

Abstract
Understanding the genetic basis of complex traits is a fundamental goal of evolutionary genetics. Yet, the genetics controlling complex traits in many important species such as hemp (Cannabis sativa) remain poorly investigated. Because hemp’s change in legal status with the 2014 and 2018 U.S. Federal Farm Bills, interest in the genetics controlling its numerous agriculturally important traits has steadily increased. To better understand the genetics of agriculturally important traits in hemp, we developed an F2 population by crossing two phenotypically distinct hemp cultivars (Carmagnola and USO31). Using whole-genome sequencing, we mapped quantitative trait loci (QTL) associated with variation in numerous agronomic and biochemical traits. A total of 69 loci associated with agronomic (34) and biochemical (35) trait variation were identified. We found that most QTL co-localized, suggesting that the phenotypic distinctions between Carmagnola and USO31 are largely controlled by a small number of loci. We identified TINY and olivetol synthase as candidate genes underlying co-localized QTL clusters for agronomic and biochemical traits, respectively. We functionally validated the olivetol synthase candidate by expressing the alleles in yeast. Gas chromatography-mass spectrometry assays of extracts from these yeast colonies suggest that the USO31 olivetol synthase is functionally less active and potentially explains why USO31 produces lower cannabinoids compared to Carmagnola. Overall, our results help modernize the genomic understanding of complex traits in hemp.
Keywords: genetic architecture, hemp, olivetol synthase
Introduction
A long-term goal of genetics and evolutionary genetics is to understand the genetic basis of complex traits. In the past, studies have approached this goal by using molecular markers to investigate fundamental questions such as: for any given trait, how many loci control variation; are these loci dominant; and is variation additive, or do epistatic interactions explain a large proportion of phenotypic variance (Hill 2010)? Despite nearly 30 years of using molecular markers, our understanding of complex trait genetics remains incomplete because of the limited capacity for high-resolution mapping of loci (MacKay et al. 2009). Now in the genomics era, with the ease of sequencing whole genomes, this long-term goal is more feasible since studies have an improved ability to dissect the genetic architecture of complex traits (Mackay et al. 2009). As a result, it is becoming increasingly common for studies to combine whole-genome sequencing (WGS) with bi-parental mapping populations to identify quantitative trait loci (QTL) controlling variation in complex traits in numerous species (Mojica et al. 2016; Yang et al. 2017; Burga et al. 2019).
Historically, plants have been widely used to study fundamental questions related to the genetics of complex traits because of the possibility for extensive experimental design and control of environments (Speed and Balding 2012). In many staple crop species such as Maize and rice, the genetic understanding of complex traits has improved steadily in recent years because of the well-established genetic resources and dense volume of literature. For less studied crop species such as industrial hemp (Cannabis sativa), the availability of genetic resources and literature is narrow, which limits the capacity to understand complex traits. To help establish a basic quantitative genetic understanding in crops such as industrial hemp, studies that investigate fundamental questions regarding the genetics of these crop’s complex traits are needed. Hemp is a scientifically interesting plant and valuable crop, producing a high yield of plant biomass including stalks, bast fibers (used in building materials, composites, and textiles) and a high protein and lipid grain with unique nutritional properties. C. sativa is also the sister species of hops (Humulus lupulusKovalchuk et al. 2020) and similarly produces an array of secondary metabolites that have numerous potential uses. In addition, hemp is interesting because it is in a clade where it evolved dioecy and an annual habit from progenitors which were monoecious and perennial (Kovalchuk et al. 2020).
Since its initial change in legal status in the 2014 Federal Farm Bill and subsequent broadening of those rules in the 2018 Federal Farm Bill (U.S. Govt. 2014, 2018), interest in cultivating and researching industrial hemp has steadily increased in the United States. In Canada, the European Union, and the United States, hemp is legally defined as C. sativa plants with a total tetrahydrocannabinol (THC) content less than 0.3%. Cultivars of C. sativa with a total THC content above 0.3% are federally illegal and such plants are scheduled as a controlled substance known as marijuana. Given its importance for regulation, there have been a number of studies focused on trying to understand the biochemistry of THC synthesis (Sirikantaramas et al. 2004, 2005; Zirpel et al. 2018). THC is not the only biochemical trait that is important in hemp. There is a growing market for terpenes (e.g., alpha-pinene) and other cannabinoids [e.g., cannabidiol (CBD)] outside of THC, that have value for their medicinal and therapeutic properties, use as chemosensory additives, natural pesticides, and other potential uses (Russo 2011; Gallily et al. 2015). Agronomic traits like grain yield and plant biomass are also key breeding targets to make hemp a competitive grain and fiber crop. Despite a number of agronomic studies on industrial hemp (Van der Werf et al. 1995a, 1995b; Struik et al. 2000), literature that investigates the genetic factors that contribute to the variation in these traits has only recently begun to emerge (Petit et al. 2020a, 2020b, 2020c). Petit et al. (2020a, 2020b, 2020c) used a diversity mapping panel of hemp cultivars to perform genome-wide associations to identify additive loci and genotype by environment interactions associated with variation in numerous fiber quality and flowering traits.
To date, two studies have utilized bi-parental mapping populations to identify QTL associated with variation in biochemical traits in C. sativa. Both of these studies utilized the same mapping population derived from a cross between hemp and marijuana cultivars (Weiblen et al. 2015; Grassa et al. 2021). Weiblen et al. (2015) identified a single large-effect QTL associated with chemotype ratios for THC and CBD (Weiblen et al. 2015). In a follow-up study using the same mapping population, Grassa et al. (2021) mapped for loci associated with cannabinoid variation and were able to identify two candidate genes possibly linked to their QTL.
In this study, QTL contributing to the variation of important agronomic and of biochemical traits were characterized in C. sativa utilizing an F2 mapping population derived from a cross between two foundational hemp cultivars bred for different markets as well as developed in different countries: Carmagnola and USO31. Carmagnola is a dioecious fiber cultivar developed in Italy and USO31 is a monoecious dual-purpose cultivar developed in Ukraine, bred for both grain and fiber production (Salentijn et al. 2015). Carmagnola produces late-flowering, tall plants that are typical of fiber cultivars, while USO31 is an early maturing, shorter-statured cultivar that is more suitable for grain cropping. We characterized several QTL by using WGS to identify segregating variants that span the C. sativa genome and phenotyping of numerous agronomic and biochemical traits. Our results identify numerous QTL of varying effect size, co-located QTL, and candidate genes underlying two co-located QTL clusters.
Materials and methods
Mapping population creation
Seed of the cultivars of industrial hemp Carmagnola and USO31 were imported from Italy to Colorado in 2015 as part of a set of variety trials (Campbell et al. 2019). Seeds of Carmagnola and USO31 were sown in Promix potting soil (Premier Horticulture, Quakertown, PA, USA), in a Conviron E8 growth chamber at the Colorado State University greenhouse. Growth chambers were initially set to a 20:4 h (light: dark) regimen in order to keep plants in the vegetative stage. During flowering, the light regimen was changed to a 12:12 h regimen. Daytime and nighttime temperatures were kept at 23°C and relative humidity was kept at 40%. Growth chamber light intensity was kept at 330 μmol m−2 s−1. Plants were watered with a full strength (1-1-1) vegetative nutrient solution (General Hydroponics Flora Series, Sebastopol, CA, USA). Healthy and representative plants of Carmagnola and USO31 were chosen as parents of a bi-parental QTL mapping population. Pollen from a monoecious USO31 plant was crossed to a female Carmagnola plant. F1 seed was grown using the same methods as for the parent plants. A single monoecious healthy F1 plant was then self-fertilized to produce the F2 mapping population. Propagated clones were taken from the original parent and F1 plants to use later in the field experiment by taking cuttings. These clonal cuttings were then dipped into cloning solution (Olivia’s Solutions Cloning Solution, Calistoga, CA, USA), planted in Promix potting soil, kept under humidity domes and watered as needed with a full strength (1-1-1) vegetative nutrient solution (General Hydroponics Flora Series, Sebastopol, CA, USA). These clones were kept in their vegetive stage in the same growth chamber using the 20:4 regime. F2 seed was germinated within rockwool plugs (Grodan, Roermond, the Netherlands) in the Colorado State University greenhouse in May 2017. No light supplementation occurred during the 4 weeks that F2 seed were in the greenhouse. In order to have replication of F2 lines, clones were taken from each seedling so that each line could be replicated three times in the experiment. Clonal propagation of the F2 plants was conducted using the same method as for the parent and F1 plants. Once three clonal propagations of each F2 plant were obtained, the parent, F1 and F2 plants were transplanted to a field located at the Colorado State University Agricultural Research and Education Center (ARDEC).
Field experiment
The experiment was conducted at ARDEC located in Fort Collins, CO, USA. Prior to transplanting clones, glyphosate (RoundUp, Powermax, Monsanto) and dicamba (Sterling Blue, Winfield United) were applied (15.70 gallons per acre) to clear the field of any existing weeds. Clones representing 372 F2 lines along with clones of the parents and F1 were transplanted from the greenhouse into the field by hand in June 2017. Plants were spaced 1.5 m apart in both directions to avoid interplant competition. A Latinized row-column design was utilized to minimize spatial bias. The experiment was replicated three times, with one clone of each F2 line and 3 clones of each parental and F1 line represented in each replicate block. The plots were 1.5 m in length and width with a single plant in the center of the plot. Weed pressure was controlled manually and no pesticides were applied during the growing season. For calculating precipitation, the growing season was defined as the date of transplant into the field until the harvest of the last plot. The trial received a total of 157 mm of precipitation as rainfall and an additional 254 mm was applied as irrigation. Plots were hand watered with a hose due to breakage of the overhead linear irrigation sprinkler system.
The date that each plant reached initiation of maturity, was noted and the number of days that elapsed between when the clones were propagated and when the initiation of that stage was noted and were calculated. Plant maturity was considered as seed maturity, i.e., when bracts began to dehisce and darkening of the seed coat was visible as described by Campbell et al. (2019). Mature plants were harvested within 3 days.
To measure leaf water content, one fully expanded and undamaged leaf was randomly selected from the middle of the primary stem of each plant at a single time point during the vegetative growth stage of the plant and placed in airtight containers. The leaves were weighed, lyophilized, and then weighed again. The calculated difference in mass is reported as leaf water content.
Before harvest, plant height was measured as the vertical distance from the soil surface to the tallest naturally occurring part of a plant.
Plants were cut at the soil surface and air-dried for a minimum of 30 days. Total plant biomass (dry biomass) was measured as the mass of the aboveground portion of the plant material. Stems were weighed separately after threshing to determine stem biomass. The dried stems were measured at the widest part of the base with digital calipers to determine stem diameter.
Grain was separated from inflorescences by hand and seed was cleaned using a column blower (Agriculex, Guelph, ON, Canada). Grain was air-dried to approximately 8–10% seed moisture, as determined by a GAC 500XT grain moisture tester (Dickey-John, Auburn, IL, USA). A subsample of 50 seeds was counted from each sample to extrapolate Thousand Seed Mass.
Biochemical trait analysis
Biochemical traits were analyzed from female flowers collected after plants were dried. Seeds were removed from the flowers by hand and composite samples were made with the flower chaff. Cannabinoid and terpene profiles were analyzed using ultra-high-pressure liquid chromatography (Waters UPLC) and gas chromatography (Shimadzu GC-2014) with flame ionization detector (GC-FID) by ProVerde Labs (Milford, MA, USA). Sample preparation for the analysis of cannabinoid profiles was performed by extraction of the cannabinoids in organic solvent. Approximately 300 mg of homogenized plant material was extracted with 4 ml of isopropanol with sonication for 20 min. The resulting extract was filtered with a syringe filter, and further diluted with 71% acetonitrile (ACN) to the appropriate concentration for LC analysis, and transferred to an auto-sampler vial.
The liquid chromatographic analyses were performed using an ultra-high-pressure liquid chromatographic system (Waters UPLC) with Photo Diode Array, UV Detection (PDA), with a Cortecs C18 column (2.7 µm, 2.1 mm × 100 mm) (Waters Corporation, MA, USA). Mobile phases were water (A) and acetonitrile (B), both acidified with 0.1% formic acid. Separation was achieved under gradient conditions of 59–100% mobile phase B over 2.5 min at a flow rate of 0.56 mL min−1 at 40°C. Samples were introduced with a 3.5 µl injection, with chromatographic data collected at 225 nm. Cannabinoid certified reference standards (Cerilliant, Sigma-Aldrich, and Cayman Chemicals) were used for peak identification and generation of calibration curves used for quantitation, and included: THC acid (THCa), CBD acid (CBDa), cannabigerolic acid (CBGa), and cannabichromene (CBC). Data were recorded and processed using Empower Software (Version 3, Waters Corporation).
Analysis of terpene profiles was performed using Full Evaporative Technique GC-FID Chromatography (FET-GC-FID) which is a form of head-space sampling, for which standards or samples are placed and sealed directly in a head space vial. The sealed vial was equilibrated at elevated temperatures to vaporize volatile compounds for head-space sampling. For these evaluations, samples were homogenized and sealed directly into the head-space vials, then equilibrated for 30 min at 140°C prior to injection using a Hewlett Packard head-space autosampler (HP G1290A).
Gas chromatography was performed using Shimadzu GC-2014 gas chromatograph with Flame Ionization Detection (FID), with a Rxi-624Sil MS column (30 m × 0.25 mm × 1.4 µm) (Restek, Bellefonte, PA, USA). Samples were introduced directly from the head-space auto sampler via a transfer line held at 160°C to prevent condensation of sample vapors prior to injection.
Nitrogen was used as the GC carrier gas at a flow rate of ∼80 mL min−1. Hydrogen and compressed air were used as the combustion gases. The following instrument parameters were employed: air, 50 psi; hydrogen, 70 psi; nitrogen, 60 psi; linear velocity flow control, 33 cm s−1; split ratio, 20:1; injector temperature, 250°C; detector temperature, 320°C; oven program, 75°C (hold 0.4 min) to 160°C at 8°C min−1; ramped to 250°C at 20°C min−1; ramped to 300°C at 12.5°C min−1 (hold 3 min); run time, 22.2 min. Terpene certified reference materials (Restek CRMs #34095 and 34096) were used for peak identification and generation of calibration curves used for quantitation. Data were recorded and processed using Clarity Software (Version 5.0.4.158).
Whole-genome sequencing
DNA was extracted using the Qiagen DNeasy Plant Mini Kit (Valencia, CA, USA) and then quantified using a Qubit Fluorometer (ThermoFisher Scientific). A total of 375 samples were whole-genome sequenced (2 × 150 bp paired-end reads) using Illumina Nextera library preparation system. Sequencing efforts aimed for 30x, 15x, and 7x coverage of the parents, F1 and F2 progeny, respectively. All samples were sequenced at the University of Colorado Anschutz Medical Campus using an Illumina NovaSeq.
Raw sequence data were evaluated with FastQC (Andrews 2010, version 0.11.8) to assess read quality and adapter contamination. Trimmomatic (Bolger et al. 2014, version 0.39) was then used with default parameters to remove low-quality reads and any adapter contamination identified in the FastQC report. The trimmed sequence reads were then aligned to version 2 of the Finola reference genome (Laverty et al. 2019, GenBank assembly accession ID = GCA_003417725.2) using BWA-MEM with the default settings (Li 2013, version 0.7.17) . Samtools (Li et al. 2009, version 1.9) was then used to sort sequence alignment files and mark duplicate reads. BCFtools (Narasimhan et al. 2016, version 1.9) was then used with default parameters to identify genetic variants using both the “mpileup” and “call” functions to produce three separate variant call files (VCFs) for the Carmagnola/USO31 parents, the F1, and F2, respectively.
BCFtools were used to filter the F2 VCF to contain biallelic single nucleotide polymorphisms (SNPs) that possessed a genotyping rate of ≥75% across individuals, quality of ≥30, base quality bias of ≥0.8, base position bias of ≥0.8, and mapping quality of ≥60. VCFtools (Danecek et al. 2011, version 0.1.16) was then used to filter the F2 VCF to contain loci with genotype frequencies resembling 1:2:1 Mendelian segregation ratios by incorporating an exact test with a P-value threshold of 0.05 followed by a minor allele frequency filter of 0.4. The BCFtools command “isec” was then used to extract the filtered F2 VCF loci from the Carmagnola/USO31 parent and F1 VCF files. The parent, F1 and F2 VCF files were then filtered again with BCFtools to contain only loci where Carmagnola and USO31 possessed alternate homozygous SNPs with quality ≥200, read depth of ≥50 and phred-scaled genotype quality ≥99 for which the F1 was also heterozygous. All three VCF files were then merged to contain a total of 1827 SNPs across all samples. Using the “VariantsToTable” command from the Genome Analysis Toolkit (McKenna et al. 2010, version 4.1.4.0), the merged VCF was then exported to a tab-separated file format. This tab-separated file was read into excel and the F2 genotypes were manually converted to the “a” (Carmagnola), “b” (USO31), and “h” (heterozygote) genetic linkage map format.
Quantitative trait loci mapping and trait correlations
A genetic linkage map was created using JoinMap 4 (Van Ooijen 2006), with markers assigned to linkage groups based on a recombination frequency threshold of 0.25. We identified 10 linkage groups, corresponding to the 10 chromosomes from Laverty et al. (2019), whose numbering convention we used. The markers on the 10 linkage groups were mapped using the regression mapping algorithm and Kosambi mapping function. A total of 10 duplicate markers were identified and removed, for a remaining total of 1817 markers in the genetic map. All QTL mapping was conducted in R/qtl (Broman et al. 2003, version 1.44 - 9). Recombination frequencies calculated from JoinMap and the R package “qtlTools” (Lovell 2019, version 1.2.0) were used to estimate the 1.5 log of odds (LOD) QTL location confidence intervals.
We calculated the simple means of each F2 line’s phenotype to use for downstream analyses. Raw mean phenotype data were assessed for normality using the Shapiro–Wilk test base R function. Because no trait’s distribution passed the Shapiro–Wilk normality test, the raw phenotype data were quantile normalized to better-fit assumptions of normality. For traits reported, there was no substantial differences in QTL between raw and normalized data. Multiple QTL models for normally distributed traits were selected using the STEPWISE.QTL(max.qtl = 6) function with penalties based on 1000 permutations. QTL models for traits that could not be adequately transformed were constructed using the significant peak locations based on 1000 permutations identified from the SCANONE output. QTL peak positions obtained from SCANONE were further refined using REFINEQTL. Significance and effect sizes of QTL in models were validated using FITQTL. QTL that did not explain a significant proportion of variance (P > 0.05) were removed from models.
To test for significant correlations (P < 0.05) among measured traits, we used the “rstatix” package (Kassambara 2020, version 0.6.0) in R (R Core Team 2019, version 3.6.0) to obtain Spearman’s rank correlation coefficient (ρ). Correlation coefficients were then organized into a matrix and plotted using ggplot2 (Wickham 2016, version 3.3.2).
QTL candidate gene identification
To identify candidate genes of agronomic traits, we focused on identifying genes underlying QTL with narrow 1.5 LOD intervals spanning ∼50,000 base pairs or less. To investigate these narrow QTL intervals and their close surrounding regions, we extracted the reference sequences contained in these LOD intervals plus an extra 15,000 base pairs on each flanking end. Since there was no gene annotation available for version 2 of the Finola assembly, we used the AUGUSTUS (Stanke et al. 2004) output from BUSCO (Simão et al. 2015) to predict the models of potential genes located within these extracted regions. Basic local alignment search tool (BLAST) was then used to identify homologs to the predicted gene model sequences. Predicted gene model sequences that showed strong homolog matches (100% coverage and ≥95% identity) were then further investigated for sequence variation between Carmagnola and USO31 in the VCF. The predicted gene model sequences were then annotated with identified SNPs using Geneious Prime (Kearse et al. 2012, version 2020.1.2).
To identify candidate genes of biochemical traits, we focused on identifying genes underlying the co-located QTL clusters. Using published coding sequences of genes involved in terpene and cannabinoid biosynthesis, we used BLAST to identify alignments within the biochemical trait QTL intervals. Gene sequences that had high coverage, identity, and functional relevance to traits included in QTL intervals were then investigated for genetic differences between Carmagnola and USO31 within the VCF. Sequence variation identified in the VCF were then confirmed in Carmagnola and USO31 using Sanger sequencing. Gene sequences with confirmed genetic variation were then annotated with SNPs using Geneious Prime (Kearse et al. 2012, version 2020.1.2).
Olivetol synthase functional assay
Olivetol synthase coding sequences from Carmagnola and USO31 were synthesized by Twist Bioscience with codon optimization for Saccharomyces cerevisiae and addition of flanking BamHI (5’) and NotI (3’) restriction sites. We used the published and functionally validated olivetol synthase coding sequence from Taura et al. (2009) as the basis for sites where Carmagnola and USO31 did not genetically differ. Only overlapping genetic variation between Carmagnola and USO31 from our Illumina and Sanger sequence data were incorporated into the sequences. Thus, we denoted these two olivetol synthase alleles as “Carmagnola-derived” and “USO31-derived.” At the amino acid scale, the USO31-derived sequence was identical to the Taura et al. (2009) OLS while the Carmagnola-derived OLS differed by 9 amino acids. Synthesized genes were cloned into the BamHI/NotI sites of the pYES2 expression vector containing a GAL1 promoter for galactose-inducible expression of the inserted genes. The resulting constructs and the empty pYES2 vector were introduced into S. cerevisiae BY4741 cells using lithium acetate/polyethylene glycol transformation with the Frozen-EZ Yeast Transformation II Kit (Zymo). Colonies selected on -uracil (-URA) dropout media were grown in 3 ml liquid cultures in media lacking uracil and containing raffinose as a noninductive carbon source [0.17% (w/v) yeast nitrogen base without amino acids, 0.08% (w/v) CSM-URA, 0.5% (w/v) ammonium sulfate, 2% (w/v) raffinose]. For induction, raffinose was substituted with galactose [2% (w/v)], and cultures were initiated in 50 ml of media in 250 ml Erlenmeyer flasks at a density of 0.1 optical density. Cells were maintained at 25°C with shaking (130 rpm). Hexanoic acid (NuChek Prep) was added at a concentration of 1 mM to cultures after 24 h of growth. Cultures were sampled in 6 ml aliquots at 2-day intervals over a 6-day time course. The results provided are from an experiment with three independent cultures for each treatment, and experiments were repeated three times with similar trends.
For olivetol analyses, pelleted cells were extracted in 2 ml of chloroform with 30 min of incubation in a sonicating water bath (Branson 2800). Following sonication, tubes were centrifuged at 16,000 x g for 10 min. The solvent was transferred to a glass screw cap tube, dried under N2, and dissolved in 100 µl of chloroform. Olivetol in extracts was identified and quantified by analysis on an Agilent 7890 A gas chromatograph (GC) interfaced with an Agilent 5975 C mass selective detector fitted with an Agilent HP-5 column (30 m length × 0.25 mm outer diameter, 0.25 µm film thickness). The inlet temperature was 250°C and a 9 ml/min flow rate of H2 carrier with the oven programmed for 90°C for 1 min followed by a 30°C/min temperature ramp to 300°C. The olivetol product was identified by the 124 m/z diagnostic ion fragment and 180 m/z molecular ion and by retention time and mass spectrum identical to those of an authentic olivetol standard (Sigma Aldrich). Olivetol production was quantified using a standard curve derived from the olivetol standard.
Results
Trait values and correlations
In our field experiment, we measured a total of eight agronomic and seventeen biochemical traits. A summary of trait values for the parent, F1, and F2 plants can be found in Supplementary Table S1. For nearly all traits measured, Carmagnola exhibited higher trait values compared to USO31. Since the parents of the population we developed were traditional fiber and seed industrial hemp cultivars, their production of biochemical traits was modest compared to cultivars that have been specifically bred for cannabinoid and terpene production. We also note that pollination and seed set may have also reduced production of biochemical traits (Mehmedic et al. 2010). F2 population mean trait values were generally intermediate relative to the Carmagnola and USO31 parents. The range for most F2 traits extended beyond the mean trait values of Carmagnola and USO31. Some biochemical traits, such as citronellol and geraniol produced no detectible quantities in the parents or F1 but did show a distribution of detectible quantities among the F2 population. As described in the methods, F2 genotypes were replicated by vegetative propagation, and then clones were transplanted into an agricultural field, where survival was low. In total 256, 170, and 238 F2 plants were phenotyped in replicate blocks 1, 2, and 3, respectively. Transplant death appeared to be random with respect to genotype and thus resulted in many F2 lines having reduced replication or only a single observation. While this did not prevent us from detecting QTL and fitting polygenic models for most traits, our experiment’s power was reduced and thus may have inhibited our ability to detect a larger number small-effect QTL.
Significant (P < 0.05) and positive correlations were observed among all agronomic traits measured (Figure 1 and Supplementary Table S2). Dry biomass and stem biomass exhibited the strongest correlation strength (ρ = 0.95). Correlations between agronomic and biochemical traits were low, with the strongest between days to maturity and CBC (ρ = −0.23, Figure 1 and Supplementary Table S2).
Figure 1.
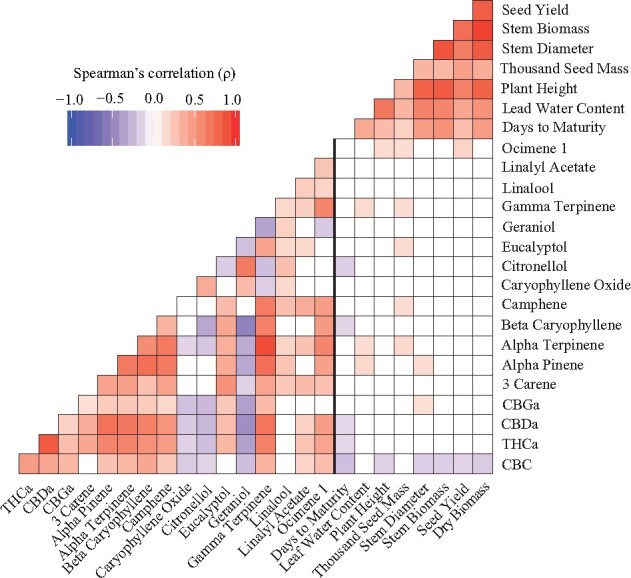
Trait correlations of measured phenotypes. Correlation plot depicting Spearman’s rank correlation coefficient (ρ), between measured phenotypes in F2 population. Red colors indicate positive correlations while blue colors indicate negative correlations. The vertical bolded black line separates biochemical from agronomic traits. Only significant correlations (P < 0.05) are shown.
Most of the 17 biochemical traits were positively correlated (Figure 1 and Supplementary Table S2). Of the cannabinoids measured, CBDa and THCa were the most associated (ρ = 0.85, Figure 1). Correlations between terpenes were largely positive with gamma and alpha terpinene being the most associated (ρ = 0.90, Figure 1) among all biochemical traits. Citronellol, geraniol, and caryophyllene oxide, which were inversely correlated with most other biochemical traits, displayed significant positive correlations.
QTL mapping
Our genetic map identified 10 linkage groups consistent with the 10 chromosomes identified by Laverty et al. (2019), which we used for naming our linkage groups. Mapping of QTL identified a total of 69 loci associated with measurable variation in agronomic (34) and biochemical traits (35). Of these 69 QTL identified, we found that numerous agronomic and biochemical QTL co-localized across our linkage map. In total, four agronomic and two biochemical QTL co-localized clusters (Figure 2, A and B) were identified. Henceforth, we refer to each of these co-located QTL clusters by their linkage group name followed by their average genetic position (e.g., LG3.60, Figure 2A).
Figure 2.

Linkage map and QTL intervals. (A) Linkage map showing boxplots depicting the 1.5 LOD confidence intervals of QTL for the 8 measured agronomic traits. (B) Linkage map showing boxplots depicting the 1.5 LOD confidence intervals of QTL for the 17 measured biochemical traits. Black bars within each boxplot indicate the location of the peak LOD values while box colors indicate QTL identified for the respectively colored trait.
Individual QTL models for agronomic traits are shown in Table 1. Agronomic trait QTL models were largely additive with the exception of stem diameter which had the most complex model that included a significant interaction between QTL on linkage groups 3 and 9 (Supplementary Figure S1). Leaf water content and plant height exhibited the simplest models consisting of three QTL. For 31 of the 34 agronomic QTL identified, F2 plants homozygous for the Carmagnola allele exhibited higher trait values (Supplementary Table S3). F2 plants homozygous for the USO31 allele at two QTL (SY.1 and DTM.4) had higher trait values (Supplementary Figures S2 and S3). F2 plants that were heterozygous at QTL generally exhibited either additivity or dominance among alleles at each agronomic QTL. The only exception to this pattern was a single QTL for thousand seed mass (TSM.2) which displayed genotype-phenotype patterns suggestive of overdominance (Supplementary Figure S4). Agronomic co-located QTL clusters explained ranges of 5.22–22.35% (LG3.60), 3.91–5.92% (LG4.50), 12.92–34.27% (LG5.05), and 3.81–5.65% (LG10.25) of variance across traits with a detected QTL in these clusters (Supplementary Table S4).
Table 1.
QTL models, locations, and effect sizes for agronomic traits
| Phenotype | QTL | Linkage group | Marker (bp) | Genetic position (cM) | LOD | Variance explained (%) |
|---|---|---|---|---|---|---|
| Leaf water content (g) | LWC.1 | 3 | 50,700,151 | 61.73 | 14.65 | 16.25 |
| LWC.2 | 5 | 23,530,617 | 3.00 | 12.37 | 13.48 | |
| LWC.3 | 10 | 58,605,089 | 8.27 | 4.23 | 4.32 | |
| Plant height (cm) | PLHT.1 | 3 | 46,248,568 | 60.33 | 19.41 | 22.35 |
| PLHT.2 | 5 | 31,242,637 | 9.30 | 14.01 | 15.38 | |
| PLHT.3 | 10 | 74,603,632 | 5.88 | 4.40 | 4.45 | |
| Thousand seed mass (g) | TSM.1 | 3 | 89,444,283 | 74.00 | 8.44 | 8.29 |
| TSM.2 | 5 | 31,294,490 | 10.00 | 17.22 | 18.16 | |
| TSM.3 | 8 | 14,446,033 | 32.62 | 6.93 | 6.73 | |
| TSM.4 | 10 | 16,666,160 | 13.12 | 5.78 | 5.56 | |
| Stem diameter (cm) | SD.1 | 3 | 65,433,658 | 59.44 | 18.86 | 15.71 |
| SD.2 | 4 | 86,599,640 | 53.04 | 6.38 | 4.80 | |
| SD.3 | 5 | 41,915,966 | 12.00 | 23.29 | 20.13 | |
| SD.4 | 5 | 84,759,484 | 109.77 | 4.45 | 3.30 | |
| SD.5 | 9 | 1,825,282 | 38.22 | 8.42 | 6.44 | |
| SD.6 | 10 | 16,666,160 | 13.12 | 5.11 | 3.81 | |
| SD.1:SD.5 | 5.84 | 4.38 | ||||
| Stem biomass (g) | SB.1 | 3 | 22,597,724 | 58.82 | 11.24 | 9.01 |
| SB.2 | 4 | 15,873,733 | 32.36 | 7.60 | 5.92 | |
| SB.3 | 5 | 31,242,637 | 9.30 | 32.48 | 31.05 | |
| SB.4 | 5 | 84,871,972 | 102.83 | 4.46 | 3.39 | |
| SB.5 | 10 | 24,849,063 | 37.50 | 7.26 | 5.65 | |
| Seed yield (g) | SY.1 | 2 | 94,185,707 | 98.47 | 12.59 | 11.35 |
| SY.2 | 3 | 41,816,369 | 61.18 | 6.72 | 5.78 | |
| SY.3 | 4 | 84,450,770 | 49.52 | 4.92 | 4.17 | |
| SY.4 | 5 | 23,530,617 | 3.00 | 14.15 | 12.92 | |
| SY.5 | 5 | 57,357,245 | 36.00 | 4.04 | 3.41 | |
| Dry biomass (g) | DB.1 | 3 | 22,597,724 | 58.82 | 8.30 | 8.04 |
| DB.2 | 4 | 86,642,457 | 54.30 | 4.17 | 3.91 | |
| DB.3 | 5 | 31,242,637 | 9.30 | 22.29 | 24.19 | |
| DB.4 | 9 | 8,629,041 | 8.00 | 4.17 | 3.91 | |
| Days to maturity | DTM.1 | 3 | 26,862,649 | 57.70 | 6.30 | 5.22 |
| DTM.2 | 4 | 86,599,640 | 53.00 | 6.05 | 5.01 | |
| DTM.3 | 5 | 23,530,617 | 3.00 | 33.10 | 34.27 | |
| DTM.4 | 9 | 3,462,161 | 43.60 | 5.24 | 4.31 |
Markers are reported as the physical base pair position of the linkage map marker closest to the LOD peak of the respective QTL. Genetic positions (centiMorgans), LOD values, and variance estimates of QTL have been rounded to two decimal places. Colons indicate interactions between the specified QTL.
Individual QTL models for all biochemical traits are shown in Table 2. Biochemical trait QTL models were overall more complex with multiple significant QTL interactions identified. Alpha-pinene had the most complex model that consisted of 6 additive QTL and two separate interactions. CBC, citronellol, geraniol, linalool, and linalyl acetate exhibited the simplest models consisting of a single QTL. Of the 35 biochemical QTL, 30 exhibited a pattern similar to agronomic QTL where F2 plants homozygous for the Carmagnola allele produced greater trait values (Supplementary Table S5). Models for CBGa, citronellol, geraniol, ocimene 1, and caryophyllene oxide all contained at least one QTL for which F2 plants homozygous for the USO31 allele exhibited greater trait values. F2 plants that were heterozygous at the QTL exhibited evidence for either additivity or dominance among alleles at each biochemical QTL. Biochemical co-located QTL clusters explained ranges of 9.35–28.21% (LG6.35) and 10.23–44.79% (LG9.40) of variance across traits with a detected QTL in these clusters (Supplementary Table S4). These co-located QTL clusters were comprised of both cannabinoids and terpenes. For 8 of the 17 measured biochemical traits, we detected a significant interaction between QTL in LG6.35 and LG9.40 which explained a range of 3.89–15.11% of variance observed across these 8 phenotypes. Figure 3 shows this interaction between QTL within LG6.35 and LG9.40 for CBDa production.
Table 2.
QTL models, locations, and effect sizes for biochemical traits
| Phenotype | QTL | Linkage group | Marker (bp) | Genetic position (cM) | LOD | Variance explained (%) |
|---|---|---|---|---|---|---|
| Alpha-pinene (ppm) | AP.1 | 1 | 730,085 | 20.00 | 12.11 | 9.90 |
| AP.2 | 2 | 1,811,866 | 15.80 | 5.94 | 4.54 | |
| AP.3 | 6 | 65,509,552 | 41.60 | 15.76 | 13.43 | |
| AP.4 | 7 | 71,316,711 | 56.49 | 4.84 | 3.65 | |
| AP.5 | 8 | 3,907,307 | 24.00 | 8.64 | 6.80 | |
| AP.6 | 9 | 2,778,113 | 43.00 | 32.09 | 31.04 | |
| AP.1:AP.5 | 6.71 | 5.16 | ||||
| AP.3:AP.6 | 9.61 | 7.64 | ||||
| Alpha terpinene (ppm) | AT.1 | 6 | 19,863,635 | 39.10 | 10.17 | 15.70 |
| AT.2 | 9 | 2,941,221 | 44.00 | 14.31 | 23.15 | |
| AT.1:AT.2 | 3.51 | 5.04 | ||||
| Beta caryophyllene (ppm) | BC.1 | 2 | 2,134,594 | 13.00 | 9.86 | 9.20 |
| BC.2 | 8 | 47,143,084 | 58.32 | 6.37 | 5.72 | |
| BC.3 | 9 | 2,323,644 | 41.28 | 35.06 | 43.88 | |
| Camphene (ppm) | CAM.1 | 6 | 17,849,490 | 36.29 | 10.51 | 17.36 |
| CAM.2 | 9 | 2,941,221 | 44.00 | 11.40 | 19.01 | |
| CAM.1:CAM.2 | 4.65 | 7.19 | ||||
| Caryophyllene oxide (ppm) | CO.1 | 9 | 2,992,284 | 44.38 | 11.75 | 20.42 |
| CO.2 | 10 | 34,580,106 | 93.00 | 10.59 | 18.18 | |
| CO.1:CO.2 | 6.91 | 11.38 | ||||
| CBC (%) | CBC.1 | 9 | 2,778,113 | 43.00 | 6.56 | 12.57 |
| CBDa (%) | CBDa.1 | 6 | 29,867,178 | 33.73 | 32.56 | 28.21 |
| CBDa.2 | 9 | 2,453,237 | 41.10 | 44.85 | 44.79 | |
| CBDa.1:CBDa.2 | 13.64 | 9.59 | ||||
| CBGa (%) | CBGa.1 | 6 | 45,639,112 | 33.37 | 20.32 | 26.46 |
| CBGa.2 | 9 | 2,453,237 | 41.10 | 25.53 | 35.21 | |
| CBGa.1:CBGa.2 | 7.87 | 8.97 | ||||
| Citronellol (ppm) | CIT.1 | 9 | 2,083,967 | 37.46 | 12.41 | 23.44 |
| Eucalyptol (ppm) | EUC.1 | 5 | 72,066,134 | 53.98 | 3.52 | 6.39 |
| EUC.2 | 9 | 2,941,221 | 44.00 | 5.52 | 10.23 | |
| Gamma terpinene (ppm) | GT.1 | 6 | 19,863,635 | 39.10 | 7.64 | 11.03 |
| GT.2 | 9 | 2,270,995 | 39.39 | 17.44 | 28.12 | |
| GT.1:GT.2 | 2.84 | 3.89 | ||||
| Geraniol (ppm) | GE.1 | 9 | 2,083,904 | 36.00 | 26.39 | 43.33 |
| Linalool (ppm) | LI.1 | 7 | 35,578,586 | 31.65 | 6.12 | 12.34 |
| Linalyl acetate (ppm) | LA.1 | 6 | 5,006,390 | 22.00 | 4.56 | 9.35 |
| Ocimene 1 (ppm) | OC.1 | 1 | 2,554,340 | 34.00 | 8.52 | 13.11 |
| OC.2 | 6 | 39,795,404 | 27.98 | 6.42 | 9.65 | |
| OC.3 | 9 | 2,941,221 | 44.00 | 7.67 | 11.68 | |
| THCa (%) | THCa.1 | 6 | 39,794,190 | 28.90 | 20.58 | 24.54 |
| THCa.2 | 9 | 2,453,237 | 41.10 | 25.56 | 32.10 | |
| THCa.1:THCa.2 | 8.04 | 8.36 | ||||
| 3 Carene (ppm) | 3C.1 | 6 | 55,569,529 | 37.18 | 16.34 | 26.70 |
| 3C.2 | 9 | 3,462,212 | 43.48 | 15.23 | 24.59 | |
| 3C.1:3C.2 | 9.94 | 15.11 |
Markers are reported as the physical base pair position of the linkage map marker closest to the LOD peak of the respective QTL. Genetic positions (centiMorgans), LOD values, and variance estimates of QTL have been rounded to two decimal places. Colons indicate interactions between the specified QTL.
Figure 3.
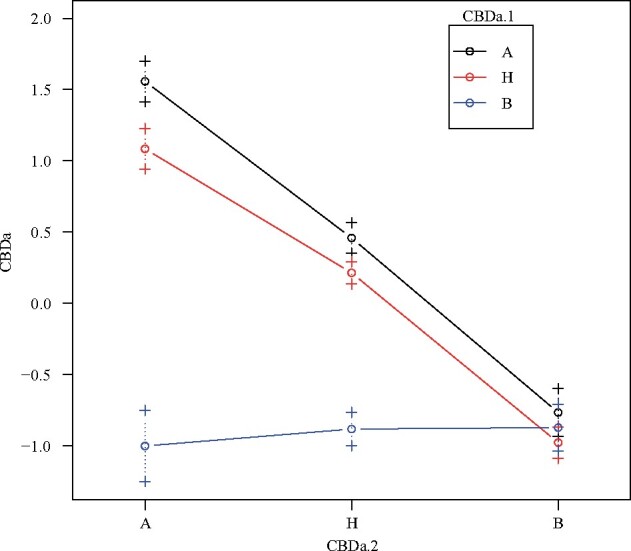
Epistatic interaction of the QTL clusters LG6.35 (CBDa.1) and LG9.40 (CBDa.2). Line colors indicate F2 plant genotype at CBDa.1 while x-axis positions indicate F2 plant genotype at CBDa.2 (A, Carmagnola; B, USO31). y-axis values indicate the mean (± standard error) quantile normalized CBDa quantities. Lesser y-axis values indicate lower quantities while higher y-axis values indicate greater quantities of CBDa produced.
QTL candidate gene identification
Of the 34 agronomic QTL identified, SB.3 was the only QTL with a narrow 1.5 LOD interval which spanned ∼50,000 base pairs (Figure 2A). Our BUSCO analysis on the region covering this 1.5 LOD interval and its nearby surrounding sequence identified models for five predicted genes. BLAST analyses of the five predicted gene sequences resulted in one of these predicted genes to have a homolog match to the ethylene-responsive transcription factor TINY (Wilson et al. 1996; Xie et al. 2019) with 100% coverage and 99.7% sequence identity. Sequences of the other four predicted gene models did not result in any homolog matches. Further inspection of the Carmagnola and USO31 TINY gene sequences identified a single nonsynonymous homozygous SNP (Figure 4A) not represented in the genotype matrix used to create the linkage map because of hard SNP filtering. Extraction and subsequent analysis of variance (ANOVA) of this nonsynonymous SNP revealed significant differences (P < 0.05) among all F2 agronomic trait means (Supplementary Table S6 and Figure S5).
Figure 4.

Models for candidate genes underlying LG5.05 and LG9.40. Shown are the physical locations of the TINY (A) and OLS (B) genes with annotations depicting the three letter codes for amino acid substitution(s) identified between Carmagnola (blue) and USO31 (red).
Using the published cDNA sequence from Taura et al. (2009), we identified olivetol synthase (OLS) as a candidate gene for LG9.40 which aligned inside this region with 100% coverage and 99.00% sequence identity. Comparison of the USO31 and Carmagnola OLS gene sequences revealed 9 nonsynonymous homozygous SNPs in the coding regions (Figure 4B). While no candidate gene linked to LG6.35 could be identified, we note that the partial isopentenyl-diphosphate delta-isomerase (synthesizes the substrate used for geranyl pyrophosphate synthesis) coding sequence from Booth et al. (2017) exhibited the greatest homology to a genomic region within LG6.35.
Functional validation of olivetol synthase alleles
We tested whether the variation in OLS alleles between the parents could affect biochemical production by expressing the alleles in S. cerevisiae (yeast) supplemented with the substrate hexanoic acid to measure olivetol production. Olivetol standard calibrations using GC-MS displayed a retention time of ∼6.40 min, a diagnostic ionization fragment of 124 m/z and molecular ion of 180 m/z, which were used to identify olivetol produced by yeast colonies expressing the Carmagnola-derived and USO31-derived OLS alleles (Figure 5, C and D). At each sampling time point, yeast colonies expressing the Carmagnola-derived OLS allele produced significantly more (P < 0.05) olivetol quantities compared to yeast colonies expressing the USO31-derived OLS allele (Figure 5B), consistent with the effect of genotype at the QTL. Yeast colonies transformed with empty pYES2 vector did not produce detectible amounts of olivetol.
Figure 5.

Olivetol production in media supplemented with 1 mM hexanoic acid. (A) Synthesis of olivetol from hexanoic acid and hexanoyl-Coenzyme A (CoA) as described by Taura et al. (2009) and Kovalchuk et al. (2020). (B) Shown are bar plots comparing the mean (± standard deviation) olivetol production for biological triplicates of yeast colonies expressing the Carmagnola-derived OLS (black) and USO31-derived OLS (white). Yeast colonies transformed with empty pYES2 vector is shown in gray. Single asterisks indicate a t-test P-value less than 0.05 while double asterisks indicate a t-test P-value less than 0.01. The dashed horizontal line is used to indicate no detectible (ND) quantity of olivetol for colonies transformed with empty pYES2 vectors. (C) Mass spectra showing the major diagnostic ionization fragment and molecular ion (M+) for olivetol for the standard (top) and Carmagnola-derived OLS (bottom). (D) Chromatograms showing the retention time (min) for the peak containing the major diagnostic ionization fragment for olivetol for the olivetol standard, Carmagnola-derived OLS, and USO31-derived OLS.
Discussion
Using hemp as a system to study fundamental questions regarding the genetics of complex traits, our results are relevant to both theoretical and applied quantitative genetics. Theory predicts that variation of complex traits is often attributed to many loci across the genome that can act in an additive, dominant, or epistatic manner (Lynch and Walsh 1998). In the context of C. sativa, for which genetic studies have largely focused on understanding the inheritance of cannabinoids, the literature has only demonstrated instances of dominance and additivity associated with these traits’ variation (de Meijer et al. 2003; Weiblen et al. 2015; Campbell et al. 2020; Petit et al. 2020a, 2020b; Grassa et al. 2021). Our study builds upon the current literature by suggesting that while epistasis is also a prevalent genetic factor for both cannabinoids and terpenes, epistatic interactions explain considerably less phenotypic variance (range = 3.89–15.11%, Table 2) than additive genetic effects. Thus, while our results suggest that the effect sizes of epistatic interactions are low for biochemical traits, they explain a significant amount of trait variation. Identification of specific QTL has been applied for genetic improvement in numerous crop species such as Maize and rice (Yousef and Juvik 2002; Luo et al. 2014; Kumar et al. 2017). In C. sativa, traits of importance such as THCa and CBDa can be improved by identifying QTL that predict trait values (Toth et al. 2020; Wenger et al. 2020). The numerous instances of epistasis identified here suggest that breeding for enhanced biochemical phenotypes in C. sativa may require a more complex selection process in order to take advantage of these epistatic interactions (Holland 2001). Overall, our results demonstrate how classical quantitative genetics approaches can be used to better understand complex trait genetic architecture in a nonmodel species and furthermore they provide evidence for the causal genetics that potentially underlie the numerous trait differences between two industrial hemp cultivars.
Phenotype correlations
The presence of co-located QTL suggests trait correlations (Luo et al. 2017). While many of the QTL identified in this study did co-localize, this was only observed for QTL associated with traits of the same class type (agronomic or biochemical, Figure 2, A and B). Indeed, traits of the same class type exhibited significant correlations (P < 0.05) that reflect their co-located QTL (Figure 1). The absence of co-located QTL and significant correlation between trait classes however suggests that the loci associated with variation in agronomic traits are largely independent of loci associated with variation in biochemical traits. Whether or not the lack of association between agronomic and biochemical traits is population-specific is currently not possible to determine since no other whole-genome mapping population data exist for C. sativa.
Quantitative genetic architecture of C. sativa agronomic traits
Our mapping efforts identified a total of 34 QTL associated with measurable variation among the eight measured agronomic traits. Although most agronomic QTL co-localized into four distinct clusters (Figure 2A), single QTL were identified for seed yield, thousand seed mass, and dry biomass. Overall, the observed QTL clustering patterns suggest that much of the agronomic differences between Carmagnola and USO31 are controlled by a few pleiotropic large-effect loci. We note however that the summation of most agronomic trait’s total variance explained in Table 1 are ∼50% which may indicate the existence of additional small effect QTL that our experiment did not have the power to identify. Interestingly, each agronomic trait model contained a QTL located within LG3.60 and LG5.05. This suggests that the function of the genes linked with LG3.60 and LG5.05 may control overall plant growth.
Using the narrow 1.5 LOD interval of SB.3 within LG5.05, we identified a predicted gene with high sequence homology to TINY, a dehydrin response element which has been shown to affect overall plant growth in Arabidopsis thaliana (Wilson et al. 1996; Xie et al. 2019). After further analyzing variation in this gene’s sequence, we identified a single nonsynonymous homozygous SNP within the Carmagnola and USO31 TINY coding region (Figure 4A) for which the F2 genotypic classes were significantly different for all agronomic traits (Supplementary Table S6, and Figure S5). While it is possible that stronger genotype-phenotype associations may exist in regulatory regions, our limited ability to annotate the LOD interval of SB.3 only allowed us to identify the coding sequence of TINY. Thus, we hypothesize that the single nonsynonymous SNP in the TINY coding sequence underlies QTL at LG5.05 across all eight agronomic traits. Future studies will need to utilize functional genetics to validate the phenotypic effect of the single nonsynonymous SNP identified between Carmagnola and USO31.
Quantitative genetic architecture of C. sativa biochemical traits
Our mapping efforts identified a total of 35 QTL associated with measurable variation among the 17 measured biochemical traits. While our model for alpha-pinene contained 6 QTL that spanned our linkage map, most other trait models contained QTL that co-localized on linkage groups 6 and 9. Similar to agronomic traits, the extensive clustering patterns of biochemical QTL at LG6.35 and LG9.40 suggest that most of the cannabinoid and terpene trait differences between the Carmagnola and USO31 parents are controlled by a small number of regions of the C. sativa genome. Again, however, we note the power limitation of our experimental design to identify additional small effect QTL which could explain the residual variance of biochemical traits (Table 2). Interestingly, an epistatic interaction between QTL within LG6.35 and LG9.40. was identified across models of three cannabinoids and five terpenes (Table 2, Figure 3). Our ability to detect these numerous instances of epistasis across biochemical traits is reflective of their greater heritability compared to agronomic traits in C. sativa (Campbell et al. 2019).
The common epistatic interaction identified for numerous biochemical trait models suggests two possible hypotheses for the genes linked to the QTL within LG6.35 and LG9.40. First, we hypothesize that these instances of epistasis may indicate the locations of genes that synthesize precursor molecules to all biochemical traits measured. Alternatively, we hypothesize that these shared epistatic interactions may suggest the presence of genes involved in the interacting biosynthesis pathways for terpenes and cannabinoids (Booth and Bohlmann 2019). In general, terpenes are synthesized through either the plastidial methylerythritol phosphate (MEP) or the cytosolic mevalonate (MEV) pathways while cannabinoids are synthesized through the polyketide pathway (Kovalchuk et al. 2020). The MEP, MEV, and polyketide pathways all utilize geranyl pyrophosphate (GPP) as a substrate to produce their downstream compounds (Szkopińska and Płochocka 2005; Chizzola 2013; Booth et al. 2017; Booth and Bohlmann 2019; Kovalchuk et al. 2020). Evidence also supports that the MEP pathway synthesizes both classes of terpenes in the glandular trichomes and flower tissue (McCaskill and Croteau 1995; Dudareva et al. 2005; Wölwer-Rieck et al. 2014). The polyketide pathway, via CBGa synthase (CBGAS), uses GPP derived from the MEP pathway and olivetolic acid as substrates to form CBGa which is the precursor molecule to all downstream cannabinoids (Fellermeier et al. 2001; Gagne et al. 2012; Kovalchuk et al. 2020).
Using published sequences of genes involved in the MEP, MEV, and polyketide synthesis pathways, we identified olivetol synthase (OLS, Taura et al. 2009) as the candidate gene underlying LG9.40 (Figure 4B). We hypothesize that OLS is the gene underlying LG9.40 because of its critical step in the cannabinoid synthesis pathway and the interaction between OLS and the MEP synthesis pathway. OLS acts in conjunction with olivetolic acid cyclase to produce olivetolic acid, the compound that subsequently combines with MEP-derived GPP to form CBGa via CBGAS (Taura et al. 2009; Gagne et al. 2012; Kovalchuk et al. 2020). With OLS’s critical involvement in the cannabinoid synthesis pathway, variation in the quantity or efficiency of OLS is likely to greatly affect production of cannabinoids (Gagne et al. 2012). We identified nine homozygous amino acid substitutions segregating within the F2 OLS coding sequence which may be responsible for the highly contrasting quantities of cannabinoids produced between the Carmagnola and USO31 parents through alteration of their OLS enzyme function (Supplementary Table S1). Our functional assays in yeast show that the divergent OLS alleles between Carmagnola and USO31 underlie their differences in cannabinoid quantities which suggests that the USO31 OLS may be less efficient at converting hexanoyl-CoA to olivetol (Figure 5, A and B). If the USO31 OLS is less efficient, this would reduce the quantity of cannabinoids produced by USO31 compared to Carmagnola which was a trend we observed in our field experiment (Supplementary Table S1).
Although the OLS functional assay explains why F2 plants with the Carmagnola allele at LG9.40 produced significantly more cannabinoids, it is not clear why the genotype at this QTL cluster also causes differences in terpene production. At LG9.40 F2 plants possessing the Carmagnola allele generally produced more of each terpene compared to individuals with the USO31 allele (Supplementary Table S5). Additional whole-genome data from C. sativa mapping populations is needed to determine whether or not this phenomenon for cannabinoid and terpenes at LG9.40 is population-specific. While general schematics of the MEP, MEV, and polyketide pathways have been described in C. sativa (Fellermeier et al. 2001; Taura et al. 2009; Gagne et al. 2012; Booth et al. 2017; Booth and Bohlmann 2019; Kovalchuk et al. 2020), much still remains uncertain about the nuances of their exact mechanisms. These data at LG9.40 suggest two possible hypotheses. First, we hypothesize that these data may indicate complex interactions between cannabinoid and terpene biosynthesis pathways which have not been previously described. Alternatively, we hypothesize that these data may reflect a pleiotropic regulatory mechanism controlling both cannabinoid and terpene biosynthesis pathways in C. sativa as suggested by Zager et al. (2019). Thus, the observed patterns of terpene production at LG9.40 necessitate more investigation into the genetic mechanisms of the cannabinoid and terpene biosynthesis pathways.
In conclusion, understanding the genetics of complex traits remains a formidable challenge. This is because complex traits can vary considerably ranging from traits controlled by a few QTL of large effect to other traits controlled by a number of loci of small effect. Other factors such as epistasis and dominance add additional complications which further inhibit our ability to fully understand the genetics of complex traits. However, in the past decade, strides have been made in model species by combining WGS with bi-parental mapping populations to identify numerous QTL associated with variation in complex traits such as drug resistance in Caenorhabditis elegans (Burga et al. 2019), ethanol tolerance in yeast (Swinnen et al. 2012), water use physiology in A. thaliana (Mojica et al. 2016), and nitrogen use efficiency in rice (Yang et al. 2017). In less characterized species, genetic understanding of complex traits lags far behind since these species lack many of the well-established genetic resources available to model species. Therefore, it is necessary that for less characterized species such as industrial hemp, investigations of fundamental questions regarding the genetics of complex traits are conducted because they provide the foundations for understanding these species genetic architectures. Despite the prominence of hemp and its numerous uses in society, the genetics of agriculturally important traits in hemp have been seldom investigated. To evaluate the genetic architecture of complex traits in hemp, we used a classical quantitative genetics approach paired with WGS for high-resolution mapping of QTL and heterologous expression in yeast to functionally validate a candidate gene. Rather than adhering to the additive model whereby traits are controlled by numerous loci of small effect (Fisher 1919), we show that the phenotypic distinctions between Carmagnola and USO31 are attributed to a small number of loci of relatively large effect. While additional steps remain necessary to: (1) validate the parental alleles for TINY and (2) resolve the mechanisms of the cannabinoid and terpene biosynthesis pathways, the results discussed here demonstrate the exploration of fundamental complex trait questions in a nonmodel species, improving upon the current understanding of the genetics controlling agriculturally important traits in hemp.
Data availability
The F2 linkage map and phenotype data used for all analyses (QTL mapping, phenotype correlations, and so on.) have been made available on the GSA figshare portal. Supplementary File_S1 contains the raw F2 phenotype data. Supplementary File_S2 contains the quantile normalized F2 phenotype data used for mapping. Supplementary File_S3 contains the F2 linkage map and genotype information. Yeast strains and plasmids are available upon request. Raw fastq files have been deposited to NCBI’s short read archive under BioProject Accession number: PRJNA723060. Supplementary material is available at figshare: https://doi.org/10.25386/genetics.14079962.
Supplementary material is available at GENETICS online.
Author Contributions
B.J.C. created the mapping population and performed the field experiment; T.J.N. assayed transformed yeast colonies by GC-MS and assisted with paper writing; E.B.C. organized, constructed yeast expression vectors, designed yeast assays, and assisted with paper writing; P.W. wrote the paper, performed all computational steps, and analysis of data; J.L.M. calculated the genetic map and assisted with paper writing; J.K.M. designed the experiment, provided manuscript edits and research advice throughout the duration of this study.
Funding
The authors thank the Colorado State University Agricultural Experiment Station for funding this research.
Conflicts of interest
J.K.M. is the Chief Scientific Officer for New West Genetics, Inc. and holder of U.S. Patent 10,499,584 B2.
Acknowledgments
The authors thank Kevin Lehner for helpful comments and Kyle Evans and Anne Howard for assistance with DNA extraction and help in the field. They thank D.B. Sloan, W.C. Funk, and R.A. Hufbauer for their early reviews of this manuscript. They would also like to thank Stefano Amaducci and the Federation Nationale des Producteurs de Chanvre for providing seed of Carmagnola and USO31, respectively.
Literature cited
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- BLAST. 2009. Nucleotide BLAST: Search nucleotide databases using a nucleotide query.
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth JK, Bohlmann J.. 2019. Terpenes in Cannabis sativa – From plant genome to humans. Plant Sci. 284: 67–72. doi:10.1016/j.plantsci.2019.03.022. [DOI] [PubMed] [Google Scholar]
- Booth JK, Page JE, Bohlmann J.. 2017. Terpene synthases from Cannabis sativa. PLoS One. 12:e0173911. doi:10.1371/journal.pone.0173911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen Ś, Churchill GA.. 2003. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 19:889–890. doi:10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- Burga A, Ben-David E, Lemus Vergara T, Boocock J, Kruglyak L.. 2019. Fast genetic mapping of complex traits in C. elegans using millions of individuals in bulk. Nat Commun. 10:2680. doi:10.1038/s41467-019-10636-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell BJ, Berrada AF, Hudalla C, Amaducci S, McKay JK.. 2019. Genotype × environment interactions of industrial hemp cultivars highlight diverse responses to environmental factors. Agrosyst Geosci Environ. 2:1. doi:10.2134/age2018.11.0057. [Google Scholar]
- Campbell LG, Dufresne J, Sabatinos SA.. 2020. Cannabinoid inheritance relies on complex genetic architecture. Cannabis Cannabinoid Res. 5:105–116. doi:10.1089/can.2018.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizzola R. 2013. Regular monoterpenes and sesquiterpenes (essential oils). In: Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes. doi:10.1007/978-3-642-22144-6_130.
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, et al. 2011. The variant call format and VCFtools. Bioinformatics. 27:2156–2158. doi:10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meijer EPM, Bagatta M, Carboni A, Crucitti P, Moliterni VMC, et al. 2003. The inheritance of chemical phenotype in Cannabis sativa L. Genetics. 163:335–346. doi:10.1093/genetics/163.1.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudareva N, Andersson S, Orlova I, Gatto N, Reichelt M, et al. 2005. The nonmevalonate pathway supports both monoterpene and sesquiterpene formation in snapdragon flowers. Proc Natl Acad Sci USA. 102:933–938. doi:10.1073/pnas.0407360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellermeier M, Eisenreich W, Bacher A, Zenk MH.. 2001. Biosynthesis of cannabinoids. Eur J Biochem. 268:1596–1604. doi:10.1046/j.1432-1327.2001.02030.x. [DOI] [PubMed] [Google Scholar]
- Fisher RA. 1919. XV.—The correlation between relatives on the supposition of mendelian inheritance. Trans R Soc Edinb. 52:399–433. doi:10.1017/S0080456800012163. [Google Scholar]
- Gagne SJ, Stout JM, Liu E, Boubakir Z, Clark SM, et al. 2012. Identification of olivetolic acid cyclase from Cannabis sativa reveals a unique catalytic route to plant polyketides. Proc Natl Acad Sci USA. 109:12811–12816. doi:10.1073/pnas.1200330109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallily R, Yekhtin Z, Hanuš LO.. 2015. Overcoming the bell-shaped dose-response of cannabidiol by using cannabis extract enriched in cannabidiol. Pharmacol Pharm. 6:75–85. doi:10.4236/pp.2015.62010. [Google Scholar]
- Grassa CJ, Weiblen GD, Wenger JP, Dabney C, Poplawski SG, et al. 2021. A new Cannabis genome assembly associates elevated cannabidiol (CBD) with hemp introgressed into marijuana. New Phytol. 230:1665–1679. doi:10.1111/nph.17243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG. 2010. Understanding and using quantitative genetic variation. Philos Trans R Soc B. 365:73–85. doi:10.1098/rstb.2009.0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland JB. 2001. Epistasis and plant breeding. Plant Breed Rev. 21:27–92. doi:10.1002/9780470650196.ch2. [Google Scholar]
- Kassambara A. 2020. rstatix: Pipe-Friendly Framework for Basic Statistical Tests. R package version 0.6.0. https://CRAN.R-project.org/package=rstatix
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649. doi:10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk I, Pellino M, Rigault P, van Velzen R, Ebersbach J, et al. 2020. The genomics of Cannabis and its close relatives. Annu Rev Plant Biol. 71:713–739. doi:10.1146/annurev-arplant-081519-040203. [DOI] [PubMed] [Google Scholar]
- Kumar J, Gupta DS, Gupta S, Dubey S, Gupta P, et al. 2017. Quantitative trait loci from identification to exploitation for crop improvement. Plant Cell Rep. 36:1187–1213. doi:10.1007/s00299-017-2127-y. [DOI] [PubMed] [Google Scholar]
- Laverty KU, Stout JM, Sullivan MJ, Shah H, Gill N, et al. 2019. “A physical and genetic map of Cannabis sativa identifies extensive rearrangements at the THC/CBD acid synthase loci”. Genome Res. 29:146–156. doi:10.1101/gr.242594.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 1–3. https://arxiv.org/abs/1303.3997
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. 2009. The sequence Alignment/Map format and SAMtools. Bioinformatics. 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JT. 2019. qtlTools: Data Processing and Plotting in Association with R/qtl. R package version 1.2.0.
- Luo H, Ren X, Li Z, Xu Z, Li X, et al. 2017. Co-localization of major quantitative trait loci for pod size and weight to a 3.7 cM interval on chromosome A05 in cultivated peanut (Arachis hypogaea L.). BMC Genomics. 18:58. doi:10.1186/s12864-016-3456-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Zakaria S, Basyah B, Ma T, Li Z, et al. 2014. Marker-assisted breeding of Indonesia local rice variety Siputeh for semi-dwarf phonetype, good grain quality and disease resistance to bacterial blight. Rice. 7:33. doi:10.1186/s12284-014-0033-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Walsh B.. 1998. Genetics and Analysis of Quantitative Traits. Sunderland: Sinauer Associates. [Google Scholar]
- MacKay TFC, Stone EA, Ayroles JF.. 2009. The genetics of quantitative traits: challenges and prospects. Nat Rev Genet. 10:565–577. doi:10.1038/nrg2612. [DOI] [PubMed] [Google Scholar]
- McCaskill D, Croteau R.. 1995. Monoterpene and sesquiterpene biosynthesis in glandular trichomes of peppermint (Mentha x Piperita) rely exclusively on plastid-derived isopentenyl diphosphate. Planta. 197: 49–56. doi:10.1007/BF00239938. [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. doi:10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehmedic Z, Chandra S, Slade D, Denham H, Foster S, et al. 2010. Potency trends of Δ9-THC and other cannabinoids in confiscated Cannabis preparations from 1993 to 2008. J Forensic Sci. 55:1209–1217. doi:10.1111/j.1556-4029.2010.01441.x. [DOI] [PubMed] [Google Scholar]
- Mojica JP, Mullen J, Lovell JT, Monroe JG, Paul JR, et al. 2016. Genetics of water use physiology in locally adapted Arabidopsis thaliana. Plant Sci. 251:12–22. doi:10.1016/j.plantsci.2016.03.015. [DOI] [PubMed] [Google Scholar]
- Narasimhan V, Danecek P, Scally A, Xue Y, Tyler-Smith C, et al. 2016. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics. 32:1749–1751. doi:10.1093/bioinformatics/btw044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit J, Salentijn EMJ, Paulo MJ, Denneboom C, Trindade LM.. 2020a. Genetic architecture of flowering time and sex determination in hemp (Cannabis sativa L.): a genome-wide association study. Front Plant Sci. 11:569958doi:10.3389/fpls.2020.569958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit J, Salentijn EMJ, Paulo MJ, Denneboom C, van Loo EN, et al. 2020b. Elucidating the genetic architecture of fiber quality in hemp (Cannabis sativa L.) using a genome-wide association study. Front Genet. 11:566314. doi:10.3389/fgene.2020.566314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit J, Salentijn EMJ, Paulo MJ, Thouminot C, van Dinter BJ, et al. 2020c. Genetic variability of morphological, flowering, and biomass quality traits in hemp (Cannabis sativa L.). Front Plant Sci. 11:102. doi:10.3389/fpls.2020.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2019. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria.
- Russo EB. 2011. Taming THC: potential Cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br J Pharmacol. 163:1344–1364. doi:10.1111/j.1476-5381.2011.01238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentijn EMJ, Zhang Q, Amaducci S, Yang M, Trindade LM.. 2015. New developments in fiber hemp (Cannabis sativa L.) breeding. Industrial Crops Products. 68:32–41. doi:10.1016/j.indcrop.2014.08.011. [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31:3210–3212. doi:10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Sirikantaramas S, Morimoto S, Shoyama Y, Ishikawa Y, Wada Y, et al. 2004. The gene controlling marijuana psychoactivity. Molecular cloning and heterologous expression of Δ1-tetrahydrocannabinolic acid synthase from Cannabis sativa L. J Biol Chem. 279:39767–39774. doi:10.1074/jbc.M403693200. [DOI] [PubMed] [Google Scholar]
- Sirikantaramas S, Taura F, Tanaka Y, Ishikawa Y, Morimoto S, et al. 2005. Tetrahydrocannabinolic acid synthase, the enzyme controlling marijuana psychoactivity, is secreted into the storage cavity of the glandular trichomes. Plant Cell Physiol. 46:1578–1582. doi:10.1093/pcp/pci166. [DOI] [PubMed] [Google Scholar]
- Speed D, Balding DJ.. 2012. Understanding complex traits: from farmers to pharmas. Genome Med. 4:59. doi:10.1186/gm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Steinkamp R, Waack S, Morgenstern B.. 2004. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32:W309–W312. doi:10.1093/nar/gkh379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struik PC, Amaducci S, Bullard MJ, Stutterheim NC, Venturi G, et al. 2000. Agronomy of fibre hemp (Cannabis sativa L.) in Europe. Industrial Crops Products. 11:107–118. doi:10.1016/S0926-6690(99)00048-5. [Google Scholar]
- Swinnen S, Schaerlaekens K, Pais T, Claesen J, Hubmann G, et al. 2012. Identification of novel causative genes determining the complex trait of high ethanol tolerance in yeast using pooled-segregant whole-genome sequence analysis. Genome Res. 22:975–984. doi:10.1101/gr.131698.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szkopińska A, Płochocka D.. 2005. Farnesyl diphosphate synthase; regulation of product specificity. Acta Biochim Pol. 52:45–55. doi:10.18388/abp.2005_3485. [PubMed] [Google Scholar]
- Taura F, Tanaka S, Taguchi C, Fukamizu T, Tanaka H, et al. 2009. Characterization of olivetol synthase, a polyketide synthase putatively involved in cannabinoid biosynthetic pathway. FEBS Lett. 583:2061–2066. doi:10.1016/j.febslet.2009.05.024. [DOI] [PubMed] [Google Scholar]
- Toth JA, Stack GM, Cala AR, Carlson CH, Wilk RL, et al. 2020. Development and validation of genetic markers for sex and cannabinoid chemotype in Cannabis sativa L. GCB Bioenergy. 12:213–222. doi:10.1111/gcbb.12667. [Google Scholar]
- U.S. Govt. 2014. Print. Farm Bill – Section 7606 (enacted). Print.
- U.S. Govt. 2018. Print. Farm Bill – Sections 7125, 7401, 7415, 7501,7605, 10111, 10113, 11101, 11106, 11112, 11120, 11121, 12608, 12619 (enacted). Print.
- Van der Werf HMG, Brouwer K, Wijlhuizen M, Withagen JCM.. 1995a. The effect of temperature on leaf appearance and canopy establishment in fibre hemp (Cannabis sativa L.). Ann Appl Biol. 126:551–561. doi:10.1111/j.1744-7348.1995.tb05389.x. [Google Scholar]
- Van der Werf HMG, van Geel WCA, van Gils LJC, Haverkort AJ.. 1995b. Nitrogen fertilization and row width affect self-thinning and productivity of fibre hemp (Cannabis sativa L.). Field Crops Res. 42:27–37. doi:10.1016/0378-4290(95)00017-K. [Google Scholar]
- Van Ooijen JW. 2006. Kyazma. JoinMap ® 4. JoinMap. https://www.kyazma.nl/index.php/JoinMap/
- Weiblen GD, Wenger JP, Craft KJ, ElSohly MA, Mehmedic Z, et al. 2015. Gene duplication and divergence affecting drug content in Cannabis sativa. New Phytol. 208:1241–1250. doi:10.1111/nph.13562. [DOI] [PubMed] [Google Scholar]
- Wenger JP, Dabney CJ, ElSohly MA, Chandra S, Radwan MM, et al. 2020. Validating a predictive model of cannabinoid inheritance with feral, clinical, and industrial Cannabis sativa. Am J Bot. 107:1423–1432. doi:10.1002/ajb2.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2016. ggplot2 Elegant Graphics for Data Analysis. J R Stat Soc Ser A (Stat Soc). 2:1–260. doi:10.1007/978-3-319-24277-4. [Google Scholar]
- Wilson K, Long D, Swinburne J, Coupland G.. 1996. A dissociation insertion causes a semidominant mutation that increases expression of TINY, an Arabidopsis gene related to APETALA2. Plant Cell. 8:659–671. doi:10.2307/3870342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wölwer-Rieck U, May B, Lankes C, Wüst M.. 2014. Methylerythritol and mevalonate pathway contributions to biosynthesis of mono-, sesqui-, and diterpenes in glandular trichomes and leaves of Stevia rebaudiana Bertoni. J Agric Food Chem. 62:2428–2435. doi:10.1021/jf500270s. [DOI] [PubMed] [Google Scholar]
- Xie Z, Nolan T, Jiang H, Tang B, Zhang M, et al. 2019. The AP2/ERF transcription factor TINY modulates brassinosteroid-regulated plant growth and drought responses in Arabidopsis. Plant Cell. 31:1788–1806. doi:10.1105/tpc.18.00918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Xia X, Zhang Z, Nong B, Zeng Y, et al. 2017. QTL mapping by whole genome re-sequencing and analysis of candidate genes for nitrogen use efficiency in rice. Front Plant Sci. 8:1634. doi:10.3389/fpls.2017.01634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef GG, Juvik JA.. 2002. Enhancement of seedling emergence in sweet corn by marker-assisted backcrossing of beneficial QTL. Crop Sci. 42:96. doi:10.2135/cropsci2002.0096. [DOI] [PubMed] [Google Scholar]
- Zager JJ, Lange I, Srividya N, Smith A, Lange BM.. 2019. Gene networks underlying cannabinoid and terpenoid accumulation in Cannabis. Plant Physiol. 180:1877–1897. doi:10.1104/pp.18.01506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirpel B, Kayser O, Stehle F.. 2018. Elucidation of structure-function relationship of THCA and CBDA synthase from Cannabis sativa L. J Biotechnol. 284:17–26. doi:10.1016/j.jbiotec.2018.07.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The F2 linkage map and phenotype data used for all analyses (QTL mapping, phenotype correlations, and so on.) have been made available on the GSA figshare portal. Supplementary File_S1 contains the raw F2 phenotype data. Supplementary File_S2 contains the quantile normalized F2 phenotype data used for mapping. Supplementary File_S3 contains the F2 linkage map and genotype information. Yeast strains and plasmids are available upon request. Raw fastq files have been deposited to NCBI’s short read archive under BioProject Accession number: PRJNA723060. Supplementary material is available at figshare: https://doi.org/10.25386/genetics.14079962.
Supplementary material is available at GENETICS online.