacteriophage T7 RNA polymerase promoter binding and preferred transcriptional initiation sequence at their 5′ end are indicated with the prefix T7. The target-complementary portions of primers P1 and P2 have been characterized by Cromeans et al. (6). The remaining primers were selected from published nucleotide sequences of the genes encoding VP1 and VP2 (GenBank, accession number 14707). Digoxigenin (DIG)-labeled oligonucleotide probes internal to the nucleotide regions defined by the different primer pairs were synthesized for the detection of amplified target nucleic acid sequences by solid phase hybridization as described below.
TABLE 1.
Nucleotide sequences of oligonucleotide primers and probes used in this study
| Primer or probe | Sequencea | Location | Reference |
|---|---|---|---|
| P1 | 5′-GTTTTGCTCCTCTTTATCATGCTATG-3′ | 2167–2192 | 6 |
| P2 | 5′-GGAAATGTCTCAGGTACTTTCTTTG-3′ | 2414–2389 | 6 |
| P probe | 5′-TCAACAACAGTTTCTACAGA-3′ | 2232–2251 | 6 |
| T7P2 | 5′-AATTCTAATACGACTCACTATAGGGAGAGGAAATGTCTCAGGTACTTTCTTTG-3′ | 2414–2389 | This study |
| BB1 | 5′-CAGATTGGCTTACTACACA-3′ | 1000–1018 | This study |
| T7BB2 | 5′-AATTCTAATACGACTCACTATAGGGAGACATGCAACTCCAAATCTGT-3′ | 1446–1428 | This study |
| BB probe | 5′-GATTGATCTGTGCTATGGTTCCTGGTGACC-3′ | 1171–1200 | This study |
| HPA | 5′-GGCAGACATTGAGGAAGAGC-3′ | 800–819 | This study |
| T7HPA | 5′-AATTCTAATACGACTCACTATAGGGAGATGGTCACCAGGAACCATAGC-3′ | 1201–1182 | This study |
| HPA probe | 5′-CTGAGGTTGGATCACACCAGGTTGAACCTT-3′ | 901–930 | This study |
| AD | 5′-TCTTCAACCTCTAATCCTCCTC-3′ | 2547–2568 | This study |
| T7AD | 5′-AATTCTAATACGACTCACTATAGGGAGAGTCTTGTCACCCAAACCATC-3′ | 2872–2853 | This study |
| AD probe | 5′-CCGTTGATACTCCTTGGGTAGAGAAGGAGT-3′ | 2701–2730 | This study |
T7 promoter sequence is underscored.
NASBA procedure.
The NASBA reactions were performed as described by Blais et al. (3) with some modifications. The principle of the NASBA reaction is shown in Fig. 1. The final reaction mixture volume was 25 μl. An 18-μl prereaction mixture was prepared to give final concentrations in 25 μl of 40 mM Tris-HCl (pH 8.5), 50 mM KCl, 12 mM MgCl2, 1 mM (each) deoxyribonucleoside triphosphate, 2 mM (each) ribonucleoside-5′-triphosphate, 10 mM dithiothreitol, 15% (vol/vol) dimethylsulphoxide, and 5 pmol of each gel-purified primer. Five microliters of purified viral RNA or crude viral lysate was added to the 18 μl of prereaction mixture in a 0.6-ml microcentrifuge tube, which was incubated for 5 min at 65°C in order to disrupt secondary structure in the target RNA. Viral lysate was prepared by heating the viral suspension at 100°C for 5 min, and viral RNA was purified using Trizol Reagent (Life Technologies) according to the manufacturer's instructions. Each tube was immediately transferred to a circulating water bath adjusted to 40°C. After 5 min, 2 μl of an enzyme mixture containing 2.6 μg of bovine serum albumin (in 50% glycerol; Roche Diagnostics, Laval, Canada), 40 U of T7 RNA polymerase (Pharmacia, Baie d'Urfé, Canada), 8 U of avian myeloblastosis virus reverse transcriptase (Seikagaku, Ijamsville, Md.), 0.2 U of RNase H (Pharmacia), and 12.5 U of RNasin (Promega, Madison, Wisc.) were added to each tube, followed by incubation at 40 ± 1°C for 90, 150, or 210 min. The amplification products were analyzed by denaturing agarose gel electrophoresis, by Northern blot or dot blot hybridization onto a nylon membrane using one of the DIG-labeled oligonucleotide probes.
FIG. 1.

Scheme for the amplification of RNA by the NASBA reaction.
Denaturing agarose gel electrophoresis and Northern blotting.
For the analysis of NASBA products by denaturing agarose gel electrophoresis, 5 μl of NASBA amplification products as well as 5 μl of an RNA molecular weight marker (Roche Diagnostics) were first prepared in loading buffer (10 μl of formamide, 4.5 μl of formaldehyde, and 2 μl of 10× running buffer [0.2 M borate buffer, pH 8.3, containing 2 mM EDTA]). Samples were heated at 65°C for 2 min and cooled on ice, and 2 μl of tracking dye (bromophenol blue and xylene cyanol in 50% glycerol) was added. Each sample was analyzed on a 1.2% denaturing agarose gel (130 V for 75 min in 1× running buffer added with 6% formaldehyde). The gel was washed several times with water and stained in ethidium bromide. After being destained overnight in water, the gel was examined under UV light. For Northern blot analysis, amplified RNA was transferred from the gel onto a positively charged nylon membrane (Roche Diagnostics) for 1 h using a vacuum transfer apparatus (PosiBlot pressure blotter; Stratagene, La Jolla, Calif.) in the presence of 10× SSC (1× SSC is 0.15 M NaCl and 15 mM Na citrate). The nylon membrane was subjected to the hybridization and detection procedures as described below.
Dot blotting.
The NASBA product was diluted 1:1 with 20× SSC, and 3-μl aliquots of the diluted product were deposited onto a strip of nylon membrane. The spots were allowed to air dry, and the RNA was cross-linked to the membrane by a 2-min exposure to UV using a UV cross-linker (model CL-1000; UVP, San Gabriel, Calif.). The membranes were subjected to the probe hybridization and detection procedure described below.
Solid phase hybridization and detection of NASBA-amplified RNA.
After Northern blotting or dot blotting, nylon membranes were prehybridized for 30 min at 55°C in RNase-free hybridization solution containing 5× SSC, 0.1% (wt/vol) N-laurylsarcosine, 0.02% (wt/vol) sodium dodecyl sulfate (SDS) and 1% (wt/vol) protein blocking reagent (Roche Diagnostics). Hybridization was carried out for 2 h at 55°C in 5 ml of the same hybridization solution containing 50 nM of the DIG-labeled oligonucleotide probe. Both prehybridization and hybridization were carried out in a hybridization oven (model 400; Robbins Scientific, Sunnyvale, Calif.). Unbound probe was removed by washing the membrane twice for 5 min at room temperature in 2× SSC containing 0.1% (wt/vol) SDS and twice for 15 min at 55°C with 0.1× SSC containing 0.1% (wt/vol) SDS. The membrane was incubated for 30 min in a blocking solution (0.1 M maleic acid and 0.15 M NaCl [pH 7.5] containing 1% [wt/vol] blocking reagent) prior to immunoenzymatic detection of the bound probe. The immunoenzymatic detection procedure consists of the incubation of membranes for 30 min at room temperature in the presence of anti-DIG-peroxidase (Roche Diagnostics) at a concentration of 75 mU/ml in the blocking solution followed by five washes in PBS-T (0.01 mmol of phosphate-buffered saline [pH 7.2]/liter plus 0.85% NaCl and 0.05% Tween 20). Bound peroxidase was assayed using 3,3′,5,5′-tetramethylbenzidine (Kirkegaard & Perry Laboratories, Gaithersburg, Md.) substrate solution. A positive result was characterized by the formation of a blue precipitate.
Comparison of the sensitivity between NASBA and RT-PCR.
To evaluate whether or not the NASBA offers higher sensitivity than RT-PCR, an experiment was designed in which 10-fold serial dilutions of purified HAV RNA starting at 400 ng/ml and 5-fold serial dilutions of crude HAV viral lysate starting at 5 × 106 PFU/ml were prepared and amplified by both techniques. For RT-PCR, primers P1 and P2 (Table 1) and amplification conditions as well as the oligoprobe hybridization conditions reported by Cromeans et al. (6) were used. For NASBA, the same primers and oligoprobe were used, except that in this case the P2 primer contained the bacteriophage T7 RNA polymerase promoter sequence (T7-P2). Three microliters of each amplified product diluted 1:1 with 20× SSC was transferred to a nylon membrane and hybridized with the respective DIG-labeled oligoprobe. This experiment was repeated twice, and the negative control sample, consisting of 5 μl of RNase-free water which did not contain any target RNA or viruses, was also considered.
Detection of HAV added to waste water.
Waste water samples were collected from a sewage treatment plant in Saint-Nicolas, Canada, at different stages of the treatment (raw waste water, waste water after aerobic digestion with activated sludge, and waste water after aerobic digestion and UV treatment). Before use, waste water samples were analyzed for total bacteria and fecal coliforms using plate count agar and violet red bile agar (Difco), respectively. Samples were experimentally inoculated with 106 PFU of HAV/ml, and 5 μl of each sample was heated to 100°C for 5 min to lyse the virus particles and subjected to the NASBA and dot blot hybridization procedure as described above. Unexperimentally inoculated waste water samples were also analyzed by NASBA for the presence of HAV background.
Simulation of food contamination by HAV using experimentally inoculated water.
Lettuce and blueberry samples obtained from a local retailer were inoculated with experimentally HAV-inoculated water as follows.
(i) for Boston lettuce, 20 μl of water containing 108 PFU of HAV/ml was spotted on a 3-cm2 area of leaf. The spot was air dried for 30 min at room temperature, and viruses were eluted therefrom by washing through repeatedly drawing and expelling in a micropipette 100 μl of the following buffers: tryptose phosphate broth (Difco), 3% beef extract (Difco), phosphate-buffered saline (PBS), PBS-Tween 20, PBS–Triton X-100, and MilliQ water. Negative control samples were handled exactly as was the experimentally inoculated sample using HAV-free water. The efficiencies of the various buffers were evaluated by determining the viral titer in the eluate using the plaque assay described above. The buffer giving the highest elution was used for the recovery of HAV from the contaminated lettuce samples. Fifty microliters of eluate was heated to 100°C for 5 min to disrupt the viral particles, and 5 μl of this sample was subjected to NASBA as described above.
(ii) For blueberries, a similar inoculation protocol was used. To elute the viruses, blueberry samples were placed in a 5-ml test tube containing 100 μl of the selected elution buffer. The eluate was recovered and heat treated as described above prior to performing the NASBA.
All experiments were performed at least twice to ensure repeatability of the results obtained.
RESULTS
Efficiencies of different primer pairs in the NASBA.
The efficiencies of four different oligonucleotide primer pairs for the amplification of different specific regions within the HAV genome by the NASBA reaction were compared. Two of these pairs target sequences within the gene encoding VP1, and the other two target sequences within the gene encoding VP2 (Table 1). The efficiency of each primer pair was assessed by the determination of the minimum detection limit using both purified RNA and crude viral lysate. The lowest detection limit was obtained with the primer pair BB and P, which enabled the detection of a minimum of 0.4 ng of purified RNA/ml compared to 4 ng/ml for HPA and 40 ng/ml for the AD primer pair (data not shown). When crude HAV viral lysate was used, a detection limit of 2 PFU (4 × 102 PFU/ml) with BB primers, compared to 50 PFU (104 PFU/ml) obtained with primer pair P and 500 PFU (105 PFU/ml) for primer pairs HPA and AD (data not shown). BB primers were selected and used for the rest of the study, except for the comparison between RT-PCR and NASBA in which P primers were used.
Analysis of NASBA products.
As shown in Fig. 1, the main product of the NASBA reaction is antisense RNA. To characterize the products of the HAV NASBA, the detection was achieved by both denaturing agarose gel electrophoresis and Northern blotting. The separation of NASBA products by denaturing agarose gel electrophoresis results in a very sharp band corresponding to the expected size for the RNA product of 474 nucleotides (Fig. 2A). No band was observed with the negative control. The specificity of the band obtained was confirmed by Northern blotting using a DIG-labeled oligoprobe. In this case, the NASBA product was electrophoretically separated on an agarose gel, transferred onto a nylon membrane, and hybridized with a DIG-labeled oligoprobe. The amplification product generated a strong hybridization signal of a molecular size corresponding to the expected value of 474 nucleotides (Fig. 2B). The smaller band evident on the membrane likely corresponds to amplification intermediates of the NASBA reaction. These results confirm that the NASBA system employing primer pair BB effectively amplified the target region of the HAV genome.
FIG. 2.

Analysis of the NASBA products by denaturing agarose gel electrophoresis (A) and by Northern blotting (B). NASBA was performed using BB primers. Lanes 1, RNA molecular weight marker III; lanes 2, NASBA product from HAV; lanes 3, NASBA negative control.
Time course for the NASBA reaction.
In the previous experiments the NASBA reaction was carried out for 90 min. In order to verify whether this incubation period was sufficient to produce the maximum signal, we studied the effect of longer incubation periods on the sensitivity of NASBA using the primer pair BB and 10-fold serial dilutions of purified HAV RNA samples. As shown in Fig. 3, the highest sensitivity was obtained when an amplification time of 210 min was used, resulting in the detection of as little as 0.4 ng/ml. Lower amplification times result in a slight loss of sensitivity, since a concentration of 4 ng of RNA/ml was needed to obtain a detectable signal. For the rest of the study, a time of 210 min was used during the amplification reaction by NASBA.
FIG. 3.

Time course of the NASBA reaction and effect on sensitivity. Samples containing 10-fold serial dilutions of HAV RNA ranging from 400 to 0.04 ng/ml were prepared and subjected to the NASBA reaction for 90, 150, and 210 min, followed by hybridization with the DIG-labeled probe as described in Materials and Methods.
Sensitivity of the NASBA system.
To determine the minimum number of PFU detectable by the NASBA system, samples containing twofold serially diluted HAV (6.4 × 104 to 1.5 × 102 PFU/ml) were prepared in duplicate and subjected to the NASBA and dot blot hybridization procedure. The NASBA system was capable of producing a detectable signal with a minimum viral concentration of 4 × 102 PFU/ml (Fig. 4). This minimum viral concentration corresponds to as low as 2 PFU if we consider that only 5 μl from each viral dilution was added to the reaction mixture and submitted to amplification by NASBA (the lowest dilution giving a detectable signal with NASBA was 4 × 102 PFU/ml [0.4 PFU/μl], and the detection limit was 2 PFU [0.4 PFU × 5 μl]).
FIG. 4.
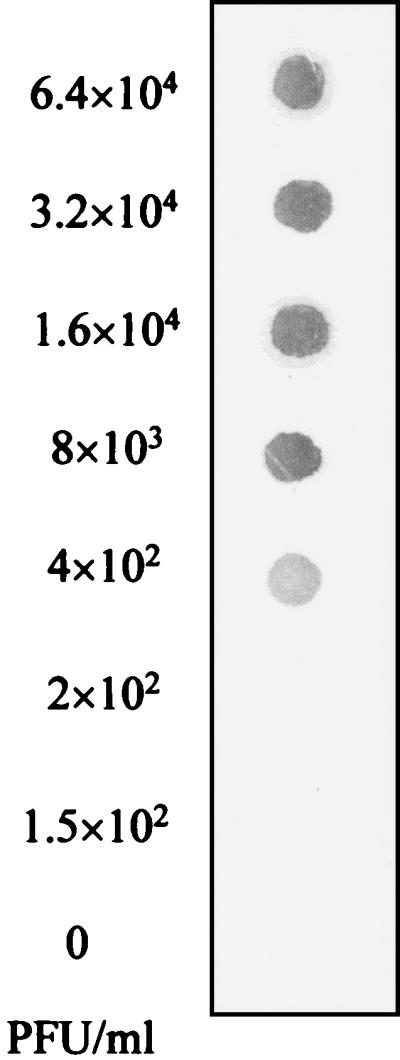
Sensitivity of the NASBA system. Samples containing twofold serially diluted HAV starting at 6.4 × 104 PFU/ml were subjected to the NASBA and dot blot hybridization procedure described in Materials and Methods. The detection limit is expressed as the minimum concentration of HAV giving a detectable signal by dot blot hybridization.
Comparison of the sensitivity of the NASBA and RT-PCR.
One of the major goals of this study was to compare the efficiency of NASBA for direct, sensitive, and rapid amplification of viral target RNA and to investigate the suitability of the NASBA technique as an alternative approach to RT-PCR. In this part of the work, the sensitivity of the NASBA was compared to that of RT-PCR using both 10-fold serial dilutions of HAV target RNA and 5-fold serial dilutions of crude HAV viral lysates. For RT-PCR, the primers and oligoprobe described by Cromeans et al. (6) were used. The same primers and probe were used for the amplification by NASBA, with the exception that the reverse primer contained the bacteriophage T7 RNA polymerase promoter sequence (Table 1). Using the NASBA technique, as little as 0.4 ng of target RNA/ml gave a detectable signal after amplification and dot blot hybridization. This is 10 times more sensitive than the RT-PCR, which allowed the detection of only 4 ng of target RNA/ml (Fig. 5).
FIG. 5.

Comparison of the sensitivity in the detection of target HAV RNA and HAV lysate between the developed NASBA using BB primers and RT-PCR using P primers. Tenfold serial dilutions of purified target RNA ranging from 400 to 0.04 ng/ml (A) and 10-fold serially diluted HAV lysate starting at 106 PFU/ml (B) were prepared and subjected to either the RT-PCR (lanes 1) or NASBA (lanes 2). The detection limit is expressed as the minimum concentration of target RNA giving a detectable signal by dot blot hybridization.
When crude HAV viral lysate was used as a template, a minimum of 50 PFU (104 PFU/ml) could be detected by NASBA, compared to a minimum of 200 PFU (4 × 104 PFU/ml) by RT-PCR (data not shown). These experiments demonstrate the superiority of the NASBA technique over RT-PCR for the amplification of viral target RNA and its great potential for the direct amplification of target RNA sequences.
Specificity of the NASBA system.
The specificity of the NASBA system was examined by carrying out amplification reactions with and without HAV in the presence of various amounts of nontarget microorganisms and nucleic acids. The presence of nontarget bacteria, such as E. coli and L. monocytogenes, which are frequently encountered in foods and related samples, did not interfere with the amplification and detection of HAV with NASBA (Table 2). When added at concentrations lower than 105 PFU/ml, rotavirus did not seem to interfere with the amplification of target nucleic acid from HAV. However, when rotavirus was added at a concentration of 106 PFU/ml or more, no specific signal was obtained even in the presence of HAV, indicating possible interference of nonhomologous RNA with the NASBA reaction. (Table 2). No interference was encountered when excess amounts of yeast tRNA or E. coli rRNA (Roche Diagnostics) were added in the NASBA mixture reaction.
TABLE 2.
The effect of nontarget microorganisms and nucleic acids on the specificity of the NASBA detection system
| Organism or nucleic acid | Concn | No. positive/No. totala |
|---|---|---|
| E. coli ATCC-11775 | 2 × 107 CFU/ml | 0/2 |
| 2 × 106 CFU/ml | 0/2 | |
| 2 × 105 CFU/ml | 0/2 | |
| HAV (106 PFU/ml) + E. coli | 2 × 107 CFU/ml | 2/2 |
| 2 × 106 CFU/ml | 2/2 | |
| 2 × 105 CFU/ml | 2/2 | |
| L. monocytogenes ATCC-43256 | 1 × 108 CFU/ml | 0/2 |
| HAV (106 PFU/ml) + L. monocytogenes | 1 × 108 CFU/ml | 2/2 |
| Rotavirus | 1 × 106 PFU/ml | 0/2 |
| 1 × 105 PFU/ml | 0/2 | |
| 1 × 104 PFU/ml | 0/2 | |
| HAV (106 PFU/ml) + rotavirus | 1 × 106 PFU/ml | 0/2 |
| 1 × 105 PFU/ml | 2/2 | |
| 1 × 104 PFU/ml | 2/2 | |
| Yeast tRNA | 50 μg/ml | 0/2 |
| HAV (106 PFU/ml) + yeast tRNA | 50 μg/ml | 2/2 |
| E. coli rRNA | 50 μg/ml | 0/2 |
| HAV (106 PFU/ml) + E. coli rRNA | 50 μg/ml | 2/2 |
No. of replicates giving positive results in the combined NASBA-dot blot procedure per total number tested.
No signal was obtained with any of the above nontarget agents when the procedure was carried out in the absence of HAV, thus demonstrating the specificity of the NASBA assay system for HAV sequences.
Application of the NASBA system to the detection of HAV in waste water and foods.
Waste water samples taken from a local municipal facility at different stages during the treatment process were analyzed for total bacterial count and fecal coliforms. Raw waste water contained 2.5 × 106 CFU of total bacteria/ml and 2 × 105 CFU of fecal coliforms/ml compared to 8.5 × 103 CFU of total bacteria/ml and 1.5 × 103 CFU of fecal coliforms/ml for waste water after digestion and 7.5 × 101 CFU of total bacteria/ml and no fecal coliforms for waste water after digestion and UV treatment. The effectiveness of the NASBA for detecting HAV in waste water and foods was investigated. The three types of waste waters were experimentally inoculated with HAV at a final concentration of 106 PFU/ml and processed by the developed NASBA procedure. A strong positive signal was obtained with waste water which was inoculated with HAV after aerobic digestion and UV treatment, although a weaker signal was obtained with the raw waste water sample (Fig. 6A).
FIG. 6.

Detection of HAV in various matrices. For panel A, waste water was sampled at different stages during the municipal treatment process and spiked with HAV to a final concentration of 106 PFU/ml. Five microliters of sample was subjected to the NASBA and dot blot hybridization procedure as described in Materials and Methods. (A) 1, spiked Rnase-free water (positive control); 2, spiked raw waste water; 3, spiked waste water after aerobic digestion before UV treatment; 4, spiked waste water after aerobic digestion and UV treatment; 5, negative control (unspiked waste water sample). For panel B, lettuce and blueberry samples were inoculated as described in Material and Methods using HAV-spiked water. Attached viruses were eluted, and 5 μl of the eluate was subjected to the NASBA and dot blot hybridization procedure. 1, signal obtained using eluate from spiked lettuce; 2, signal obtained using eluate from spiked blueberries. In this case, the negative controls consisted of eluates from lettuce and blueberries treated with uncontaminated water (data not presented).
The ability of the NASBA system to detect HAV eluted from the surfaces of food samples, such as fruits and vegetables, was also examined. All four eluants tested showed an elution recovery of 80% (data not shown). When water was used as an eluant, the analysis of the eluate by NASBA and dot blot hybridization revealed a strong specific hybridization signal (Fig. 6B), while no signal was obtained with control samples, confirming the specificity of the signal obtained.
DISCUSSION
Until recently, the RT-PCR technique has been the only molecular amplification approach put into practice for the detection of enteric viruses. In this study, we propose the NASBA technique, an amplification approach allowing direct and isothermal amplification of viral RNA, as an alternative to RT-PCR. There is no doubt that RT-PCR has provided several advantages over nucleic acid probes and immunological methods for the detection of food pathogens in general and specifically enteric viruses. However, in its present form, RT-PCR does not allow detection of low concentrations of enteric viruses unless combined with a concentration step. Moreover, RT-PCR involves two amplifications steps which are highly susceptible to contamination by inhibitors. We have demonstrated that the NASBA technique coupled with a simple dot blot hybridization assay offers a great potential for the sensitive and specific detection of HAV. We have clearly demonstrated that NASBA was more sensitive than the RT-PCR method published by Cromeans et al. (6). In contrast with RT-PCR, NASBA is performed in a single step and at one temperature (40°C), compared to two steps for RT-PCR and different temperatures for each PCR cycle. Moreover, NASBA is performed in a simple water bath and does not need any extra pieces of equipment, in contrast to RT-PCR, which requires a thermocycler. In principle, NASBA has a much higher inherent amplification capability than RT-PCR, since in the former technique each cDNA template serves to produce many RNA copies at every cycle, while in the latter, each cDNA doubles in number at each cycle. Detection limits of 0.4 ng of purified HAV RNA/ml and 2 PFU (4 × 102 PFU/ml) of HAV were obtained after only 210 min with NASBA. This incubation time probably allows the highest amplicon accumulation needed to achieve the detection limit of 4 × 102 PFU/ml. A longer amplification time was used (240 min), and more intense signals were obtained, but the detection limit remains the same. This may be due to the high yield of amplicons obtainable with NASBA, which has been reported to produce a 108-fold amplification of target nucleic acid sequences (25). The detection limit of the NASBA system was found to vary with the primer sequences selected, underscoring the importance of examining a variety of primer sequences when designing a new NASBA system. Primer pair BB, selected in this study, targets sequences located within the gene encoding VP2, which is highly conserved among differing HAV strains (4).
The specificity of the NASBA reaction for HAV was demonstrated by Northern blot analysis, which has shown a band with the expected molecular size, and by dot blot hybridization, which has shown a specific signal. No specific signal was obtained with nontarget nucleic acid sequences from various sources. The fact that HAV could be amplified even in the presence of an abundance of nontarget nucleic acid demonstrates the potential of this technique to detect HAV in samples containing high loads of extraneous microorganisms, as is routinely encountered in many foods and environmental samples.
The performance of the NASBA system was also examined by analysis of HAV inoculated in waste water, lettuce, and blueberries, which represent typical samples having the potential for contamination with HAV. In waste water, a detectable specific signal was obtained with the different samples tested, including those collected before UV treatment, which are often heavily contaminated with fecal coliforms. This result further confirms the specificity of the NASBA system and its ability to detect HAV without any interference in complex samples. When fruit and vegetable samples deliberately contaminated with HAV were tested, a strong hybridization signal was obtained with the NASBA system after elution of the attached viruses. These results represent a further confirmation of the potential of the NASBA system. The simulated contamination protocol used in our study was considered suitable for the validation of the detection method developed, since natural contamination of such food samples usually involves drying of contaminated water on the food surfaces. Similar results were obtained by Bidawid et al. (1) using RT-PCR.
Based on this study, we conclude that the NASBA technique offers great promise for the detection of HAV in foods and related samples. The NASBA process, with its integration of the RT step, is particularly well suited for the amplification of RNA viruses and should therefore be applicable to the detection of other enteric viruses. Work is now under way in our laboratories to improve the techniques for separation and concentration of HAV from complex sample matrices and for the application of the NASBA technique to the detection of other enteric viruses, such as rotavirus and the Norwalk-like viruses. More work is also necessary in order to study the performance of this approach in the assay of naturally contaminated samples.
ACKNOWLEDGMENTS
This work was supported by a grant from the Conseil de Recherches en Pêche et en Agro-Alimentaire du Québec (CORPAQ).
We thank L. M. Phillippe (Laboratory Services Division, Canadian Food Inspection Agency) for her excellent technical assistance and S. Bidawid (Bureau of Microbial Hazards, Health Canada) for providing the HAV strain.
REFERENCES
- 1.Bidawid S, Farber J M, Sattar S A. Rapid concentration and detection of hepatitis A virus from lettuce and strawberries. J Virol Methods. 2000;88:175–185. doi: 10.1016/s0166-0934(00)00186-5. [DOI] [PubMed] [Google Scholar]
- 2.Blais B W, Turner G. Detection of Listeria monocytogenes by the nucleic acid sequence-based amplification (NASBA) technique. In: Spencer J F T, Ragout de Spencer A L, editors. Methods in bio/technology: food microbiology protocols, chapter 9. Vol. 14. Totowa, N.J: Humana Press; 2000. [Google Scholar]
- 3.Blais B W, Turner G, Sooknanan R, Malek L T. A nucleic acid sequence-based amplification system for detection of Listeria monocytogenes hlyA sequences. Appl Environ Microbiol. 1997;63:310–313. doi: 10.1128/aem.63.1.310-313.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen J I, Ticehurst J R, Purcell R H, Buckler-White A, Baroudy B M. Complete nucleotide sequence of wild-type hepatitis A virus: comparison with different strains of hepatitis A virus and other picornaviruses. J Virol. 1987;61:50–59. doi: 10.1128/jvi.61.1.50-59.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Compton J. Nucleic acid sequence-based amplification. Nature (London) 1991;350:91–92. doi: 10.1038/350091a0. [DOI] [PubMed] [Google Scholar]
- 6.Cromeans T L, Nainan O V, Margolis H S. Detection of hepatitis A virus RNA in oyster meat. Appl Environ Microbiol. 1997;63:2460–2463. doi: 10.1128/aem.63.6.2460-2463.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng M Y, Day S P, Cliver D O. Detection of hepatitis A virus in environmental samples by antigen capture PCR. Appl Environ Microbiol. 1994;60:1927–1933. doi: 10.1128/aem.60.6.1927-1933.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henkel J. Food firm gets huge fine for tainted strawberry harvest. FDA Consumer. 1999;33:37–38. [PubMed] [Google Scholar]
- 9.Hernandez F, Monge R, Jimenez C, Taylor L. Rotavirus and hepatitis A virus in market lettuce (Latuca sativa) in Costa Rica. Int J Food Microbiol. 1997;37:221–223. doi: 10.1016/s0168-1605(97)00058-5. [DOI] [PubMed] [Google Scholar]
- 10.Jaykus L-A, De Leon R, Sobsey M D. A virion concentration method for detection of human enteric viruses in oysters by PCR and oligoprobe hybridization. Appl Environ Microbiol. 1996;62:2074–2080. doi: 10.1128/aem.62.6.2074-2080.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jothikumar N, Cliver D O, Mariam T W. Immunomagnetic capture PCR for rapid concentration and detection of hepatitis A virus from environmental samples. Appl Environ Microbiol. 1998;64:504–508. doi: 10.1128/aem.64.2.504-508.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kievits T, van Gemen B, van Strijp D, Schukkink R, Dircks M, Adriaanse H, Malek L, Sooknanan R, Lens R. NASBA™ isothermal enzymatic in vitro nucleic acid amplification optimized for the diagnosis of HIV-1 infection. J Virol Methods. 1991;35:273–286. doi: 10.1016/0166-0934(91)90069-c. [DOI] [PubMed] [Google Scholar]
- 13.Le Guyader F, Apaire-Marchais V, Brillet J, Billaudel S. Use of genomic probes to detect hepatitis A virus and enterovirus RNAs in wild shellfish and relationship of viral contamination to bacterial contamination. Appl Environ Microbiol. 1993;59:3963–3968. doi: 10.1128/aem.59.11.3963-3968.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis G D, Metcalf T G. Polyethylene glycol precipitation for recovery of pathogenic viruses, including hepatitis A virus and human rotavirus, from oyster, water, and sediment samples. Appl Environ Microbiol. 1988;54:1983–1988. doi: 10.1128/aem.54.8.1983-1988.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez-Sabater E I, Deng M-Y, Cliver D O. Magnetic immunoseparation PCR assay (MIPA) for detection of hepatitis A virus (HAV) in American oyster (Crassostrea virginica) Lett Appl Microbiol. 1997;24:101–104. doi: 10.1046/j.1472-765x.1997.00357.x. [DOI] [PubMed] [Google Scholar]
- 16.Mbithi J N, Springthorpe V S, Sattar S A. Effect of relative humidity and air temperature on survival of hepatitis A virus on environmental surfaces. Appl Environ Microbiol. 1991;57:1394–1399. doi: 10.1128/aem.57.5.1394-1399.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mbithi J N, Springthorpe V S, Boulet J R, Sattar S. Survival of hepatitis A virus on human hands and its transfer on contact with animate and inanimate surfaces. J Clin Microbiol. 1992;30:757–763. doi: 10.1128/jcm.30.4.757-763.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niu M T, Polish L B, Robertson B H, Khanna B K, Woodruff B A, Shapiro C N, Miller M A, Smith J D, Gedrose J K, Alter M J, Margolis H S. Multistate outbreak of hepatitis A associated with frozen strawberries. J Infect Dis. 1992;166:518–524. doi: 10.1093/infdis/166.3.518. [DOI] [PubMed] [Google Scholar]
- 19.Payment P, Trudel M. Detection and quantitation of human enteric viruses in waste waters: increased sensitivity using human immune serum globulin-immunoperoxidase assay on MA-104 cells. Can J Microbiol. 1987;33:568–570. doi: 10.1139/m87-097. [DOI] [PubMed] [Google Scholar]
- 20.Reid T M S, Robinson H G. Frozen raspberries and hepatitis A. Epidemiol Infect. 1987;98:109–112. doi: 10.1017/s095026880006177x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richards G P. Outbreaks of shellfish-associated enteric viruses illness in the United States: requisite for development of viral guidelines. J Food Prot. 1985;48:815–823. doi: 10.4315/0362-028X-48.9.815. [DOI] [PubMed] [Google Scholar]
- 22.Rosenblum L S, Mirkin I R, Allen D T, Safford S, Halder S C. A multifocal outbreak of hepatitis A traced to commercially distributed lettuce. Am J Public Health. 1990;80:1075–1079. doi: 10.2105/ajph.80.9.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sattar S A, Springthorpe V S, Karim Y, Loro P. Chemical disinfection of non-porous inanimate surfaces experimentally contaminated with four human pathogenic viruses. Epidemiol Infect. 1989;102:493–505. doi: 10.1017/s0950268800030211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sobseys M D. Methods for recovering viruses from shellfish, seawater, and sediments. In: Berg G, editor. Methods for recovering viruses from the environment. Boca Raton, Fla: CRC Press, Inc.; 1987. pp. 77–108. [Google Scholar]
- 25.Sooknanan R L, Malek X I, Wang T, Siebert T, Keating A. Detection and direct sequence identification of BCR-ACL mRNA in Ph+ chronic myeloid leukemia. Exp Hematol (New York) 1993;21:1719–1724. [PubMed] [Google Scholar]
- 26.Uyttendaele M, Schukkink R, van Gemen B, Debevere J. Identification of Campylobacter jejuni, Campylobacter coli and Campylobacter lari by the nucleic acid amplification system NASBA. J Appl Bacteriol. 1994;77:694–701. doi: 10.1111/j.1365-2672.1994.tb02821.x. [DOI] [PubMed] [Google Scholar]
- 27.van der Vliet G M E, Schukkink R A F, van Gemen B, Schepers P, Klatser P R. Nucleic acid sequence-based amplification (NASBA) for the identification of mycobacteria. J Gen Microbiol. 1993;139:2423–2429. doi: 10.1099/00221287-139-10-2423. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Y J, Estes M K, Jiang X, Metcalf T G. Concentration and detection of hepatitis A virus and rotavirus from shellfish by hybridization tests. Appl Environ Microbiol. 1991;57:2963–2968. doi: 10.1128/aem.57.10.2963-2968.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]