

Abstract
BACKGROUND
In a randomized, placebo controlled clinical trial, bamlanivimab, a SARS-CoV-2 neutralizing monoclonal antibody, given in combination with remdesivir, did not improve outcomes among hospitalized COVID-19 patients based on an early futility assessment.
OBJECTIVE
Evaluate the a priori hypothesis that there is greater benefit of bamlanivimab in patients without detectable endogenous neutralizing antibody levels at study entry compared to those with antibodies, especially if viral levels are high.
DESIGN
Randomized, placebo-controlled trial.
SETTING
Multicenter trial.
PATIENTS
Hospitalized COVID-19 patients without end organ failure.
INTERVENTION
Bamlanivimab (7000mg) or placebo.
MEASUREMENTS
Antibody, antigen and viral RNA levels were centrally measured on stored specimens collected at baseline. Patients were followed for 90 days for sustained recovery (defined as discharged home for 14 consecutive days) and a composite safety outcome (death, serious adverse events, organ failure or serious infections).
RESULTS
Among 314 participants (163 on bamlanivimab and 151 on placebo), the median time to sustained recovery was 19 days and did not differ between the bamlanivimab and placebo groups, sub-hazard ratio (sHR) =0.99 (95% CI: 0.79–1.22) (sHR> 1 favors bamlanivimab). At entry, 50% evidenced production of anti-spike neutralizing antibodies (nAbs); 50% had SARS-CoV-2 nucleocapsid plasma antigen levels ≥ 1,000 ng/L. Among those without and with nAbs at study entry, the sHRs were 1.24 (95% CI: 0.90–1.70) and 0.74 (95% CI: 0.54–1.00), respectively (nominal p=0.018 for interaction). The sHR was also >1 for those with plasma antigen or nasal viral RNA levels above (versus below) median level at entry and was greatest for those without antibodies and with elevated antigen or viral RNA levels: 1.48 (95% CI: 0.99–2.23), and 1.89 (1.23, 2.91), respectively. Hazard ratios for the composite safety outcome (< 1 favors bamlanivimab) also differed by serostatus at entry; 0.67 (0.37–1.20) for those without and 1.79 (0.92–3.48) for those with nAbs.
LIMITATIONS
Subgroup analysis of a trial prematurely stopped because of futility. Small sample size. Multiple subgroups analyzed.
CONCLUSIONS
Efficacy and safety of bamlanivimab may differ depending on whether an endogenous neutralizing antibody response has been mounted or not. The limited sample size of the study does not allow firm conclusions based on these findings and further independent trials are required assessing other types of passive immune therapies in the same patient setting.
REGISTRATION
ClinicalTrials.gov number, NCT04501978.
PRIMARY FUNDING SOURCE
US Government Operation Warp Speed and National Institute of Allergy and Infectious Diseases.
Introduction
SARS-CoV-2 neutralizing monoclonal antibodies reduce hospitalization risk among outpatients with early COVID-19 and appear to accelerate viral load decline in the nasopharynx (1–6). The US Food and Drug Administration has issued emergency use authorization for several such products (7–10).
Preliminary results of the Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV)-3 (11) (called: Therapeutics for Inpatients With COVID-19 (TICO) (12)) trial of bamlanivimab in hospitalized patients with COVID-19 were reported after enrollment was terminated because futility guidelines were met (13). While other neutralizing monoclonal products, convalescent plasma, and hyperimmune immunoglobulin products have also not provided overall clinical benefit for inpatients (14–16), a preprint from the RECOVERY trial found clinical benefit in seronegative hospitalized patients given casirivimab/imdevimab versus standard of care alone (17).
We report here the final results of the ACTIV-3/TICO bamlanivimab trial and an a priori defined subgroup analysis that addresses the hypothesis that patients without endogenous neutralizing antibodies at entry would benefit more from bamlanivimab than those with antibodies, and that benefit would be greatest in nAb-negative patients with high plasma antigen levels or with high nasal viral RNA levels. This subgroup hypothesis was tested using biological material collected prospectively as part of the clinical trial and analyzed at the conclusion of the trial.
Methods
Design and Treatments
As previously described, this randomized placebo controlled study compared bamlanivimab (7000mg) to placebo, administered as a single intravenous infusion over a 1-hour period on the day of randomization (12, 13).
The infusion was prepared by an unblinded pharmacist. All other site personnel, and study participants, were blinded to treatment assignment.
Patients
We enrolled adult hospitalized patients with documented SARS-CoV-2 infection without invasive mechanical ventilation who had symptoms attributable to COVID-19 for ≤ 12 days. Detailed information regarding eligibility criteria, exclusions, human subjects protections, and written consent have been reported previously (13) and are provided in the supplementary appendix. All patients received study supplied remdesivir except if contraindications existed.
Outcomes
Two ordinal outcomes termed “pulmonary” and “pulmonary-plus” assessed at day 5 following infusion were used to assess futility after at least 300 patients. These outcomes are defined in the supplementary appendix; results for the pulmonary outcome are shown in Figure S1 (13).
The primary endpoint for agents studied in TICO is time to sustained recovery, where “sustained recovery” is achieved when a participant was discharged to home and remaining at home for at least 14 days for the first time, assessed over the first 90 days of follow-up. The shortest possible time from randomization to sustained recovery is 14 days.
Other outcomes included composite safety outcomes assessed through days 5, 28, and 90, and mortality. A composite safety outcome assessed through Day 90 included death, serious adverse events, end organ disease, and serious infections. An expanded composite outcome, assessed through Day 28, also included grade 3 and 4 adverse events (see supplementary appendix for further details).
Serologic and Virologic Assays on Stored Samples
Details concerning the a priori hypotheses that were developed regarding antibody and viral levels and details of laboratory methods are provided in the supplementary appendix. Briefly, SARS-CoV-2 viral RNA levels were measured from a mid-turbinate nasal swab. Next generation sequencing was performed using an IlluminaHiSeq2000 machine. Sequences were aligned to SARS-CoV-2 reference (Genbank) and assigned Nextstrain clades and PANGO lineages. Plasma samples collected at study entry and at days 1, 3 and 5 were used to measure anti-spike receptor binding domain neutralizing antibodies (nAbs) in a surrogate viral neutralization test (SVNT), as well as anti-nucleocapsid (anti-N) binding antibody levels. Qualitative plasma SARS-CoV-2 N antigen was measured using a microbead-based immunoassay. Stored samples were used to measure plasma levels of interleukin-6 (IL-6) and D-dimer and serum levels of C-reactive protein (CRP).
Statistical Analyses
The analyses in this report are based on the same population described in our preliminary report (13). This modified intention-to-treat analysis population was restricted to randomized patients who received any volume of bamlanivimab or placebo (Figure S2).
Antibody levels and status (percent seronegative or seropositive) were summarized for nAbs and for anti-N antibodies at baseline and at days 1, 3, and 5; nAb seropositive was defined as percent binding inhibition ≥30%, and anti-N Ab seropositive as specimen ratio ≥1. Data for each antibody assay are summarized for the overall cohort and are reported for two groups, seronegative and seropositive at the time of study entry. Antigen levels are described as the percentage negative, < 3 ng/L (the lower level of quantification), at each follow-up timepoint. Treatment differences in the percentage negative at days 1, 3, and 5 were estimated with 95% confidence intervals (CIs). Additional analyses describing antibody and antigen levels are described in the supplemental appendix.
Time to sustained recovery was analyzed using methods that account for the competing risk of death; correspondingly, the cumulative incidence of “sustained recovery” through day 90 was estimated using the Aalen-Johansen method (18) and sub-hazard ratios (sHR) comparing bamlanivimab versus placebo were estimated with 95% CIs using Fine-Gray models, stratified by study site pharmacy (19, 20). To address the stated a priori hypotheses, the heterogeneity of sHRs for sustained recovery across subgroups defined by antibody status (negative versus positive for nAb at entry) and dichotomized by median levels of viral replication markers (plasma antigen (< 1000, the approximate median, versus ≥ 1000 ng/L) or viral RNA (<10,000, the approximate median, versus ≥ 10,000 copies/mL)), and combinations of these subgroups were assessed by including interaction terms between treatment and subgroup indicators in expanded Fine-Gray models. These subgroup analyses are described further in the supplemental appendix.
Subgroups defined by nAb status were also examined for the day 90 composite safety outcome and for all-cause mortality. Proportional hazards regression models were used to estimate hazard ratios (HRs) and 95% CIs. Additional subgroups are summarized in the supplemental appendix.
Statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC) and R version 4.0 (R Foundation for Statistical Computing) (21). Nominal 2-sided p-values, which are given for selected comparisons, need to be interpreted with caution since the trial was stopped for futility and several subgroup analyses are presented. The statistical analysis plan, including the supplemental plan developed for the measurements made on specimens, are included as appendices.
Role of Funder
The Division for Clinical Research at the National Institute of Allergy and Infectious Diseases funded this project. All analyses of biological material were done in a blinded manner at laboratories affiliated with the funder; data were sent to the statistical and data management center at the University of Minnesota for linkage to the trial database. Several representatives from the funder are part of the writing group for the manuscript.
Results:
Study Participants
Of the 326 patients randomized between August 5 and October 13, 2020, 314 were infused and are included in the analysis cohort (Figure S2); 163 received bamlanivimab and 151 received placebo. All but 2 patients, one in each treatment group, were infused on the day of randomization; two-thirds of patients were randomized on the day of hospital admission or one day following admission.
Characteristics at study entry, overall and by nAb status at entry are shown in Table 1. At enrollment, 50% were seropositive for endogenous nAbs, and 59% had antibodies against nucleocapsid (anti-N Abs). At enrollment, 95% had plasma antigen levels ≥ 3 ng/L, and 50% had antigen levels ≥ 1,000 ng/L.
Table 1:
Baseline Characteristics by nAb Status
| Overall (n=314) | nAb Negative (n=153) | nAb Positive (n=152) | ||
|---|---|---|---|---|
|
|
||||
| Age | Median (IQR) – yr | 61 (49, 71) | 65 (50, 73) | 58 (47, 68) |
| Female sex | No. (%) | 137 (44%) | 69 (45%) | 64 (42%) |
| Pre-COVID-19 residence: | No. (%) | |||
| independent/community dwelling | 299 (95%) | 148 (97%) | 143 (94%) | |
| Race/ethnicity | No. (%) | |||
| White | 147 (47%) | 76 (50%) | 67 (44%) | |
| Hispanic | 74 (24%) | 29 (19%) | 43 (28%) | |
| Black | 67 (21%) | 35 (23%) | 29 (19%) | |
| Other | 26 (8%) | 13 (8%) | 13 (9%) | |
| Body mass index; | No. (%) | |||
| ≥ 30 | 164 (52%) | 78 (51%) | 82 (54%) | |
| ≥ 40 | 42 (13%) | 21 (14%) | 18 (12%) | |
| History of: | No. (%) | |||
| Hypertension requiring medication | 158 (50%) | 83 (54%) | 70 (46%) | |
| Diabetes requiring medication | 90 (29%) | 43 (28%) | 44 (29%) | |
| Renal impairment | 33 (11%) | 22 (14%) | 9 (6%) | |
| Asthma and/or COPD | 45 (14%) | 23 (15%) | 21 (14%) | |
| At least one co-morbidity | 217 (69%) | 109 (71%) | 101 (66%) | |
| Days since symptom onset | Median (IQR) | 7 (5, 9) | 7 (4, 8) | 8 (5, 10) |
| Current use of: | No. (%) | |||
| Remdesivir | 126 (40%) | 57 (37%) | 66 (43%) | |
| Antibacterial | 93 (30%) | 35 (23%) | 55 (36%) | |
| Glucocorticoid | 159 (51%) | 71 (46%) | 85 (56%) | |
| Antiplatelets/anticoagulation | 207 (66%) | 92 (60%) | 109 (72%) | |
| ACE inhibitor or ARB | 72 (23%) | 41 (27%) | 31 (20%) | |
| NSAID | 33 (11%) | 13 (8%) | 16 (11%) | |
| Oxygen Requirement | No. (%) | |||
| Not receiving supplemental oxygen | 86 (27%) | 54 (35%) | 30 (20%) | |
| Supplemental oxygen < 4 L/min | 116 (37%) | 52 (34%) | 61 (40%) | |
| Supplemental oxygen ≥ 4 L/min | 64 (20%) | 28 (18%) | 36 (24%) | |
| Non-invasive ventilation or HFNC | 48 (15%) | 19 (12%) | 25 (16%) | |
| Invasive ventilation or ECMO | 0 (0%) | 0 (0%) | 0 (0%) | |
| Laboratory assessments | ||||
| Plasma nucleocapside antigen | Median (IQR) – ng/L | 1037 (141, 3578) | 2130 (796, 5060) | 297 (36.9, 1924) |
| ≥ 3 ng/L (positive) | No. (%) | 290 (95%) | 152 (99%) | 138 (91%) |
| Nasal swab fluid viral RNA | No. (%) positive | 243 (80%) | 137 (90%) | 106 (71%) |
| Viral load if RNA positive | Mean±SD log10 copies/mL | 4.53 ± 1.61 | 4.82 ± 1.65 | 4.15 ± 1.48 |
| nAb | No. (%) positive | 152 (50%) | 0 (0%) | 152 (100%) |
| Anti-N antibodies | No. (%) positive | 180 (59%) | 49 (32%) | 131 (86%) |
| Interleukin-6* | Median (IQR) – ng/L | 6.7 (2.7, 15.0) | 7.7 (3.1, 14.8) | 5.7 (2.4, 15.2) |
| D-dimer* | Median (IQR) – mg/L | 0.90 (0.63, 1.38) | 0.84 (0.57, 1.37) | 0.98 (0.71, 1.39) |
| C-reative protein* | Median (IQR) – mg/L | 46 (25, 83) | 40 (20, 70) | 54 (29, 89) |
| B-Lymphocytes† | Median (IQR) – 109 | 0.80 (0.55, 1.12) | 0.77 (0.54, 1.05) | 0.83 (0.56, 1.21) |
Upper limit of normal for Interleukin-6 (1.8 ng/L), D-dimer (0.5 mg/L) and CRP (10 mg/L).
Lower limit of normal for B-lymphocytes is 1×109/L.
Among 303 participants with available nasal swab material at entry, viral RNA was detected in 243 (80%); median (IQR) RNA viral level among those RNA positives was 30,513 (1,296, 355,325) copies/mL. The rank correlation between viral RNA levels and antigen levels was 0.14.
Among those who were nAb positive, median (IQR) plasma antigen and viral RNA levels were 297 (37, 1,924) ng/L and 14,164 (649, 131,794) copies/mL respectively; for those nAb negative these levels were 2,130 (796, 5,060) ng/L and 70,953 (2,880, 850,202) copies/mL.
Viral RNA sequences from 255 participants (average depth: 4393; 204 with 75% or more genome coverage; Figure S3A and S3B) identified no concerning mutations in codons 417, 452 or 484 in the spike protein; virus from six participants had deletions in codon 69–70. All genomes contained the D614G mutation, and one person had strain B.1.1.7 (i.e. alpha variant). No patient was infected with the delta variant.
Baseline characteristics by treatment group, overall and by nAb status, are given in Tables S1–S3.
Antibody and Antigen Changes through 5 Days of Follow-up Analysed by nAb Status at Entry
Overall, nAb levels increased following the infusion of bamlanivimab (Figure S4A). As anticipated, nearly all (97%) seronegative patients given the neutralizing monoclonal antibody bamlanivimab were nAb seropositive on day 1 post infusion, compared with 32% on placebo (p<0.001). Subsequently, the placebo group progressively seroconverted (Figure S4B). Among those who were nAb seropositive at entry, little difference between treatment groups was observed (Figure S4C).
Overall, the percentage with plasma antigen levels < 3 ng/L at day 5 comprised 44% and 43% of the bamlanivimab and placebo groups, respectively (p=0.95); the percentage with levels < 1000 ng/L at day 5 was 98% for both the bamlanivimab and placebo groups (Figure S4D and Table S4). Among those who were nAb seronegative at study entry, fewer patients achieved antigen levels < 3 ng/L at day 5 in both the bamlanivimab and placebo groups(Figure S4E; 26% and 27%, respectively) compared to those who were nAb positive (Figure S4F; 57% and 58%, respectively).
Additional summaries of nAb titers, antigen levels, and anti-N titers are given in the supplemental appendix (Figures S5–S8).
Sustained Recovery
Sustained recovery by day 90 was achieved by 144 patients (88%) in the bamlanivimab group and 136 patients (90%) in the placebo group; sHR=0.99 (95% CI:0.79–1.22; p=0.89) (Figures 1A and 1B); median days to sustained recovery was 19 days, and 80% in each treatment group achieved sustained recovery by day 28.
Figure 1:
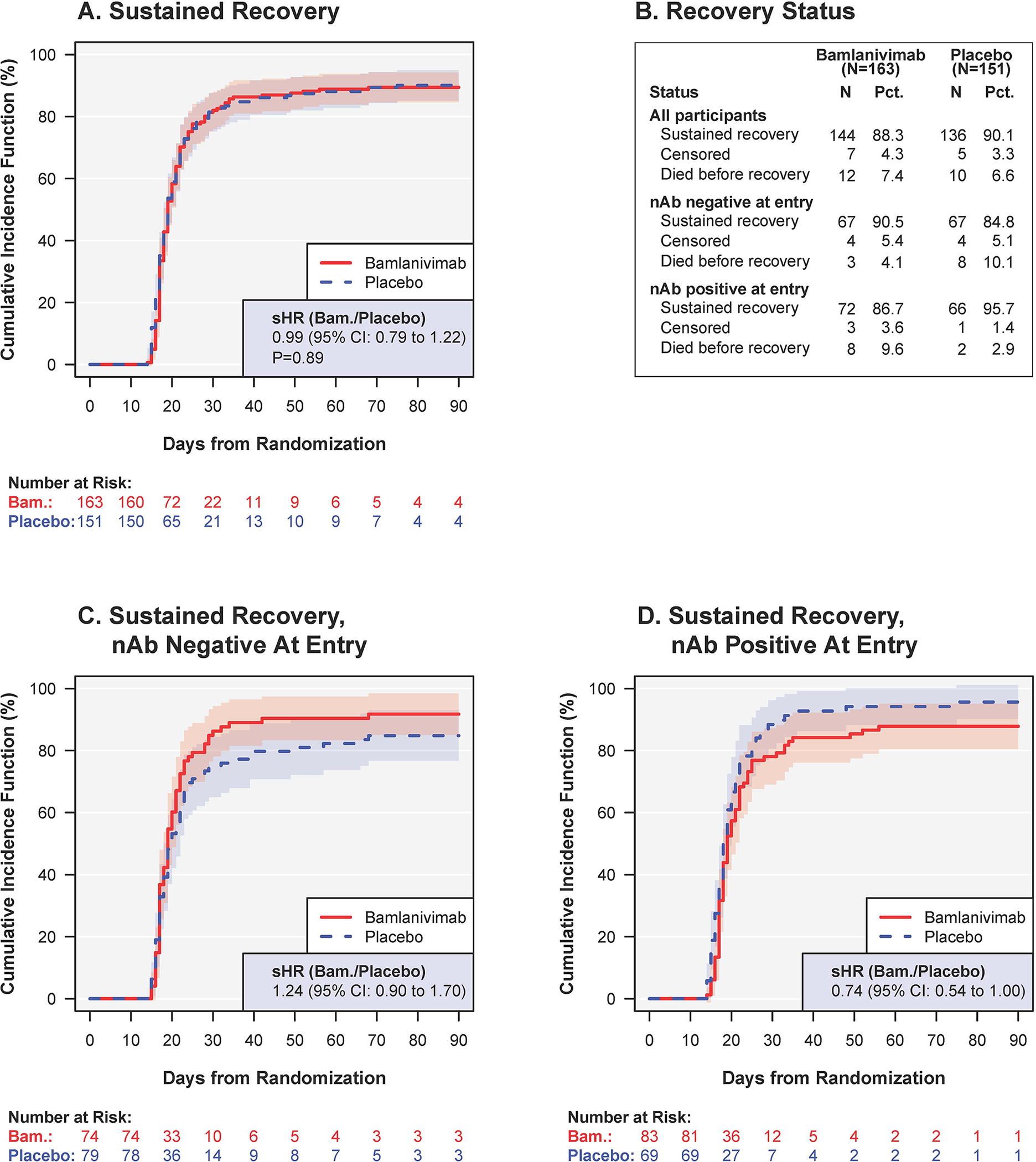
Sustained recovery for bamlanivimab versus placebo: overall (A), and according to neutralizing antibody (nAb) status at entry: (C) nAb negative and (D) nAb positive.* (B) summarizes the recovery status at day 90 for cohorts displayed in (A), (C) and (D).
* The sub-hazard ratios in (C) were nominally different from those in (D) (p=0.018 for difference).
Abbreviations: nAB= neutralising antibody status from a surrogate viral neutralization test; sHR= sub-hazard ratio for time to sustained recovery (also called “recovery rate ratio”).
Among patients who were nAb negative and positive at entry, the sHRs comparing bamlanivimab versus placebo were 1.24 (95% CI: 0.90–1.70) and 0.74 (95% CI: 0.54–1.00), respectively (nominal p=0.018 for interaction) (Figures 1B, 1C and Figure 2). Among nAb negative patients, 91% in the bamlanivimab group achieved sustained recovery by day 90, compared with 85% for the placebo group; these percentages were 87% and 96%, respectively, for those who were nAb positive.
Figure 2.
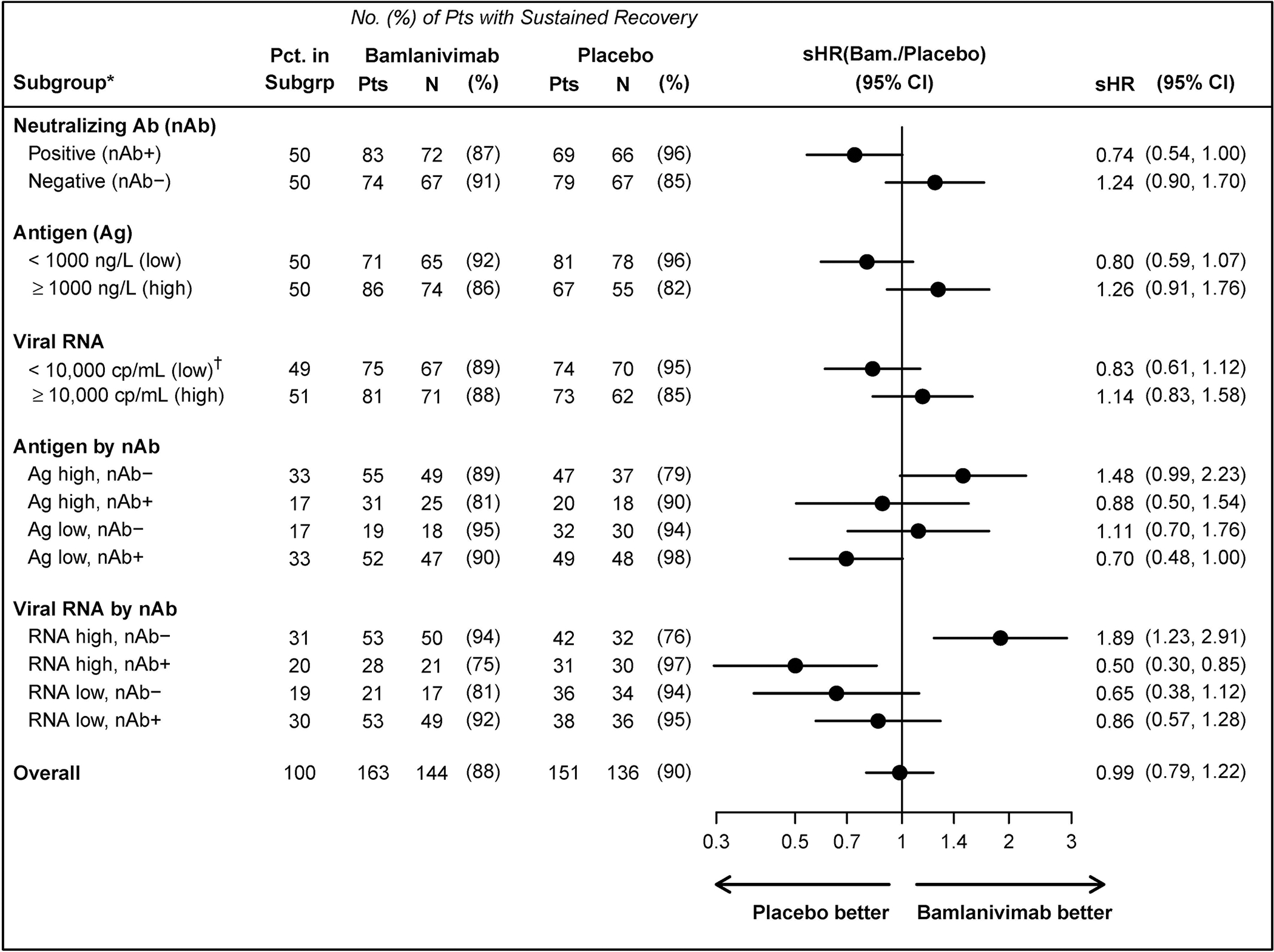
Sustained recovery according to subgroups at study entry: nAb status and levels of viral measures (plasma antigen and nasal viral RNA)+.
* Nominal P-values for differences in the treatment effect across subgroups (interactions between the subgroup indicator and treatment group indicator) are as follows: subgroups by nAb status, P=0.018; antigen level by nAb status, P=0.038; viral RNA level by nAb status, P<0.001.
† low viral RNA = viral RNA level < 10,000 cp/mL, negative, or indeterminate
+ See Appendix Figures S9–S11 for other subgroupings.
Abbreviations: Ag = plasma nucleocapside antigen levels at study entry; nAb = neutralising antibody status from a surrogate viral neutralization test at study entry; sHR=sub-hazard ratio for time to sustained recovery (also called “recovery rate ratio”); viral RNA = quantification of viral copies in fluid from nasal swab at study entry.
We next compared sustained recovery in subgroups according to median entry levels of plasma antigen and viral RNA, and then, further subdividing these subgroups, according to nAb status at entry. In both treatment groups higher antigen and viral RNA levels were associated with lower percentages who achieved recovery. As hypothesized, among those who were nAb negative, the difference between bamlanivimab and placebo was more evident if plasma antigen or nasal-swab viral RNA were above the median entry levels (sHRs=1.48; 95% CI: 0.99–2.23 and 1.89; 95% CI: 1.23–2.91, respectively) (Figure 2).
Additional subgroups for sustained recovery and for the day 5 pulmonary ordinal outcome are summarized in Figures S9–S12.
Safety Outcomes
Safety outcomes, overall and by nAb status at entry, are described below and summarized in Figure 3, Tables S5–S7.
Figure 3.
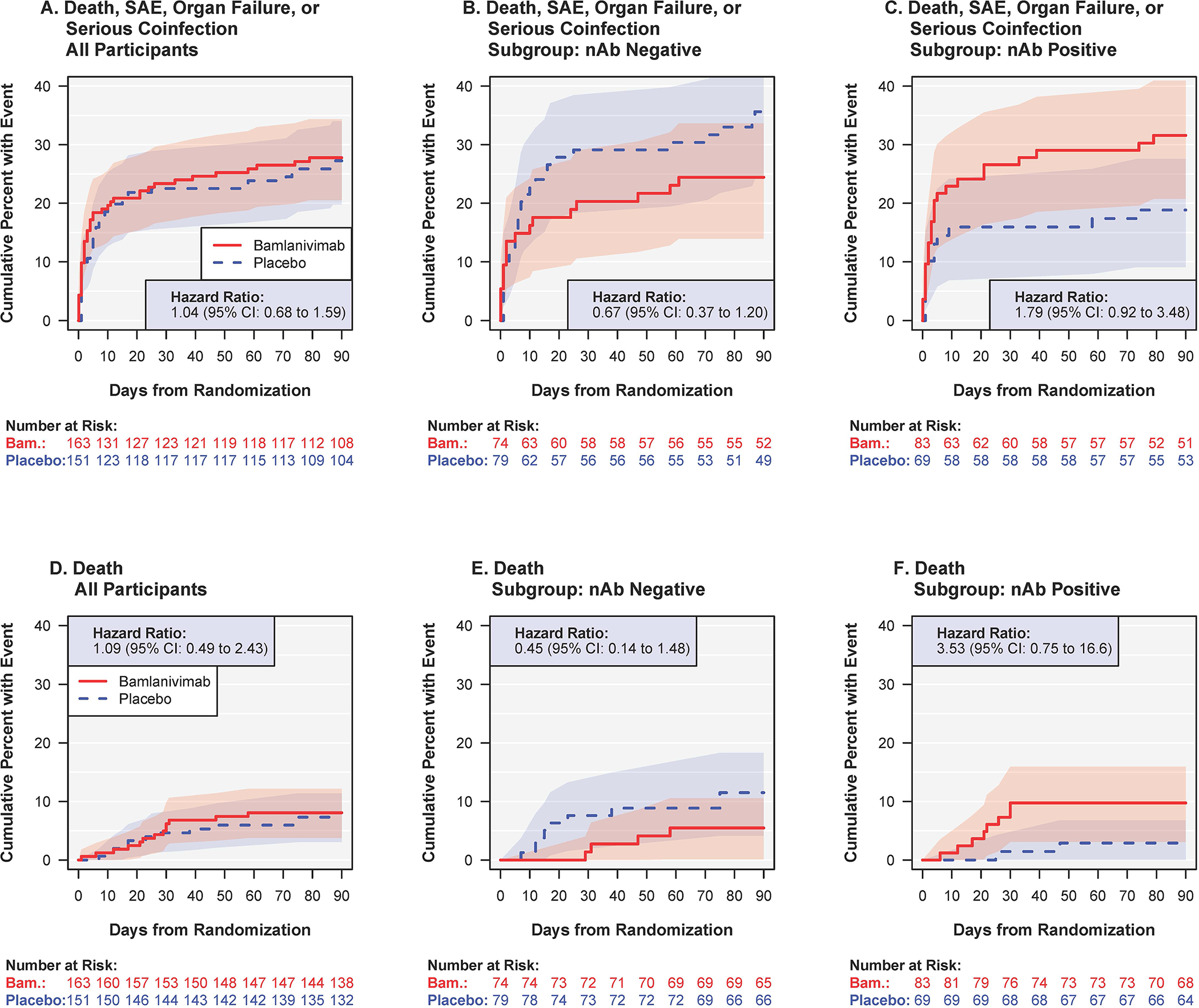
A composite safety outcome (death, serious adverse event, organ failure or serious infection) (top row, A-C) and death (bottom row, D-F) for bamlanivimab versus placebo: overall (left column, A, D), and according to neutralizing antibody (nAb) status at entry: (middle column, B, E) negative and (right column C, F) positive.
The hazard ratios in (B) were different from those in (C) (nomial p=0.03 for difference), as were the those in (E) versus (F) (nominal p=0.04). See table S7 for overview of all predefined safety outcomes.
Abbreviations: nAb=neutralising antibody status from a surrogate viral neutralization test at study entry; SAE=serious adverse event.
By day 5, the expanded composite outcome of mortality, organ failure, serious infections, serious adverse events, or grade 3 or 4 adverse events occurred in 45 patients (28%) in the bamlanivimab group and 28 (19%) patients in the placebo group (OR=1.83 (95% CI: 1.01–3.29); p=0.05). By day 28, 52 patients (32%) in the bamlanivimab group and 42 patients (28%) in the placebo group had developed this composite outcome (p=0.35) (Table S5).
The composite outcome of mortality, organ failure, serious infections, or serious adverse events through day 90 occurred in 45 (28%) patients in the bamlanivimab group and in 41 patients (27%) in the placebo group (p=0.83) (Figure 3A and Table S5). The most frequent component of the composite at day 90 was organ failure (68 events) (Table S7); respiratory failure accounted for 41 of the 68 organ failure events. Through day 90, 13 deaths (8%) occurred in the bamlanivimab group and 11 deaths (7%) occurred in the placebo group (hazard ratio = 1.09; 95% CI: 0.49–2.43) (Table S5 and Figure 3D). Twenty-two of the 24 patients who died did so before sustained recovery (see Figure 1B).
For the day 90 composite safety outcome, the HRs (bamlanivimab versus placebo) for those who were nAb negative and positive at entry, were 0.67 (95% CI: 0.37–1.20) (Figure 3B) and 1.79 (95% CI: 0.92–3.48) (Figure 3C), respectively (see also Table S6).
For death, HRs for bamlanivimab versus placebo were 0.45 (95% CI: 0.14–1.48) for nAb negative patients (Figure 3E) and 3.53 (95% CI: 0.75–16.6) for those who were nAb positive (Figure 3F). Treatment differences followed similar patterns with respect to nAb status at entry for each of the components of the day 90 composite safety outcome (Table S7).
Inflammatory and Coagulation Markers through 5 Days of Follow-up
Average log2 transformed levels of IL-6 (Figure S13A–S13C) and CRP (Figure S13D–S13F) declined in both treatment groups from entry through day 5, overall and for subgroups defined by nAb status at entry. The decline in D-dimer for both treatment groups appeared more modest than for IL-6 and CRP (Figure S14A–S14C). Overall and for each subgroup, there was no evidence for a difference between treatment groups with respect to any of those markers at days 1, 3, or 5.
Discussion
In support of a pre-specified hypothesis, we found that treatment benefit and harm from bamlanivimab compared to placebo differed according to the presence of neutralizing antibodies at study entry, with possible benefit in seronegative patients with high plasma antigen or high nasal viral RNA levels. Additionally, administration of 7 grams of bamlanivimab may cause harm in patients already having mounted an endogenous antibody response. We note that while these findings are based on a specific a priori hypothesis, several other subgroup analyses have been conducted. This, along with the limited sample size of the study, limits our ability to draw firm conclusions. Further studies on passive immune therapies in the same patient setting are required.
Overall, we found that the cumulative incidence of sustained recovery was comparable for those randomized to bamlanivimab versus placebo. This contrasts with the benefit observed from using various neutralizing monoclonal antibody preparations in early COVID-19 (7–10, 22, 23). Before reviewing unblinded laboratory results, we considered whether this null-effect could be due to enrolling patients who had already started to mount an effective neutralizing antibody response, compared to patients earlier in the course of their infection, and that infusion of an additional exogenous neutralizing antibody could be redundant in hospitalized patients.
In support of such an explanation, we found a positive trend for recovery for the bamlanivimab arm among the subgroup of patients (50% of total) without nAbs at study entry and a trend in the opposite direction for those with nAbs. A similar pattern was observed across subgroups according to total nucleocapsid antigen antibodies.
Consistent with these observations, a recent preprint reported preliminary data comparing open label use of two neutralizing monoclonal antibodies (casirivimab and imdevimab) versus usual care in inpatients from the RECOVERY study (17). The antibody treatment reduced the risk of death among hospitalized patients who were seronegative for anti-SARS-CoV-2 antibodies at study entry, but not overall or for those who were seropositive at entry (17). Those results and ours support using serological screening at hospital admission to identify the subgroup with a possible clinical benefit.
In the RECOVERY trial, remdesivir was not standard-of-care; it was in our trial. Here, most participants had COVID-19 pneumonia, but none had pulmonary failure requiring mechanical ventilation. Remdesivir showed clinical benefit in the population not requiring invasive ventilation, in an earlier randomized, placebo-controlled trial (24). Of note, 28-day mortality in the usual care arm of RECOVERY was 21%; in our trial 90-day mortality in the placebo group was 7%. Regardless, given the two trials’ consistent findings by antibody status, possible benefit from neutralizing monoclonal antibodies in those without a current neutralizing antibody response may be expected, whether or not combined with remdesivir.
Neutralizing monoclonal antibodies function as antiviral agents. Identification of patients in whom SARS-CoV-2 is replicating and advancing the disease course should be the goal—this is the population where benefit seems likely. We assessed two viral replication markers, plasma antigen and nasal-swab viral RNA levels. Higher plasma antigen and viral RNA levels at entry were associated with lower chance of achieving sustained recovery. Higher levels of either or both viral replication markers at entry were associated with reduced chance of sustained recovery from the disease; this confirms findings from several smaller studies assessing viral markers and outcomes (25–28). Higher levels of either viral marker, among patients not yet having mounted endogenous neutralizing antibodies at entry may identify a subgroup for which there is clinical utility of passive immune therapy.
The correlation between plasma antigen and nasal viral RNA levels at entry was minimal. This suggests that viral replication in the nasal cavity and elsewhere in the body is partly uncoupled.
Plasma antigen levels decreased rapidly over time and in 40% of the population were cleared after 5 days of follow-up; the clearance rate was comparable for the two treatment groups, providing some reassurance that bamlanivimab does not lead to enhanced viral replication. Additionally, as some patients experienced progression of their COVID-19 disease post day 5, despite the rapid lowering of viral replication, other deleterious immunopathology such as hyperinflammation may be at play.
The neutralising effect of bamlanivimab in vitro is markedly reduced by certain mutations (including those encoding residues K417N/T, L452R and E484K) in the spike protein (29, 30). However, these viral variants are not present in our patient cohort. The cohort was recruited between August and October 2020, before escape variants with these signature mutations were widely observed. Deep sequencing of 255 viral isolates retrieved at study entry failed to identify any of these mutations. None of the participants were infected with the delta variant of the virus.
Infusion of high concentrations of neutralizing antibodies, as was done in this trial, may have led to undesired and harmful reactions in some patients, which may have nullified our ability to find a beneficial effect of the intervention here. We cannot verify this possibility, but among participants with endogenous neutralizing antibody production, and hence a low chance of benefit from infused neutralizing antibodies, the treatment differences in sustained recovery, in the composite safety outcome of organ failure, serious adverse events, serious infections or death, and in mortality all favored placebo. This information requires independent confirmation in larger cohorts. A mortality trend, albeit not statistically significant, favoring standard-of-care among seropositive patients was also seen in the RECOVERY trial (17). Hence, infused neutralizing antibodies may cause harm in hospitalized patients who have already mounted an endogenous antibody response. There is an urgent need for point of care antibody tests that reliably assess serostatus and identify patients at risk of being harmed. Further research is needed into appropriate management of patients that experience breakthrough infections after vaccination where antibody tests may be positive due to the vaccine.
Trajectories of three host biomarkers, IL-6, CRP and D-dimer, though the first 5 days of follow-up, did not differ substantially according to randomized treatment. However, the limited sample size does not allow us to exclude an adverse inflammatory effect from bamlanivimab overall and, more specifically, in those patients with endogenous antibody production. Additional studies of these and other biomarkers in larger cohorts assessing passive immunotherapy, is underway.
The TICO platform will proceed with clinical evaluation of additional COVID-19 treatments. Two other neutralizing antibody agents, VIR-7831 and Brii Bio 196/198, failed TICO’s futility assessment (31). A fourth neutralizing antibody product (AZD7442) (32), genetically modified to remove binding to FcγR and complement proteins (which none of the other previously tested products were), is currently under late stage evaluation. Those cohorts are being characterized for antibody and antigen status at entry in order to permit similar analyses to those reported here.
Supplementary Material
Support:
The trial was primarily funded by Operation Warp Speed, with the support of NIAID, NIH Grant U01-AI136780, and the Division of Clinical Research and Leidos Biomedical Research, Inc., Contract HHSN261200800001E, for the International Network for Strategic Initiatives in Global HIV Trials (INSIGHT) Network, and NHLBI and the Research Triangle Institute for the PETAL and CTSN Networks (NIH Agreement 10T2HL156812-01). Other funding and support were provided by the U.S. Departments of Veterans Affairs, and the governments of Denmark (National Research Foundation; grant no 126), Australia (National Health and Medical Research Council), and U.K. (Medical Research Council, MRC_UU_12023/23). Study medications were donated by Gilead Sciences, and Eli Lilly.
The members of the writing group (Jens D. Lundgren, M.D., D.M.Sc. [protocol chair, Copenhagen International Network for Strategic Initiatives in Global HIV Trials (INSIGHT) International Coordinating Center (ICC) lead], Birgit Grund, Ph.D. [lead unblinded statistician], Christina E. Barkauskas, M.D., Thomas L. Holland, M.D., Robert L. Gottlieb, M.D., Ph.D., Uriel Sandkovsky, M.D., M.Sc., Samuel M. Brown, M.D. M.Sc., Kirk U. Knowlton, M.D., Wesley H. Self, M.D., M.P.H., D. Clark Files, M.D., Mamta K. Jain, M.D., M.P.H., Thomas Benfield, M.D., D.M.Sc., Michael E. Bowdish, M.D., M.Sc., Bradley G. Leshnower, M.D., Jason V. Baker, M.D., M.Sc., Jens-Ulrik Jensen, M.D., Ph.D., Edward M. Gardner, M.D., Adit A. Ginde, M.D., M.P.H., Estelle S. Harris, M.D., Isik S. Johansen, M.D., D.M.Sc., Norman Markowitz, M.D., Michael A. Matthay, M.D., Lars Østergaard, M.D., Ph.D., D.M.Sc., Christina C. Chang, M.D., Ph.D., Anna L. Goodman, F.R.C.P., D.Phil., Weizhong Chang, Ph.D., Robin L. Dewar, Ph.D., Norman P. Gerry, Ph.D., Elizabeth S. Higgs, M.D., Helene Highbarger, M.S., Daniel D. Murray, Ph.D., Thomas A. Murray, Ph.D., Ven Natarajan, Ph.D., Roger Paredes, M.D., Ph.D., Mahesh K.B. Parmar, Ph.D., Andrew N. Phillips, Ph.D., Cavan Reilly, Ph.D., Adam W. Rupert, B.S., MT(ASCP), Shweta Sharma, M.S., Kathryn Shaw-Saliba, Ph.D., Brad T. Sherman, M.S., Marc Teitelbaum, M.D., M.S., Deborah Wentworth, M.P.H., Huyen Cao, M.D., Paul Klekotka, M.D., Ph.D., Abdel G. Babiker, Ph.D. [London INSIGHT ICC lead], Victoria J. Davey, Ph.D., M.P.H. [VA ICC lead], Annetine C. Gelijns, Ph.D. [CTSN ICC lead], Virginia L. Kan, M.D. [Washington INSIGHT ICC lead], Mark N. Polizzotto, M.D., Ph.D. [Sydney INSIGHT ICC lead], B. Taylor Thompson, M.D. [PETAL ICC lead], H. Clifford Lane, M.D. [DCR, NIAID lead], and James D. Neaton, Ph.D. [INSIGHT PI]) of the ACTIV-3/TICO Study Group assume responsibility for the overall content and integrity of this article. The affiliations of the members of the writing group are as follows:
CHIP Centre of Excellence for Health, Immunity, and Infections, Department of Infectious Diseases, Rigshospitalet, Copenhagen, Denmark (J.D.L., J.-U.J. and D.D.M.), Division of Biostatistics, School of Public Health, University of Minnesota, Minneapolis (B.G., T.A.M, C.R., S.S., D.W., J.D.N.), Division of Pulmonary, Allergy, and Critical Care Medicine (C.E.B.) and Division of Infectious Diseases (T.L.H.), Department of Medicine, Duke University, Durham, Baylor University Medical Center, Dallas (R.L.G., U.S.), Intermountain Medical Center, Murray (S.M.B.), University of Utah, Salt Lake City (S.M.B., E.S.H., K.U.K.), Department of Emergency Medicine, Vanderbilt University Medical Center, Nashville (W.H.S), Department of Internal Medicine, Section on Pulmonary, Critical Care, Allergy, and Immunology, Wake Forest School of Medicine, Winston-Salem (D.C.F.), UT Southwestern Medical Center, Dallas (M.K.J), Department of Infectious Diseases, Copenhagen University Hospital, Amager and Hvidovre, Denmark (T.B), Department of Surgery, Keck School of Medicine, University of Southern California, Los Angeles (M.E.B.), Division of Cardiothoracic Surgery Emory University School of Medicine, Atlanta (B.G.L.), Hennepin Healthcare Research Institute, Minneapolis and University of Minnesota (J.V.B.), Department of Internal Medicine, Respiratory Medicine Section, Herlev and Gentofte Hospital, University of Copenhagen, Hellerup, Denmark and Department of Infectious Diseases, Rigshospitalet, Copenhagen, Denmark (J.-U.J.), Denver Public Health, Denver Health and Hospital Authority, Denver (E.M.G.), Department of Emergency Medicine, University of Colorado School of Medicine, Aurora (A.A.G.) Department of Infectious Diseases, Odense University Hospital, Odense (I.S.J.), Department of Infectious Diseases, Henry Ford Hospital, Detroit (N.M.), Department of Medicine, Department of Anesthesia, The University of California, San Francisco (M.A.M.), Aarhus University Hospital, Skejby (L.Ø.), The Kirby Institute, University of New South Wales, Sydney (C.C.C., M.N.P.), U.S. Department of Veterans Affairs, Washington, D.C. (V.J.D.), Medical Research Council Clinical Trials Unit at UCL, University College London (A.L.G., A.G.B., M.K.B.P.), Guy’s & St. Thomas’ NHS Foundation Trust, London (A.G.), National Institute of Allergy and Infectious Diseases, Bethesda (E.S.H., K.S.S., H.C.L.), Infectious Diseases Department & irsiCaixa AIDS Research Institute, Hospital Universitari Germans Trias i Pujol, Catalonia (R.P.), Institute for Global Health, University College London (A.N.P.), Leidos Biomedical Research, Inc., Frederick (R.L.D., M.T., H.H.), Advanced Biomedical Laboratories, LLC., Cinnaminson (N.P.G.), Laboratory of Human Retrovirology and Immunoinformatics (W.C. and B.T.S.), Laboratory of Molecular Cell Biology (V.N.) and AIDS Monitoring Laboratory (A.W.R, H.H.), all Frederick National Laboratory for Cancer Research, Frederick, Gilead Sciences, Foster City (H.C.), Eli Lilly and Company, Indianapolis (P.K.), Department of Population Health Science and Policy, Icahn School of Medicine at Mount Sinai, New York (A.C.G.), Veteran Affairs Medical Center, Washington, D.C. (V.L.K.), George Washington University School of Medicine and Health Sciences, Washington (V.L.K.), St Vincent’s Hospital, Sydney (M.N.P.), Division of Pulmonary and Critical Care, Department of Medicine, Massachusetts General Hospital, Boston (B.T.T.), Harvard Medical School Boston (B.T.T.)
Footnotes
A complete list of members in the ACTIV-3/TICO Bamlanivimab Study Group is provided in the Supplementary Appendix.
Disclaimer:
This is the prepublication, author-produced version of a manuscript accepted for publication in Annals of Internal Medicine. This version does not include post-acceptance editing and formatting. The American College of Physicians, the publisher of Annals of Internal Medicine, is not responsible for the content or presentation of the author-produced accepted version of the manuscript or any version that a third party derives from it. Readers who wish to access the definitive published version of this manuscript and any ancillary material related to this manuscript (e.g., correspondence, corrections, editorials, linked articles) should go to Annals.org or to the print issue in which the Annals of Internal Medicine © 2022 American College of Physicians Document Publication Date: 04/19/2022 PAGE 18 article appears. Those who cite this manuscript should cite the published version, as it is the official version of record.
Preprint:
A preprint of this manuscript was released on 22nd July 2021 to the medRxiv server: doi: https://doi.org/10.1101/2021.07.19.21260559.
References
- 1.Gottlieb RL, Nirula A, Chen P, Boscia J, Heller B, Morris J, et al. Effect of Bamlanivimab as Monotherapy or in Combination With Etesevimab on Viral Load in Patients With Mild to Moderate COVID-19: A Randomized Clinical Trial. Jama. 2021;325(7):632–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gupta A, Gonzalez-Rojas Y, Juarez E, Casal MC, Moya J, Falci DR, et al. Early Covid-19 Treatment With SARS-CoV-2 Neutralizing Antibody Sotrovimab. medRxiv. 2021:2021.05.27.21257096. [Google Scholar]
- 3.Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with Covid-19. N Engl J Med. 2021;384(3):238–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen MS, Nirula A, Mulligan MJ, Novak RM, Marovich M, Yen C, et al. Effect of Bamlanivimab vs Placebo on Incidence of COVID-19 Among Residents and Staff of Skilled Nursing and Assisted Living Facilities: A Randomized Clinical Trial. Jama. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Brien MP, Forleo Neto E, Chen KC, Isa F, Heirman I, Sarkar N, et al. Casirivimab with Imdevimab antibody cocktail for COVID-19 prevention: interim results. CROI 2021. [Google Scholar]
- 6.Dougan M, Nirula A, Gottlieb RL, Azizad M, Mocherla B, Chen P, et al. Bamlanivimab+Etesevimab for treatment of COVI-19 in high-risk ambulatory patients. CROI 2021. [Google Scholar]
- 7.U.S. Food & Drug Administration. Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibody for Treatment of COVID-19, Nov 09, 2020. Online, accessed on Oct 20, 2021 at https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibody-treatment-covid-19. [Google Scholar]
- 8.U.S. Food & Drug Administration. Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19, Nov 21, 2020. Online, accessed on Oct 20, 2021 at https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19. [Google Scholar]
- 9.U.S. Food & Drug Administration. Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19, Feb 09, 2021. Online, accessed on Oct 20, 2021 at https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19-0. [Google Scholar]
- 10.U.S. Food & Drug Administration. Coronavirus (COVID-19) Update: FDA Authorizes Additional Monoclonal Antibody for Treatment of COVID-19, May 26, 2021. Online, accessed on Oct 20, 2021 at https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-additional-monoclonal-antibody-treatment-covid-19. [Google Scholar]
- 11.National Institute of Health 2020; Online, accessed on Oct 20, 2021 at https://www.nih.gov/research-training/medical-research-initiatives/activ.
- 12.Murray DD, Babiker AG, Baker JV, Barkauskas CE, Brown SM, Chang C, et al. Design and implementation of the multi-arm, multi-stage Therapeutics for Inpatients with COVID-19 (TICO) platform master protocol: An Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) initiative. medRxiv. 2020. [Google Scholar]
- 13.Lundgren JD, Grund B, Barkauskas CE, Holland TL, Gottlieb RL, Sandkovsky U, et al. A Neutralizing Monoclonal Antibody for Hospitalized Patients with Covid-19. N Engl J Med. 2021;384(10):905–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Donnell MR, Grinsztejn B, Cummings MJ, Justman JE, Lamb MR, Eckhardt CM, et al. A randomized double-blind controlled trial of convalescent plasma in adults with severe COVID-19. J Clin Invest. 2021;131(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piechotta V, Iannizzi C, Chai KL, Valk SJ, Kimber C, Dorando E, et al. Convalescent plasma or hyperimmune immunoglobulin for people with COVID-19: a living systematic review. Cochrane Database Syst Rev. 2021;5(5):Cd013600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.RECOVERY Collaborative Group. Convalescent plasma in patients admitted to hospital with COVID-19 (RECOVERY): a randomised controlled, open-label, platform trial. Lancet. 2021;397(10289):2049–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horby PW, Mafham M, Peto L, Campbell M, Pessoa-Amorim G, Spata E, et al. Casirivimab and imdevimab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. medRxiv. 2021:2021.06.15.21258542. [Google Scholar]
- 18.Aalen OO, Johansen S. An Empirical Transition Matrix for Non-Homogeneous Markov Chains Based on Censored Observations. Scandinavian Journal of Statistics. 1978;5(3):141–50. [Google Scholar]
- 19.Fine JP, Gray RJ. A Proportional Hazards Model for the Subdistribution of a Competing Risk. Journal of the American Statistical Association. 1999;94(446):496–509. [Google Scholar]
- 20.Zhou B, Latouche A, Rocha V, Fine J. Competing risks regression for stratified data. Biometrics. 2011;67(2):661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.; 2020. [Google Scholar]
- 22.Jones BE, Brown-Augsburger PL, Corbett KS, Westendorf K, Davies J, Cujec TP, et al. The neutralizing antibody, LY-CoV555, protects against SARS-CoV-2 infection in nonhuman primates. Sci Transl Med. 2021;13(593). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor PC, Adams AC, Hufford MM, de la Torre I, Winthrop K, Gottlieb RL. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat Rev Immunol. 2021;21(6):382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the Treatment of Covid-19 - Final Report. N Engl J Med. 2020;383(19):1813–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fajnzylber J, Regan J, Coxen K, Corry H, Wong C, Rosenthal A, et al. SARS-CoV-2 viral load is associated with increased disease severity and mortality. Nat Commun. 2020;11(1):5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hogan CA, Stevens BA, Sahoo MK, Huang C, Garamani N, Gombar S, et al. High Frequency of SARS-CoV-2 RNAemia and Association With Severe Disease. Clin Infect Dis. 2021;72(9):e291–e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L, Tan C, Zeng J, Luo C, Hu S, Peng Y, et al. Analysis of viral load in different specimen types and serum antibody levels of COVID-19 patients. J Transl Med. 2021;19(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shan D, Johnson JM, Fernandes SC, Suib H, Hwang S, Wuelfing D, et al. N-protein presents early in blood, dried blood and saliva during asymptomatic and symptomatic SARS-CoV-2 infection. Nat Commun. 2021;12(1):1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Starr TN, Greaney AJ, Dingens AS, Bloom JD. Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep Med. 2021;2(4):100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen RE, Winkler ES, Case JB, Aziati ID, Bricker TL, Joshi A, et al. In vivo monoclonal antibody efficacy against SARS-CoV-2 variant strains. Nature. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.National Institute of Health 2021; Online, accessed on Oct 20, 2021 at https://www.nih.gov/news-events/news-releases/nih-sponsored-activ-3-clinical-trial-closes-enrollment-into-two-sub-studies.
- 32.Dong J, Zost SJ, Greaney AJ, Starr TN, Dingens AS, Chen EC, et al. Genetic and structural basis for recognition of SARS-CoV-2 spike protein by a two-antibody cocktail. bioRxiv. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.