

Abstract
Dysregulation of life and death at the cellular level leads to a variety of diseases. In the nervous system, aberrant neuronal death is an outstanding feature of neurodegenerative diseases. Since the discovery of the caspase family of proteases, much effort has been made to determine how caspases function in disease, including neurodegenerative diseases. Although many papers have been published examining caspases in neuronal death and disease, the pathways have not been fully clarified. In the present review, we examine the potential players in the death pathways, the current tools for examining these players and the models for studying neurological disease. Alzheimer’s disease, the most common neurodegenerative disorder, and cerebral ischaemia, the most common cause of neurological death, are used to illustrate our current understanding of death signalling in neurodegenerative diseases. A better understanding of the neuronal death pathways would provide targets for the development of therapeutic interventions for these diseases.
Keywords: Alzheimer’s disease, Bcl-2 family, caspase, cerebral ischaemia, inhibitor of apoptosis protein (IAP), stroke
INTRODUCTION
The balance of life and death at the cellular level is critical from development through maturity. Developmental neuronal death shapes the nervous system, but, after development, the majority of the post-mitotic neurons should live for the life of the organism. In neurodegenerative diseases, aberrant neuronal death occurs and, although much has been postulated about the mechanisms of the death in various diseases, we still have no clear understanding of these processes. From a mechanistic viewpoint, it is important to first understand what the potential players may be and then how the mechanisms can be elucidated. In the present review, we discuss the molecules involved in death pathways, potential mechanisms of neuronal loss, and the tools to study neuronal death. We illustrate the current understanding of neuronal death employing two diseases of neuronal loss, AD (Alzheimer’s disease), which is the most common chronic neurodegenerative disorder, and cerebral ischaemia/stroke, the third most common cause of death in the Western world, which is an acute neuronal disorder with chronic sequelae. Evaluation of the relevance of data requires an appreciation of the pathology of the disease and the fidelity of models under study to the disease itself.
DEATH MOLECULES
Caspases
Central to apoptotic death is the caspase family of proteases. There are many studies of caspases in neuronal death and disease (for a recent review, see [1]). In order to evaluate this literature, first we should consider how to approach the study of caspases. Caspases are a multi-membered family that is conserved from nematode worms through mammals. However, although there is only one death-related caspase in Caenorhabditis elegans, there are 13 different mammalian caspases, as shown in Figure 1. This multiplicity of caspases raises the questions of why there are so many and whether they have unique functions. Specific non-redundant functions for individual caspases would provide potential therapeutic targets. In order to address the question of the function of individual caspases, appropriate tools must be used. Our understanding of caspases and the manner in which to study them has been constantly evolving.
Figure 1. Mammalian caspases.
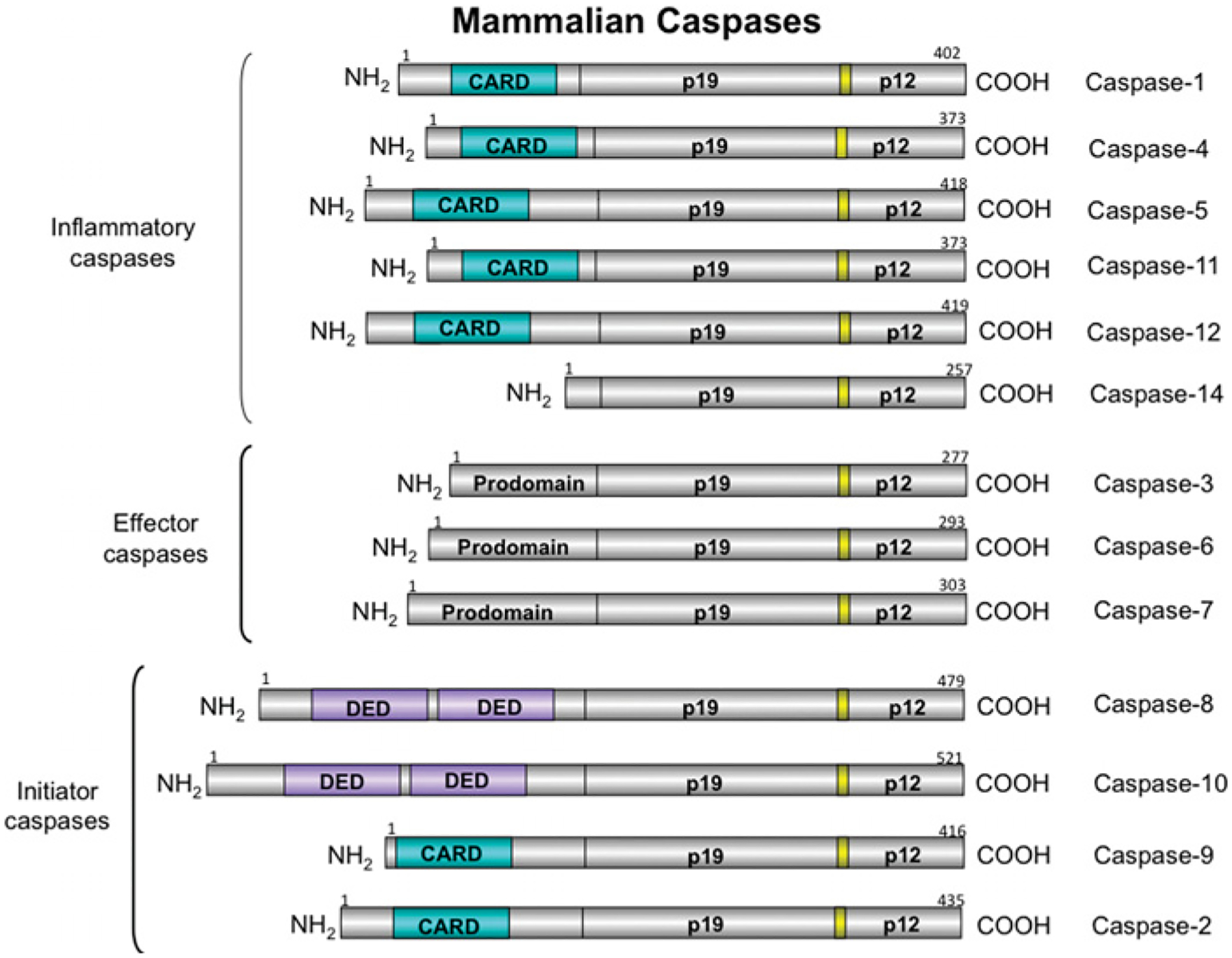
Caspases are cysteine-aspartate proteases critically involved in apoptosis. There are 13 mammalian caspases and they can be divided into three groups according to their functions: inflammatory caspases (1, 4, 5, 11, 12 and 14), effector caspases (3, 6 and 7), and initiator caspases (2, 8, 9 and 10). CARD, caspase recruitment domain; DED, death effector domain.
The first of these proteases, caspase 1, was identified in 1992 as the IL (interleukin)-1β-cleaving enzyme (ICE) [2,3]. Within a few months, an enzyme with significant homology with ICE was identified in C. elegans and was found to execute apoptosis [4]. Over the next few years, many more mammalian homologues were identified, and the term ‘caspase’, for cysteine dependent, aspartate-specific protease, was agreed upon for the mammalian family. The 13 different mammalian caspases can be divided into two groups based on their structure, and three groups based on putative actions (Figure 1). Structurally, caspases segregate into those with short prodomains (caspases 3, 6, 7 and 14) and those with long prodomains (caspases 1, 2, 4, 5, 8, 9, 10, 11 and 12). Caspase 14 is probably involved in keratinocyte differentiation [5] and will not be discussed further. Caspases 3, 6 and 7 are the effectors of apoptosis. Of the caspases with long prodomains, caspases 2, 8, 9 and 10 are initiators of apoptosis, although caspase 2 may act as both an initiator and effector. Caspases 1, 4, 5 and 11 process cytokines: caspases 4 and 5 are found only in humans, whereas caspase 11 is present only in rodents. The generation of cytokines, as discussed below, can also lead to death, often by an autocrine mechanism. Thus, although these caspases may not be direct apoptosis initiators, their activity can lead to death. Caspase 12 may be involved in cytokine processing or in death; its function is unclear, as is its expression: although the gene for caspase 12 is expressed in rodents and humans, the protein is expressed in rodents, but only in a small number of humans [6]. The most recent studies of caspase 12 suggest that it acts as a dominant-negative regulator of caspase 1 activity and that the caspase 12 proteolytic activity results only in autoprocessing [7].
Caspases are also grouped on the basis of their mechanism of activation (Figure 2). For effector caspases, cleavage of the intersubunit linker is essential for activation [8]. For initiator caspases, the initial step is dimerization with a structural change that provides for formation of an active site [9,10]. The longer intersubunit linker allows flexibility, leading to formation of an active site. Most of the initiators exist as monomers in the zymogen form; however, the caspase 2 zymogen is a dimer [11]. Interaction of the specific adaptor protein with the prodomain leads to dimerization and to activation. Once the zymogen is activated, there is limited proteolysis, best studied for caspase 9. For a review of the apoptosome, the caspase 9 activation platform (Figure 3), see [12]. After caspase 9 is incorporated into the apoptosome, there is autocleavage to a p35 fragment. There is also cleavage of caspase 9 to p37 by caspase 3; this cleavage does not activate caspase 9, but potentiates activation by preventing XIAP (X-linked inhibitor of apoptosis protein) inhibition of caspase 9 [13], as described below. The activation platform for caspase 2 is proposed to be the PIDDosome, a complex of PIDD (p53-inducible protein with a death domain), RAIDD {RIP (receptor-interacting protein)-associated ICH-1 [ICE/CED-3 (cell-death determining 3) homologue 1] protein with a death domain} and caspase 2 [14]. Recent data suggest that, in primary neurons, this complex may exist in healthy cells and that knockdown of PIDD leads to activation of caspase 2 [15]. Other studies on the PIDD complex in tumour cell lines showed that overexpression of PIDD induces RAIDD-dependent cell death [16] and that PIDD function in those cells depends on the cleavage state of PIDD [17]. Partially cleaved PIDD, PIDD-C, complexes with NEMO [NF-κB (nuclear factor κB) essential modulator] and leads to NF-κB activation and survival signalling. Further cleavage of PIDD produces a fragment, PIDD-CC, that complexes with RAIDD and leads to cell death (Figure 4). A recent study examined three isoforms of PIDD [18]: isoform 1 contains full-length PIDD and can be processed to PIDD-C and PIDD-CC; isoform 2 has a deletion of 146 amino acids at the N-terminus and an 11-amino-acid deletion at position 580; and isoform 3 has a 17-amino-acid deletion at position 705. Isoform 2 cannot form the PIDD-CC fragment. Expression of these isoforms is tissue-and cell-type-specific: isoform 2 is present mainly in transformed cell lines and in normal liver, pancreas and leucocytes, whereas isoforms 1 and 3 are found in normal tissue, with the exception of skeletal muscle. Isoform 1 is also expressed in transformed cell lines, and isoforms 1 and 2 are found in several tumours. It is possible that PIDD function is different in tumour cell lines, which are mitotic and transformed, and in primary neurons, which are post-mitotic. This would have interesting implications for the regulation of caspase 2 function in the different cell types. In general, it is important to consider the context in which data have been obtained. Much of our knowledge of caspase function is from studies of tumour cell lines. By definition, tumour cell lines are immortalized cells, and thus the regulation of life and death is not necessarily the same as it is in post-mitotic non-transformed cells.
Figure 2. Caspase activation.
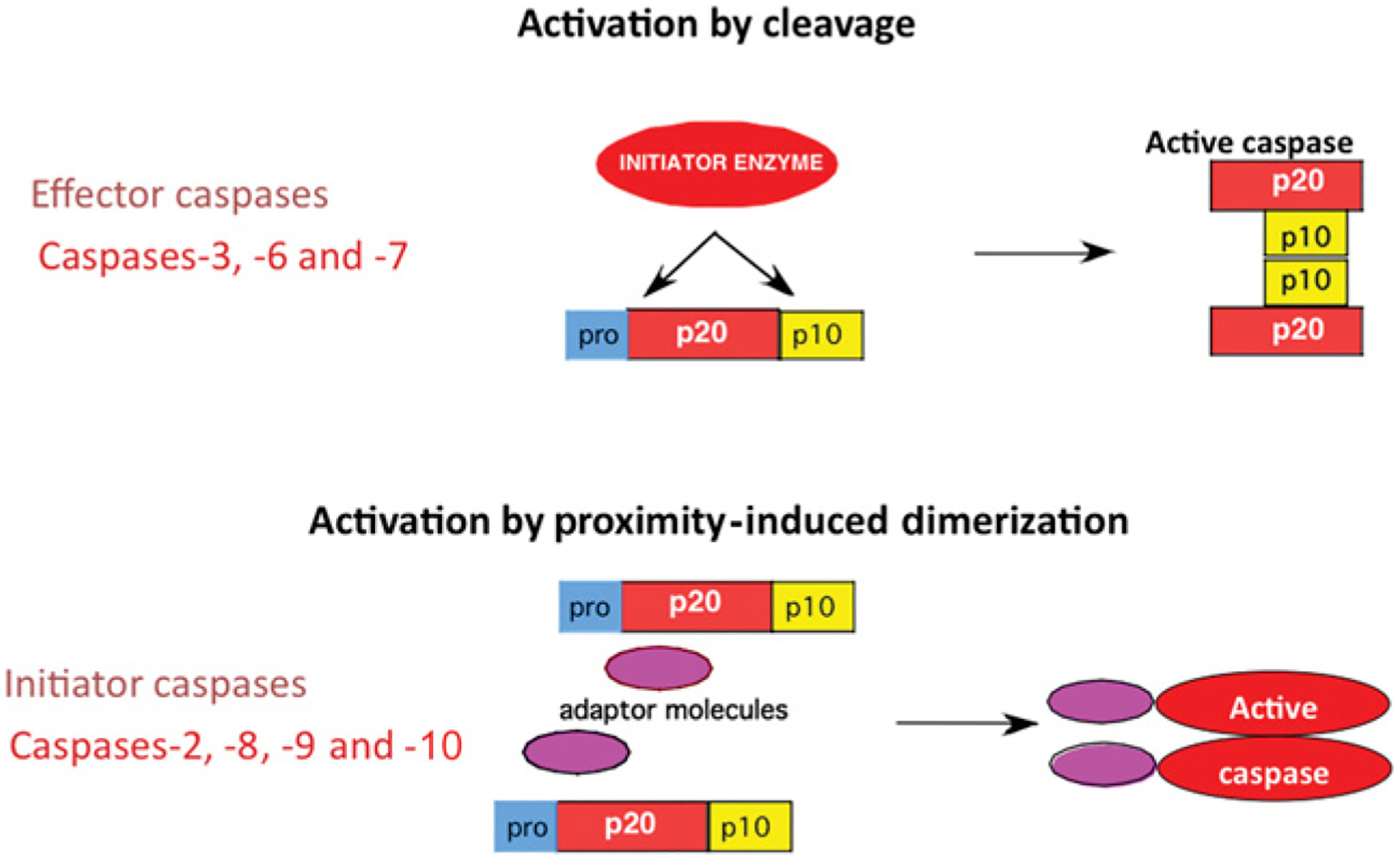
Initiator caspases differ from effector caspases not only at the structural level, but also in the mechanism of activation. Whereas effector caspases require cleavage for their activation, initiator caspases become activated by proximity-induced dimerization, leading to a conformational change that generates the active site.
Figure 3. Intrinsic and extrinsic death pathways.
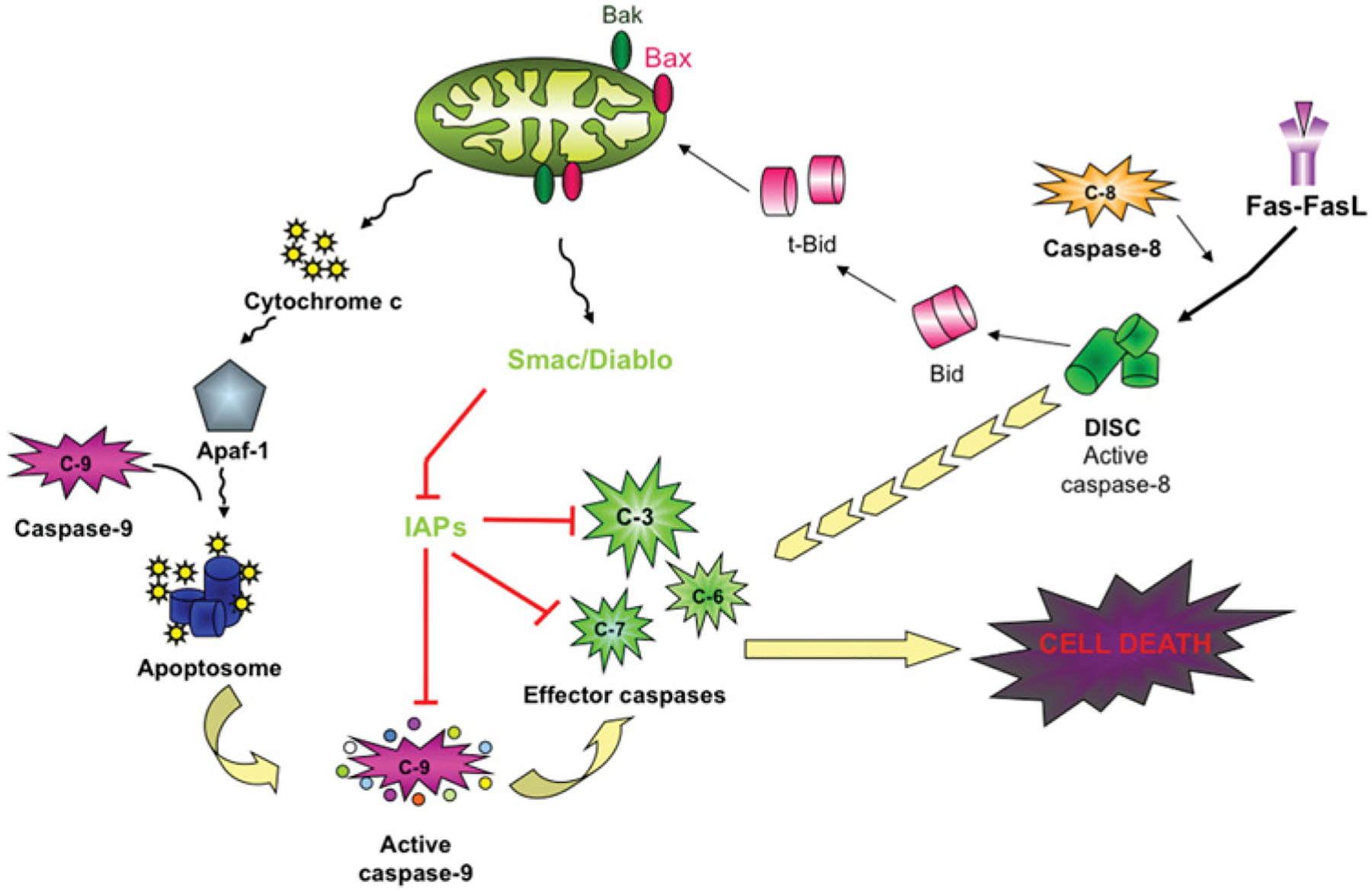
The intrinsic pathway is illustrated on the left-hand side of the Figure. This pathway is mediated by permeabilization of mitochondria, followed by release of cytochrome c, which, in an ATP-dependent fashion, assembles Apaf-1 (apoptotic protease-activating factor 1; the specific adaptor of caspase 9) and pro-caspase 9 into the apoptosome where caspase 9 dimerizes, resulting in a conformational change and caspase 9 activation. On the right-hand side of the Figure is the extrinsic pathway. Death receptor ligands, illustrated here with Fas ligand (FasL), bind the appropriate receptor and recruit death adaptor proteins [FADD (Fas-associated death domain)], which dimerize and activate caspase 8 in the DISC. Active caspase 8 and active caspase 9 cleave and activate effector caspases, leading to cell death. The DISC can potentiate the intrinsic pathway through the cleavage of BID to t-Bid, which enhances mitochondrial permeabilization. The cytosolic IAPs can inhibit caspases 3, 7 and 9. IAPs can be inhibited by Smac/Diablo, which is released from permeabilized mitochondria. C-, caspase.
Figure 4. Caspase 2 activation: the PIDDosome.
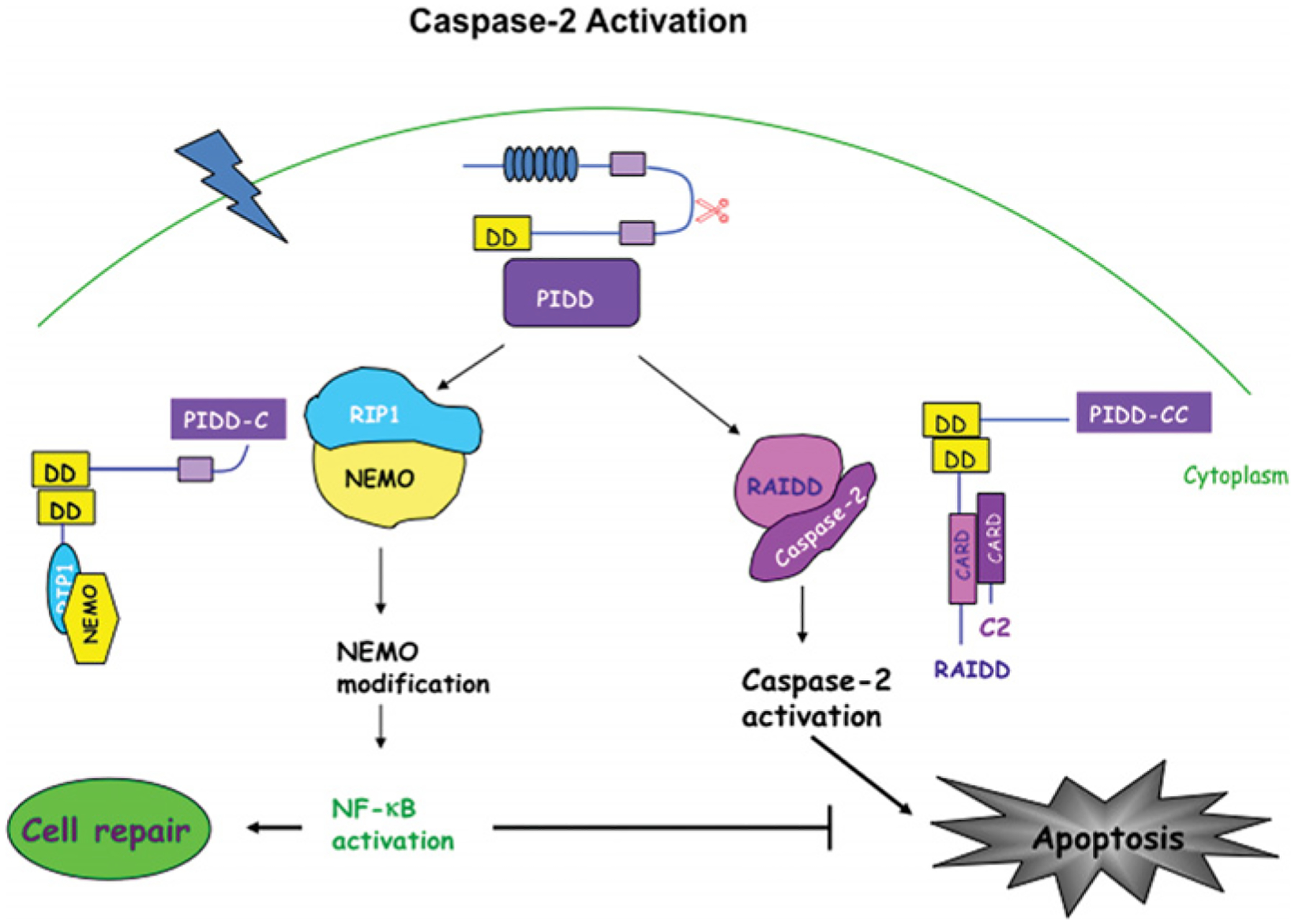
Recently, an activating complex for caspase 2 has been identified comprising caspase 2, RAIDD and PIDD, called the PIDDosome. In this complex, RAIDD is the specific adaptor for caspase 2 and contains a CARD (caspase recruitment domain) at the N-terminal region through which it interacts and recruits caspase 2. RAIDD also has a death domain (DD) at the C-terminal region and via this domain RAIDD establishes a homotypic interaction with PIDD, a p53-inducible protein containing a death domain. PIDD can also interact with RIP1 (receptor interacting protein 1) and NEMO activating the NF-κB pathway and promoting cell survival. Different binding partners associate with different PIDD fragments. PIDD C-terminus forms the complex with NEMO leading to cell survival, whereas PIDD-CC complexes with RAIDD and caspase 2, modulating cell death. These two different complexes suggest a dual role for PIDD in the modulation of the balance between cell survival and death.
Caspase 8 (found in humans and rodents) and caspase 10 (found only in humans) are activated in the canonical extrinsic pathway, where there is a receptor-mediated event leading to assembly of the DISC (death-inducing signalling complex). After incorporation into the DISC, caspase 8 undergoes autocleavage [19] (Figure 3). Recent work suggests that TNFα (tumour necrosis factor α) induces two different caspase 8-activation pathways, a RIPK1 (receptor-interacting serine/threonine protein kinase 1)-dependent and a RIPK1-independent pathway [20]. There is also phosphorylation-mediated regulation of caspase 8 activity [21].
Endogenous inhibitors of caspases, the IAP (inhibitor of apoptosis protein) family
The IAP family is classified by the presence of BIR (baculovirus IAP repeat) domains (Figure 5). In humans, eight IAPs have been identified (see [22] for a recent review). cIAP (cellular IAP) 1, cIAP2 and XIAP, have been shown to bind to caspases (Figure 3). Of these three, XIAP may be the only one to inactivate caspases [23]. Other IAPs do inhibit cell death, but not by inhibiting caspase activity directly. XIAP is the best studied of the caspase-binding IAPs and the most potent inhibitor of cell death. XIAP binds to and inactivates active caspases 3 and 7 via the BIR2 linker region. cIAP1 and cIAP2 can also bind to active caspases 3 and 7, but lack the region necessary for inactivating the caspases [24]. However, binding to the caspases may alter cellular localization and block access of the caspases to their substrates. XIAP–BIR3 binds to and inactivates caspase 9, binding to the neo-epitope generated by autocleavage of caspase 9 at position 315. XIAP cannot bind to the caspase 9 neo-epitope generated by caspase 3 cleavage of caspase 9 at position 330. Thus caspase 3 cleavage promotes activity of caspase 9 by preventing the inhibition of caspase 9 by XIAP. The BIR3 domains of cIAP1 and cIAP2 contain only one of the four residues required to inactivate caspase 9 [23]. Caspase 8 also has an endogenous inhibitor, c-FLIP (cellular FLICE [FADD (Fas-associated death domain)-like ICE]-inhibitory protein), which is caspase 8 without the active site [25]. c-FLIP dimerizes with caspase 8, preventing its activation.
Figure 5. Mammalian IAP family.
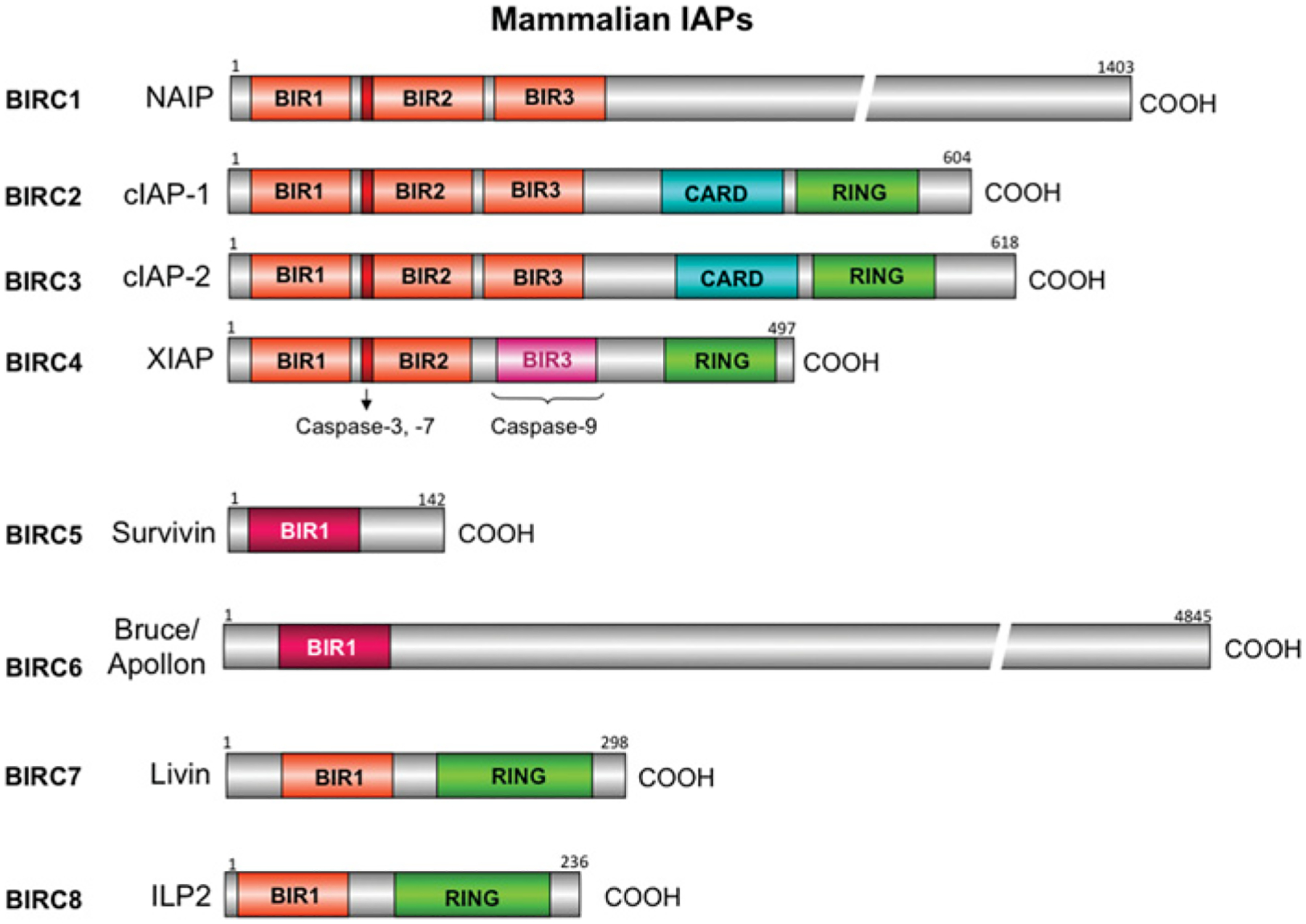
There are eight human IAPs and they are characterized by the presence of BIR domains. BIRs can be divided into two groups: Type I bind to and inhibit caspases, and Type II bind to caspases, but also have a role in cell-cycle regulation (Survivin/BIRC5, BIRC6/Bruce/Apollon). The RING (really interesting new gene) domain located on cIAP1, cIAP2, Livin and ILP2 (IAP-like protein 2) is an E3 ubiquitin ligase. cIAP1 and cIAP2 also contain a CARD (caspase recruitment domain) at the C-terminal region of unknown function since the typical CARD exhibiting protein–protein interactions with other CARD-containing proteins is located at the N-terminal region. The common name as well as the BIRC number are given for each IAP.
Endogenous inhibitors of IAPs
There are several endogenous inhibitors of IAPs. In the mitochondria of healthy cells, there are Smac (second mitochondrial-derived activator of caspase)/Diablo (direct IAP-binding protein with low pI) [26,27] and HtrA2/Omi [28], the N-terminal amino acids of these proteins form the IBM (IAP-binding motif). These proteins are released when the mitochondria are permeabilized during death and interfere with the IAP inhibition of caspases, resulting in potentiation of cell death (Figure 3). Another endogenous inhibitor of IAPs is XAF1 (XIAP-associated factor 1) [29]. Originally identified as a specific inhibitor of XIAP, it is now reported that XAF1 can also bind to cIAP1 and cIAP2 [30]. As the function of cIAP1 and cIAP2 is not quite clear, it is also not clear what the function of XAF1 binding to these molecules would be.
Regulation of mitochondria in apoptosis
Permeabilization of the mitochondrial membrane leads to selective release of compounds such as cytochrome c, Smac/Diablo, HtrA2/Omi and AIF (apoptosis-inducing factor). The Bcl-2 family of proteins has multiple members that can regulate events at the mitochondria [10]. Bcl-2 family members are identified by the presence of a BH (Bcl-2 homology) domain and are grouped into anti-apoptotic which contain four BH domains, pro-apoptotic containing three BH domains, and pro-apoptotic containing only a BH3 domain. Some members contain transmembrane domains and are found in the mitochondrial membrane, whereas others are cytoplasmic. There is also evidence that caspase 2 may regulate mitochondrial permeability in some death pathways [31].
Measurement of caspase activation/activity
There is a distinction between activation and activity that is not always observed in studies of caspases. Activation can lead to activity, but an activated caspase is not necessarily active because it can be inhibited by an IAP. This is an important distinction from a functional perspective. Methods used to measure caspase activity and activation include Western blotting, immunocytochemistry and synthetic peptide substrate/inhibitor assays. Western blotting with specific antibodies reveals changes in levels of the caspase and appearance of fragments generated by proteolytic processing. As noted above, proteolytic processing of the zymogen is required only for effector caspases (3, 6 and 7) and thus appearance of the p18/20 and p10/12 fragments indicates that these caspases have been activated. However, caspases 3, 7 or 9 may have been inhibited by an IAP in situ, but lysis of the cells for Western blotting disrupts the IAP–caspase complex. Thus activated but inhibited caspases cannot be distinguished from active caspases using Western blotting of cell lysates. For initiator caspases, interpretation of Western blotting data is even more problematic. Both caspase 8 and caspase 9 undergo autoproteolysis when incorporated into their respective activation complexes, although the full-length caspase has activity as well [13,32,33]. Caspase 2 may also undergo autoproteolysis [11]. Western blotting has also been used in some studies to compare the relative amounts of zymogen and cleaved fragments in a given sample. These are not valid measures. The antigen presentation of the different protein and protein fragments does not always yield the same amount of signal, thus different protein fragments cannot be quantified relative to one another, but quantification and comparison of amounts of a particular band across different samples is a valid measure. Immunocytochemistry can be used to examine changes in the subcellular localization of a caspase or appearance of cleaved caspase fragments using antibodies which detect the neo-epitopes, with the same caveats noted above for Western blotting.
A great deal of work has been published utilizing synthetic peptide pseudosubstrate caspase inhibitors/substrates, despite work from over a decade ago showing that these reagents are not specific for individual caspases [34,35]. A recent study has evaluated what these reagents measure and found that all of the so-called specific caspase inhibitors/substrates are best at detecting caspase 3, and some do not even detect the targeted caspase [36]. This means that data acquired using these reagents need to be re-evaluated. To date, the best measure of the activity of a specific caspase is with active site affinity ligands, such as b-VAD-fmk (biotin-valyl-alanyl-aspartyl-fluoromethylketone), where there is irreversible binding of a biotinylated substrate to the active caspase in the cell. Once bound, the caspase is no longer active, so this is a measure of which caspases are active at the time at which the b-VAD-fmk is present; downstream events will be inhibited. Thus the time of applying the b-VAD-fmk will determine which caspases are detected. Tu et al. [37] showed that pre-loading cells with b-VAD-fmk detected initiator caspases, activated in a death-stimulus-specific manner; addition of b-VAD-fmk when the cells were harvested detected active effector caspases. The biotinylated affinity ligand–caspase complex is isolated with streptavidin and separated by SDS/PAGE, and the specific caspase bound is detected using Western blotting for individual caspases. This method has shown that the initiator caspases that bind the affinity ligand are uncleaved, while the effectors are cleaved, supporting the proximity-induced dimerization model of activation (no cleavage necessary) for initiator caspases. Adaptation of this method to animal studies will enable the detection of which caspases are active in vivo. Functional measures of activity are also important. Molecular knockdown/knockout of individual caspases provides specific data about the functional relevance of the targeted caspase. Abrogation of death shows that specific caspases are critical for execution of the death pathway under study. Thus, as measures of caspase activation, Western blotting for cleaved caspases and immunocytochemistry for neo-epitopes will provide data about activation of caspases 3, 6 and 7. The only measure of active caspases is the affinity ligand method.
Determining mechanisms of neuronal death in neurodegenerative diseases
It is clear that there are distinct death pathways, mobilizing different combinations of death molecules. What is not clear is how the specificity is determined: by stimulus, by cell type or by both. Caspases can cleave many proteins; cleavage is limited (cleavage at one or two sites), as illustrated by the activation of effector caspases by initiator caspases. There have been multiple reports of substrate cleavage changing the activity of the substrate, that is converting a pro-apoptotic protein into an anti-apoptotic one, or vice versa. Such events serve to amplify the signalling pathways leading to death or survival. Although predictions have been made, careful time courses of substrate cleavage events, combined with specific blockade of these events will be required to determine the function of such cleavage in individual death pathways. The study of diseases requires the use of models of the disease. We present two diseases that are among the most highly prevalent human neurological disorders of aging and consider what the appropriate models are and what the current understanding of the death pathways is in each disease.
ALZHEIMER’S DISEASE
Lifespan in industrialized countries has increased an average of 25 years during the last century. With this increased life expectancy, the age of the population has shifted, with more people living beyond 80 years of age. AD affects 2–3% of people at the age of 65 years, but the incidence of this disease doubles for every 5 years of age afterwards, affecting approx. 50% of individuals over the age of 85 years. There are approx. 4.5 million cases of AD in the U.S.A. [38]. Because of increasing life expectancy and expanding population, it is estimated that by 2050, the number of individuals with AD will be approx. 14 million in the U.S.A. and over 100 million worldwide [38]. AD is the most common form of dementia among the elderly population and represents the fourth cause of death in industrialized countries [39,40].
First described in 1907 by Alois Alzheimer, AD is characterized by a progressive impairment of cognitive functions. The most frequent symptom is gradual loss of memory, mainly of recent events, called episodic memory. As the disease progresses, there is impairment in several brain regions controlling language, cognition, reason and temporospatial orientation, together with other changes in mood and personality. The initial clinical symptoms are followed by continuous and progressive decline characterized by mutism, vegetative state, inanition and finally death. This period lasts 5–20 years after the detection of the first symptoms of the disease. A definite diagnosis of AD can only be obtained from brain tissue, thus the clinical diagnosis can be of possible or probable AD, as biopsies are rarely done and tissue is usually obtained post-mortem. The two main histopathological hallmarks required for the definitive diagnosis of AD are [41]: (i) senile plaques, extracellular congophyllic deposits composed mainly of Aβ (amyloid-β peptide), and (ii) NFTs (neurofibrillary tangles), intraneuronal filamentous aggregates composed of hyperphosphorylated tau proteins and PHFs (paired helical filaments).
The lack of a clear-cut clinical diagnosis, the chronic nature of the disease and the insidious onset of symptoms have not allowed a clear understanding of the order of events in the development of the disease. It has been postulated that an important factor in AD is the progressive loss of synapses and neurons in limbic and cortical areas, which is translated as a disconnection between different brain areas, thus leading to the manifestation of the clinical symptoms [42,43]. At the beginning of the disease, this initial disconnection does not seem to have repercussions on behaviour if we keep in mind that the gradual loss of synapses and neurons seems to start 20–40 years before the manifestation of the first symptoms [44,45]. The NFT distribution correlates better than the amyloid plaque burden with the severity of dementia and neuronal death [46–50]. However, as there are no studies of progressive pathological analysis of single patients over time, all of the human data with regards to molecular mechanisms need to be considered as correlative, but not definitive.
Genetics of AD
Although neither the aetiology of the disease nor the molecular mechanisms underlying AD is clear, it is accepted that AD is a complex multifactorial disease in which both genetic and environmental factors are involved and modulate the progression of the disease. In approx. 10% of AD cases the first symptoms occur before the age of 60–65 years. This type of AD is known as EOAD (early-onset AD). The remaining 90% is associated with cases in which the clinical presentation takes place after the age of 65 years and is referred to as LOAD (late-onset AD). Approx. 50% of EOAD shows autosomal dominant Mendelian inheritance [51]. To date, both sporadic and familial forms of the disease have similar clinical presentation and manifestations suggesting common pathogenic mechanisms.
Genetic analysis from families has allowed the identification of three genes linked to AD: APP (amyloid precursor protein) [52], located on chromosome 21, PSEN1 (presenilin 1) [53] and PSEN2 [54] located on chromosomes 14 and 1 respectively. Some 85% of familial EAOD cases correspond to a mutation in the PSEN1 gene, whereas mutations in PSEN2 or APP are much less frequent [55,56] (Figure 6). Today, 157 mutations in PSEN1 and ten in PSEN2 have been described worldwide. There are families with a clear genetic component that have not been associated with mutations in any of these three genes. This raises the idea that other genes may be involved. Only one gene, from a long list of susceptibility genes, has been confirmed as a risk factor for sporadic LOAD: the APOE (apolipoprotein E) gene located on chromosome 19 [57]. However, less than 50% of the sporadic LOAD cases are carriers for ApoEε4 [58]. We must emphasize that risk factors function as predisposition markers for the disease; they have not been shown to be causal agents and by themselves cannot induce the disease. They modify the risk of developing disease, increasing it compared with the non-carrier population. Genes involved in AD, as causative genes or as or risk factors, are summarized in Table 1. Many of the proposed susceptibility genes are proteolytic enzymes, membrane receptors or growth factors. Most of them have been implicated in the proteolytic processing of APP and/or the degradation/clearance of Aβ [59–62].
Figure 6. Genetics in AD.
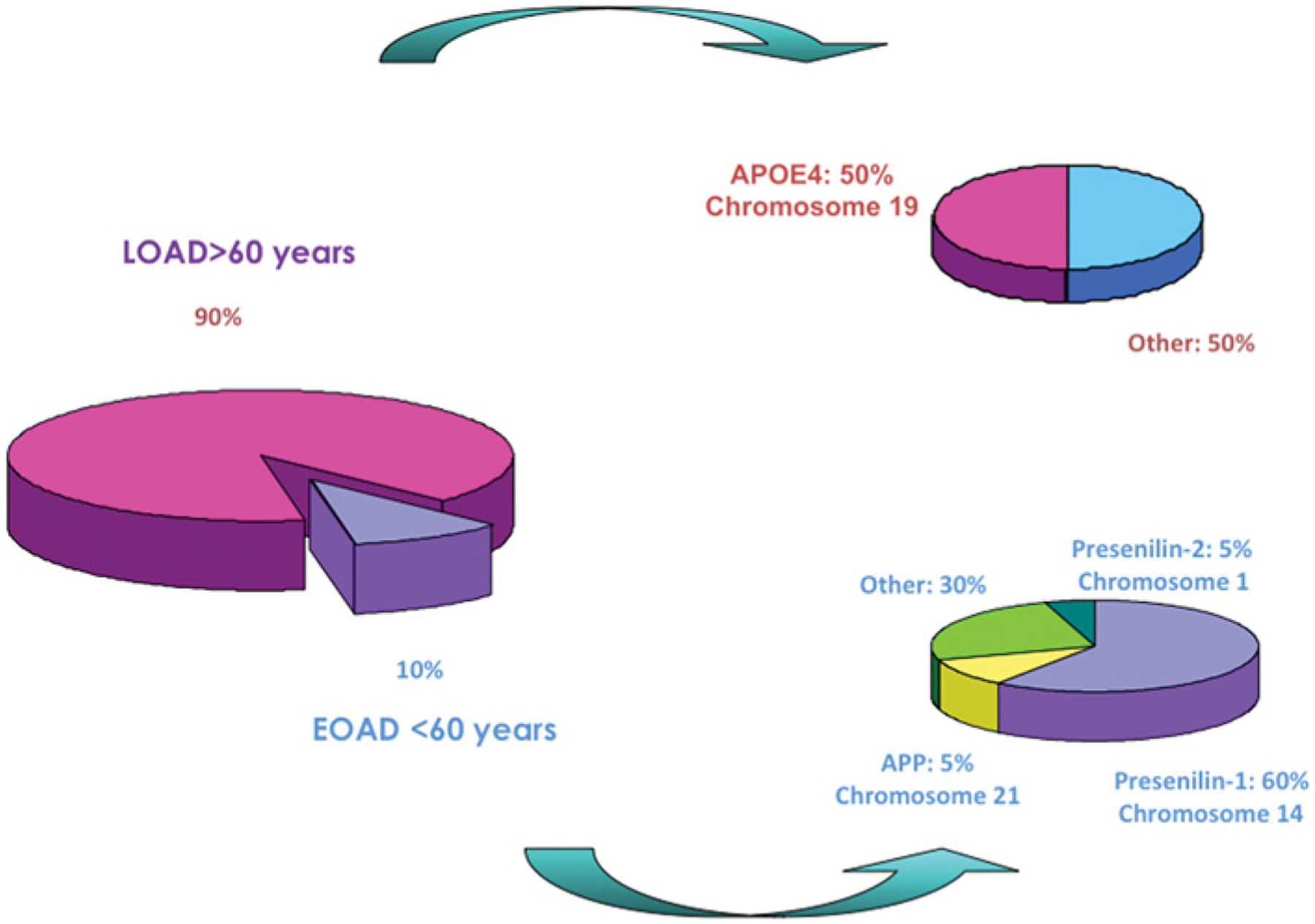
Schematic representation of the impact of LOAD compared with EOAD. Over 90% of total AD cases are late-onset and only ApoEε4 has been identified and confirmed as a susceptibility gene which modifies the risk of developing LOAD. Approx. 50% of LOAD cases are carriers for ApoEε4. Although the majority of the AD cases are of late-onset, approx. 10% of AD cases are of early-onset. To date, three genes have been identified as causative genes: APP located on chromosome 21 (5% of EOAD cases), PSEN1 on chromosome 14 (60% cases) and PSEN2 on chromosome 1 (5% cases).
Table 1. Causative and susceptibility genes involved in AD.
ACT, α1-antichymotrypsin gene; A2M, α2-macroglobulin gene; BCHE, butyrylcholinesterase gene.
| Gene | Locus | Onset | Status | Involvement |
|---|---|---|---|---|
| APP | 21q21.3-q22.05 | Early | Familial | Demonstrated |
| PSEN1 | 14q24.3 | Early | Familial | Demonstrated |
| PSEN2 | 1q31-q42 | Early | Familial | Demonstrated |
| APOE | 19q32.2 | Late | Familial/sporadic | Demonstrated |
| ACT | 14q32.1 | Late | Sporadic | Suggested |
| A2M | 12p | Late | Sporadic | Suggested |
| BCHE | 3q26.1-q26.2 | Late | Sporadic | Suggested |
Mouse models in AD: pros and cons
After the identification of genetic mutations in AD, mouse models were generated for the study of AD. Several transgenic mice lines overexpressing human mutant APP reproduce several histopathological hallmarks of AD such as amyloid deposits, abnormal tau hyperphosphorylation, gliosis, and memory and learning impairments [63–69]. Figure 7 shows the location of the majority of the identified APP mutations. However, despite the presence of hyperphosphorylated tau, the brains of these mice do not exhibit NFTs or neuronal loss. Owing to the lack of tau pathology and neuronal loss in those APP-overexpressing mice, several new models overexpressing several tau isoforms and mutants have emerged in order to recapitulate the remaining pathological features lacking in the previous models [70–80]. More recently, transgenic mice have been crossed with other transgenic mice with the intention of affecting several genes involved in AD. Table 2 summarizes the salient features of several of the different types of mouse model. These new tools provide the advantage of allowing the study of pathological interactions that exist between several genetic factors involved in the disease and may also increase the understanding of the order in which these neuropathological events take place in the human AD cases. However, some of these models overexpress mutant genes that are not even found in AD cases, so the results extracted from these models should be treated with caution. It is also important to consider whether the phenotype observed in the mouse represents a real phenocopy of the disease. However, although these mice do not exhibit every single hallmark of the disease, they are a valuable tool for: (i) understanding the interaction between genetic factors able to modify the in vivo production and deposition of Aβ; (ii) the identification of molecular targets; and (iii) the evaluation of new therapies designed to stop or slow down the pathological and clinical manifestations of this devastating disease.
Figure 7. Mutations in APP.
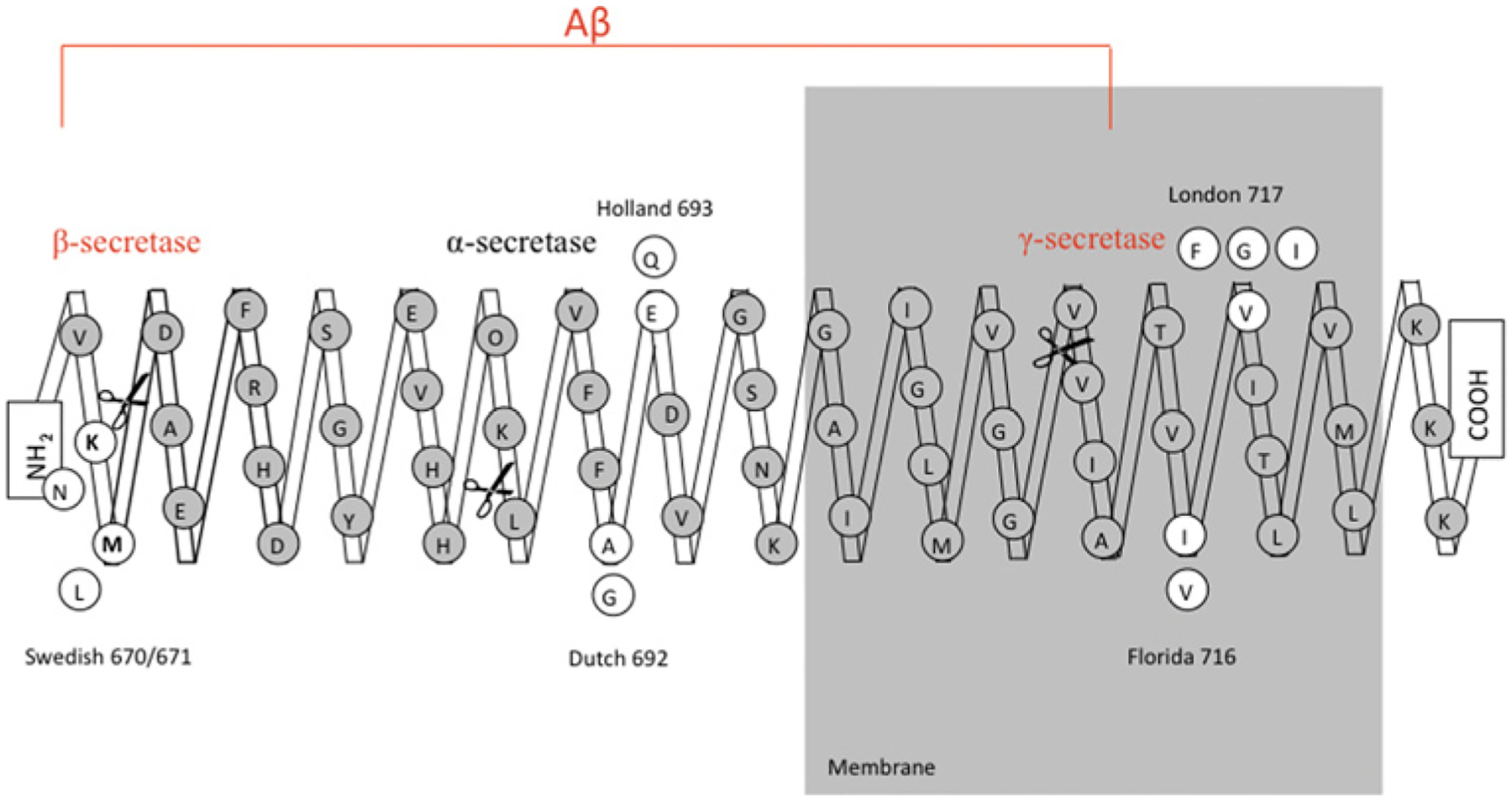
Location of the most relevant mutations causing EOAD. The majority of the mutations are located close to the secretase cleavage sites. One example is the Swedish mutation (APP670/671), located on the N-terminal region of Aβ and close to the β-secretase cleavage site. Another example is the London mutation (APP717) located on the C-terminal region and in the area surrounding the γ-secretase site.
Table 2. Transgenic models of AD.
Several of the many transgenic models of AD are presented to illustrate the pathology found in these animals. CA1, hippocampal CA1 region; EC, entorrhinal cortex; PDGFβ, platelet-derived growth factor β; PrP, prion protein.
| Transgenic line | Mutations | Promoter | Age of onset (months) | Tau pathology | Synaptic dysfunction | Neuronal death |
|---|---|---|---|---|---|---|
| PDAPP [63] | APPV717F | Hu PDGFβ | 6–9 | No | Yes | No |
| Tg2756 [64] | APPswe | Mu PrP | 9–12 | No | Yes | No |
| 3XTg [197] | APPswe, TauP301L, PS1M146V | Mu Thy1 | 6 | Yes, 12 months | Yes | No |
| Tg2756XVLW [198] | APPswe, TauG272V,P301L,R406W | Mu PrP Mu Thy1 | 12 | Yes, 16 months | Yes | Yes, 9 months (CA1/EC) |
In vitro models of AD: testing the amyloid hypothesis
In vitro models have been used to decipher the pathological mechanisms underlying the disease. Primary neuronal cultures provide the opportunity to decipher molecular mechanisms. Cell cultures have been used to test how mutations in certain genes affect Aβ metabolism, secretion and/or degradation [81]. In vitro models of familial AD showed an enhancement of the APP processing via alterations in the γ -secretase complex [82,83] and an increase in the intracellular concentration of Aβ species [84]. These modifications in the proteolytic processing led to the alteration in the Aβ40/Aβ42 ratio [85–87]. Cultured cells have been used to determine the cellular compartment in which APP is processed and how Aβ is generated and transported along different cellular compartments [88]. Thus in vitro models are a critical tool in the study of the secretases. Primary cultures and hippocampal slices have been widely used in order to determine the influence of Aβ species and for electrophysiological purposes. But probably the most relevant use of in vitro models is to unravel the neurotoxic species triggering the progression of the disease. The relevant role of Aβ and the so-called amyloid hypothesis still remains questionable in the aetiology of AD, mainly because there is little correlation between the amyloid burden and the clinical progression of the disease. However, this idea may be readdressed since the discovery that early memory impairment and synaptic dysfunction correlate better with the levels of oligomeric Aβ, and not with the fibrillar species. In order to reproduce the neuronal degeneration observed in AD brains, researchers have used a wide variety of cell systems, different lengths of peptides and several degrees of aggregation. An important fact to keep in mind is that Aβ is in active equilibrium with its environment: the assemblies are highly dynamic. This property makes it very difficult to ascribe specific toxic characteristics to a certain type of aggregation state. However, there are several studies that try to identify the relationship between toxicity and aggregation. This idea was established in 1998 with the characterization of non fibrillar oligomeric species, ADDLs (Aβ-diffusible-derived ligands), and their neurotoxic properties [89]. ADDLs caused neuronal death in primary hippocampal neuron cultures and inhibited LTP (long-term potentiation) in cultured slices from rat brains. However, when 500 nM ADDLs were incubated with organotypic mouse brain slices for 1 h, no overt neuronal loss was detected, although the blockage of the LTP was highly dramatic [89,90]. The latest modification of the in vitro systems employs naturally produced cell-derived Aβ oligomers [91,92]. 7PA2 cells [CHO (Chinese-hamster ovary) cells] were modified to overexpress human mutant APPV717F. These modified cells generated SDS-stable oligomers that generate dimers, trimers and occasionally tetramers [92]. Intracerebral injection of culture medium containing oligomeric Aβ species into rat brain showed inhibition of LTP [92]. Oligomers as large as dodecamers have been isolated from AD brains [93] and from transgenic mice [94]; these species are also toxic in primary neuronal cultures and impair LTP in hippocampal slices [95]. Taken together, it seems that oligomeric Aβ species alter hippocampal potentiation and synaptic plasticity both in vitro and in vivo.
In vitro systems play a crucial role in unravelling the cytotoxic properties not only of Aβ, but also of the fragments derived from the proteolysis of its precursor APP, particularly the CTFs (C-terminal fragments). For many years, it was thought that the CTFs derived from γ- and β-secretases were inert, but it has been shown that expression of C99 is toxic to neurons [96] and that expression of C99 in rodent brains induced Aβ deposition, neurodegeneration, alterations in behaviour and synaptic deficits [97]. The mechanism underlying these effects is still unknown, but previous findings suggest that the cytoplasmic tail of APP can be cleaved by caspases at Asp664 (APP695), generating a new CTF of 31 residues named C31 [97,98]. Since expression of C31 alone is cytotoxic to the cell, it is believed that subsequent cleavage of CTFs to generate C31 may potentiate susceptibility to apoptosis [99]. Moreover, mutations in APP to abolish the caspase cleavage site significantly attenuate the cytotoxic effects of C99 [98]. PDAPP mice carrying the cleavage mutation [PDAPP(D664A) mice] deposit Aβ, but do not develop behavioural deficits, supporting a role for C31 in the potentiation of AD deficits [100].
Death molecules in AD
The detection of cleaved caspases and the accumulation of cleaved caspase substrates in post-mortem AD brain tissue support the hypothesis that apoptosis may play a role in the subsequent neuronal loss found in AD brains [101,102]. There is evidence demonstrating that caspases are increased in brains from patients with AD. Analysis from AD brain tissue showed elevated mRNA expression of caspases 1, 2, 3, 5, 6, 7, 8 and 9 in the brain of AD patients compared with controls [103]. Pyramidal neurons from vulnerable regions involved in the disease showed an increase in activated caspase 3 and 6 [104,105]. Synaptosomes prepared from AD brain frontal cortices showed an enrichment in caspase 9 compared with non-demented controls [99]. APP contains caspase cleavage sites and it has been described that caspases 3, 6, 7 and 8 can cleave APP [102,104,106]. Cultures of neuronal cells deprived of serum showed activation of caspase 6 and subsequent processing of APP by caspase 6 generating a 6.5 kDa fragment that contains Aβ [104]. In this context, caspases may play an active role in Aβ-induced neurotoxicity. Amyloid plaques are enriched in caspase-cleaved APP [102]. However, caspases do not only process APP, as caspase cleavage of tau enhances tau filament polymerization in vitro [107,108]. Neurons from caspase 2-knockout mice and from caspase 12-knockout mice are resistant to Aβ [109,110]. As noted above, caspase 12 protein is expressed in only a subset of people and there is no increase of AD in this population [111]. These data demonstrate that caspases are very closely linked to the degeneration process found in AD brains, but that the specific caspases involved have not been adequately defined. Table 3 summarizes the current evidence of a role for each caspase in AD.
Table 3. Caspases implicated in AD.
Multiple caspases have been implicated in the progression of AD. Measure indicates the method of detecting caspase changes. As we can appreciate, the majority of the studies are based on alterations at the mRNA levels between AD cases and control non-demented subjects for the caspase(s) under study. siRNA, small interfering RNA.
| Caspase | Paradigm | Measure | Reference |
|---|---|---|---|
| 1 | AD brains | Increased mRNA | [101] |
| 2 | AD brains | Increased mRNA | [101] |
| 2 | Aβ42 treatment of primary hippocampal neurons | Death blocked by antisense to C2; C2-null neurons | [109] |
| 3 | AD brain pyramidal neurons | Increased activated caspase 3 | [105] |
| 3 | AD brains | Increased mRNA | [101] |
| 4 | Aβ25–35 treatment of SK-N-SH cells | Death blocked by siRNA to C4 | [199] |
| 5 | AD brains | Increased mRNA | [101] |
| 6 | AD brains | Increased cleaved caspase 6 | [104] |
| 6 | AD brains | Increased mRNA | [101] |
| 7 | AD brains | Increased mRNA | [101] |
| 8 | AD brains | Increased mRNA | [101] |
| 9 | AD brains | Increased mRNA | [101] |
| 12 | Aβ40 treatment of primary cortical neurons | Death blocked by antisense to C12; C12-null neurons | [110] |
Although caspases are the main players involved in apoptosis, there are other molecules involved in the progression of the apoptotic cascade that are relevant to human AD. The Bcl-2 family of proteins has members that regulate apoptosis by forming mitochondrial permeability transition pores, contributing to the release of calcium and controlling the release of mitochondrial apoptogenic factors into the cytosol [112–114]. It is known that mutations in PSEN1 sensitize cells to undergo apoptosis, and it has been reported that PSEN1 and Bcl-2 interact in cultured cells [115] and also that PSEN1 interacts with calcium-binding proteins [116–118]. In this context, the interaction of these two proteins may contribute to the maintenance of calcium intracellular levels and help to maintain the integrity of the cell. Not only Bcl-2, but also Bcl-XL, an anti-apoptotic member of the Bcl-2 family, has been found in association with PSEN1 and PSEN2 [119]. Moreover, this group described that presenilins may enhance the pro-apoptotic effects of Bax and influence the release of cytochrome c [119]. Other studies support the idea that the Bcl-2 family members are linked to AD, since it has been shown that Bcl-2 and Bax active sites highly co-localize in frontal cortex from AD patients, whereas the levels of co-localization in control cases is non-significant [120]. A recent study showed that crossing 3XTg AD mice with mice overexpressing Bcl-2 showed a decrease in caspase cleavage of tau, a decrease in APP cleavage and decreased Aβ deposition [121]. In vitro studies have shown that Aβ induces an up-regulation of pro-apoptotic molecules such as Bax and down-regulation of anti-apoptotic proteins such as Bcl-2, Bcl-XL or Bcl-w [122]. But Bcl-2 has also been reported to be up-regulated in vivo in transgenic models of amyloidosis [123]. The striking result is that only those regions that exhibit high amyloid load exhibited the elevated expression of Bcl-2, suggesting that Aβ deposition is triggering the Bcl-2 overexpression. Another member of the Bcl-2 family has been implicated in the pathophysiology of AD. Bim (Bcl-2-interacting mediator of cell death), a pro-apoptotic protein, is induced in both cortical and hippocampal neuronal cultures after Aβ exposure and its induction is essential for the neurotoxic effects of Aβ [124]. Moreover, analysis of post-mortem human AD brains showed a marked up-regulation of Bim immunoreactivity in entorhinal cortex, but not in cerebellum, of AD cases compared with non-demented controls [124]. In conclusion, it seems that several levels of regulation are required in order to control the apoptotic cascade and to prevent neurons from dying. Misregulation of this pathway at any level will activate the apoptotic machinery finally leading to neural death, the most critical hallmark of AD. Figure 8 shows a possible pathway for AD.
Figure 8. Proposed pathway of neuronal dysfunction/death in AD.
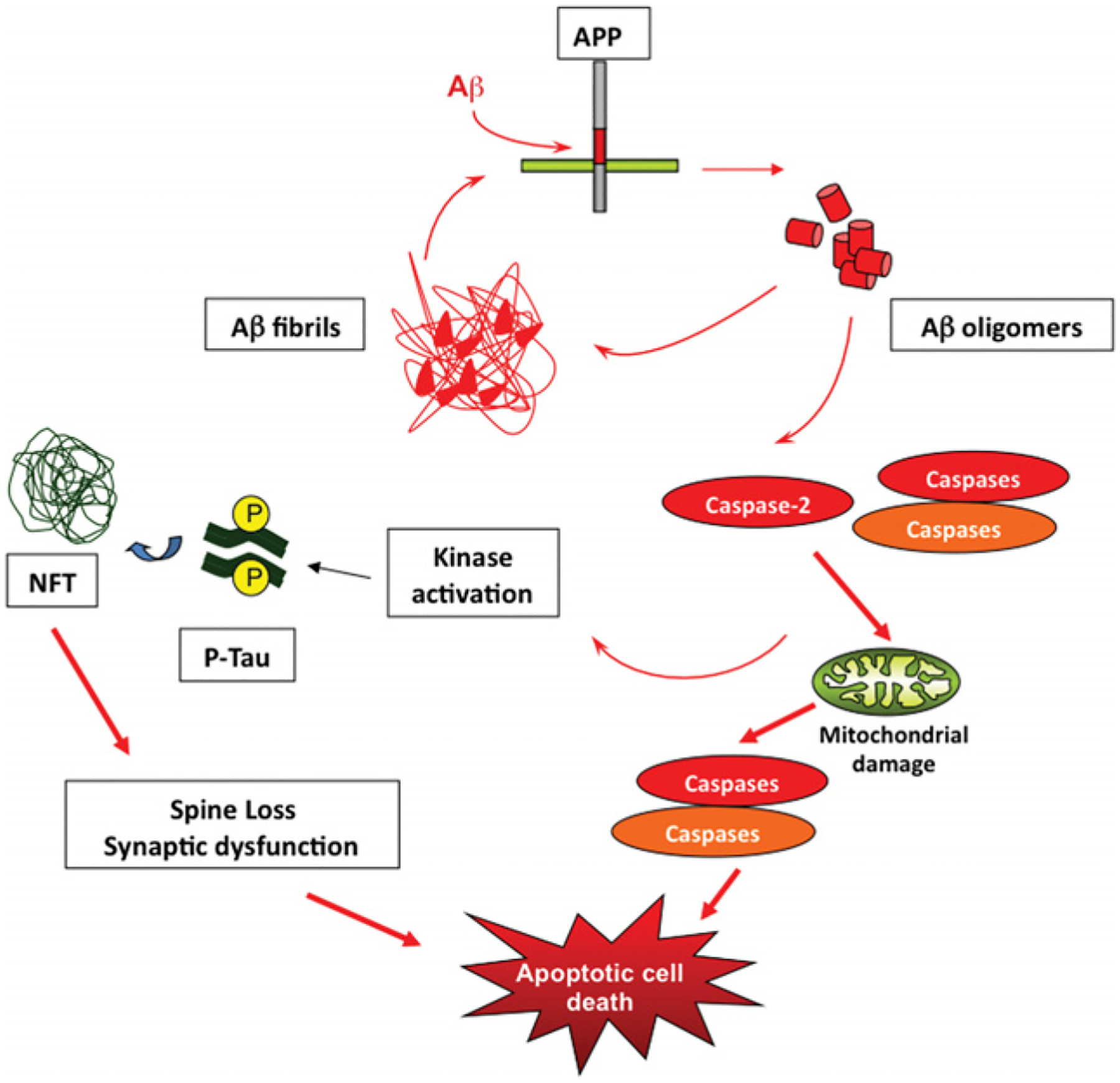
Although during the last decade, our knowledge of AD has increased greatly, the correct pathway and the order of the events leading to the neurodegeneration process associated with this disease remain unclear. Aβ is generated by proteolytic processing from APP, and Aβ is then released into the extracellular space. Aβ aggregates and will deposit into fibrils forming a ‘seeding’ or nucleation core. Continuing production of Aβ increases deposition around the core forming the so-called amyloid plaque. There is a dynamic equilibrium between Aβ fibrils, oligomers and monomers. Although the critical species involved in the aetiology of AD remains unknown, there is increasing evidence that oligomers, not fibrils, are critical in activating caspases leading to kinase activation, tau hyperphosphorylation and NFTs. Moreover, Aβ by itself is able to activate kinases leading to NFT formation potentiating these effects. NFTs enhance the process generated by Aβ contributing to microtubule destabilization and axonal transport impairment leading to synaptic dysfunction and spine loss. Caspases will trigger mitochondrial damage which in turn will activate downstream effectors and finally the death of the compromised cell. An animated version of this Figure can be seen at http://www.BiochemJ.org/bj/415/0165/bj4150165add.htm.
CEREBRAL ISCHAEMIA
Stroke is an acute neurodegenerative disorder that is one of the leading causes of death and disability in major industrialized countries [125]. Most strokes (approx. 85%) are ischaemic and result from an occlusion of a major cerebral artery by a thrombus, which leads to a focal loss of blood flow. Less commonly, stroke results from the absence of blood flow to the entire brain due to cardiac arrest or systemic haemorrhage. Without recovery of normal blood flow within a short period of time, death of all cells within the ischaemic territory is typically the final outcome; however, the pathway of destruction remains unclear.
Ischaemic stroke initiates a complex series of events, both spatial and temporal, that evolve over hours and days. They include inflammation and altered microvascular permeability, which produces tissue oedema, in addition to direct effects on cells that triggers glutamatergic excitotoxicity, ionic imbalance and free-radical reactions, among others. Over the last few years, the awareness of the relevance of dynamic interactions of the neurovascular unit (the complex of neurons, the microvessels that supply them, and the supportive cells: astroglia and microglia, and other resident inflammatory cells) has increased. The relative proportions of the components of this unit may vary in different brain areas and between species. This variation could account for morphological and functional differences in the reaction to ischaemia in different areas of the brain and between species [126]. The neurovascular unit places stroke in the context of an integrative tissue response disease with a significant vascular component where some risk factors are diabetes, elevated homocysteine blood levels, elevated arterial blood pressure or artheriosclerosis (for a review, see [127]). In addition to the loss of oxygen and glucose, disruption of the BBB (blood–brain barrier) allows entry of vascular inflammatory cells and proteins that are toxic to neurons.
Traditionally, the cell death following cerebral ischaemia was considered to be necrotic in nature, but research in the last decade has revealed that there is a pattern of combined necrotic and apoptotic cell death. In cerebral ischaemia, there is complete energy depletion in the central lesion, the core, and a gradient from the core towards the bordering zone, the penumbra. The core has been considered to be necrotic [128,129], whereas, in the penumbra, apoptotic mechanisms are activated [130–132]. However, there is still much debate about this. Benchoua et al. [133] have suggested that the initial events in the core are apoptotic, and secondary necrosis results from a rapid failure to fully develop the apoptotic programme because of the maintained depletion of apoptosis-requiring energy stores in the core.
The morphological manifestations of apoptosis as initially described by Kerr et al. [134] are controversial when considering cell death in the adult brain. Hypoxic/ischaemic insults during the neonatal period can cause typical morphological changes of apoptosis, although these morphological changes are less consistent in adult brain, perhaps because neurons in adult brains are equipped with anti-apoptotic molecules that raise the apoptotic threshold [135]. However, despite this apparent contradiction, there is a large body of evidence that there is caspase-mediated cell death after ischaemia [136]. Furthermore, there are a growing number of studies that implicate apoptotic pathways after ischaemia [137]. Martin et al. [138] suggest that cell death in the CNS following injury can coexist as apoptosis, necrosis, and hybrid forms along an apoptosis–necrosis continuum. Although some caspase-dependent apoptosis might occur in the adult brain, there are also alternative mechanisms of death that result in different morphologies that might rely on either caspases or calpains as the dominant execution proteases [139,140]. An alternative explanation is offered by the attractive concept that, under anoxic/ischaemic conditions, apoptosis may be masked by necrosis [141]. In vitro studies have provided several examples of shifts towards apoptosis from necrosis, resulting from abrupt environmental perturbations [142].
Another important aspect in the death mechanisms in stroke is the reperfusion damage that is associated with the sudden return of blood flow to the oxygen-starved energetically compromised tissue [143]. It is held that much of the injury produced by transient ischaemia, such as the disruption of the BBB and formation of brain oedema, results from reperfusion itself [144].
Animal models of stroke
The present understanding of stroke pathogenesis has been derived from studies involving cell lines, primary and organotypic cultures under anoxia, hypoxia, and oxygen- and glucose-deprivation conditions. These models allow the dissection of the pathways that may take place under a particular insult, such as excitotoxicity, oxidative stress, ionic imbalance or the implication of a particular molecule, either in one cellular type or in a mixture of them representing the relationship between the neurovascular unit elements. However, animal models are mainly used to mimic human stroke. Unlike other neurodegenerative diseases, there are excellent animal models of stroke that provide evidence for mechanisms, prevention and treatment efficacy in vivo that allow for control of collateral factors.
Models of cerebral ischaemia can be performed in small and large animals, but rodents are most widely used. Despite the differences in anatomy and functionality between rodent and human brains, these models permit a careful dissection of mechanisms of injury and neuroprotection and allow examination of events that occur from seconds to days after the ischaemic event (for a review, see [145]). Unlike other neurodegenerative diseases, in stroke, we know the primary insult that triggers the pathology and the actual models provide a good approach to recreate this acute insult.
There are a variety of models that reflect the assortment of insults in humans. These can be divided into three subgroups: global ischaemia, focal ischaemia and haemorrhagic infarct. Global models involve occluding the major blood vessels that supply the forebrain, but these models are considered to mimic consequences of cardiac arrest rather than stroke [146]. Focal models occlude a specific vessel, usually the MCA (middle cerebral artery), because the majority of human ischaemic strokes result from an occlusion in the region of the MCA [146]. MCAo (MCA occlusion) models are either permanent (pMCAo) or transient (tMCAo), allowing examination of the damage that results from both ischaemia and reperfusion.
In the last decade, the failure of neuroprotectants against stroke in clinical trials had brought into question the value of the animal models in pre-clinical research. A proof of confidence in the value of animal models is that changing several physiological parameters, such as reperfusion, hyperglycaemia, hyperthermia or blood pressure, has similar effects on the outcome of stroke in humans and in animal models (for a review, see [146]). Nevertheless, although animal models mimic quite well the onset of the stroke, small animal brains are quite different from human brains in function and morphology, and this may influence the efficacy of the therapies. This phenomenon was represented recently in an in silico model which considers the differences in the percentage of white matter and glia between rodents and humans [147]. If this hypothesis is correct, the efficacy of the therapies should be apparent when animal models with a more similar brain composition to that of humans are used.
Since there is a logistical problem in treating patients at early time points, clinical investigators should focus on mechanisms that occur later in the ischaemic cascade. However, there are also clinical settings where the risk of stroke is increased, such as cardiac surgery, where preventative treatments would be useful. Additionally, it is important to consider how much protection is achieved in pre-clinical trials, a reduction of at least 50% of the ischaemic damage has been the goal, as well as to develop appropriate behavioural tests. There are complex mechanisms occurring simultaneously and sequentially during stroke, so the best outcome may be achieved by a combination of neuroprotective agents that interfere at different points in the ischaemic pathways. The administration of these compounds should be at the appropriate time to prevent the targeted event. Therefore it is important to know the chronology and the relevance of the different mechanisms that are occurring as a consequence of ischaemia and reperfusion.
In vitro models of ischaemia
In vitro models have been utilized for over two decades in the study of cerebral ischaemia [148]. The OGD (oxygen/glucose deprivation) model is believed to most resemble the in vivo ischaemia setting [149,150] although hypoxia alone has also been studied in detail [151–153]. OGD is normally simulated by incubating cells in glucose-free medium and placing them in an anoxic gas chamber [142,149,154] or by infusing nitrogen into the medium [155]. Typical insults last for 30–90 min and are followed immediately by reperfusion, i.e. addition of glucose to the medium and return to normoxic conditions [142,149]. Additionally, acute ischaemic insults (10–20 min) are used for electrophysiological analysis in hippocampal neurons [156,157] as well as spiny/aspiny striatal neurons [158,159]. The discussion below gives a brief overview of work in hippocampal primary cultures or brain slices, unless stated otherwise.
Early evidence with the OGD model suggested that ischaemic injury is mainly necrotic and mediated by glutamate excitotoxicity [160–162]. These reports proposed that apoptosis was not a major cause of neuron loss in ischaemia. However, Gwag et al. [142] showed that apoptosis does occur in OGD in primary cultures, but it is masked by glutamate receptor activation. Additionally, apoptosis in primary cultures is a delayed event (~24–36 h post-insult), whereas glutamate-mediated necrosis was observed as early as 2 h after the insult [149]. However, hippocampal brain slices exhibit different death behaviour with OGD compared with primary cultures. In slices, apoptosis occurs early after the ischaemic episode (~3 h post-insult) [153] and coincides with glutamate-dependent necrotic tissue injury [153,163,164]. Therefore necrosis and apoptosis are not mutually exclusive in organotypic slice cultures. These discrepancies could result from differences in the preparation of the cultures or other procedural artefacts. Alternatively, they could signify the importance of the neuron–glia context in studying ischaemia in vitro.
The role of caspases has been studied in limited detail in OGD [153,164–167]. These studies show that caspase 3 is activated by OGD, and can be regulated by pan-caspase inhibitors. OGD cells treated with NMDA (N-methyl-d-aspartate) receptor antagonists undergo apoptotic death that can be inhibited by a pan-caspase inhibitor, z-VAD-fmk (benzyloxycarbonyl-valyl-alanyl-aspartyl-fluoromethylketone) [153,165]. Another caspase inhibitor, Ac-YVAD-cmk (acetyl-tyrosyl-valyl-alanyl-aspartyl-chloromethylketone) exhibited similar neuroprotection in OGD brain slices [164]. These inhibitors are not specific for individual caspases [36], therefore these studies do not clarify which caspases are important in promoting death in this ischaemia model. Knockout of caspase 3 is also neuroprotective against OGD [168]. It has also been shown that cleaved caspase 7 and caspase 3 are found exclusively in microglia and neurons, and not astrocytes in OGD in mixed primary cultures [166], again suggesting that the neuron–glia interactions are important in modelling ischaemia.
Another cellular model that may be useful in deciphering the molecular mechanisms of ischaemia is the down-regulation of SOD1 (superoxide dismutase 1). In this model, knockdown of SOD1, the cytoplasmic SOD, leads to neuronal death [169,170]. This death is mediated by activation of caspase 1, secretion of IL-1β, and generation of peroxynitrite. These molecules have been shown to be important in the progression of ischaemia in vivo [171–174], and thus this model may provide a cellular model for investigation of the mechanism of neuronal death in ischaemia.
Caspases in stroke
Ischaemic stroke was the first neurodegenerative disease in which activation of a caspase was documented [175]. A considerable amount of research has been done since then that has suggested a very important role of caspases in ischaemic cell death. Although there are some contradictory studies in the literature, caspases have been established as key molecules in the death mechanisms after either permanent or transient ischaemia. Nevertheless, this activation is more consistent following a transient ischaemic injury as a consequence of the reperfusion injury [136,143].
Schielke et al. [176] have shown that mice deficient in the caspase 1 gene exhibited reduced ischaemic damage. Indeed, several lines of evidence point to an important role for this caspase. The pro-inflammatory cytokine IL-1β, which is processed by caspase 1 into its active form, is rapidly increased within the brains of rats subjected to focal/global ischaemic [177]. But more significant is the almost 80% protection in IL-1β-null mice after the ischaemic damage [173]. Thornton et al. [178] have shown that IL-1β neurotoxicity is mediated by glial cells, but only in the presence of caspases and ROS (reactive oxygen species), supporting the importance of considering the interconnection of the neurovascular unit components as a consequence of the ischaemic damage.
Caspase 3 is present in the ischaemic penumbra [179], and its deletion renders mice more resistant to ischaemic injury [168]. However, although caspase activation is generally described in the penumbra of focal infarcts, immunohistochemical analysis in MCAo models has detected neurons containing caspase 3 in the infarct core [180,181]. In humans, although there is limited information available, there is a pro-caspase 3 increase within hours resulting from permanent arterial occlusion [182], and activated caspase 3 and cleaved PARP [poly(ADP-ribose) polymerase] have been detected in some neurons several days after cardiac arrest with reperfusion [136].
Harrison et al. [183] reported a strict transcriptional regulation of caspase expression following pMCAo in rats. There is a pattern of expression with an increase of caspase 1, 3, 6, 7, 8 and 11 mRNA and a decrease of caspase 9 was detected at different time points. Caspase 2 mRNA shows no changes. But in this study, the cellular types expressing these proteases are not examined. An increase in pro-caspase 1, 3, 8 and 9 immunoreactivity in a tMCAo model in adult rats has also been reported [184]. The knowledge of the pathways and molecules that interconnect these caspases is still elusive and more research has to be carried out in this area. Table 4 summarizes the evidence for a role for specific caspases in stroke.
Table 4. Caspases implicated in ischaemia.
Several caspases have been implicated in both pMCAo and tMCAo animal models of stroke and in the cellular model of OGD. The time of occlusion in the pMCAo model or the time of analysis post-reperfusion in the tMCAo model are indicated in parentheses. Measure indicates the method of detecting caspase changes. Ac-IETD-AFC, acetyl-isoleucyl-glutamyl-threonyl-aspartyl-7-amino-4-trifluoromethyl coumarin; Ac-YVAD-AFC, acetyl-tyrosyl-valyl-alanyl-aspartyl-7-amino-4-trifluoromethyl coumarin; IHC, immunohistochemistry; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling.
| Caspase | Paradigm | Measure | Reference |
|---|---|---|---|
| 1 | tMCAo (3 h) | Knockout mice | [176] |
| 1 | tMCAo (3 h) | Dominant-negative caspase 1 | ([200], but see [201]) |
| 1 | pMCAo (30 min, 1 and 12 h) | Ac-YVAD-AFC | [133] |
| 1 | pMCAo (at 24 h) | mRNA levels | [183] |
| 3 | tMCAo (2 h) | Knockout mice | [168] |
| 3 | pMCAo | Western blot, cleaved caspase 3 | [180] |
| 3 | pMCAo (1 and 12 h) | Ac-DEVD-AFC, Western blot, IHC | [133] |
| 3 | pMCAo (at 24 h) | mRNA levels | [183] |
| 3 | OGD | Western blot, cleaved caspase 3 | [166] |
| 6 | pMCAo (only at 6 h) | mRNA levels | [183] |
| 7 | pMCAo (from 6 h) | mRNA levels | [183] |
| 8 | pMCAo (at 24 h) | mRNA levels | [183] |
| 8 | pMCAo (30 min, 1 and 12 h) | Ac-IETD-AFC, Western blot, IHC | [133] |
| 8 | pMCAo | Western blot, cleaved caspase 8 | [180] |
| 9 | tMCAo (1 and 4 h) | IHC, caspase 9 | [184] |
| 9 | tMCAo canine (2 h) | Release of caspase 9 from mitochondria | [202] |
| 11 | pMCAo (from 6 h) | mRNA levels | [183] |
| 11 | pMCAo knockout | Mice (TUNEL-positive cells) | [203] |
Additionally, several in vivo studies have shown that the IAPs, the endogenous caspase inhibitors, are involved in the ischaemic process. In a tMCAo model, there is an increase in XIAP in the core and in the penumbra. Interestingly, the overexpression of this molecule promotes the attenuation of the ischaemic damage by the down-regulation of caspase 3 expression [185]. Indeed, after MCAo, there is an interaction between XIAP and Smac/Diablo [186]. How this occurs has been postulated in a SOD-1-overexpression model, where, as a consequence of the reduction of oxidative stress, there is a down-regulation of caspase 9 expression, as well as of Smac/Diablo and XIAP [187]. Overexpression of NAIP (neuronal apoptosis inhibitor protein) also reduces ischaemic damage in the hippocampus [188]. There is also evidence that ischaemic preconditioning may increase cIAP2 and thus prevent activated caspase 3 from killing cells [189].
Pro-apoptotic Bcl-2 members are implicated in apoptosis in ischaemia/reperfusion models. Overexpression of Bcl-2 with a herpes simplex virus vector protected against tMCAo through the inhibition of caspase activity [190]. This protection was produced in different models of ischaemia, either transient [190–192] or permanent [193]. Bax is translocated from the cytosol to the mitochondria in ischaemic brain [194]. The ‘BH-3 only’ factor Bid has been considered an important component of hypoxia-induced apoptosis [195]. Again, using a model of tMCAo, Bid-null mice show a reduced infarct volume. It is postulated that caspase 8 is responsible for Bid cleavage to truncated Bid (t-Bid), which is then translocated to the mitochondria, activating a death response [196]. It is interesting to address here that almost all of these Bcl-2 member proteins are implicated in ischaemic/reperfusion models, suggesting that the processes where they are implicated are a secondary consequence of reperfusion. Figure 9 illustrates potential mechanisms of ischaemic death.
Figure 9. Proposed pathway of events in cerebral ischaemia.
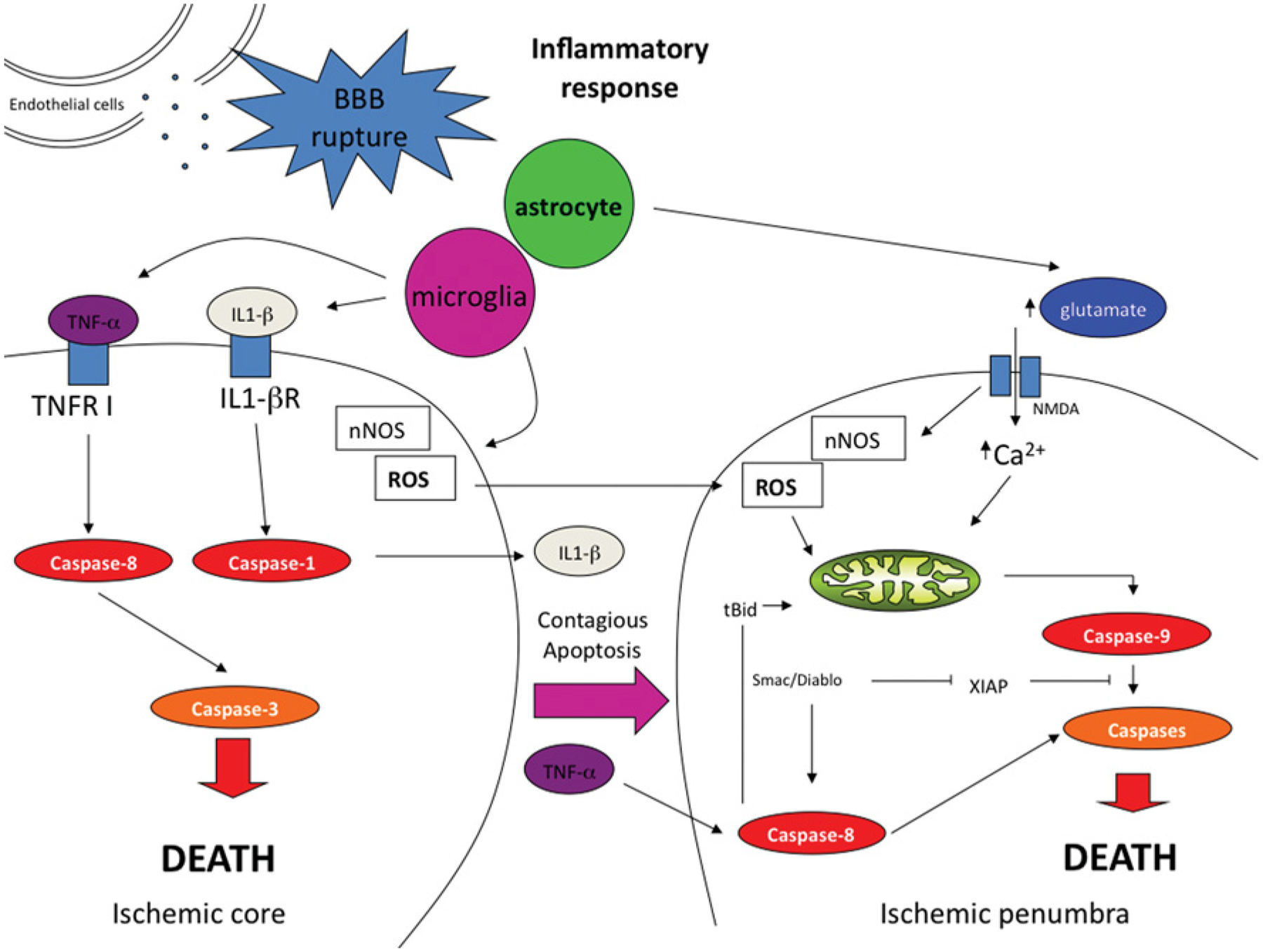
During cerebral ischaemia, a depletion of energy combined with the disruption of the BBB triggers both inflammatory and excitotoxic responses to different degrees in the ischaemic core and the penumbra. Whereas the core has traditionally been considered necrotic, recent data reveal the implication of caspases in this region as well as in the penumbra. The secretion of diffusible factors by damaged cells triggers an apoptotic cascade which affects surrounding healthy cells increasing the overall response to the ischaemic event. IL-1βR, IL-1β receptor; NMDA, N-methyl-d-aspartate; nNOS, neuronal nitric oxide synthase; ROS, reactive oxygen species; TNF-α, tumour necrosis factor α; TNFR I, TNF-α receptor I.
FUTURE DIRECTIONS
Although it is clear that apoptotic death mechanisms play an important role in neuronal death in neurological disease, much still remains to be elucidated about the specific pathways that operate in these diseases. Although many of the previous studies have provided circumstantial evidence for caspase involvement in neuronal diseases, much of the previous work was hampered by the use of measures of caspases that it is now clear did not actually provide data about specific caspases. In order to delineate the death pathways, it is important that appropriate tools and interpretations of data be used. Although this may sound obvious, a quick look at the literature will show how many studies still use the pseudopeptide inhibitors/substrates as evidence for function of specific caspases, and still equate caspase cleavage, as is evident from Western blotting, with activity. The literature will continue to be confusing until appropriate tools for measuring caspase function are used. At present, the best tools are molecular manipulation and affinity ligands. The next issue in the study of human disease is the models available. In the case of cerebral ischaemia, the in vivo models mimic the human disease quite accurately and offer excellent systems for studying death mechanisms. The cellular models of ischaemia, while providing the advantages of cell-based models in the study of biochemical and molecular events, do not offer the complexity of the various elements clearly involved in evolution of the ischaemic event. For AD, the animal models available still do not provide an accurate model of the human disease. Very few of the animal models display neuronal death, thus making the study of death mechanisms in these animals difficult. The animals that do display neuronal death express mutant genes not found in humans with AD, providing concern that signalling mechanisms in these animals may not replicate those found in humans. Thus, at present, the cell models and the hippocampal slice systems offer the best models for studying death mechanisms in AD. Much work is ongoing to produce animal models that more accurately replicate AD, and these are eagerly awaited.
Abbreviations used:
- Aβ
amyloid β-peptide
- AD
Alzheimer’s disease
- ADDL
Aβ-diffusible-derived ligand
- ApoE
apolipoprotein E
- APP
amyloid precursor protein
- BBB
blood–brain barrier
- BH
Bcl-2 homology
- Bim
Bcl-2-interacting mediator of cell death
- b-VAD-fmk
biotin-valyl-alanyl-aspartyl-fluoromethylketone
- cIAP
cellular inhibitor of apoptosis protein
- CTF
C-terminal fragment
- DISC
death-inducing signalling complex
- EOAD
early-onset Alzheimer’s disease
- IAP
inhibitor of apoptosis protein
- BIR
baculovirus IAP repeat
- Diablo
direct IAP-binding protein with low pI
- IL
interleukin
- ICE
IL-1β-cleaving enzyme
- c-FLIP
cellular FLICE [FADD (Fas-associated death domain)-like ICE]-inhibitory protein
- LOAD
late-onset Alzheimer’s disease
- LTP
long-term potentiation
- MCA
middle cerebral artery
- MCAo
MCA occlusion
- NF-κB
nuclear factor κB
- NEMO
NF-κB essential modulator
- NFT
neurofibrillary tangle
- OGD
oxygen/glucose deprivation
- PHF
paired helical filament
- PIDD
p53-inducible protein with a death domain
- pMCAo
permanent MCAo
- PSEN
presenilin
- RAIDD
RIP (receptor-interacting protein)-associated ICH-1 [ICE/CED-3 (cell-death determining 3) homologue 1] protein with a death domain
- RIPK1
receptor-interacting serine/threonine protein kinase 1
- Smac
second mitochondrial-derived activator of caspase
- SOD1
superoxide dismutase 1
- t-Bid
truncated Bid
- tMCAo
temporary MCAo
- XIAP
X-linked inhibitor of apoptosis protein
- XAF1
XIAP-associated factor 1
REFERENCES
- 1.Ribe Garrido E, Heidt L, Beaubier N and Troy CM (2008) Molecular mechanisms of neuronal death. Handb. Neurochem. Mol. Neurobiol, in the press [Google Scholar]
- 2.Miura M, Zhu H, Rotello R, Hartwieg EA and Yuan J (1993) Induction of apoptosis in fibroblasts by IL-1β-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell 75, 653–660 [DOI] [PubMed] [Google Scholar]
- 3.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J et al. (1992) A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature 356, 768–774 [DOI] [PubMed] [Google Scholar]
- 4.Yuan J, Shaham S, Ledoux S, Ellis HM and Horvitz HR (1993) The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1β-converting enzyme. Cell 75, 641–652 [DOI] [PubMed] [Google Scholar]
- 5.Eckhart L, Ban J, Fischer H and Tschachler E (2000) Caspase-14: analysis of gene structure and mRNA expression during keratinocyte differentiation. Biochem. Biophys. Res. Commun 277, 655–659 [DOI] [PubMed] [Google Scholar]
- 6.Fischer H, Koenig U, Eckhart L and Tschachler E (2002) Human caspase 12 has acquired deleterious mutations. Biochem. Biophys. Res. Commun 293, 722–726 [DOI] [PubMed] [Google Scholar]
- 7.Roy S, Sharom JR, Houde C, Loisel TP, Vaillancourt JP, Shao W, Saleh M and Nicholson DW (2008) Confinement of caspase-12 proteolytic activity to autoprocessing. Proc. Natl. Acad. Sci. U.S.A 105, 4133–4138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boatright KM and Salvesen GS (2003) Caspase activation. Biochem. Soc. Symp 70, 233–242 [DOI] [PubMed] [Google Scholar]
- 9.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR and Salvesen GS (2003) A unified model for apical caspase activation. Mol. Cell 11, 529–541 [DOI] [PubMed] [Google Scholar]
- 10.Adams JM and Cory S (2007) The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26, 1324–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baliga BC, Read SH and Kumar S (2004) The biochemical mechanism of caspase-2 activation. Cell Death Differ 11, 1234–1241 [DOI] [PubMed] [Google Scholar]
- 12.Riedl SJ and Salvesen GS (2007) The apoptosome: signalling platform of cell death. Nat. Rev 8, 405–413 [DOI] [PubMed] [Google Scholar]
- 13.Denault JB, Eckelman BP, Shin H, Pop C and Salvesen GS (2007) Caspase 3 attenuates XIAP (X-linked inhibitor of apoptosis protein)-mediated inhibition of caspase 9. Biochem. J 405, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tinel A and Tschopp J (2004) The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 304, 843–846 [DOI] [PubMed] [Google Scholar]
- 15.Ribe E and Troy CM (2007) Activation of caspase-2 in neuronal death: functional implications for Alzheimer’s Disease, Neuroscience 2007 Meeting, San Diego, CA, U.S.A., 3–7 November 2007, Abstract J22 [Google Scholar]
- 16.Berube C, Boucher LM, Ma W, Wakeham A, Salmena L, Hakem R, Yeh WC, Mak TW and Benchimol S (2005) Apoptosis caused by p53-induced protein with death domain (PIDD) depends on the death adapter protein RAIDD. Proc. Natl. Acad. Sci. U.S.A 102, 14314–14320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tinel A, Janssens S, Lippens S, Cuenin S, Logette E, Jaccard B, Quadroni M and Tschopp J (2007) Autoproteolysis of PIDD marks the bifurcation between pro-death caspase-2 and pro-survival NF-κB pathway. EMBO J 26, 197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuenin S, Tinel A, Janssens S and Tschopp J (2008) p53-induced protein with a death domain (PIDD) isoforms differentially activate nuclear factor-κB and caspase-2 in response to genotoxic stress. Oncogene 27, 387–396 [DOI] [PubMed] [Google Scholar]
- 19.Muzio M, Stockwell BR, Stennicke HR, Salvesen GS and Dixit VM (1998) An induced proximity model for caspase-8 activation. J. Biol. Chem 273, 2926–2930 [DOI] [PubMed] [Google Scholar]
- 20.Wang L, Du F and Wang X (2008) TNF-α induces two distinct caspase-8 activation pathways. Cell 133, 693–703 [DOI] [PubMed] [Google Scholar]
- 21.Jia SH, Parodo J, Kapus A, Rotstein OD and Marshall JC (2008) Dynamic regulation of neutrophil survival through tyrosine phosphorylation or dephosphorylation of caspase-8. J. Biol. Chem 283, 5402–5413 [DOI] [PubMed] [Google Scholar]
- 22.Srinivasula SM and Ashwell JD (2008) IAPs: what’s in a name? Mol. Cell 30, 123–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckelman BP, Salvesen GS and Scott FL (2006) Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep 7, 988–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eckelman BP and Salvesen GS (2006) The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J. Biol. Chem 281, 3254–3260 [DOI] [PubMed] [Google Scholar]
- 25.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C et al. (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388, 190–195 [DOI] [PubMed] [Google Scholar]
- 26.Du C, Fang M, Li Y, Li L and Wang X (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102, 33–42 [DOI] [PubMed] [Google Scholar]
- 27.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ and Vaux DL (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102, 43–53 [DOI] [PubMed] [Google Scholar]
- 28.Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, DuBois G, Lazebnik Y, Zervos AS, Fernandes-Alnemri T and Alnemri ES (2002) Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein–caspase interaction. J. Biol. Chem 277, 432–438 [DOI] [PubMed] [Google Scholar]
- 29.Liston P, Fong WG, Kelly NL, Toji S, Miyazaki T, Conte D, Tamai K, Craig CG, McBurney MW and Korneluk RG (2001) Identification of XAF1 as an antagonist of XIAP anti-caspase activity. Nat. Cell Biol 3, 128–133 [DOI] [PubMed] [Google Scholar]
- 30.Arora V, Cheung HH, Plenchette S, Micali OC, Liston P and Korneluk RG (2007) Degradation of survivin by the X-linked inhibitor of apoptosis (XIAP)–XAF1 complex. J. Biol. Chem 282, 26202–26209 [DOI] [PubMed] [Google Scholar]
- 31.Bonzon C, Bouchier-Hayes L, Pagliari LJ, Green DR and Newmeyer DD (2006) Caspase-2-induced apoptosis requires bid cleavage: a physiological role for bid in heat shock-induced death. Mol. Biol. Cell 17, 2150–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang DW, Xing Z, Capacio VL, Peter ME and Yang X (2003) Interdimer processing mechanism of procaspase-8 activation. EMBO J 22, 4132–4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pop C, Fitzgerald P, Green DR and Salvesen GS (2007) Role of proteolysis in caspase-8 activation and stabilization. Biochemistry 46, 4398–4407 [DOI] [PubMed] [Google Scholar]
- 34.Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP et al. (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B: functional relationships established for key mediators of apoptosis. J. Biol. Chem 272, 17907–17911 [DOI] [PubMed] [Google Scholar]
- 35.Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD and Wong WW (1997) Substrate specificities of caspase family proteases. J. Biol. Chem 272, 9677–9682 [DOI] [PubMed] [Google Scholar]
- 36.McStay GP, Salvesen GS and Green DR (2008) Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ 15, 322–331 [DOI] [PubMed] [Google Scholar]
- 37.Tu S, McStay GP, Boucher LM, Mak T, Beere HM and Green DR (2006) In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat. Cell Biol 8, 72–77 [DOI] [PubMed] [Google Scholar]
- 38.Turner RS (2006) Alzheimer’s disease. Semin. Neurol 26, 499–506 [DOI] [PubMed] [Google Scholar]
- 39.Pappolla MA, Chyan YJ, Poeggeler B, Frangione B, Wilson G, Ghiso J and Reiter RJ (2000) An assessment of the antioxidant and the antiamyloidogenic properties of melatonin: implications for Alzheimer’s disease. J. Neural Transm 107, 203–231 [DOI] [PubMed] [Google Scholar]
- 40.Blennow K, de Leon MJ and Zetterberg H (2006) Alzheimer’s disease. Lancet 368, 387–403 [DOI] [PubMed] [Google Scholar]
- 41.Small GW, Rabins PV, Barry PP, Buckholtz NS, DeKosky ST, Ferris SH, Finkel SI, Gwyther LP, Khachaturian ZS, Lebowitz BD et al. (1997) Diagnosis and treatment of Alzheimer disease and related disorders: consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. JAMA, J. Am. Med. Assoc 278, 1363–1371 [PubMed] [Google Scholar]
- 42.Terry RD, Peck A, DeTeresa R, Schechter R and Horoupian DS (1981) Some morphometric aspects of the brain in senile dementia of the Alzheimer type. Ann. Neurol 10, 184–192 [DOI] [PubMed] [Google Scholar]
- 43.Scott SA, DeKosky ST, Sparks DL, Knox CA and Scheff SW (1992) Amygdala cell loss and atrophy in Alzheimer’s disease. Ann. Neurol 32, 555–563 [DOI] [PubMed] [Google Scholar]
- 44.Khachaturian ZS (2000) Toward a comprehensive theory of Alzheimer’s disease: challenges, caveats, and parameters. Ann. N.Y. Acad. Sci 924, 184–193 [DOI] [PubMed] [Google Scholar]
- 45.Khachaturian ZS (2000) Neurobiology of aging: Alzheimer’s clinical review series. Bridging bench to bedside: clinical problems in search of basic solution. Neurobiol. Aging 21, 843. [DOI] [PubMed] [Google Scholar]
- 46.Arnold SE, Hyman BT, Flory J, Damasio AR and Van Hoesen GW (1991) The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb. Cortex 1, 103–116 [DOI] [PubMed] [Google Scholar]
- 47.Braak H and Braak E (1991) Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol 1, 213–216 [DOI] [PubMed] [Google Scholar]
- 48.Braak H and Braak E (1991) Morphological changes in the human cerebral cortex in dementia. J. Hirnforsch 32, 277–282 [PubMed] [Google Scholar]
- 49.Arriagada PV, Marzloff K and Hyman BT (1992) Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology 42, 1681–1688 [DOI] [PubMed] [Google Scholar]
- 50.Arriagada PV, Growdon JH, Hedley-Whyte ET and Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639 [DOI] [PubMed] [Google Scholar]
- 51.Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev 81, 741–766 [DOI] [PubMed] [Google Scholar]
- 52.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L et al. (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706 [DOI] [PubMed] [Google Scholar]
- 53.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K et al. (1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760 [DOI] [PubMed] [Google Scholar]
- 54.Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, Bird TD and Schellenberg GD (1995) A familial Alzheimer’s disease locus on chromosome 1. Science 269, 970–973 [DOI] [PubMed] [Google Scholar]
- 55.Kowalska A, Pruchnik-Wolinska D, Florczak J, Modestowicz R, Szczech J, Kozubski W, Rossa G and Wender M (2004) Genetic study of familial cases of Alzheimer’s disease. Acta Biochim. Pol 51, 245–252 [PubMed] [Google Scholar]
- 56.Kowalska A, Pruchnik-Wolinska D, Florczak J, Szczech J, Kozubski W, Rossa G and Wender M (2004) Presenilin 1 mutations in Polish families with early-onset Alzheimer’s disease. Folia Neuropathol 42, 9–14 [PubMed] [Google Scholar]
- 57.Lusis AJ, Heinzmann C, Sparkes RS, Scott J, Knott TJ, Geller R, Sparkes MC and Mohandas T (1986) Regional mapping of human chromosome 19: organization of genes for plasma lipid transport (APOC1, -C2, and -E and LDLR) and the genes C3, PEPD, and GPI. Proc. Natl. Acad. Sci. U.S.A 83, 3929–3933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL and Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 [DOI] [PubMed] [Google Scholar]
- 59.Ma J, Yee A, Brewer HB Jr, Das S and Potter H (1994) Amyloid-associated proteins α1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature 372, 92–94 [DOI] [PubMed] [Google Scholar]
- 60.Das S and Potter H (1995) Expression of the Alzheimer amyloid-promoting factor antichymotrypsin is induced in human astrocytes by IL-1. Neuron 14, 447–456 [DOI] [PubMed] [Google Scholar]
- 61.Mattila KM, Rinne JO, Roytta M, Laippala P, Pietila T, Kalimo H, Koivula T, Frey H and Lehtimaki T (2000) Dipeptidyl carboxypeptidase 1 (DCP1) and butyrylcholinesterase (BCHE) gene interactions with the apolipoprotein E ε4 allele as risk factors in Alzheimer’s disease and in Parkinson’s disease with coexisting Alzheimer pathology. J. Med. Genet 37, 766–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, Adamson J, Ronald J, Blangero J, Hutton M and Younkin SG (2000) Linkage of plasma Aβ42 to a quantitative locus on chromosome 10 in late-onset Alzheimer’s disease pedigrees. Science 290, 2303–2304 [DOI] [PubMed] [Google Scholar]
- 63.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F et al. (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 373, 523–527 [DOI] [PubMed] [Google Scholar]
- 64.Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D et al. (1995) Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron 15, 1203–1218 [DOI] [PubMed] [Google Scholar]
- 65.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F and Cole G (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274, 99–102 [DOI] [PubMed] [Google Scholar]
- 66.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D et al. (1997) Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A 94, 1550–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA et al. (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. U.S.A 94, 13287–13292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Terai K, Iwai A, Kawabata S, Tasaki Y, Watanabe T, Miyata K and Yamaguchi T (2001) β-Amyloid deposits in transgenic mice expressing human β-amyloid precursor protein have the same characteristics as those in Alzheimer’s disease. Neuroscience 104, 299–310 [DOI] [PubMed] [Google Scholar]
- 69.Callahan MJ, Lipinski WJ, Bian F, Durham RA, Pack A and Walker LC (2001) Augmented senile plaque load in aged female β-amyloid precursor protein-transgenic mice. Am. J. Pathol 158, 1173–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gotz J, Probst A, Spillantini MG, Schafer T, Jakes R, Burki K and Goedert M (1995) Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J 14, 1304–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gotz J, Chen F, Barmettler R and Nitsch RM (2001) Tau filament formation in transgenic mice expressing P301L tau. J. Biol. Chem 276, 529–534 [DOI] [PubMed] [Google Scholar]
- 72.Brion JP, Tremp G and Octave JN (1999) Transgenic expression of the shortest human tau affects its compartmentalization and its phosphorylation as in the pretangle stage of Alzheimer’s disease. Am. J. Pathol 154, 255–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A et al. (1999) Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am. J. Pathol 155, 2153–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Probst A, Gotz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, Hong M, Ishihara T, Lee VM, Trojanowski JQ et al. (2000) Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol 99, 469–481 [DOI] [PubMed] [Google Scholar]
- 75.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Murphy MP, Baker M, Yu X et al. (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet 25, 402–405 [DOI] [PubMed] [Google Scholar]
- 76.Tanemura K, Akagi T, Murayama M, Kikuchi N, Murayama O, Hashikawa T, Yoshiike Y, Park JM, Matsuda K, Nakao S et al. (2001) Formation of filamentous tau aggregations in transgenic mice expressing V337M human tau. Neurobiol. Dis 8, 1036–1045 [DOI] [PubMed] [Google Scholar]
- 77.Tanemura K, Murayama M, Akagi T, Hashikawa T, Tominaga T, Ichikawa M, Yamaguchi H and Takashima A (2002) Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J. Neurosci 22, 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lim F, Hernandez F, Lucas JJ, Gomez-Ramos P, Moran MA and Avila J (2001) FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and Tau filaments in forebrain. Mol. Cell. Neurosci 18, 702–714 [DOI] [PubMed] [Google Scholar]
- 79.Tatebayashi Y, Miyasaka T, Chui DH, Akagi T, Mishima K, Iwasaki K, Fujiwara M, Tanemura K, Murayama M, Ishiguro K et al. (2002) Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc. Natl. Acad. Sci. U.S.A 99, 13896–13901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gotz J, Chen F, van Dorpe J and Nitsch RM (2001) Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Aβ42 fibrils. Science 293, 1491–1495 [DOI] [PubMed] [Google Scholar]
- 81.De Jonghe C, Esselens C, Kumar-Singh S, Craessaerts K, Serneels S, Checler F, Annaert W, Van Broeckhoven C and De Strooper B (2001) Pathogenic APP mutations near the γ-secretase cleavage site differentially affect Aβ secretion and APP C-terminal fragment stability. Hum. Mol. Genet 10, 1665–1671 [DOI] [PubMed] [Google Scholar]
- 82.Golde TE and Janus C (2005) Homing in on intracellular Aβ? Neuron 45, 639–642 [DOI] [PubMed] [Google Scholar]
- 83.Golde TE (2005) The Aβ hypothesis: leading us to rationally-designed therapeutic strategies for the treatment or prevention of Alzheimer disease. Brain Pathol 15, 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kowalska A (2004) Genetic aspects of amyloid β-protein fibrillogenesis in Alzheimer’s disease. Folia Neuropathol 42, 235–237 [PubMed] [Google Scholar]
- 85.Theuns J, Marjaux E, Vandenbulcke M, Van Laere K, Kumar-Singh S, Bormans G, Brouwers N, Van den Broeck M, Vennekens K, Corsmit E et al. (2006) Alzheimer dementia caused by a novel mutation located in the APP C-terminal intracytosolic fragment. Hum. Mutat 27, 888–896 [DOI] [PubMed] [Google Scholar]
- 86.Theuns J, Brouwers N, Engelborghs S, Sleegers K, Bogaerts V, Corsmit E, De Pooter T, van Duijn CM, De Deyn PP and Van Broeckhoven C (2006) Promoter mutations that increase amyloid precursor-protein expression are associated with Alzheimer disease. Am. J. Hum. Genet 78, 936–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Walker ES, Martinez M, Brunkan AL and Goate A (2005) Presenilin 2 familial Alzheimer’s disease mutations result in partial loss of function and dramatic changes in Aβ 42/40 ratios. J. Neurochem 92, 294–301 [DOI] [PubMed] [Google Scholar]
- 88.Duyckaerts C, Potier MC and Delatour B (2008) Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol 115, 5–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL et al. (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A 95, 6448–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA and Trommer BL (2002) Soluble oligomers of β amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res 924, 133–140 [DOI] [PubMed] [Google Scholar]
- 91.Walsh DM, Tseng BP, Rydel RE, Podlisny MB and Selkoe DJ (2000) The oligomerization of amyloid β-protein begins intracellularly in cells derived from human brain. Biochemistry 39, 10831–10839 [DOI] [PubMed] [Google Scholar]
- 92.Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ and Selkoe DJ (2002) Amyloid-β oligomers: their production, toxicity and therapeutic inhibition. Biochem. Soc. Trans 30, 552–557 [DOI] [PubMed] [Google Scholar]
- 93.Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, Bahr M, Schmidt M, Bitner RS, Harlan J et al. (2005) Globular amyloid β-peptide oligomer: a homogenous and stable neuropathological protein in Alzheimer’s disease. J. Neurochem 95, 834–847 [DOI] [PubMed] [Google Scholar]
- 94.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M and Ashe KH (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 [DOI] [PubMed] [Google Scholar]
- 95.Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U and Bruehl C (2008) Amyloid β oligomers (Aβ(1–42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J. Neurosci 28, 788–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McPhie DL, Golde T, Eckman CB, Yager D, Brant JB and Neve RL (2001) β-Secretase cleavage of the amyloid precursor protein mediates neuronal apoptosis caused by familial Alzheimer’s disease mutations. Mol. Brain Res 97, 103–113 [DOI] [PubMed] [Google Scholar]
- 97.Koo EH (2002) The β-amyloid precursor protein (APP) and Alzheimer’s disease: does the tail wag the dog? Traffic 3, 763–770 [DOI] [PubMed] [Google Scholar]
- 98.Zheng H and Koo EH (2006) The amyloid precursor protein: beyond amyloid. Mol. Neurodegener 1, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH and Bredesen DE (2000) A second cytotoxic proteolytic peptide derived from amyloid β-protein precursor. Nat. Med 6, 397–404 [DOI] [PubMed] [Google Scholar]
- 100.Galvan V, Zhang J, Gorostiza OF, Banwait S, Huang W, Ataie M, Tang H and Bredesen DE (2008) Long-term prevention of Alzheimer’s disease-like behavioral deficits in PDAPP mice carrying a mutation in Asp664. Behav. Brain Res 191, 246–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cotman CW and Anderson AJ (1995) A potential role for apoptosis in neurodegeneration and Alzheimer’s disease. Mol. Neurobiol 10, 19–45 [DOI] [PubMed] [Google Scholar]
- 102.Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS et al. (1999) Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-β precursor protein and amyloidogenic Aβ peptide formation. Cell 97, 395–406 [DOI] [PubMed] [Google Scholar]
- 103.Pompl PN, Yemul S, Xiang Z, Ho L, Haroutunian V, Purohit D, Mohs R and Pasinetti GM (2003) Caspase gene expression in the brain as a function of the clinical progression of Alzheimer disease. Arch. Neurol 60, 369–376 [DOI] [PubMed] [Google Scholar]
- 104.LeBlanc A, Liu H, Goodyer C, Bergeron C and Hammond J (1999) Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer’s disease. J. Biol. Chem 274, 23426–23436 [DOI] [PubMed] [Google Scholar]
- 105.Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Bruck W, Jellinger K and Lassmann H (1999) Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease: evidence for apoptotic cell death. Am. J. Pathol 155, 1459–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pellegrini L, Passer BJ, Tabaton M, Ganjei JK and D’Adamio L (1999) Alternative, non-secretase processing of Alzheimer’s β-amyloid precursor protein during apoptosis by caspase-6 and −8. J. Biol. Chem 274, 21011–21016 [DOI] [PubMed] [Google Scholar]
- 107.Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N et al. (2003) Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A 100, 10032–10037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT and Cotman CW (2004) Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J. Clin. Invest 114, 121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K and Shelanski ML (2000) Caspase-2 mediates neuronal cell death induced by β-amyloid. J. Neurosci 20, 1386–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA and Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403, 98–103 [DOI] [PubMed] [Google Scholar]
- 111.Saleh M, Vaillancourt JP, Graham RK, Huyck M, Srinivasula SM, Alnemri ES, Steinberg MH, Nolan V, Baldwin CT, Hotchkiss RS et al. (2004) Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature 429, 75–79 [DOI] [PubMed] [Google Scholar]
- 112.Reed JC (1994) Bcl-2 and the regulation of programmed cell death. J. Cell Biol 124, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP and Wang X (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275, 1129–1132 [DOI] [PubMed] [Google Scholar]
- 114.Kluck RM, Bossy-Wetzel E, Green DR and Newmeyer DD (1997) The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275, 1132–1136 [DOI] [PubMed] [Google Scholar]
- 115.Alberici A, Moratto D, Benussi L, Gasparini L, Ghidoni R, Gatta LB, Finazzi D, Frisoni GB, Trabucchi M, Growdon JH et al. (1999) Presenilin 1 protein directly interacts with Bcl-2. J. Biol. Chem 274, 30764–30769 [DOI] [PubMed] [Google Scholar]
- 116.Shinozaki K, Maruyama K, Kume H, Tomita T, Saido TC, Iwatsubo T and Obata K (1998) The presenilin 2 loop domain interacts with the μ-calpain C-terminal region. Int. J. Mol. Med 1, 797–799 [DOI] [PubMed] [Google Scholar]
- 117.Buxbaum JD, Choi EK, Luo Y, Lilliehook C, Crowley AC, Merriam DE and Wasco W (1998) Calsenilin: a calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat. Med 4, 1177–1181 [DOI] [PubMed] [Google Scholar]
- 118.Keller JN, Guo Q, Holtsberg FW, Bruce-Keller AJ and Mattson MP (1998) Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J. Neurosci 18, 4439–4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Passer BJ, Pellegrini L, Vito P, Ganjei JK and D’Adamio L (1999) Interaction of Alzheimer’s presenilin-1 and presenilin-2 with Bcl-XL: a potential role in modulating the threshold of cell death. J. Biol. Chem 274, 24007–24013 [DOI] [PubMed] [Google Scholar]
- 120.Lu G, Kwong WH, Li Q, Wang X, Feng Z and Yew DT (2005) bcl2, bax, and nestin in the brains of patients with neurodegeneration and those of normal aging. J. Mol. Neurosci 27, 167–174 [DOI] [PubMed] [Google Scholar]
- 121.Rohn TT, Vyas V, Hernandez-Estrada T, Nichol KE, Christie LA and Head E (2008) Lack of pathology in a triple transgenic mouse model of Alzheimer’s disease after overexpression of the anti-apoptotic protein Bcl-2. J. Neurosci 28, 3051–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yao M, Nguyen TV and Pike CJ (2005) β-Amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J. Neurosci 25, 1149–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Karlnoski R, Wilcock D, Dickey C, Ronan V, Gordon MN, Zhang W, Morgan D and Taglialatela G (2007) Up-regulation of Bcl-2 in APP transgenic mice is associated with neuroprotection. Neurobiol. Dis 25, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Biswas SC, Shi Y, Vonsattel JP, Leung CL, Troy CM and Greene LA (2007) Bim is elevated in Alzheimer’s disease neurons and is required for β-amyloid-induced neuronal apoptosis. J. Neurosci 27, 893–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Warlow CP (1998) Epidemiology of stroke. Lancet 352, (Suppl. 3), SIII1–SIII4 [DOI] [PubMed] [Google Scholar]
- 126.del Zoppo GJ (2006) Stroke and neurovascular protection. N. Engl. J. Med 354, 553–555 [DOI] [PubMed] [Google Scholar]
- 127.Lo EH, Dalkara T and Moskowitz MA (2003) Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci 4, 399–415 [DOI] [PubMed] [Google Scholar]
- 128.Garcia JH, Liu KF and Ho KL (1995) Neuronal necrosis after middle cerebral artery occlusion in Wistar rats progresses at different time intervals in the caudoputamen and the cortex. Stroke 26, 636–642 [DOI] [PubMed] [Google Scholar]
- 129.Lipton P (1999) Ischemic cell death in brain neurons. Physiol. Rev 79, 1431–1568 [DOI] [PubMed] [Google Scholar]
- 130.Linnik MD, Zobrist RH and Hatfield MD (1993) Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke 24, 2002–2008 [DOI] [PubMed] [Google Scholar]
- 131.Charriaut-Marlangue C, Margaill I, Represa A, Popovici T, Plotkine M and Ben-Ari Y (1996) Apoptosis and necrosis after reversible focal ischemia: an in situ DNA fragmentation analysis. J. Cereb. Blood Flow Metab 16, 186–194 [DOI] [PubMed] [Google Scholar]
- 132.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J et al. (1995) Mice deficient in IL-1β-converting enzyme are defective in production of mature IL-1β and resistant to endotoxic shock. Cell 80, 401–411 [DOI] [PubMed] [Google Scholar]
- 133.Benchoua A, Guegan C, Couriaud C, Hosseini H, Sampaio N, Morin D and Onteniente B (2001) Specific caspase pathways are activated in the two stages of cerebral infarction. J. Neurosci 21, 7127–7134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kerr JF, Wyllie AH and Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mattson MP (2000) Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol 1, 120–129 [DOI] [PubMed] [Google Scholar]
- 136.Love S (2003) Apoptosis and brain ischaemia. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 267–282 [DOI] [PubMed] [Google Scholar]
- 137.Ferrer I (2006) Apoptosis: future targets for neuroprotective strategies. Cerebrovasc. Dis 21, (Suppl. 2), 9–20 [DOI] [PubMed] [Google Scholar]
- 138.Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE and Portera-Cailliau C (1998) Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res. Bull 46, 281–309 [DOI] [PubMed] [Google Scholar]
- 139.Leist M and Jaattela M (2001) Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol 2, 589–598 [DOI] [PubMed] [Google Scholar]
- 140.Zeng YS and Xu ZC (2000) Co-existence of necrosis and apoptosis in rat hippocampus following transient forebrain ischemia. Neurosci. Res 37, 113–125 [DOI] [PubMed] [Google Scholar]
- 141.Choi DW (1996) Ischemia-induced neuronal apoptosis. Curr. Opin. Neurobiol 6, 667–672 [DOI] [PubMed] [Google Scholar]
- 142.Gwag BJ, Lobner D, Koh JY, Wie MB and Choi DW (1995) Blockade of glutamate receptors unmasks neuronal apoptosis after oxygen-glucose deprivation in vitro. Neuroscience 68, 615–619 [DOI] [PubMed] [Google Scholar]
- 143.Pan J, Konstas AA, Bateman B, Ortolano GA and Pile-Spellman J (2007) Reperfusion injury following cerebral ischemia: pathophysiology, MR imaging, and potential therapies. Neuroradiology 49, 93–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R, Neumann-Haefelin T and Brandes RP (2007) NADPH oxidase plays a central role in blood–brain barrier damage in experimental stroke. Stroke 38, 3000–3006 [DOI] [PubMed] [Google Scholar]
- 145.Traystman RJ (2003) Animal models of focal and global cerebral ischemia. ILAR J 44, 85–95 [DOI] [PubMed] [Google Scholar]
- 146.Richard Green A, Odergren T and Ashwood T (2003) Animal models of stroke: do they have value for discovering neuroprotective agents? Trends Pharmacol. Sci 24, 402–408 [DOI] [PubMed] [Google Scholar]
- 147.Dronne MA, Grenier E, Chapuisat G, Hommel M and Boissel JP (2008) A modelling approach to explore some hypotheses of the failure of neuroprotective trials in ischemic stroke patients. Prog. Biophys. Mol. Biol 97, 28–39 [DOI] [PubMed] [Google Scholar]
- 148.Rothman SM (1983) Synaptic activity mediates death of hypoxic neurons. Science 220, 536–537 [DOI] [PubMed] [Google Scholar]
- 149.Goldberg MP and Choi DW (1993) Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J. Neurosci 13, 3510–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Noraberg J, Poulsen FR, Blaabjerg M, Kristensen BW, Bonde C, Montero M, Meyer M, Gramsbergen JB and Zimmer J (2005) Organotypic hippocampal slice cultures for studies of brain damage, neuroprotection and neurorepair. Curr. Drug Targets CNS Neurol. Disord 4, 435–452 [DOI] [PubMed] [Google Scholar]
- 151.Goldberg MP, Monyer H, Weiss JH and Choi DW (1988) Adenosine reduces cortical neuronal injury induced by oxygen or glucose deprivation in vitro. Neurosci. Lett 89, 323–327 [DOI] [PubMed] [Google Scholar]
- 152.Goldberg MP, Weiss JH, Pham PC and Choi DW (1987) N-methyl-d-aspartate receptors mediate hypoxic neuronal injury in cortical culture. J. Pharmacol. Exp. Ther 243, 784–791 [PubMed] [Google Scholar]
- 153.Moroni F, Meli E, Peruginelli F, Chiarugi A, Cozzi A, Picca R, Romagnoli P, Pellicciari R and Pellegrini-Giampietro DE (2001) Poly(ADP-ribose) polymerase inhibitors attenuate necrotic but not apoptotic neuronal death in experimental models of cerebral ischemia. Cell Death Differ 8, 921–932 [DOI] [PubMed] [Google Scholar]
- 154.Laake JH, Haug FM, Wieloch T and Ottersen OP (1999) A simple in vitro model of ischemia based on hippocampal slice cultures and propidium iodide fluorescence. Brain Res. Protoc 4, 173–184 [DOI] [PubMed] [Google Scholar]
- 155.Frantseva MV, Carlen PL and El-Beheiry H (1999) A submersion method to induce hypoxic damage in organotypic hippocampal cultures. J. Neurosci. Methods 89, 25–31 [DOI] [PubMed] [Google Scholar]
- 156.Schurr A, Payne RS, Miller JJ and Rigor BM (1999) Study of cerebral energy metabolism using the rat hippocampal slice preparation. Methods 18, 117–126 [DOI] [PubMed] [Google Scholar]
- 157.Schurr A, Tseng MT, West CA and Rigor BM (1987) Taurine improves the recovery of neuronal function following cerebral hypoxia: an in vitro study. Life Sci 40, 2059–2066 [DOI] [PubMed] [Google Scholar]
- 158.Calabresi P, Centonze D and Bernardi G (2000) Cellular factors controlling neuronal vulnerability in the brain: a lesson from the striatum. Neurology 55, 1249–1255 [DOI] [PubMed] [Google Scholar]
- 159.Pedersen JZ, Bernardi G, Centonze D, Pisani A, Rossi L, Rotilio G and Calabresi P (1998) Hypoglycemia, hypoxia, and ischemia in a corticostriatal slice preparation: electrophysiologic changes and ascorbyl radical formation. J. Cereb. Blood Flow Metab 18, 868–875 [DOI] [PubMed] [Google Scholar]
- 160.Benveniste H, Drejer J, Schousboe A and Diemer NH (1984) Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J. Neurochem 43, 1369–1374 [DOI] [PubMed] [Google Scholar]
- 161.Choi DW and Rothman SM (1990) The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci 13, 171–182 [DOI] [PubMed] [Google Scholar]
- 162.Simon RP, Swan JH, Griffiths T and Meldrum BS (1984) Blockade of N-methyl-d-aspartate receptors may protect against ischemic damage in the brain. Science 226, 850–852 [DOI] [PubMed] [Google Scholar]
- 163.Meli E, Pangallo M, Baronti R, Chiarugi A, Cozzi A, Pellegrini-Giampietro DE and Moroni F (2003) Poly(ADP-ribose) polymerase as a key player in excitotoxicity and post-ischemic brain damage. Toxicol. Lett 139, 153–162 [DOI] [PubMed] [Google Scholar]
- 164.Ray AM, Owen DE, Evans ML, Davis JB and Benham CD (2000) Caspase inhibitors are functionally neuroprotective against oxygen glucose deprivation induced CA1 death in rat organotypic hippocampal slices. Brain Res 867, 62–69 [DOI] [PubMed] [Google Scholar]
- 165.Gottron FJ, Ying HS and Choi DW (1997) Caspase inhibition selectively reduces the apoptotic component of oxygen-glucose deprivation-induced cortical neuronal cell death. Mol. Cell. Neurosci 9, 159–169 [DOI] [PubMed] [Google Scholar]
- 166.Malagelada C, Xifro X, Minano A, Sabria J and Rodriguez-Alvarez J (2005) Contribution of caspase-mediated apoptosis to the cell death caused by oxygen-glucose deprivation in cortical cell cultures. Neurobiol. Dis 20, 27–37 [DOI] [PubMed] [Google Scholar]
- 167.Taranukhin AG, Taranukhina EY, Saransaari P, Djatchkova IM, Pelto-Huikko M and Oja SS (2008) Taurine reduces caspase-8 and caspase-9 expression induced by ischemia in the mouse hypothalamic nuclei. Amino Acids 34, 169–174 [DOI] [PubMed] [Google Scholar]
- 168.Le DA, Wu Y, Huang Z, Matsushita K, Plesnila N, Augustinack JC, Hyman BT, Yuan J, Kuida K, Flavell RA and Moskowitz MA (2002) Caspase activation and neuroprotection in caspase-3- deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc. Natl. Acad. Sci. U.S.A 99, 15188–15193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Troy CM, Derossi D, Prochiantz A, Greene LA and Shelanski ML (1996) Downregulation of Cu/Zn superoxide dismutase leads to cell death via the nitric oxide-peroxynitrite pathway. J. Neurosci 16, 253–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Davidson TJ, Harel S, Arboleda VA, Prunell GF, Shelanski ML, Greene LA and Troy CM (2004) Highly efficient small interfering RNA delivery to primary mammalian neurons induces microRNA-like effects before mRNA degradation. J. Neurosci 24, 10040–10046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fujimura M, Morita-Fujimura Y, Noshita N, Sugawara T, Kawase M and Chan PH (2000) The cytosolic antioxidant copper/zinc-superoxide dismutase prevents the early release of mitochondrial cytochrome c in ischemic brain after transient focal cerebral ischemia in mice. J. Neurosci 20, 2817–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kondo T, Reaume AG, Huang TT, Carlson E, Murakami K, Chen SF, Hoffman EK, Scott RW, Epstein CJ and Chan PH (1997) Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J. Neurosci 17, 4180–4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y and Rothwell NJ (2001) Role of IL-1α and IL-1β in ischemic brain damage. J. Neurosci 21, 5528–5534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rothwell N (2003) Interleukin-1 and neuronal injury: mechanisms, modification, and therapeutic potential. Brain Behav. Immun 17, 152–157 [DOI] [PubMed] [Google Scholar]
- 175.Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, MacDonald G, Fishman MC, Greenberg AH, Moskowitz MA and Yuan J (1997) Expression of a dominant negative mutant of interleukin-1β converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J. Exp. Med 185, 933–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Schielke GP, Yang GY, Shivers BD and Betz AL (1998) Reduced ischemic brain injury in interleukin-1β converting enzyme-deficient mice. J. Cereb. Blood Flow Metab 18, 180–185 [DOI] [PubMed] [Google Scholar]
- 177.Allan SM, Tyrrell PJ and Rothwell NJ (2005) Interleukin-1 and neuronal injury. Nat. Rev. Immunol 5, 629–640 [DOI] [PubMed] [Google Scholar]
- 178.Thornton P, Pinteaux E, Gibson RM, Allan SM and Rothwell NJ (2006) Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. J. Neurochem 98, 258–266 [DOI] [PubMed] [Google Scholar]
- 179.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J and Moskowitz MA (1998) Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J. Neurosci 18, 3659–3668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Velier JJ, Ellison JA, Kikly KK, Spera PA, Barone FC and Feuerstein GZ (1999) Caspase-8 and caspase-3 are expressed by different populations of cortical neurons undergoing delayed cell death after focal stroke in the rat. J. Neurosci 19, 5932–5941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Guegan C and Sola B (2000) Early and sequential recruitment of apoptotic effectors after focal permanent ischemia in mice. Brain Res 856, 93–100 [DOI] [PubMed] [Google Scholar]
- 182.Love S, Barber R, Srinivasan A and Wilcock GK (2000) Activation of caspase-3 in permanent and transient brain ischaemia in man. NeuroReport 11, 2495–2499 [DOI] [PubMed] [Google Scholar]
- 183.Harrison DC, Davis RP, Bond BC, Campbell CA, James MF, Parsons AA and Philpott KL (2001) Caspase mRNA expression in a rat model of focal cerebral ischemia. Mol. Brain Res 89, 133–146 [DOI] [PubMed] [Google Scholar]
- 184.Ferrer I and Planas AM (2003) Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J. Neuropathol. Exp. Neurol 62, 329–339 [DOI] [PubMed] [Google Scholar]
- 185.Trapp T, Korhonen L, Besselmann M, Martinez R, Mercer EA and Lindholm D (2003) Transgenic mice overexpressing XIAP in neurons show better outcome after transient cerebral ischemia. Mol. Cell. Neurosci 23, 302–313 [DOI] [PubMed] [Google Scholar]
- 186.Saito A, Hayashi T, Okuno S, Ferrand-Drake M and Chan PH (2003) Interaction between XIAP and Smac/DIABLO in the mouse brain after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab 23, 1010–1019 [DOI] [PubMed] [Google Scholar]
- 187.Saito A, Hayashi T, Okuno S, Nishi T and Chan PH (2004) Oxidative stress is associated with XIAP and Smac/DIABLO signaling pathways in mouse brains after transient focal cerebral ischemia. Stroke 35, 1443–1448 [DOI] [PubMed] [Google Scholar]
- 188.Xu D, Bureau Y, McIntyre DC, Nicholson DW, Liston P, Zhu Y, Fong WG, Crocker SJ, Korneluk RG and Robertson GS (1999) Attenuation of ischemia-induced cellular and behavioral deficits by X chromosome-linked inhibitor of apoptosis protein overexpression in the rat hippocampus. J. Neurosci 19, 5026–5033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tanaka H, Yokota H, Jover T, Cappuccio I, Calderone A, Simionescu M, Bennett MV and Zukin RS (2004) Ischemic preconditioning: neuronal survival in the face of caspase-3 activation. J. Neurosci 24, 2750–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhao H, Yenari MA, Sapolsky RM and Steinberg GK (2004) Mild postischemic hypothermia prolongs the time window for gene therapy by inhibiting cytochrome c release. Stroke 35, 572–577 [DOI] [PubMed] [Google Scholar]
- 191.Antonawich FJ, Federoff HJ and Davis JN (1999) BCL-2 transduction, using a herpes simplex virus amplicon, protects hippocampal neurons from transient global ischemia. Exp. Neurol 156, 130–137 [DOI] [PubMed] [Google Scholar]
- 192.Zhao H, Yenari MA, Cheng D, Sapolsky RM and Steinberg GK (2003) Bcl-2 overexpression protects against neuron loss within the ischemic margin following experimental stroke and inhibits cytochrome c translocation and caspase-3 activity. J. Neurochem 85, 1026–1036 [DOI] [PubMed] [Google Scholar]
- 193.Linnik MD, Zahos P, Geschwind MD and Federoff HJ (1995) Expression of bcl-2 from a defective herpes simplex virus-1 vector limits neuronal death in focal cerebral ischemia. Stroke 26, 1670–1674 [DOI] [PubMed] [Google Scholar]
- 194.Cao G, Minami M, Pei W, Yan C, Chen D, O’Horo C, Graham SH and Chen J (2001) Intracellular Bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J. Cereb. Blood Flow Metab 21, 321–333 [DOI] [PubMed] [Google Scholar]
- 195.Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Qiu J, Chiarugi A, Thomas SS, Kohane DS, Korsmeyer SJ and Moskowitz MA (2001) BID mediates neuronal cell death after oxygen/glucose deprivation and focal cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A 98, 15318–15323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yin XM, Luo Y, Cao G, Bai L, Pei W, Kuharsky DK and Chen J (2002) Bid-mediated mitochondrial pathway is critical to ischemic neuronal apoptosis and focal cerebral ischemia. J. Biol. Chem 277, 42074–42081 [DOI] [PubMed] [Google Scholar]
- 197.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y and LaFerla FM (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron 39, 409–421 [DOI] [PubMed] [Google Scholar]
- 198.Ribe EM, Perez M, Puig B, Gich I, Lim F, Cuadrado M, Sesma T, Catena S, Sanchez B, Nieto M et al. (2005) Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol. Dis 20, 814–822 [DOI] [PubMed] [Google Scholar]
- 199.Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y and Tohyama M (2004) Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J. Cell Biol 165, 347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Friedlander RM, Brown RH, Gagliardini V, Wang J and Yuan J (1997) Inhibition of ICE slows ALS in mice. Nature 388, 31. [DOI] [PubMed] [Google Scholar]
- 201.Erratum (1998) Nature 392, 560. [DOI] [PubMed] [Google Scholar]
- 202.Krajewski S, Krajewska M, Ellerby LM, Welsh K, Xie Z, Deveraux QL, Salvesen GS, Bredesen DE, Rosenthal RE, Fiskum G and Reed JC (1999) Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A 96, 5752–5757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kang SJ, Wang S, Hara H, Peterson EP, Namura S, Amin-Hanjani S, Huang Z, Srinivasan A, Tomaselli KJ, Thornberry NA et al. (2000) Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J. Cell Biol 149, 613–622 [DOI] [PMC free article] [PubMed] [Google Scholar]