

Abstract

Transition metal catalysis is of utmost importance for the development of sustainable processes in academia and industry. The activity and selectivity of metal complexes are typically the result of the interplay between ligand and metal properties. As the ligand can be chemically altered, a large research focus has been on ligand development. More recently, it has been recognized that further control over activity and selectivity can be achieved by using the “second coordination sphere”, which can be seen as the region beyond the direct coordination sphere of the metal center. Hydrogen bonds appear to be very useful interactions in this context as they typically have sufficient strength and directionality to exert control of the second coordination sphere, yet hydrogen bonds are typically very dynamic, allowing fast turnover. In this review we have highlighted several key features of hydrogen bonding interactions and have summarized the use of hydrogen bonding to program the second coordination sphere. Such control can be achieved by bridging two ligands that are coordinated to a metal center to effectively lead to supramolecular bidentate ligands. In addition, hydrogen bonding can be used to preorganize a substrate that is coordinated to the metal center. Both strategies lead to catalysts with superior properties in a variety of metal catalyzed transformations, including (asymmetric) hydrogenation, hydroformylation, C–H activation, oxidation, radical-type transformations, and photochemical reactions.
1. Introduction
Homogeneous catalysis using metal complexes provides tools for effective and selective chemical transformations, which are crucial for the chemical process industry from both an economic and sustainability point of view. The field of homogeneous catalysis has been developed to an impressive level in the past 50 years, underscored by three awards for the Nobel prize in 2001,1 2005,2 and 20103 as well as the many applications of homogeneous catalysts found in industry.4−6 As the demands for the chemical industry are continuously changing, by the pressure to make the chemical industry more sustainable and based on renewable feedstocks as well as stimulated by product innovations in society, the demand for new catalysts is very high. As such, the research field of homogeneous catalysis is very active and many new catalytic conversions and novel concepts have been reported in recent years.
The properties of metal complexes that are used as catalysts are controlled, to a large extend, by the ligands that are coordinated to them, and therefore, traditionally a strong focus has been on ligand development. To aid the rational development of metal catalysts, many ligand parameters have been developed over the years.7 Rational development of catalysts started7 with primitive models to describe the electronic properties (Tolman’s χ-parameter)8 and size of the ligand (Tolman’s cone angle)8 and, somewhat later, the ligand bite angle.9 Nowadays, very sophisticated models can be used with the available computational tools.10 Currently, new machine learning strategies are also being developed to further aid the development of novel catalysts.11−13
In developing new metal complexes for catalysis, activity, selectivity, and stability are crucial parameters. Especially the selectivity that a catalyst displays can be hard to control. Both enantioselectivity and regioselectivity can be very difficult to achieve, as precise control of the reaction pathways is required. Energy differences in the competing pathways induced by ligand effects as small as 3 kcal·mol–1 already lead to sufficient selectivity, and such effects are, thus, very subtle. Although computational techniques are far advanced, rational design of ligands for selective catalytic processes, in general, remains challenging, although some interesting examples have been reported.14 As a result, the search for selective catalysts often relies on trial-and-error approaches, which are facilitated by high throughput experimentation.15 The availability of catalyst libraries of sufficient size and diversity is required to allow rapid screening methodologies, which means that ligands preferably need to be prepared in a modular fashion using relatively simple steps.16,17 In practice, the search for new catalysts may be based on a combined combinatorial and rational design approach, depending on the respective challenge to be solved. What all these approaches have in common is that the properties of the catalyst are controlled via the “first coordination sphere”, i.e. tuning the properties of the ligands that coordinate to the metal, as illustrated in Figure 1. The combination of different metals from the d-block of the periodic table and a variety of ligands that are diverse in electronic properties and steric size already gives enormous potential to control catalyst properties, hence the success of metal complexes in homogeneous catalysis. Despite the successes, there remain many challenges that have not been solved. As such, new tools to control catalyst properties are continuously being developed.
Figure 1.

Changing catalyst parameters via the “first coordination sphere” implies changing the properties of the ligands that are coordinated to the metal complex (top) and control of catalyst properties via hydrogen bonds in the second coordination sphere (bottom). M = transition metal, L = ligand, S = substrate.
One of the more recent strategies to control catalyst properties focuses on the “second coordination sphere”, i.e. the interactions beyond metal–ligand coordination that are important. These include noncovalent interactions between the ligands themselves, interactions with the ligand(s) and the substrate(s) (Figure 1), and/or interactions with the environment. Arguably, this approach is inspired by natural systems as enzymatic conversions also rely, to a very large extent, on control via residues and cofactors not directly involved in the chemistry of a catalytic cycle. Substrates that are docked in the pocket of enzymes near the active center are well preorganized to give the proper activation and selectivity.18 In redox enzymes, electrons, protons, and substrates are usually preorganized to facilitate rapid conversions.19 Such preorganization strategies have been intensively explored in the field of proton reduction catalysis by using ligands with internal basic functions,20 a topic that will be covered in this thematic issue by others. In the field of supramolecular chemistry, which mostly developed in parallel with the field of metal-based catalysis, enzymes have been a source of inspiration in the development of supramolecular catalysts.21−26 However, supramolecular catalysis has traditionally been very focused on relatively simple conversions, such as hydrolysis and Diels–Alder reactions, to prove that some of the concepts found in nature can be mimicked by synthetic analogues. More recently, the implementation of supramolecular strategies into homogeneous catalysis with metal complexes has been explored and demonstrated to be very powerful.27,28 This has resulted in novel tools to control metal catalyzed processes via the second coordination sphere, including the self-assembly of bidentate ligands using ligand building blocks, substrate orientation at the metal center via additional supramolecular interactions, and the use of molecular cages around the metal center to control catalytic reactions.
This review focuses on the application of hydrogen bonds (HBs) to control catalyst properties via the second coordination sphere (Figure 1). The organization of this review is as follows: In section 2, we provide some fundamental information on HBs and factors that influence their utility. Next, in section 3, we review the use of self-assembled bidentate ligands via HBs. Section 4 reviews the use of hydrogen bonding between the substrate and the catalyst as a way to control catalytic conversions. In transfer hydrogenation reactions, as pioneered by Noyori,29 the reaction can also proceed via an outer sphere mechanism in which hydrogen bonding is crucial. In these types of reaction mechanisms the HB donor typically also actively participates in the reaction by delivering the proton to the substrate. As this is a different use of the HB, it is beyond the scope of this review, and we refer the interested reader to well-established and recently published reviews.30−41 Also the HB-assisted activation of small molecules in redox reactions, such as the reduction of oxygen and carbon dioxide, is beyond the scope of the current review, yet we want to stress that also for these types of conversions HB interactions in the second coordination sphere can greatly affect the performance of the catalyst.42 Finally, we provide a summary and an outlook (section 5).
2. Fundamentals of Hydrogen Bonding
2.1. Definitions and Characteristics of Simple HBs
The concept of a hydrogen bonding interaction is more than a century old,43,44 has a well-documented history,45,46 and has been extensively reviewed.46−49 Some of the key characteristics of simple HBs are summarized in Table 1. A HB is typically understood as an attractive interaction between a covalently bound and positively polarized hydrogen atom and an electronegative entity. In its most simple form, a HB can be written as X–H···Y, where X stands for the donor atom and Y for the acceptor.46,50 In order for the hydrogen atom to be positively polarized, the donor atom must be more electronegative, which is the case for most main group elements. The acceptor moiety has to be electron rich and typically involves a lone pair of electrons from an atom or anion.46,50−52 Other less-typical53,54 HB acceptors include π-electrons,55−60 some transition metals,61−64 and even hydrides.65
Table 1. Some Guideline Characteristics of a Single [X–H···Y] Hydrogen Bonding Interaction Based on the Review in Section 2.1 and Adapted from Ref (48).
| Strong | Moderate | Weak | |
|---|---|---|---|
| ΔE (kcal·mol–1)a | 15–45 | 4–15 | <4 |
| Directionality | Strong | Moderate | Weak |
| X–H vs H···Y distanceb | X–H ≈ H···Y | X–H < H···Y | X–H ≪ H···Y |
| X–H···Y angle (deg)c | 170–180 | >130 | >90 |
| IR Δν̃X–Hred-field (% of cm–1) | >25 | 10–25 | <10 |
| 1H NMR Δδdownfield (ppm) | 14–22 | <14 | |
| Typical driving force | Orbital and/or electrostatic interaction | Electrostatics | Dispersion |
Estimated values from calculations in the gas phase, roughly synonymous with the enthalpy of formation, ΔH (entropy is often ignored; see also section 2.2).
Exact values of these distances are highly dependent on the van der Waals radii of the elements involved.
Values only apply to singular HBs, not bifurcated and more complex structures.
The strength of a simple HB thus depends on the nature of X and Y, and the bond is strengthened when X is more electron withdrawing and/or when Y is more electron dense. Charge-assisted HBs, where X–H is cationic and/or Y is anionic, are particularly strong.66,67 It is thus no surprise that the interaction energies of simple hydrogen bonding interactions can vary greatly. The interaction energy between the very weakly polarized C–H of methane and the π-bond of, e.g., ethylene is approximately −0.7 kcal·mol–1.48,55,68 Such weak interactions represent the under-boundary of what can be interpreted as a hydrogen bonding interaction and are typically driven by dispersion.55,68−70 While an energy of −0.7 kcal·mol–1 is very small, it must be noted that a difference of 0.5 kcal·mol–1 in a transition state has been reported to impact selectivity in catalysis.71 HBs with more polarized hydrogens, such as in amines, amides, and alcohols, are much stronger and more common. For example, the interaction energy of the water dimer is about −5 kcal·mol–1.72,73 Such interactions are typically driven by electrostatics, and their strength can also be anticipated by a simple inspection of the molecular electrostatic potential maps (MEPs) of the HB donor and acceptor.74−78 The MEP of a molecule can even be obtained with computationally very cheap semiempirical calculations for a qualitative estimation, and such calculations have been correlated to more accurate interaction energies.79 Electrostatic interactions are often dominant in HBs, and HBs have often been recognized as a particular type of “σ-hole” interaction,75,80,81 much like halogen-,82 chalcogen-,83 pnictogen-,84 and tetrel bonding85 interactions.
In its most extreme form, the outcome of a hydrogen bonding interaction can be a proton transfer reaction, where the donor donates a proton to the acceptor via a formally hypervalent [X–H–Y] species.86,87 In the end result of such a reaction, the original donor has become the acceptor and vice versa. As can thus be expected, the pKa of the donor and pKb of the acceptor are correlated with the energy of the HB formed between them.79,88 The proton transfer component also rationalizes the linear directionality of HBs58,59,74,89−95 as well as the elongation of the X–H and shortening of the H···Y bond in strong HBs observed in crystal structures.96−101 The orbital component of HBs can be seen as donation of the electron density of a lone-pair (n) on Y into the antibonding orbital of an X–H σ bond, typically written as n → σ*.81,86
In rare cases where the HB is very strong and symmetrical, the hypervalent species might actually be a stable compound. For example, the [F–H–F]− anion has a “hydrogen bonding” interaction energy of approximately −40 kcal·mol–1,86 which is on par with a weak covalent bond such as the peroxide σ bond in (CH3)3CO–OH (−47 kcal·mol–1)102 and represents the upper-boundary of a single HB.
The bonding strength of a simple HB actually correlates well with the covalent character of the interaction, which can be deduced from the electron density of the H···Y bond.46,70,103−107 This density can be calculated and is often expressed by using the density of bond critical points of Baders’ quantum theory of atoms in molecules (QTAIM),108 which indeed correlates well with the binding energy of a HB.95,109−111
Information about HBs can be gathered using various computational tools (energies and geometries, mostly in the gas phase),70,74,86,87,112 and data from crystal structures provide direct evidence for geometric characteristics and directional preferences.58,59,68,90,91,94,113−117 In solution, which is most relevant to catalysis, infrared (IR)48,60,118−122 and nuclear magnetic resonance (NMR)48,119,122−129 spectroscopy are common techniques to evaluate the presence and binding strength of a HB. The IR spectroscopy stretching vibration of the X–H bond (ν̃X–H) is particularly informative and typically displays a red-shift, broadening and/or intensifying when involved in a HB. In strong HBs, the shift of ν̃X–H can be as large as 2,500 cm–1.118,120 Formation of a HB also has a significant effect on the NMR spectroscopic properties of a proton (and the atoms it is in contact with, X and Y). 1H NMR spectroscopy is, therefore, routinely used to evaluate hydrogen bonding, and downfield shifts exceeding Δδ = 20 ppm have been observed for strong HBs.125
2.2. Factors Influencing the Utility of a HB
A most obvious manner to influence the interaction energy of a simple X–H···Y HB is to adjust the electronic properties of X and/or Y.46,50 In general, for a stronger HB, X has to be more electron withdrawing and Y more electron rich. The properties of X and Y can partially be tuned by choosing the atoms; a nitrogen is more polarizing than a carbon. The hybridization of the donor atom and its further chemical context can also have a large effect on the electrostatic potential on H in the X–H donor. For example, amides are far better HB donors than amines, and carbonyls are superior HB acceptors compared to alcohols.130−133 Using electron withdrawing groups (e.g., a nearby positive charge) can even render an otherwise fairly unpolarized C–H bond into a functional HB donor.56,57,134−143
Computations under idealized gas-phase conditions indicate that the interaction energies of a single HB can be up to −45 kcal·mol–1.48,144 However, such computed interaction energies are best seen as de facto enthalpies (ΔH), not Gibbs free energies (ΔG). Indeed, solution-phase experimental evidence reveals that the ΔG of a single HB is at most approximately −10 kcal·mol–1.122 This 4–5 factor difference between computed (gas-phase) interaction energies and observed Gibbs free energies can in part be understood by the role of the solvent on binding.79,145−149 For example, charges that strengthen a HB in the gas phase are more diffuse when in solution, with the consequence that gas-phase calculations overestimate the reinforcement of a charge on the enthalpy of a HB.150−154 Furthermore, in solution both the HB donor and acceptor will be solvated and association into the intended HB complex will have to overcome this solvation. For example, the interaction energy of perfluoro-tert-butanol hydrogen bonded with tri-n-butylphosphine oxide in vacuo can be calculated at −19 kcal·mol–1 (DFT/B3LYP-D3/def2-TZVPPD), while ΔG has been measured as merely −4.68 kcal·mol–1 in CDCl3 (Ka = 2700 M–1).145 Hydrogen-bonded complexes are thus typically strongest in apolar aprotic solvents such as alkanes and weakest in polar protic media such as water or methanol.79,149
In addition to the solvent effects on the enthalpy, the entropy component (−TΔS) can have a profound impact on the Gibbs free energy. There is an obvious translational entropy penalty of bringing two entities together to form a HB complex, and the magnitude of such a bimolecular association has been estimated to be about 3–9 kcal·mol–1 for simple molecules in solution.79,155−157 Moreover, the conformational freedom can be expected to diminish when a HB complex is tightly bound.158,159 Such a reasoning can explain the often observed inverse proportional relationship between the enthalpy and entropy of formation measured for an adduct.158−164 While often observed, this “enthalpy–entropy compensation” cannot be considered a general feature of molecular associations.165−168 In some instances, entropy can be a substantial driving force of binding, especially when a guest can replace several entropically confined solvent molecules from a binding site.169−172 A similar rationale can be applied to the entropy component of the hydrophobic effect.170,173,174
The entropy component of a HB can be markedly different when a HB is established within the same molecule, as there is no loss of translational entropy.175−177 It is thus unsurprising that intramolecular HBs can be stronger than intermolecular HBs with a similar donor and acceptor, especially if the donor and acceptor are nearby in a conformationally rigid molecule.178−181 Entropy aside, intramolecular HBs have very similar characteristics as their more frequently studied intermolecular counterparts. For example, solvation also tends to weaken intermolecular HBs,147,182 they display similar directionality,95 and computational analysis has shown that the electronic density of the H···Y bond correlates with the strength of the bond.110,183
A useful feature of intramolecular HBs is the possibility to program the conformation of a molecule to steer its structural (pre)organization. This option has been copiously exploited by nature in, for example, protein folding57,184,185 and in the stabilization of transition states that often make proteins such good catalysts.22,24,186−189 The concept of intramolecular preorganization with HBs has also been utilized in crystal engineering,182 in medicinal chemistry,190 and in the design of receptor binding pockets.67,177,191−193 For example, as is illustrated in Scheme 1a, the amides in isophthalamide (1) can be preorganized by intramolecular O–H···O HBs (in blue).193−195 This preorganized structure was shown to bind an order of magnitude more strongly to halide anions compared to an analogue that lacks the two alcohols and is thus not preorganized.193
Scheme 1. Example of Intramolecular HBs (in Blue) and Multipronged HB Donor Groups: (a) an Isophthalamide Derivative (1) Where Both Amides Have Been Preorganized by an Intramolecular O–H···O HB;193−195; (b) General Structure of a Urea Bifurcated HB Donor (2);196,197 (c) General Structure of a Squareamide Bifurcated HB Donor (3);198,199 (d) Diamide 4a, Which Might Be a Bifurcated HB Donor Similar to a Urea but Where an Intramolecular HB Will Preorganize the Diamide into a Different Energy Minimum Conformer (4b)140.

The isophthalamide can also be seen as a trifurcated HB donor (include the CH).
R can be any substituent. Y stands for the acceptor.
The isophthalamide structure shown in Scheme 1a also illustrates that multipronged200 HBs will lead to more stable adducts compared to a simple single X–H···Y HB.48,201−205 In this instance, isophthalamide (1) can be seen as a “bifurcated” HB donor when counting only the amidic H’s but as a “trifurcated” HB donor when also counting the central CH as a HB donor. Other well-known and often used examples of bifurcated HB donors are the ureas (2)196,197 and squareamides (3)198,199 shown in Scheme 1b and c, respectively.
It must be noted that preorganization using intramolecular HBs can also be a disadvantage. For example, as is illustrated in Scheme 1d, one might envisage that a decent bifurcated HB donor such as 4a—very similar to a urea—can be obtained, if two amides are N-linked by a methylene. However, such a motif will result in structure 4b, which is stabilized by intramolecular hydrogen bonding, and the anticipated bifurcated motif will not be the most stable conformer.140 The detrimental effect that intramolecular hydrogen bonding can have on the preorganization of a binding pocket is well-documented, for example in cholic acid derived anion binders.206
For many multipronged HBs as well as for some intramolecular HBs, it is possible to envisage tautomers based on simple Lewis structures. It has been noted that the possibility of resonance structures can have a stabilizing effect on inter-207 and intramolecular HBs.208 For example, drawing tautomers of 2-hydroxy-N-methylbenzamide (5) (Scheme 2a) can rationalize why the intramolecular O–H···O=C HB conformer (top) is about 6.4 kcal·mol–1 more stable198 than the N–H···OH HB conformer (bottom): in the latter, proton transfer would lead to a species with a formal separation of charges.70Intermolecular resonance-assisted HBs include carboxylic acid dimers (6) (Scheme 2b).207,209 The phenomenon has also been described as a stabilizing factor for the secondary structure of proteins210 and in base pairs.211,212
Scheme 2. Examples of Resonance Stabilized HBs: (a) 2-Hydroxy-N-methylbenzamide (5) Is Stabilized by an Intramolecular Resonance-Assisted HB;198 (b) an Interesting Feature of Both Intermolecular and Multipronged HBs Is That They Form a Ringlike Structure as Found for Carboxylic Acids (6).

When multiple HB donors and/or acceptors form an array, such as for carboxylic acid dimers (6) (Scheme 2b), there can be secondary electrostatic interactions70,213 between adjacent donor (D) and acceptor (A) moieties. Note that such arrays are distinct from multipronged HBs, as each HB donor is complemented by one HB acceptor. This is illustrated for the guanine-cytosine base pairs (7–8)213,214 in Figure 2a, where the repulsive secondary interactions are indicated with red arrows (D↔D, but could also be A↔A) and attractive interactions with green arrows (D↔A). The concept of secondary interactions has been used to design synthetic heterodimeric215−217 and homodimeric218,219 systems (e.g., self-complementary AADD218 or ADAD219 HB arrays). As an example, Figure 2b shows an exceptionally stable AAAA–DDDD quadruple HB array (9–10) (Ka > 1012 M–1 in CD2Cl2), where all the secondary interactions are attractive.217
Figure 2.

HB arrays with indication of attractive (D↔A) and repulsive (can be D↔D or A↔A) secondary interactions (a) found in nature between the base pairs guanidine (7) and cytosine (8)213,214 or (b) as implemented in a synthetic system from 9 and 10 with exceptionally high binding affinity exceeding 1012 M–1.217
Cooperativity can be defined as the interplay between two or more interactions that cause a system as a whole to behave differently than what might have been anticipated based on the properties of isolated individual interactions.220 Several elements of cooperativity relevant for a binding site consisting of HBs have already been introduced: multipronged HBs, resonance stabilization, HB arrays, and secondary interactions. However, when a molecule consists of multiple separate binding sites, the molecule can be described as multivalent.221 Multivalency can lead to an additional type of cooperativity, which has been referred to as “chelate-cooperativity”.220,222−224 A most basic example is illustrated in Scheme 3 and involves a bivalent self-complementary molecule that can form a dimer or polymer, depending on the preference for intra- or intermolecular bonding of the second binding event (K2).220−222 The space in between the two binding sites is known to have a large effect on the intra- versus intermolecular association by virtue of the enlarged “effective concentration”221 that the second binding event enjoys.221,224 When designing a binding pocket, such self-complementarity is best avoided. Multivalent cooperativity has been used to generate a large variety of structures based on HB assemblies,225,226 and it has been exploited to make supramolecular polymers.218,227−229 The cooperativity of multivalent binding is copiously exploited in nature,230 also using HBs, such as in the canonical double helical Watson–Crick structure of DNA,207 in protein folding,57,184,185,189 and in the cooperative effects that have been noticed in water clusters.231
Scheme 3. Illustration of So-Called “Chelate-Cooperativity” That Occurs in Multivalent Molecules.

2.3. HBs to Control the Second Coordination Sphere
In this section we have detailed the characteristics of basic HBs and the factors that influence their utility, including more complicated HB structures. The utility of HBs to control the catalyst properties in the second coordination sphere will be detailed in the next two sections. From these surveys, it will become apparent that most examples deploy rather typical HBs of a classical type.
That this presents a clear opportunity is underscored by the fact that some of the most successful examples, whether intended or not, actually rely on one or more of the fine-tuning parameters highlighted in the previous section. For example, the success of P=O bonds (e.g., section 3.1.1, Figure 6, and Scheme 39) can be seen as an example of electronic tuning as P=O bonds are highly polarized and are thus among the best known (neutral) HB acceptor groups.79,145 The impact of solvation and entropy on the utility of HBs to control the second coordination sphere is reflected in the typical choice for noncompetitive (apolar aprotic) solvents such as dichloromethane (DCM), tetrahydrofuran (THF), and alkanes found throughout this review. At the same time, some systems can operate in much more competitive solvents and at higher temperatures (e.g.,Scheme 301, and section 3.2.2.1). It is no surprise that such systems actually rely on strong intramolecular HBs. Similarly unsurprising is the success of catalyst control using multipronged HB donor units derived from ureas (section 3.2.1 and Figure 16), “DIMPhos” (Figure 9), and acyl guanidine (Figure 15). The utility of HB arrays is evident by the examples highlighted throughout this review (e.g., Schemes 22, 23, 29, 33–38, and 62 and Figures 20 and 21), although it must be noted that most of the secondary interactions in these examples are repulsive (which provides a clear opportunity to redesign these structures). Resonance stabilized structures have been used, although sparingly, such as the “6-DPPon” structure (135 shown in Scheme 29). The concepts of structure preorganization with HBs, multivalency, and cooperativity do not yet seem to have been utilized in the catalyst control of the second coordination sphere.
Figure 6.

Formation of the substrate coordinated complex, in which the hydroxy function of substrate 175 forms two HBs with the functional groups of the ligands, as indicated by the dotted lines in the schematic representation of the DFT calculated structure.
Scheme 39. Redesign of the Catalyst, Using Phosphine Oxide-Containing Functional Groups That Are Strong HB Acceptors, Leading to More Active and Selective Catalysts.

A schematic picture is shown displaying how the HBs are formed during the transition state.
Scheme 30. Rh/6-DPPon Catalyst Used in (a) n-Alkene Hydroformylations, (b) 1,1-Disubstituted Allene Hydroformylation, and (c) Alkyne Hydroformylation. (d) The Used Ligands (135 and 135b) Are Shown Together with a Covalent Bidentate Ligand That Was Used for Comparison (BiPhePhos).

Figure 16.

(a) Concept of HB directed selective C–H activation and (b) urea functionalized bipy ligand that forms an iridium complex that displays regioselectivity in C–H borylation reactions.
Figure 9.
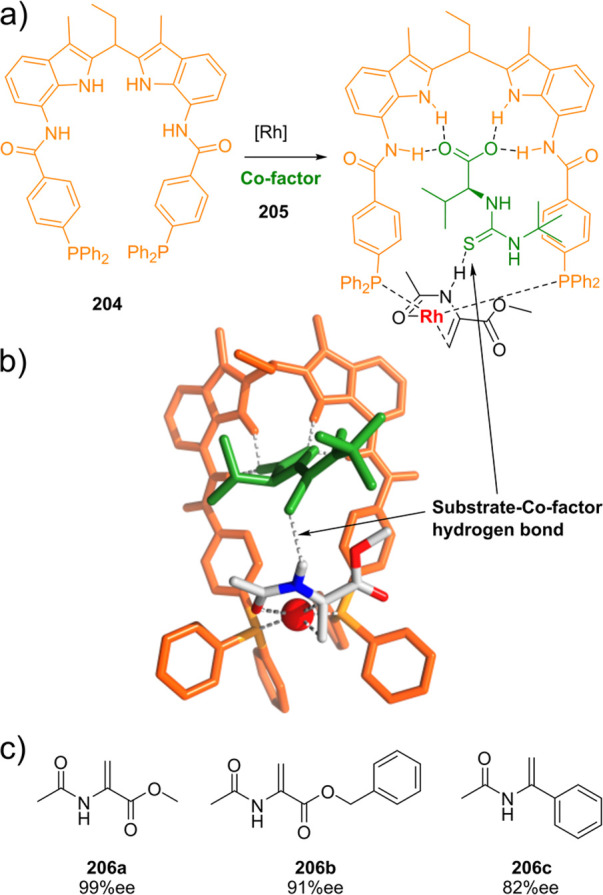
(a) Use of DIMPhos (204, in orange) with a chiral cofactor (green) for rhodium catalyzed hydrogenation. (b) Computed structure showing the HB established between the substrate and the bound cofactor, which is of crucial importance to obtain high selectivity in this reaction. (c) Three substrates that were converted with good to high enantioselectivity using the same cofactor. Adapted with permission from ref (346). Copyright 2014 RSC under a CC-BY-NC license [https://pubs.rsc.org/en/content/articlelanding/2014/SC/C3SC53505C].
Figure 15.
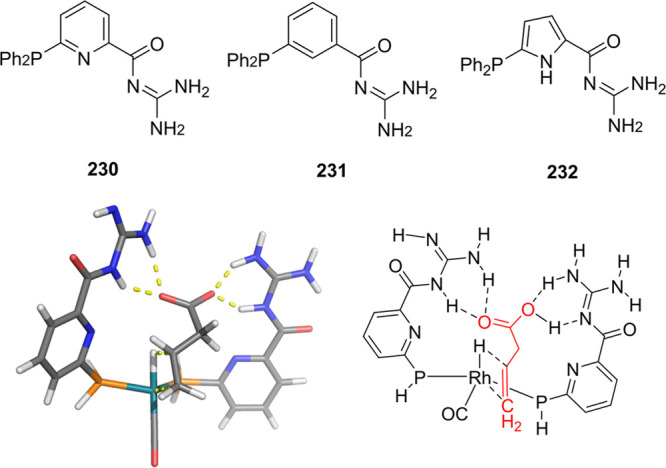
Acyl guanidine functionalized phosphine ligands (230–232) which form HBs to carboxylic acid functionalized substrates. The DFT calculated structure based on 230 (with in silico PPh2 to PH2 mutation) shows that the substrate is preorganized at the rhodium complex by hydrogen bonding, to control the subsequent selectivity determining hydride migration step (see also Figure 10b).
Scheme 22. General Design of Phthalaphos Ligands (110) That Form Supramolecular Bidentate Ligands by Hydrogen Bonding When Coordinated to a Metal (e.g., Rh).

Scheme 23. (a) Concept of the SupraPhanePhos (112). (b) Different Peptidyl Phosphine and Phosphite Ligand Building Blocks Used in the Rhodium Catalyzed Asymmetric Hydrogenation.

Scheme 29. Tautomeric Structure of 6-DPPon (135) and Self-Assembly in the Absence and Presence of a Transition Metal.

Scheme 33. Atropoisomeric Supramolecular Hydrogen-Bonded 6-DPPon-Based Ligands (157) and Their Application in Rhodium Catalyzed Asymmetric Hydrogenation of Methyl Acetamidomethacrylate (158).

Scheme 38. General Structure of SupraPeptiPhos (173), a Supramolecular Bidentate Ligand Based on a Peptide Chain That Mimics the Antiparallel β-Sheet Structures.

Scheme 62. (a) Organic Dyes with a Hydrogen Bonding Site (336a–c) Developed in the Lab of Bach; (b) Intramolecular [2 + 2] Photocycloaddition of Quinoline (337) Proceeds with Higher Enantioselectivity with the More Rigid Xanthone (336b) Compared to Benzoquinone Dye (336a); (c) Substrates Bind to the Lactam Binding Motif by the DA HB Array.

Figure 20.

Crabtree’s dinuclear Mn oxidation catalyst containing a H-bond recognition site.
Figure 21.

Stereoselective C–H oxidation mediated by directional substrate recognition in the second coordination sphere as reported by Bach and co-workers.
The presence of HBs in a metal complex, between the ligand building blocks or between the ligand and the substrate, can be established by a variety of techniques, including X-ray analysis and NMR and IR spectroscopy. Typically, the signals of the hydrogen atoms involved in hydrogen bonding are shifted in both the NMR spectrum and the IR spectrum, and the same holds for the HB acceptor. These signals can be used as a probe, and from titration studies, the HB strength under the used conditions can be established. To what extent the HB in the second coordination sphere controls the activity and the selectivity is more difficult to probe, and typically this information is obtained by comparison of catalysis results with proper control experiments. Job plot analysis is frequently used in supramolecular chemistry to reveal the stoichiometry of the complex, and this could also be used to get insight into HB-containing metal complexes. Extension to information on the active species has been explored by kinetic Job plots, in which the reaction rate is plotted against the fraction of components.232 To the best of our knowledge, such an approach has not been used to evaluate supramolecular bidentate ligands (section 3) or supramolecular catalyst systems that operate via substrate orientation (section 4), but such experiments may provide additional information. Related experiments that have been reported involve a supramolecular bidentate ligand, in which one of the two components is added in increasing amounts, probing the activity and selectivity. These experiments showed in this particular case that the supramolecular ligand was present, even in the presence of an excess of one of the building blocks.233
The above identified examples (detailed in the sections to come) underscore that there is much potential to improve the manipulation and study of hydrogen bonding phenomena in the second coordination sphere. It is thus interesting to keep in mind while reading the next sections that the full potential of hydrogen bonding interactions, using all the known tricks that influence their utility, has not been used yet. For example, HB arrays such as those displayed in Figure 2 could be used to generate supramolecular bidentate ligands. Such motifs will lead to strong bonds between the ligand building blocks, and it is anticipated that this will lead to robust systems that can be applied in polar competitive solvents. The synthesis of such building blocks, however, may be challenging.
3. Hydrogen-Bonded Supramolecular Multidentate Ligands
Bidentate ligands hold a privileged place in most homogeneous transition metal catalyzed reactions as they often yield higher activity and selectivity. However, preparation of large and diverse libraries of bidentate ligands often requires tedious synthetic efforts. To circumvent this challenge, supramolecular bidentate ligands can be used which are functionalized monodentate ligands that self-assemble in situ into a bidentate ligand using noncovalent interactions. Among the different noncovalent interactions, such as coordination and ionic bonds, hydrogen bonding has been frequently used, as it has several favorable characteristics, including (a) predictability, (b) directionality, (c) dynamic bonding, (d) tunability, and (e) various HB donor and acceptor synthons being synthetically accessible.
All those factors led to the rapid development of supramolecular bidentate ligands based on hydrogen bonding in the last decades. Systems based on both single HBs and HB arrays have been reported. This part of the review deals with hydrogen-bonded supramolecular bidentate ligands used in transition metal catalysis. We focus exclusively on reports where experimental evidence is provided for the relevance of hydrogen bonding in a precatalytic state or during catalysis. It is also worth mentioning that sometimes no clear distinction between a hydrogen-bonded supramolecular bidentate ligand and substrate orientation by hydrogen bonding (section 4) can be drawn, as in some specific cases the substrate intercalates into the ligand’s HB network during the catalytic cycle. Those cases will be reviewed in both parts in line with the focus of the section.
3.1. Supramolecular Ligands Using a Single HB
3.1.1. Secondary Phosphine Oxides
Secondary phosphine oxides (SPOs) (11a) are weak acids and subject to tautomerism (see Scheme 4). SPO (11a) is a pentavalent phosphorus oxide, while its tautomer is a trivalent phosphinous acid (PA) (11b). The equilibrium depends on the electronic properties of the SPO, and strongly electron withdrawing substituents on the phosphorus shift the equilibrium toward the trivalent PA form. As the pentavalent oxide form normally predominates, the SPO ligands are air and moisture stable. At the same time, metal coordination can shift the equilibrium toward the trivalent PA tautomer, forming metal–phosphinous acid complexes with a metal-to-phosphorus bond of comparable strength as typical metal–phosphine bonds. The ability to coordinate with both the phosphorus and oxygen creates a rich coordination chemistry of mononuclear and multinuclear complexes of this class of compounds.234 Structural diversification of this class of ligands is possible by exchanging the alkyl or aryl groups, for alkoxy or amide groups which are referred to as heteroatom secondary phosphine oxide (HASPO) ligands (12).
Scheme 4. Tautomerism of (HA)SPO Ligands: (11) R1, R2 = alkyl or aryl; (12) X = O or NR.

One particular feature of (HA)SPO ligands is that they form hydrogen-bonded supramolecular bidentate and tridentate ligands, as first reported in 1975.235 In such a case, both SPO ligands are in the P(III) state and, therefore, have a lone pair of electrons to coordinate to the metal center.236 The addition of 1 equiv of base deprotonates one P–OH, leading to the formation of the H-bond acceptor for the anionic supramolecular bidentate (see Scheme 5). Although different Lewis structures are reported, even within single reports, X-ray crystal structures and DFT optimized structures show that both SPO ligands are in a P(III) state.237−241 For clarity, throughout this review the covalent bond is indicated with a solid line and the HB is indicated by a dashed line. In an anionic supramolecular bidentate of SPO ligands, the HB (and the covalent O–H bond) exchanges between the two oxygen atoms.242 The SPO–bidentate complexes (13a–c) [M{(PR2O)2H}] can be formed by the three different routes detailed in Scheme 5: (a) mixing 2 equiv of SPO (11a) and a metal precursor in the presence of base, (b) mixing 2 equiv of SPO (11a) and a metal precursor containing a basic ligand (such as acetate or methoxide), or (c) mixing a low valent metal precursor with 2 equiv of SPO (11a). For routes a and b, the metal valency remains unchanged, while in method c, the metal center is oxidized, with the concomitant formation of a catalytically relevant metal-hydride species.243 Such a reaction does not require any additional anions, providing additional stability of these complexes. The participation of the P–O–H···O–P six-membered cycle in bonding and reaction mechanisms shows the bifunctional nature of these catalysts, which has been reviewed elsewhere.244
Scheme 5. Formation of Supramolecular Hydrogen-Bonded Bidendate Ligands Using SPO Ligands.

Synthetic approaches involve (a) the use of an external base to deprotonate one SPO, (b) the use of an internal base, or (c) formation of an anionic SPO via oxidative addition. X– can be OAc– or OMe–. In structures 13a–c, the HB exchanges between the two oxygen atoms.
3.1.1.1. Hydroformylation
The first application of SPO ligands in metal catalyzed reactions dates back to the 1980s when van Leeuwen and co-workers reported the Pt/SPO-hydride catalyzed hydroformylation reaction. The active catalyst (15) is formed by mixing Ph2POH and Pt(COD)2 (COD = 1,5-cylooctadiene) under hydroformylation conditions, yielding [PtH(PPh2OH){(PPh2O)2H}] (15, see Scheme 6). Complex 15 catalyzes the conversion of 1- and 2-heptenes to the corresponding linear aldehydes with selectivities of 90% and 60%, respectively.245 These regioselectivities are in the range of traditional bidentate ligand systems such as Xantphos and demonstrate that the hydrogen-bonded bidentate structure remains intact at the reaction temperature 100 °C. Calculations indeed confirm the additional stabilization by the hydrogen-bonded bidentate by 13.0 kcal·mol–1.246 The hydroformylation of ethylene with 15 produces the typical product propanal but also, unexpectedly, the hydroacylated product pentan-3-one, formed by a second ethylene insertion into the Pt–acyl bond. The authors pointed out that the SPO ligands have peculiar properties, capable of assisting in the activation of dihydrogen, which is considered to be the bottleneck in Pt catalyzed hydroformylation (Scheme 6).244,247 Detailed DFT studies suggest that the supramolecular bidentate ligand is maintained throughout the catalytic cycle and that the proton in the P–O–H···O–P six-membered cycle readily migrates between the two oxygen atoms, providing fine-tuning of the electron density in the catalytic cycle at each reaction step.244,248
Scheme 6. SPO-Assisted Hydrogen Activation.

Much later, the interest in rhodium/SPO catalyzed hydroformylation was initiated by the work of Börner and co-workers.249 They studied a limited library of SPO ligands with different electronic properties. The supramolecular bidentate ligand was formed by mixing [Rh(COD)(acac)] (16) with 2 equiv of ligand (11a–e) in which the acac ligand acts as an internal base (Scheme 7, acac = acetylacetonato). SPO ligands with electron withdrawing substituents react rapidly (at −78 °C), while the electron-rich di(tert-Bu)phosphine oxide yields only traces of the [Rh(COD){(tert-Bu2PO)2H}] complex at 80 °C, reflecting the tautomeric equilibrium (not shown in Scheme 7).250 Hydroformylation reactions with complexes 17a–d were performed using cyclohexene and 1-octene. The substrate cyclohexene was readily converted to the aldehyde with yields up to 50%. These results are superior to the benchmark Rh/PPh3 catalyst, yielding only 18% under identical conditions with cyclohexene as substrate. In contrast to traditional hydroformylation catalysts that are more active when the ligands are electron poor, more electron-poor SPO ligands resulted in only 7% conversion. Using 1-octene as substrate, the aldehydes were produced in 88% yield, albeit with a moderate selectivity for linear aldehydes due to considerable olefin isomerization. The rhodium-HASPO catalyst (17e) was evaluated in the hydroformylation of 1-octene, providing 91% conversion to the aldehydes, of which 38% favor the linear aldehyde (in THF, 100 °C, 50 bar syngas (CO/H2 = 1:1), Rh/olefin = 1:8000, b/1 = 1:4).251
Scheme 7. Formation of Rh-(HA)SPO Catalysts (17a–e) Used for the Hydroformylation of 1-Octene.

Due to the acidic P–H, (HA)SPOs can add to olefins or aldehydes via their PA tautomer to produce α-hydroxyphosphine oxides by an Abramov or Pudovik reaction which is reversible at elevated temperatures and thus forms a reservoir of SPO ligands (11) (Scheme 8). It was shown that the SPO ligands are liberated during product distillation and act as a stabilizing ligand for the rhodium catalyst, improving the thermal stability and recyclability of the precious metal catalyst.252
Scheme 8. Reaction between Aldehydes 18 and SPO 11 (as the Acid Tautomer) to Produce α-Hydroxyphosphine Oxides 19.

3.1.1.2. Hydrogenation
The hydrogenation of aldehydes by metal-SPO catalysts has been a topic for both academic and industrial groups. An early patent by Shell describes the use of [PtH(PPh2OH){(PPh2O)2H}] catalyst (15) for the hydrogenation of a range of aldehyde and ketone substrates with high activity and high chemoselectivity.253tert-Amyl aldehyde 20 can be efficiently converted to the corresponding alcohol 21 with a turnover frequency (TOF) of 9000 molsubstrate/molcatalyst·h–1. Interestingly, both the hydroformylation reaction and the aldehyde hydrogenation reaction are catalyzed by the same catalyst (15), and this potentially opens the pathway to perform the sequential hydroformylation/aldehyde reduction in a one-pot procedure (Scheme 9).
Scheme 9. Aldehyde Reduction by Catalyst 15 (See Also Scheme 6).

Iridium-SPO catalysts have also been applied in the chemoselective aldehyde hydrogenation. Treatment of the (COD)(methoxy)iridium(I) dimer with 4 equiv of tert-butyl(phenyl) phosphine oxide and 2 equiv of water in THF at room temperature affords supramolecular bidentate complex [Ir(COD){(P(t-Bu)PhO)2H}] (22, Scheme 10).254 This complex is the precursor to a catalyst that forms under 5 bar hydrogen atmosphere leading to a mixture of monohydride, diastereomeric dihydrides, and three bridging dihydride dimer complexes.255 Interestingly, the oxidative addition of dihydrogen to the Ir(I)-SPO complex (which contains the achiral SPO) is highly stereoselective, as all generated Ir(III) hydride complexes are homochiral and no meso isomers are detected.
Scheme 10. Aldehyde Reduction by Ir-SPO Catalyst (22).

As is shown in Scheme 10, deploying 22 as catalyst precursor gave highly chemoselective conversions of a variety of aldehydes using mild reaction conditions (25 °C and 5 bar hydrogen pressure). The reduction of the aromatic α,β-unsaturated cinnamaldehyde (23a) to the cinnamyl alcohol (24a) could even be established, thus leaving the C=C double bond untouched, with >99% selectivity and a TOF > 2000 molsubstrate/molcatalyst·h–1. p-Nitrobenzaldehyde (23b) is selectively converted to nitrobenzyl alcohol (24b) with perfect retention of the nitro group (selectivity >99%). Also, other groups such as nitrile (23c) and ester groups (23d) are well tolerated, as these substrates are converted with nearly perfect chemoselectivity. Aliphatic α,β-unsaturated aldehydes are also readily reduced to their alcohols (24e–g), as is the furan analogue (24h). The notoriously difficult to hydrogenate substrate 2-thiophenecarboxaldehyde (23i), a known poison to homogeneous catalysts, is even converted to the corresponding alcohol (24i), and this shows the versatility of this catalyst system. The mild reaction conditions and the absence of base invoke that the supramolecular anionic bidentate ligand is involved in ligand-assisted hydrogen splitting.
3.1.1.3. Hydrogen Transfer Reactions
Van Leeuwen and co-workers reported the rhodium(III) catalyzed transfer hydrogenation of ketones in isopropanol using SPOs and HASPOs, and although this is not covered in general in this review, it is briefly mentioned here in the context of this class of ligands.238 Out of an initial catalyst screening using various metal salts and the diphenylphosphine oxide ligand, using RhCl3 resulted in the highest activity using cyclohexanone and benzophenone as benchmark substrates (Scheme 11). Under optimal conditions, cyclohexanone was reduced with a 92% conversion and a TOF of 1825 molsubstrate/molcatalyst·h–1. It turned out that the Rh/SPO ratio is crucial for good catalytic activity and was therefore optimized for every substrate. Spectroscopic studies confirmed that the neutral dinuclear complex (25) was formed, which interestingly bears a supramolecular bidentate ligand on one rhodium center and a supramolecular tridentate ligand on the other. The corresponding hydride complex (28) can be obtained by a reaction with butoxide as base. The asymmetric transfer hydrogenation of acetophenone was also studied using the chiral HASPO ligands 26 and 27, yielding the product with an enantiomeric excess of 89%.
Scheme 11. Formation of a Neutral Dimeric Complex by Reaction of Diphenyl Phosphine Oxide (11a) with RhCl3 and Subsequent Generation of the Rhodium-Hydride Species.

3.1.1.4. Nitrile Hydration
The selective hydration of nitriles is an atom-economical and green way to produce N-unsubstituted amides which represent an important class of compounds. Parkins and Ghaffar showed that the platinum-hydride complex of dimethylphosphine oxide [PtH(PMe2OH){(PMe2O)2H}] (29a) and the corresponding chloride complex [PtCl(PMe2OH){(PMe2O)2H}] (29b) are active catalysts for the hydration of nitriles.256−258 As is illustrated in Scheme 12, in complexes such as 29 and 30 two SPO ligands are coordinated as hydrogen-bonded bidentate and one ligand is coordinated in a monodentate fashion which are in fast exchange at room temperature on the NMR spectroscopy time scale.
Scheme 12. Fast Exchange between the Supramolecular Hydrogen-Bonded Bidentate and Monodentate Phosphinous Acid Ligands in Complexes Such as 29a (X = H) and 29b (X = Cl; See Also Scheme 13 for 29c with X = OH).

A plausible mechanism of nitrile hydration is given in Scheme 13 starting from 29c where X = OH. The mechanism involves the initial coordination of the nitrile moiety (32) to the cationic platinum center (31), after which the nitrile is susceptible to nucleophilic attack.256,259 A key step in the suggested mechanism is the intramolecular nucleophilic attack of the hydroxy group of the coordinated phosphinous acid on the coordinated nitrile to give a five-membered ring (34). After attack of a water molecule, the N-unsubstituted amide is liberated and the cationic Pt species reforms, which completes the catalytic cycle. The size of the substituents on phosphorus has a large effect on the catalytic activity, and the highest activity was obtained with the ligand with the least sterically hindered groups studied, i.e, dimethyl phosphine oxide.
Scheme 13. Hydration of Nitriles as Proposed by Parkins256,259.

As is shown in Scheme 14, the [PtH(PMe2OH){(PMe2O)2H}] catalyst (29a) selectively hydrates different nitrile substrates in water, aqueous ethanol, or aqueous THF at 70–90 °C, yielding the amides as the only product in high yield with no further hydrolysis of the corresponding acids. Interestingly, nitrile hydration of acrylonitrile by 29a proceeds smoothly with perfect chemoselectivity, leaving the carbon–carbon double bond untouched. The reaction only required 0.0013% catalyst, giving an impressive turnover number (TON) of 77.000. Also, sterically hindered tertiary nitriles and nitriles containing acid- or base-sensitive functional groups can be converted with excellent yields and chemoselectivities (see Scheme 14).260,261 Even the sensitive d-amygdalin (36p) was converted to the amide in 98% yield without racemization of any of the stereogenic centers in the sugar moieties.
Scheme 14. Substrate Scope of the Nitrile Hydration by the [PtH(PMe2OH){(PMe2O)2H}] Catalyst (29a)256−258,260−263.

Due to the exceptionally high functional group compatibility combined with the high activity, nitrile hydration has been a catalyst of choice in the synthesis of a large number of natural and biologically active products of elaborate structure.259 From the many examples reported in the open and patent literature summarized in Table 2, it is clear that the catalyst can tolerate a multitude of protecting groups but also tolerates strained ring systems (cyclopropyl) and activated C=C double bonds prone to Wacker oxidation and that even oxiranes are tolerated. Interestingly, the synthesis of 8-azabicyclo[3.2.1]octyl-2-hydroxybenzamide (57) includes a rare example of the hydration of thiocyanate (entry 17 in Table 2).
Table 2. Overview of Natural Product Synthesis Using Parkins’ Hydration Catalyst [PtH(PMe2OH){(PMe2O)2H}] (29a).




Attempts have been made for the nitrile hydration in a kinetic resolution of 2-phenyl proprionitrile using (R)-(+)-(t-Bu)PhP(H)O (60) as chiral ligand (see Scheme 15, top).261 Analysis of the reaction shows that only the racemic product is formed, as the chiral SPO-ligand (60) was racemized under the reaction conditions. Interestingly, van Leeuwen showcased the possibility of kinetic resolution in the hydration of the racemic 1,1′-binaphthalene-2,2′-dicarbonitrile (61) in tert-amyl alcohol, illustrated in Scheme 15, bottom.285 The cationic platinum catalyst [Pt(PR2OH){(PR2O)2H}(solvent)] could be prepared in situ from either Pt(PPh3)4/(27) or Pt(COD)Cl2/AgNO3/(27) (for ligand 27 see also Scheme 11). In the hydrolysis reaction of 61, only three compounds were observed in the reaction mixtures: enantioenriched dinitrile 61, mononitrile-monoamide intermediate 62, and the diamide 63. With the Pt(PPh3)4/27 catalyst at 83% conversion, the enantiomeric ratios for the three products are 2/98, 86/14, and 76/24, respectively.
Scheme 15. Attempted Kinetic Resolution in the Hydration of 2-Phenyl Proprionitrile Using PtCl2/60 (Top) and in the Hydration of 1,1′-Binaphthalene-2,2′-dicarbonitrile (61) Using Pt(PPh3)4/27 (Bottom).

Another interesting class of nitrile substrates to hydrolyze is the cyanohydrins, as it affords an atom-economical route to high value α-hydroxyamides, α-hydroxycarboxylic acids, and α-hydroxycarboxylic esters. An acid-free catalytic process is desirable as it reduces the number of side reactions and eliminates the stoichiometric formation of salts or alkyl chlorides.
Although the overall reactivity of [PtCl(PMe2OH){(PMe2O)2H}] (29b) in the hydrolysis of cyanohydrins is low, 29b outperforms previously reported nitrile hydration catalyst Cp2Mo(OH)(OH)2 (64).286 The low reactivity of cyanohydrins is due to the liberation of hydrogen cyanide (HCN) by an undesired background reaction, which commonly leads to catalyst deactivation (cyanide coordination). As illustrated in Scheme 16, Brown and Konopelski bypassed this issue in their route to the total synthesis of Psymberin/Irciniastatin A by immediately protecting a cyanohydrin derived from 65 with a tert-butyldiphenylsilyl group in 66.287 Subsequent nitrile hydrolysis of 66 using Parkins’ catalyst gave the separable diastereoisomeric amides 67 and 68 in 73% yields.
Scheme 16. Syn-Selective Cyanide Induction of 65 to Produce the Unstable Cyanohydrin Which Was Immediately Protected by TBDPSCl (tert-Butyl(chloro)diphenylsilane) to Give 66, Which Was Subjected to Nitrile Hydrolysis Producing 67 and 68.

The nitrile hydration has also been extended to one-pot sequential reaction methodologies. In this light, de Vries and co-workers reported the direct conversion of a number of unactivated nitriles to N-substituted amides with both primary and secondary amines, as exemplified in Scheme 17.288 The reaction is initially very fast and can be performed with catalyst amounts as low as 0.02 mol %. However, the ammonia produced by a reaction of the primary unsubstituted amide product with the amine leads to an appreciable reduction of the reaction rate as the reaction progresses. Primary and secondary amines work equally well; albeit, more forcing conditions are required for high conversions. The reaction of succinonitrile with pyrrolidine and water in DME at 160 °C catalyzed by [PtH(PMe2OH)(PMe2O)2H] (29a) gave the corresponding substituted amide in 89% isolated yield after 24 h.
Scheme 17. Direct Conversion of an Unactivated Dinitrile to Provide N-Substituted Amides Using 29a (See Scheme 12).

The next example of the sequential reaction methodology that involves the nitrile hydration is the facile formation of 1-alkoxyisoquinolines and (2H)-isoquinolones by an intramolecular 6-endo-dig cyclization of o-alkynylbenzonitriles catalyzed by [PtH(PMe2OH){(PMe2O)2H}] (29a).289 An overview of the scope of this reaction is provided in Table 3. Substrates bearing both electron donating and electron withdrawing substituents at the para-position with respect to the alkenylphenyl moiety (R1 in Table 3) gave the cyclized products with comparable yields (entries 2 (72b) and 3 (72c) of Table 3) whereas substituents in the ortho-position hampered the cyclization process. In these reactions both the 1-alkoxyisoquinolines (73a–j) and the isoquinolones (74a–j) were isolated and the former could be efficiently converted into the isoquinolones in a subsequent reaction with HBr in acetic acid. Finally, cyano pyridines (X or Y = nitrogen) can be converted under the used reaction conditions. This approach has also been applied for the synthesis of heterocyclic antiviral compounds patented by Hoffmann-La Roche.290
Table 3. Scope of the Cyclization of o-Alkynylbenzonitriles (X and Y Can Be CH or N) and Amides Using 29a (See Scheme 12).

| Entry | Compound | R1 | R2 | Yield of 73a–l (%) | Yield of 74a–l (%) |
|---|---|---|---|---|---|
| 1 | 72a | Ph | H | 48 | 15 |
| 2 | 72b | p-MeO-C6H4 | H | 31 | 6 |
| 3 | 72c | p-F-C6H4 | H | 40 | 5 |
| 4 | 72d | o-CH3-C6H4 | H | 0 | 0 |
| 5 | 72e | o-Cl-C6H4 | H | 0 | 5 |
| 6 | 72f | 3-Pyridyl | H | 30 | 12 |
| 7 | 72g | 3-Thiophene | H | 49 | 14 |
| 8 | 72h | Cyclohexyl | H | 43 | 19 |
| 9 | 72i | Ph | 3-F | 42 | ∼5 |
| 10 | 72j | Ph | 5-CF3 | 37 | 11 |
| 11 | 72k | Ph | 5-MeO | 16 | <5 |
| 12 | 72l | Ph | 4-Me | 40 | 16 |
3.1.1.5. Oxidation
Nuel, Giordano, and co-workers reported a Pd(II) catalyzed oxidation of alcohols using catalyst (75) with regeneration of the active Pd species through hydrogen transfer to an alkene (see Scheme 18).291 This so-called “hydrogen abstracting methodology” (HAM)292 is performed under relatively mild conditions at neutral pH and allows for the wide substrate scope displayed in Scheme 18. Benzylic alcohols 76a–g and 76n, aliphatic cyclic alcohols 76h–m, o, u, and v, and acyclic secondary alcohols 76p–t, including sterically congested substrates, are oxidized to the corresponding ketones. Interestingly, under the applied reaction conditions the nitrile group remained unaffected, which is evidenced by the conversion of 76r to 77r in 93% yield. Also, other strongly coordinated moieties did not hamper the oxidation, and the alcoholic substrates containing secondary sulfide, sulfoxide, and morpholine moieties are readily oxidized, showing the potential of the approach. Also, diols containing both primary and secondary alcohol functions were converted with high chemoselectivity; that is, the secondary alcohol was converted to the corresponding ketone while the primary alcohol remained untouched (76o).
Scheme 18. Scope of Pd Catalyzed (75) Alcohol Oxidation Reaction Showing Reaction Products.

Ketones 77x–y were produced using a similar Pt catalyst ([Pt(OH){(P(t-Bu)(Ph)O)2H}] (78a).
After initial hydrolysis of complex 75, the proposed mechanism starts with the catalytically active monomeric [Pd(OH)(P(t-Bu)PhO)2H] (78a), which reacts with the substrate alcohol, thereby producing the ketone and yielding the corresponding [Pd(H){(P(t-Bu)PhO)2H}] complex 79. This complex then reacts with the hydrogen acceptor (methyl vinyl ketone) 80 in a stepwise 1,4-addition to produce methylethyl ketone 84 and concomitantly reform the [Pd(OH){(P(t-Bu)PhO)2H}] (78a) which closes the catalytic cycle (Scheme 19).
Scheme 19. Proposed Mechanism of the Hydrogen Abstracting Methodology.

Similar reactivity has been found for platinum complexes. For example, in situ formed hydroxy-platinum [Pt(OH){(P(t-Bu)(Ph)O)2H}] catalyst 78b was found to be a superior catalyst for the aerobic/anaerobic oxidation of challenging substrates such as N-alkyl-2,2,6,6-tetramethylpiperidin-4-ols (76x) and analogues, which are smoothly oxidized to the corresponding ketone (77) at room temperature in good to excellent yields (67–99%).239 The corresponding palladium complexes were not active under the applied reaction conditions. This study was further extended to the oxidative defragmentation of N-alkyl-2,2,6,6-tetramethylpiperidin-4-ols (76x), which could be accomplished in a two-step one-pot process.293 The [Pt(OH){(P(t-Bu)(Ph)O)2H}] catalyst (78b) plays a dual role, and the supramolecular hydrogen-bonded ligand acts as a hydrogen source and the cationic metal center as Lewis acidic site. Under optimized reaction conditions the substrate N-benzyl-2,2,6,6-tetramethylpiperidin-4-ol (76y) was oxidized by the in situ formed [Pt(OH){(P(t-Bu)PhO)2H}] to the corresponding ketone N-benzyl-2,2,6,6-tetramethylpiperidin-4-one (77y) under basic conditions within 4 h at 105 °C. Subsequent addition of 5 equiv of acetic acid and hydrogen acceptor trans-phenylbut-3-en-2-one followed by another 5 h of reaction time lead to the fragmentation of the ketone 77y to obtain phorone 2,6-dimethylhepta-2,5-dien-4-one (79) and benzylamine. The goal of the authors was to isolate the liberated alkyl amine, which was isolated conveniently by a simple acid/base extraction in 86% yield.
With this Pt catalyst system (78b), also challenging substrates such as 1,2- or 1,3-diols can be converted into α- or β-hydroxy ketones in moderate to good yields (44–89%). The relatively mild reaction conditions also allow the oxidation of substrates containing base-sensitive functional groups such as esters. For example, ethyl 3-hydroxycyclohexane-1-carboxylate (85) is readily oxidized to the corresponding ketone.239 The platinum-SPO catalyzed oxidation proceeds via a proposed mechanism similar to the palladium catalyzed oxidation (vide supra).
3.1.1.6. Cross-coupling
The palladium- and, to a lesser extent, the nickel catalyzed cross-coupling chemistry have been extensively studied using SPO ligands, and the literature has been reviewed previously.236,294 One of the earliest examples is provided by Li and co-workers,294 who demonstrated that the coupling of aryl chlorides and tert-butyl acrylate in the presence of K2CO3 in N,N-dimethylformamide (DMF) could be accomplished using 1.5 mol % of the palladium dimer catalyst (86), resulting in α,β-unsaturated esters in good yields. A proposed mechanism for this reaction is shown in Scheme 20. Further research has shown that this catalyst is active in a wide variety of C–C, C–N, and C–S bond forming processes involving aryl chlorides. The high activity toward the aryl chlorides is attributed to the cleavage of the palladium(II) dimer 86 and deprotonation of both phoshinous acid ligands, yielding an electron-rich phosphine-containing anionic complex (87 or 90), which accelerates the rate determining oxidative addition of aryl chlorides in the catalytic cycle (from 87/90 to form 88/89). Although the results show the potential of SPO-containing catalysts for cross-coupling reactions, the supramolecular bidentate structure is not maintained during catalysis.
Scheme 20. Proposed Mechanism for Pd-SPO (86) Cross-coupling Chemistry by Li et al.294.

3.1.2. Phosphine-Phosporamidite Single HB Systems
Reek and co-workers reported in 2009 the formation of supramolecular heterobidentate ligands formed by a single HB.295,296 As is shown in Figure 3a, leucine-based phosphoramidite ligand 93 contains a strongly polarized N–H that forms a HB with the carbonyl group of the urea functionalized phosphine ligand (94) when coordinated to a rhodium center. The existence of the HB is also clear from the X-ray structure (see Figure 3c). This HB was also observed by IR spectroscopy in the [Rh]BF4 complex and confirmed by DFT calculations to be more stable than the N–H···O=Curea HB in the absence of the metal. In the IR spectroscopic data, the IR band corresponding to the ester moiety remains unchanged whereas the band corresponding to the carbonyl of the urea shifts from 1703 to 1681 cm–1, indicating that the urea is involved in a HB as a proton acceptor. The phosphoramidite ligand building block (93) as well as the phosphine building block (94) can be varied by changing the electronic and steric properties, and various combinations were studied in the rhodium catalyzed hydrogenation of methyl 2-hydroxymethyl acrylate (96) (Roche ester precursor).295 Among the combinations, the supramolecular bidentate ligand based on (S,S)-Leuphos (93) and the monourea triphenylphosphine 94 gave the highest enantioselectivity (99%+ e.e.), while only (S,S)-Leuphos (93) as ligand showed only 31% e.e., as is shown in Figure 3b. Further detailed investigation of this catalytic system led to the observation that substrate orientation by hydrogen bonding between the functional group of the substrate and the ligands is important in achieving high selectivity, and this will be discussed in section 4.1.
Figure 3.

(a) Ligand building blocks that form single hydrogen-bonded bidentate ligands when coordinated to rhodium. (b) Rhodium catalyzed asymmetric hydrogenation of Roche ester precursor by supramolecular bidentate ligand. (c) HB is indicated by a dotted line in the X-ray structure.
3.2. Supramolecular Ligands Using Multiple HBs
Ding and co-workers explored the use of phosphoramidite (DPenphos)-type ligands in the asymmetric hydrogenation of (Z)-methyl α-(acetoxy)acrylates and (E)-α-aryl itaconate derivatives containing various substituents.297 Catalysts based on (R,R)-DPenPhos-H (99) provided full conversion and excellent enantioselectivity (94–99%) (see Scheme 21). The enantiomeric excess induced by the Rh((R,R)-DPenPhos-H systems was not majorly affected by the substituent on the phosphoramidite nitrogen atom. DFT calculations and NMR spectroscopic studies confirmed the presence of N–H ···O HBs between two coordinated DPenphos ligands (see Figure 4 and Scheme 211) exhibiting the relatively small interligand bite angle of 89.9°. The proximity of the two ligands to the substrate as programmed by the HBs subtly influences catalyst structure and reactivity, and according to the authors, the hydrogen-bonded bidentate structure is maintained under the hydrogenation conditions (employing nonpolar solvents). Remarkably, the closely related (R,R)-DPenPhos-Me and Monophos did not display any reactivity under the applied reaction conditions; both are based on the dimethylamino-phosphoramidite and, thus, are not able to form the hydrogen bonding bidentate structure. Later, the substrate scope was further extended to the asymmetric hydrogenation of (E)- or (Z)-β-substituted dehydro-β-amino acid esters, and it was again reported that the N–H moiety in the phosphoramidite ligand is critically important for achieving high activity, and catalysts based on ligand 99f provided 92–96% e.e.298 The versatility of the Rh/DPenphos-H catalyst system was further demonstrated by the asymmetric hydrogenation of α- or β-acyloxy α,β-unsaturated phosphonates (100) and α- and β-enamido phosphonates (101).299−301 During these studies, it surfaced that the ligand (R,R)-DPenPhos-H (99) provided superior enantioselectivities (96–99+% e.e.) for a large substrate scope consisting of 50 entries and asymmetric hydrogenation of enol esters (100, 102, 104, and 108) with enantioselectivities between 87 and 95% e.e. Besides benchmark reactions, Rh/(99b) has also been successfully applied in the synthesis of biologically active compounds such as the Danshensu–cysteine conjugate, which has considerable interest due to its various biological activities, such as antioxidative compounds and daidzein derivatives.302,303
Scheme 21. (R,R)-DPenPhos-Me (98) and (R,R)-DPenPhos-H (99) Ligands Developed by Ding et al. and Applied in the Asymmetric Hydrogenation of Different Substrate Classes.

Figure 4.

Structure of Rh[DPenPhos-H (99b)]2 (left-hand side) and its B3LYP/6-31G(d) optimized structure using a structural mimic (right-hand side). For both, the COD ligand is omitted for clarity. Adapted with permission from ref (297). Copyright 2006 American Chemical Society.
In the 2010s, Gennari and co-workers reported Phthalaphos ligands, which are binol-derived phosphites linked to a phthalic amide moiety (110); see Scheme 22.304,305 These ligands form a supramolecular bidentate complex based on HBs between two phthalic amides, as is shown on the right-hand side of Scheme 22. The authors investigated the hydrogen bonding of a Phthalaphos rhodium precatalyst by 1H NMR and IR spectroscopy and confirmed the supramolecular bidentate behavior in the precatalyst complex. A library of these ligands consisting of 19 ligands was studied in the rhodium catalyzed hydrogenation of several prochiral dehydroamino esters and enamides, yielding excellent e.e.’s of up to 99% for the benchmark substrates (e.g., methyl 2-acetamido acrylate, methyl (Z)-2-acetamido cinnamate, and N-(1-phenylvinyl)acetamide). Also, challenging and industrially relevant substrates such as N-(3,4-dihydronaphthalen-1-yl)-acetamide and methyl (E)-2-(acetamidomethyl)-3-phenyl acrylate could be converted with 96% and 99% e.e., respectively. DFT calculations suggest that the phthalic acid amide is also involved in the substrate preorganization, at the expense of the supramolecular bidentate ligand, and is responsible for the observed high selectivities. The substrate preorganization will be discussed in more detail in section 4.
Another explored strategy using amides as HB motif was developed by Breit and co-workers in 2008.306 As is illustrated in Scheme 23, “SupraPhanePhos” (112) is a motif structurally similar to regular “PhanePhos” (111) (a well-known covalently linked bidentate ligand) that is held together by HBs with its peptidic fragment. X-ray spectroscopic analysis of a complex formed after SupraPhanePhos coordination to PtCl2 revealed a helical conformation of the hydrogen-bonded peptidyl chains. 1H NMR spectroscopic studies further confirm that this conformation remained intact in solution. Screening of binary mixtures of SupraPhanePhos (112) ligands in the rhodium catalyzed asymmetric hydrogenation of a number of benchmark substrates (e.g., methyl N-acetyl dehydro-amino acids and dimethyl itaconate) showed that the peptidyl phosphite-based complexes gave a selectivity of up to 99%, which is higher than that obtained using phosphine analogues. The enantioselectivities are moderate (up to 51%), yet these results are interesting because the chiral centra in SupraPhanePhos complexes are at least seven atoms away from the active metal site. The authors observed a match/mismatch effect by changing the binol enantiomer in combination with the chiral peptide. Also, for one of the reactions the complex based on the ligand without any peptidyl chain gave the highest e.e., suggesting a negative effect of the supramolecular chelate ligand.
The chirality transfer between the distant chiral peptidyl chain and the metal center, also referred to as “backdoor induction” but perhaps better described by the “chirality relay mechanism”, was further developed by Kirin and co-workers using a slightly modified SupraPhanePhos ligand design. Most notably, in their approach, Kirin and co-workers installed the phosphine group para with respect to the peptidyl chain. This ensures that the ligand is relatively insensitive to rotation around the phenyl–peptidic C–C bond. Another feature of Kirin’s design is that the peptidyl chains are terminated by an amide group for a more extended hydrogen bonding array (Kirin set 1, see Scheme 24).307 Asymmetric hydrogenation of methyl 2-acetamidoacrylate using Rh complexes of their ligands resulted in moderate e.e.’s, which were nonetheless an improvement over an analogous complex reported by the Breit group (from 51% to 68% e.e.). The strategy was extended to bispetidyl phosphine ligands with amino acids (117a) and dipeptidyl (117b) and tripeptidyl chains (117c). After complexation to [Rh(COD)2]BF4, these ligands form intermolecular hydrogen-bonded β- or γ-turns.308 Both unary and binary mixtures were studied in the Rh catalyzed asymmetric hydrogenation of a number of benchmark substrates yielding up to 80% e.e.
Scheme 24. Chirality Induction by Distant Chiral (Peptidyl) Groups as Developed by Kirin and Coworkers.

Designs using a more rigid backbone based on cyclic diamine (Kirin set 2; see Scheme 24) provided catalysts that induced higher e.e. in the conversion of the same substrate classes (up to 92% e.e.) and up to 97% e.e. in the case of the phenyl derivative.309 Characterization of the hydrogen bonding of the ligand building block by NMR and IR spectroscopy indicated that only one amide N–H was involved in the interligand hydrogen bonding and one amide proton is not hydrogen bonded. For the square planar Pt and Rh complexes, circular dichroism showed a metal–ligand absorption around 435 nm corroborating the relay of chirality to the metal complex.
3.2.1. Urea-Based Supramolecular Bidentate Ligands
Reek and co-workers reported supramolecular bidentate P-donor ligands based on ureas as a self-complementary hydrogen bonding motif. The targeted synthons to build such ligands are selected to be either commercially available or easily prepared. This makes the approach modular, and with it, it is easy to build a large and diverse ligand library that easily exceeds 100,000 members. These “UREAPhos” ligands are easily accessible by simple “click”-type reactions allowing parallel automated synthesis of 32 new ligand building blocks in a single day, highlighting the potential of this approach.310 Assembly of two of such phosphine ligand building blocks allowed the formation of hydrogen-bonded chelating structures in the presence of Pd and Rh complexes, as illustrated in Figure 5 and evidenced by detailed NMR spectroscopic studies.311
Figure 5.

Self-assembly of UREAphos ligand systems to form supramolecular bidentate ligands in the presence of palladium or rhodium precursors (M).
In two separate studies the use of a significant library of 18 structurally related ligands (summarized in Scheme 25) was explored in the asymmetric hydrogenation of industrially relevant prochiral substrates, revealing that the products were formed in high selectivities.310,312−314 Importantly, small changes in the spacer unit between the donor atom and the urea motif resulted in large variation in the catalytic performance of the related rhodium complex, making variation in the UREAphos structure relevant. The observed enantioselectivities are good to high, also for the inherently difficult substrates (119e) and the tetrasubstituted substrate (119c), which were hydrogenated to form the product in 97% and 87% e.e., respectively. For 119e, a substrate–ligand interaction has been invoked based on NMR and IR spectroscopic studies.314 These results support the significance of generating large and diverse catalyst libraries for lead discovery. After optimization of the reaction conditions for two successful catalysts, it was demonstrated that the catalyst activity is easily improved by varying the temperature. As the UREAPhos ligands are prepared by simple reactions steps, scale-up of the catalysts is relatively easy.
Scheme 25. Asymmetric Hydrogenation Reaction of a Series of Substrates (119) to Useful Products (120) Using a Library of Rhodium Catalysts Based on Self-Assembled Bidentate UREAPhos Ligands (121).

Chikkali and co-workers further extended the approach to P-chiral UREAPhos ligand building blocks to form complex 122 shown in Scheme 26.315 Formation of 122 upon coordination with [Rh(COD)2]BF4 was confirmed by 1H NMR and IR spectroscopy, and upon application in the hydrogenation of dimethylitaconate, the complex gave only 33% e.e. whereas N-acetyldehydrophenylalanine was converted with 99% e.e.. Further investigations suggest that with this substrate’s HBs between the substrate and the urea moiety of complex 122 are responsible for the high enantioselectivity observed.311
Scheme 26. P-Chiral UREAphos and the Application in the Rhodium Catalyzed Hydrogenation of Dimethylitaconate and N-Acetyldehydrophenylalanine.

Supramolecular hydrogen-bonded bidentate ligands using the urea binding motif were also exploited to generate the self-assembled hetero-bidentate ligands illustrated in Scheme 27. Using UREAphos ligands 121t or 121u and anionic urea-containing ligands 127a–b, a small library of palladium complexes was generated (128a–c and 129a–c), forming a supramolecular SHOP-like Pd complex.316 The existence of the hydrogen-bonded bidentate structure was confirmed by an array of spectroscopic and DFT studies. Catalysts 128 (with DMSO as solvent) were applied in the Pd catalyzed ethylene polymerization, and 128a was found to be the most active catalyst, leading to the production of highly branched polyethylene with a molecular weight of 55.700 g/mol and melting temperature of 112 °C.
Scheme 27. Self-Assembly of Anionic Urea-Containing Ligands and UREAPhos Ligands to Generate a Small Library of Palladium Complexes (128a–c and 129a–c) (Solvent Is DMSO or Pyridine, Respectively).

Further diversification of the coordination moiety of the urea-based ligand building blocks was done by Piarulli and co-workers by generating the urea-based oxazoline SupraBox ligands illustrated in Scheme 28. They applied these in the copper catalyzed asymmetric benzoylation of vic-diols.317 Variable temperature 1H NMR spectroscopy and dilution studies of [Pd(132a)2Cl2] in CD2Cl2 were used to probe the supramolecular interaction between the two monomers and confirmed the hydrogen bonding between the urea moieties within the complex. The application of these self-assembled ligands in asymmetric benzoylation of vic-diols gave interesting enantioselectivities (up to 86% with ligand 132c) but lower than the traditional bidentate complex (R,R)-PhBox (99% e.e.). Nonetheless, the importance of the hydrogen bonding motif was underlined by the generation of racemic product when a monomer (134) lacking hydrogen bonding ability was used.
Scheme 28. Overview of Some Suprabox Ligands Used in the Copper Catalyzed Asymmetric Benzoylation of vic-Diols317.

3.2.2. 6-DDPon Ligands
In 2003, Breit and co-workers described the formation of supramolecular bidentate ligands using 6-DPPon (135) shown in Scheme 29, which is based on the pyridone/hydroxypyridine tautomers (135)318 that can form a dimer by a DA/AD HB array (see also section 2.2, Figure 2). The formation of the supramolecular bidentate ligands is driven by coordination of the monomers to the metal center, and the supramolecular bidentate structure was initially confirmed by X-ray structure analysis of cis-[PtCl2(6-DPPon)2] (136). Later, also a rhodium complex was characterized by X-ray analysis, showing the hydrogen bonding between the ligand building blocks.319 Based on NMR spectroscopic experiments, enthalpic stabilization through hydrogen bonding was found to contribute 14–15 kcal·mol–1 to the complex formation.320 With bite angles close to 105° and being relatively electron poor, these ligands have found application in multiple catalytic transformations.
3.2.2.1. Hydroformylation
The self-assembled bidentate ligands were evaluated in the rhodium catalyzed hydroformylation of 1-octene (and other n-alkenes), and these catalysts displayed high linear/branch selectivity (l:b ratio 97/3). These catalysts were significantly more active than known bidentate phosphine ligands such as the tert-Bu-Xantphos ligand, which is a well-known wide bite angle ligand that can form both cis and trans spanning coordination complexes.321 Interestingly, the catalytic system kept its selectivity up to 100 °C, suggesting that the HBs hold at elevated temperatures. A lower selectivity (similar to that of complexes based on PPh3) was obtained when the complex was used at even higher temperatures or when methanol was used as solvent, indicating that under these conditions the HB motif is disrupted. A detailed study of the hydroformylation reaction using ESI-MS identified crucial intermediates of the Heck–Breslow catalytic cycle in which the RO–H···O=C HB remains intact.322 Interestingly, in these gas-phase reactions, oxidative addition of the H–N moiety provides additional pyridine coordinated intermediates which are involved in the hydrogen activation process. DFT calculations of the parent system complex [HRh(CO)(6-DPPon)2] indeed confirm that the O–H···O=C HB is much stronger than the N–H···N hydrogen bonding interaction.319 The weaker N–H···N is thus disrupted to a great extent in the course of the catalytic cycle, making the H–N moiety accessible for the proposed oxidative addition.
The high activity/selectivity allowed hydroformylation to be effectively performed at room temperature and just 1 bar syngas pressure, alleviating the need for high pressure equipment.323 This practicality was displayed by converting a large substrate scope consisting of 30 n-alkenes, as illustrated in Scheme 30a. The alkenes were equipped with many important functional groups, and the aldehyde products were obtained in high yields with l/b selectivities between 91/9 and >99/1. Direct comparison of the complexes based on 6-DPPon (135) and “BiPhePhos” (Scheme 30d) showed that at similar conversion and selectivity, the 6-DPPon (135)-based system displayed lower isomerization than the BiPhePhos (8% vs 54%) at 22 °C and 1 atm of syngas (H2/CO = 1:1). Interestingly, similar reactivity and selectivity were obtained in aqueous hydroformylation reactions in the presence of an emulsifier, performed at 1 bar syngas pressure and room temperature.324
In a separate study, the Rh/(135) system was found to be active and selective in the hydroformylation of 1,1-disubstituted allenes (140), as shown in Scheme 30b.325 Recently, the Rh/(135) catalyst system was also applied to the sequential double hydroformylation of butadiene by Subramaniam and co-workers.326 The traditional bidentate ligand DIOP gave the best results in the first hydroformylation step, providing a maximum 4-pentenal selectivity of 48%. The use of 135 as ligand showed the best performance for the subsequent 4-pentenal hydroformylation step with adipaldehyde selectivity exceeding 93%.
The slightly modified Rh/(135b) catalyst system in which 3,5-(trifluoromethyl)phenyl substituents are placed on the phosphine ligand also proved to be efficient in the hydroformylation of internal alkynes (142), achieving high selectivity and activity (see Scheme 30c).327 The observed reactivity and selectivity were somehow lower using alkyne substrates with electron-poor aryl groups (R1 and R2 = 4-CF3Ar, conversion 83%, yield 58%). The conversion of aromatic terminal alkynes was also reported.
Interestingly, the Rh/6-DPPon system was found suitable for the low temperature domino hydroformylation/l-proline catalyzed cross-adol addition shown in Scheme 31a. To prevent a buildup of the primary hydroformylation aldehyde product (leading to undesired homocoupling of the aldehyde), the rate of the hydroformylation reaction was attenuated by using 135c in the domino hydroformylation/aldol condensation reaction. When salicylaldehyde was used as the acceptor aldehyde, the corresponding lactols were isolated. The reaction was performed in DMF, but the authors did not provide any information on the stability of the HB network of the catalytic system under these conditions.328
Scheme 31. 6-DPPon (135) Ligand System Used in (a) Domino Hydroformylation-Aldol Condensation, (b) Tandem n-Alkene Hydroformylation-Hydrogenation, (c) and the Regioselective Hydroformylation-Decarboxylative Knoevenagel Reaction.

The tandem rhodium catalyzed hydroformylation–hydrogenation of alkenes illustrated in Scheme 31b was also reported using the combination of 4-fluorophenyl-6-DPPon (135d) and an acyl-guanidine decorated ligand (144) (the latter catalytic system will be further described in section 4.1).329 The Rh/6-DPPon catalyst is responsible for the stereoselective hydroformylation and the Rh/(144) catalyst for the aldehyde hydrogenation. Fine-tuning of the electronic properties was necessary to match the relative rates of hydroformylation and aldehyde hydrogenation, of which 135d/144 was the most efficient combination.
As illustrated in Scheme 31c, the combination of regioselective hydroformylation and decarboxylative Knoevenagel condensation allows for two-step, one-pot C3 homologation of terminal alkenes to (E)-α,β-unsaturated acids and esters, (E)-β,γ-unsaturated acids, (E)-α-cyano acrylic acids, and α,β-unsaturated nitriles.330 The scope of the reaction was illustrated by 30 examples in which the initially formed aldehydes (by hydroformylation) were reacted with malonic acid, malonic acid methyl ester, or nitrilacetic acid in the presence of pyridine or DBU (1,8-diazabicyclo[5.4.0]undec-7-ene). The overall yield varied between 48 and 99%, and the regioselectivity ranged between 87/13 and 99/1, strongly depending on the base used.
3.2.2.2. Hydrogenation
Rh/(135) systems based on chiral phospholane analogues have been evaluated in the asymmetric hydrogenation of a number of benchmark substrates.331 As is depicted in the top of Scheme 32, the different building blocks vary in steric/size at the phospholane unit. Ligands (150a–c) which are incapable of forming hydrogen-bonded bidentates were included as reference. Application of the self-assembled bidentate ligands (135e–g) in the rhodium catalyzed hydrogenation of methyl acetamidomethacrylate (151), methyl acetamidocynnamate (153), and dimethylitaconate (155) showed that the (135g) performed best for all three substrates in terms of enantioselectivity (with 91%, 94%, and 99% e.e., respectively). A noticeable loss of enantioselectivity in the conversion of methyl acetamidocynnamate and dimethylitaconate was observed when the reaction was performed in methanol instead of DCM, suggesting loss of the HB bidentate structure. DFT calculations on ligands 135e–g show that phospholane-substituted 2-hydroxypyridines are lower in Gibbs free energy and are therefore the dominant tautomers in the gas phase. Compared to the parent 2-pyridone/2-hydroxypyridine system, the equilibria for 135e and 135f are shifted to the 2-hydroxypyridine side, while that of 135g changes only slightly. Calculations of Rh/(135g)2, with a relatively large steric bulk, favor the formation of the supramolecular bidentate ligand, compared to Rh/(135e)2 and Rh/(135f)2 (with ΔG of 14.3, 7.9, and 3.2 kcal·mol–1, respectively).
Scheme 32. Phospholane-Based 6-DPPon Ligands (135e–g) and Ligands (150a–c) Used in the Rhodium Catalyzed Asymmetric Hydrogenation.

Recently, the library of chiral 6-DPPon building blocks was extended to analogues with a chiral functional group at the 5 position of the pyridine ring, as illustrated in Scheme 33.332 These self-assembled bidentate ligands are not planar but twisted around the HB motif, leading to atropisomeric structures, and the presence of additional chiral groups results in the formation of diastereotopic species. These diastereoisomers interconvert rapidly at room temperature but can be spectroscopically resolved at low temperature (−40 °C), showing the presence of a dominant species. Application of this ligand system (157) in asymmetric hydrogenation of methyl acetamidomethacrylate (158) gave interesting enantioselectivity (52% e.e.) at room temperature in 1,2-dichloroethane which can be further improved to 86% e.e. when performing the hydrogenation reaction at −30 °C. By changing the solvent to dichlorobenzene, an e.e. of 90% could even be obtained. The low activity and selectivity observed for this catalytic system in THF and MeOH are in line with the low amplitude of the circular dichroism spectra observed in those solvents compared to the circular dichroism spectra observed in 1,2-dichloroethane. This example shows the potential of chirality induction in the catalytic system through the HB network.
3.2.3. Heterobidentate Ligands 6-DPPAP and 3-DPPICon
In analogy to the adenine-thymine DNA base paring illustrated in Scheme 34, the supramolecular heterobidentate ligands based on aminopyridine (160)/isoquinolone (161) have been developed by Breit and co-workers.333
Scheme 34. Supramolecular Heterobidentate Ligands Based on Complementary Hydrogen-Bonded Ligand Building Blocks, Inspired by the A-T DNA Pairs.

A combinatorial library of 4 × 4 supramolecular heterobidentate ligands becomes accessible based on a 4 + 4 building block.
Mixing of 1 equiv of 6-diphenylphosphino-N-pivaloyl-2-aminopyridine (6-DPPAP; 160a) with 1 equiv of 3-diphenylphosphinoisoquinolone (3-DPPICon; 161a) in the presence of [PtCl2(COD)] yielded the heteroleptic cis-[PtCl2(160a)(161a)] in quantitative yield. The ligands 160 and 161 were evaluated in the rhodium catalyzed hydroformylation of terminal alkenes and showed that only the combination of 6-DPPAP (160a) and 3-DPPICon (161a) displayed a much higher linear/branched selectivity compared to the reaction employing either 160a or 161a alone. Exploration of a ligand library showed that the activity expressed in TOF varied between 1040 and 8643 molsubstrate/molcatalyst·h–1. The fastest catalyst was based on 160d and 161d, and very small changes in the selectivity were noted, as detailed in Table 4. In addition, LyondellBasell has patented the Rh/6-DPPAP/3-DPPICon system for the selective hydroformylation of allyl alcohol into 4-hydroxybutyraldehyde.334 The combination Rh/160e/161e gave the highest aldehyde yield (99%) and highest linear selectivity (linear/branched ratio 23.1). Interestingly, the combination Rh/160f/161f mainly produced the linear alcohol by a hydroformylation–hydrogenation sequence (alcohol yield 79%, l:b ratio 9.7).
Table 4. Rhodium Catalyzed Hydroformylation Using a 4 × 4 Library of 6-DPPAP (160a–d) and 3-DPPICon (161a–d) Derivatives (for Structures See Scheme 34)a with the Activity in TOFb and Selectivity in l:b Ratio Providedc.
| 3-DPPIcon
monomer |
||||
|---|---|---|---|---|
| 6-DPPAP monomer | 161a (R = Ph) | 161b (R = (4-MeOPh) | 161c (R = 3,5-bis-CF3Ph) | 161d (R = 4-FPh) |
| 160a (R = Ph) | 2425 | 1040 | 2732 | 2559 |
| 94:6 | 94:6 | 96:4 | 95:5 | |
| 160b (R = 4-MeOPh) | 2033 | 1058 | 1281 | 1772 |
| 93:7 | 92:8 | 96:4 | 94:6 | |
| 160c (R = m-tolyl) | 3537 | 1842 | 1808 | 2287 |
| 94:6 | 93:7 | 96:4 | 94:6 | |
| 160d (R = 3,5-bis-CF3Ph) | 7439 | 2695 | 7465 | 8643 |
| 94:6 | 95:5 | 94:6 | 94:6 | |
Reaction conditions: [Rh(CO)2(acac)], [Rh]:(160):(161):1-octene = 1:10:10:7500, 10 bar H2/CO (1:1), toluene (c0(1-octene) = 2.91 M), 5 h. Catalyst preformation: 5 bar CO/H2 (1:1), 30 min, RT to 80 °C.
Turnover frequency (TOF) in molsubstrate/molcatalyst·h–1.
Regioselectivity in linear (l) to branched (b) (l:b) ratio.
Ruthenium complexes based on the supramolecular heterobidentate ligands 6-DPPAP (160) and 3-DPPICon (161) were also investigated in the ruthenium catalyzed hydration of 1-nonyne.335 In a preliminary study, different [RuCp(L)2(MeCN)] complexes with L2 being traditional and supramolecular bidentate ligands were evaluated. The complexes based on the traditional ligand and the homosupramolecular bidentate 6-DPPon (135) performed poorly, leading to formation of the Markovnikov product, while the self-assembled heterobidentate complex [RuCp(160)(161)(MeCN)] gave high conversion and perfect selectivity for the aldehyde (anti-Markovnikov product; see Scheme 35).
Scheme 35. [RuCp(160)(161)(MeCN)] (165) Catalyzed anti-Markovnikov Hydration of Terminal Alkynes (166).

As is illustrated in Scheme 36 (left), the selectivity arises from both the bidentate character of the ligand and an interaction of the functional groups of the complex with the water substrate.336 DFT calculations correlated with previous experimental data resulted in the authors proposing a mechanism involving an initial alkyne–vinylidene tautomerism, which occurs via a ligand-assisted proton shuttle mechanism. The HB between the pivaloyl moiety and the isoquinolone remains intact while a water molecule intercalates in the other HB (see 169 in Scheme 36). This example highlights once more the various roles that the functional groups of the HB motif can have, such as leading to the formation of a supramolecular bidentate ligand interacting with the substrate during the catalytic cycle.
Scheme 36. HB Involvement with a Supramolecular Bidentate Ligand during a Catalytic Cycle in (a) Activation of a Water Molecule in the Hydration of Alkynes (See Also Scheme 35) and (b) Activation of Allyl Alcohol for Allylation Reactions.

A similar hydrogen bond activation of the unreactive allyl alcohol substrate is shown on the right side of Scheme 36 and has been proposed for the palladium catalyzed allylation of indoles and pyrroles with allyl alcohol.337 A ligand library of 6-DPPon (135), a heterobidentate combination using 6-DPPAP (160), and 3-DPPICon (161) was evaluated. The library was extended with 6-DPAIND (171) and 2-DPPAT (172), which also have complementary HB motifs. The catalyst system consisting of [Pd(η3-allyl)(COD)]/(135a)2 provided the most efficient catalyst, yielding the 3-subsituted allyl indoles in 58–91% yield. The allyl alcohol activation is proposed to be based on a HB between the alcohol and the HB motif of the supramolecular bidentate ligand (see Scheme 36). The application of the bidentate ligand based on 6-DPPAP and 3-DPPICon was also explored in the ruthenium catalyzed nitrile hydration.338 The highest activity was observed for [Ru(160a)(161a)(acac)2] and was rationalized based on electronic factors. Furthermore, it was hypothesized that the nucleophilic attack of water may be facilitated by hydrogen bonding with the ligands, as was also found for Ru complexes used in the hydration of alkynes (see Scheme 35).
A catalyst library consisting of aminopyridine (160a–b, d, and g) ligands and isoquinolone ligands (161a–d and g) was also evaluated in the nickel hydrocyanation of styrene.339 Several complexes based on supramolecular heterobidentate ligands were found catalytically active, and the performance depends more on the nature of the isoquinolone than on the aminopyridine building block. The nickel complex based on 160b/161b gave yields greater than 95% and branched/linear ratios up to 97:3.
A small library of ligand building blocks with chiral phospholane units that self-assemble into supramolecular homobidentate ligands was explored in the palladium catalyzed Tsuji–Trost reaction and compared to ligand building blocks that are protected and thus does not form self-assembled ligands. Both the self-assembled ligands and the building blocks that are coordinated as monodentate ligands form complexes that induce enantioselectivity. However, the smaller ligand based on E (see Scheme 37) that does not form self-assembled bidentate ligands provided the highest e.e.’s (99% e.e.).341
Scheme 37. Overview of the Different Ligand Building Blocks with Complementary HB Motifs. Top: Core Structures of Ligands 160, 161, 171, and 172. Combination with Phosphorus Groups A–G (Bottom) Leads to a Library of 120 Supramolecular Hetereobidentate Ligands That Was Used in the Combinatorial Iterative Library Deconvolution Strategy. In Addition, Core Structure 150 Has Been Combined with Substituents H–M.

The same HB motif was used to make a library with chiral phosphine and phosphinite ligand building blocks, providing supramolecular bidentate ligands that were explored in rhodium catalyzed asymmetric hydrogenation (see, for example, the ligand library in Scheme 37). Formation of RhL1/L2COD complexes based on these supramolecular bidentate ligands was confirmed by NMR and MS spectroscopy.340 The library of chiral self-assembled catalysts consisting of chiral phosphines and phosphonites was formed by just mixing the components and explored in asymmetric hydrogenation. The phosphonite systems based on the BINOL skeleton resulted in the catalysts that induce the highest selectivity. For example, high enantioselectivities (>99% e.e.) and excellent catalyst activities were observed in the asymmetric hydrogenation of methyl acetamidoacrylate using [Rh(COD)2]BF4/160a/161a as catalyst. In a follow-up study, the solvent was found to have an influence, but no correlation could be found to the HB characters of the ligands.
The full potential of the approach was demonstrated by the generation of a 10 × 12 library of supramolecular bidentate ligands.342 The ligand building blocks were based on the acceptor–donor hydrogen bonding units (in red) 6-DPPAP (160, with A–G) and 171 (with A–G) and the donor–acceptor hydrogen bonding units (in blue) 3-DPPICon (161, A–G) and 172 (A–E) as illustrated in Scheme 37. By just mixing these building blocks, a large library of 120 self-assembled bidentate ligands was available that could be evaluated individually. Next to individual testing, the screening for the best catalyst was also exploited through an iterative library deconvolution strategy.
The combinatorial iterative library deconvolution strategy involves the division of the catalysts into subgroups that are evaluated as a mixture, thereby accelerating the evaluation of the entire library’s potential. As the ligand building blocks form the supramolecular bidentate ligands in situ, such mixtures are easy to prepare. With this concept in mind, the activity and selectivity of mixtures of catalysts were probed in the rhodium catalyzed asymmetric hydrogenation of methyl acetamidomethacrylate. The mixture of catalysts that provided the highest selectivity was used in the next set of experiments, in which the mixtures consisted of a smaller number of catalysts, based on the building blocks present in the winner subset of the first round. In the final set of experiments, four reactions were performed with pure catalyst solutions to determine which supramolecular bidentate ligand gave the complexes that produced the product in the highest enantioselectivity. In this particular example, the strategy identified three catalysts that yield full conversion and 99% e.e. of the product. Importantly, only 17 reactions were required using this iterative library deconvolution strategy to identify the best catalyst from the set of 120. This combinatorial strategy was further applied to other substrates with similar results.
3.2.4. Ligands Based on Other HB Motifs
The supramolecular bidentate ligands based on one or two HBs were demonstrated to provide selective catalysts for a variety of transformations. Extension to motifs based on a larger array of HBs is interesting, as this would make the system more rigid. The motifs based on AAAA-DDDD arrays would provide the strongest interaction (see also Figure 2); however, synthetic procedures to access such building blocks seem complicated. To form supramolecular bidentate ligands based on a larger HB array, the application of peptidic chains has been explored. The peptide sequence chosen is shown in Scheme 38 and mimics the typical β-sheet structures found in proteins.343 This set of ligand building blocks that form bidentate ligands was coined SupraPeptiPhos (173). Self-assembly of PtCl2 with 173 was monitored by 1H NMR spectroscopy, which indeed revealed the formation of a β-sheet-type structure. Moreover, 173 was applied in the rhodium catalyzed asymmetric hydroformylation of styrene, resulting in an e.e. of up to 38%. While this is modest, these data do provide evidence of chirality transfer from the peptidic chain to the product via the metal center, as highlighted earlier for the work of Kirin (see Scheme 24).
4. Substrate Orientation by Hydrogen Bonding
Metal catalyzed transformations are typically described as a sequence of elementary steps that occur at the metal center.344 Depending on the specific details, the selectivity of the reaction is determined during one of these steps. In many reactions, a migration step is involved in the selectivity determining step, such as in hydrogenation and hydroformylation reactions. The orientation of the substrate at the metal center plays an important role in such a selectivity determining step. The substrate orientation at the metal center depends on the ligand environment around the metal center and can also be realized via coordination of a “directing group” to a metal center.345−350 Recently, hydrogen bonding between the functional groups of the substrate and the functional groups of the catalyst has been used as a strategy to achieve the proper orientation of the substrate and, hence, to control the selectivity of the reaction. In this manner, the interactions in the second coordination sphere are of crucial importance. In analogy to the substrate orientation effects found in natural systems, such control of the second coordination sphere may further guide the rational design of selective catalysts. The hydrogen bonding interactions between the substrate and the functional groups of the catalyst are ideal for substrate orientation, and in this section we will review examples of this concept in various different reactions.
4.1. Asymmetric Hydrogenation
The asymmetric hydrogenation reaction is a powerful asymmetric transformation, as it provides a general strategy to create chiral centers in organic molecules. This relevance was underscored by the Nobel prize in 2001 awarded to Knowles and Noyori.1,29 Kagan and Knowles reported chiral bidentate phosphine ligands that formed rhodium complexes displaying significant e.e. in asymmetric hydrogenation (up to 70% e.e.).351,352 These bidentate ligands were further developed to get catalysts displaying exceptionally high enantioselectivities for a large set of substrates. In the early nineties it was also demonstrated that complexes based on monodentate ligands can induce high enantioselectivity. In the early 2000s, the concept of supramolecular bidentate ligands, that is the generation of bidentate ligands based on the self-assembly of ligand building blocks, was reported. These ligands feature the benefit of easy synthesis typically for monodentates, yet they display control over the coordination sphere like bidentate ligands. Also, supramolecular bidentate ligands are ideal for the generation of large catalyst libraries, as the number of catalysts grows exponentially with the number of synthesized building blocks (see also section 3). Our group has developed ligand building blocks that can form supramolecular bidentate ligands based on a single HB between a urea C=O and a phosphoramidite N–H (see section 3.1.2 and Figure 6).295,353 Rhodium complexes of these ligands were shown to convert methyl 2-hydroxymethacrylate and its derivatives in very high enantioselectivities, in contrast to substrates that did not have the hydroxy functional group (methyl or methyl ester). This difference in reactivity suggests that hydrogen bonding between the catalyst and the hydroxy group of the substrate plays an important role.
Detailed spectroscopic studies, in combination with kinetic analysis and DFT calculations, revealed the consequence of the HB formed in the second coordination sphere.354 Addition of (E)-methyl 2-(hydroxymethyl)-3-phenyl acrylate to a solution of the precomplex (under either hydrogen or the solvato complex prepared from the COD precomplex) results in a structure in which the alkene coordinates to the rhodium center and the hydroxyl group of the substrate binds via hydrogen bonding to the functional groups of the ligands. The complex was observed during catalysis by in situ NMR spectroscopy and, therefore, was identified to be the resting state of the reaction. According to DFT calculations, the HBs established between the substrate and the ligands of the complex stay intact throughout the whole catalytic cycle. As illustrated in Figure 7, the reaction pathway deploying a ligand that can form this HB (blue) involves lower overall energy barriers of transition states (TS1, TS2, and TS3) compared to the use of a ligand that cannot use this HB (red, TS1′, TS2′, and TS3′). Detailed analysis of all reaction pathways displayed by the various diastereomeric complexes showed that those forming HBs in the starting complex are favored. As such, these complexes operate via an anti-Halpern model, in which the most energetically favored structure (by hydrogen bonding) is the one that is most productive.
Figure 7.

Comparing relative energies of intermediates along the reaction path of a substrate that can form a HB interaction (blue) to that of a substrate that cannot (red).
The reaction follows Michaelis–Menten kinetics, and a stronger association (precomplex formation) is due to the HBs between the substrate and the ligands. In addition, these HBs also give a higher Vmax, which is in line with the lower transition state barrier. As the HBs formed in the second coordination sphere play a crucial role, another catalyst was designed by only modifying the potential for such hydrogen bonding.355 This catalyst is illustrated in Scheme 39 (176, left) and consists of a supramolecular bidentate ligand where the urea part in the previous catalyst (see Scheme 38) was replaced by a phosphine oxide, which is a much stronger HB acceptor. A mechanistic study demonstrates that also for this catalyst two HB interactions between the catalyst and the substrate are involved, leading to stabilization of a catalyst–substrate complex intermediate shown at the bottom in Scheme 39 (177).
DFT calculations of the reaction pathway show that the stronger HB interactions between the catalyst and the substrate result in a lower energy barrier by transition state stabilization. In line with this, it was found that the second generation catalyst indeed provides higher rates (factor 4). In addition, the product is also generated in higher selectivity (>99% e.e.) and the catalyst is more robust, as demonstrated by its performance at elevated temperatures. Chikkali and co-workers used urea functionalized P-chiral ligand building blocks to form supramolecular bidentate ligands that were used in asymmetric hydrogenation (Scheme 26). For these systems it was also proposed that a HB interaction between the substrate and the urea of the ligand was of crucial importance to steer the outcome of catalysis.311
The group of Zhang developed a different strategy for substrate organization by hydrogen bonding in the second coordination sphere. They developed the ligand Zhaophos (179) shown in Figure 8, which is based on a ferrocene diphosphine (red) and a thio-urea HB motif (blue).356 The ditrifluorophenyl group on the thiourea makes the HB donors a bit more acidic and, as such, even stronger HB donors. Initial studies demonstrated that the Rh–bisphosphine–thiourea complex was an excellent catalyst for the rhodium catalyzed asymmetric hydrogenation of challenging β,β-disubstituted nitroalkenes, providing the product in 99% e.e. for the benchmark methyl-phenyl analogue and above 86% for all analogues reported in the initial study. Control experiments show that the binding site is important, and later spectroscopic and DFT evidence supports this (not reported in the initial study).357
Figure 8.

Design of the bidentate phosphine ligand Zhaophos (179) with a thio-urea binding site as a handle to organize substrates at the metal center via HBs in the second coordination sphere. Initial studies showed that β,β-disubstituted nitroalkenes (180) were converted with high selectivity.
After the initial results using Zhaophos (179) in β,β-disubstituted nitroalkenes (180), the substrate scope for the rhodium catalyzed asymmetric hydrogenation was extended to challenging α,β-unsaturated ketones (182)358 and α,β-unsaturated esters (184)359 such as those shown in Scheme 40. These include substrates containing silicon (184a)514 and boron (184b)515 functional groups that provide handles for further functionalization. Also β-thio-α,β-unsaturated esters were converted with high selectivity using rhodium complexes based on Zhaophos (179).360
Scheme 40. Part of the Extended Substrate Scope That Was Explored Using Zhaophos (179).

The asymmetric hydrogenation of β-cyano-α,β-unsaturated esters (187) was also explored using rhodium complexes based on Zhaophos. While the product was obtained already in high e.e., the selectivity could be further improved by using an analogue of Zhaophos (186) in which the least acidic NH of the thio-urea functional group was methylated (see Scheme 41).357 A Job plot analysis of a the binding study indicated that the substrate and ligand bind in a 1:1 ratio. DFT calculations suggest that the single HB formed between the substrate and the ligand leads to a stronger binding compared to that based on the two HBs formed between the substrate and the original Zhaophos (179). Also, the complex features a broad substrate scope of β-cyano-α,β-unsaturated esters, and the reaction was performed at gram scale to demonstrate the applicability.
Scheme 41. (a) NH-Methylated Analogue (186) of Zhaophos; (b) Asymmetric Hydrogenation of β-Cyano-α,β-unsaturated Esters (187) Using 186 to Yield the Product in 99% Selectivity; (c) Proposed Intermediate Structure (189) towards the Product.

The substrate scope for Zhaophos and analogues was further explored in the rhodium catalyzed asymmetric hydrogenation and appeared extremely versatile, as is summarized in Scheme 42. For many different subclasses of substrates, conditions were found in which the alkenes were hydrogenated in high e.e. (often >95% was reported), including exocyclic α,β-unsaturated lactones361 and lactams, (E)-2-chorman-4-ylidene)acetates,362 α,β-unsaturated primary amides,359 acylpyrazoles,363 five-membered α,β-unsaturated lactams,364 maleinimides, maleic anhydrides,365 2-substituted benzo[b][1,4]dioxines, and benzo[b]thiophene 1,1,dioxides.366
Scheme 42. Further Extended Substrate Scope for Selective Rhodium Catalyzed Asymmetric Hydrogenation Using Zhaophos.

The reaction scope was further extended to the iridium catalyzed asymmetric hydrogenation shown in Scheme 43. Various challenging tetrasubstituted α-fluoro-β-enamino esters were converted by the Ir/ZhaoPhos system, leading to products with two adjacent tertiary stereocenters. Excellent diastereoselectivities/enantioselectivities were reported (73%–99% yields, >25:1 diastereomeric ratio, 91%–>99% e.e.),367 with a very high activity using a relatively low catalyst loading (TON ≤ 8.600). Deuterium labeling studies suggest that the substrate does not isomerize to the imine before it is reduced. Also, different analogues of Zhaophos show low selectivity in this iridium catalyzed transformation, indicating also that for this system substrate orientation via the thiourea plays a crucial role.
Scheme 43. Iridium Catalyzed Asymmetric Hydrogenation Using Zhaophos (179).

The Zhang group combined an SPO (see also section 3.1.1) with a normal phosphine in a bidentate ligand, coined SPO-Wudaphos (201), as illustrated in Scheme 44.368 When P coordinated to a metal, SPO-Wudaphos was tautomerized to its phosphinous acid and the resulting PO-H HB donor was demonstrated to give a HB interaction with substrates such as α-methylene-γ-keto carboxylic acids. Rhodium complexes based on the SPO-Wudaphos ligand converted this type of substrates with very high e.e. DFT calculations and control experiments demonstrated that the P-OH hydrogen bonded with the ketone, while the carboxylic acid function formed an amine salt ion pair with the amine function, and both were found to be important for the induction of high selectivity.
Scheme 44. (a) Rhodium Catalyzed Asymmetric Hydrogenation Using SPO-Wudaphos (201); (b) Upon Coordination of SPO-Wudaphos (201), the P-OH Group Forms That Can Preorganize the Substrate by Hydrogen Bonding to the Ketone of 202 (Formation of a P–OH···O=C HB).

Interestingly, the functional group for substrate orientation and the bidentate ligand do not necessarily need to be covalently linked, but similar cooperativity can also be generated via cofactor binding in the second coordination sphere. To demonstrate this concept, Reek and co-workers have explored a bidentate ligand with an integrated binding site for anions, coined DIMPhos (204) (see Figure 9).369 Chiral cofactors such as 205, containing a carboxylate function, are bound to DIMPhos (204) rather strongly because of the formation of four HBs. The nonchiral bidentate DIMPhos (204) was explored in the rhodium catalyzed asymmetric hydrogenation of methyl 2-acetamidoacrylate (206a) in the presence of various chiral cofactors. Importantly, under these conditions the cofactor was the only source of chirality. From the 18 cofactors evaluated, one provided a supramolecular complex that converted methyl-2-acetamido-acrylate in very high enantioselectivity (99% e.e.), whereas other cofactors resulted in low to moderate selectivity (≤61% e.e.). Two other amidoacrylates (206b and 206c) were converted with high enantioselectivity using the same cofactor. Control experiments and DFT calculations suggest that also for this catalyst a HB between the substrate that is coordinated to the rhodium metal center and the cofactor is of crucial importance. Interestingly, similar to the Zhang system (Figure 8), a thiourea-containing cofactor appeared to be important for selectivity, but in this system the HB is formed between the sulfur and the amide NH of the substrate, rather than between the urea NH and the carbonyl of the substrate (Figure 9). The substrate scope was not extensively explored, but considering the results of Zhaophos, it could well be that this system is also widely applicable. Importantly, the above systems all show that hydrogen bonding between the substrate and the catalyst in the second coordination sphere, next to substrate coordination, can play an important role in achieving highly selective catalysis. Thus, hydrogen bonding is a powerful tool to guide the rational design of hydrogenation catalysts.
4.2. Hydroformylation Catalysis
The hydroformylation reaction typically involves a metal catalyzed addition of CO and H2 to an alkene as is shown in Figure 10a. This reaction was discovered by serendipity in the thirties of the last century and has further resulted in many industrial applications.370,371 Fundamental studies have provided detailed insight in the reaction mechanism,372,373 and many issues regarding selectivity and activity have been solved. Yet, there are still several challenges left that, when successfully sorted out, can lead to new industrial applications. These challenges mainly involve selectivity issues, including the branched selective hydroformylation, the selective hydroformylation of internal alkenes, and the selective hydroformylation of tri- and tetrasubstituted alkenes. Also, the asymmetric hydroformylation of terminal disubstituted alkenes is a largely unsolved problem. Whereas the typical approach to control activity and selectivity in rhodium catalyzed hydroformylation involves changes in the ligand structure (electronic, steric, and the bite angle), more recently it has been demonstrated by the groups of Reek374 and Breit375 that hydrogen bonding between the substrate and the catalyst in the second coordination sphere can also be used to obtain selective hydroformylation catalysts.
Figure 10.

(a) Hydroformylation reaction in which alkene groups are converted to aldehyde products by the addition of CO and H2; (b) Generally accepted mechanism, proceeding via alkene coordination (212 to form 213), hydride migrations (213 to form 214/218), CO coordination and insertion (214/218 to form 216/220), and hydrogenolysis (via oxidative addition/reductive elimination or metathesis; 216/220 to form 222); (c) molecular model of the selectivity determining hydride species (213) to illustrate that hydrogen bonding can control this step by preventing substrate rotation (clockwise or counterclockwise), thus determining the reaction pathway (to 214 or 218 resulting in linear or branched products, respectively).
The generally accepted mechanism for rhodium complexes with phosphorus ligands is displayed in Figure 10b.372,373 For complexes based on phosphine ligands, the resting state is usually the rhodium(I) hydrido complex (211). The cycle starts with CO dissociation and subsequent alkene coordination to form the alkene–rhodium hydride complex (213), of which a model is presented in Figure 10c. The hydride migration step that follows to either C2 (to give 214) or C1 (to give 218) leads the path to form the linear or the branched product, respectively. During this selectivity determining migration step, the alkene rotates clockwise or anticlockwise, depending on the carbon atom to which the hydride migrates. As such, fixation of the substrate by hydrogen bonding in the second coordination sphere may block some of these rotations and can thus lead to more selective hydroformylation catalysis.
Reek and co-workers employed bidentate DIMPhos ligands (204a–c) such as those shown in Scheme 45 for the selective rhodium catalyzed hydroformylation of alkene substrates containing carboxylate or phosphate functional groups.374 These ligands have an integrated HB binding site for carboxylate (and phosphate) functional groups (in blue) that can be used to orient a substrate containing these groups. Under hydroformylation conditions, the ligands bind to a rhodium center in a bidentate fashion forming the typical rhodium-hydride bis-carbonyl complexes that are active in hydroformylation. Binding studies monitored by IR and NMR spectroscopy showed that the metal complexes strongly bind to acetate groups in the NH-rich binding pocket of DIMPhos (blue), while leaving the coordination geometry around the metal unaffected. Exploration of a series of substrates with carboxylate functional groups in hydroformylation catalysis demonstrated that substrate preorganization results in unprecedented selectivities. A broad range of terminal and internal alkenes functionalized with an anionic carboxylate (or phosphate) group has been used.
Scheme 45. Three DIMPhos Ligands with Aryl Phosphines (DIMPhos1, 204a and 204b) and with Phosphites (DIMPhos2, 204c) with the Binding Site Displayed in Blue and the Ligand in Red.

The three DIMPhos ligands (204a–c) shown in Scheme 45 were used in the rhodium catalyzed hydroformylation of terminal unsaturated carboxylates. As is summarized in Figure 11, 4-pentenoate up to 10-undecenoate are converted to the aldehyde with high selectivities for the linear product when DIMPhos1a is used (204a). The methyl ester analogues of these substrates, which have no significant affinity to the binding pocket, are converted with low selectivity, confirming the need for HB formation. Detailed kinetic analysis shows that these complexes convert carboxylate substrates via a mechanism that follows Michaelis–Menten kinetics, with product inhibition.376 The Michaelis–Menten constant is the same as the product inhibition constant, reflecting the carboxylate binding in the DIMPhos pocket. The authors concluded from detailed experiments that first the substrate is bound in the pocket, experiencing competition from the product, and then the alkene is converted to the aldehyde. Importantly, although the mechanism is described with a model that includes product inhibition, the reaction rates are high (and accelerated by binding) and full conversion can easily be reached. The use of DIMPhos1b (204b) also gave high selectivities for the larger substrates, but the trend could not be clearly explained. The use of the phosphite analogue DIMPhos2 (204c) also resulted in the formation of the 1-aldehyde in high selectivity, also for the smallest substrate (n = 1). Whereas the distance between the alkene and the carboxylate in this substrate is too small to simultaneously coordinate to rhodium with the alkene and to the DIM pocket with the carboxylate in complexes formed by DIMPhos1a (204a), the extra flexibility in the phosphite analogue allows this.
Figure 11.

Selective hydroformylation of terminal alkenes (222) to linear aldehydes (223) using various DIMPhos ligands (204a–c) that can form HBs with the substrate. The l:b ratio as a function of the number of carbon atoms (n) is displayed.
The concept of substrate orientation by hydrogen bonding was further supported by DFT calculations on the hydride migration step as is illustrated in Figure 12.374−376 Only the carbon atom close to the hydride is available for the migration because of the carboxylate binding in the pocket, and in the precomplex, the alkene is already rotated toward the transition state of hydride migration. The hydride migration that leads to the intermediate that forms the branched aldehyde requires the alkene to rotate clockwise (about 180°). This is, however, not possible without breaking the HBs between the substrate and the HB pocket of DIMPhos. From these calculations it is also clear that 3-butenoate (n = 1 in Figure 11) is too short to bind simultaneously to the receptor moiety and the metal center in complexes based on DIMPhos1a (204a).
Figure 12.

DFT calculated structures of the hydride migration that leads to the linear aldehyde. The pathway to the branched product would require the breakage of the HBs or proceed via other intermediates that are even higher in energy (not displayed).
In order to also address the small 3-butenoate substrate with phosphine-based ligands, DIMPhos1a (204a) was redesigned by repositioning the -PPh2 moieties from the para position to the ortho position in OrthoDIMPhos (204d). The DFT calculated structures displayed in Figure 13 show that this alteration shortens the distance between the HB binding site and the rhodium complex from 10 to 7.8 Å, the ideal distance to ditopically bind 3-butenoate. Application of this ligand in the rhodium catalyzed hydroformylation of 3-butenoate indeed demonstrated selectivity to the linear product in record high selectivity (l/b = 84).377
Figure 13.

Redesign of DIMPhos1a (204a) to provide OrthoDIMPhos (204d), an analogue with a shorter distance between the Rh-complex and the DIM pocket. Adapted with permission from ref (377). Copyright 2019 Wiley under a CC-BY-NC license [https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cctc.201900487].
The concept of supramolecular substrate orientation by hydrogen bonding was also explored for internal alkenes. These substrates are less reactive, and the selectivity is harder to control, as there is generally no electronic bias with the consequence that the two aldehydes are typically produced in a ratio close to 1:1. Rhodium complexes based on the phosphine-based ligand DIMPhos1a (204a) are too unreactive to convert internal alkenes under mild conditions. In contrast, rhodium complexes based on a phosphite ligand are generally more reactive.373 Indeed, deploying DIMPhos2 (204c) to support a Rh complex led to conversion of internal alkenes under mild conditions.376 The application of a rhodium complex based on DIMPhos2 (204c) on the series of carboxylate functionalized substrates listed in Scheme 46 showed that these were converted to aldehydes with very high selectivity. In the products that are formed, CO is inserted in the carbon atom furthest away from the carboxylate, in line with the selectivity obtained for terminal alkenes. For some substrates, exceptionally high selectivities are observed with a quotient of external/internal aldehyde of 78. Substrates with different distances between the alkene and the carboxylate were selectively converted with the highest selectivity obtained for the internal alkene on the 4-position.
Scheme 46. Selective Hydroformylation of Internal Alkenes Using Rhodium Complexes Based on DIMPhos2 (204c) with the Indicated Quotients of External (a)/Internal (b) Aldehydes.

Using the same ligand, it was demonstrated that even reversal of selectivity can be obtained by organization of the substrate by hydrogen bonding. The linear aldehyde is usually the disfavored product in the hydroformylation of vinyl 2- and 3-carboxyarenes, but by application of DIMPhos2 (204c), chemo- and regioselectivities up to 100% can be achieved, as summarized in Scheme 47. The catalyst proved to be selective for a wide scope of substrates, could be applied at low catalyst loading, and worked well at ambient pressure.378−380
Scheme 47. Styrene Derivatives That Have Been Converted with High Selectivity Using DIMPhos2.

The catalyst was still selective at temperatures up to 120 °C, and very high reaction rates were observed at these temperatures. Follow-up reactions on the formed products demonstrated the wide possible applicability, paving the way to designing new synthetic routes for biorelevant compounds. Also, the most challenging substrates with internal double bonds, such as methylstyrene derivatives and the cyclic analogues thereof, were converted with exceptional selectivity. Kinetic studies and in situ spectroscopy revealed that the active species involve complex equilibria including dormant species. The reaction kinetics is described by a model including both product inhibition and substrate inhibition due to binding of the carboxylate to the binding site and to the metal center. Nonetheless, efficient formation of the desired product is observed with TOF’s as high as 2000 molsubstrate/molcatalyst·h–1.
The hydroformylation of natural monounsaturated fatty acids (MUFAs) in a regioselective fashion requires a catalyst with a larger distance between the rhodium center and the HB binding pocket of the DIMPhos ligand. With this in mind, the phenyl “linker” between the HB binding pocket and the -PPh2 moieties was extended to a biphenyl in 204e, as shown in Figure 14a.381 DFT calculations suggest that 9-decanoate spans the distance between the HB binding pocket and the rhodium complex reasonably well, making it a reasonable model for fatty acids. The hydroformylation catalyst based on the extended DIMPhos ligand (204e) converts substrates with high regioselectivity, including monounsaturated fatty acids (MUFAs) and their model substrates. For example, as shown in Figure 14b, the natural fatty acid cis-myristoleic acid could selectively be hydroformylated with a 10-formyl/9-formyl ratio of 2.51. This, in fact, is the first selective hydroformylation catalyst for this biobased compound.
Figure 14.

(a) Extended DIMPhos ligand (204e) for substrate preorganization by hydrogen bonding of larger substrates; (b) modeling picture showing the binding of 9-decanoate as a model for natural fatty acids such as 228. (c) 228 is selectively converted to aldehydes such as 229. Adapted with permission from ref (381). Copyright 2020 Wiley.
The results obtained with different versions of the DIMPhos ligand demonstrate that substrate preorganization via hydrogen bonding allows the redesign of catalysts to provide selective conversions for substrates of different sizes.
Substrate orientation can also be achieved by using monodentate ligands with functional groups. Breit and co-workers explored the use of the acyl guanidinium functionalized phosphine ligands (230–232) shown in Figure 15, in which the guanidine can form HBs to unsaturated carboxylic acids.375
These HBs between substrate and ligand lead to substrate preorganization of the alkene at the metal center. 3-Butenoic acid is converted by a rhodium catalyst based on ligand 230 with a high selectivity for the linear product (l:b = 41 under optimized conditions). In experiments in which competitive guests (with carboxylic acid functional groups) are present, the substrates are converted with lower selectivity and activity in line with the substrate preorganization model. As is exemplified for 3-butenoic acid in the bottom of Figure 15, DFT calculations show the Rh(H)CO–substrate complex is most stable when two P ligands are coordinated to Rh and the carboxylic acid moiety of the substrate forms HBs with the guanidines. In this example, one guanidine deprotonates the acid to give a carboxylate that can form four HBs, very similar to the case observed for DIMPhos. No substrate–ligand interaction was observed in complexes with only one ligand coordinated to the metal center, and as such, the bis-coordinated species is proposed to be the most likely intermediate responsible for the high selectivity.382 Analysis of the calculated structures indicates that preceding the hydride migration step, the alkene is already rotated toward the hydride as a result of constraints imposed by the HBs between the guanidinium moieties and carboxylic acid moiety of the substrate.
The catalyst also converts internal alkenes such as 3-pentenoic acid with high selectivity for aldehyde introduction on the unsaturated carbon atom furthest away from the carboxylic acid (with ratio 18:1). The selectivity was found to be highly dependent on the distance between the acid moiety and the alkene function. 4-Pentenoic acid was converted with comparable selectivity to levels typically found for triphenyl phosphine-based catalysts. This clearly shows that for this catalyst system the alkene–acid distance must be precise in order to control selectivity through hydrogen bonding. The high selectivity for the 3-pentenoic acid was only obtained for the cis configurated alkene. In a following paper383 the authors demonstrated that the use of an electron-poor acylguanidine ligand (252) provided complexes that could also convert β-alkynoic acids to provide similar products.
This substrate selectivity can actually be exploited for substrates containing two alkenes at different distances from the carboxylate, as shown in Scheme 48a. As the alkene closer to the carboxylic acid better matches the guanidine–rhodium distance, this alkene is converted at a higher rate with a ratio of 8.8:1. In addition, this alkene is converted with higher selectivity for the linear aldehyde with l:b = 32, compared to an l:b ratio of merely 0.3 for the other alkene moiety.382
Scheme 48. (a) Site-Selective Hydroformylation by Substrate Organization via Hydrogen Bonding Containing Multiple Olefinic Sites, (b) Decarboxylative Hydroformylation of α,β Unsaturated Carboxylic Acids by Substrate Organization, (c) Tandem Hydroformylation–Hydrogenation Sequence Converting 1-Octene into 1-Nonanol, (d) Tandem Decarboxylative Hydroformylation–Hydrogenation of α,β Unsaturated Carboxylic Acids, and (e) Tandem Supramolecular Rh Catalyzed Hydroformylation of α-Alkynoic Acids Followed by Michael Addition and Decarboxylation.

Next to regular hydroformylation, also decarboxylative hydroformylation of α,β-unsaturated carboxylic acids was explored (see Scheme 48b).384 Using substrate preorganization, the alkene was selectively functionalized, leading to the aldehyde intermediate, which after decarboxylation, results in the final linear aldehyde product. Complexes based on triphenylphosphine did not lead to this linear aldehyde but instead only gave reduction of the double bond.
Modifications on the ligand building blocks were explored. When instead of the pyridine-containing ligand (230), the benzene (231) or a pyrrole (232) analogue was used as ligand (see Figure 15), aldehyde hydrogenation is observed.385 Complexes based on 231 and 232 were thus used in a tandem hydroformylation–hydrogenation sequence converting 1-octene into 1-nonanol (Scheme 48c). The selectivity for the linear alcohol can be enhanced by using a catalyst based on the pyrrole analogue of the guanidium ligand, in combination with the 2-pyridone/2-hydroxypyridine hydrogen-bonded bidentate (6-DPPon (135), Scheme 29). This indeed provided a highly selective hydroformylation–hydrogenation reaction of 1-octene to 1-nonanol. Furthermore, combining the decarboxylative hydroformylation catalyst with a supramolecular aldehyde hydrogenation catalyst yields an effective system for tandem decarboxylative hydroformylation–hydrogenation (Scheme 48d).386 The yield for the alcohol can be 99% when 230 is used or when a mixture of ligands is used. Finally, the supramolecular approach was used for the Rh catalyzed hydroformylation of α-alkynoic acids followed by Michael addition and decarboxylation, using an electron-poor phosphorus ligand equipped with the acyl guanidine moiety (252) (see Scheme 48e).387 This domino reaction is triggered by the Rh catalyzed hydroformylation of α-alkynoic acids, requiring the hydrogen bonding interaction between the ligand and the substrate. Consecutive Michael addition of arenes as nucleophiles lead to an intermediate which after decarboxylation of the carboxyl function leads to the β-aryl aldehyde products. In this sequence the carboxyl function is a transient and traceless directing group for the introduction of the aldehyde function. The strategy has been used for the preparation of a key intermediate for the synthesis of Avitriptan.
4.3. C–H Activation
Transition metal catalyzed C–H bond activation allows the introduction of functional groups at a late stage of a synthesis protocol and, as such, is an increasingly applied tool.388−394 The direct C–H borylation is of particular interest as it installs a boron functional group, which allows further modification by, for example, Suzuki coupling reactions, amination, hydroxylation, and halogenation.395−399 Controlling the selectivity of a C–H activation reaction is particularly challenging, as typically there are several similar C–H bonds present in a molecule and they are not electronically activated. Recently, the use of hydrogen bonding between the substrate and the ligand of a metal complex has been explored to control the selectivity. These approaches resulted in regioselective catalysts for C–H borylation for a diversity of substrates.
The hydrogen bonding approach was pioneered by Kanai and co-workers, who explored the use of urea functionalized 2,2′-bipyridine (bpy) ligands exemplified in Figure 16.4001H NMR spectroscopic experiments established that HBs form between the amide functional groups of the substrate and the urea motifs. In the hydrogen-bonded complex, the substrate is preorganized for meta-selective C–H activation. Application of N,N-dihexylbenzylamide as a substrate in the iridium catalyzed borylation afforded the meta product in high selectivity (meta/para = 8.3 under standard conditions, 27 in p-xylene as solvent). Importantly, control experiments in which the urea functional groups were not properly positioned resulted in unselective borylation reactions. The thiourea analogue was also used; however, the iridium complex of this ligand did not provide any product. It was also shown that the hydrogen bonding resulted in faster catalysis and further optimization of the ligand was possible by the presence of functional groups at the bipy part of the ligand.401 The approach was followed up by Phipps and co-workers, who used an iridium catalyst containing sulfonated anionic bpy ligands that were active for meta-selective borylation.402 In these systems HBs were proposed to be formed with the anionic sulfonate group. Using a similar preorganization strategy as reported by Kanai, Chattopadhyay, and co-workers made a bpy ligand functionalized with a naftapyrildone functional group, which also gave highly meta-selective borylation catalysts.403,404 Part of the substrate scope that could be achieved with urea-functionalized ligands is shown in Scheme 49, demonstrating that the reaction can tolerate a variety of different functional groups.
Scheme 49. HB Directed meta-Selective C–H Activation Using the Urea Functionalized Ligand (253) (See Figure 16).

Part of the substrate scope is displayed with meta/ortho > 7.
In order to have access to ortho-borylated compounds (256), a new ligand was designed that also operates via HB-assisted substrate orientation. A bipyridine ligand was functionalized with an indole amide functional group, coined BAIPy (257) (Figure 17c), which has the HB donors closer to the metal in the iridium complex.405 DFT calculations of the C–H activation step show that three HBs form between the substrate and the catalyst. The two anticipated HBs form between the indole amide and the carbonyl of the substrate, while a third unexpected HB was identified between the N–H moiety of the substrate and an oxygen of a Bpin group attached to iridium. Experiments employing the model substrate show high selectivity for ortho-selective C–H borylation. In contrast, in the control experiment with the parent bis-pyridyl ligated iridium complex, only meta and para borylated product are formed, and the ortho borylated product is not formed at all (Figure 17a and b). Interestingly, using N,N-dimethylbenzylamide as substrate gave substantially lower ortho selectivity (1:1), indicating that the substrateN–H···OBpin HB identified in the complex with N-methylbenzylamide (Figure 17c) is of importance.
Figure 17.

(a) Ir catalyzed C–H borylation of 259 to 260; (b) selectivity patterns obtained using Ir catalysts based on BAIPy (257) or bpy (258). (c) DFT modeling of the substrate–BAIPy (257)–trisboryl–Ir catalyst complex shows that three HBs form between the substrate and the catalyst (purple dashed lines). The indole amide functional group in 257 thus orients the substrate by hydrogen bonding with secondary aromatic amide substrates, leading to ortho-selective C–H borylation. The model reaction using complexes based on bipyridine as the ligand (bpy, 258) does not display this selectivity. Adapted with permission from ref (405). Copyright 2019 Wiley under a CC-BY-NC license [https://onlinelibrary.wiley.com/doi/10.1002/anie.201907366].
In addition to the high selectivity achieved, hydrogen bonding between the substrate and ligand also enabled faster catalytic reactions. In the substrate scope illustrated in Scheme 50, more than 26 examples of N-methylbenzamides and aromatic amides were reported, including peptide-based analogues. This large substrate variety demonstrates that this supramolecular catalyst is compatible with a plethora of functional groups, featuring the catalyst’s general applicability. The supramolecular iridium catalyst has been applied at gram scale with high conversion and selectivity at elevated temperature. The ligand is easily prepared at large scale, facilitating the application of catalysts that operate via hydrogen bonding in C–H borylation. These two examples show that supramolecular substrate orientation by hydrogen bonding is a powerful approach to control the regioselectivity in challenging C–H borylation reactions.
Scheme 50. Part of the Substrate Scope of Selective Borylation Using Iridium Complexes of BAIPy (257).

The strategy of substrate orientation by hydrogen bonding was further extended by the group of Sawamura to asymmetric C–H borylation of aliphatic amides and esters.406 To this end, they used chiral iridium complexes (262) based on a chiral Bisnaphtol-based bulky phosphite ligand (263) and a phenyl urea functionalized pyridyl ligand (264) as shown in Scheme 51a. Chiral information is provided by the phosphorus ligand (263), whereas the urea moiety on the pyridyl ligand (264) preorganizes the substrate with respect to the iridium center. Secondary amides and esters were converted in high e.e.’s with the borylation occurring at the gamma position of the substrate. Variation of the pyridyl ligand (247) in which the nitrogen is placed in the para or ortho position with respect to the phenyl urea group also resulted in borylation at the gamma position but with lower selectivity and yield. When the reaction was carried out at elevated temperature, the enantioselectivity dropped only slightly (from 99 to 87% at 80 °C). The substrate scope was very broad, as a variety of substrates with different functionalities at the amide were converted. Also, the reaction was shown to be tolerant to variation at the aliphatic tail, and larger substrates and substrates with alkenes in the chain were still selectively converted (Scheme 51c).
Scheme 51. (a) Iridium Catalysts (262) with a Chiral Ligand (263) and a Ligand for Substrate Orientation (264); (b) Leading to Remote C–H Borylation Producing the Product 266 in High Enantioselectivity (up to 99% e.e.); (c) Part of the Substrate Scope with Aliphatic Chains for Asymmetric Remote C–H Borylation.

For substrates 267 the borylated intermediate product was directly converted to the alcohol.
4.4. Radical-Type Carbene and Nitrene Transfer Reactions
Metallocarbene and nitrene radicals are important intermediates in a variety of radical-type carbene and nitrene transfer reactions mediated by cobalt(II) catalysts. Implementing hydrogen bonding interactions between structural motifs in a catalyst with the substrate has been shown to be a powerful method to control both the activity and (enantio)selectivity of these reactions. In particular, chiral porphyrin complexes with HB donors attached to the ligand framework have been shown to be powerful catalysts for enantioselective radical-type reactions. Such reactions typically rely on a combination of the unique electronic structures of the key intermediates and supramolecular interactions with HB moieties in the second coordination sphere of the catalysts.407−411 Instead of traditional (Fischer-type) carbene and nitrene complexes of cobalt(II), unique cobalt(II)-carbene and nitrene radicals are formed by single electron transfer from cobalt to the carbene or nitrene moiety. Computational and EPR studies have shown that the spin densities of the carbene and nitrene complexes are almost entirely located on a p-orbital of the carbene or nitrene moiety, as is illustrated in Figure 18.412 The resulting substrate radical species are key intermediates in a broad range of catalytic group transfer reactions to C–H, C=C, and C≡C bonds. A unique feature of these systems is the fact that intramolecular electron transfer from cobalt to the substrate occurs simultaneously with generation of the carbene or nitrene moiety. This leads to one-electron reduced Fischer-type carbenes or nitrenes, resulting in lower electrophilicity of activated substrates, rendering them partially nucleophilic and imposing radical reactivity together with a unique selectivity (Figure 18).
Figure 18.

Formation of radical-type carbenes and nitrenes in cobalt(II) catalyzed group transfer catalysis.
Increased electron density at bound substrates also provides novel opportunities to control the enantioselectivity of these radical-type reactions using hydrogen bonding interactions in the second coordination sphere. The high activity and enantioselectivity of such reactions can be explained by combined transition state stabilization and substrate orientation. This is illustrated in Figure 19 for carbene radical formation.407 In the presence of chiral amide HB donors in the second coordination sphere, formation of the carbene radical intermediate shows a lower barrier than without these HBs. Electron transfer from cobalt to the carbene moiety results in a stronger HB to the hypovalent carbene moiety than in the precursor, which explains the lower transition state barrier. At the same time, these interactions bring the chiral information of the catalyst close to the reactive substrate, which can lead to high enantioselectivities of the follow-up carbene transfer reactions. Similar effects play a role in radical-type nitrene transfer reactions with cobalt catalysts.410,411
Figure 19.

Hydrogen bonding leading to transition state stabilization in cobalt(II) catalyzed group transfer catalysis.
Shown in Scheme 52 is a selection of cobalt(II) porphyrins that have been studied in radical-type group transfer catalysis. The nonchiral [CoII(P1)] (270) (P1 = meso-tetraphenylporphyrin) has been used extensively as a workhorse for the synthesis of several cyclic and noncyclic products via metalloradical catalyzed carbene transfer. The cobalt–carbene radical intermediates involved in these reactions undergo stepwise controlled radical addition or hydrogen atom abstraction (HAA) and then transform to various interesting structures in a mild and efficient manner. The redox-active carbene substrates thereby give access to the [CoII(P1)] catalyzed formation of, e.g., cyclopropanes,413−418 2H-chromenes,419,420 furans,421 indenes,422 ketenes,423,424 butadienes,425 dihydronaphthalenes,425 piperidines,426 pyrrolidines,427 dibenzocyclooctenes,428,429 and monobenzocyclooctadienes.429 Carbene precursors for these reactions are typically diazo compounds (R2C=N2) and N-tosylhydrazones. With some exceptions,417,430−432 enantioselective carbene transfer reactions typically rely on catalysts capable of providing additional hydrogen bonding interactions in the second coordination sphere (Figure 19 and Scheme 52).
Scheme 52. Selection of Cobalt(II) Porphyrins Used in Radical-Type Group Transfer Catalysis.

Scheme 53 shows examples of asymmetric cyclopropanation of aromatic, aliphatic, electron-rich, and electron-deficient olefins under mild reaction conditions.415,418,433−444 These cyclopropanation reactions are dominated by reactions involving mostly styrenes and alkenes bearing electron withdrawing and radical-stabilizing substituents. With some exceptions,434,438,445−447 most of these reactions involve diazo compounds (or their tosylhydrazone precursors) containing a single substituent at the carbenoid carbon atom. Catalyst [CoII(P2)] (271) and closely related analogs were also successfully applied in the asymmetric cyclopropanation of allyl α-diazoacetates and α-formyldiazoacetates, which were obtained in high yields and with good e.e.’s.447−449 Complex [CoII(P3)] (272) provides a more rigid structure due to intramolecular O···H–N hydrogen bonding interactions in the ligand backbone (indicated in red), which enables trans-cyclopropanation of styrene with HC(N2)(p-toluenesulfonyl) with excellent enantioselectivity.415
Scheme 53. Asymmetric Cyclopropanation of Alkenes Catalyzed by Catalysts of the Types [CoII(P2)] (271) and [CoII(P3)] (272) and Analogs (See Also Scheme 52).

Next to cyclopropanation, several (enantioselective) cyclization reactions proceeding via carbene insertion into (activated) C–H bonds have been disclosed. Hydrogen bonding interactions between the substrate and the catalyst again play a crucial role in some of these reactions, both to facilitate activation of the carbene precursor and, in particular, to control the enantioselectivity. For example, as depicted in Scheme 54, the chiral amidoporphyrin analogs of [CoII(P2)] (i.e., [CoII(P4)]) are also capable of asymmetric intramolecular 1,5-C–H alkylation/cyclization of α-methoxycarbonyl-α-diazosulfones to form sulfolanes in high yields and with good diastereo- and enantioselectivities.450
Scheme 54. [CoII(Px)] Catalyzed (See Also Scheme 52) Asymmetric C–H Alkylation Leading to Cyclization of α-Methoxycarbonyl-α-Diazosulfones.

R = aryl, triazole, alkenes, and allene.
Related protocols have also been developed for the synthesis of pyrrolidines (Scheme 55a, top),447 piperidines (Scheme 55b),426 and indolines (Scheme 55c).451,452
Scheme 55. [CoII(Px)] Catalyzed (Asymmetric) C–H Alkylation Cyclization Reactions for the Synthesis of (Chiral) Pyrrolidines (a), Piperidines (b), and Indolines (c).

Catalysts [Co(Px)] are of the type [Co(P1)] (270), [Co(P2)] (271), [Co(P3)] (272), and analogs.
More recent examples from the Zhang group revealed that four-membered cyclic ketones also can be constructed in an enantioselective manner by a metalloradical catalyzed HAA and rebound sequence as shown in Scheme 56a.453 The related radical cascade reaction also allows for the enantioselective synthesis of bicyclic compounds illustrated in Scheme 56b.454 The enantioselectivities of these reactions again seem to be largely controlled by the hydrogen bonding interactions between the substrate(s) and chiral HB donors in the second coordination sphere of the catalyst.
Scheme 56. (a) [CoII(Px)] Catalyzed Asymmetric Cyclization Reactions Leading to Chiral Cyclobutanones (b) and Bicyclic Compounds via Cascade Radical Catalysis.

Catalysts [CoII(Px)] are of the type [CoII(P2)] (271), [CoII(P4)] (273a), [CoII(P5)] (273b), and analogs.
Just like radical-type carbene transfer reactions, radical-type nitrene transfer reactions can also be effectively controlled by hydrogen bonding interactions in the periphery of the catalyst binding site. As such, several (enantioselective) cobalt catalyzed aziridination reactions have been reported, as exemplified by those shown in Scheme 57. With few exceptions,455−457 the amide moiety of the macrocyclic ligand in the catalyst acts as the HB donor, while the substrate is a HB acceptor in the form of a (nitrene radical generated from) reactive, preactivated organic azide such as RSO2N3, (RO)2P(=O)N3, or ROC(=O)CN3, as shown in Scheme 57.411,443,458−468
Scheme 57. Selected Examples of [CoII(Px)] Catalyzed HB-Assisted (Asymmetric) Aziridination Reactions.

Catalysts [CoII(Px)] are of the type [CoII(P2)] (271), [CoII(P3)] (272), [CoII(P4)] (273a), [CoII(P5)] (273b), and analogs.
The nitrene radical species illustrated in Scheme 58 were unambiguously detected and characterized with ESI-MS spectrometry and EPR, UV/vis, IR, and XAS spectroscopy (supported by DFT), and IR and VCD studies clearly revealed hydrogen bonding interactions between the amide moieties of the catalyst and the substrate for these types of substrates.469 DFT studies showed that the barriers for activation of the azide substrate are lowered by these same hydrogen bonding interactions.411
Scheme 58. Hydrogen Bonding Interaction between the Chiral Amide Moiety of the Ligand, the Preactivated Azide, and the Reactive Nitrene-Radical Substrate.

Several cyclization reactions to form five- and six-membered ring compounds proceeding via nitrene insertion into C–H bonds have also been developed (Scheme 59), with most reactions involving preactivated HB acceptor containing organic azides as the nitrene (radical) source.465,470−473
Scheme 59. [CoII(Px)] Catalyzed Cyclization Reaction via Formal Nitrene Insertion into a C–H Bond of the Substrate.

Catalysts [CoII(Px)] are of the type [CoII(P2)] (271), [CoII(P3)] (272), [CoII(P4)] (273a), [CoII(P5)] (273b), and (nonchiral) analogs.
HB interactions between the catalyst and the substrate seem to play an essential role in most of these reactions, but conversions are mostly nonenantioselective. However, some of the more recently developed D2-symmetric catalysts bearing HB donors give surprisingly high e.e.’s,474−476 in particular for catalysts bearing tethered side groups of the types [CoII(P4)], [CoII(P5)], and their analogs (see Scheme 52).
A recent report by the Zhang group is particularly noteworthy.477 In that study the authors have shown that racemic alkylsulfamoyl azide substrates can be converted with cobalt(II) catalysts of the type [CoII(P2)] in an enantioconvergent manner in order to produce chiral six-membered ring products. As is shown in Scheme 60, the reactions proceed via a HAA step converting the chiral center of the racemic substrate into a planar carbon radical, followed by an enantioselective radical-rebound step controlled by the chiral catalyst to produce the product.
Scheme 60. Enantioconvergent Cyclization of Racemic Alkylsulfamoyl Azides with Complexes of Type [CoII(P2)], Proceeding via Formal Nitrene Insertion into a C–H Bond of the Substrate.

In addition to the examples given above where substrate orientation is achieved by a HB, this principle can also be combined with the effects of steric interactions and van der Waals forces in the second coordination sphere. For example, as illustrated in Scheme 61, enantioselective intramolecular C–H amination can be steered to produce pyrrolidines and related five- and six-membered ring compounds from unprotected and non-preactivated aliphatic azides.478
Scheme 61. Synthesis of Pyrrolidines and Related Ring Compounds from Unprotected, Non-preactivated Aliphatic Azides.

Some intermolecular nitrene insertion reactions into benzylic C–H bonds have also been disclosed,479,480 although thus far, only a few examples involve HB assistance from the ligand.481 To our best knowledge, only a single example of a HB-assisted enantioselective intermolecular nitrene C–H insertion reaction has been disclosed.482
4.5. Hydrogen Bonding in Oxidation Catalysis
Hydrogen bonding interactions have also been shown to be useful in enabling site-specific reactivity in oxidation catalysis. An illustrative example is the supramolecular manganese catalyst developed by Crabtree and Brudvig (Figure 20), which is equipped with a carboxylic acid that forms hydrogen bonding interactions with the substrate to position the targeted benzylic position close to the reactive dinuclear Mn-oxo site.483 The terpyridine ligand of the Mn catalyst contains a rigid U-shaped motif with a carboxylic acid moiety acting as the H-bonding recognition site for substrates containing complementary carboxylic acids for H-bonding. The resulting supramolecular catalytic systems are capable of regioselective oxidation of the benzylic C–H bonds of ibuprofen with oxone (∼70% conversion, with 97% selectivity). The selectivity for the remote benzylic position is enforced by H-bonding interactions between the substrate and the catalyst, which was proven by comparison with the results with a similar complex without the substrate recognition site producing oxidized product at both benzylic positions in only a 3:1 ratio. Other control experiments using the ester variant of the substrate and performing the reaction in the presence of acetic acid also led to a loss in selectivity. The C–H bonds of the distant tertiary carbon atoms of trans-4-methylcyclohexyl acetic acid can also be selectively oxidized with this catalyst, but the conversions and yields are lower for this substrate. A mixture of cis- and trans-4-methylcyclohexyl acetic acid leads to selective conversion of the trans isomer, which has been proposed to be the result of steric blocking of the reactive site from access to the unbound substrate.484
Similar approaches were explored by Bach and co-workers (Figure 21). They used a [Ru(porphyrin)] complex as the oxidation catalyst, equipped with a lactam binding motif as the DA HB array. A series of substrates containing a complementary DA HB array could be oxidized stereoselectively.485 As such a high enantiomeric ratio was achieved (95:5 e.r.; 20% yield), the yield could be improved to 70% by addition of an auxiliary oxidant. Interestingly, alkylation of the N–H bond of the substrate repressed the yields substantially, thus showing the importance of H-bonding recognition during catalysis.
Enantioselective oxidation of the benzylic C–H bonds of 3,4-dihydroquinolones was also successful.486 For this purpose Ru was replaced by Mn, resulting in a more efficient catalyst with a decreased tendency for overoxidation. High enantioselectivities (up to 99% e.e.) with preference for the (S)-alcohol were obtained for a broad range of substituents. Again, the importance of H-bonding was shown by alkylation of the N–H bond, leading to a close to racemic mixture. This work was extended to dirhodium complexes utilized with the same binding motif for substrate orientation for the enantioselective C–H amination reaction.487,488 In a related approach, the group of Costas designed a supramolecular (White-type) aminopyridine Mn catalyst for site-selective oxidation of ammonium salts, using H2O2 as the oxidant.489 The catalyst is equipped with an 18-benzocrown-6 ether in the second coordination sphere that interacts with the ammonium ion functionality of the substrate and, as such, positions the C(8)–H and C(9)–H bonds close to the Mn=O site for hydroxylation (Figure 22). A selectivity of 81% for site-selective C–H hydroxylation of the C8 and C9 position in a series of linear alkyl ammonium salts with different chain lengths (C6 to C14) could be achieved. Control experiments revealed the importance of H-bonding between the ammonium group and the crown ether moiety: Addition of blocking agents to the crown ether, such as Ba(II), or alkylation of the N–H bonds of the substrate leads to loss of selectivity for oxidation of the C8/C9 position.
Figure 22.

Site-selective oxidation of the C8 and C9 positions of linear ammonium salts directed by H-bonding interactions with a crown ether in the second coordination sphere of the Mn catalyst reported by Costas and co-workers.
The above examples clearly show that hydrogen bonding is a broad and effective tool to achieve selectivity in C–H bond oxidation reactions, and future studies are likely to reveal many more applications.
4.6. Photocatalysis
Photochemical reactions such as photoinduced electron transfer (PET)490 and, more recently, also triplet energy transfer (EnT)491 have gained increasing interest in organic synthesis in the past years. Enantioselective photocatalysis is especially intriguing, as photochemical reactions such as photocycloadditions may generate multiple stereocenters in one step. A major challenge in this respect is that photocatalysis involves highly reactive intermediates, which typically follow unimolecular relaxation pathways, leading to rapid deactivation. In the past, photocatalysts have been combined with a second chiral catalyst in order to control the stereochemistry of the reaction.492 The short lifetime of excited states limits enantioselective catalysis in biomolecular systems, since deactivation is often faster than diffusion, and thus, chirality transfer from the chiral auxiliary is not compatible. In order to achieve better enantioselectivity by chiral transfer reagents, novel concepts are required that are able to cope with this challenge. Meggers has demonstrated that chiral-at-metal photocatalysts can serve as bifunctional catalysts providing high enantioselectivity due to “enantioface separation”.493,494 As was detailed in the previous sections, hydrogen bonding serves as a general, highly potent tool to preorganize substrates and catalysts. In the following, recent advances will be discussed where enantioselective photocatalysis is achieved by hydrogen bonding strategies with a focus on transition metal-based catalysts.
Research on hydrogen bonding in photocatalysis has initially focused on organic dyes as photocatalysts. In the first proof-of-principle study, Krische and co-workers combined a hydrogen bonding motif for substrate binding with benzoquinone photosensitizer to catalyze intramolecular [2 + 2] photocycloaddition reactions of quinolones.495 Even though the enantioselectivity was relatively low in this system (22% e.e.), this study inspired further research in this direction. After this, hydrogen bonding as a strategy in photocatalysis has been mainly developed by the groups of Bach and Yoon. Several recent reviews on the topic have been published;490,496,497 therefore, we will only briefly mention selected examples of organic photocatalysts and focus our discussion on transition metal-based photocatalysts.
Bach developed a lactam-binding motif attached via a 1,5,7-trimethyl-3-azabicyclo[3.3.1]nonan-2-one backbone to a series of organic dyes (336a–c) shown in Scheme 62a.496 Substrates containing lactam units bind in a complementary fashion to this site via a DA HB array (see also Figure 2) as illustrated in Scheme 62c. For intramolecular [2 + 2] photocycloaddition of quinolones such as 337, benzoquinone-based photocatalyst 336a showed lower enantioselectivity compared to xanthone-based catalyst 336b because it is not planar and substrates do not bind sufficiently strongly.498 More rigid and planar xanthone (336b) and thioxanthone (336c)-based catalysts yield overall better enantioselectivities in such reactions. Both catalysts (336b and 336c), however, feature low stability in solvents which are prone to hydrogen abstraction.499 Therefore, low reaction temperature and relatively nonpolar solvents, such as trifluorotoluene (PhCF3), are required. Under these optimized conditions, a variety of reactions are catalyzed by 336b and 336c, featuring high yields and excellent enantioselectivities (Scheme 62b), including intra- and intermolecular [2 + 2] photocycloadditions500−502 as well as deracemization reactions.503
In comparison to organic dyes, many transition metal-based photosensitizers feature superior chemical stability and longer excited state lifetimes. For instance, iridium(III)- and ruthenium(II)-based photosensitizers display high activity and stability at low catalyst loading and, therefore, are both highly potent photocatalysts for PET and photosensitizers for EnT due to their high energy and long-lived triplet states.491
Yoon and co-workers developed the bifunctional iridium(III) polypyridyl complex (339) shown in Scheme 63, bearing a pyridylpyrazole hydrogen bonding moiety for hydrogen bonding in its ligand.504 This complex acts as a triplet photosensitizer in the asymmetric intramolecular [2 + 2] photocycloaddition of quinolones. As illustrated in Scheme 63, quinolone (340) binds via a DA HB array to the pyrazol ligand. Excitation of the iridium photosensitizer results in triplet energy transfer from the photosensitizer to the bound substrate following a Dexter energy transfer mechanism (e.g., simultaneous transfer of the excited electron from the sensitizer to the LUMO of the substrate and electron transfer from the HOMO of the substrate to the photosensitizer). The product (341) contains four new stereocenters, and due to the hydrogen bonding site, the reaction proceeds with high yield and more than 80% e.e. for 13 different substrates. The reaction scope is, however, limited to intramolecular reactions of substrates containing a lactam moiety.
Scheme 63. Intramolecular [2 + 2] Photocycloaddition Catalyzed by Iridium(III) Photosensitizer 339 with a Hydrogen Bonding Motif in the Ligand.

Energy transfer proceeds via the Dexter mechanism.
Slight modification of the photosensitizer to iridium(III) complex 342 shown in Scheme 64 also enabled intermolecular photocycloaddition of 3-alkoxyquinolones 343 and maleimide 344.505 In contrast to the previous example where energy transfer was directed from the sensitizer to the bound substrate, a combination of kinetic, spectroscopic, and computational studies showed that in this case, the reaction proceeds to nonbound maleimide 344, yielding 3maleimide (a triplet). Substrate preorganization of hydrogen-bonded 3-alkoxyquinoline 343 enables rapid intermolecular cycloaddition to the activated maleimide, leading to excellent overall yield and enantioselectivity. This mechanism was supported by a variety of experiments. First, titration experiments monitored by NMR spectroscopy showed that the ground-state hydrogen bonding interaction of the catalyst is negligible with maleimide 344 compared to its interaction with quinolone 343. Even though 344 does not bind to the photocatalyst, luminescence measurements suggested that it plays a critical role in the photocatalytic mechanism. Stern–Volmer quenching studies with photocatalyst and each of the reaction partners showed significantly larger quenching with maleimide 344 compared to quinolone 343. Transient absorption spectroscopy further supported the assumption that the reaction proceeds via energy transfer to 344: When iridium(III) complex 342 was excited in the presence of 343, the lifetime of the excited state was not significantly altered. However, in the presence of 344, the excited state lifetime was significantly reduced from 4.3 to 0.5 μs. Interestingly, variation of the structure of the photosensitizer showed that the ppy ligand has a larger effect on the reaction than the hydrogen bonding ligand. In addition, the identity of the 3-alkoxy substituent on substrate 343 has a large impact on the stereoselectivity.
Scheme 64. Intermolecular [2 + 2] Photocycloaddition with Modified Iridium(III) Photosensitizer 342.

Energy transfer proceeds to the nonbound maleimide 344.
Following up on their work on organic photosensitizers, Bach and co-workers developed transition metal-based chiral supramolecular catalysts where the metal center and chiral unit are covalently linked but spatially separated. In analogy to their organic photosensitizers containing the lactam unit for a DA HB array of interaction complementary substrates, they synthesized iridium(III) catalyst 346 shown in Scheme 65 with the same motif linked to the bipyridine ligand of the complex. As before, the lactam DA HB array enables hydrogen bonding with other lactams. The resulting complexes are kinetically labile, with lifetimes of 10–100 ns. Two different linkers between the photosensitizer and the hydrogen bonding motif were examined: alkynyl (346a) and enthano (346b). The investigation of the substrate and reaction scope was started with prochiral halide substrates, which were expected to form radicals upon reduction and C–X cleavage, that should then rapidly perform C–C coupling to form the cyclization product. For these substrates, instead of the desired cyclization product, only hydrodebromination products were observed. The authors suggested that the problem might lie in the reaction pathway of photoredox reactions: many photoredox mechanisms proceed via radical chain processes instead of via closed reaction cycles. Therefore, even substrates that are not kept close to the metal center (via hydrogen bonding) can be reduced eventually. The authors, therefore, turned their focus to reactions involving a triplet energy transfer (EnT) mechanism from the sensitizer to the substrate. Epoxide rearrangement of spirooxindole substrate 347 was studied. The ethano version of the iridium photocatalyst (346b) showed better conversion and overall yield (to both isomers) compared to the alkynyl-linked catalyst (346a). However, the enantioselectivity remained low (29% e.e. at best). The low enantioselectivity was explained by the fact that the enantioselectivity does not only depend on the steric bias of the respective catalyst but is also influenced by the rate of intermediate dissociation from the catalyst. In the case that the complex dissociation is faster than the selectivity determining step (epoxide rearrangement), the e.e. remains low. A similar observation has been made for the organic xanthone photocatalyst (336b) in previous studies.506
Scheme 65. Epoxide rearrangement catalyzed by iridium(III) photosensitizer 346b containing a lactam binding motif proceeds in high yields and moderate enantioselectivity.

Turning back to bimolecular catalyst systems, Bach and co-workers merged an achiral photoredox catalyst (349) and chiral hydrogen bonding template (350) as shown in Scheme 66, which works very well as a chiral auxiliary in nonpolar solvent at low temperatures. It should be noted that in this system, the hydrogen bonding motif is not included in the second coordination sphere of the metal complex but as part of the organic cocatalyst. In analogy to previous studies, substrates containing a lactam binding motif bind to the template using the DA HB array provided by the lactam. In this study, radicals were generated via PET from the excited photosensitizer to trimethylsilyl (TMS) methyl-substituted amines (351). 3-Alkylidene indolin-2-ones such as 352 are readily bound to chiral template 350 and, thus, shielded from one side by the tetrahydro-1-oxa-3-azacyclopenta[b]naphthalene motif. This hydrogen-bonded complex is attacked by radicals that are formed via photoinduced electron transfer from photosensitizer 349 to nonbound substrate 351. Overall moderate to good enantioselectivities were observed.
Scheme 66. Dual System by Bach with Photocatalyst 349 and Chiral Template 350 for Radical Addition Reactions.

Here, the hydrogen bonding motif is not in the second coordination sphere of the metal complex but in the organic cocatalyst.
4.7. Allylic Substitution
The palladium catalyzed allylic substitution reaction is a key reaction for organic synthesis and has been explored for decades, leading to many different protocols using a large variety of ligand scaffolds.507,508 In a typical reaction, illustrated in Scheme 67, the palladium allyl complex (352) is generated from a palladium(0) complex and an alkene susbstrate (351) with a proper leaving group. Subsequent outer sphere nucleophilic attack leads to the formation of the substituted product (353). The application of a chiral ligand in this reaction can result in the formation of the product with very high enansioselectivity, making it a versatile tool for organic synthesis. The outer sphere nucleophilic attack typically plays an important role in controlling the enantioselectivity. If unsymmetric 1,3-disubstituted substrates are used, that is when R1 and R2 are different (Scheme 67); the nucleophilic attack also determines the regioselectivity of the reaction. It has been demonstrated that hydrogen bonding between the functional groups of the ligand and the nucleophile can orient the nucleophile and with that a better control of the selectivity can be achieved.
Scheme 67. General Scheme for the Allylic Substitution Reaction.

The bis(sulfoxide)phosphine ligand BiSO-P (354)509 shown in Scheme 68 forms palladium complexes in which the ligand coordinates in a P–S bidentate mode with a dangling sulfoxide. The complex was explored in the Pd catalyzed dynamic kinetic resolution of racemic unsymmetrically 1,3-disubstituted allylic acetates with indoles, providing a high level of stereocontrol. The high selectivity of this catalyst was explained by the presence the sulfoxide as a HB acceptor, directing the indole as a nucleophile by hydrogen bonding. NMR studies and Job plot analysis show that the indole indeed HBs with the complex, leading to supramolecular complexes with a 1:1 stochiometry.
Scheme 68. BiSO-P Ligand That Forms a Bidentate Chelated Palladium Complex, with a Dangling Sulfoxide That Can HB to the Indole Nucleophile.

In an early paper by Hayashi et al., a diphosphine ferrocene-based ligand was reported with a dangling hydroxy group for nucleophile preorganization (355, Figure 23).510 This ligand provided a palladium complex that induced significant enantioselectivity (81% e.e.) in the allylation of 1,3-dicarbonyl compounds, which was proposed to be a result of hydrogen bonding between the OH of the ligand and the nucleophile. In a few other papers, hydrogen bonding between the ligand and the nucleophile has been proposed to play a role in controlling allylic substitution reactions.511,512
Figure 23.

DPPF-based ligand with a dangling hydroxyl group proposed to direct the nucleophile by HB formation.
5. Summary and Outlook
Traditionally, the development of homogeneous catalysis involves the preparation of novel transition metal complexes that can be used for a certain chemical transformation. The activity and selectivity that such complexes possess are a result of the interplay between the ligand and metal properties in the complex. A large focus has, therefore, been on ligand development and descriptors for ligands to facilitate a more rational approach to catalyst development. High throughput experimentation and combinatorial techniques can speed up the search for catalysts for specific conversions, provided that libraries of sufficient size and diversity can be generated for the specific conversion at stake. More recently, additional strategies to control catalyst properties have been explored that involve the second coordination sphere, which is beyond the direct coordination sphere of the metal center. HBs appear to be very useful interactions in this context, as they typically have sufficient strength and directionality (section 2). In this review we have summarized the use of HBs to bridge two ligands that are coordinated to a metal center to effectively lead to supramolecular bidentate ligands (section 3), as well as the use of HBs to preorganize a substrate (section 4).
Supramolecular bidentate ligands have typical bidentate behavior, leading to larger control over the first coordination sphere. Concurrently, the ligand building blocks are generally easier to prepare. This is particularly the case for heterobidentate ligands (i.e., two different donor atoms), where the number of possible bidentates grows exponentially with the number of monodentate building block, thus enabling the generation of larger libraries of bidentates.
Utilizing HBs between ligand systems and the substrate has allowed for a more precise orientation of the substrate with respect to a metal center, which was shown to be of great benefit for the preparation of selective catalysts. In particular, HBs can be used to block (altered selectivity) or facilitate (higher activity) certain reaction pathways that a substrate could naturally undergo as determined by the electronic and/or steric biases of the substrate. Indeed, such reprogramming of selectivity and reactivity could be demonstrated in asymmetric hydrogenations (section 4.1), hydroformylations (section 4.2), C–H activations (section 4.3), radical-type reactions (section 4.4), oxidations (section 4.5), photochemical reactions (section 4.6), and allylic substitutions (section 4.7). The utilization of HBs in the second coordination sphere provide many examples of selectivities that are not reached with traditional catalysts. Moreover, the large variety of conditions that have been deployed, such as elevated temperatures and polar solvents, show that using HBs in this manner is a more powerful strategy than one may intuitively anticipate.
These advances notwithstanding, a more rational and systematic design approach of catalysts that make use of the full plethora of tricks to manipulate hydrogen bonding effects outlined in section 2.2 is an obvious next step. For example, very strong HB arrays with optimal secondary interactions (e.g., AAA/DDD or AAAA/DDDD) are relatively rare and have not been explored in the context of second coordination sphere control for catalysis. Moreover, the design of a catalyst from scratch addressing an unsolved selectivity issue in catalysis would truly demonstrate the power of using HBs in the second coordination sphere. Finally, the extension of the concepts reviewed here to include the deployment of other noncovalent interactions71,345,513 besides HBs (e.g., π–π stacking and halogen- or tetrel-bonding interactions) would broaden the scope of the approach. We are convinced that these types of approaches will become more standard in the design of the next generation of efficient catalysts with unique selectivities.
Biographies
Joost N. H. Reek obtained his Ph.D. at the University of Nijmegen under the supervision of Prof. R. J. M. Nolte in the area of supramolecular chemistry. After a postdoc with Prof. Crossley in Sydney, he moved to the University of Amsterdam in 1998 to start as a Lecturer in the group of Prof. P. W. N. M. van Leeuwen. He was promoted to Full Professor in 2006 (Chair Homogeneous and Supramolecular Catalysis) and to Distinguished Faculty Professor in 2017. In 2005 he was elected a young member of the Royal Netherlands Academy of Arts and Sciences (KNAW), and in 2015 he was elected a member of the KNAW. In 2013 he was elected as a new member of the Royal Holland Society of Sciences and Humanities (KHMW), in 2018 he was an honoree member of the Israel Chemical Society, and in 2019 he became an elected member of the European Academy of Science. He received numerous personal grants including a VICI grant and TOP grants of the National Research Funding Agency NWO and an ERC advanced grant. His research interests include homogeneous catalysis and supramolecular chemistry, and he is exploring new concepts in supramolecular transition metal catalysis. In addition, he has a research program on catalysis for green energy applications, aiming at solar to fuel devices based on molecular components, also including supramolecular approaches.
Bas de Bruin obtained his Ph.D. at the University of Nijmegen (group Gal, Metal–Organic Chemistry) in the area of metal-mediated olefin oxygenation reactions. He did his postdoc in the group of Wieghardt at the Max-Planck Institut für Bioanorganische Chemie (Mülheim a/d Ruhr; Alexander-von-Humboldt fellowship). After his postdoc he returned to the University of Nijmegen as an Assistant Professor in Inorganic Chemistry (Metal–Organic Chemistry), where he was involved in several research activities ranging from olefin oxygenation to radical organometallic chemistry to EPR spectroscopy to catalysis to modelling to DFT calculations to synthesis of new materials based on highly functionalized polymers. In November 2005 he moved to the University of Amsterdam, where he was promoted to Associate Professor (UHD) in 2008 and to Full Professor in 2013. He received several prestigious personal grants, including an NWO-VIDI grant in 2005, an ERC Grant in 2008, an NWO-VICI grant in 2012, and an NWO-TOP grant in 2015. He is an elected member of the ARC-CBBC (January 2016), and he was elected as UvA Teacher of the Year 2015, in a university-broad competition organized by the students of the University of Amsterdam. In 2020 he was elected Chemistry Europe Fellow. Bas de Bruin presently focuses on the fundamental development of homogeneous catalysis with metals in unconventional oxidation states and with unconventional ligands, specifically aiming at the development of new catalytic reactions.
Sonja Pullen is an Assistant Professor in homogeneous, supramolecular, and bioinspired catalysis at the Van‘t Hoff Institute for Molecular Sciences at the University of Amsterdam (The Netherlands). She holds a Bachelor Degree from WWU Münster (Germany) and a Master Degree in chemistry from Uppsala University (Sweden). From 2012–2017 she performed her doctoral research at Uppsala University with Sascha Ott, where she developed strategies to mimic the outer coordination sphere in [FeFe]-hydrogenase active site models. From 2018 she worked as a Postdoctoral Researcher in the group of Guido Clever at TU Dortmund, funded by a Marie Sklodowska-Curie fellowship of the European Union’s Horizon 2020 research and innovation programme. There she investigated the potential of coordination cages for light-driven processes. Her current research focuses on supramolecular systems such as metal–organic frameworks (MOFs) and discrete coordination cages with applications in artificial photosynthesis and organic photoredox catalysis.
Tiddo J. Mooibroek studied chemistry at the University of Leiden and philosophy at the University of Leiden and the Free University of Amsterdam. He obtained his Ph.D. in 2011 from the University of Leiden under the supervision of Elisabeth Bouwman and Eite Drent (Shell), working on the Pd-diphosphane catalyzed reductive carbonylation of nitroaromatics. Tiddo then went to the University of Bristol for a 3-year postdoctoral research project under the auspices of Anthony Davis in the area of (pyrene-bases) artificial carbohydrate receptors. He then worked for NovoNordisk as a STAR research fellow on their glucose-responsive insulin project with Thomas Hoeg-Jensen in collaboration with Anthony Davis at the University of Bristol. In 2016, Tiddo obtained a NWO-VIDI grant that he spent at the University of Amsterdam, where he developed novel caging molecules for the supramolecular binding of carbohydrates. Throughout his career, Tiddo has also been interested in the directional character of weak/unconventional noncovalent interactions and has collaborated in this area with Jan Reedijk, Patrick Gamez, Antonio Frontera, and many others. From October 2021 to March 2022 he has been working for the Dutch Scientific Research Council (NWO) before being appointed as a Patent Examiner at the European Patent Office in the Hague.
Alexander (Sander) M. Kluwer studied chemistry at the University of Leiden under the supervision of Prof. E. Bouwman and Prof. E. Drent (Shell) in the area of nickel hydrogenase functional models. He obtained his Ph.D. from the University of Amsterdam under the supervision of Prof. C. J. Elsevier, on the topic of palladium catalyzed semihydrogenation of alkynes in common solvents and supercritical CO2, which was awarded the Backer prize in 2005. He did his postdoc in the group of Prof. K. Woelk at the University van Missouri, Rolla (USA), studying diffusion ordered spectroscopy (DOSY) edge enhancement, followed by a postdoc at the University of Amsterdam in the area of light-driven catalysis. In 2008, he joined the UvA spin-off company Catfix as a Scientific Researcher, developing technology for the capture, recycle, and reuse of homogeneous catalysts using supramolecular strategies. In 2009, he joined InCatT as Commercial Research Manager, where he was working on high throughput screening and development of new catalyst systems for the pharmaceutical, flavor and fragrance, and agrochemical industries. Since November 2013, he has been the CEO of InCatT B.V., and while working on further growing the company, he is also advising the scientists of InCatT and guiding Ph.D. students in joint projects. His research interests include homogeneous and heterogeneous catalysis, supramolecular chemistry, and bringing new catalytic technologies into commercial practice.
Xavier Caumes obtained his Ph.D. from the Université Pierre et Marie Curie (nowadays Sorbonne Univiversité) in 2016 under the supervision of Dr. L. Bouteiller and Dr. M. Raynal, working on hydrogen-bonded supramolecular polymers as dynamic scaffolds for catalysis. He then joined the group of Prof. J. N. H. Reek in 2017 to pursue research on chirality amplification in supramolecular cages and their application in transition metal catalysis. After this, he joined InCatT as a Senior Researcher in catalyst screening and development for the pharmaceutical, flavor and fragrance, and agrochemical industries.
The authors declare the following competing financial interest(s): A.M.K. and X.C. work for InCatT, which is an independent catalyst screening and optimization company.
References
- Adam D.Chemistry Nobel 2001. Nature 2001, 10.1038/news011011-17. [DOI] [Google Scholar]
- Ball P.Nobel Winners made Carbon Dance. Nature 2005, 10.1038/news051003-7. [DOI] [Google Scholar]
- Croft L.Prestige for Palladium. Nature 2010, 10.1038/nchem.897. [DOI] [PubMed] [Google Scholar]
- Montreux A.; Petit F.. Industrial Applications of Homogeneous Catalysis; Springer, 1988. [Google Scholar]
- Bahaduri S.; Mekesh D.. Homogeneous Catalysis: Mechanisms and Industrial Application, 2 ed.; Wiley, 2014. [Google Scholar]
- Cornils B.; Herrmann W. A.. Applied Homogeneous Catalysis with Organometallic Compounds, 2 ed.; Wiley, 2002. [Google Scholar]
- van Leeuwen P. W. N. M.; Kamer P. C. J.. Phosphorus (III) Ligands in Homogeneous Catalysis Design and Synthesis; John Wiley & Sons, Ltd., 2012; pp 405–426. [Google Scholar]
- Tolman C. A. Steric Effects of Phosphorus Ligands in Organometallic Chemistry and Homogeneous Catalysis. Chem. Rev. 1977, 77, 313–348. 10.1021/cr60307a002. [DOI] [Google Scholar]
- van Leeuwen P. W. N. M.; Kamer P. C. J.; Reek J. N. H.; Dierkes P. Ligand Bite Angle Effects in Metal-Catalyzed C-C Bond Formation. Chem. Rev. 2000, 100, 2741–2770. 10.1021/cr9902704. [DOI] [PubMed] [Google Scholar]
- Durand D. J.; Fey N. Computational Ligand Descriptors for Catalyst Design. Chem. Rev. 2019, 119, 6561–6594. 10.1021/acs.chemrev.8b00588. [DOI] [PubMed] [Google Scholar]
- Milo A.; Neel A. J.; Toste F. D.; Sigman M. S. A Data-Intensive Approach to Mechanistic Elucidation Applied to Chiral Anion Catalysis. Science 2015, 347, 737–743. 10.1126/science.1261043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigman M. S.; Harper K. C.; Bess E. N.; Milo A. The Development of Multidimensional Analysis Tools for Asymmetric Catalysis and Beyond. Acc. Chem. Res. 2016, 49, 1292–1301. 10.1021/acs.accounts.6b00194. [DOI] [PubMed] [Google Scholar]
- Kulik H. J.; Sigman M. S. Advancing Discovery in Chemistry with Artificial Intelligence: From Reaction Outcomes to New Materials and Catalysts. Acc. Chem. Res. 2021, 54, 2335–2336. 10.1021/acs.accounts.1c00232. [DOI] [PubMed] [Google Scholar]
- Donoghue P. J.; Helquist P.; Norrby P.-O.; Wiest O. Prediction of Enantioselectivity in Rhodium Catalyzed Hydrogenations. J. Am. Chem. Soc. 2009, 131, 410–411. 10.1021/ja806246h. [DOI] [PubMed] [Google Scholar]
- Jäkel C.; Paciello R. High-Throughput and Parallel Screening Methods in Asymmetric Hydrogenation. Chem. Rev. 2006, 106, 2912–2942. 10.1021/cr040675a. [DOI] [PubMed] [Google Scholar]
- Renom-Carrasco M.; Lefort L. Ligand Libraries for High Throughput Screening of Homogeneous Catalysts. Chem. Soc. Rev. 2018, 47, 5038–5060. 10.1039/C7CS00844A. [DOI] [PubMed] [Google Scholar]
- Reetz M. T. Combinatorial Transition-Metal Catalysis: Mixing Monodentate Ligands to Control Enantio-, Diastereo-, and Regioselectivity. Angew. Chem.-Int. Ed. 2008, 47, 2556–2588. 10.1002/anie.200704327. [DOI] [PubMed] [Google Scholar]
- Ringe D.; Petsko G. A. How Enzymes Work. Science 2008, 320, 1428–1429. 10.1126/science.1159747. [DOI] [PubMed] [Google Scholar]
- Schilter D.; Camara J. M.; Huynh M. T.; Hammes-Schiffer S.; Rauchfuss T. B. Hydrogenase Enzymes and Their Synthetic Models: The Role of Metal Hydrides. Chem. Rev. 2016, 116, 8693–8749. 10.1021/acs.chemrev.6b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M. L.; Stewart M. P.; Bullock R. M.; DuBois M. R.; DuBois D. L. A Synthetic Nickel Electrocatalyst with a Turnover Frequency Above 100,000 s–1 for H2 Production. Science 2011, 333, 863–866. 10.1126/science.1205864. [DOI] [PubMed] [Google Scholar]
- Vriezema D. M.; Aragones M. C.; Elemans J.; Cornelissen J.; Rowan A. E.; Nolte R. J. M. Self-Assembled Nanoreactors. Chem. Rev. 2005, 105, 1445–1489. 10.1021/cr0300688. [DOI] [PubMed] [Google Scholar]
- Hooley R. J.; Rebek J. Chemistry and Catalysis in Functional Cavitands. Chem. Biol. 2009, 16, 255–264. 10.1016/j.chembiol.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa M.; Klosterman J. K.; Fujita M. Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts. Angew. Chem.-Int. Ed. 2009, 48, 3418–3438. 10.1002/anie.200805340. [DOI] [PubMed] [Google Scholar]
- Raynal M.; Ballester P.; Vidal-Ferran A.; van Leeuwen P. W. N. M. Supramolecular Catalysis. Part 2: Artificial Enzyme Mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. 10.1039/C3CS60037H. [DOI] [PubMed] [Google Scholar]
- Brown C. J.; Toste F. D.; Bergman R. G.; Raymond K. N. Supramolecular Catalysis in Metal-Ligand Cluster Hosts. Chem. Rev. 2015, 115, 3012–3035. 10.1021/cr4001226. [DOI] [PubMed] [Google Scholar]
- Leenders S.; Gramage-Doria R.; de Bruin B.; Reek J. N. H. Transition Metal Catalysis in Confined Spaces. Chem. Soc. Rev. 2015, 44, 433–448. 10.1039/C4CS00192C. [DOI] [PubMed] [Google Scholar]
- Meeuwissen J.; Reek J. N. H. Supramolecular Catalysis Beyond Enzyme Mimics. Nat. Chem. 2010, 2, 615–621. 10.1038/nchem.744. [DOI] [PubMed] [Google Scholar]
- Raynal M.; Ballester P.; Vidal-Ferran A.; van Leeuwen P. W. N. M. Supramolecular Catalysis. Part 1: Non-Covalent Interactions as a Tool for Building and Modifying Homogeneous Catalysts. Chem. Soc. Rev. 2014, 43, 1660–1733. 10.1039/C3CS60027K. [DOI] [PubMed] [Google Scholar]
- Noyori R.; Yamakawa M.; Hashiguchi S. Metal-Ligand Bifunctional Catalysis: A Nonclassical Mechanism for Asymmetric Hydrogen Transfer between Alcohols and Carbonyl Compounds. J. Org. Chem. 2001, 66, 7931–7944. 10.1021/jo010721w. [DOI] [PubMed] [Google Scholar]
- Hale L. V. A.; Szymczak N. K. Hydrogen Transfer Catalysis beyond the Primary Coordination Sphere. ACS Catal. 2018, 8, 6446–6461. 10.1021/acscatal.7b04216. [DOI] [Google Scholar]
- Dub P. A.; Gordon J. C. Metal Ligand Bifunctional Catalysis: The “Accepted” Mechanism, the Issue of Concertedness, and the Function of the Ligand in Catalytic Cycles Involving Hydrogen Atoms. ACS Catal. 2017, 7, 6635–6655. 10.1021/acscatal.7b01791. [DOI] [Google Scholar]
- Belkova N. V.; Epstein L. M.; Filippov O. A.; Shubina E. S. Hydrogen and Dihydrogen Bonds in the Reactions of Metal Hydrides. Chem. Rev. 2016, 116, 8545–8587. 10.1021/acs.chemrev.6b00091. [DOI] [PubMed] [Google Scholar]
- Abdur-Rashid K.; Clapham S. E.; Hadzovic A.; Harvey J. N.; Lough A. J.; Morris R. H. Mechanism of the Hydrogenation of Ketones Catalyzed by trans-Dihydrido(diamine)ruthenium(II) Complexes. J. Am. Chem. Soc. 2002, 124, 15104–15118. 10.1021/ja016817p. [DOI] [PubMed] [Google Scholar]
- Khusnutdinova J. R.; Milstein D. Metal-Ligand Cooperation. Angew. Chem.-Int. Ed. 2015, 54, 12236–12273. 10.1002/anie.201503873. [DOI] [PubMed] [Google Scholar]
- Kuwata S.; Ikariya T. Metal-Ligand Bifunctional Reactivity and Catalysis of Protic N-Heterocyclic Carbene and Pyrazole Complexes Featuring β-NH Units. Chem. Commun. 2014, 50, 14290–14300. 10.1039/C4CC04457F. [DOI] [PubMed] [Google Scholar]
- Blum Y.; Czarkie D.; Rahamim Y.; Shvo Y. (Cyclopentadienone)ruthenium Carbonyl Complexes - a new Class of Homogeneous Hydrogenation Catalysts. Organometallics 1985, 4, 1459–1461. 10.1021/om00127a027. [DOI] [Google Scholar]
- Warner M. C.; Casey C. P.; Backvall J. E.. Shvo’s Catalyst in Hydrogen Transfer Reactions. In Bifunctional Molecular Catalysis; Ikariya T., Shibasaki M., Eds.; Springer-Verlag Berlin: Berlin, 2011; Vol. 37, pp 85–125. [Google Scholar]
- Noyori R.; Sandoval C. A.; Muniz K.; Ohkuma T. Metal-Ligand Bifunctional Catalysis for Asymmetric Hydrogenation. Philos. Trans. R. Soc. A 2005, 363, 901–912. 10.1098/rsta.2004.1536. [DOI] [PubMed] [Google Scholar]
- Hartmann R.; Chen P. Noyori’s Hydrogenation Catalyst Needs a Lewis Acid Cocatalyst for High Activity. Angew. Chem. -Int. Ed. 2001, 40, 3581–3585. . [DOI] [PubMed] [Google Scholar]
- Shvo Y.; Czarkie D.; Rahamim Y.; Chodosh D. F. A new Group of Ruthenium Complexes: Structure and Catalysis. J. Am. Chem. Soc. 1986, 108, 7400–7402. 10.1021/ja00283a041. [DOI] [Google Scholar]
- Conley B. L.; Pennington-Boggio M. K.; Boz E.; Williams T. J. Discovery, Applications, and Catalytic Mechanisms of Shvo’s Catalyst. Chem. Rev. 2010, 110, 2294–2312. 10.1021/cr9003133. [DOI] [PubMed] [Google Scholar]
- Borovik A. S. Bioinspired Hydrogen Bond Motifs in Ligand Design: The Role of Noncovalent Interactions in Metal Ion Mediated Activation of Dioxygen. Acc. Chem. Res. 2005, 38, 54–61. 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- Latimer W. M.; Rodebush W. H. Polarity and Ionization From the Standpoint of the Lewis Theory of Valance. J. Am. Chem. Soc. 1920, 42, 1419–1433. 10.1021/ja01452a015. [DOI] [Google Scholar]
- Goymer P. 100 Years of the Hydrogen Bond. Nat. Chem. 2012, 4, 863–864. 10.1038/nchem.1482. [DOI] [PubMed] [Google Scholar]
- Smith D. A.A Brief History of the Hydrogen Bond. In Modeling the Hydrogen Bond; Smith D. A., Ed.; American Chemical Society: Chicago, IL, 1994; Vol. 569. [Google Scholar]
- Arunan E.; Desiraju G. R.; Klein R. A.; Sadlej J.; Scheiner S.; Alkorta I.; Clary D. C.; Crabtree R. H.; Dannenberg J. J.; Hobza P.; Kjaergaard H. G.; Legon A. C.; Mennucci B.; Nesbitt D. J. Defining the Hydrogen Bond: An Account (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 1619–1636. 10.1351/PAC-REP-10-01-01. [DOI] [Google Scholar]
- Hüttermann A.The Hydrogen Bond: A Bond for Life; De Gruyter: Boston, 2019. [Google Scholar]
- Steiner T. The Hydrogen Bond in the Solid State. Angew. Chem.-Int. Ed. 2002, 41, 48–76. . [DOI] [PubMed] [Google Scholar]
- Buckingham A. D.; Del Bene J. E.; McDowell S. A. C. The Hydrogen Bond. Chem. Phys. Lett. 2008, 463, 1–10. 10.1016/j.cplett.2008.06.060. [DOI] [Google Scholar]
- Arunan E.; Desiraju G. R.; Klein R. A.; Sadlej J.; Scheiner S.; Alkorta I.; Clary D. C.; Crabtree R. H.; Dannenberg J. J.; Hobza P.; Kjaergaard H. G.; Legon A. C.; Mennucci B.; Nesbitt D. J. Definition of the Hydrogen Bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. 10.1351/PAC-REC-10-01-02. [DOI] [Google Scholar]
- Gale P. A. Anion Receptor Chemistry. Chem. Commun. 2011, 47, 82–86. 10.1039/C0CC00656D. [DOI] [PubMed] [Google Scholar]
- Kavallieratos K.; Bertao C. M.; Crabtree R. H. Hydrogen Bonding in Anion Recognition: A Family of Versatile, Nonpreorganized Neutral and Acyclic Receptors. J. Org. Chem. 1999, 64, 1675–1683. 10.1021/jo982382l. [DOI] [PubMed] [Google Scholar]
- Janotti A.; Van de Walle C. G. Hydrogen Multicentre Bonds. Nat. Mater. 2007, 6, 44–47. 10.1038/nmat1795. [DOI] [PubMed] [Google Scholar]
- Alkorta I.; Rozas I.; Elguero J. Non-conventional Hydrogen Bonds. Chem. Soc. Rev. 1998, 27, 163–170. 10.1039/a827163z. [DOI] [Google Scholar]
- Nishio M. The CH/π Hydrogen Bond in Chemistry. Conformation, Supramolecules, Optical Resolution and Interactions Involving Carbohydrates. Phys. Chem. Chem. Phys. 2011, 13, 13873–13900. 10.1039/c1cp20404a. [DOI] [PubMed] [Google Scholar]
- Nishio M.; Umezawa Y.; Fantini J.; Weiss M. S.; Chakrabarti P. CH-π Hydrogen Bonds in Biological Macromolecules. Phys. Chem. Chem. Phys. 2014, 16, 12648–12683. 10.1039/C4CP00099D. [DOI] [PubMed] [Google Scholar]
- Hudson K. L.; Bartlett G. J.; Diehl R. C.; Agirre J.; Gallagher T.; Kiessling L. L.; Woolfson D. N. Carbohydrate-Aromatic Interactions in Proteins. J. Am. Chem. Soc. 2015, 137, 15152–15160. 10.1021/jacs.5b08424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio M. CH/π Hydrogen Bonds in Crystals. CrystEngComm 2004, 6, 130–158. 10.1039/b313104a. [DOI] [Google Scholar]
- Mooibroek T. J.; Gamez P. How Directional are D-H···Phenyl Interactions in the Solid State (D = C, N, O)?. CrystEngComm 2012, 14, 8462–8467. 10.1039/c2ce26205c. [DOI] [Google Scholar]
- Suzuki S.; Green P. G.; Bumgarner R. E.; Dasgupta S.; Goddard W. A.; Blake G. A. Benzene Forms Hydrogen Bonds with Water. Science 1992, 257, 942–944. 10.1126/science.257.5072.942. [DOI] [PubMed] [Google Scholar]
- Brookhart M.; Green M. L. H.; Wong L. L. Carbon-Hydrogen Transition-Metal Bonds. Prog. Inorg. Chem. 1988, 36, 1–124. 10.1002/9780470166376.ch1. [DOI] [Google Scholar]
- Brookhart M.; Green M. L. H. Carbon-Hydrogen-Transition Metal Bonds. J. Organomet. Chem. 1983, 250, 395–408. 10.1016/0022-328X(83)85065-7. [DOI] [Google Scholar]
- Belkova N. V.; Shubina E. S.; Epstein L. M. Diverse World of Unconventional Hydrogen Bonds. Acc. Chem. Res. 2005, 38, 624–631. 10.1021/ar040006j. [DOI] [PubMed] [Google Scholar]
- Brookhart M.; Green M. L. H.; Parkin G. Agostic Interactions in Transition Metal Compounds. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6908–6914. 10.1073/pnas.0610747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree R. H.; Siegbahn P. E. M.; Eisenstein O.; Rheingold A. L. A New Intermolecular Interaction: Unconventional Hydrogen Bonds with Element-Hydride Bonds as Proton Acceptor. Acc. Chem. Res. 1996, 29, 348–354. 10.1021/ar950150s. [DOI] [PubMed] [Google Scholar]
- Emsley J. Very Strong Hydrogen Bonding. Chem. Soc. Rev. 1980, 9, 91–124. 10.1039/cs9800900091. [DOI] [Google Scholar]
- Busschaert N.; Caltagirone C.; Van Rossom W.; Gale P. A. Applications of Supramolecular Anion Recognition. Chem. Rev. 2015, 115, 8038–8155. 10.1021/acs.chemrev.5b00099. [DOI] [PubMed] [Google Scholar]
- Mooibroek T. J. DFT and IsoStar Analyses to Assess the Utility of σ- and π-Hole Interactions for Crystal Engineering. ChemPhysChem 2021, 22, 141–153. 10.1002/cphc.202000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su P.; Li H. Energy Decomposition Analysis of Covalent Bonds and Intermolecular Interactions. J. Chem. Phys. 2009, 131, 014102. 10.1063/1.3159673. [DOI] [PubMed] [Google Scholar]
- van der Lubbe S. C. C.; Guerra C. F. The Nature of Hydrogen Bonds: A Delineation of the Role of Different Energy Components on Hydrogen Bond Strengths and Lengths. Chem.-Asian J. 2019, 14, 2760–2769. 10.1002/asia.201900717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel A. J.; Hilton M. J.; Sigman M. S.; Toste F. D. Exploiting Non-Covalent π Interactions for Catalyst Design. Nature 2017, 543, 637–646. 10.1038/nature21701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andric J. M.; Janjic G. V.; Ninkovic D. B.; Zaric S. D. The Influence of Water Molecule Coordination to a Metal Ion on Water Hydrogen Bonds. Phys. Chem. Chem. Phys. 2012, 14, 10896–10898. 10.1039/c2cp41125c. [DOI] [PubMed] [Google Scholar]
- Reinhardt P.; Piquemal J. P. New Intermolecular Benchmark Calculations on the Water Dimer: SAPT and Supermolecular Post-Hartree-Fock Approaches. Int. J. Quantum Chem. 2009, 109, 3259–3267. 10.1002/qua.22299. [DOI] [Google Scholar]
- Shields Z. P.; Murray J. S.; Politzer P. Directional Tendencies of Halogen and Hydrogen Bonds. Int. J. Quantum Chem. 2010, 110, 2823–2832. 10.1002/qua.22787. [DOI] [Google Scholar]
- Politzer P.; Murray J. S. Electrostatics and Polarization in σ- and π-Hole Noncovalent Interactions: An Overview. ChemPhysChem 2020, 21, 579–588. 10.1002/cphc.201900968. [DOI] [PubMed] [Google Scholar]
- Spackman M. A.; McKinnon J. J.; Jayatilaka D. Electrostatic Potentials Mapped on Hirshfeld Surfaces Provide Direct Insight into Intermolecular Interactions in Crystals. CrystEngComm 2008, 10, 377–388. 10.1039/b715227b. [DOI] [Google Scholar]
- Brinck T.; Murray J. S.; Politzer P. Surface Electrostatic Potentials of Halogenated Methanes as Indicators of Directional Intermolecular Interactions. Int. J. Quantum Chem. 1992, 44, 57–64. 10.1002/qua.560440709. [DOI] [Google Scholar]
- Politzer P.; Murray J. S.; Peralta-Inga Z. Molecular Surface Electrostatic Potentials in Relation to Noncovalent Interactions in Biological Systems. Int. J. Quantum Chem. 2001, 85, 676–684. 10.1002/qua.1706. [DOI] [Google Scholar]
- Hunter C. A. Quantifying Intermolecular Interactions: Guidelines for the Molecular Recognition Toolbox. Angew. Chem.-Int. Ed. 2004, 43, 5310–5324. 10.1002/anie.200301739. [DOI] [PubMed] [Google Scholar]
- Scheiner S. Understanding Noncovalent Bonds and Their Controlling Forces. J. Chem. Phys. 2020, 153, 140901. 10.1063/5.0026168. [DOI] [PubMed] [Google Scholar]
- Grabowski S. J. Hydrogen and Halogen Bonds are Ruled by the Same Mechanisms. Phys. Chem. Chem. Phys. 2013, 15, 7249–7259. 10.1039/c3cp50537e. [DOI] [PubMed] [Google Scholar]
- Desiraju G. R.; Ho P. S.; Kloo L.; Legon A. C.; Marquardt R.; Metrangolo P.; Politzer P.; Resnati G.; Rissanen K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. 10.1351/PAC-REC-12-05-10. [DOI] [Google Scholar]
- Aakeroy C. B.; Bryce D. L.; Desiraju G.; Frontera A.; Legon A. C.; Nicotra F.; Rissanen K.; Scheiner S.; Terraneo G.; Metrangolo P.; Resnati G. Definition of the Chalcogen Bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. 10.1515/pac-2018-0713. [DOI] [Google Scholar]
- Scheiner S. The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 2013, 46, 280–288. 10.1021/ar3001316. [DOI] [PubMed] [Google Scholar]
- Bauza A.; Mooibroek T. J.; Frontera A. Tetrel Bonding Interactions. Chem. Rec. 2016, 16, 473–487. 10.1002/tcr.201500256. [DOI] [PubMed] [Google Scholar]
- Crabtree R. H. Hypervalency, Secondary Bonding and Hydrogen Bonding: Siblings Under the Skin. Chem. Soc. Rev. 2017, 46, 1720–1729. 10.1039/C6CS00688D. [DOI] [PubMed] [Google Scholar]
- Bauza A.; Mooibroek T. J.; Frontera A. The Bright Future of Unconventional σ/π-Hole Interactions. ChemPhysChem 2015, 16, 2496–2517. 10.1002/cphc.201500314. [DOI] [PubMed] [Google Scholar]
- Gilli P.; Pretto L.; Bertolasi V.; Gilli G. Predicting Hydrogen-Bond Strengths from Acid-Base Molecular Properties. The pKa Slide Rule: Toward the Solution of a Long-Lasting Problem. Acc. Chem. Res. 2009, 42, 33–44. 10.1021/ar800001k. [DOI] [PubMed] [Google Scholar]
- Taylor R.; Kennard O. Crystallographic Evidence for the Existence of C-H···O, C-H···N, and C-H···Cl Hydrogen-Bonds. J. Am. Chem. Soc. 1982, 104, 5063–5070. 10.1021/ja00383a012. [DOI] [Google Scholar]
- Hay B. P.; Dixon D. A.; Bryan J. C.; Moyer B. A. Crystallographic Evidence for Oxygen Acceptor Directionality in Oxyanion Hydrogen Bonds. J. Am. Chem. Soc. 2002, 124, 182–183. 10.1021/ja0173775. [DOI] [PubMed] [Google Scholar]
- Platts J. A.; Howard S. T.; Bracke B. R. F. Directionality of Hydrogen Bonds to Sulfur and Oxygen. J. Am. Chem. Soc. 1996, 118, 2726–2733. 10.1021/ja952871s. [DOI] [Google Scholar]
- Mooibroek T. J.; Gamez P. Halogen···Phenyl Supramolecular Interactions in the Solid State: Hydrogen versus Halogen Bonding and Directionality. CrystEngComm 2013, 15, 1802–1805. 10.1039/c2ce26853a. [DOI] [Google Scholar]
- Mooibroek T. J.; Gamez P. Halogen Bonding versus Hydrogen Bonding: What Does the Cambridge Database Reveal?. CrystEngComm 2013, 15, 4565–4570. 10.1039/c3ce40285a. [DOI] [Google Scholar]
- Wood P. A.; Allen F. H.; Pidcock E. Hydrogen-Bond Directionality at the Donor H Atom - Analysis of Interaction Energies and Database Statistics. CrystEngComm 2009, 11, 1563–1571. 10.1039/b902330e. [DOI] [Google Scholar]
- Majerz I. Directionality of Inter- and Intramolecular OHO Hydrogen Bonds: DFT Study Followed by AIM and NBO Analysis. J. Phys. Chem. A 2012, 116, 7992–8000. 10.1021/jp300942n. [DOI] [PubMed] [Google Scholar]
- Steiner T.; Saenger W. Lenghtening of the Covalent O-H Bond in O-H···O Hydrogen Bonds Re-examined From Low-Temperature Neutron-Diffraction Data of Organic Compounds. Acta Crystallogr. Sect. B-Struct. Sci. Cryst. Eng. Mater. 1994, 50, 348–357. 10.1107/S0108768193011966. [DOI] [Google Scholar]
- Steiner T. Lengthening of the N-H Bond in N-H···N Hydrogen Bonds - Preliminary Structural Data and Implications of the Bond Valence Concept. J. Chem. Soc.-Chem. Commun. 1995, 1331–1332. 10.1039/C39950001331. [DOI] [Google Scholar]
- Steiner T. Weak Hydrogen-Bonding. 1. Neutron-Diffracion Data of Amino-Acid Cα-H Suggesting Lengthening of the Covalent C-H Bond in the C-H···O Interaction. J. Chem. Soc.-Perkin Trans. 2 1995, 1315–1319. 10.1039/P29950001315. [DOI] [Google Scholar]
- Steiner T. Lengthening of the Covalent X-H Bond in Heteronuclear Hydrogen Bonds Quantified From Organic and Organometallic Neutron Crystal Structures. J. Phys. Chem. A 1998, 102, 7041–7052. 10.1021/jp981604g. [DOI] [Google Scholar]
- Steiner T.; Majerz I.; Wilson C. C. First O-H···N Hydrogen Bond With a Centered Proton Obtained by Thermally Induced Proton Migration. Angew. Chem.-Int. Ed. 2001, 40, 2651–2654. . [DOI] [PubMed] [Google Scholar]
- Nakamoto K.; Margoshes M.; Rundle R. E. Stretching Frequencies as a Function of Distances in Hydrogen Bonds. J. Am. Chem. Soc. 1955, 77, 6480–6486. 10.1021/ja01629a013. [DOI] [Google Scholar]
- Blanksby S. J.; Ellison G. B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255–263. 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- Koch U.; Popelier P. L. A. Characterization of C-H-O Hydrogen-Bonds on the Basis of Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. 10.1021/j100024a016. [DOI] [Google Scholar]
- Espinosa E.; Molins E.; Lecomte C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. 10.1016/S0009-2614(98)00036-0. [DOI] [Google Scholar]
- Pendas A. M.; Blanco M. A.; Francisco E. The Nature of the Hydrogen Bond: A Synthesis From the Interacting Quantum Atoms Picture. J. Chem. Phys. 2006, 125, 184112. 10.1063/1.2378807. [DOI] [PubMed] [Google Scholar]
- Vener M. V.; Egorova A. N.; Churakov A. V.; Tsirelson V. G. Intermolecular Hydrogen Bond Energies in Crystals Evaluated Using Electron Density Properties: DFT Computations With Periodic Boundary Conditions. J. Comput. Chem. 2012, 33, 2303–2309. 10.1002/jcc.23062. [DOI] [PubMed] [Google Scholar]
- Mata I.; Alkorta I.; Espinosa E.; Molins E. Relationships Between Interaction Energy, Intermolecular Distance and Electron Density Properties in Hydrogen Bonded Complexes Under External Electric Fields. Chem. Phys. Lett. 2011, 507, 185–189. 10.1016/j.cplett.2011.03.055. [DOI] [Google Scholar]
- Bader R. F. W. Atoms in Molecules. Acc. Chem. Res. 1985, 18, 9–15. 10.1021/ar00109a003. [DOI] [Google Scholar]
- Lane J. R.; Contreras-Garcia J.; Piquemal J. P.; Miller B. J.; Kjaergaard H. G. Are Bond Critical Points Really Critical for Hydrogen Bonding?. J. Chem. Theory Comput. 2013, 9, 3263–3266. 10.1021/ct400420r. [DOI] [PubMed] [Google Scholar]
- Fuster F.; Grabowski S. J. Intramolecular Hydrogen Bonds: the QTAIM and ELF Characteristics. J. Phys. Chem. A 2011, 115, 10078–10086. 10.1021/jp2056859. [DOI] [PubMed] [Google Scholar]
- Parthasarathi R.; Subramanian V.; Sathyamurthy N. Hydrogen Bonding Without Borders: An Atoms-In-Molecules Perspective. J. Phys. Chem. A 2006, 110, 3349–3351. 10.1021/jp060571z. [DOI] [PubMed] [Google Scholar]
- Grabowski S. J. What Is the Covalency of Hydrogen Bonding?. Chem. Rev. 2011, 111, 2597–2625. 10.1021/cr800346f. [DOI] [PubMed] [Google Scholar]
- Battle G. M.; Allen F. H. Learning About Intermolecular Interactions from the Cambridge Structural Database. J. Chem. Educ. 2012, 89, 38–44. 10.1021/ed200139t. [DOI] [Google Scholar]
- Sobolev V.; Sorokine A.; Prilusky J.; Abola E. E.; Edelman M. Automated Analysis of Interatomic Contacts in Proteins. Bioinformatics 1999, 15, 327–332. 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- Bernstein J.; Davis R. E.; Shimoni L.; Chang N. L. Patterns in Hydrogen Bonding - Functionality and Graphset Analysis in Crystals. Angew. Chem.-Int. Ed. 1995, 34, 1555–1573. 10.1002/anie.199515551. [DOI] [Google Scholar]
- Bruno I. J.; Cole J. C.; Lommerse J. P. M.; Rowland R. S.; Taylor R.; Verdonk M. L. IsoStar: A library of Information About Nonbonded Interactions. J. Comput.-Aided Mol. Des. 1997, 11, 525–537. 10.1023/A:1007934413448. [DOI] [PubMed] [Google Scholar]
- Allen F. H.; Cruz-Cabeza A. J.; Wood P. A.; Bardwell D. A. Hydrogen-Bond Landscapes, Geometry and Energetics of Squaric Acid and its Mono- and Dianions: a Cambridge Structural Database, IsoStar and Computational Study. Acta Crystallogr. Sect. B-Struct. Sci. Cryst. Eng. Mater. 2013, 69, 514–523. 10.1107/S2052519213020277. [DOI] [PubMed] [Google Scholar]
- Iogansen A. V. Direct Proportionality of the Hydrogen Bonding Energy and the Intensification of the Stretching ν(XH) Vibration in Infrared Spectra. Spectroc. Acta Pt. A-Mol. Biomol. Spectr. 1999, 55, 1585–1612. 10.1016/S1386-1425(98)00348-5. [DOI] [Google Scholar]
- Hobza P.; Havlas Z. Blue-Shifting Hydrogen Bonds. Chem. Rev. 2000, 100, 4253–4264. 10.1021/cr990050q. [DOI] [PubMed] [Google Scholar]
- Rozenberg M.; Loewenschuss A.; Marcus Y. An Empirical Correlation Between Stretching Vibration Redshift and Hydrogen Bond Length. Phys. Chem. Chem. Phys. 2000, 2, 2699–2702. 10.1039/b002216k. [DOI] [Google Scholar]
- Hobza P. The H-index Unambiguously Discriminates Between Hydrogen Bonding and Improper Blue-Shifting Hydrogen Bonding. Phys. Chem. Chem. Phys. 2001, 3, 2555–2556. 10.1039/b103068j. [DOI] [Google Scholar]
- Biedermann F.; Schneider H. J. Experimental Binding Energies in Supramolecular Complexes. Chem. Rev. 2016, 116, 5216–5300. 10.1021/acs.chemrev.5b00583. [DOI] [PubMed] [Google Scholar]
- Wagner G.; Pardi A.; Wuthrich K. Hydrogen-Bond Length and 1H-NMR Chemical-Shifts in Proteins. J. Am. Chem. Soc. 1983, 105, 5948–5949. 10.1021/ja00356a056. [DOI] [Google Scholar]
- Wishart D. S.; Sykes B. D.; Richards F. M. Relationship Between Nuclear Magnetic Resonance Chemical Shift and Protein Secundary Structure. J. Mol. Biol. 1991, 222, 311–333. 10.1016/0022-2836(91)90214-Q. [DOI] [PubMed] [Google Scholar]
- Smirnov S. N.; Golubev N. S.; Denisov G. S.; Benedict H.; SchahMohammedi P.; Limbach H. H. Hydrogen/Deuterium Isotope Effects on the NMR Chemical Shifts and Geometries of Intermolecular Low-Barrier Hydrogen-Bonded Complexes. J. Am. Chem. Soc. 1996, 118, 4094–4101. 10.1021/ja953445+. [DOI] [Google Scholar]
- Benedict H.; Shenderovich I. G.; Malkina O. L.; Malkin V. G.; Denisov G. S.; Golubev N. S.; Limbach H. H. Nuclear Scalar Spin-Spin Couplings and Geometries of Hydrogen Bonds. J. Am. Chem. Soc. 2000, 122, 1979–1988. 10.1021/ja9907461. [DOI] [Google Scholar]
- Fielding L. Determination of Association Constants (Ka) from Solution NMR Data. Tetrahedron 2000, 56, 6151–6170. 10.1016/S0040-4020(00)00492-0. [DOI] [Google Scholar]
- Cavalli A.; Salvatella X.; Dobson C. M.; Vendruscolo M. Protein Structure Determination From NMR Chemical Shifts. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 9615–9620. 10.1073/pnas.0610313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor A.; Martinez-Viviente E. NMR Spectroscopy in Coordination Supramolecular Chemistry: A Unique and Powerful Methodology. Coord. Chem. Rev. 2008, 252, 2314–2345. 10.1016/j.ccr.2008.01.025. [DOI] [Google Scholar]
- Rablen P. R.; Lockman J. W.; Jorgensen W. L. Ab Initio Study of Hydrogen-Bonded Complexes of Small Organic Molecules with Water. J. Phys. Chem. A 1998, 102, 3782–3797. 10.1021/jp980708o. [DOI] [Google Scholar]
- Ren P. Y.; Wu C. J.; Ponder J. W. Polarizable Atomic Multipole-Based Molecular Mechanics for Organic Molecules. J. Chem. Theory Comput. 2011, 7, 3143–3161. 10.1021/ct200304d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavezzotti A.; Filippini G. Geometry of the Intermolecular X-H···Y (X, Y = N, O) Hydrogen Bond and the Calibration of Empirical Hydrogen-Bond Potentials. J. Phys. Chem. 1994, 98, 4831–4837. 10.1021/j100069a010. [DOI] [Google Scholar]
- Infantes L.; Motherwell W. D. S. Hydrogen Bond Competition Between Chemical Groups: New Methodology and the Cambridge Structural Database. Z. Kristallogr. 2005, 220, 333–339. 10.1524/zkri.220.4.333.61617. [DOI] [Google Scholar]
- Tresca B. W.; Hansen R. J.; Chau C. V.; Hay B. P.; Zakharov L. N.; Haley M. M.; Johnson D. W. Substituent Effects in CH Hydrogen Bond Interactions: Linear Free Energy Relationships and Influence of Anions. J. Am. Chem. Soc. 2015, 137, 14959–14967. 10.1021/jacs.5b08767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- August D. P.; Nichol G. S.; Lusby P. J. Maximizing Coordination Capsule-Guest Polar Interactions in Apolar Solvents Reveals Significant Binding. Angew. Chem.-Int. Ed. 2016, 55, 15022–15026. 10.1002/anie.201608229. [DOI] [PubMed] [Google Scholar]
- Lewis J. E. M.; Crowley J. D. Exo- and Endo-Hedral Interactions of Counteranions with Tetracationic Pd2L4 Metallosupramolecular Architectures. Supramol. Chem. 2014, 26, 173–181. 10.1080/10610278.2013.842644. [DOI] [Google Scholar]
- Timmer B. J. J.; Mooibroek T. J. Anion Binding Properties of a Hollow PdL-Cage. Chem. Commun. 2021, 57, 7184. 10.1039/D1CC02663A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L. P.; Sun Q. F. A Self-Assembled Pd2L4 Cage That Selectively Encapsulates Nitrate. Chem. Commun. 2015, 51, 16767–16770. 10.1039/C5CC07306E. [DOI] [PubMed] [Google Scholar]
- Sekiya R.; Fukuda M.; Kuroda R. Anion-Directed Formation and Degradation of an Interlocked Metallohelicate. J. Am. Chem. Soc. 2012, 134, 10987–10997. 10.1021/ja303634u. [DOI] [PubMed] [Google Scholar]
- Li Y. J.; Flood A. H. Pure C-H Hydrogen Bonding to Chloride Ions: A Preorganized and Rigid Macrocyclic Receptor. Angew. Chem.-Int. Ed. 2008, 47, 2649–2652. 10.1002/anie.200704717. [DOI] [PubMed] [Google Scholar]
- Desiraju G. R. The C-H···N Hydrogen Bond: Structural Implications and Supramolecular Design. Acc. Chem. Res. 1996, 29, 441–449. 10.1021/ar950135n. [DOI] [PubMed] [Google Scholar]
- Horowitz S.; Trievel R. C. Carbon-Oxygen Hydrogen Bonding in Biological Structure and Function. J. Biol. Chem. 2012, 287, 41576–41582. 10.1074/jbc.R112.418574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas R.; Garza J.; Dixon D. A.; Hay B. P. How Strong is the CαH···O = C Hydrogen Bond?. J. Am. Chem. Soc. 2000, 122, 4750–4755. 10.1021/ja993600a. [DOI] [Google Scholar]
- Steed J. W.; Atwood J. L.. Supramolecular Chemistry, 2nd ed.; Wiley, 2009. [Google Scholar]
- Cook J. L.; Hunter C. A.; Low C. M. R.; Perez-Velasco A.; Vinter J. G. Solvent Effects on Hydrogen Bonding. Angew. Chem.-Int. Ed. 2007, 46, 3706–3709. 10.1002/anie.200604966. [DOI] [PubMed] [Google Scholar]
- Abraham M. H. Scales of Solute Hydrogen-Bonding: Their Construction and Application to Physicochemical and Biochemical Processes. Chem. Soc. Rev. 1993, 22, 73–83. 10.1039/cs9932200073. [DOI] [Google Scholar]
- Bouchy A.; Rinaldi D.; Rivail J. L. Solvent Effect on Intramolecular Hydrogen Bonds in Push-Pull Conjugated Molecules. Int. J. Quantum Chem. 2004, 96, 273–281. 10.1002/qua.10569. [DOI] [Google Scholar]
- Robertson C. C.; Wright J. S.; Carrington E. J.; Perutz R. N.; Hunter C. A.; Brammer L. Hydrogen Bonding vs. Halogen Bonding: The Solvent Decides. Chem. Sci. 2017, 8, 5392–5398. 10.1039/C7SC01801K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein E.; Ferrand Y.; Barwell N. P.; Davis A. P. Solvent Effects in Carbohydrate Binding by Synthetic Receptors: Implications For the Role of Water in Natural Carbohydrate Recognition. Angew. Chem.-Int. Ed. 2008, 47, 2693–2696. 10.1002/anie.200704733. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Schuurmann G. COSMO - a New Approach to Dielectric Screening in Solvents With Explicit Expressions for the Screening Energy and its Gradient. J. Chem. Soc.-Perkin Trans. 2 1993, 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Mathew K.; Sundararaman R.; Letchworth-Weaver K.; Arias T. A.; Hennig R. G. Implicit Solvation Model for Density-Functional Study of Nanocrystal Surfaces and Reaction Pathways. J. Chem. Phys. 2014, 140, 084106. 10.1063/1.4865107. [DOI] [PubMed] [Google Scholar]
- Cramer C. J.; Truhlar D. G. Implicit Solvation Models: Equilibria, Structure, Spectra, and Dynamics. Chem. Rev. 1999, 99, 2161–2200. 10.1021/cr960149m. [DOI] [PubMed] [Google Scholar]
- Schneider H. J. Binding Mechanisms in Supramolecular Complexes. Angew. Chem.-Int. Ed. 2009, 48, 3924–3977. 10.1002/anie.200802947. [DOI] [PubMed] [Google Scholar]
- Camara-Campos A.; Musumeci D.; Hunter C. A.; Turega S. Chemical Double Mutant Cycles for the Quantification of Cooperativity in H-Bonded Complexes. J. Am. Chem. Soc. 2009, 131, 18518–18524. 10.1021/ja9083495. [DOI] [PubMed] [Google Scholar]
- Cabot R.; Hunter C. A. Non-Covalent Interactions Between Iodo-Perfluorocarbons and Hydrogen Bond Acceptors. Chem. Commun. 2009, 2005–2007. 10.1039/b822284c. [DOI] [PubMed] [Google Scholar]
- Dunitz J. D. Win Some, Lose Some: Enthalpy-Entropy Compensation in Weak Intermolecular Interactions. Chem. Biol. 1995, 2, 709–712. 10.1016/1074-5521(95)90097-7. [DOI] [PubMed] [Google Scholar]
- Calderone C. T.; Williams D. H. An Anthalpic Component in Cooperativity: The Relationship Between Enthalpy, Entropy, and Noncovalent Structure in Weak Associations. J. Am. Chem. Soc. 2001, 123, 6262–6267. 10.1021/ja003016y. [DOI] [PubMed] [Google Scholar]
- Williams D. H.; Westwell M. S. Aspects of Weak Interactions. Chem. Soc. Rev. 1998, 27, 57–63. 10.1039/a827057z. [DOI] [Google Scholar]
- Searle M. S.; Williams D. H. The Cost of Conformaional Order: Entopy Changes in Molecular Associations. J. Am. Chem. Soc. 1992, 114, 10690–10697. 10.1021/ja00053a002. [DOI] [Google Scholar]
- Lumry R.; Rajender S. Enthalpy-Entropy Compensation Phenomena in Water Solutions of Proteins and Small Molecules: a Ubiquitous Property of Water. Biopolymers 1970, 9, 1125–1227. 10.1002/bip.1970.360091002. [DOI] [PubMed] [Google Scholar]
- Rekharsky M.; Inoue Y. Chiral Recognition Thermodynamics of β-Cyclodextrin: The Thermodynamic Origin of Enantioselectivity and the Enthalpy-Entropy Compensation Effect. J. Am. Chem. Soc. 2000, 122, 4418–4435. 10.1021/ja9921118. [DOI] [Google Scholar]
- Bustamante P.; Romero S.; Pena A.; Escalera B.; Reillo A. Enthalpy-Entropy Compensation for the Solubility of Drugs in Solvent Mixtures: Paracetamol, Acetanilide, and Nalidixic Acid in Dioxane-Water. J. Pharm. Sci. 1998, 87, 1590–1596. 10.1021/js980149x. [DOI] [PubMed] [Google Scholar]
- Chodera J. D.; Mobley D. L.. Entropy-Enthalpy Compensation: Role and Ramifications in Biomolecular Ligand Recognition and Design. In Annual Review of Biophysics; Dill K. A., Ed.; Annual Reviews: Palo Alto, 2013; Vol. 42, pp 121–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryde U. A Fundamental View of Enthalpy-Entropy Compensation. MedChemComm 2014, 5, 1324–1336. 10.1039/C4MD00057A. [DOI] [Google Scholar]
- Korth M. A Quantum Chemical View of Enthalpy-Entropy Compensation. MedChemComm 2013, 4, 1025–1033. 10.1039/c3md00038a. [DOI] [Google Scholar]
- DeLorbe J. E.; Clements J. H.; Teresk M. G.; Benfield A. P.; Plake H. R.; Millspaugh L. E.; Martin S. F. Thermodynamic and Structural Effects of Conformational Constraints in Protein-Ligand Interactions. Entropic Paradoxy Associated with Ligand Preorganization. J. Am. Chem. Soc. 2009, 131, 16758–16770. 10.1021/ja904698q. [DOI] [PubMed] [Google Scholar]
- Kang J. M.; Rebek J. Entropically Driven Binding in a Self-Assembling Molecular Capsule. Nature 1996, 382, 239–241. 10.1038/382239a0. [DOI] [PubMed] [Google Scholar]
- Biedermann F.; Nau W. M.; Schneider H. J. The Hydrophobic Effect Revisited - Studies with Supramolecular Complexes Imply High-Energy Water as a Noncovalent Driving Force. Angew. Chem.-Int. Ed. 2014, 53, 11158–11171. 10.1002/anie.201310958. [DOI] [PubMed] [Google Scholar]
- Mooibroek T. J.; Casas-Solvas J. M.; Harniman R. L.; Renney C.; Carter T. S.; Crump M. P.; Davis A. P. A Threading Receptor for Polysaccharides. Nat. Chem. 2016, 8, 69–74. 10.1038/nchem.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios P.; Carter T. S.; Mooibroek T. J.; Crump M. P.; Lisbjerg M.; Pittelkow M.; Supekar N. T.; Boons G. J.; Davis A. P. Synthetic Receptors for the High-Affinity Recognition of O-GlcNAc Derivatives. Angew. Chem.-Int. Ed. 2016, 55, 3387–3392. 10.1002/anie.201510611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow R. Hydrophobic Effects on Simple Organic Reactions in Water. Acc. Chem. Res. 1991, 24, 159–164. 10.1021/ar00006a001. [DOI] [Google Scholar]
- Southall N. T.; Dill K. A.; Haymet A. D. J. A View of the Hydrophobic Effect. J. Phys. Chem. B 2002, 106, 521–533. 10.1021/jp015514e. [DOI] [Google Scholar]
- Doig A. J.; Williams D. H. Binding Energy of an Amide-Amide Hydrogen Bond in Aqueous and Nonpolar-Solvents. J. Am. Chem. Soc. 1992, 114, 338–343. 10.1021/ja00027a044. [DOI] [Google Scholar]
- Mammen M.; Shakhnovich E. I.; Deutch J. M.; Whitesides G. M. Estimating the Entropic Cost of Self-Assembly of Multiparticle Hydrogen-Bonded Aggregates Based on the Cyanuric Acid · Melamine Lattice. J. Org. Chem. 1998, 63, 3821–3830. 10.1021/jo970944f. [DOI] [Google Scholar]
- Molina P.; Zapata F.; Caballero A. Anion Recognition Strategies Based on Combined Noncovalent Interactions. Chem. Rev. 2017, 117, 9907–9972. 10.1021/acs.chemrev.6b00814. [DOI] [PubMed] [Google Scholar]
- Wendler K.; Thar J.; Zahn S.; Kirchner B. Estimating the Hydrogen Bond Energy. J. Phys. Chem. A 2010, 114, 9529–9536. 10.1021/jp103470e. [DOI] [PubMed] [Google Scholar]
- Jablonski M.; Kaczmarek A.; Sadlej A. J. Estimates of the Energy of Intramolecular Hydrogen Bonds. J. Phys. Chem. A 2006, 110, 10890–10898. 10.1021/jp062759o. [DOI] [PubMed] [Google Scholar]
- Mountford A. J.; Lancaster S. J.; Coles S. J.; Horton P. N.; Hughes D. L.; Hursthouse M. B.; Light M. E. Intramolecular and Intermolecular N-H···F-C Hydrogen-Bonding Interactions in Amine Adducts of Tris(pentafluorophenyl)borane and Tris(pentafluorophenyl)alane. Inorg. Chem. 2005, 44, 5921–5933. 10.1021/ic050663n. [DOI] [PubMed] [Google Scholar]
- Nagy P. I. Competing Intramolecular vs. Intermolecular Hydrogen Bonds in Solution. Int. J. Mol. Sci. 2014, 15, 19562–19633. 10.3390/ijms151119562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolasi V.; Gilli P.; Ferretti V.; Gilli G. Intramolecular O-H···O Hydrogen Bonds Assisted by Resonance. Correlation Between Crystallographic Data and 1H NMR Chemical Shifts. J. Chem. Soc.-Perkin Trans. 2 1997, 945–952. 10.1039/a606862f. [DOI] [Google Scholar]
- Grabowski S. J. An Estimation of Strength of Intramolecular Hydrogen Bonds - Ab Initio and AIM Studies. J. Mol. Struct. 2001, 562, 137–143. 10.1016/S0022-2860(00)00863-2. [DOI] [Google Scholar]
- Kabsch W.; Sander C. Dictionary of Protein Secondary Structure: Pattern-Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Bolen D. W.; Rose G. D. Structure and Energetics of the Hydrogen-Bonded Backbone in Protein Folding. Annu. Rev. Biochem. 2008, 77, 339–362. 10.1146/annurev.biochem.77.061306.131357. [DOI] [PubMed] [Google Scholar]
- Leatherbarrow R. J.; Fersht A. R.; Winter G. Transition-State Stabilization in the Mechanism of Tyrosyl-tRNA Synthetase Revealed by Protein Engineering. P. Natl. Acad. Sci. USA 1985, 82, 7840–7844. 10.1073/pnas.82.23.7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C.; Frechet J. M. J. Applying Key Concepts from Nature: Transition State Stabilization, Pre-Concentration and Cooperativity Effects in Dendritic Biomimetics. Prog. Polym. Sci. 2005, 30, 385–402. 10.1016/j.progpolymsci.2005.01.004. [DOI] [Google Scholar]
- Wulff G.; Liu J. Q. Design of Biomimetic Catalysts by Molecular Imprinting in Synthetic Polymers: The Role of Transition State Stabilization. Acc. Chem. Res. 2012, 45, 239–247. 10.1021/ar200146m. [DOI] [PubMed] [Google Scholar]
- Perutz M. F. Mechanism of Cooperativity and Allosteric Regulation in Proteins. Q. Rev. Biophys. 1989, 22, 139–236. 10.1017/S0033583500003826. [DOI] [PubMed] [Google Scholar]
- Kuhn B.; Mohr P.; Stahl M. Intramolecular Hydrogen Bonding in Medicinal Chemistry. J. Med. Chem. 2010, 53, 2601–2611. 10.1021/jm100087s. [DOI] [PubMed] [Google Scholar]
- Alvarez J.; Gomez-Kaifer M. Importance of Intramolecular Hydrogen Bonding for Preorganization and Binding of Molecular Guests by Water-Soluble Calix[6]Arene Hosts. Chem. Commun. 1998, 1455–1456. 10.1039/A800846I. [DOI] [Google Scholar]
- Merckx T.; Verwilst P.; Dehaen W. Preorganization in Bistriazolyl Anion Receptors. Tetrahedron Lett. 2013, 54, 4237–4240. 10.1016/j.tetlet.2013.05.133. [DOI] [Google Scholar]
- Santacroce P. V.; Davis J. T.; Light M. E.; Gale P. A.; Iglesias-Sanchez J. C.; Prados P.; Quesada R. Conformational Control of Transmembrane Cl– Transport. J. Am. Chem. Soc. 2007, 129, 1886–1887. 10.1021/ja068067v. [DOI] [PubMed] [Google Scholar]
- Gale P. A. From Anion Receptors to Transporters. Acc. Chem. Res. 2011, 44, 216–226. 10.1021/ar100134p. [DOI] [PubMed] [Google Scholar]
- Davis J. T.; Gale P. A.; Okunola O. A.; Prados P.; Iglesias-Sanchez J. C.; Torroba T.; Quesada R. Using Small Molecules to Facilitate Exchange of Bicarbonate and Chloride Anions Across Liposomal Membranes. Nat. Chem. 2009, 1, 138–144. 10.1038/nchem.178. [DOI] [PubMed] [Google Scholar]
- Custelcean R. Crystal Engineering with Urea and Thiourea Hydrogen-Bonding Groups. Chem. Commun. 2008, 295–307. 10.1039/B708921J. [DOI] [PubMed] [Google Scholar]
- Amendola V.; Fabbrizzi L.; Mosca L. Anion Recognition by Hydrogen Bonding: Urea-Based Receptors. Chem. Soc. Rev. 2010, 39, 3889–3915. 10.1039/b822552b. [DOI] [PubMed] [Google Scholar]
- Malerich J. P.; Hagihara K.; Rawal V. H. Chiral Squaramide Derivatives are Excellent Hydrogen Bond Donor Catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. 10.1021/ja805693p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X. H.; Wang W. Hydrogen-Bond-Mediated Asymmetric Catalysis. Chem.-Asian J. 2008, 3, 516–532. 10.1002/asia.200700415. [DOI] [PubMed] [Google Scholar]
- In a multipronged HB either there can be multiple X-H fragments in contact with the same acceptor (Y) or there can be several acceptors interacting with a single hydrogen atom.
- Karas L. J.; Wu C. H.; Das R.; Wu J. I. C. Hydrogen Bond Design Principles. WIRES Comput. Mol. Sci. 2020, 10, e1477. 10.1002/wcms.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aakeroy C. B.; Seddon K. R. The Hydrogen Bond and Crystal Engineering. Chem. Soc. Rev. 1993, 22, 397–407. 10.1039/CS9932200397. [DOI] [Google Scholar]
- Taylor R.; Kennard O.; Versichel W. Geometry of the N-H···O = C Hydrogen Bond. 2. Three-Center (“Bifurcated“) and four-center (“Trifurcated“) Bonds. J. Am. Chem. Soc. 1984, 106, 244–248. 10.1021/ja00313a047. [DOI] [Google Scholar]
- Rozas I.; Alkorta I.; Elguero J. Bifurcated Hydrogen Bonds: Three-Centered Interactions. J. Phys. Chem. A 1998, 102, 9925–9932. 10.1021/jp9824813. [DOI] [Google Scholar]
- Thomas S. P.; Pavan M. S.; Row T. N. G. Charge Density Analysis of Ferulic Acid: Robustness of a Trifurcated C-H···O Hydrogen Bond. Cryst. Growth Des. 2012, 12, 6083–6091. 10.1021/cg301211h. [DOI] [Google Scholar]
- Davis A. P. Anion Binding and Transport by Steroid-Based Receptors. Coord. Chem. Rev. 2006, 250, 2939–2951. 10.1016/j.ccr.2006.05.008. [DOI] [Google Scholar]
- Kurczab R.; Mitoraj M. P.; Michalak A.; Ziegler T. Theoretical Analysis of the Resonance Assisted Hydrogen Bond Based on the Combined Extended Transition State Method and Natural Orbitals for Chemical Valence Scheme. J. Phys. Chem. A 2010, 114, 8581–8590. 10.1021/jp911405e. [DOI] [PubMed] [Google Scholar]
- Grosch A. A.; van der Lubbe S. C. C.; Guerra C. F. Nature of Intramolecular Resonance Assisted Hydrogen Bonding in Malonaldehyde and Its Saturated Analogue. J. Phys. Chem. A 2018, 122, 1813–1820. 10.1021/acs.jpca.7b12635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck J. F.; Mo Y. R. How Resonance Assists Hydrogen Bonding Interactions: An Energy Decomposition Analysis. J. Comput. Chem. 2007, 28, 455–466. 10.1002/jcc.20523. [DOI] [PubMed] [Google Scholar]
- Bertolasi V.; Gilli P.; Ferretti V.; Gilli G. Evidence for Resonance-Assisted Hydrogen-Bonding. 2. Intercorrelation Between Crystal-Structure and Spectroscopic Parameters in Eight Intramolecular Hydrogen-Bonded 1,3-Diaryl-1,3-propanedione Enols. J. Am. Chem. Soc. 1991, 113, 4917–4925. 10.1021/ja00013a030. [DOI] [Google Scholar]
- Sanz P.; Mo O.; Yanez M.; Elguero J. Resonance-Assisted Hydrogen Bonds: A Critical Examination. Structure and Stability of the Enols of β-Diketones and β-Enaminones. J. Phys. Chem. A 2007, 111, 3585–3591. 10.1021/jp067514q. [DOI] [PubMed] [Google Scholar]
- Guillaumes L.; Simon S.; Fonseca Guerra C. The Role of Aromaticity, Hybridization, Electrostatics, and Covalency in Resonance-Assisted Hydrogen Bonds of Adenine-Thymine (AT) Base Pairs and Their Mimics. ChemistryOpen 2015, 4, 318–327. 10.1002/open.201402132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Pranata J. Importance of Secondary Interactions in Triply Hydrogen-Bond complexes: Guanine-Cytosine vs Uracil-2,6-diaminopyridine. J. Am. Chem. Soc. 1990, 112, 2008–2010. 10.1021/ja00161a061. [DOI] [Google Scholar]
- Sponer J.; Jurecka P.; Hobza P. Accurate Interaction Energies of Hydrogen-Bonded Nucleic Acid Base Pairs. J. Am. Chem. Soc. 2004, 126, 10142–10151. 10.1021/ja048436s. [DOI] [PubMed] [Google Scholar]
- Djurdjevic S.; Leigh D. A.; McNab H.; Parsons S.; Teobaldi G.; Zerbetto F. Extremely Strong and Readily Accessible AAA-DDD Triple Hydrogen Bond Complexes. J. Am. Chem. Soc. 2007, 129, 476–477. 10.1021/ja067410t. [DOI] [PubMed] [Google Scholar]
- Blight B. A.; Camara-Campos A.; Djurdjevic S.; Kaller M.; Leigh D. A.; McMillan F. M.; McNab H.; Slawin A. M. Z. AAA-DDD Triple Hydrogen Bond Complexes. J. Am. Chem. Soc. 2009, 131, 14116–14122. 10.1021/ja906061v. [DOI] [PubMed] [Google Scholar]
- Blight B. A.; Hunter C. A.; Leigh D. A.; McNab H.; Thomson P. I. T. An AAAA-DDDD Quadruple Hydrogen-Bond Array. Nat. Chem. 2011, 3, 244–248. 10.1038/nchem.987. [DOI] [PubMed] [Google Scholar]
- Sijbesma R. P.; Beijer F. H.; Brunsveld L.; Folmer B. J. B.; Hirschberg J.; Lange R. F. M.; Lowe J. K. L.; Meijer E. W. Reversible Polymers Formed From Self-Complementary Monomers Using Quadruple Hydrogen Bonding. Science 1997, 278, 1601–1604. 10.1126/science.278.5343.1601. [DOI] [PubMed] [Google Scholar]
- Beijer F. H.; Kooijman H.; Spek A. L.; Sijbesma R. P.; Meijer E. W. Self-Complementarity Achieved Through Quadruple Hydrogen Bonding. Angew. Chem.-Int. Ed. 1998, 37, 75–78. . [DOI] [Google Scholar]
- Hunter C. A.; Anderson H. L. What is Cooperativity?. Angew. Chem.-Int. Ed. 2009, 48, 7488–7499. 10.1002/anie.200902490. [DOI] [PubMed] [Google Scholar]
- Mulder A.; Huskens J.; Reinhoudt D. N. Multivalency in Supramolecular Chemistry and Nanofabrication. Org. Biomol. Chem. 2004, 2, 3409–3424. 10.1039/b413971b. [DOI] [PubMed] [Google Scholar]
- Ercolani G.; Schiaffino L. Allosteric, Chelate, and Interannular Cooperativity: A Mise au Point. Angew. Chem.-Int. Ed. 2011, 50, 1762–1768. 10.1002/anie.201004201. [DOI] [PubMed] [Google Scholar]
- Hunter C. A.; Misuraca M. C.; Turega S. M. Solvent Effects on Chelate Cooperativity. Chem. Sci. 2012, 3, 589–601. 10.1039/C1SC00635E. [DOI] [Google Scholar]
- Jiang W.; Nowosinski K.; Low N. L.; Dzyuba E. V.; Klautzsch F.; Schafer A.; Huuskonen J.; Rissanen K.; Schalley C. A. Chelate Cooperativity and Spacer Length Effects on the Assembly Thermodynamics and Kinetics of Divalent Pseudorotaxanes. J. Am. Chem. Soc. 2012, 134, 1860–1868. 10.1021/ja2107096. [DOI] [PubMed] [Google Scholar]
- Prins L. J.; Reinhoudt D. N.; Timmerman P. Noncovalent Synthesis Using Hydrogen Bonding. Angew. Chem.-Int. Ed. 2001, 40, 2382–2426. . [DOI] [PubMed] [Google Scholar]
- Mateos-Timoneda M. A.; Crego-Calama M.; Reinhoudt D. N. Supramolecular Chirality of Self-Assembled Systems in Solution. Chem. Soc. Rev. 2004, 33, 363–372. 10.1039/b305550g. [DOI] [PubMed] [Google Scholar]
- Hirschberg J.; Brunsveld L.; Ramzi A.; Vekemans J.; Sijbesma R. P.; Meijer E. W. Helical Self-Assembled Polymers From Cooperative Stacking of Hydrogen-Bonded Pairs. Nature 2000, 407, 167–170. 10.1038/35025027. [DOI] [PubMed] [Google Scholar]
- Lange R. F. M.; Van Gurp M.; Meijer E. W. Hydrogen-Bonded Supramolecular Polymer Networks. J. Polym. Sci., Polym. Chem. 1999, 37, 3657–3670. . [DOI] [Google Scholar]
- De Greef T. F. A.; Smulders M. M. J.; Wolffs M.; Schenning A.; Sijbesma R. P.; Meijer E. W. Supramolecular Polymerization. Chem. Rev. 2009, 109, 5687–5754. 10.1021/cr900181u. [DOI] [PubMed] [Google Scholar]
- Whitty A. Cooperativity and Biological Complexity. Nat. Chem. Biol. 2008, 4, 435–439. 10.1038/nchembio0808-435. [DOI] [PubMed] [Google Scholar]
- Xantheas S. S. Cooperativity and Hydrogen Bonding Network in Water Clusters. Chem. Phys. 2000, 258, 225–231. 10.1016/S0301-0104(00)00189-0. [DOI] [Google Scholar]
- Renny J. S.; Tomasevich L. L.; Tallmadge E. H.; Collum D. B. Method of Continuous Variations: Applications of Job Plots to the Study of Molecular Associations in Organometallic Chemistry. Angew. Chem.-Int. Ed. 2013, 52, 11998–12013. 10.1002/anie.201304157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slagt V. F.; Roder M.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Reek J. N. H. Supraphos: A Supramolecular Strategy to Prepare Bidentate Ligands. J. Am. Chem. Soc. 2004, 126, 4056–4057. 10.1021/ja038955f. [DOI] [PubMed] [Google Scholar]
- Gallen A.; Riera A.; Verdaguer X.; Grabulosa A. Coordination Chemistry and Catalysis with Secondary Phosphine Oxides. Catal. Sci. Technol. 2019, 9, 5504–5561. 10.1039/C9CY01501A. [DOI] [Google Scholar]
- Beaulieu W. B.; Rauchfuss T. B.; Roundhill D. M. Interconversion Reactions Between Substituted Phosphinous Acid-Phosphinito Complexes Of Platinum(II) and Their Capping Reactions With Boron Trifluoride-Diethyl Etherate. Inorg. Chem. 1975, 14, 1732–1734. 10.1021/ic50149a065. [DOI] [Google Scholar]
- Shaikh T. M.; Weng C.-M.; Hong F.-E. Secondary Phosphine Oxides: Versatile Ligands in Transition Metal-Catalyzed Cross-Coupling Reactions. Coord. Chem. Rev. 2012, 256, 771–803. 10.1016/j.ccr.2011.11.007. [DOI] [Google Scholar]
- Ghorai D.; Muller V.; Keil H.; Stalke D.; Zanoni G.; Tkachenko B. A.; Schreiner P. R.; Ackermann L. Secondary Phosphine Oxide Preligands for Palladium-Catalyzed C-H (Hetero)Arylations: Efficient Access to Pybox Ligands. Adv. Synth. Catal. 2017, 359, 3137–3141. 10.1002/adsc.201700663. [DOI] [Google Scholar]
- Castro P. M.; Gulyás H.; Benet-Buchholz J.; Bo C.; Freixa Z.; van Leeuwen P. W. N. M. SPOs as New Ligands in Rh(III) Catalyzed Enantioselective Transfer Hydrogenation. Catal. Sci. Technol. 2011, 1, 401–407. 10.1039/c0cy00022a. [DOI] [Google Scholar]
- Membrat R.; Vasseur A.; Martinez A.; Giordano L.; Nuel D. Phosphinous Acid Platinum Complex as Robust Catalyst for Oxidation: Comparison with Palladium and Mechanistic Investigations. Eur. J. Org. Chem. 2018, 2018, 5427–5434. 10.1002/ejoc.201801040. [DOI] [Google Scholar]
- Chang Y. C.; Chang C. H.; Wang L. W.; Liang Y. H.; Hu D. F.; Weng C. M.; Mao K. C.; Hong F. E. Imidazolio-Substituted Secondary Phosphine Oxides as Potential Carbene Reagents. Polyhedron 2015, 100, 382–391. 10.1016/j.poly.2015.08.041. [DOI] [Google Scholar]
- Lhermet R.; Moser E.; Jeanneau E.; Olivier-Bourbigou H.; Breuil P. A. R. Outer-Sphere Reactivity Shift of Secondary Phosphine Oxide-Based Nickel Complexes: From Ethylene Hydrophosphinylation to Oligomerization. Chem.—Eur. J. 2017, 23, 7433–7437. 10.1002/chem.201701414. [DOI] [PubMed] [Google Scholar]
- Rafter E.; Gutmann T.; Low F.; Buntkowsky G.; Philippot K.; Chaudret B.; van Leeuwen P. W. N. M. Secondary Phosphine Oxides as Pre-Ligands for Nanoparticle Stabilization. Catal. Sci. Technol. 2013, 3, 595–599. 10.1039/C2CY20683H. [DOI] [Google Scholar]
- Bennett M. A.; Mitchell T. R. B. Oxidative Addition of Dialkylphosphites to Comples of Iridium(I) and Rhodium(I). J. Org. Chem. 1983, 250, 499–508. 10.1016/0022-328X(83)85073-6. [DOI] [Google Scholar]
- van Leeuwen P. W. N. M.; Cano I.; Freixa Z. Secondary Phosphine Oxides: Bifunctional Ligands in Catalysis. ChemCatChem. 2020, 12, 3982–3994. 10.1002/cctc.202000493. [DOI] [Google Scholar]
- van Leeuwen P. W. N. M.; Roobeek C. F.; Wife R. L.; Frijns J. H. G. Platinum Hydroformylation Catalysis Containing Diphenylphosphine Oxide Ligands. J. Chem. Soc. Chem. Comm. 1986, 31–33. 10.1039/c39860000031. [DOI] [Google Scholar]
- Ustynyuk Y. A.; Babin Y. V.; Savchenko V. G. Proton Trigger” in Catalytic Hydroformylation of Alkenes on Platinum Complexes with Hydrophosphoryl Ligands. Dokl. Phys. Chem. 2010, 430, 25–28. 10.1134/S001250161002003X. [DOI] [Google Scholar]
- van Leeuwen P. W. N. M.; Roobeek C. F.; Frijns J. H. G.; Orpen A. G. Characterization of the Intermediates in the Hydroformylation Reaction Catalyzed by Platinum Diphenylphosphinous Acid Complexes. Organometallics 1990, 9, 1211–1222. 10.1021/om00118a049. [DOI] [Google Scholar]
- Ustynyuk Y. A.; Babin Y. V. Tautomerism of Hydrophosphoryl Compounds and Their Features as Ligands in Metal Complex Catalysis. Quantum-Chemical Simulations by the Density Functional Method. Russ. J. Gen. Chem. 2008, 78, 822–832. 10.1134/S1070363208040415. [DOI] [Google Scholar]
- Christiansen A.; Li C.; Garland M.; Selent D.; Ludwig R.; Spannenberg A.; Baumann W.; Franke R.; Börner A. On the Tautomerism of Secondary Phosphane Oxides. Eur. J. Org. Chem. 2010, 2010, 2733–2741. 10.1002/ejoc.201000037. [DOI] [Google Scholar]
- Christiansen A.; Selent D.; Spannenberg A.; Baumann W.; Franke R.; Boerner A. Reaction of Secondary Phosphine Oxides with Rhodium(I). Organometallics 2010, 29, 3139–3145. 10.1021/om100171p. [DOI] [Google Scholar]
- Christiansen A.; Selent D.; Spannenberg A.; Koeckerling M.; Reinke H.; Baumann W.; Jiao H.; Franke R.; Boerner A. Heteroatom-Substituted Secondary Phosphine Oxides (HASPOs) as Decomposition Products and Preligands in Rhodium-Catalysed Hydroformylation. Chem.—Eur. J. 2011, 17, 2120–2129. 10.1002/chem.201002823. [DOI] [PubMed] [Google Scholar]
- Matsumoto M.; Tamura M. Rhodium-Catalyzed Low-Pressure Hydroformylation of Higher α-Olefins - New, Thermally stable Rhodium Catalysts by Reaction of RhH(CO)PPh3)3 with Phosphinous Acids. J. Mol. Catal. 1983, 19, 365–376. 10.1016/0304-5102(83)80008-X. [DOI] [Google Scholar]
- van Leeuwen P. W. N. M.; Roobeek C. F.. Process for the Reduction of Carbonyl Compounds. European Patent: EP0349087 A1, 1990.
- Cano I.; Martinez-Prieto L. M.; Chaudret B.; van Leeuwen P. W. N. M. Iridium versus Iridium: Nanocluster and Monometallic Catalysts Carrying the Same Ligand Behave Differently. Chem.—Eur. J. 2017, 23, 1444–1450. 10.1002/chem.201605352. [DOI] [PubMed] [Google Scholar]
- Cano I.; Martinez-Prieto L. M.; Vendier L.; van Leeuwen P. W. N. M. An Iridium-SPO Complex as Bifunctional Catalyst for the Highly Selective Hydrogenation of Aldehydes. Catal. Sci. Technol. 2018, 8, 221–228. 10.1039/C7CY01953J. [DOI] [Google Scholar]
- Ghaffar T.; Parkins A. W. A New Homogeneous Platinum-Containing Catalyst for the Hydrolysis of Nitriles. Tetrahedron Lett. 1995, 36, 8657–8660. 10.1016/0040-4039(95)01785-G. [DOI] [Google Scholar]
- Ghaffar T.; Parkins A. W. The Catalytic Hydration of Nitriles to Amides Using a Homogeneous Platinum Phosphinito Catalyst. J. Mol. Catal. A-Chem. 2000, 160, 249–261. 10.1016/S1381-1169(00)00253-3. [DOI] [Google Scholar]
- Akisanya J.; Parkins A. W.; Steed J. W. A Synthesis of Atenolol Using a Nitrile Hydration Catalyst. Org. Process Res. Dev. 1998, 2, 274–276. 10.1021/op9800313. [DOI] [Google Scholar]
- Cadierno V. Synthetic Applications of the Parkins Nitrile Hydration Catalyst [PtH{(PMe2O)2H}(PMe2OH)]: A Review. Appl. Sci. -Basel 2015, 5, 380–401. 10.3390/app5030380. [DOI] [Google Scholar]
- Yasuno Y.; Yoshida Y.; Nishimura A.; Ohfune Y.; Shinada T. A Facile Synthesis of (2R/S, 5R)-1-tert-Butyl 2-Methyl-5-(((tert-Butyldimethylsilyl)Oxo)Methyl)Pyrrolidine-1,2-Dicarboxylate. Heterocycles 2015, 91, 2377–2385. 10.3987/COM-15-13347. [DOI] [Google Scholar]
- Jiang X. B.; Minnaard A. J.; Feringa B. L.; de Vries J. G. Platinum-Catalyzed Selective Hydration of Hindered Nitriles and Nitriles with Acid- or Base-Sensitive Groups. J. Org. Chem. 2004, 69, 2327–2331. 10.1021/jo035487j. [DOI] [PubMed] [Google Scholar]
- Tafesse L.Substituted Pyridines Useful for Treating Pain. United States Patent: US8389549 B2, 2013.
- Saito D. R.; Long D. D.; van Dyke P.; Church T. J.; Jiang L.; Frieman B.. 8-Azabicyclo[3.2.1]Octyl-2-Hydroxybenzamide Compounds as μ-Opioid Receptor Antagonists. Brazilian Patent: PI08157979 A2, 2009.
- Fujieda H.; Kogami M.; Sakairi M.; Kato N.; Makino M.; Takahashi N.; Miyazawa T.; Harada S.; Yamashita T. Discovery of a Potent Glucokinase Activator With a Favorable Liver and Pancreas Distribution Pattern for the Treatment of Type 2 Diabetes Mellitus. Eur. J. Med. Chem. 2018, 156, 269–294. 10.1016/j.ejmech.2018.06.060. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Zhang L.; Lam T. M. Synthesis of the Aminocyclitol Core of Jogyamycin via an Enantioselective Pd-Catalyzed Trimethylenemethane (TMM) Cycloaddition. Org. Lett. 2018, 20, 3938–3942. 10.1021/acs.orglett.8b01518. [DOI] [PubMed] [Google Scholar]
- Mai C.-K.; Sammons M. F.; Sammakia T. A Concise Formal Synthesis of Diazonamide A by the Stereoselective Construction of the C10 Quaternary Center. Angew. Chem.-Int. Ed. 2010, 49, 2397–2400. 10.1002/anie.200906318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews R. S.; Becker J. J.; Gagné M. R. A Photoflow Reactor for the Continuous Photoredox-Mediated Synthesis of C-Glycoamino Acids and C-Glycolipids. Angew. Chem.-Int. Ed. 2012, 51, 4140–4143. 10.1002/anie.201200593. [DOI] [PubMed] [Google Scholar]
- Tun M. K. M.; Wüstmann D.-J.; Herzon S. B. A Robust and Scalable Synthesis of the Potent Neuroprotective Agent (−)-Huperzine A. Chem. Sci. 2011, 2, 2251–2253. 10.1039/c1sc00455g. [DOI] [Google Scholar]
- Mercado-Marin E. V.; Sarpong R. Unified Approach to Prenylated Indole Alkaloids: Total Syntheses of (−)-17-Hydroxy-Citrinalin B, (+)-Stephacidin A, and (+)-Notoamide I. Chem. Sci. 2015, 6, 5048–5052. 10.1039/C5SC01977J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado-Marin E. V.; Garcia-Reynaga P.; Romminger S.; Pimenta E. F.; Romney D. K.; Lodewyk M. W.; Williams D. E.; Andersen R. J.; Miller S. J.; Tantillo D. J.; Berlinck R. G. S.; Sarpong R. Total Synthesis and Isolation of Citrinalin and Cyclopiamine Congeners. Nature 2014, 509, 318–324. 10.1038/nature13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Jiang X.; De Brabander J. K. Studies Toward the Unique Pederin Family Member Psymberin: Full Structure Elucidation, Two Alternative Total Syntheses, and Analogs. J. Am. Chem. Soc. 2012, 134, 17083–17093. 10.1021/ja3057612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.; Nguyen V. T.; Dang H. T.; Nguyen D. P.; Arman H. D.; Larionov O. V. Photoinduced Carboborative Ring Contraction Enables Regio- and Stereoselective Synthesis of Multiply Substituted Five-Membered Carbocycles and Heterocycles. J. Am. Chem. Soc. 2017, 139, 11365–11368. 10.1021/jacs.7b07128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M. J. R.; Schneider M.; Brandstatter M.; Krautwald S.; Carreira E. M. Total Synthesis of (−)-Mitrephorone A. J. Am. Chem. Soc. 2018, 140, 16704–16710. 10.1021/jacs.8b09685. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Williams N.; De Brabander J. K. Synthesis of Psymberin Analogues: Probing a Functional Correlation With the Pederin/Mycalamide Family of Natural Products. Org. Lett. 2007, 9, 227–230. 10.1021/ol062656o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan T.; Kawamoto Y.; Asakawa T.; Furuta T.; Fukuyama T. Synthetic Studies on Altemicidin: Stereocontrolled Construction of the Core Framework. Org. Lett. 2008, 10, 169–171. 10.1021/ol701940f. [DOI] [PubMed] [Google Scholar]
- Hirooka Y.; Ikeuchi K.; Kawamoto Y.; Akao Y.; Furuta T.; Asakawa T.; Inai M.; Wakimoto T.; Fukuyama T.; Kan T. Enantioselective Synthesis of SB-203207. Org. Lett. 2014, 16, 1646–1649. 10.1021/ol5002973. [DOI] [PubMed] [Google Scholar]
- Jones R. A.; Krische M. J. Asymmetric Total Synthesis of the Iridoid β-Glucoside (+)-Geniposide via Phosphine Organocatalysis. Org. Lett. 2009, 11, 1849–1851. 10.1021/ol900360h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez F. d. J.; Sarpong R. Ga(III)-Catalyzed Cycloisomerization Approach to (±)-Icetexone and (±)-epi-Icetexone. Org. Lett. 2010, 12, 1428–1431. 10.1021/ol902959v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers A. G.; Herzon S. B.; Wulff J. E.; Siegrist R.; Svenda J.; Zajac M. A.. Synthesis of Avrainvillamide, Strephacidin B, and Analogues Thereof. United States Patent: US7902196 B2, 2011.
- Greshock T. J.; Funk R. L. An Approach to the Total Synthesis of Welwistatin. Org. Lett. 2006, 8, 2643–2645. 10.1021/ol0608799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers A. G.; Herzon S. B.; Wulff J. E.; Siegrist R.; Svenda J.; Zajac M. A.. Synthesis of Avrainvillamide, Strephacidin B, and Analogues Thereof. Worldwide Patent: WO2006102097 A2, 2006.
- Finch H.; Fox C.; Sajad M.. Respiratory Disease Treatment. Worldwide Patent: WO2010015818 A1, 2010.
- Park J. H.; Tafesse L.. Nitrogen Containing Morphinan Derivatives and the Use Thereof. Worldwide Patent: WO2014091298 A2, 2014.
- Jiang X.; Bender C. F.; Visnick M.; Hotema M. R.; Sheldon Z. S.; Lee C.; Caprathe B. W.; Bolton G.; Kornberg B.. Pyrimidine Tricyclic Enone Derivatives for Inhibition of RORγ and Other Issues. Worldwide Patent: WO 2018111315 A1, 2018.
- Gulyás H.; Rivilla I.; Curreli S.; Freixa Z.; van Leeuwen P. W. N. M. Highly Active, Chemo- and Enantioselective Pt-SPO Catalytic Systems for the Synthesis of Aromatic Carboxamides. Catal. Sci. Technol. 2015, 5, 3822–3828. 10.1039/C5CY00627A. [DOI] [Google Scholar]
- Ahmed T. J.; Fox B. R.; Knapp S. M. M.; Yelle R. B.; Juliette J. J.; Tyler D. R. Investigation of the Reactivity of Pt Phosphinito and Molybdocene Nitrile Hydration Catalysts With Cyanohydrins. Inorg. Chem. 2009, 48, 7828–7837. 10.1021/ic900734d. [DOI] [PubMed] [Google Scholar]
- Brown L. E.; Landaverry Y. R.; Davies J. R.; Milinkevich K. A.; Ast S.; Carlson J. S.; Oliver A. G.; Konopelski J. P. Progress Toward the Total Synthesis of Psymberin/Irciniastatin A. J. Org. Chem. 2009, 74, 5405–5410. 10.1021/jo9009003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobley C. J.; van den Heuvel M.; Abbadi A.; de Vries J. G. Platinum Catalysed Hydrolytic Amidation of Unactivated Nitriles. Tetrahedron Lett. 2000, 41, 2467–2470. 10.1016/S0040-4039(00)00181-7. [DOI] [Google Scholar]
- Li J.; Chen L.; Chin E.; Lui A. S.; Zecic H. Platinum(II)-Catalyzed Intramolecular Cyclization of Alkynylbenzonitriles: Synthesis of 1-Alkoxyisoquinolines and Isoquinolones. Tetrahedron Lett. 2010, 51, 6422–6425. 10.1016/j.tetlet.2010.09.136. [DOI] [Google Scholar]
- Brameld K. A.; Carter D. S.; Chin E.; De Vicente J.; Li J.; Schoenfeld R. C.; Sjogren E. B.; Talamas F. X.. Heterocyclic Antiviral Compounds. United States Patent: US20100021423 A1, 2010.
- Vasseur A.; Membrat R.; Gatineau D.; Tenaglia A.; Nuel D.; Giordano L. Secondary Phosphine Oxides as Multitalented Preligands En route to the Chemoselective Palladium-Catalyzed Oxidation of Alcohols. ChemCatChem. 2017, 9, 728–732. 10.1002/cctc.201601261. [DOI] [Google Scholar]
- Muzart J. Pd-Catalyzed Hydrogen-Transfer Reactions from Alcohols to C = C, C = O, and C = N Bonds. Eur. J. Org. Chem. 2015, 2015, 5693–5707. 10.1002/ejoc.201500401. [DOI] [Google Scholar]
- Membrat R.; Vasseur A.; Moraleda D.; Michaud-Chevallier S.; Martinez A.; Giordano L.; Nuel D. Platinum-(Phosphinito-Phosphinous Acid) Complexes as Bi-Talented Catalysts for Oxidative Fragmentation of Piperidinols: An Entry to Primary Amines. RSC Adv. 2019, 9, 37825–37829. 10.1039/C9RA08709E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G. Y.; Marshall W. J. Air-Stable Phosphine Sulfide Ligand Precursors for Nickel-Catalyzed Cross-Coupling Reactions of Unactivated Aryl Chlorides with Aryl Grignard Reagents. Organometallics 2002, 21, 590–591. 10.1021/om010828+. [DOI] [Google Scholar]
- Breuil P.-A. R.; Patureau F. W.; Reek J. N. H. Singly Hydrogen Bonded Supramolecular Ligands for Highly Selective Rhodium-Catalyzed Hydrogenation Reactions. Angew. Chem.-Int. Ed. 2009, 48, 2162–2165. 10.1002/anie.200806177. [DOI] [PubMed] [Google Scholar]
- Breuil P.-A. R.; Reek J. N. H. Amino Acid Based Phosphoramidite Ligands for the Rhodium-Catalyzed Asymmetric Hydrogenation. Eur. J. Org. Chem. 2009, 2009, 6225–6230. 10.1002/ejoc.200901044. [DOI] [Google Scholar]
- Liu Y.; Sandoval C. A.; Yamaguchi Y.; Zhang X.; Wang Z.; Kato K.; Ding K. Hydrogen Bonding Makes a Difference in the Rhodium-Catalyzed Enantioselective Hydrogenation Using Monodentate Phosphoramidites. J. Am. Chem. Soc. 2006, 128, 14212–14213. 10.1021/ja063350f. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Wang Z.; Ding K. L. DpenPhos/Rh(I) Catalyzed Asymmetric Hydrogenation of Dehydro-β-Amino Acid Esters. Acta Chim. Sin. 2012, 70, 1464–1470. 10.6023/A12030050. [DOI] [Google Scholar]
- Zhang J. Z.; Li Y.; Wang Z.; Ding K. L. Asymmetric Hydrogenation of α-and β-Enamido Phosphonates: Rhodium(I)/Monodentate Phosphoramidite Catalyst. Angew. Chem.-Int. Ed. 2011, 50, 11743–11747. 10.1002/anie.201104912. [DOI] [PubMed] [Google Scholar]
- Zhang J. Z.; Dong K. W.; Wang Z.; Ding K. L. Asymmetric Hydrogenation of α- or β-Acyloxy α,β-Unsaturated Phosphonates Catalyzed by a Rh(I) Complex of Monodentate Phosphoramidite. Org. Biomol. Chem. 2012, 10, 1598–1601. 10.1039/c1ob06835k. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Wang Z.; Ding K. L. Rh(I)/DpenPhos Catalyzed Asymmetric Hydrogenation of Enol Esters and Potassium E-3-Cyano-5-Methylhex-3-Enoate. Tetrahedron 2012, 68, 7581–7585. 10.1016/j.tet.2012.05.096. [DOI] [Google Scholar]
- Pan L. L.; Wang J.; Jia Y. L.; Zheng H. M.; Wang Y.; Zhu Y. Z. Asymmetric Synthesis and Evaluation of Danshensu-Cysteine Conjugates as Novel Potential Anti-Apoptotic Drug Candidates. Int. J. Mol. Sci. 2015, 16, 628–644. 10.3390/ijms16010628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zhu Y.; Liu X.; Jia Y.; Pan L.; Yang H.. A Method for Preparing Danshensu Derivatives and the Application of the Danshensu Derivatives in Pharmaceuticals (loosely translated). Chinese Patent: CN103202831 B, 2012.
- Pignataro L.; Carboni S.; Civera M.; Colombo R.; Piarulli U.; Gennari C. PhthalaPhos: Chiral Supramolecular Ligands for Enantioselective Rhodium-Catalyzed Hydrogenation Reactions. Angew. Chem.-Int. Ed. 2010, 49, 6633–6637. 10.1002/anie.201002958. [DOI] [PubMed] [Google Scholar]
- Pignataro L.; Boghi M.; Civera M.; Carboni S.; Piarulli U.; Gennari C. Rhodium-Catalyzed Asymmetric Hydrogenation of Olefins with PhthalaPhos, a New Class of Chiral Supramolecular Ligands. Chem.—Eur. J. 2012, 18, 1383–1400. 10.1002/chem.201102018. [DOI] [PubMed] [Google Scholar]
- Laungani A. C.; Breit B. Supramolecular PhanePhos-Analogous Ligands Through Hydrogen-Bonding for Asymmetric Hydrogenation. Chem. Commun. 2008, 844–846. 10.1039/B716529C. [DOI] [PubMed] [Google Scholar]
- Kokan Z.; Kirin S. I. The Application of “Backdoor Induction” in Bioinspired Asymmetric Catalysis. RSC Adv. 2012, 2, 5729–5737. 10.1039/c2ra20598j. [DOI] [Google Scholar]
- Kokan Z.; Kirin S. I. Backdoor Induction” of Chirality in Asymmetric Hydrogenation with Rhodium(I) Complexes of Amino Acid Substituted Triphenylphosphane Ligands. Eur. J. Org. Chem. 2013, 2013, 8154–8161. 10.1002/ejoc.201301011. [DOI] [Google Scholar]
- Opačak S.; Kokan Z.; Glasovac Z.; Peric B.; Kirin S. I. Backdoor Induction” of Chirality: Trans-1,2-cyclohexanediamine as Key Building Block for Asymmetric Hydrogenation Catalysts. Eur. J. Org. Chem. 2019, 2019, 2115–2128. 10.1002/ejoc.201801647. [DOI] [Google Scholar]
- Sandee A. J.; van der Burg A. M.; Reek J. N. H.. Process for the Production of Phosphorus Compounds. United States Patent: US20090082595, 2009.
- Koshti V. S.; Sen A.; Shinde D.; Chikkali S. H. Self-Assembly of P-Chiral Supramolecular Phosphines on Rhodium and Direct Evidence for Rh-Catalyst-Substrate Interactions. Dalton Trans. 2017, 46, 13966–13973. 10.1039/C7DT02923C. [DOI] [PubMed] [Google Scholar]
- Sandee A. J.; van der Burg A. M.; Reek J. N. H. UREAphos: Supramolecular Bidentate Ligands for Asymmetric Hydrogenation. Chem. Commun. 2007, 864–866. 10.1039/B614571J. [DOI] [PubMed] [Google Scholar]
- Reek J. N. H.; Chen R.; Kamer P. C. J.; Slagt V. F.; van Leeuwen P. W. N. M.. Coordination Complex System Comprising Building Blocks. United States Patent: US20060258858 A1, 2006.
- Meeuwissen J.; Kuil M.; van der Burg A. M.; Sandee A. J.; Reek J. N. H. Application of a Supramolecular-Ligand Library for the Automated Search for Catalysts for the Asymmetric Hydrogenation of Industrially Relevant Substrates. Chem.—Eur. J. 2009, 15, 10272–10279. 10.1002/chem.200901110. [DOI] [PubMed] [Google Scholar]
- Koshti V. S.; Mote N. R.; Gonnade R. G.; Chikkali S. H. Highly Enantioselective Pd-Catalyzed Synthesis of P-Stereogenic Supramolecular Phosphines, Self-Assembly, and Implication. Organometallics 2015, 34, 4802–4805. 10.1021/acs.organomet.5b00664. [DOI] [Google Scholar]
- Mote N. R.; Patel K.; Shinde D. R.; Gaikwad S. R.; Koshti V. S.; Gonnade R. G.; Chikkali S. H. H-Bonding Assisted Self-Assembly of Anionic and Neutral Ligand on Metal: A Comprehensive Strategy To Mimic Ditopic Ligands in Olefin Polymerization. Inorg. Chem. 2017, 56, 12448–12456. 10.1021/acs.inorgchem.7b01923. [DOI] [PubMed] [Google Scholar]
- Durini M.; Russotto E.; Pignataro L.; Reiser O.; Piarulli U. SupraBox: Chiral Supramolecular Oxazoline Ligands. Eur. J. Org. Chem. 2012, 2012, 5451–5461. 10.1002/ejoc.201200516. [DOI] [Google Scholar]
- Breit B.; Seiche W. Hydrogen Bonding as a Construction Element for Bidentate Donor Ligands in Homogeneous Catalysis: Regioselective Hydroformylation of Terminal Alkenes. J. Am. Chem. Soc. 2003, 125, 6608–6609. 10.1021/ja0348997. [DOI] [PubMed] [Google Scholar]
- Gellrich U.; Seiche W.; Keller M.; Breit B. Mechanistic Insights into a Supramolecular Self-Assembling Catalyst System: Evidence for Hydrogen Bonding during Rhodium-Catalyzed Hydroformylation. Angew. Chem.-Int. Ed. 2012, 51, 11033–11038. 10.1002/anie.201203768. [DOI] [PubMed] [Google Scholar]
- Gellrich U.; Huang J.; Seiche W.; Keller M.; Meuwly M.; Breit B. Ligand Self-Assembling through Complementary Hydrogen-Bonding in the Coordination Sphere of a Transition Metal Center: The 6-Diphenylphosphanylpyridin-2(1H)-one System. J. Am. Chem. Soc. 2011, 133, 964–975. 10.1021/ja108639e. [DOI] [PubMed] [Google Scholar]
- Kamer P. C. J.; van Leeuwen P. W. N. M.; Reek J. N. H. Wide Bite Angle Diphosphines: Xantphos Ligands in Transition Metal Complexes and Catalysis. Acc. Chem. Res. 2001, 34, 895–904. 10.1021/ar000060+. [DOI] [PubMed] [Google Scholar]
- Beierlein C. H.; Breit B.; Schmidt R. A. P.; Plattner D. A. Online Monitoring of Hydroformylation Intermediates by ESI-MS. Organometallics 2010, 29, 2521–2532. 10.1021/om100131t. [DOI] [Google Scholar]
- Seiche W.; Schuschkowski A.; Breit B. Bidentate Ligands by Self-Assembly Through Hydrogen Bonding: A General Room Temperature/Ambient Pressure Regioselective Hydroformylation of Terminal Alkenes. Adv. Synth. Catal. 2005, 347, 1488–1494. 10.1002/adsc.200505174. [DOI] [Google Scholar]
- Straub A. T.; Otto M.; Usui I.; Breit B. Room Temperature Ambient Pressure (RTAP)-Hydroformylation in Water Using a Self-Assembling Ligand. Adv. Synth. Catal. 2013, 355, 2071–2075. 10.1002/adsc.201300258. [DOI] [Google Scholar]
- Köpfer A.; Breit B. Rhodium-Catalyzed Hydroformylation of 1,1-Disubstituted Allenes Employing the Self-Assembling 6-DPPon System. Angew. Chem.-Int. Ed. 2015, 54, 6913–6917. 10.1002/anie.201502086. [DOI] [PubMed] [Google Scholar]
- Yu S. M.; Snavely W. K.; Chaudhari R. V.; Subramaniam B. Butadiene Hydroformylation to Adipaldehyde with Rh-based Catalysts: Insights into Ligand Effects. Mol. Catal. 2020, 484, 110721. 10.1016/j.mcat.2019.110721. [DOI] [Google Scholar]
- Agabekov V.; Seiche W.; Breit B. Rhodium-Catalyzed Hydroformylation of Alkynes Employing a Self-Assembling Ligand System. Chem. Sci. 2013, 4, 2418–2422. 10.1039/c3sc50725d. [DOI] [Google Scholar]
- Abillard O.; Breit B. Domino Hydroformylation/Enantioselective Cross-Aldol Addition. Adv. Synth. Catal. 2007, 349, 1891–1895. 10.1002/adsc.200700216. [DOI] [Google Scholar]
- Fuchs D.; Rousseau G.; Diab L.; Gellrich U.; Breit B. Tandem Rhodium-Catalyzed Hydroformylation-Hydrogenation of Alkenes by Employing a Cooperative Ligand System. Angew. Chem.-Int. Ed. 2012, 51, 2178–2182. 10.1002/anie.201108946. [DOI] [PubMed] [Google Scholar]
- Kemme S. T.; Smejkal T.; Breit B. Combined Transition-Metal- and Organocatalysis: An Atom Economic C3 Homologation of Alkenes to Carbonyl and Carboxylic Compounds. Chem.—Eur. J. 2010, 16, 3423–3433. 10.1002/chem.200903223. [DOI] [PubMed] [Google Scholar]
- Birkholz M.-N.; Dubrovina N. V.; Jiao H.; Michalik D.; Holz J.; Paciello R.; Breit B.; Boerner A. Enantioselective Hydrogenation with Self-Assembling Rhodium Phosphane Catalysts: Influence of Ligand Structure and Solvent. Chem.—Eur. J. 2007, 13, 5896–5907. 10.1002/chem.200601607. [DOI] [PubMed] [Google Scholar]
- Wenz K. M.; Leonhardt-Lutterbeck G.; Breit B. Inducing Axial Chirality in a Supramolecular Catalyst. Angew. Chem.-Int. Ed. 2018, 57, 5100–5104. 10.1002/anie.201801048. [DOI] [PubMed] [Google Scholar]
- Breit B.; Seiche W. Self-Assembly of Bidentate Ligands for Combinatorial Homogeneous Catalysis Based on an A-T Base-Pair Model. Angew. Chem.-Int. Ed. 2005, 44, 1640–1643. 10.1002/anie.200462499. [DOI] [PubMed] [Google Scholar]
- White D. F.; Boogaerts I.; Cole-Hamilton D. J.. Hydroformylation Process. Worldwide Patent: WO2011084268 A2, 2011.
- Chevallier F.; Breit B. Self-Assembled Bidentate Ligands for Ru-Catalyzed Anti-Markovnikov Hydration of Terminal Alkynes. Angew. Chem.-Int. Ed. 2006, 45, 1599–1602. 10.1002/anie.200503826. [DOI] [PubMed] [Google Scholar]
- Breit B.; Gellrich U.; Li T.; Lynam J. M.; Milner L. M.; Pridmore N. E.; Slattery J. M.; Whitwood A. C. Mechanistic Insight Into the Ruthenium-Catalysed anti-Markovnikov Hydration of Alkynes Using a Self-Assembled Complex: a Crucial Role for Ligand-Assisted Proton Shuttle Processes. Dalton Trans. 2014, 43, 11277–11285. 10.1039/C4DT00712C. [DOI] [PubMed] [Google Scholar]
- Usui L.; Schmidt S.; Keller M.; Breit B. Allylation of N-Heterocycles with Allylic Alcohols Employing Self-Assembling Palladium Phosphane Catalysts. Org. Lett. 2008, 10, 1207–1210. 10.1021/ol800073v. [DOI] [PubMed] [Google Scholar]
- Šmejkal T.; Breit B. Self-Assembled Bidentate Ligands for Ruthenium-Catalyzed Hydration of Nitriles. Organometallics 2007, 26, 2461–2464. 10.1021/om0611047. [DOI] [Google Scholar]
- de Greef M.; Breit B. Self-Assembled Bidentate Ligands for the Nickel-Catalyzed Hydrocyanation of Alkenes. Angew. Chem.-Int. Ed. 2009, 48, 551–554. 10.1002/anie.200805092. [DOI] [PubMed] [Google Scholar]
- Weis M.; Waloch C.; Seiche W.; Breit B. Self-Assembly of Bidentate Ligands for Combinatorial Homogeneous Catalysis: Asymmetric Rhodium-Catalyzed Hydrogenation. J. Am. Chem. Soc. 2006, 128, 4188–4189. 10.1021/ja058202o. [DOI] [PubMed] [Google Scholar]
- Birkholz M.-N.; Dubrovina N. V.; Shuklov I. A.; Holz J.; Paciello R.; Waloch C.; Breit B.; Boerner A. Enantioselective Pd-Catalyzed Allylic Amination with Self-Assembling and Non-Assembling Monodentate Phosphine Ligands. Tetrahedron Asymmetr. 2007, 18, 2055–2060. 10.1016/j.tetasy.2007.08.026. [DOI] [Google Scholar]
- Wieland J.; Breit B. A Combinatorial Approach to the Identification of Self-Assembled Ligands for Rhodium-Catalysed Asymmetric Hydrogenation. Nat. Chem. 2010, 2, 832–837. 10.1038/nchem.800. [DOI] [PubMed] [Google Scholar]
- Laungani A. C.; Slattery J. M.; Krossing I.; Breit B. Supramolecular Bidentate Ligands by Metal-Directed in situ Formation of Antiparallel β-Sheet Structures and Application in Asymmetric Catalysis. Chem.—Eur. J. 2008, 14, 4488–4502. 10.1002/chem.200800359. [DOI] [PubMed] [Google Scholar]
- van Leeuwen P. W. N. M.Homogeneous Catalysis: Understanding the Art; Kluwer Academic Publishers: Dordrecht, 2004. [Google Scholar]
- Davis H. J.; Phipps R. J. Harnessing Non-Covalent Interactions to Exert Control over Regioselectivity and Site-Selectivity in Catalytic Reactions. Chem. Sci. 2017, 8, 864–877. 10.1039/C6SC04157D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dydio P.; Reek J. N. H. Supramolecular Control of Selectivity in Transition-Metal Catalysis Through Substrate Preorganization. Chem. Sci. 2014, 5, 2135–2145. 10.1039/C3SC53505C. [DOI] [Google Scholar]
- Daugulis O.; Do H.-Q.; Shabashov D. Palladium- and Copper-Catalyzed Arylation of Carbon-Hydrogen Bonds. Acc. Chem. Res. 2009, 42, 1074–1086. 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Engle K. M.; Wang D.-H.; Yu J.-Q. Palladium(II)-Catalyzed C-H Activation/C-C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chem.-Int. Ed. 2009, 48, 5094–5115. 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leow D.; Li G.; Mei T.-S.; Yu J.-Q. Activation of Remote meta-C-H Bonds Assisted by an End-On Template. Nature 2012, 486, 518–522. 10.1038/nature11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.-H.; Engle K. M.; Shi B.-F.; Yu J.-Q. Ligand-Enabled Reactivity and Selectivity in a Synthetically Versatile Aryl C-H Olefination. Science 2010, 327, 315–319. 10.1126/science.1182512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang T. P.; Kagan H. B. The Asymmetric Synthesis of Hydratropic Acid and Amino-Acids by Homogeneous Catalytic Hydrogenation. J. Chem. Soc. D Chem. Commun. 1971, 7, 481–481. 10.1039/c29710000481. [DOI] [Google Scholar]
- Knowles W. S.; Sabacky M. J.; Vineyard B. D.; Weinkauff D. J. Asymmetric Hydrogenation with a Complex of Rhodium and a Chiral Bisphosphine. J. Am. Chem. Soc. 1975, 97, 2567–2568. 10.1021/ja00842a058. [DOI] [Google Scholar]
- Daubignard J.; Detz R. J.; Jans A. C. H.; de Bruin B.; Reek J. N. H. Rational Optimization of Supramolecular Catalysts for the Rhodium-Catalyzed Asymmetric Hydrogenation Reaction. Angew. Chem.-Int. Ed. 2017, 56, 13056–13060. 10.1002/anie.201707670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubignard J.; Lutz M.; Detz R. J.; de Bruin B.; Reek J. N. H. Origin of the Selectivity and Activity in the Rhodium-Catalyzed Asymmetric Hydrogenation Using Supramolecular Ligands. ACS Catal. 2019, 9, 7535–7547. 10.1021/acscatal.9b01809. [DOI] [Google Scholar]
- Daubignard J.; Detz R. J.; de Bruin B.; Reek J. N. H. Phosphine Oxide Based Supramolecular Ligands in the Rhodium-Catalyzed Asymmetric Hydrogenation. Organometallics 2019, 38, 3961–3969. 10.1021/acs.organomet.9b00484. [DOI] [Google Scholar]
- Zhao Q.; Li S.; Huang K.; Wang R.; Zhang X. A Novel Chiral Bisphosphine-Thiourea Ligand for Asymmetric Hydrogenation of β,β-Disubstituted Nitroalkenes. Org. Lett. 2013, 15, 4014–4017. 10.1021/ol401816y. [DOI] [PubMed] [Google Scholar]
- Li X.; You C.; Yang Y.; Yang Y.; Li P.; Gu G.; Chung L. W.; Lv H.; Zhang X. Rhodium-Catalyzed Asymmetric Hydrogenation of β-Cyanocinnamic Esters with the Assistance of a Single Hydrogen Bond in a Precise Position. Chem. Sci. 2018, 9, 1919–1924. 10.1039/C7SC04639A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G.; Yin C.; Yang X.; Li A.; Wang M.; Zhang X.; Dong X.-Q. Highly Chemo- and Enantioselective Rh-Catalyzed Hydrogenation of β-Sulfonyl-α,β-unsaturated Ketones: Access to Chiral γ-Ketosulfones. Org. Lett. 2021, 23, 19–24. 10.1021/acs.orglett.0c03517. [DOI] [PubMed] [Google Scholar]
- Wen J.; Jiang J.; Zhang X. Rhodium-Catalyzed Asymmetric Hydrogenation of α,β-Unsaturated Carbonyl Compounds via Thiourea Hydrogen Bonding. Org. Lett. 2016, 18, 4451–4453. 10.1021/acs.orglett.6b01812. [DOI] [PubMed] [Google Scholar]
- Octa-Smolin F.; Thiele M.; Yadav R.; Platzek A.; Clever G. H.; Niemeyer J. Chiral Receptors for Lysine Based on Covalently Linked Bis- and Tris-binaphthylphosphoric Acids. Org. Lett. 2018, 20, 6153–6156. 10.1021/acs.orglett.8b02619. [DOI] [PubMed] [Google Scholar]
- Yang J.; Li X.; You C.; Li S.; Guan Y.-Q.; Lv H.; Zhang X. Rhodium-Catalyzed Asymmetric Hydrogenation of Exocyclic α,β-Unsaturated Carbonyl Compounds. Org, Biomol. Chem. 2020, 18, 856–859. 10.1039/C9OB02536G. [DOI] [PubMed] [Google Scholar]
- Tao L.; Zhao Q. Y.; Zhang X. M.; Dong X. Q. Facile Access to Chiral 4-Substituted Chromanes Through Rh-Catalyzed Asymmetric Hydrogenation. Chin. Chem. Lett. 2020, 31, 1859–1862. 10.1016/j.cclet.2020.01.001. [DOI] [Google Scholar]
- Li P.; Hu X.; Dong X.-Q.; Zhang X. Rhodium/Bisphosphine-Thiourea-Catalyzed Enantioselective Hydrogenation of α,β-Unsaturated N-Acylpyrazoles. Chem. Commun. 2016, 52, 11677–11680. 10.1039/C6CC04987G. [DOI] [PubMed] [Google Scholar]
- Lang Q.; Gu G.; Cheng Y.; Yin Q.; Zhang X. Highly Enantioselective Synthesis of Chiral γ-Lactams by Rh-Catalyzed Asymmetric Hydrogenation. ASC Catal. 2018, 8, 4824–4828. 10.1021/acscatal.8b00827. [DOI] [Google Scholar]
- Wang L.; Xie Y.-B.; Huang N.-Y.; Yan J.-Y.; Hu W.-M.; Liu M.-G.; Ding M.-W. Catalytic aza-Wittig Reaction of Acid Anhydride for the Synthesis of 4H-Benzo[d][1,3]oxazin-4-ones and 4-Benzylidene-2-aryloxazol-5(4H)-ones. ACS Catal. 2016, 6, 4010–4016. 10.1021/acscatal.6b00165. [DOI] [Google Scholar]
- Liu G.; Zhang H.; Huang Y.; Han Z.; Liu G.; Liu Y.; Dong X.-Q.; Zhang X. Efficient synthesis of chiral 2,3-dihydro-benzo[b]thiophene 1,1-dioxides via Rh-catalyzed hydrogenation. Chem. Sci. 2019, 10, 2507–2512. 10.1039/C8SC05397A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z.; Guan Y.-Q.; Liu G.; Wang R.; Yin X.; Zhao Q.; Cong H.; Dong X.-Q.; Zhang X. Iridium-Catalyzed Asymmetric Hydrogenation of Tetrasubstituted α-Fluoro-β-enamino Esters: Efficient Access to Chiral α-Fluoro-β-amino Esters with Two Adjacent Tertiary Stereocenters. Org. Lett. 2018, 20, 6349–6353. 10.1021/acs.orglett.8b02503. [DOI] [PubMed] [Google Scholar]
- Chen C. Y.; Zhang Z. F.; Jin S. C.; Fan X. R.; Geng M. Y.; Zhou Y.; Wen S. W.; Wang X. R.; Chung L. W.; Dong X. Q.; Zhang X. M. Enzyme-Inspired Chiral Secondary-Phosphine-Oxide Ligand with Dual Noncovalent Interactions for Asymmetric Hydrogenation. Angew. Chem.-Int. Ed. 2017, 56, 6808–6812. 10.1002/anie.201701394. [DOI] [PubMed] [Google Scholar]
- Dydio P.; Rubay C.; Gadzikwa T.; Lutz M.; Reek J. N. H. Cofactor”-Controlled Enantioselective Catalysis. J. Am. Chem. Soc. 2011, 133, 17176–17179. 10.1021/ja208589c. [DOI] [PubMed] [Google Scholar]
- Franke R.; Selent D.; Boerner A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. 10.1021/cr3001803. [DOI] [PubMed] [Google Scholar]
- Gusevskaya E. V.; Jimenez-Pinto J.; Boerner A. Hydroformylation in the Realm of Scents. ChemCatChem. 2014, 6, 382–411. 10.1002/cctc.201300474. [DOI] [Google Scholar]
- Heck R. F.; Breslow D. S. The Reaction of Cobalt Hydrotetracarbonyl with Olefins. J. A. Chem. Soc. 1961, 83, 4023–4027. 10.1021/ja01480a017. [DOI] [Google Scholar]
- van Leeuwen P. W. N. M.; Claver C.. Rhodium Catalyzed Hydroformylation; Springer Science & Business Media, 2006. [Google Scholar]
- Dydio P.; Dzik W. I.; Lutz M.; de Bruin B.; Reek J. N. H. Remote Supramolecular Control of Catalyst Selectivity in the Hydroformylation of Alkenes. Angew. Chem.-Int. Ed. 2011, 50, 396–400. 10.1002/anie.201005173. [DOI] [PubMed] [Google Scholar]
- Šmejkal T.; Breit B. A Supramolecular Catalyst for Regioselective Hydroformylation of Unsaturated Carboxylic Acids. Angew. Chem.-Int. Ed. 2008, 47, 311–315. 10.1002/anie.200703192. [DOI] [PubMed] [Google Scholar]
- Dydio P.; Detz R. J.; Reek J. N. H. Precise Supramolecular Control of Selectivity in the Rh-Catalyzed Hydroformylation of Terminal and Internal Alkenes. J. Am. Chem. Soc. 2013, 135, 10817–10828. 10.1021/ja4046235. [DOI] [PubMed] [Google Scholar]
- Bai S.-T.; Sinha V.; Kluwer A. M.; Linnebank P. R.; Abiri Z.; de Bruin B.; Reek J. N. H. Rational Redesign of a Regioselective Hydroformylation Catalyst for 3-Butenoic Acid by Supramolecular Substrate Orientation. ChemCatChem. 2019, 11, 5322–5329. 10.1002/cctc.201900487. [DOI] [Google Scholar]
- Dydio P.; Reek J. N. H. Supramolecular Control of Selectivity in Hydroformylation of Vinyl Arenes: Easy Access to Valuable beta-Aldehyde Intermediates. Angew. Chem.-Int. Ed. 2013, 52, 3878–3882. 10.1002/anie.201209582. [DOI] [PubMed] [Google Scholar]
- Dydio P.; Detz R. J.; de Bruin B.; Reek J. N. H. Beyond Classical Reactivity Patterns: Hydroformylation of Vinyl and Allyl Arenes to Valuable beta- and gamma-Aldehyde Intermediates Using Supramolecular Catalysis. J. Am. Chem. Soc. 2014, 136, 8418–8429. 10.1021/ja503033q. [DOI] [PubMed] [Google Scholar]
- Dydio P.; Reek J. N. H. Scalable and Chromatography-free Synthesis of 2-(2-Formylalkyl) Arenecarboxylic Acid Derivatives Through the Supramolecularly Controlled Hydroformylation of Cinylarene-2-Carboxylic Acids. Nat. Protoc. 2014, 9, 1183–1191. 10.1038/nprot.2014.077. [DOI] [PubMed] [Google Scholar]
- Linnebank P. R.; Ferreira S. F.; Kluwer A. M.; Reek J. N. H. Regioselective Hydroformylation of Internal and Terminal Alkenes via Remote Supramolecular Control. Chem.—Eur. J. 2020, 26, 8214–8219. 10.1002/chem.202000620. [DOI] [PubMed] [Google Scholar]
- Šmejkal T.; Gribkov D.; Geier J.; Keller M.; Breit B. Transition-State Stabilization by a Secondary Substrate-Ligand Interaction: A New Design Principle for Highly Efficient Transition-Metal Catalysis. Chem.—Eur. J. 2010, 16, 2470–2478. 10.1002/chem.200902553. [DOI] [PubMed] [Google Scholar]
- Fang W. W.; Breit B. Tandem Regioselective Hydroformylation-Hydrogenation of Internal Alkynes Using a Supramolecular Catalyst. Angew. Chem.-Int. Ed. 2018, 57, 14817–14821. 10.1002/anie.201809073. [DOI] [PubMed] [Google Scholar]
- Smejkal T.; Breit B. A Supramolecular Catalyst for the Decarboxylative Hydroformylation of α,β-Unsaturated Carboxylic Acids. Angew. Chem.-Int. Ed. 2008, 47, 3946–3949. 10.1002/anie.200800956. [DOI] [PubMed] [Google Scholar]
- Diab L.; Šmejkal T.; Geier J.; Breit B. Supramolecular Catalyst for Aldehyde Hydrogenation and Tandem Hydroformylation-Hydrogenation. Angew. Chem.-Int. Ed. 2009, 48, 8022–8026. 10.1002/anie.200903620. [DOI] [PubMed] [Google Scholar]
- Diab L.; Gellrich U.; Breit B. Tandem Decarboxylative Hydroformylation-Hydrogenation Reaction of α,β-Unsaturated Carboxylic Acids Toward Aliphatic Alcohols Under Mild Conditions Employing a Supramolecular Catalyst System. Chem. Commun. 2013, 49, 9737–9739. 10.1039/c3cc45547e. [DOI] [PubMed] [Google Scholar]
- Fang W. W.; Bauer F.; Dong Y. X.; Breit B. A Domino Reaction for Generating β-Aryl Aldehydes from Alkynes by Substrate Recognition Catalysis. Nat. Commun. 2019, 10, 4868. 10.1038/s41467-019-12770-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Engle K. M.; Wang D.-H.; Yu J.-Q. Palladium(II)-Catalyzed C-H Activation/C-C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chem.-Int. Ed. 2009, 48, 5094–5115. 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree R. H.; Lei A. Introduction: CH Activation. Chem. Rev. 2017, 117, 8481–8482. 10.1021/acs.chemrev.7b00307. [DOI] [PubMed] [Google Scholar]
- Gandeepan P.; Mueller T.; Zell D.; Cera G.; Warratz S.; Ackermann L. 3d Transition Metals for C-H Activation. Chem. Rev. 2019, 119, 2192–2452. 10.1021/acs.chemrev.8b00507. [DOI] [PubMed] [Google Scholar]
- Godula K.; Sames D. C-H bond Functionalization in Complex Organic Synthesis. Science 2006, 312, 67–72. 10.1126/science.1114731. [DOI] [PubMed] [Google Scholar]
- Labinger J. A. Platinum-Catalyzed C-H Functionalization. Chem. Rev. 2017, 117, 8483–8496. 10.1021/acs.chemrev.6b00583. [DOI] [PubMed] [Google Scholar]
- Mkhalid I. A. I.; Barnard J. H.; Marder T. B.; Murphy J. M.; Hartwig J. F. C-H Activation for the Construction of C-B Bonds. Chem. Rev. 2010, 110, 890–931. 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- Wencel-Delord J.; Glorius F. C-H bond Activation Enables the Rapid Construction and Late-Stage Diversification of Functional Molecules. Nat. Chem. 2013, 5, 369–375. 10.1038/nchem.1607. [DOI] [PubMed] [Google Scholar]
- Haldar C.; Hogue M. E.; Bisht R.; Chattopadhyay B. Concept of Ir-Catalyzed C-H Bond Activation/Borylation by Noncovalent Interaction. Tetrahedron Lett. 2018, 59, 1269–1277. 10.1016/j.tetlet.2018.01.098. [DOI] [Google Scholar]
- Hartwig J. F. Regioselectivity of the Borylation of Alkanes and Arenes. Chem. Soc. Rev. 2011, 40, 1992–2002. 10.1039/c0cs00156b. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F. Borylation and Silylation of C-H Bonds: A Platform for Diverse C-H Bond Functionalizations. Acc. Chem. Res. 2012, 45, 864–873. 10.1021/ar200206a. [DOI] [PubMed] [Google Scholar]
- Ishiyama T.; Takagi J.; Kamon A.; Miyaura N. Palladium-Catalyzed Cross-Coupling Reaction of Bis(Pinacolato)Diboron with Vinyl Triflates β-Substituted by a Carbonyl Group: Efficient Synthesis of β-Boryl-α,β-Unsaturated Carbonyl Compounds and their Synthetic Utility. J. Organomet. Chem. 2003, 687, 284–290. 10.1016/S0022-328X(03)00611-9. [DOI] [Google Scholar]
- Ros A.; Fernandez R.; Lassaletta J. M. Functional Group Directed C-H Borylation. Chem. Soc. Rev. 2014, 43, 3229–3243. 10.1039/C3CS60418G. [DOI] [PubMed] [Google Scholar]
- Kuninobu Y.; Ida H.; Nishi M.; Kanai M. A meta-Selective C-H Borylation Directed by a Secondary Interaction Between Ligand and Substrate. Nat. Chem. 2015, 7, 712–717. 10.1038/nchem.2322. [DOI] [PubMed] [Google Scholar]
- Lu X.; Yoshigoe Y.; Ida H.; Nishi M.; Kanai M.; Kuninobu Y. Hydrogen Bond-Accelerated meta-Selective C-H Borylation of Aromatic Compounds and Expression of Functional Group and Substrate Specificities. ACS Catal. 2019, 9, 1705–1709. 10.1021/acscatal.8b05005. [DOI] [Google Scholar]
- Davis H. J.; Mihai M. T.; Phipps R. J. Ion Pair-Directed Regiocontrol in Transition-Metal Catalysis: A meta-Selective C-H Borylation of Aromatic Quaternary Ammonium Salts. J. Am. Chem. Soc. 2016, 138, 12759–12762. 10.1021/jacs.6b08164. [DOI] [PubMed] [Google Scholar]
- Bisht R.; Hoque M. E.; Chattopadhyay B. Amide Effects in C-H Activation: Noncovalent Interactions with L-Shaped Ligand for meta Borylation of Aromatic Amides. Angew. Chem.-Int. Ed. 2018, 57, 15762–15766. 10.1002/anie.201809929. [DOI] [PubMed] [Google Scholar]
- Hoque M. E.; Bisht R.; Haldar C.; Chattopadhyay B. Noncovalent Interactions in Ir-Catalyzed C-H Activation: L-Shaped Ligand for para-Selective Borylation of Aromatic Esters. J. Am. Chem. Soc. 2017, 139, 7745–7748. 10.1021/jacs.7b04490. [DOI] [PubMed] [Google Scholar]
- Bai S.-T.; Bheeter C. B.; Reek J. N. H. Hydrogen Bond Directed ortho-Selective C-H Borylation of Secondary Aromatic Amides. Angew. Chem.-Int. Ed. 2019, 58, 13039–13043. 10.1002/anie.201907366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes R. L.; Sato M.; Iwai T.; Suzuki K.; Maeda S.; Sawamura M. Asymmetric Remote C-H Borylation of Aliphatic Amides and Esters with a Modular Iridium Catalyst. Science 2020, 369, 970–974. 10.1126/science.abc8320. [DOI] [PubMed] [Google Scholar]
- Dzik W. I.; Xu X.; Zhang X. P.; Reek J. N. H.; de Bruin B. ‘Carbene Radicals’ in Co(II)-por-Catalyzed Olefin Cyclopropanation. J. Am. Chem. Soc. 2010, 132, 10891–10902. 10.1021/ja103768r. [DOI] [PubMed] [Google Scholar]
- Dzik W. I.; Zhang X. P.; de Bruin B. Redox Noninnocence of Carbene Ligands: Carbene Radicals in (Catalytic) C-C Bond Formation. Inorg. Chem. 2011, 50, 9896–9903. 10.1021/ic200043a. [DOI] [PubMed] [Google Scholar]
- Lu H.; Dzik W. I.; Xu X.; Wojtas L.; de Bruin B.; Zhang X. P. Experimental Evidence for Cobalt(III)-Carbene Radicals: Key Intermediates in Cobalt(II)-Based Metalloradical Cyclopropanation. J. Am. Chem. Soc. 2011, 133, 8518–8521. 10.1021/ja203434c. [DOI] [PubMed] [Google Scholar]
- Lyaskovskyy V.; Suarez A. I. O.; Lu H.; Jiang H.; Zhang X. P.; de Bruin B. Mechanism of Cobalt(II) Porphyrin-Catalyzed C-H Amination with Organic Azides: Radical Nature and H-Atom Abstraction Ability of the Key Cobalt(III)-Nitrene Intermediates. J. Am. Chem. Soc. 2011, 133, 12264–12273. 10.1021/ja204800a. [DOI] [PubMed] [Google Scholar]
- Suarez A. I. O.; Jiang H.; Zhang X. P.; de Bruin B. The Radical Mechanism of Cobalt(II) Porphyrin-Catalyzed Olefin Aziridination and the Importance of Cooperative H-Bonding. Dalton Trans. 2011, 40, 5697–5705. 10.1039/c1dt10027k. [DOI] [PubMed] [Google Scholar]
- van Leest N. P.; de Bruin B. Revisiting the Electronic Structure of Cobalt Porphyrin Nitrene and Carbene Radicals with NEVPT2-CASSCF Calculations: Doublet versus Quartet Ground States. Inorg. Chem. 2021, 60, 8380–8387. 10.1021/acs.inorgchem.1c00910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intrieri D.; Caselli A.; Gallo E. Cyclopropanation Reactions Mediated by Group 9 Metal Porphyrin Complexes. Eur. J. Inorg. Chem. 2011, 2011, 5071–5081. 10.1002/ejic.201100664. [DOI] [Google Scholar]
- Chirila A.; Das B. G.; Paul N. D.; De Bruin B. Diastereoselective Radical-Type Cyclopropanation of Electron-Deficient Alkenes Mediated by the Highly Active Cobalt(II) Tetramethyltetraaza[14]annulene Catalyst. ChemCatChem. 2017, 9, 1413–1421. 10.1002/cctc.201601568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Ruppel J. V.; Lu H.; Wojtas L.; Zhang X. P. Cobalt-Catalyzed, Asymmetric Cyclopropanation with Diazosulfones: Rigidification and Polarization of Ligand Chiral Environment via Hydrogen Bonding and Cyclization. J. Am. Chem. Soc. 2008, 130, 5042–5043. 10.1021/ja7106838. [DOI] [PubMed] [Google Scholar]
- Goswami M.; de Bruin B.; Dzik W. I. Difluorocarbene Transfer From a Cobalt Complex to an Electron-Deficient Alkene. Chem. Commun. 2017, 53, 4382–4385. 10.1039/C7CC01418J. [DOI] [PubMed] [Google Scholar]
- Huang L. Y.; Chen Y.; Gao G. Y.; Zhang X. P. Diastereoselective and Enantioselective Cyclopropanation of Alkenes Catalyzed by Cobalt Porphyrins. J. Org. Chem. 2003, 68, 8179–8184. 10.1021/jo035088o. [DOI] [PubMed] [Google Scholar]
- Lee W.-C. C.; Wang D.-S.; Zhang C.; Xie J.; Li B.; Zhang X. P. Asymmetric Radical Cyclopropanation of Dehydroaminocarboxylates: Stereoselective Synthesis of Cyclopropyl α-Amino Acids. Chem. 2021, 7, 1588–1601. 10.1016/j.chempr.2021.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul N. D.; Mandal S.; Otte M.; Cui X.; Zhang X. P.; de Bruin B. Metalloradical Approach to 2H-Chromenes. J. Am. Chem. Soc. 2014, 136, 1090–1096. 10.1021/ja4111336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar N.; Paul N. D.; Mandal S.; de Bruin B.; Wulff W. D. Catalytic Synthesis of 2H-Chromenes. ACS Catal. 2015, 5, 2329–2366. 10.1021/acscatal.5b00026. [DOI] [Google Scholar]
- Cui X.; Xu X.; Wojtas L.; Kim M. M.; Zhang X. P. Regioselective Synthesis of Multisubstituted Furans via Metalloradical Cyclization of Alkynes with α-Diazocarbonyls: Construction of Functionalized α-Oligofurans. J. Am. Chem. Soc. 2012, 134, 19981–19984. 10.1021/ja309446n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B. G.; Chirila A.; Tromp M.; Reek J. N. H.; de Bruin B. Co(III)-Carbene Radical Approach to Substituted 1H-Indenes. J. Am. Chem. Soc. 2016, 138, 8968–8975. 10.1021/jacs.6b05434. [DOI] [PubMed] [Google Scholar]
- Paul N. D.; Chirila A.; Lu H.; Zhang X. P.; de Bruin B. Carbene Radicals in Cobalt(II)-Porphyrin-Catalysed Carbene Carbonylation Reactions; A Catalytic Approach to Ketenes. Chem.—Eur. J. 2013, 19, 12953–12958. 10.1002/chem.201301731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirila A.; van Vliet K. M.; Paul N. D.; de Bruin B. Co(MeTAA) Metalloradical Catalytic Route to Ketenes via Carbonylation of Carbene Radicals. Eur. J. Inorg. Chem. 2018, 2018, 2251–2258. 10.1002/ejic.201800101. [DOI] [Google Scholar]
- te Grotenhuis C.; Das B. G.; Kuijpers P. F.; Hageman W.; Trouwborst M.; de Bruin B. Catalytic 1,2-Dihydronaphthalene and E-Aryl-Diene Synthesis via Co(III)-Carbene Radical and o-Quinodimethane Intermediates. Chem. Sci. 2017, 8, 8221–8230. 10.1039/C7SC03909C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankelma M.; Olivares A. M.; de Bruin B. Co(TPP)-Catalyzed Formation of Substituted Piperidines. Chem.—Eur. J. 2019, 25, 5658–5663. 10.1002/chem.201900587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Wen X.; Cui X.; Zhang X. P. Enantioselective Radical Cyclization for Construction of 5-Membered Ring Structures by Metalloradical C-H Alkylation. J. Am. Chem. Soc. 2018, 140, 4792–4796. 10.1021/jacs.8b01662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Grotenhuis C.; van den Heuvel N.; van der Vlugt J. I.; de Bruin B. Catalytic Dibenzocyclooctene Synthesis via Cobalt(III)-Carbene Radical and ortho-Quinodimethane Intermediates. Angew. Chem.-Int. Ed. 2018, 57, 140–145. 10.1002/anie.201711028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Lankelma M.; van der Vlugt J. I.; de Bruin B. Catalytic Synthesis of 8-Membered Ring Compounds via Cobalt(III)-Carbene Radicals. Angew. Chem.-Int. Ed. 2020, 59, 11073–11079. 10.1002/anie.202002674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Zhang X. P. Vitamin B-12 Derivatives as Natural Asymmetric Catalysts: Enantioselective Cyclopropanation of Alkenes. J. Org. Chem. 2004, 69, 2431–2435. 10.1021/jo049870f. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Gao G. Y.; Zhang X. P. Palladium-Mediated Synthesis of Novel meso-Chiral Porphyrins: Cobalt-Catalyzed Cyclopropanation. Tetrahedron Lett. 2005, 46, 4965–4969. 10.1016/j.tetlet.2005.05.089. [DOI] [Google Scholar]
- Fields K. B.; Engle J. T.; Sripothongnak S.; Kim C.; Zhang X. P.; Ziegler C. J. Cobalt Carbaporphyrin-Catalyzed Cyclopropanation. Chem. Commun. 2011, 47, 749–751. 10.1039/C0CC03894F. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Fields K. B.; Zhang X. P. Bromoporphyrins as Versatile Synthons for Modular Construction of Chiral Porphyrins: Cobalt-Catalyzed Highly Enantioselective and Diastereoselective Cyclopropanation. J. Am. Chem. Soc. 2004, 126, 14718–14719. 10.1021/ja044889l. [DOI] [PubMed] [Google Scholar]
- Zhu S.; Xu X.; Perman J. A.; Zhang X. P. A General and Efficient Cobalt(II)-Based Catalytic System for Highly Stereoselective Cyclopropanation of Alkenes with α-Cyanodiazoacetates. J. Am. Chem. Soc. 2010, 132, 12796–12799. 10.1021/ja1056246. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Zhang X. P. Trans Effect on Cobalt Porphyrin Catalyzed Asymmetric Cyclopropanation. Synthesis-Stuttgart 2006, 2006, 1697–1700. 10.1055/s-2006-926450. [DOI] [Google Scholar]
- Chen Y.; Zhang X. P. Asymmetric Cyclopropanation of Styrenes Catalyzed by Metal Complexes of D2-Symmetrical Chiral Porphyrin: Superiority of Cobalt over Iron. J. Org. Chem. 2007, 72, 5931–5934. 10.1021/jo070997p. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Ruppel J. V.; Zhang X. P. Cobalt-Catalyzed Symmetric Cyclopropanation of Electron-Deficient Olefins. J. Am. Chem. Soc. 2007, 129, 12074–12075. 10.1021/ja074613o. [DOI] [PubMed] [Google Scholar]
- Zhu S.; Perman J. A.; Zhang X. P. Acceptor/Acceptor-Substituted Diazo Reagents for Carbene Transfers: Cobalt-Catalyzed Asymmetric Z-Cyclopropanation of Alkenes with α-Nitrodiazoacetates. Angew. Chem.-Int. Ed. 2008, 47, 8460–8463. 10.1002/anie.200803857. [DOI] [PubMed] [Google Scholar]
- Ruppel J. V.; Gauthier T. J.; Snyder N. L.; Perman J. A.; Zhang X. P. Asymmetric Co(II)-Catalyzed Cyclopropanation with Succinimidyl Diazoacetate: General Synthesis of Chiral Cyclopropyl Carboxamides. Org. Lett. 2009, 11, 2273–2276. 10.1021/ol9005882. [DOI] [PubMed] [Google Scholar]
- Zhu S.; Cui X.; Zhang X. P. Ligand Effect on Cobalt(II)-Catalyzed Asymmetric Cyclopropanation with Diazosulfones - Approaching High Stereoselectivity through Modular Design of D2-Symmetric Chiral Porphyrins. Eur. J. Inorg. Chem. 2012, 2012, 430–434. 10.1002/ejic.201101023. [DOI] [Google Scholar]
- Jin C.; Decker A. M.; Huang X.-P.; Gilmour B. P.; Blough B. E.; Roth B. L.; Hu Y.; Gill J. B.; Zhang X. P. Synthesis, Pharmacological Characterization, and Structure-Activity Relationship Studies of Small Molecular Agonists for the Orphan GPR88 Receptor. ACS Chem. Neurosci. 2014, 5, 576–587. 10.1021/cn500082p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Wen X.; Cui X.; Wojtas L.; Zhang X. P. Asymmetric Radical Cyclopropanation of Alkenes with in situ Generated Donor-Substituted Diazo Reagents via Co(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc. 2017, 139, 1049–1052. 10.1021/jacs.6b11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Lang K.; Tao J.; Marshall M. K.; Cheng Q.; Cui X.; Wojtas L.; Zhang X. P. Next-Generation D2-Symmetric Chiral Porphyrins for Cobalt(II)-Based Metalloradical Catalysis: Catalyst Engineering by Distal Bridging. Angew. Chem.-Int. Ed. 2019, 58, 2670–2674. 10.1002/anie.201812379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Ke J.; Zhu Y.; Deb A.; Xu Y.; Zhang X. P. Asymmetric Radical Process for General Synthesis of Chiral Heteroaryl Cyclopropanes. J. Am. Chem. Soc. 2021, 143, 11121–11129. 10.1021/jacs.1c04655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X.; Xu X.; Lu H.; Zhu S.; Wojtas L.; Zhang X. P. Enantioselective Cyclopropenation of Alkynes with Acceptor/Acceptor-Substituted Diazo Reagents via Co(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc. 2011, 133, 3304–3307. 10.1021/ja111334j. [DOI] [PubMed] [Google Scholar]
- Xu X.; Zhu S.; Cui X.; Wojtas L.; Zhang X. P. Cobalt(II)-Catalyzed Asymmetric Olefin Cyclopropanation with α-Ketodiazoacetates. Angew. Chem.-Int. Ed. 2013, 52, 11857–11861. 10.1002/anie.201305883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Wang Y.; Cui X.; Wojtas L.; Zhang X. P. Metalloradical Activation of α-Formyldiazoacetates for the Catalytic Asymmetric Radical Cyclopropanation of Alkenes. Chem. Sci. 2017, 8, 4347–4351. 10.1039/C7SC00658F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppel J. V.; Cui X.; Xu X.; Zhang X. P. Stereoselective Intramolecular Cyclopropanation of α-Diazoacetates via Co(II)-Based Metalloradical Catalysis. Org. Chem. Front. 2014, 1, 515–520. 10.1039/C4QO00041B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Lu H.; Ruppel J. V.; Cui X.; de Mesa S. L.; Wojtas L.; Zhang X. P. Highly Asymmetric Intramolecular Cyclopropanation of Acceptor-Substituted Diazoacetates by Co(II)-Based Metalloradical Catalysis: Iterative Approach for Development of New-Generation Catalysts. J. Am. Chem. Soc. 2011, 133, 15292–15295. 10.1021/ja2062506. [DOI] [PubMed] [Google Scholar]
- Cui X.; Xu X.; Jin L.-M.; Wojtas L.; Zhang X. P. Stereoselective Radical C-H Alkylation With Acceptor/Acceptor-Substituted Diazo Reagents via Co(II)-Based Metalloradical Catalysis. Chem. Sci. 2015, 6, 1219–1224. 10.1039/C4SC02610A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karns A. S.; Goswami M.; de Bruin B. Catalytic Synthesis of Indolines by Hydrogen Atom Transfer to Cobalt(III)-Carbene Radicals. Chem.—Eur. J. 2018, 24, 5253–5258. 10.1002/chem.201704626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X.; Wang Y.; Zhang X. P. Enantioselective Radical Process for Synthesis of Chiral Indolines by Metalloradical Alkylation of Diverse C(sp3)-H bonds. Chem. Sci. 2018, 9, 5082–5086. 10.1039/C8SC01476K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Xu P.; Zhu Y.; Wang J.; Lee W.-C. C.; Zhang X. P. New Catalytic Radical Process Involving 1,4-Hydrogen Atom Abstraction: Asymmetric Construction of Cyclobutanones. J. Am. Chem. Soc. 2021, 143, 11670–11678. 10.1021/jacs.1c04968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Wang D.-S.; Lee W.-C. C.; McKillop A. M.; Zhang X. P. Controlling Enantioselectivity and Diastereoselectivity in Radical Cascade Cyclization for Construction of Bicyclic Structures. J. Am. Chem. Soc. 2021, 143, 11130–11140. 10.1021/jacs.1c04719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenini S.; Gallo E.; Penoni A.; Ragaini F.; Tollari S. Amination of Benzylic C-H Bonds by Aryl Azides Catalysed by Co(II)-Porphyrin Complexes. A New Reaction Leading to Secondary Amines and Imines. Chem. Commun. 2000, 2265–2266. 10.1039/b006136k. [DOI] [Google Scholar]
- Intrieri D.; Zardi P.; Caselli A.; Gallo E. Organic Azides: “Energetic Reagents’’ for the Intermolecular Amination of C-H Bonds. Chem. Commun. 2014, 50, 11440–11453. 10.1039/C4CC03016H. [DOI] [PubMed] [Google Scholar]
- Gao G.-Y.; Harden J. D.; Zhang X. P. Cobalt-Catalyzed Efficient Aziridination of Alkenes. Org. Lett. 2005, 7, 3191–3193. 10.1021/ol050896i. [DOI] [PubMed] [Google Scholar]
- Gao G.-Y.; Jones J. E.; Vyas R.; Harden J. D.; Zhang X. P. Cobalt-Catalyzed Aziridination with Diphenylphosphoryl Azide (DPPA): Direct Synthesis of N-Phosphorus-Substituted Aziridines from Alkenes. J. Org. Chem. 2006, 71, 6655–6658. 10.1021/jo0609226. [DOI] [PubMed] [Google Scholar]
- Ruppel J. V.; Jones J. E.; Huff C. A.; Kamble R. M.; Chen Y.; Zhang X. P. A Highly Effective Cobalt Catalyst for Olefin Aziridination With Azides: Hydrogen Bonding Guided Catalyst Design. Org. Lett. 2008, 10, 1995–1998. 10.1021/ol800588p. [DOI] [PubMed] [Google Scholar]
- Jones J. E.; Ruppel J. V.; Gao G.-Y.; Moore T. M.; Zhang X. P. Cobalt-Catalyzed Asymmetric Olefin Aziridination with Diphenylphosphoryl Azide. J. Org. Chem. 2008, 73, 7260–7265. 10.1021/jo801151x. [DOI] [PubMed] [Google Scholar]
- Subbarayan V.; Ruppel J. V.; Zhu S.; Perman J. A.; Zhang X. P. Highly Asymmetric Cobalt-Catalyzed Aziridination of Alkenes with Trichloroethoxysulfonyl Azide (TcesN3). Chem. Commun. 2009, 4266–4268. 10.1039/b905727g. [DOI] [PubMed] [Google Scholar]
- Jin L.-M.; Xu X.; Lu H.; Cui X.; Wojtas L.; Zhang X. P. Effective Synthesis of Chiral N-Fluoroaryl Aziridines through Enantioselective Aziridination of Alkenes with Fluoroaryl Azides. Angew. Chem.-Int. Ed. 2013, 52, 5309–5313. 10.1002/anie.201209599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J.; Jin L.-M.; Zhang X. P. Synthesis of Chiral N-Phosphoryl Aziridines Through Enantioselective Aziridination of Alkenes with Phosphoryl Azide via Co(II)-Based Metalloradical Catalysis. Beilstein J. Org. Chem. 2014, 10, 1282–1289. 10.3762/bjoc.10.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbarayan V.; Jin L.-M.; Cui X.; Zhang X. P. Room Temperature Activation of Aryloxysulfonyl Azides by Co(II)(TPP) for Selective Radical Aziridination of Alkenes via Metalloradical Catalysis. Tetrahedron Lett. 2015, 56, 3431–3434. 10.1016/j.tetlet.2015.01.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Lang K.; Jiang H.; Wojtas L.; Zhang X. P. Intramolecular 1,5-C(sp3)-H Radical Amination via Co(II)-Based Metalloradical Catalysis for Five-Membered Cyclic Sulfamides. Chem. Sci. 2016, 7, 6934–6939. 10.1039/C6SC02231F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H.; Lang K.; Lu H.; Wojtas L.; Zhang X. P. Intramolecular Radical Aziridination of Allylic Sulfamoyl Azides by Cobalt(II)-Based Metalloradical Catalysis: Effective Construction of Strained Heterobicyclic Structures. Angew. Chem.-Int. Ed. 2016, 55, 11604–11608. 10.1002/anie.201605238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H.; Lang K.; Lu H.; Wojtas L.; Zhang X. P. Asymmetric Radical Bicyclization of Allyl Azidoformates via Cobalt(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc. 2017, 139, 9164–9167. 10.1021/jacs.7b05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riart-Ferrer X.; Sang P.; Tao J.; Xu H.; Jin L.-M.; Lu H.; Cui X.; Wojtas L.; Zhang X. P. Metalloradical Activation of Carbonyl Azides for Enantioselective Radical Aziridination. Chem. 2021, 7, 1120–1134. 10.1016/j.chempr.2021.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami M.; Lyaskovskyy V.; Domingos S. R.; Buma W. J.; Woutersen S.; Troeppner O.; Ivanovic-Burmazovic I.; Lu H.; Cui X.; Zhang X. P.; Reijerse E. J.; DeBeer S.; van Schooneveld M. M.; Pfaff F. F.; Ray K.; de Bruin B. Characterization of Porphyrin-Co(III)-’Nitrene Radical’ Species Relevant in Catalytic Nitrene Transfer Reactions. J. Am. Chem. Soc. 2015, 137, 5468–5479. 10.1021/jacs.5b01197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppel J. V.; Kamble R. M.; Zhang X. P. Cobalt-Catalyzed Intramolecular C-H Amination with Arylsulfonyl Azides. Org. Lett. 2007, 9, 4889–4892. 10.1021/ol702265h. [DOI] [PubMed] [Google Scholar]
- Lu H.; Tao J.; Jones J. E.; Wojtas L.; Zhang X. P. Cobalt(II)-Catalyzed Intramolecular C-H Amination with Phosphoryl Azides: Formation of 6- and 7-Membered Cyclophosphoramidates. Org. Lett. 2010, 12, 1248–1251. 10.1021/ol100110z. [DOI] [PubMed] [Google Scholar]
- Lu H.; Hu Y.; Jiang H.; Wojtas L.; Zhang X. P. Stereoselective Radical Amination of Electron-Deficient C(sp3)-H Bonds by Co(II)-Based Metalloradical Catalysis: Direct Synthesis of α-Amino Acid Derivatives via α-C-H Amination. Org. Lett. 2012, 14, 5158–5161. 10.1021/ol302511f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Jiang H.; Hu Y.; Wojtas L.; Zhang X. P. Chemoselective Intramolecular Allylic C-H Amination versus C = C Aziridination Through Co(II)-Based Metalloradical Catalysis. Chem. Sci. 2011, 2, 2361–2366. 10.1039/c1sc00366f. [DOI] [Google Scholar]
- Li C.; Lang K.; Lu H.; Hu Y.; Cui X.; Wojtas L.; Zhang X. P. Catalytic Radical Process for Enantioselective Amination of C(sp3)-H Bonds. Angew. Chem.-Int. Ed. 2018, 57, 16837–16841. 10.1002/anie.201808923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang K.; Torker S.; Wojtas L.; Zhang X. P. Asymmetric Induction and Enantiodivergence in Catalytic Radical C-H Amination via Enantiodifferentiative H-Atom Abstraction and Stereoretentive Radical Substitution. J. Am. Chem. Soc. 2019, 141, 12388–12396. 10.1021/jacs.9b05850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Lang K.; Li C.; Gill J. B.; Kim I.; Lu H.; Fields K. B.; Marshall M.; Cheng Q.; Cui X.; Wojtas L.; Zhang X. P. Enantioselective Radical Construction of 5-Membered Cyclic Sulfonamides by Metalloradical C-H Amination. J. Am. Chem. Soc. 2019, 141, 18160–18169. 10.1021/jacs.9b08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang K.; Li C.; Kim I.; Zhang X. P. Enantioconvergent Amination of Racemic Tertiary C-H Bonds. J. Am. Chem. Soc. 2020, 142, 20902–20911. 10.1021/jacs.0c11103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers P. F.; Tiekink M. J.; Breukelaar W. B.; Broere D. L. J.; van Leest N. P.; van der Vlugt J. I.; Reek J. N. H.; de Bruin B. Cobalt-Porphyrin-Catalysed Intramolecular Ring-Closing C-H Amination of Aliphatic Azides: A Nitrene-Radical Approach to Saturated Heterocycles. Chem.—Eur. J. 2017, 23, 7945–7952. 10.1002/chem.201700358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harden J. D.; Ruppel J. V.; Gao G.-Y.; Zhang X. P. Cobalt-Catalyzed Intermolecular C-H Amination with Bromamine-T as Nitrene Source. Chem. Commun. 2007, 4644–4646. 10.1039/b710677g. [DOI] [PubMed] [Google Scholar]
- Lu H.; Subbarayan V.; Tao J.; Zhang X. P. Cobalt(II)-Catalyzed Intermolecular Benzylic C-H Amination with 2,2,2-Trichloroethoxycarbonyl Azide (TrocN3). Organometallics 2010, 29, 389–393. 10.1021/om900916g. [DOI] [Google Scholar]
- Jin L.-M.; Lu H.; Cui Y.; Lizardi C. L.; Arzua T. N.; Wojtas L.; Cui X.; Zhang X. P. Selective Radical Amination of Aldehydic C(sp2)-H Bonds with Fluoroaryl Azides via Co(II)-Based Metalloradical Catalysis: Synthesis of N-Fluoroaryl Amides From Aldehydes Under Neutral and Nonoxidative Conditions. Chem. Sci. 2014, 5, 2422–2427. 10.1039/C4SC00697F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L.-M.; Xu P.; Xie J.; Zhang X. P. Enantioselective Intermolecular Radical C-H Amination. J. Am. Chem. Soc. 2020, 142, 20828–20836. 10.1021/jacs.0c10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S.; Incarvito C. D.; Crabtree R. H.; Brudvig G. W. Molecular Recognition in the Selective Oxygenation of Saturated C-H Bonds by a Dimanganese Catalyst. Science 2006, 312, 1941–1943. 10.1126/science.1127899. [DOI] [PubMed] [Google Scholar]
- Das S.; Brudvig G. W.; Crabtree R. H. High Turnover Remote Catalytic Oxygenation of Alkyl Groups: How Steric Exclusion of Unbound Substrate Contributes to High Molecular Recognition Selectivity. J. Am. Chem. Soc. 2008, 130, 1628–1637. 10.1021/ja076039m. [DOI] [PubMed] [Google Scholar]
- Frost J. R.; Huber S. M.; Breitenlechner S.; Bannwarth C.; Bach T. Enantiotopos-Selective C-H Oxygenation Catalyzed by a Supramolecular Ruthenium Complex. Angew. Chem.-Int. Ed. 2014, 54, 691–695. 10.1002/anie.201409224. [DOI] [PubMed] [Google Scholar]
- Burg F.; Gicquel M.; Breitenlechner S.; Pothig A.; Bach T. Site- and Enantioselective C-H Oxygenation Catalyzed by a Chiral Manganese Porphyrin Complex with a Remote Binding Site. Angew. Chem.-Int. Ed. 2018, 57, 2953–2957. 10.1002/anie.201712340. [DOI] [PubMed] [Google Scholar]
- Hoke T.; Herdtweck E.; Bach T. Hydrogen-Bond Mediated Regio- and Enantioselectivity in a C-H Amination Reaction Catalysed by a Supramolecular Rh(II) Complex. Chem. Commun. 2013, 49, 8009–8011. 10.1039/c3cc44197k. [DOI] [PubMed] [Google Scholar]
- Zhong F. R.; Bach T. Enantioselective Construction of 2,3-Dihydrofuro [2,3-b]quinolines through Supramolecular Hydrogen Bonding Interactions. Chem.—Eur. J. 2014, 20, 13522–13526. 10.1002/chem.201404617. [DOI] [PubMed] [Google Scholar]
- Olivo G.; Farinelli G.; Barbieri A.; Lanzalunga O.; Di Stefano S.; Costas M. Supramolecular Recognition Allows Remote, Site-Selective C-H Oxidation of Methylenic Sites in Linear Amines. Angew. Chem.-Int. Ed. 2017, 56, 16347–16351. 10.1002/anie.201709280. [DOI] [PubMed] [Google Scholar]
- Prentice C.; Morrisson J.; Smith A. D.; Zysman-Colman E. Recent Developments in Enantioselective Photocatalysis. Beilstein J. Org. Chem. 2020, 16, 2363–2441. 10.3762/bjoc.16.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strieth-Kalthoff F.; Glorius F. Triplet Energy Transfer Photocatalysis: Unlocking the Next Level. Chem. 2020, 6, 1888–1903. 10.1016/j.chempr.2020.07.010. [DOI] [Google Scholar]
- Brimioulle R.; Lenhart D.; Maturi M. M.; Bach T. Enantioselective Catalysis of Photochemical Reactions. Angew. Chem.-Int. Ed. 2015, 54, 3872–3890. 10.1002/anie.201411409. [DOI] [PubMed] [Google Scholar]
- Huang X.; Meggers E. Asymmetric Photocatalysis with Bis-Cyclometalated Rhodium Complexes. Acc. Chem. Res. 2019, 52, 833–847. 10.1021/acs.accounts.9b00028. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Meggers E. Stereogenic-Only-at-Metal Asymmetric Catalysts. Chem.-Asian. J. 2017, 12, 2335–2342. 10.1002/asia.201700739. [DOI] [PubMed] [Google Scholar]
- Cauble D. F.; Lynch V.; Krische M. J. Studies on the Enantioselective Catalysis of Photochemically Promoted Transformations: “Sensitizing Receptors” as Chiral Catalysts. J. Org. Chem. 2003, 68, 15–21. 10.1021/jo020630e. [DOI] [PubMed] [Google Scholar]
- Burg F.; Bach T. Lactam Hydrogen Bonds as Control Elements in Enantioselective Transition-Metal-Catalyzed and Photochemical Reactions. J. Org. Chem. 2019, 84, 8815–8836. 10.1021/acs.joc.9b01299. [DOI] [PubMed] [Google Scholar]
- Großkopf J.; Kratz T.; Rigotti T.; Bach T. Enantioselective Photochemical Reactions Enabled by Triplet Energy Transfer. Chem. Rev. 2022, 122, 1626. 10.1021/acs.chemrev.1c00272. [DOI] [PubMed] [Google Scholar]
- Müller C.; Bauer A.; Bach T. Light-Driven Enantioselective Organocatalysis. Angew. Chem.-Int. Ed. 2009, 48, 6640–6642. 10.1002/anie.200901603. [DOI] [PubMed] [Google Scholar]
- Cavaleri J. J.; Prater K.; Bowman R. M. An Investigation of the Solvent Dependence on the Ultrafast Intersystem Crossing Kinetics of Xanthone. Chem. Phys. Lett. 1996, 259, 495–502. 10.1016/0009-2614(96)00782-8. [DOI] [Google Scholar]
- Müller C.; Bauer A.; Maturi M. M.; Cuquerella M. C.; Miranda M. A.; Bach T. Enantioselective Intramolecular [2 + 2] Photocycloaddition Reactions of 4-Substituted Quinolones Catalyzed by a Chiral Sensitizer with a Hydrogen-Bonding Motif. J. Am. Chem. Soc. 2011, 133, 16689–16697. 10.1021/ja207480q. [DOI] [PubMed] [Google Scholar]
- Alonso R.; Bach T. A Chiral Thioxanthone as an Organocatalyst for Enantioselective [2 + 2] Photocycloaddition Reactions Induced by Visible Light. Angew. Chem.-Int. Ed. 2014, 53, 4368–4371. 10.1002/anie.201310997. [DOI] [PubMed] [Google Scholar]
- Maturi M. M.; Bach T. Enantioselective Catalysis of the Intermolecular [2 + 2] Photocycloaddition between 2-Pyridones and Acetylenedicarboxylates. Angew. Chem.-Int. Ed. 2014, 53, 7661–7664. 10.1002/anie.201403885. [DOI] [PubMed] [Google Scholar]
- Plaza M.; Jandl C.; Bach T. Photochemical Deracemization of Allenes and Subsequent Chirality Transfer. Angew. Chem.-Int. Ed. 2020, 59, 12785–12788. 10.1002/anie.202004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skubi K. L.; Kidd J. B.; Jung H.; Guzei I. A.; Baik M.-H.; Yoon T. P. Enantioselective Excited-State Photoreactions Controlled by a Chiral Hydrogen-Bonding Iridium Sensitizer. J. Am. Chem. Soc. 2017, 139, 17186–17192. 10.1021/jacs.7b10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J.; Swords W. B.; Jung H.; Skubi K. L.; Kidd J. B.; Meyer G. J.; Baik M.-H.; Yoon T. P. Enantioselective Intermolecular Excited-State Photoreactions Using a Chiral Ir Triplet Sensitizer: Separating Association from Energy Transfer in Asymmetric Photocatalysis. J. Am. Chem. Soc. 2019, 141, 13625–13634. 10.1021/jacs.9b06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maturi M. M.; Wenninger M.; Alonso R.; Bauer A.; Poethig A.; Riedle E.; Bach T. Intramolecular 2 + 2 Photocycloaddition of 3-and 4-(But-3-enyl)oxyquinolones: Influence of the Alkene Substitution Pattern, Photophysical Studies, and Enantioselective Catalysis by a Chiral Sensitizer. Chem.—Eur. J. 2013, 19, 7461–7472. 10.1002/chem.201300203. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Crawley M. L. Asymmetric Transition-Metal-Catalyzed Allylic Alkylations: Applications in Total Synthesis. Chem. Rev. 2003, 103, 2921–2943. 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]
- Pamies O.; Margalef J.; Canellas S.; James J.; Judge E.; Guiry P. J.; Moberg C.; Backvall J. E.; Pfaltz A.; Pericas M. A.; Dieguez M. Recent Advances in Enantioselective Pd-Catalyzed Allylic Substitution: From Design to Applications. Chem. Rev. 2021, 121, 4373–4505. 10.1021/acs.chemrev.0c00736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L.; Cao P.; Xing J. W.; Lou Y. Z.; Jiang L. Y.; Li L. C.; Liao J. Hydrogen-Bond-Promoted Palladium Catalysis: Allylic Alkylation of Indoles with Unsymmetrical 1,3-Disubstituted Allyl Acetates Using Chiral Bis(sulfoxide) Phosphine Ligands. Angew. Chem.-Int. Ed. 2013, 52, 4207–4211. 10.1002/anie.201209485. [DOI] [PubMed] [Google Scholar]
- Hayashi T.; Yamamoto A.; Hagihara T.; Ito Y. Modification of Optically Active Ferrocenylphosphine Ligands for Palladium-Catalyzed Asymmetric Allylic Alkylation. Tetrahedron Lett. 1986, 27, 191–194. 10.1016/S0040-4039(00)83974-X. [DOI] [Google Scholar]
- Mahadik G. S.; Knott S. A.; Szczepura L. F.; Peters S. J.; Standard J. M.; Hitchcock S. R. β-Amino Alcohol Derived β-Hydroxy- and β-(o-Diphenylphosphino)benzoyloxy(o-diphenylphosphino)benzamides: An Ester-Amide Ligand Structural Model for the Palladium-Catalyzed Allylic Alkylation Reaction. J. Org. Chem. 2009, 74, 8164–8173. 10.1021/jo9016474. [DOI] [PubMed] [Google Scholar]
- Xu J. X.; Ye F.; Bai X. F.; Cui Y. M.; Xu Z.; Zheng Z. J.; Xu L. W. A Mechanistic Study on Multifunctional Fei-Phos ligand-Controlled Asymmetric Palladium-Catalyzed Allylic Substitutions. RSC Adv. 2016, 6, 70624–70631. 10.1039/C6RA15665G. [DOI] [Google Scholar]
- Fanourakis A.; Docherty P. J.; Chuentragool P.; Phipps R. J. Recent Developments in Enantioselective Transition Metal Catalysis Featuring Attractive Noncovalent Interactions between Ligand and Substrate. ACS Catal. 2020, 10, 10672–10714. 10.1021/acscatal.0c02957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Han Z.; Gu G.; Dong X.-Q.; Zhang X. Enantioselective Synthesis of Chiral 3-Substituted-3-silylpropionic Esters via Rhodium/Bisphosphine-Thiourea-Catalyzed Asymmetric Hydrogenation. Adv. Synth. Catal. 2017, 359, 2585–2589. 10.1002/adsc.201700355. [DOI] [Google Scholar]
- Liu G.; Li A.; Qin X.; Han Z.; Dong X.-Q.; Zhang X. Efficient Access to Chiral β-Borylated Carboxylic Esters via Rh- Catalyzed Hydrogenation. Adv. Synth. Catal. 2019, 361, 2844–2848. 10.1002/adsc.201900161. [DOI] [Google Scholar]