

Abstract
Despite improvements in outcomes for children with B- and T-cell acute lymphoblastic leukemia (B-ALL and T-ALL), patients with resistant or relapsed disease fare poorly. Previous studies have demonstrated the essential role of cyclin D3 in T-ALL disease initiation and progression and that targeting of the CDK4/6-cyclin D complex can suppress T-ALL proliferation, leading to efficient cell death in animal models. Studies in leukemia and other malignancies, suggest that schedule is important when combining CDK4/6 inhibitors (CDKi) with cytotoxic agents. Based on these observations, we broadened evaluation of two CDKi, palbociclib (PD-0332991, Pfizer) and ribociclib (LEE011, Novartis) in B- and T-ALL as single agent and in combination with conventional cytotoxic chemotherapy, using different schedules in preclinical models. As monotherapy, CDKi caused cell cycle arrest with a significant decrease in S phase entry and were active in vivo across a broad number of patient-derived xenograft samples. Prolonged monotherapy induces resistance, for which we identified a potential novel mechanism using transcriptome profiling. Importantly, simultaneous but not sequential treatment of CDKi with conventional chemotherapy (dexamethasone, L-asparaginase and vincristine) led to improved efficacy compared to monotherapy in vivo. We provide novel evidence that combining CDKi and conventional chemotherapy can be safe and effective. These results led to the rational design of a clinical trial.
Introduction
Acute lymphoblastic leukemia (ALL), the most common pediatric malignancy, is a biologically heterogeneous disease with multiple different subtypes characterized by genetic alterations that result in deregulation of hematopoietic transcription factors, epigenetic modifiers, cytokine receptors and tyrosine kinases.1 While the overall prognosis for children and young adults with ALL has improved, outcomes for patients with some ALL subtypes remain poor despite intensification of cytotoxic chemotherapy.
Deregulated cell cycle progression is a hallmark feature of cancer cells. Cyclin-dependent kinases, CDK4 and CDK6 (CDK4/6) compose the core machinery governing the progression through the early G1 phase of the cell cycle. In complex with cyclin D, CDK4/6 promote cell cycle progression through at least two functions: hyper-phosphorylation of the retinoblastoma protein (RB) thereby uncoupling the CDK4/6-cyclin D complex from the E2F transcription factors; and interaction with cell cycle inhibitors, p21cip1 and p27kip, which promotes activation of the CDK2/cyclin E complex.2 The catalytic activity of CDK4 or CDK6 regulates a critical checkpoint for the G1-S transition and the commitment to cell division. As a consequence, gain of function of the G1-S checkpoint’s cyclin-CDK complexes is a major driver in a large number of human cancers. CDK4/6 inhibition has been explored as a means to bypass the deregulated cell cycle progression common in cancer pathogenesis. While CDK4/6 is rarely mutated in human cancers, aberrations that lead to the loss of proliferative controls include overexpression of CDK4 itself, amplification of the D-type cyclins, downregulation of p16INK4A, mutations in CDK4 that prevent p16INK4A binding to the enzyme, and deletion or mutation of RB1 itself.3-6 The frequency of these alterations implies that abrogation of the G1 checkpoint or progression through the CDK4/cyclin D pathway provides a distinct advantage to cancer cell proliferation and survival. Deletions of CDKN2A/B and RB1 are common in ALL, with CDKN2A/B deletions occurring in 70% of T-ALL and 36% of B-ALL. Moreover, RB1 suppressor gene deletions occur in 12% in T-ALL and 8% in B-ALL.7-1 1 Deletions involving CDKN2A/B are also common in BCR-ABL1+ ALL, and genetic alterations involving the RB1 gene are hallmarks of low hypodiploid ALL.12,13
Based on these observations, a number of CDK4/6 selective inhibitors have been developed, including ribociclib (LEE011) and palbociclib (PD0332991). Both are more selective for the ATP-binding pocket of the kinase domain compared to previous non-selective cell cycle inhibitors, with significantly less off-target effects and fewer dose-limiting toxicities.14,15 Palbociclib has progressed furthest in the clinic, having received Food and Drug Administration approval for estrogen receptor-positive (ER-positive), HER2-negative post-menopausal breast cancer patients in combination with endocrine therapy.16 Previous studies suggest induction of early G1 arrest by CDK inhibitors (CDKi) requires retinoblastoma (Rb), and this arrest is reversible in vitro and in vivo.4,17 Therefore, amplification of CDK6 and cyclin E1 are able to bypass dependency on cyc-lin D1/CDK4 signaling, while RB1 loss provides intrinsic resistance to CDKi in preclinical studies. Overall, the selectivity and reversibility of cell-cycle inhibition by CDKi and their clinical efficacy, provide a safe and effective approach to target specific phases of the cell cycle in cancer. Since the majority of conventional chemotherapeutics target actively cycling cells, some studies suggest combinations of CDKi with cytotoxic chemotherapy could be antagonistic.14,18-20 Thus, it is crucial to determine efficacy of CDKi, and understand the optimal schedule in order to combine them safely and effectively with chemotherapy. We investigated the activity of CDKi in ALL preclinical models. We found that B-ALL and T-ALL blasts were sensitive to these inhibitors via concentration-dependent cell cycle arrest in vitro. We then tested various dosing schedules of CDKi with conventional cytotoxic chemotherapy agents typically used in upfront therapy for B-ALL and TALL, including glucocorticoids, vincristine and L-asparaginase in vivo using patient-derived xenograft (PDX) models. Importantly, we provide divergent evidence from prior in vitro studies evaluating timing of CDKi and conventional chemotherapy and find that CDKi enhance combination therapy in ALL when administered concurrently rather than in sequence. These data prompted the phase I clinical trial (clinicaltrials gov. Identifier: NCT03792256) evaluating palbociclib in combination with chemotherapy for children with relapsed ALL or lymphoblastic lymphoma.
Methods
Reagents and cell lines
Human cell lines were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany) and maintained in RPMI 1640 supplemented with 10-20% heat-inactivated fetal bovine serum (Gibco, Thermo-Scientific). All cell lines were frozen and low passage cells were used for all experiments.
Western bloting
Cells were lysed and 10-15 mg were prepared,22 resolved by electrophoresis on NuPAGE 4-12% SDS-PAGE gradient gels (Invitrogen) and transferred to polyvinylidene fluoride membranes (Invitrogen). Immunoblotting was performed with primary antibodies: RB, phosphorylated RB (pRB)S807/801; pRBS807/810; cyclin D3, p21 (Cell Signaling Technologies). Bound antibody was detected using Western Chemiluminescence Reagent (PerkinElmer).
Compounds
Ribociclib (LEE011) was generously provided by Novartis Inc. (Basel, Switzerland). Palbociclib was provided by Pfizer Inc. (New York, NY). Clinical grade chemotherapy agents were purchased from commercial vendors: vincristine, Teva (Petah Tikva, Israel); L-asparaginase, Jazz pharmaceuticals (Dublin, Ireland); dexamethasone, Fresenius Kabi (Bad Homburg vor der Hohe, Germany); and everolimus, Novartis. Vincristine, L-asparaginase and dexamethasone dosing per previous studies.21,22 Everolimus was dosed at 2.5 mg/kg and 5 mg/kg by oral gavage based on prior reports in preclinical models of ALL.23
Flow cytometry
Cells were treated with vehicle versus palbociclib or ribociclib for 48-72 hours, labeled with BrdU for 1 hour and processed for flow cytometry, on an Accuri CSampler flow cytometer (BD Accuri Cytometers), and analysis was performed with FlowJo software (Tree Star, Ashland, OR). Engraftment and serial disease evaluation were determined by flow cytometric analysis of blood using antibodies against human CD19 and CD45 for B-ALL and CD45 for T-ALL.24
Xenograf therapeutic trials
Patient samples
Banked diagnostic specimens (lymphoblasts from peripheral blood or bone marrow) were obtained from patients enrolled on the Children’s Oncology Group (COG) study P9906 or from the Stem Cell and Leukemia Core at the Children’s Hospital of Philadelphia or Columbia Presbyterian Hospital. All samples were obtained after informed consent was given in accordance with the Declaration of Helsinki.24,25
Patient-derived xenografts
PDX models using non-obese diabetic/severe combined immunodeficiency (NOD/SCID/Il2rgtm1wjl/SzJ) (NSG) mice were developed as previously described.24-26 All experiments were conducted in accordance to protocols approved by the Institutional Animal Care and Use Committee and Institutional Review Board of the Children’s Hospital of Philadelphia (CHOP) and New York University School of Medicine (NYU). Samples were derived from patients with de novo ALL.
Figure 1.

CDK inhibitors induce G1 arrest in B-cell acute lymphoblastic leukemia cell lines in vitro. (A) We performed western blot analysis of a panel of B-cell acute lymphoblastic leukemia (B-ALL) cell lines (labeled in blue) and primary patient samples (labeled in green), which demonstrate high protein expression of CDK4, CDK6 and CCND3. Inhibitor p21 is also highly expressed. A T-ALL cell line, Jurkat (labeled in black), was used as a positive control, given prior published data, however, it is known to be negative for p21.40 (B) Samples were treated for a minimum of 48 hours with either palbociclib (B) or ribociclib (C) prior to harvesting for flow cytometry. Percentage of cells arrested in G1 with varying doses of CDK inhibitors (CDKi) in 3 B-ALL cell lines: REH, MHH CALL4, and KOPN8. (D) Corresponding dose-dependent inhibition by CDKi reflects loss of phosphorylated retinoblastoma (p-RB), with no effect on total retinoblastoma (Rb). Phosphorylated S6, a downstream signaling marker was differentially affected with CDK inhibition in REH and KOPN8, but not MHH CALL4.
RNA sequencing and data analysis
RNA sequencing (RNA-seq) of native Jurkat and palbociclib-resistant Jurkat cells were performed at the NYU School of Medicine Genome Technology Core. STAR 2.4.2a was applied to align the RNA-seq samples to the reference mouse genome (mm9) and count the number of reads that map to each gene in the ensemble GRCm38.80 gene model. R (v.3.5.1) (http://www.R-project.org/) and DESeq2 package (v1.10.0) were used to perform differential gene expression analysis among different sample groups.
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 7. Results were analyzed for statistical significance using repeated measures 2-way analysis of variance (ANOVA) of peripheral blast counts (P-values represent the interaction between time and treatment). Spleen blast counts were analyzed by two-sided t-test.
Results
CDK4/6 signaling is hyperactive in B-cell acute lymphoblastic leukemia
In order to confirm that genomic aberrations translate to aberrant CDK4/6 signaling within the cyclin D/CDK4/CDK6/RB pathway, we examined the activation status of RB in a comprehensive panel of well-characterized human B-ALL cell lines and patient samples. As shown in Figure 1A, robust phosphorylation of RB at serines 780 and 807/810 – residues directly targeted by CDK4 and CDK6 – was observed in all tested cell lines, and protein-level expression of CDK4, CDK6 and cyclin D3 (CCND3) occurred in the majority of cell lines and patient samples. Jurkat, a T-ALL cell line was used as a positive control given previously published data demonstrating TALL sensitivity to CDKi.20
B-cell acute lymphoblastic leukemia is sensitive to CDK4/6 inhibition in vitro
Given the observation that CDK4/6 signaling is highly active in B-ALL, thus maintaining hyperphosphorylated RB and supporting cell cycle progression through the G1-S checkpoint, we evaluated the effect of selective CDK4/6 inhibition on B-ALL cell lines in vitro (Table 1). Palbociclib (Figure 1B) and ribociclib (Figure 1C) induce significant growth arrest in G1 across a broad range of concentrations (0.5 to 4.0 mM) in a panel of cell lines (REH, MHH CALL4, and KOPN8) (Figure 1B and C; Online Supplementary Figure S1). Jurkat was used as a positive control. In spite of robust phosphorylated RB expression, the sensitivity to CDK4/6 inhibition was variable in cell lines.
In order to confirm that the growth inhibition observed in sensitive cell lines reflected targeted impairment of CDK4/6 signaling, we analyzed the levels of phosphorylated RB following treatment with either palbociclib or ribociclib (Figure 1D). Correspondingly, the amount of phosphorylated RB decreased with increasing concentrations of palbociclib or ribociclib. While both CDKi induced cell cycle arrest via inhibition of phosporylated RB, palbociclib is more potent at lower concentrations compared to ribociclib in vitro. However, based on expected comparable PK/PD in humans, these agents are considered equivalent in efficacy at their respective appropriate doses.27 Further, these two drugs are similar in their selectivity of CDK4/6 inhibition.
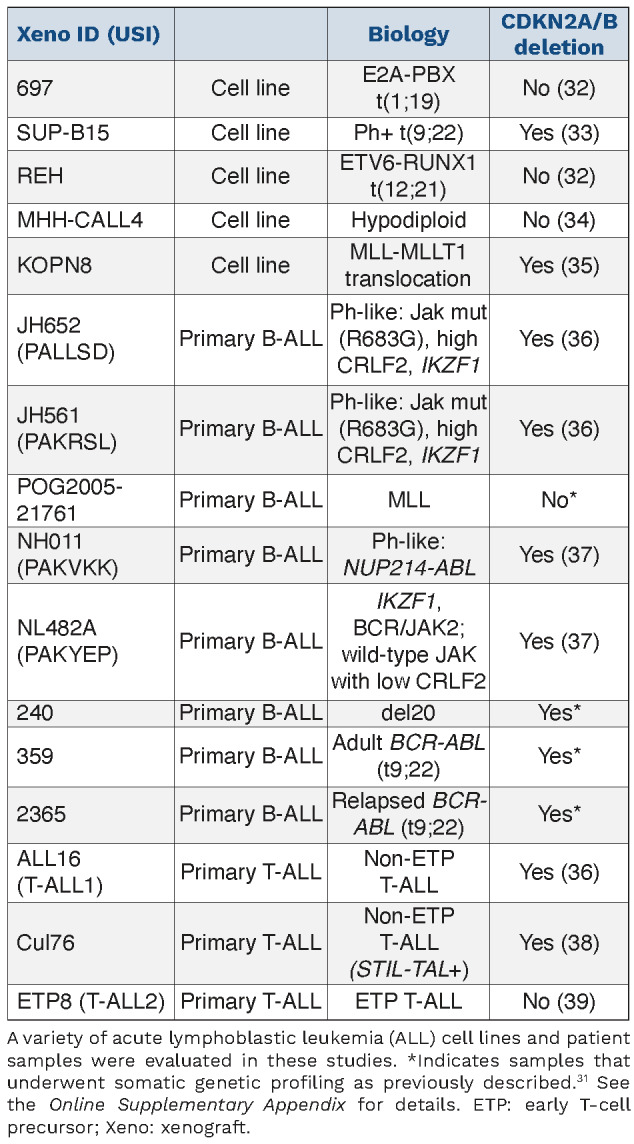
Table 1.
Panel of B-cell and T-cell acute lymphoblastic leukemia cell lines and patient samples included in this study.
CDK4/6 inhibition causes growth inhibition in vivo
We assayed for single agent in vivo efficacy using PDX models of T-ALL and B-ALL. In the two T-ALL samples, CDKi treatment markedly decreased the number of human CD45+ (hCD45+) cells (Figure 2C) with a correlated prolonged overall survival in comparison to vehicle (Figure 2E). Similar to in vitro studies, this treatment regimen appropriately induced profound cell cycle arrest of hCD45+ leukemic cells in vivo (Figure 2D). Of note, the difference in palbociclib dosing was related to increased diarrhea in the B-ALL PDX models that was not seen in the T-ALL PDX.
In B-ALL, leukemia progression was significantly inhibited in some of the samples but not all (Figure 3). In six of the seven unique PDX samples tested, tumor growth was significantly impacted in the spleen and/or blood. The seven PDX represented a heterogeneous group of high-risk B-ALL patient samples (Table 1). All of the tested samples had high levels of Rb therefore did not vary in the activation status of cyclin D, nor had any obvious biomarker to predict efficacy. In both subtypes (B- and T-ALL xenografts), the dosing strategy was well tolerated with no evidence of weight loss of other signs of toxicity.
Cyclin D3 overexpression confers palbociclib resistance
In order to explore potential de novo mechanisms of resistance to palbociclib treatment, we treated a human TALL cell line with low dose (0.1 mM) palbociclib in vitro, but gradually escalated to a final dose of 2.8 mM, a process that lasted 52 weeks, generating palbociclib resistant T-ALL cells (Figure 4A). Strikingly, resistant cells have higher rates of cycling cells (Figure 4B). This finding suggests that the resistant cells are adapting to treatment by identifying alternative ways to boost cell cycle progression. In agreement with this notion, we were able to demonstrate that resistant cells do not decrease levels of phosphorylated RB in response to palbociclib treatment (Figure 4C). In fact, we detected significantly elevated basal levels of cyclin D3 protein (CCND3), which suggests a potential mechanism of resistance. In order to further probe for such resistance mechanisms we performed RNA-seq of sensitive and resistant Jurkat cells at steady-state and after palbociclib treatment (Figure 4D). We found sensitive Jurkat cells at baseline (steady-state) had lower levels of cell cycle related genes as compared with resistant cells. Moreover, we found a significant downregulation of cell cycle related genes including cyclins A2, B1, B2 and E2; CDK1, 2 and 4 and E2F transcription factors (E2F1, 2, 7), in sensitive cells, similar to previous work in other malignancies.28 In resistant cells, expression of cell cycle related genes was only modestly decreased when treated with palbociclib. These experiments suggest that palbociclib-resistant cells have adapted to treatment by boosting cell cycle entry and progression through the upregulation of a number of essential regulators, including CCND3, CDK and members of the E2F family.
Figure 2.

CDK inhibitor suppress human primary T-cell acute lymphoblastic leukemia xenograf proliferation in vivo. (A) Schematic representation of CDK inhibitor (CDKi) treatment course with single agent palbociclib at 75 mg/kg gavage vs. vehicle. Dosing determined based on prior studies.28 (B) A representation of flow cytometric analysis of T-cell acute lymphoblastic leukemia xenograft populations (hCD45+) in peripheral blood following 3 weeks of CDKi treatment. (C) Flow cytometry analysis of T-ALL xenograft populations (hCD45+) in peripheral blood over the course of treatment. (D) DAPI staining of 2 different human xenografts (ALL16 and ETP8) following 48-hour treatment with CDKi in vivo. (E) Kaplan-Meier survival graph of the 2 T-ALL xenografts treated with CDKi.
Figure 3.

CDK inhibitors suppress human primary B-cell acute lymphoblastic leukemia xenograf proliferation in vivo. (A) Schematic representation of CDK inhibitor (CDKi) administration with single agent palbociclib at 35 mg/kg or ribociclib at 150 mg/kg, gavage vs. vehicle. Ribociclib dosing determined based on prior studies, and palbociclib dosing based on internal communication with Pfizer.28,41,42 (B) Mice (5 mice per treatment group) were randomized to palbociclib (red hashed bars), or to ribociclib (solid red bars) vs. vehicle (blue bars), for 5 consecutive days per week until sacrifice (21-28 days). Peripheral blood blast cell (PBC) counts are shown in the left panel. Splenic blast burden is shown in the right panel, with corresponding P-values. Two patient-derived xenograft (PDX) models (2,365 and 240), demonstrated significant inhibition of disease progression in blood and spleen, two PDX (359, JH561), demonstrated significant inhibition of disease progression in the peripheral blood but not in the spleen; and two PDX, JH562 and POG/MLL demonstrated significant disease inhibition in the spleen but not in the blood; 1 PDX model (NL482A) had no response. hB-ALL: human B-cell acute lymphoblastic leukemia.
Combination therapy of palbociclib was enhanced with concurrent administration of standard chemotherapy compared to sequential treatment
Given the suggestion that timing may be a critical component of efficacy with combination therapy, we tested pretreatment of palbociclib versus concurrent administration on disease burden in two high risk B-ALL PDX (JH561 and NH011) and one T-ALL xenograft (Cul76). These samples were chosen given their known risk for having inferior prognosis and modest response to conventional cytotoxic chemotherapy.
The treatment schema for combinations in B-ALL is shown in Figure 5. In order to closely mimic agents typically used in conventional therapy for B-ALL we tested dexamethasone, vincristine and L-asparaginase. Single agent vincristine (1 mg/kg) or dexamethasone (7.5 mg/kg) were too effective on their own to delineate a differential impact of combination therapy, when they were given at a dose that mirrors the PK/PD of the dose used in children with ALL.
Figure 4.

Cell cycle regulators are upregulated in CDK inhibitor-resistant T-cell acute lymphoblastic leukemia. (A) Schematic representation of the method of generating de novo CDK inhibitor (CDKi)‐resistant cells using palbociclib dose escalation. (B) Cell cycle analysis of 1 mM palbociclib (palbo) treated parental and palbociclib-resistant cells. (C) Western blotting of pRB and Cyclin D3 (CCND3) in parental and palbociclib-resistant cells. (D) Differential expression of cell cycle genes in treatment of parental and CDKi-resistant cells.
Therefore, we performed a titration of single drug in vivo, determining the concentration where single agent decreased disease burden by about 50% (half maximal inhibitory concentration [IC50]) of the maximum dose in the peripheral blood. Average IC50 of three independent determinations were performed (vincrisitine at 0.5, 0.25, 0.1 and 0.05 mg/kg; dexamethasone at 4, 2 and 1 mg/kg; data not shown). IC50 doses for each drug include vincristine at 0.1 mg/kg intraperitoneally (IP) weekly, dexamethasone at 2 mg/kg IP daily and L-asparaginase at 5,000 IU/kg IP weekly. As shown in Figure 6 (and Online Supplementary Figure S2), simultaneous administration of combination therapy (cytotoxic agent + palbociclib) was superior to monotherapy with any of the individual cytotoxics (dexamethasone, vincristine, or L-asparaginase) or palbociclib alone in both B-ALL PDX models. In contrast, sequential treatment with palbociclib or vehicle followed by vincristine or L-asparaginase or vehicle did not exhibit an augmented disease response in combination. However, palbociclib followed by dexamethasone did show superiority of the combination over either single agent.
In order to verify the benefit of the simultaneous administration of palbociclib plus cytotoxics in T-ALL, we set up four cohorts of human T-ALL xenografted animals that received combination treatment for 4 weeks (Figure 7A). We found that combination therapy of palbociclib concurrently with vincristine led to a significant difference in disease burden (Figure 7B) and survival (Figure 7C) compared to monotherapy.
Combination CDK4/6 and mTOR inhibitors is not effective in vivo against B-cell acute lymphoblastic leukemia
The PI3K/AKT/mTOR pathway is a key upstream regulator of cyclin D-CDK4/6 activity. Thus, agents that target PI3K/Akt/mTOR signaling including mTOR inhibitors have been studied in a number of malignancies.19 CDK4/6 inhibition has been demonstrated to overcome resistance to PI3K inhibition and endocrine therapy, providing a strong rationale for the combination of an mTOR inhibitor with CDKi. Recent in vitro data suggest the combination of everolimus and ribociclib may be more effective than either agent alone in ALL cell lines.20 We tested simultaneous combination therapy with palbociclib or ribociclib with everolimus in four different B-ALL PDX models (JH561, 240, POG/MLL, and NL482B) (Online Supplementary Figure S3). Inhibition of the mTOR pathway did not improve the response to CDK4/6 inhibition in any of the models.
Discussion
We provide evidence of single agent efficacy of palbociclib and ribociclib, two selective CDK4/6 inhibitors, in a heterogeneous range of B- and T-ALL samples, in vitro and in vivo. We show that RB phosphorylation via CDK4/6 signaling is prevalent in B-ALL cell lines and primary patient samples, reflected by high expression of CDK4, CDK6 and CCND1, but there are likely other mechanisms of CDK4/6 hyperactivation. We demonstrate growth inhibition reflects G1 arrest and depletion of phosphorylated RB. Similar to other studies, RB expression appeared to be essential for sensitivity. However, each sample had varying levels of sensitivity suggesting there may be additional relevant biomarkers yet to be determined, to predict sensitivity to CDKi in B-ALL. Genomic alterations in the CDK pathway are common in ALL, with deletions in CDKN2A/B occurring with an incidence of 30-50% in childhood and adult ALL.29 The prognostic value has been widely investigated but the results remain unclear. While there is some suggestion that CDKN2A/B deletions have an adverse impact on overall survival (OS), these studies have been unable to determine if CDK alterations are independently prognostic of minimal residual disease (MRD) or other sentinel genetic lesions, such as BCR-ABL, ETV6-RUNX1, in multi-variant analyses. In an attempt to associate CDKN2A/B deletion status to CDKi sensitivity (Table 1; Figure 3B), we did not identify a correlation, however were likely underpowered to make any definitive conclusions. We did identify a novel mechanism of acquired resistance to palbociclib treatment, related to upregulation of CCND3, CDK and members of the E2F family. Future studies will need to investigate the mechanisms of intrinsic resistance to CDKi. We believe large scale genomic testing embedded in future clinical trials will be necessary to confirm and/or identify additional relevant biomarkers that confer sensitivity to these agents, and to determine if CDK pathway lesions are independently prognostic when considering other clinical and biologic variables.
Not surprisingly, a number of clinical studies across malignancies suggest single agent CDKi fail to provide durable responses, indicating combination therapies are needed. Efficacy was more pronounced in T-ALL as compared to B-ALL in our preclinical models, suggesting CDKi may have single agent activity in T-ALL, but should only be tested in combination with other drugs in B-ALL in clinical trials. In fact, we noted higher baseline expression of CCND3 in B-ALL, which may partly explain the increased sensitivity of T-ALL blasts to monotherapy with CDKi as compared with B-ALL. Pikman et al. provided recent evidence in T-ALL of synergistic combinations between ribociclib and corticosteroids and everolimus, while demonstrating ribociclib antagonizes a number of cytotoxic chemotherapy agents, including mercaptopurine and asparaginase in in vitro studies of T-ALL cell lines.20,30 Importantly, our data highlight the need to investigate drug schedules using in vivo models, as in vitro models cannot replicate the complexities of drug metabolism, drug-drug interactions, bioavailability, and the microenvironment.
Figure 5.

Treatment schema of pretreatment versus concurrent regimens in vivo. Treatment schema depicting the timing of each drug (vincristine, dexamethasone or L-asparaginase and/or CDK inhibitor [CDKi]) vs. vehicle in the pretreatment and concurrent schema. Veh: vehicle; chemo: chemotherapy; hB-ALL: human B-cell acute lymphoblastic leukemia; palbo: palbociclib.
In contrast to other groups using in vitro models, we found simultaneous administration of CDKi with three different cytotoxic agents was superior to single agent alone, yet a similar benefit was not seen when CDKi were administered prior to cytotoxic chemotherapeutics. Importantly in order to determine if there was a benefit to combination therapy, the doses of some of the cytotoxic agents had to be reduced since they were too effective as single agents. This reflects one constraint of murine models as myelosuppression cannot be abrogated with supportive care such as transfusion, limiting dose intensity. Future studies should also include combinations of CDKi with other cytotoxic agents, including DNA damaging agents such as anthracyclines.
Figure 6.

Simultaneous and not sequential treatment of palbociclib with cytotoxic chemotherapy significantly inhibits disease burden in two Philadelphia chromosome-like B-cell acute lymphoblastic leukemia samples in vivo. Pretreatment with palbociclib (palbo) vs. vehicle (veh) for 7 days by oral gavage, followed by treatment by three common chemotherapy agents: vincristine (VCR, green), L-asparaginase (Asp, blue) or dexamethasone (Dex, red). Concurrent treatment of palbo vs. veh with chemotherapy agents: VCR, Asp or Dex for the duration of the trial of 21 days. Peripheral blood blast count (PBC) over time in two B-cell acute lymphoblastic leukemia (B-ALL) patient-derived xenograft (PDX) (JH561 and NH011). Graphed are means and standard error of PBCx106 per mL on the vertical axis and days of treatment on the X axis. Samples following the pretreatment schema are depicted in the left column, while concurrent treatment is shown in the right column. P-values were calculated using ANOVA to detect the difference between single agent either chemotherapy or CDK inhibitors compared to the combination of both drugs.
Figure 7.

Simultaneous and not sequential treatment of palbociclib with cytotoxic chemotherapy significantly inhibits disease burden in one T-cell acute lymphoblastic leukemia sample in vivo. (A) Schematic representation of CDK inhibitors (CDKi) and vincristine (VCR) combination regimen in a T-cell acute lymphoblastic leukemia (T-ALL) xenograft (Cul76). Peripheral blood is taken at specific time points as shown. (B) Flow cytometry analysis of peripheral blood analysis of human primary T-ALL patient-derived xenograft (PDX), Cul76, following palbociclib (palbo) treatment. (C) Kaplan-Meier survival graph of T-ALL PDX treated with the combination therapy regimen over a period of 42 days.
We also evaluated the combination of everolimus with CDKi as an alternative approach to treatment for patients with B-ALL. This is under clinical investigation in a phase I clinical trial at the Dana Farber Cancer Institute for relapsed ALL (clinicaltrials gov. Identifier: NCT03740334), based on data provided by Pikman et al.20 Importantly, their initial preclinical study evaluated the combination of these drugs in T-ALL. Our data would suggest this combination is unlikely to be of benefit in B-ALL despite preclinical efficacy of single agent CDKi or mTORi in B-ALL preclinical studies. We found CDKi influenced phosphorylated S6 expression, as seen in Figure 1D, which may explain the lack of benefit from combination therapy.
In summary, this work provides a number of novel observations that may impact clinical care and trial design. We provide novel evidence of combination therapy with CDKi to enhance cytotoxic chemotherapy when used simultaneously but not in sequence. We demonstrate the combination of CDKi and mTORi does not merit exploration in B-ALL, despite promising preclinical data in T-ALL by other groups. We identified a novel mechanism of resistance to CDKi. Finally, we found CDKi were effective in a biologically heterogeneous cohort of B-ALL preclinical models. The results of this work led to a recent early phase clinical trial for children and young adults with relapsed/refractory ALL (clinicaltrials gov. Identifier: NCT03792256) and demonstrates the importance of performing rigorous preclinical studies to inform trials that include multi-agent therapy.
Supplementary Material
Funding Statement
Funding: This work was supported by an LLS SCOR grant (to WLC, EAR and DTT), and grants: R01CA193776, R01CA264837, R03CA256550, X01HD100702-01 and UG1CA233249, Children’s Oncology Group, Alex’s Lemonade Stand Foundation for Childhood Cancer, CHOP Frontiers Program Immune Dysregulation Team, and Cookies for Kids Cancer (to DTT).
References
- 1.Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. 2017;35(9):975-983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18(22):2699-2711. [DOI] [PubMed] [Google Scholar]
- 3.Sellers WR, Kaelin WG. Role of the retinoblastoma protein in the pathogenesis of human cancer. J Clin Oncol. 1997;15(11):3301-3312. [DOI] [PubMed] [Google Scholar]
- 4.Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427-1438. [PubMed] [Google Scholar]
- 5.Bartkova J, Lukas J, Bartek J. Aberrations of the G1- and G1/S-regulating genes in human cancer. Prog Cell Cycle Res. 1997;3:211-220. [DOI] [PubMed] [Google Scholar]
- 6.Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res. 1996;68:67-108. [DOI] [PubMed] [Google Scholar]
- 7.Girardi T, Vicente C, Cools J, De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood. 2017;129(9):1113-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrasco Salas P, Fernandez L, Vela M, et al. The role of CDKN2A/B deletions in pediatric acute lymphoblastic leukemia. Pediatr Hematol Oncol. 2016;33(7-8):415-422. [DOI] [PubMed] [Google Scholar]
- 9.Strefford JC, Worley H, Barber K, et al. Genome complexity in acute lymphoblastic leukemia is revealed by array-based comparative genomic hybridization. Oncogene. 2007;26(29):4306-4318. [DOI] [PubMed] [Google Scholar]
- 10.Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758-764. [DOI] [PubMed] [Google Scholar]
- 11.Kuiper RP, Schoenmakers EF, van Reijmersdal SV, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21(6):1258-1266. [DOI] [PubMed] [Google Scholar]
- 12.Bernt KM, Hunger SP. Current concepts in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia. Front Oncol. 2014;4:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwab CJ, Chilton L, Morrison H, et al. Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica. 2013;98(7):1081-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin D, Tran N, Thomas N, Tran DD. Combining CDK4/6 inhibitors ribociclib and palbociclib with cytotoxic agents does not enhance cytotoxicity. PLoS One. 2019;14(10):e0223555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen P, Lee NV, Hu W, et al. Spectrum and degree of CDK drug interactions predicts clinical performance. Mol Cancer Ther. 2016;15(10):2273-2281. [DOI] [PubMed] [Google Scholar]
- 16.O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13(7):417-430. [DOI] [PubMed] [Google Scholar]
- 17.Huang X, Di Liberto M, Jayabalan D, et al. Prolonged early G(1) arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle-coupled loss of IRF4. Blood. 2012;120(5):1095-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bortolozzi R, Mattiuzzo E, Trentin L, et al. Ribociclib, a Cdk4/Cdk6 kinase inhibitor, enhances glucocorticoid sensitivity in B-acute lymphoblastic leukemia (B-All). Biochem Pharmacol. 2018;153:230-241. [DOI] [PubMed] [Google Scholar]
- 19.Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014;5(15):6512-6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pikman Y, Alexe G, Roti G, et al. Synergistic drug combinations with a CDK4/6 Inhibitor in T-cell acute lymphoblastic leukemia. Clin Cancer Res. 2017;23(4):1012-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liem NL, Papa RA, Milross CG, et al. Characterization of childhood acute lymphoblastic leukemia xenograft models for the preclinical evaluation of new therapies. Blood. 2004;103(10):3905-3914. [DOI] [PubMed] [Google Scholar]
- 22.Guo QL, Wu MS, Chen Z. Comparison of antitumor effect of recombinant L-asparaginase with wild type one in vitro and in vivo. Acta Pharmacol Sin. 2002;23(10):946-951. [PubMed] [Google Scholar]
- 23.Dubois M, Le Joncour V, Tonon MC, et al. Evaluation of the impact of the cancer therapy everolimus on the central nervous system in mice. PLoS One. 2014;9(12):e113533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maude SL, Tasian SK, Vincent T, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2012;120(17):3510-3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teachey DT, Obzut DA, Cooperman J, et al. The mTOR inhibitor CCI-779 induces apoptosis and inhibits growth in preclinical models of primary adult human ALL. Blood. 2006;107(3):1149-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suryani S, Carol H, Chonghaile TN, et al. Cell and molecular determinants of in vivo efficacy of the BH3 mimetic ABT-263 against pediatric acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2014;20(17):4520-4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang C, Boyson CA, Di Liberto M, et al. CDK4/6 inhibitor PD 0332991 sensitizes acute myeloid leukemia to cytarabine-mediated cytotoxicity. Cancer Res. 2015;75(9):1838-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sawai CM, Freund J, Oh P, et al. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell. 2012;22(4):452-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang W, Kuang P, Liu T. Prognostic significance of CDKN2A/B deletions in acute lymphoblastic leukaemia: a meta-analysis. Ann Med. 2019;51(1):28-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carroll WL, Aifantis I, Raetz E. Beating the clock in T-cell acute lymphoblastic leukemia. Clin Cancer Res. 2017;23(4):873-875. [DOI] [PubMed] [Google Scholar]
- 31.Surrey LF, MacFarland SP, Chang F, et al. Clinical utility of custom-designed NGS panel testing in pediatric tumors. Genome Med. 2019;11(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takahashi K, Inukai T, Imamura T, et al. Anti-leukemic activity of bortezomib and carfilzomib on B-cell precursor ALL cell lines. PLoS One. 2017;12(12):e0188680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mariani SA, Minieri V, De Dominici M, et al. CDKN2A-independent role of BMI1 in promoting growth and survival of Ph+ acute lymphoblastic leukemia. Leukemia. 2016;30(8):1682-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghandi M, Huang FW, Jane-Valbuena J, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569(7757):503-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quentmeier H, Pommerenke C, Dirks WG, et al. The LL-100 panel: 100 cell lines for blood cancer studies. Sci Rep. 2019;9(1):8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rokita JL, Rathi KS, Cardenas MF, et al. Genomic profiling of childhood tumor patient-derived xenograft models to enable rational clinical trial design. Cell Rep. 2019;29(6):1675-1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):153-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Furness CL, Mansur MB, Weston VJ, et al. The subclonal complexity of STIL-TAL1+ T-cell acute lymphoblastic leukaemia. Leukemia. 2018;32(9):1984-1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shenker BJ, Walker LM, Zekavat A, et al. The cell-cycle regulatory protein p21(CIP1/WAF1) is required for cytolethal distending toxin (Cdt)-induced apoptosis. Pathogens. 2020;9(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rader J, Russell MR, Hart LS, et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res. 2013;19(22):6173-6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teh JL, Aplin AE. Arrested developments: CDK4/6 inhibitor resistance and alterations in the tumor immune microenvironment. Clin Cancer Res. 2019;25(3):921-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.