

Abstract
Viral engineered chimeric antigen receptor (CAR) T cell therapies are potent, targeted cancer immunotherapies, but their permanent CAR expression can lead to severe adverse effects. Nonviral messenger RNA (mRNA) CAR T cells are being explored to overcome these drawbacks, but electroporation, the most common T cell transfection method, is limited by cytotoxicity. As a potentially safer nonviral delivery strategy, here, sequential libraries of ionizable lipid nanoparticle (LNP) formulations with varied excipient compositions were screened in comparison to a standard formulation for improved mRNA delivery to T cells with low cytotoxicity, revealing B10 as the top formulation with a 3-fold increase in mRNA delivery. When compared to electroporation in primary human T cells, B10 LNPs induced comparable CAR expression with reduced cytotoxicity while demonstrating potent cancer cell killing. These results demonstrate the impact of excipient optimization on LNP performance and support B10 LNPs as a potent mRNA delivery platform for T cell engineering.
Keywords: lipid nanoparticles, mRNA delivery, CAR T, T cell engineering
Graphical Abstract
In March 2021, Abecma became the fifth chimeric antigen receptor (CAR) T cell therapy to receive FDA approval within the past five years.1 The approved therapies include CD19 CAR T cells for the treatment of relapsed or refractory acute lymphoblastic leukemia (ALL) and B cell lymphomas and BCMA CAR T cells for the treatment of multiple myeloma.1–4 The continued clinical success of these therapies in treating hematological cancers has incited the investigation of CAR T cells to treat a variety of other cancer types and diseases including non-small cell lung cancer,5 glioblastoma,6,7 and HIV.8 To generate these autologous CAR T cell therapies, current methods require ex vivo T cell engineering, a process in which patient T cells are harvested, modified to express CAR, and reinfused into the patient. The transmembrane CAR constructs then allow T cells to specifically bind and eliminate the target cell population.9
Despite the success of CAR T cell therapy in inducing durable remissions, there are limitations to its clinical application due, in part, to its severe side effects. Nearly 70% of adults receiving the therapy experience immediate adverse reactions associated with their own immune response,10 which can include macrophage activation syndrome, neurotoxicity, and cytokine release syndrome.11–13 Though IL-6 blockers can mitigate these reactions, long-term effects, such as B cell aplasia and hypogammaglobulinemia, require additional intervention via intravenous immune globulin infusions.13,14 Further, explorations of new CAR constructs to target cell populations beyond B cells have also observed adverse events related to inflammation,15,16 emphasizing the need for safer CAR T cell options for early clinical investigation.
The longevity of these adverse effects stems, in part, from the continuous CAR expression induced by viral transduction. This permanent expression relies on genomic integration, leading to the risk of insertional mutagenesis, though this may be beneficial for CAR T cell proliferation in some instances, such as integration within the TET2 gene,17,18 and escalating the consequences of transducing off-target cell types ex vivo.13,19 In addition to safety concerns, viral transduction presents a number of limitations for CAR T cell manufacturing and in vivo translation including limited cargo capacity,20,21 elaborate transduction protocols, high cost, and in vivo immunogenicity.22,23 Thus, the viral transduction used in CAR T cell manufacturing contributes to adverse effects, imposes its own risks, and necessitates ex vivo T cell engineering, motivating investigations into alternative production strategies to generate safer, less expensive CAR T cells.
Previous investigations have found that utilizing messenger RNA (mRNA) to induce CAR expression offers a number of potential advantages over viral transduction. As mRNA is translated without genomic integration, it allows for transient CAR expression, mitigating the risks associated with long-term CAR T cell activity.24 Additionally, in vitro transcription allows customized CAR mRNA to be engineered for potent transfection and translation that results in dose-dependent CAR expression.25–28 In combination, administering transient mRNA CAR T cells and modulating their level of CAR expression provides the opportunity to optimize CAR T cell potency while minimizing adverse effects, but it often necessitates repeated dosing to achieve therapeutic efficacy.24,28 However, the potential benefits of mRNA CAR T cell therapies have led to their evaluation in various cancers including melanoma, Hodgkin’s lymphoma, and ALL, and they have been shown to reduce short-term disease burden as effectively as virally engineered CAR T ceHs.24,28,29 Further, mRNA CAR T cell therapy has been evaluated in numerous clinical trials for cancers including breast cancer,30 pancreatic ductal adenocarcinoma,31 and Hodgkin’s lymphoma.32
To produce mRNA CAR T cells, delivery strategies are required for rapidly degradable, negatively charged mRNA to enter the T cell cytosol. Currently, the most common technique for ex vivo transfection is electroporation (EP), which uses electric pulses to permeabilize cell membranes for potent mRNA delivery.24,26,33 However, this permeabilization risks loss of cytoplasmic content, altered gene expression, and high cytotoxicity without the guarantee of consistent membrane disruption across cells.34–36 One promising alternative to EP is the use of lipid- or polymer-based nanoparticles (NPs) for mRNA delivery.37,38 NPs can mitigate cytotoxicity, stabilize mRNA cargo, and enhance intracellular delivery while avoiding specialized equipment.39–41 Both polymeric NPs and ionizable lipid NPs (LNPs) have demonstrated mRNA delivery in a variety of cell types ex vivo and in vivo, including T cells, with some investigations confirming their reduced cytotoxicity compared to EP.42–49 One of these NP platforms47 was successfully used for CAR mRNA delivery to T cells and is undergoing further preclinical development via the spinout company Tidal Therapeutics, which was acquired by Sanofi.50 However, LNP platforms are more clinically advanced than their polymeric counterparts in terms of mRNA delivery, with the FDA approval of Alnylam’s Onpattro51 and emergency use authorization of LNPs for Moderna and Pfizer/BioNTech’s COVID-19 mRNA vaccines, making them a potentially promising platform for mRNA delivery to T cells.52,53
In addition to nucleic acid cargo, LNPs are typically composed of four main components: an ionizable lipid, cholesterol, phospholipid, and lipid-anchored PEG (Figure 1A), that can be varied to produce distinct formulations with differing physicochemical properties.43,54,55 The ionizable lipid allows LNPs to shift from a neutral to positive charge in acidic environments, facilitating endosomal escape, which is required for intracellular delivery38,56,57 and aiding in LNP formation by allowing the ionizable lipid to complex with negatively charged mRNA for high encapsulation efficiency.58 The remaining excipients also play vital roles, with cholesterol stabilizing the LNP while enabling membrane fusion, the phospholipid providing structural support while promoting endosomal escape, and PEG reducing aggregation and nonspecific endocytosis.42,43,59,60 The molar ratio at which these components are combined can be optimized for specific applications, to encapsulate desired cargos, interact with target cells, or alter biodistribution and protein corona formation.43,54,61–65
Figure 1.
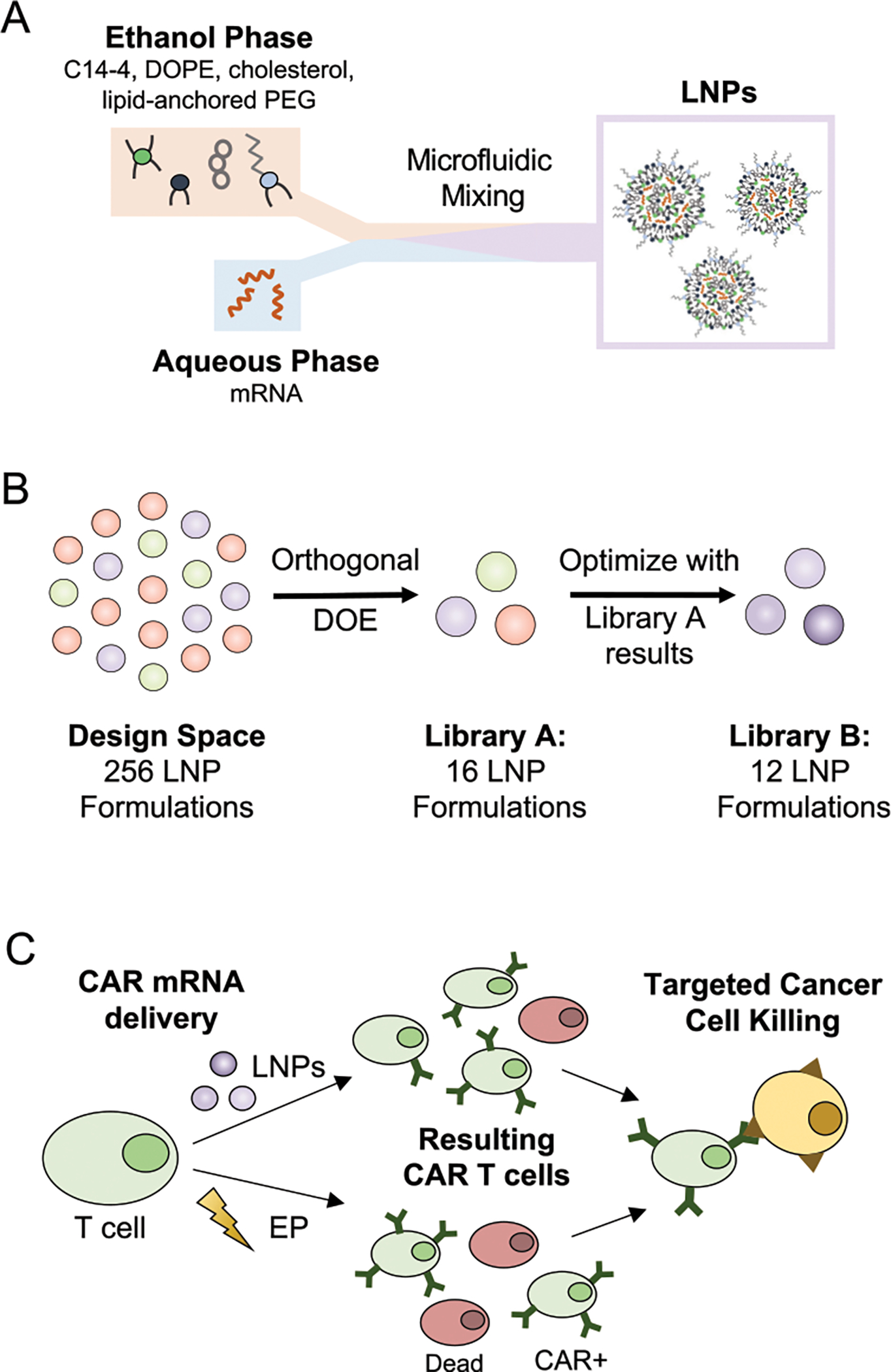
(A) Schematic of LNP synthesis including the components used to make LNPs via microfluidic mixing and the expected resulting structure. (B) Visualization of the design process used to generate libraries A and B with library A resulting from an orthogonal DOE screening a wide range of excipient molar ratios, and library B examining more formulations within a narrowed range of excipient ratios based on the results from the library A screen. (C) Schematic of the CAR T cell production utilizing either LNPs or EP for mRNA delivery to T cells. The treated T cell populations may differ in viability and CAR potency depending on the transfection method, but both are able to generate functional CAR T cells to induce targeted cancer cell killing.
In previous work, we screened a library of novel ionizable lipids in LNPs with set excipient molar ratios and identified a top-performing structure, C14–4, that achieved potent mRNA delivery to T cells with reduced cytotoxicity compared to EP.45 However, this screen used a standard excipient molar ratio for mRNA delivery.42,43 Here, we used an orthogonal design of experiments (DOE) methodology to identify novel C14–4 LNP formulations for T cell delivery via sequential library screens of C14–4 LNPs with varied excipient molar ratios (Figure 1B).54,66 This allowed a design space of 256 potential LNP formulations, generated using four molar ratios each of C14–4, cholesterol, phospholipid, and lipid-anchored PEG, to be evaluated using 16 representative LNP formulations. Library A was screened for mRNA delivery and cytotoxicity in Jurkat cells, an immortalized T cell line, and compared to the standard formulation, S2. The results indicated that increasing C14–4 and phospholipid while decreasing cholesterol led to improved mRNA delivery. These trends were explored with increased resolution, observing more formulations within a narrowed range of excipient molar ratios, in library B, which revealed B10 as the top-performing formulation. Further, to illustrate the translatability of the improved LNP platform, B10 LNPs were used to deliver CAR mRNA to primary human T cells and demonstrated lower cytotoxicity than EP (Figure 1C). The CAR T cells produced by B10 LNPs, EP, or lentivirus were compared in a killing assay with ALL cells, and all groups demonstrated potent cancer cell killing. Thus, B10 LNPs were validated as a promising platform for the ex vivo engineering of mRNA CAR T cells.
Design, Characterization, and Evaluation of LNP Libraries.
In this study, LNPs were generated using varied excipient molar ratios to investigate the impact of LNP formulation on mRNA delivery to T cells. Throughout this investigation, LNPs incorporated the C14–4 ionizable lipid as it previously demonstrated potent, nontoxic mRNA delivery to T cells.45 C14–4 was synthesized (Figure S1) and combined in ethanol with cholesterol, the phospholipid dioleoylphosphatidylethoanolamine (DOPE), and lipid-anchored PEG. To formulate LNPs, this ethanol phase and an aqueous phase containing mRNA were mixed using a microfluidic mixing device (Figure 1A).67,68 To generate a library of LNPs with varied excipient molar ratios, each ethanol phase was adjusted to contain the desired lipid components.
Previous work has optimized the ratios of ionizable lipid, phospholipid, cholesterol, and PEG in LNPs for mRNA delivery, with general trends indicating less ionizable lipid and more cholesterol and PEG may be beneficial.43,61 However, these investigations did not explore the impact of excipient ratios on delivery to T cells. This previously optimized formulation for mRNA delivery, a molar ratio of 35 ionizable lipid:46.5 cholesterol:16 DOPE:2.5 PEG, was used as a standard formulation for comparison, S2, and in combination with previous investigations exploring excipient ratios,54,65,66 it informed the initial range of excipient ratios explored in library A. Specifically, library A contained 16 formulations (Table 1) selected via an established orthogonal DOE that allowed for the four excipients to be evaluated at four molar ratios each using only 16 representative formulations (Figure 1B).54,66
Table 1.
Excipient Molar Ratios of the LNP Formulations in Libraries A and B
| Library A | ||||
|
| ||||
| LNP Name | C14–4 | DOPE | Chol | PEG |
| A1 | 15 | 10 | 20 | 0.5 |
| A2 | 15 | 20 | 25 | 1 |
| A3 | 15 | 30 | 30 | 2.5 |
| A4 | 15 | 40 | 35 | 5 |
| A5 | 25 | 10 | 25 | 2.5 |
| A6 | 25 | 20 | 20 | 5 |
| A7 | 25 | 30 | 35 | 0.5 |
| A8 | 25 | 40 | 30 | 1 |
| A9 | 35 | 10 | 30 | 5 |
| A10 | 35 | 20 | 35 | 2.5 |
| A11 | 35 | 30 | 20 | 1 |
| A12 | 35 | 40 | 25 | 0.5 |
| A13 | 45 | 10 | 35 | 1 |
| A14 | 45 | 20 | 30 | 0.5 |
| A15 | 45 | 30 | 25 | 5 |
| A16 | 45 | 40 | 20 | 2.5 |
| S2 | 35 | 16 | 46.5 | 2.5 |
| Library B | ||||
|
| ||||
| name | C14–4 | DOPE | Chol | PEG |
| B1 | 35 | 30 | 20 | 2.5 |
| B2 | 35 | 35 | 20 | 2.5 |
| B3 | 35 | 40 | 20 | 2.5 |
| B4 | 35 | 30 | 25 | 2.5 |
| B5 | 35 | 35 | 25 | 2.5 |
| B6 | 35 | 40 | 25 | 2.5 |
| B7 | 40 | 30 | 20 | 2.5 |
| B8 | 40 | 35 | 20 | 2.5 |
| B9 | 40 | 40 | 20 | 2.5 |
| B10 | 40 | 30 | 25 | 2.5 |
| B11 | 40 | 35 | 25 | 2.5 |
| B12 | 40 | 40 | 25 | 2.5 |
After formulation, all 16 LNPs were characterized by size, mRNA concentration, and pKa (Table S1). Across library A, the LNP sizes (z-average diameter) ranged from 57 to 151 nm with PDIs below 0.3, confirming homogeneous LNP formation, and mRNA concentrations ranged from 3 to 45 ng/μL. The pKa values of the LNPs, the pH at which they are 50% protonated, reflect their ability to change charge in acidic environments to facilitate endosomal escape.44,69,70 Here, the pKa values from library A ranged from 4.90 to 6.47, confirming their ionizable properties (pKa< 7.2) and indicating that these excipient ratios minimally impacted this property.
To evaluate the LNPs in library A for mRNA delivery to T cells, luciferase mRNA was used as a model cargo.43,44,71 Upon intracellular delivery, the mRNA is translated into luciferase that reacts with luciferin reagent to generate a luminescent signal, allowing luminescence as a measure of functional mRNA delivery.43,44,71 Here, the library A formulations encapsulating luciferase mRNA were used to treat Jurkat cells, an immortalized human T cell line. After 48 h, mRNA delivery was measured via luminescence, and Jurkat cell viability was quantified (Figure 2A). To assess mRNA delivery, the LNPs in library A were compared to the established S2 formulation, revealing four promising formulations, A10, A11, A12, and A16, but none with significantly enhanced mRNA delivery. To evaluate viability, the LNP formulations were normalized to untreated cells and compared to S2, which determined that only A12 significantly lowered viability (Figure 2B). As the goal of this screen was to identify LNP formulations that significantly improved mRNA delivery without decreasing viability, no formulations were considered hits, but formulations A10, A11, and A16 were noted as resulting in higher mean luminescence values without decreasing viability.
Figure 2.
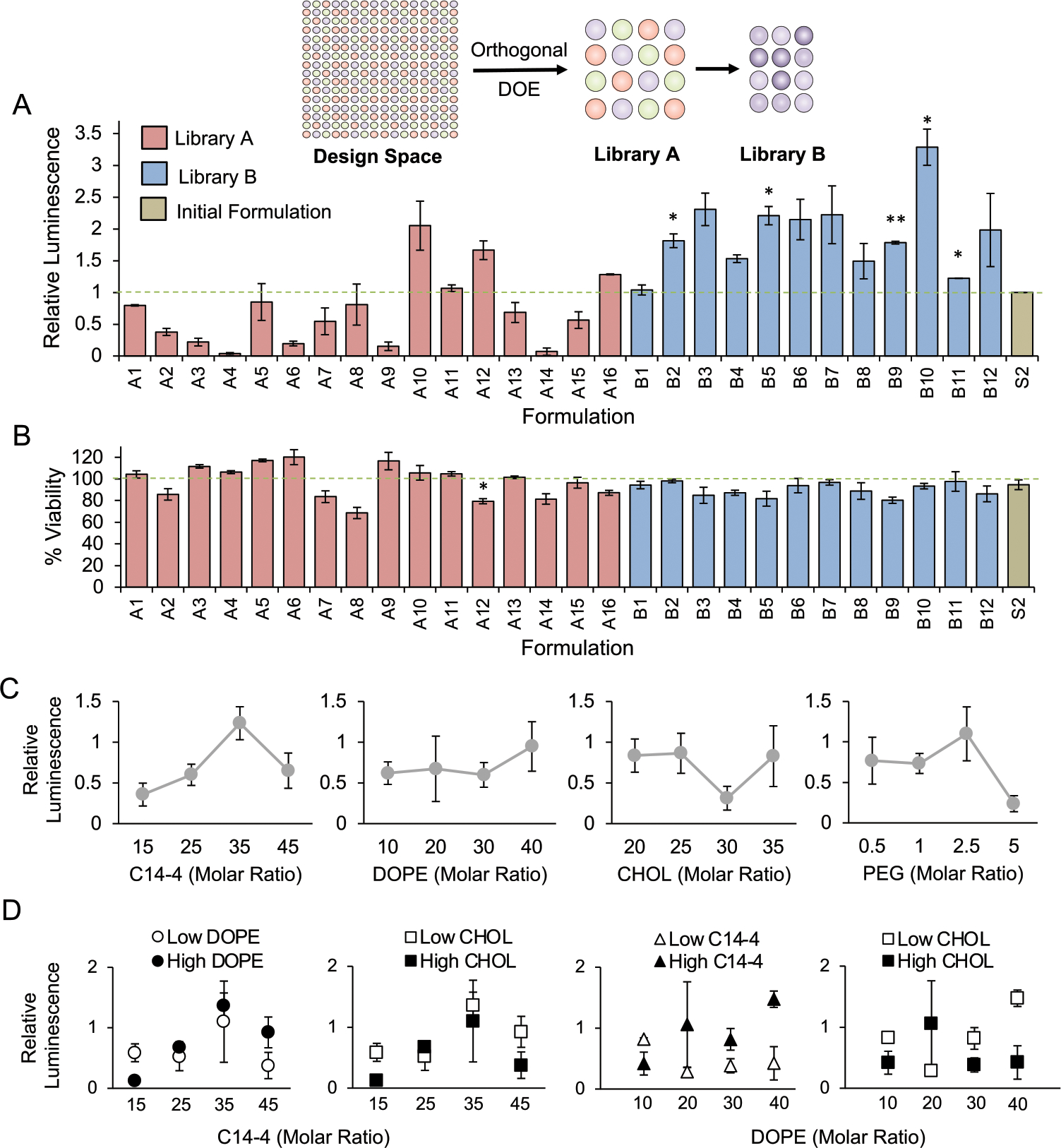
Subsequent screens of libraries A and B for luciferase mRNA delivery and toxicity in a T cell line (Jurkat) establish trends in excipient molar ratio and delivery. (A) Luciferase expression in Jurkat cells after treatment with LNP libraries and the initial formulation (S2) for 48 h at a dose of 30 ng/60 000 cells identifies formulations that achieve higher functional mRNA delivery than S2 (dashed line) and indicates that library B LNPs resulted in increased luciferase activity compared to library A. Results were normalized to cells treated with S2 and compared in a one-way ANOVA with post hoc t tests using Holm’s correction. *p < 0.05, **p < 0.01 compared to S2, n = 3 biological replicates, error bars = standard deviation. Inset schematic demonstrates the progression of library design from total potential design space with decreasing formulations in each library. (B) Viability of Jurkat cells treated with LNP libraries and S2 at 30 ng/60 000 cells for 48 h identifies formulations with increased cytotoxicity compared to S2 and reveals that library B resulted in fewer toxic LNP formulations than library A. Results were normalized to untreated cells (dashed line) and compared in a one-way ANOVA with post hoc t tests using Holm’s correction. *p < 0.05 compared to S2, n = 3 biological replicates, error bars = standard deviation. (C) To observe the effects of individual excipient ratios on mRNA delivery, each data point represents the average relative luminescence of the four LNP formulations with the given excipient molar ratio. The trends indicate that increased C14–4 and DOPE may improve delivery while moderate ratios of cholesterol or high ratios of PEG may be detrimental. error bars = standard error of the mean. (D) To observe the effects of two excipient ratios on mRNA delivery, each data point represents the average relative luminescence of two LNP formulations with the given excipient molar ratio with either the higher or lower molar ratios of the second excipient. The trends indicate that higher ratios of C14–4 and DOPE may enhance delivery when increased together while decreasing the cholesterol. Error bars = standard error of the mean.
In addition to identifying these promising formulations, we explored relationships between the molar ratio of each excipient and mRNA delivery to inform the generation of a subsequent library. To explore these trends, luminescence was compared to the molar ratios of an isolated excipient (Figure 2C). This revealed general trends of improved mRNA delivery with higher DOPE ratios, lower cholesterol ratios, and moderate ratios of C14–4 and PEG. Further, when observing cytotoxicity, it was noted that LNP formulations containing lower PEG ratios generally resulted in lower viability (Figure S3). Next, we examined the potential impacts of excipient interactions on mRNA delivery by observing the effects of two excipient molar ratios at once (Figures 2D, S2). These comparisons revealed that formulations with higher C14–4 ratios showed improved delivery with higher DOPE ratios and with lower cholesterol ratios. Similarly, formulations with higher DOPE ratios benefited from more C14–4 and less cholesterol. In all, these trends suggest that LNP formulations with increased C14–4 and DOPE ratios in combination with reduced cholesterol improve mRNA delivery to Jurkat cells, and maintaining moderate PEG ratios minimizes cytotoxicity.
These observed trends directly informed the design of library B, which featured 12 LNP formulations that explored a narrowed range of excipient molar ratios (Table 1, S3). As determined by library A, library B featured high ratios of C14–4 and DOPE while the cholesterol ratios were kept low, and the PEG ratio was held constant. Across library B, LNP sizes ranged from 71.4 to 125.1 nm with PDIs below 0.3, and mRNA concentrations ranged from 24.9 to 42.6 ng/μL, resulting in a higher average mRNA concentration for LNPs in library B compared to library A (Table S2). Library B was then screened for luciferase mRNA delivery and viability in Jurkat cells, which revealed five hit formulations featuring significantly enhanced mRNA delivery compared to S2 without decreased viability (Figure 2A,B). Further, the top-performing LNP formulation from library B, B10, resulted in >3-fold greater luminescence than S2, whereas library A only achieved a 2-fold increase over S2. Thus, the observed trends from library A successfully informed an improved library B, leading to the development of LNP formulations for potent mRNA delivery.
A16 and B10 LNPs Confirm Library Screen Results in Jurkat and Primary Human T Cells.
To confirm the results from the initial library screens, a top LNP formulation from each library was compared at varying doses in Jurkat cells. From library A, A16 was chosen for its high viability and consistently potent mRNA delivery. From library B, B10 was chosen as it achieved the highest mRNA delivery without decreasing viability. A16, B10, and S2 LNPs were used to treat Jurkat cells with luciferase mRNA, and both luminescence and viability were measured at 48 h. The results validated that B10 LNPs achieved the most potent delivery at each dose, while A16 showed a slight improvement over S2, and none of the LNPs resulted in notable toxicity (Figure 3A). While S2 is an important standard to examine the impacts of excipient molar ratios on mRNA delivery, B10 was also compared to lipofectamine, a commercially available transfection reagent (Figure 3B). B10 demonstrated enhanced mRNA delivery at two doses without decreasing viability compared to lipofectamine, confirming B10 LNPs as a promising platform for mRNA delivery to T cells.
Figure 3.
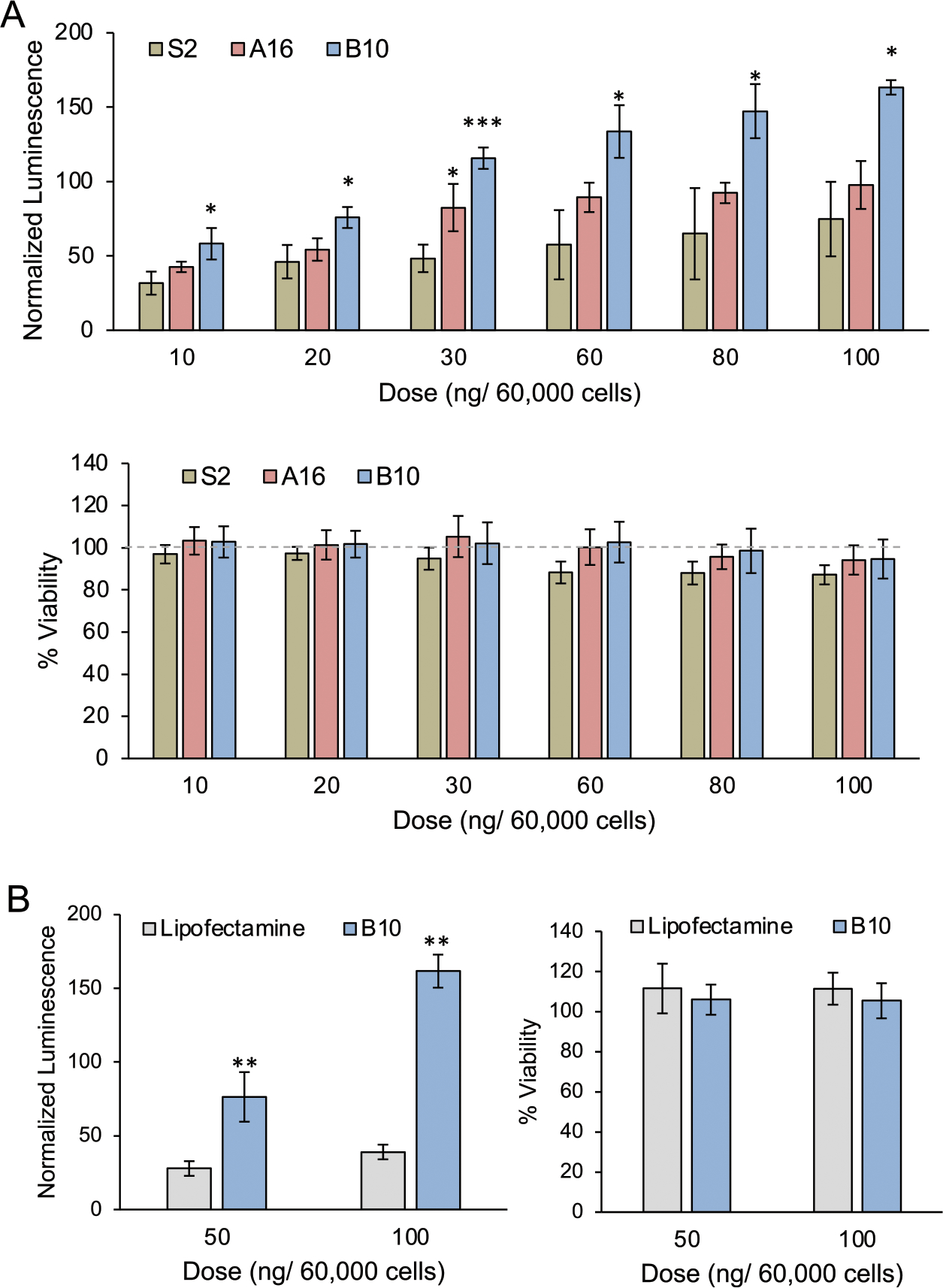
Dose–response of top-performing LNPs from libraries A and B confirm enhanced mRNA delivery to a T cell line (Jurkat) over a standard LNP formulation and lipofectamine. (A) Luciferase expression and viability of Jurkat cells treated with S2, A16, or B10 LNPs for 48 h, confirming the relative performance of each formulation. Luminescence and viability were normalized to untreated cells and compared within each dose using a one-way ANOVA with post hoc t tests using Holm’s correction. *p < 0.05, ***p < 0.001 as compared to S2, n = 3 biological replicates, error bars = standard deviation. (B) Luciferase expression and viability of Jurkat cells treated with B10 LNPs or lipofectamine for 48 h showing the ability of B10 LNPs to achieve increased mRNA delivery with no increase in cytotoxicity. Luminescence and viability were normalized to untreated cells, and the treatment groups were compared within each dose using a t test with post hoc t tests using Holm’s correction. **p < 0.01, n = 3 biological replicates, error bars = standard deviation.
As the CAR T cells used clinically are produced from patient T cells, S2, A16, and B10 LNPs were next evaluated for mRNA delivery in primary human T cells to demonstrate translatability. Briefly, CD4+ and CD8+ T cells were isolated from healthy donors, combined at a 1:1 ratio, and activated using Dynabeads. These cells were then treated with S2, A16, or B10 encapsulating luciferase mRNA, and the subsequent luminescence and viability were measured after 24 h (Figure 4). Donor-to-donor variability was observed in these results; however, this was expected as previous studies have shown variations in T cell quality across patients receiving CAR T cell therapy.72,73 Though the magnitude of luminescence varied with the donor cell population, B10 generally achieved the highest luciferase expression with S2 resulting in the lowest expression, and no differences in cytotoxicity were observed. Thus, these results from both Jurkat and primary human T cells validated the relative mRNA delivery of S2, A16, and B10, establishing B10 as the top-performing formulation.
Figure 4.
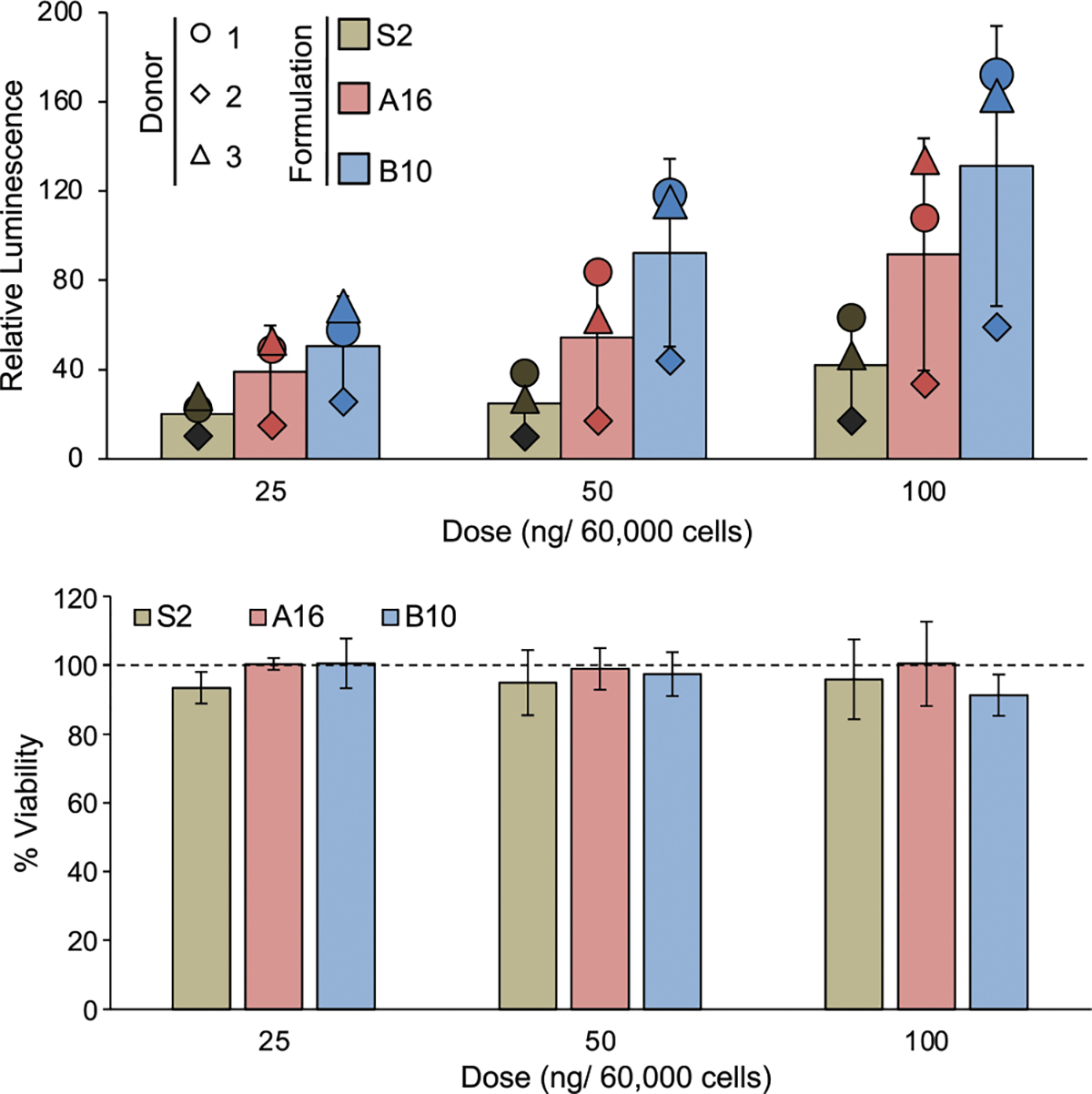
Luciferase expression and viability of primary human T cells treated with S2, A16, or B10 LNPs for 24 h, confirming trends in LNP formulation performance. The bar graphs represent an average of 3 individual donors as normalized to untreated cells. To demonstrate the donor-to-donor variability, the average relative luminescence for each donor at each treatment and dose is represented as a shape. The effects of the three treatments were compared via a one-way ANOVA at each dose, but no significance was found. However, the results from each donor demonstrate the same trends observed previously with B10 resulting in the highest luminescence and S2 resulting in the lowest. n = 3 biological replicates (bar graphs), n = 3 technical replicates (points), error bars = standard deviation.
B10 LNPs Generate Functional CAR T Cells with Low Cytotoxicity.
To further explore the translatability of B10 LNPs, this platform was used to deliver CD19-specific CAR mRNA to primary human T cells and compared to EP, the clinical standard for CAR mRNA delivery. Primary human T cells were prepared as previously described, treated with CAR mRNA for 24 h using B10 LNPs, S2 LNPs, or EP, and evaluated for CAR expression using flow cytometry (Figure 5A). As shown in the representative histogram, EP and B10 LNPs generally produced higher mean fluorescence intensity (MFI) values indicative of potent CAR expression, while S2 resulted in moderate CAR expression. When assessing the transfection rate of viable T cells after treatment with EP, B10 LNPs, or S2 LNPs across three donors, EP generally resulted in the highest percentage of CAR+ T cells, followed by B10 then S2 LNPs, but no significant difference was found across delivery methods. Further, when assessing the CD4+ and CD8+ fractions of CAR+ T cells, all treatment groups resulted in an even distribution of CAR expression across T cell types (Figure 5B), indicating these three delivery methods transfect comparable T cell populations. Though the LNPs and EP resulted in comparable transfection, EP was significantly more toxic to T cells, with B10 and S2 LNPs resulting in >70% viability compared to 55% with EP (Figure 5C). In combination, the results concerning CAR expression and viability in T cells indicate the ability of B10 LNPs to induce potent CAR expression comparable to EP with minimal toxicity, making it a promising platform for T cell transfection.
Figure 5.
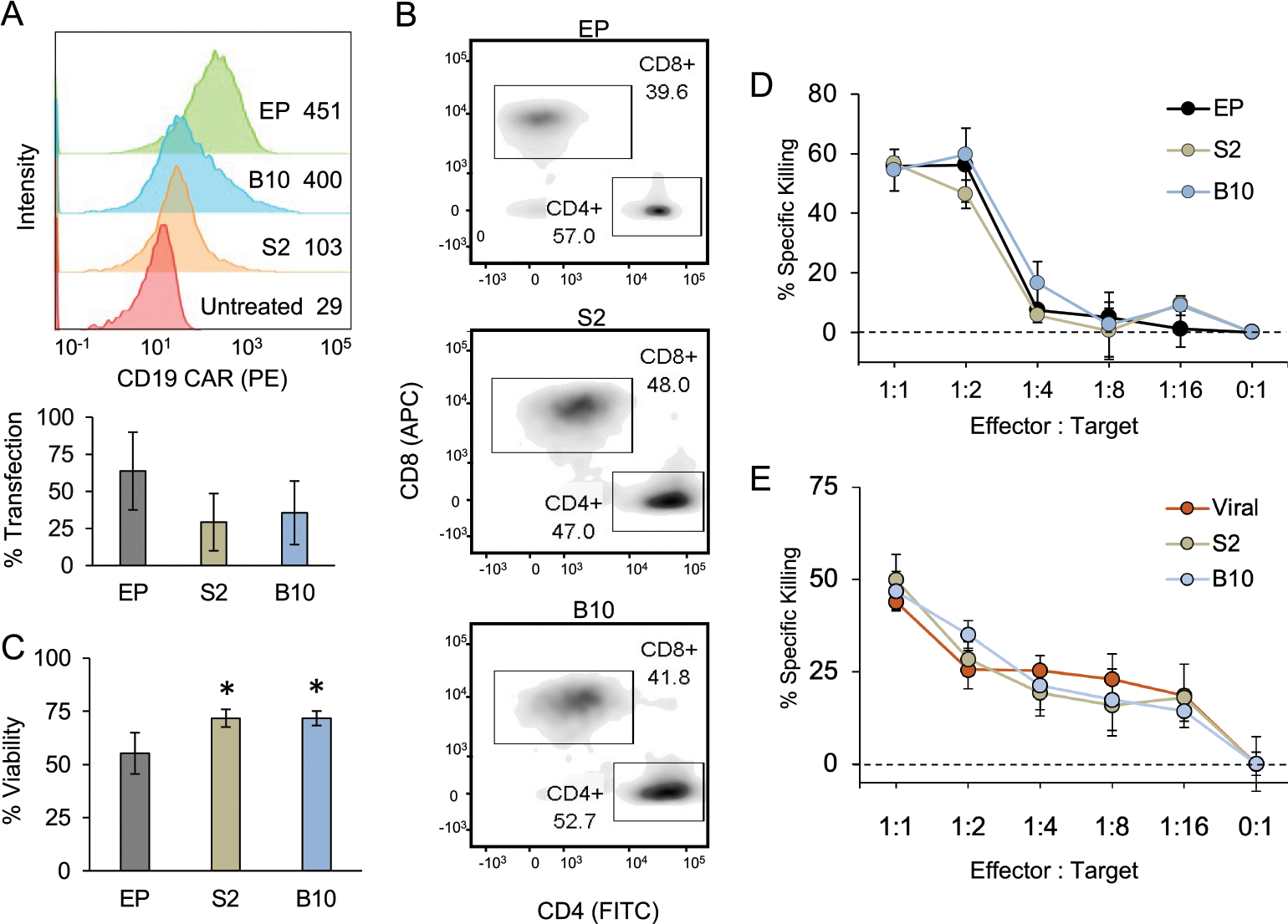
LNPs enable functional CAR mRNA delivery to primary human T cells with minimal toxicity compared to electroporation (EP). (A) Representative histogram of CAR expression (top) and average transfection rates (bottom) of primary human T cells 24 h after treatment with 300 ng of CAR mRNA per 60 000 cells using EP, S2 LNPs, or B10 LNPs. T cells were stained with a PE-labeled antibody to measure surface CAR expression with the histogram showing the mean fluorescent intensities associated with each treatment. The percent of transfection was determined as the fraction of living T cells expressing CAR. Results were compared via an ANOVA, which revealed no significant differences across the treatment groups. n = 3 biological replicates, error bars = standard deviation. (B) Representative flow cytometry data showing the CAR+ population of primary human T cells treated with EP, S2 LNPs, or B10 LNPs stained for CD8 (APC) and CD4 (FITC) surface expression. The boxes indicate the CD8+ and CD4+ T cell populations, and the percent of CAR+ T cells that fall into each population is noted. As the CAR expression is evenly split across CD8+ and CD4+ T cells for all three treatment groups, it seems the method of mRNA delivery did not impact this characteristic of the resulting CAR T cell population. (C) Viability of primary human T cells 24 h after treatment with 300 ng of CAR mRNA per 60 000 cells using EP, S2 LNPs, or B10 LNPs as compared to untreated cells. Results were compared in a one-way ANOVA with post hoc t tests using Holm’s correction. *p < 0.05 compared with EP, n = 3 biological replicates, error bars = standard deviation. (D) Representative results of CAR T and ALL cell coculture assay after 48 h at different T cell to tumor cell ratios. n = 3 wells, error bars = standard deviation. (E) Representative results of CAR T and ALL cell coculture assay comparing CAR T cells generated with lentivirus to those made with S2 or B10 LNPs. n = 4 technical replicates, error bars = standard deviation. The percent of cancer cell killing in both panels D and E was determined by comparison to ALL cells cultured without T cells as the negative control, and the results from each treatment group within each ratio were compared in a one-way ANOVA with no significance found.
After characterization, the CAR T cell function was assessed in a coculture assay to measure cancer cell killing. This commonly used assay utilized luciferase-expressing Nalm-6 ALL cells coplated with CAR T cells, which allowed for cancer cell death to be measured as decreased luminescence.45,74,75 In these assays, CAR T cells were generated using B10 LNPs, S2 LNPs, or EP, Nalm-6 and CAR T cells were plated at varied T cell to effector cell ratios, and cancer cell killing was measured at 48 h. At each T cell to effector cell ratio, there were no differences in cancer cell killing across treatment groups (Figure 5D, S4), validating that B10 LNPs generated CAR T cells with comparable functionality to those generated with EP, the clinical standard. B10 and S2 LNPs were also compared with lentiviral-generated CAR T cells in this same assay (Figure 5E), and the results demonstrated similar cancer cell killing across groups, confirming that these LNP-generated CAR T cells are comparable to current clinical standards.
In conclusion, this investigation utilized orthogonal DOE to identify new LNPs for potent mRNA delivery to T cells with lower cytotoxicity than EP. Subsequent libraries of LNP formulations with varied excipient compositions were screened for mRNA delivery and cytotoxicity in Jurkat cells, resulting in the identification of B10 as the top-performing formulation with a 3-fold increase in functional delivery compared to the standard formulation. B10 LNPs were then used to deliver CAR mRNA to primary human T cells and demonstrated CAR expression comparable to EP with less cytotoxicity. Further, in a coculture assay with ALL cells, the B10 LNP-generated CAR T cells were able to induce the same potent cancer cell killing as EP and lentiviral CAR T cells, confirming B10 LNPs as a promising delivery platform for CAR T cell engineering. Though future work should explore the mechanisms by which this altered excipient composition enhances delivery, the optimized B10 LNP platform has the potential to be utilized for a broad range of T cell engineering applications, and the B10 formulation could inform future work optimizing LNPs with various ionizable lipid components or enhancing delivery to other immune cells.
Supplementary Material
■ ACKNOWLEDGMENTS
M.J.M. acknowledges support from a Burroughs Wellcome Fund Career Award at the Scientific Interface (CASI), a US National Institutes of Health (NIH) Director’s New Innovator Award (DP2 TR002776), a grant from the American Cancer Society (129784-IRG-16-188-38-IRG), and the National Institutes of Health (NCI R01 CA241661, NCI R37 CA244911, and NIDDK R01 DK123049). M.M.B. was supported by a Tau Beta Pi Graduate Research Fellowship and an NIH Training in HIV Pathogenesis T32 Program (T32 AI007632). A.G.H. and K.L.S. were supported by National Science Foundation Graduate Research Fellowships (Award 1845298).
Footnotes
The authors declare the following competing financial interest(s): M.J.M., M.M.B., and C.H.J. are inventors on patents related to this work filed by the Trustees of the University of Pennsylvania (PCT/US20/56252 and PCT/ US20/56255).
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.nanolett.1c02503
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.nanolett.1c02503.
Materials and methods, characterization data for all LNP formulations, data from biological replicates of the coculture killing assay, and other supplementary results (PDF)
Contributor Information
Margaret M. Billingsley, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
Alex G. Hamilton, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
David Mai, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States; Abramson Cancer Center, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
Savan K. Patel, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
Kelsey L. Swingle, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
Neil C. Sheppard, Abramson Cancer Center, Perelman School of Medicine and Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
Carl H. June, Abramson Cancer Center, Perelman School of Medicine and Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
Michael J. Mitchell, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States; Abramson Cancer Center, Perelman School of Medicine, Institute for Immunology, Perelman School of Medicine, Cardiovascular Institute, Perelman School of Medicine, and Institute for Regenerative Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
■ REFERENCES
- (1).U.S. Food & Drug Administration. https://www.fda.gov/news-events/press-announcements/fda-approves-first-cell-based-gene-therapy-adult-patients-multiple-myeloma (accessed September 27, 2021).
- (2).Bouchkouj N; Kasamon YL; Claro R. A. de; George B; Lin X; Lee S; Blumenthal GM; Bryan W; McKee AE; Pazdur R FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma. Clin. Cancer Res. 2019, 25 (6), 1702–1708. [DOI] [PubMed] [Google Scholar]
- (3).FDA. FDA Approves First Cell-Based Gene Therapy For Adult Patients with Relapsed or Refractory MCL. U.S. Food Drug Adm. 2020, 1. [Google Scholar]
- (4).U.S. Food & Drug Administration. https://www.fda.gov/news-events/press-announcements/fda-approval-brings-first-gene-therapy-united-states (accessed September 27, 2021).
- (5).Li H; Huang Y; Jiang DQ; Cui LZ; He Z; Wang C; Zhang ZW; Zhu HL; Ding YM; Li LF; et al. Antitumor Activity of EGFR-Specific CAR T Cells against Non-Small-Cell Lung Cancer Cells in Vitro and in Mice. Cell Death Dis. 2018, 9 (2), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).O’Rourke DM; Nasrallah MP; Desai A; Melenhorst JJ; Mansfield K; Morrissette JJD; Martinez-Lage M; Brem S; Maloney E; Shen A; Isaacs R; Mohan S; Plesa G; Lacey SF; Navenot J-M; Zheng Z; Levine BL; Okada H; June CH; Brogdon JL; Maus MV A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9 (399), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wang D; Starr R; Chang W-C; Aguilar B; Alizadeh D; Wright SL; Yang X; Brito A; Sarkissian A; Ostberg JR; Li L; Shi Y; Gutova M; Aboody K; Badie B; Forman SJ; Barish M; Brown CE Chlorotoxin-Directed CAR T Cells for Specific and Effective Targeting of Glioblastoma. Sci. Transl. Med. 2020, 12 (533), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Maldini CR; Claiborne DT; Okawa K; Chen T; Dopkin DL; Shan X; Power KA; Trifonova RT; Krupp K; Phelps M; et al. Dual CD4-Based CAR T Cells with Distinct Costimulatory Domains Mitigate HIV Pathogenesis in Vivo. Nat. Med. 2020, 26 (11), 1776–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Benmebarek MR; Karches CH; Cadilha BL; Lesch S; Endres S; Kobold S Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20 (6), 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hartsell A Emerging Trends in Chimeric Antigen Receptor T-Cell Immunotherapy in Adults from the Vizient Clinical Database. Biol Blood Marrow Transplant. 2019, 25 (3), S336–S337. [Google Scholar]
- (11).Giavridis T; Van Der Stegen SJC; Eyquem J; Hamieh M; Piersigilli A; Sadelain M CAR T Cell-Induced Cytokine Release Syndrome Is Mediated by Macrophages and Abated by IL-1 Blockade Letter. Nat. Med. 2018, 24 (6), 731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zheng PP; Kros JM; Wang G Elusive Neurotoxicity in T Cell-Boosting Anticancer Therapies. Trends Immunol. 2019, 40 (4), 274–278. [DOI] [PubMed] [Google Scholar]
- (13).Namuduri M; Brentjens RJ Medical Management of Side Effects Related to CAR T Cell Therapy in Hematologic Malignancies. Expert Rev. Hematol 2016, 9 (6), 511–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Maude SL; Frey N; Shaw PA; Aplenc R; Barrett DM; Bunin NJ; Chew A; Gonzalez VE; Zheng Z; Lacey SF; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371 (16), 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tanyi JL; Stashwick C; Plesa G; Morgan MA; Porter D; Maus MV; June CH Possible Compartmental Cytokine Release Syndrome in a Patient with Recurrent Ovarian Cancer after Treatment with Mesothelin-Targeted CAR-T Cells. J. Immunother. 2017, 40 (3), 104–107. [DOI] [PubMed] [Google Scholar]
- (16).Siegler EL; Kenderian SS Neurotoxicity and Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy: Insights Into Mechanisms and Novel Therapies. Front. Immunol. 2020, 11 (August), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bushman FD Retroviral Insertional Mutagenesis in Humans: Evidence for Four Genetic Mechanisms Promoting Expansion of Cell Clones. Mol. Ther. 2020, 28 (2), 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nobles CL; Sherrill-Mix S; Everett JK; Reddy S; Fraietta JA; Porter DL; Frey N; Gill SI; Grupp SA; Maude SL; et al. CD19-Targeting CAR T Cell Immunotherapy Outcomes Correlate with Genomic Modification by Vector Integration. J. Clin. Invest. 2019, 130 (2), 673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ruella M; Xu J; Barrett DM; Fraietta JA; Reich TJ; Ambrose DE; Klichinsky M; Shestova O; Patel PR; Kulikovskaya I; Nazimuddin F; Bhoj VG; Orlando EJ; Fry TJ; Bitter H; Maude SL; Levine BL; Nobles CL; Bushman FD; Young RM; Scholler J; Gill SI; June CH; Grupp SA; Lacey SF; Melenhorst JJ Induction of Resistance to Chimeric Antigen Receptor T Cell Therapy by Transduction of a Single Leukemic B Cell. Nat. Med. 2018, 24 (10), 1499–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sweeney NP; Vink CA The Impact of Lentiviral Vector Genome Size and Producer Cell Genomic to Gag-Pol MRNA Ratios on Packaging Efficiency and Titre. Mol. Ther.–Methods Clin. Dev. 2021, 21 (June), 574–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Baliga UK; Dean DA Pulmonary Gene Delivery—Realities and Possibilities. Exp. Biol. Med. 2021, 246 (3), 260–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Seow Y; Wood MJ Biological Gene Delivery Vehicles: Beyond Viral Vectors. Mol. Ther. 2009, 17 (5), 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Putnam D Polymers for Gene Delivery across Length Scales. Nat. Mater. 2006, 5 (6), 439–451. [DOI] [PubMed] [Google Scholar]
- (24).Barrett DM; Zhao Y; Liu X; Jiang S; Carpenito C; Kalos M; Carroll RG; June CH; Grupp SA Treatment of Advanced Leukemia in Mice with MRNA Engineered T Cells. Hum. Gene Ther. 2011, 22 (12), 1575–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Pardi N; Hogan MJ; Porter FW; Weissman D MRNA Vaccines — a New Era in Vaccinology. Nat. Rev. Drug Discovery 2018, 17 (4), 261–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Smits E; Ponsaerts P; Lenjou M; Nijs G; Van Bockstaele DR; Berneman ZN; Van Tendeloo VFI RNA-Based Gene Transfer for Adult Stem Cells and T Cells. Leukemia 2004, 18 (11), 1898–1902. [DOI] [PubMed] [Google Scholar]
- (27).Foster JB; Barrett DM; Karikó K The Emerging Role of In Vitro -Transcribed MRNA in Adoptive T Cell Immunotherapy. Mol. Ther. 2019, 27 (4), 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Rajan TS; Gugliandolo A; Bramanti P; Mazzon E In Vitro-Transcribed Mrna Chimeric Antigen Receptor T Cell (IVT Mrna Car T) Therapy in Hematologic and Solid Tumor Management: A Preclinical Update. Int. J. Mol. Sci. 2020, 21 (18), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Foster JB; Choudhari N; Perazzelli J; Storm J; Hofmann TJ; Jain P; Storm PB; Pardi N; Weissman D; Waanders AJ; et al. Purification of MRNA Encoding Chimeric Antigen Receptor Is Critical for Generation of a Robust T-Cell Response. Hum. Gene Ther. 2019, 30 (2), 168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tchou J; Zhao Y; Levine BL; Zhang PJ; Davis MM; Melenhorst JJ; Kulikovskaya I; Brennan AL; Liu X; Lacey SF; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5 (12), 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Beatty GL; O’Hara MH; Lacey SF; Torigian DA; Nazimuddin F; Chen F; Kulikovskaya IM; Soulen MC; McGarvey M; Nelson AM; Gladney WL; Levine BL; Melenhorst JJ; Plesa G; June CH Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155 (1), 29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Svoboda J; Rheingold SR; Gill SI; Grupp SA; Lacey SF; Kulikovskaya I; Suhoski MM; Melenhorst JJ; Loudon B; Mato AR; et al. Nonviral RNA Chimeric Antigen Receptor – Modi Fi Ed T Cells in Patients with Hodgkin Lymphoma. Blood 2018, 132 (10), 1022–1027. [DOI] [PubMed] [Google Scholar]
- (33).Stewart MP; Sharei A; Ding X; Sahay G; Langer R; Jensen KF In Vitro and Ex Vivo Strategies for Intracellular Delivery. Nature 2016, 538, 183–192. [DOI] [PubMed] [Google Scholar]
- (34).DiTommaso T; Cole JM; Cassereau L; Bugge JA; Hanson JLS; Bridgen DT; Stokes BD; Loughhead SM; Beutel BA; Gilbert JB; Nussbaum K; Sorrentino A; Toggweiler J; Schmidt T; Gyuelveszi G; Bernstein H; Sharei A Cell Engineering with Microfluidic Squeezing Preserves Functionality of Primary Immune Cells in Vivo. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (46), E10907–E10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Nickoloff JA; Matthews KE; Dev SB; Toneguzzo F; Keating A Electroporation for Gene Therapy. Anim. Cell Electroporation Electrofusion Protoc. Methods Mol. Biol. 1995, 48, 273–280. [DOI] [PubMed] [Google Scholar]
- (36).Dullaers M; Breckpot K; Van Meirvenne S; Bonehill A; Tuyaerts S; Michiels A; Straetman L; Heirman C; De Greef C; Van Der Bruggen P; et al. Side-by-Side Comparison of Lentivirally Transduced and MRNA-Electroporated Dendritic Cells: Implications for Cancer Immunotherapy Protocols. Mol. Ther. 2004, 10 (4), 768–779. [DOI] [PubMed] [Google Scholar]
- (37).Hajj KA; Whitehead KA Tools for Translation: Non-Viral Materials for Therapeutic MRNA Delivery. Nature Reviews Materials. 2017, 2, 1–17. [Google Scholar]
- (38).Kauffman KJ; Webber MJ; Anderson DG Materials for Non-Viral Intracellular Delivery of Messenger RNA Therapeutics. J. Controlled Release 2016, 240, 227–234. [DOI] [PubMed] [Google Scholar]
- (39).Islam MA; Reesor EKG; Xu Y; Zope HR; Zetter BR; Shi J Biomaterials for MRNA Delivery. Biomater. Sci. 2015, 3, 1519–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zhang R; Billingsley MM; Mitchell MJ Biomaterials for Vaccine-Based Cancer Immunotherapy. J. Controlled Release 2018, 292, 256–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Mukalel AJ; Riley RS; Zhang R; Mitchell MJ Nanoparticles for Nucleic Acid Delivery: Applications in Cancer Immunotherapy. Cancer Lett. 2019, 458 (April), 102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Oberli MA; Reichmuth AM; Dorkin JR; Mitchell MJ; Fenton OS; Jaklenec A; Anderson DG; Langer R; Blankschtein D Lipid Nanoparticle Assisted MRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17 (3), 1326–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kauffman KJ; Dorkin JR; Yang JH; Heartlein MW; Derosa F; Mir FF; Fenton OS; Anderson DG Optimization of Lipid Nanoparticle Formulations for MRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15 (11), 7300–7306. [DOI] [PubMed] [Google Scholar]
- (44).Hajj KA; Ball RL; Deluty SB; Singh SR; Strelkova D; Knapp CM; Whitehead KA Branched-Tail Lipid Nanoparticles Potently Deliver MRNA In Vivo Due to Enhanced Ionization at Endosomal PH. Small 2019, 15, 1–7. [DOI] [PubMed] [Google Scholar]
- (45).Billingsley M; Singh N; Ravikumar P; Zhang R; June CH; Mitchell MJ Ionizable Lipid Nanoparticle Mediated MRNA Delivery for Human CAR T Cell Engineering. Nano Lett. 2020, 20 (3), 1578–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Olden BR; Cheng Y; Yu JL; Pun SH Cationic Polymers for Non-Viral Gene Delivery to Human T Cells Brynn. J. Controlled Release 2018, 282 (March), 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Moffett HF; Coon ME; Radtke S; Stephan SB; McKnight L; Lambert A; Stoddard BL; Kiem HP; Stephan MT Hit-and-Run Programming of Therapeutic Cytoreagents Using MRNA Nanocarriers. Nat. Commun. 2017, 8 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Parayath NN; Stephan SB; Koehne AL; Nelson PS; Stephan MT In Vitro-Transcribed Antigen Receptor MRNA Nanocarriers for Transient Expression in Circulating T Cells in Vivo. Nat. Commun. 2020, 11 (1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Tombacz I; Laczko D; Shahnawaz H; Muramatsu H; Natesan A; Yadegari A; Papp TE; Alameh M-G; Shuvaev V; Mui BL; et al. Highly Efficient CD4+ T Cell Targeting and Genetic Recombination Using Engineered CD4+ Cell-Homing MRNA-LNP. Mol. Ther. 2021, In Press. DOI: 10.1016/j.ymthe.2021.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Irvine DJ; Dane EL Enhancing Cancer Immunotherapy with Nanomedicine. Nat. Rev. Immunol. 2020, 20 (5), 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Garber K Alnylam Launches Era of RNAi Drugs. Nat. Biotechnol 2018, 36, 777. [DOI] [PubMed] [Google Scholar]
- (52).Jackson LA; Anderson EJ; Rouphael NG; Roberts PC; Makhene M; Coler RN; McCullough MP; Chappell JD; Denison MR; Stevens LJ; et al. An MRNA Vaccine against SARS-CoV-2 — Preliminary Report. N. Engl. J. Med. 2020, 383 (20), 1920–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Vogel AB; Kanevsky I; Che Y; Swanson KA; Muik A; Vormehr M; Kranz LM; Walzer KC; Hein S; Guler A; et al. BNT162b Vaccines Protect Rhesus Macaques from SARS-CoV-2. Nature 2021, 592 (7853), 283–289. [DOI] [PubMed] [Google Scholar]
- (54).Cheng Q; Wei T; Jia Y; Farbiak L; Zhou K; Zhang S; Wei Y; Zhu H; Siegwart DJ Dendrimer-Based Lipid Nanoparticles Deliver Therapeutic FAH MRNA to Normalize Liver Function and Extend Survival in a Mouse Model of Hepatorenal Tyrosinemia Type I. Adv. Mater. 2018, 30 (52), 1805308. [DOI] [PubMed] [Google Scholar]
- (55).Swingle KL; Hamilton AG; Mitchell MJ Lipid Nanoparticle-Mediated Delivery of MRNA Therapeutics and Vaccines. Trends Mol. Med. 2021, 27 (6), 616–617. [DOI] [PubMed] [Google Scholar]
- (56).Patel S; Ashwanikumar N; Robinson E; Duross A; Sun C; Murphy-Benenato KE; Mihai C; Almarsson O; Sahay G Boosting Intracellular Delivery of Lipid Nanoparticle-Encapsulated MRNA. Nano Lett. 2017, 17 (9), 5711–5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Gilleron J; Querbes W; Zeigerer A; Borodovsky A; Marsico G; Schubert U; Manygoats K; Seifert S; Andree C; Stöter M; et al. Image-Based Analysis of Lipid Nanoparticle–Mediated SiRNA Delivery, Intracellular Trafficking and Endosomal Escape. Nat. Biotechnol. 2013, 31 (7), 638–646. [DOI] [PubMed] [Google Scholar]
- (58).Kulkarni JA; Cullis PR; van der Meel R Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018, 28 (3), 146–157. [DOI] [PubMed] [Google Scholar]
- (59).Granot Y; Peer D Delivering the Right Message: Challenges and Opportunities in Lipid Nanoparticles-Mediated Modified MRNA Therapeutics—An Innate Immune System Standpoint. Semin. Immunol. 2017, 34, 68–77. [DOI] [PubMed] [Google Scholar]
- (60).Kowalski PS; Rudra A; Miao L; Anderson DG Delivering the Messenger: Advances in Technologies for Therapeutic MRNA Delivery. Mol. Ther. 2019, 27 (4), 710–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Ball RL; Hajj KA; Vizelman J; Bajaj P; Whitehead KA Lipid Nanoparticle Formulations for Enhanced Co-Delivery of SiRNA and MRNA. Nano Lett. 2018, 18 (6), 3814–3822. [DOI] [PubMed] [Google Scholar]
- (62).Blakney AK; McKay PF; Yus BI; Aldon Y; Shattock RJ Inside out: Optimization of Lipid Nanoparticle Formulations for Exterior Complexation and in Vivo Delivery of SaRNA. Gene Ther. 2019, 26 (9), 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Cheng Q; Wei T; Farbiak L; Johnson LT; Dilliard SA; Siegwart DJ Selective Organ Targeting (SORT) Nanoparticles for Tissue-Specific MRNA Delivery and CRISPR–Cas Gene Editing. Nat. Nanotechnol. 2020, IS (4), 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Monopoli MP; Aberg C; Salvati A; Dawson KA Biomolecular Coronas Provide the Biological Identity of Nanosized Materials. Nat. Nanotechnol. 2012, 7 (12), 779–786. [DOI] [PubMed] [Google Scholar]
- (65).Zhang R; El-Mayta R; Murdoch TJ; Warzecha CC; Billingsley MM; Shepherd SJ; Gong N; Wang L; Wilson JM; Lee D; et al. Helper Lipid Structure Influences Protein Adsorption and Delivery of Lipid Nanoparticles to Spleen and Liver. Biomater. Sci. 2021, 9 (4), 1449–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Li B; Luo X; Deng B; Wang J; McComb DW; Shi Y; Gaensler KML; Tan X; Dunn AL; Kerlin BA; et al. An Orthogonal Array Optimization of Lipid-like Nanoparticles for MRNA Delivery in Vivo. Nano Lett. 2015, 15 (12), 8099–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Chen D; Love K; Chen Y; Eltoukhy A; Kastrup C; Sahay G; Jeon A; Dong Y; Whitehead K; Anderson D Rapid Discovery of Potent SiRNA-Containing Lipid Nanoparticles Enabled by Controlled Microfluidic Formulation. J. Am. Chem. Soc. 2012, 134 (16), 6948–6951. [DOI] [PubMed] [Google Scholar]
- (68).Stroock AD; Dertinger SKW; Ajdari A; Mezić I; Stone HA; Whitesides GM Chaotic Mixer for Microchannels. Science (Washington, DC, U. S.) 2002, 295 (5555), 647–651. [DOI] [PubMed] [Google Scholar]
- (69).Zhang J; Fan H; Levorse DA; Crocker LS Interaction of Cholesterol-Conjugated Ionizable Amino Lipids with Biomembranes: Lipid Polymorphism, Structure Activity Relationship, and Implications for SiRNA Delivery. Langmuir 2011, 27, 9473–9483. [DOI] [PubMed] [Google Scholar]
- (70).Heyes J; Palmer L; Bremner K; Maclachlan I Cationic Lipid Saturation Influences Intracellular Delivery of Encapsulated Nucleic Acids. J. Controlled Release 2005, 107, 276–287. [DOI] [PubMed] [Google Scholar]
- (71).Love KT; Mahon KP; Levins CG; Whitehead KA; Querbes W; Dorkin JR; Qin J; Cantley W; Qin LL; Racie T; Frank-Kamenetsky M; Yip KN; Alvarez R; Sah DWY; de Fougerolles A; Fitzgerald K; Koteliansky V; Akinc A; Langer R; Anderson DG Lipid-like Materials for Low-Dose, in Vivo Gene Silencing. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (21), 1864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Tyagarajan S; Spencer T; Smith J Optimizing CAR-T Cell Manufacturing Processes during Pivotal Clinical Trials. Mol. Ther.–Methods Clin. Dev. 2020, 16 (March), 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Jiang J; Ahuja S Addressing Patient to Patient Variability for Autologous CAR T Therapies. J. Pharm. Sci. 2021, 110 (5), 1871–1876. [DOI] [PubMed] [Google Scholar]
- (74).Castella M; Boronat A; Martín-ibáñez R; Rodríguez V; Suñé G; Caballero M; Marzal B; Pérez-amill L; Martín-antonio B; Castaño J; et al. Development of a Novel Anti-CD19 Chimeric Antigen Receptor: A Paradigm for an Affordable CAR T Cell Production at Academic Institutions. Mol. Ther.–Methods Clin. Dev. 2019, 12 (March), 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Avanzi MP; Yeku O; Li X; Wijewarnasuriya DP; van Leeuwen DG; Cheung K; Park H; Purdon TJ; Daniyan AF; Spitzer MH; Brentjens RJ Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy. Cell Rep. 2018, 23 (7), 2130–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.