

Abstract
Compared to its predecessors, the Telomere-to-Telomere CHM13 genome adds nearly 200 million base pairs of sequence, corrects thousands of structural errors, and unlocks the most complex regions of the human genome for clinical and functional study. We show how this reference universally improves read mapping and variant calling for 3202 and 17 globally diverse samples sequenced with short and long reads, respectively. We identify hundreds of thousands of variants per sample in previously unresolved regions, showcasing the promise of the T2T-CHM13 reference for evolutionary and biomedical discovery. Simultaneously, this reference eliminates tens of thousands of spurious variants per sample, including reduction of false positives in 269 medically relevant genes by up to a factor of 12. Because of these improvements in variant discovery coupled with population and functional genomic resources, T2T-CHM13 is positioned to replace GRCh38 as the prevailing reference for human genetics.
Graphical Abstract
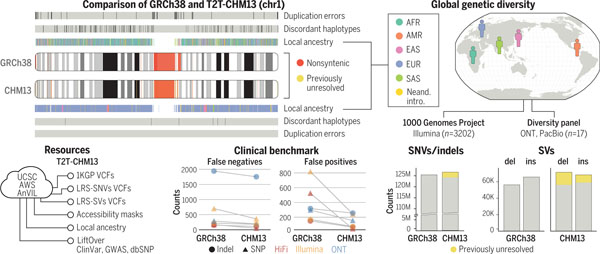
Genomic features and resources available for T2T-CHM13. Comparisons to GRCh38 reveal broad improvements in SNVs, indels, and SVs discovered across diverse human populations by means of short-read (1KGP) and long-read sequencing (LRS). These improvements are due to resolution of complex genomic loci (nonsyntenic and previously unresolved), duplication errors, and discordant haplotypes, including those in medically relevant genes.
RESEARCH ARTICLE SUMMARY
INTRODUCTION:
One of the central applications of the human reference genome has been to serve as a baseline for comparison in nearly all human genomic studies. Unfortunately, many difficult regions of the reference genome have remained unresolved for decades and are affected by collapsed duplications, missing sequences, and other issues. Relative to the current human reference genome, GRCh38, the Telomere-to-Telomere CHM13 (T2T-CHM13) genome closes all remaining gaps, adds nearly 200 million base pairs (Mbp) of sequence, corrects thousands of structural errors, and unlocks the most complex regions of the human genome for scientific inquiry.
RATIONALE:
We demonstrate how the T2TCHM13 reference genome universally improves read mapping and variant identification in a globally diverse cohort. This cohort includes all 3202 samples from the expanded 1000Genomes Project (1KGP), sequenced with short reads, as well as 17 globally diverse samples sequenced with long reads. By applying state-of-the-art methods for calling single-nucleotide variants (SNVs) and structural variants (SVs), we document the strengths and limitations of T2T-CHM13 relative to its predecessors and highlight its promise for revealing new biological insights within technically challenging regions of the genome.
RESULTS:
Across the 1KGP samples, we found more than 1 million additional high-quality variants genome-wide using T2T-CHM13 than with GRCh38. Within previously unresolved regions of the genome, we identified hundreds of thousands of variants per sample—a promising opportunity for evolutionary and biomedical discovery. T2T-CHM13 improves the Mendelian concordance rate among trios and eliminates tens of thousands of spurious SNVs per sample, including a reduction of false positives in 269 challenging, medically relevant genes by up to a factor of 12. These corrections are in large part due to improvements to 70 protein-coding genes in >9 Mbp of inaccurate sequence caused by falsely collapsed or duplicated regions in GRCh38. Using the T2T-CHM13 genome also yields a more comprehensive view of SVs genome-wide, with a greatly improved balance of insertions and deletions. Finally, by providing numerous resources for T2T-CHM13 (including 1KGP genotypes, accessibility masks, and prominent annotation databases), our work will facilitate the transition to T2T-CHM13 from the current reference genome.
CONCLUSION:
The vast improvements in variant discovery across samples of diverse ancestries position T2T-CHM13 to succeed as the next prevailing reference for human genetics. T2T-CHM13 thus offers a model for the construction and study of high-quality reference genomes from globally diverse individuals, such as is now being pursued through collaboration with the Human Pangenome Reference Consortium. As a foundation, our work underscores the benefits of an accurate and complete reference genome for revealing diversity across human populations.
For the past 20 years, the human reference genome (GRCh38) has served as the bed-rock of human genetics and genomics (1–3). One of the central applications of the human reference genome, and of reference genomes in general, has been to serve as a substrate for clinical, comparative, and population genomic analyses. More than 1 million human genomes have been sequenced to study genetic diversity and clinical relationships, and nearly all of them have been analyzed by aligning the sequencing reads from the donors to the reference genome [e.g., (4–6)]. Even when donor genomes are assembled de novo, independent of any reference, the assembled sequences are almost always compared to a reference genome to characterize variation by leveraging deep catalogs of available annotations (7, 8). Consequently, human genetics and genomics benefit from the availability of a high-quality reference genome, ideally without gaps or errors that may obscure important variation and regulatory relationships.
The current human reference genome, GRCh38, is used for countless applications, with rich resources available to visualize and annotate the sequence across cell types and disease states (3, 9–12). However, despite decades of effort to construct and refine its sequence, the human reference genome still suffers from several major limitations that hinder comprehensive analysis. Most immediately, GRCh38 contains more than 100 million nucleotides that either remain entirely unresolved (currently represented as “N” characters), such as the p-arms of the acrocentric chromosomes, or are substituted with artificial models, such as the centromeric satellite arrays (13). Furthermore, GRCh38 possesses 11.5 Mbp of unplaced and unlocalized sequences that are represented separately from the primary chromosomes (3, 14). These sequences are difficult to study, and many genomic analyses exclude them to avoid identifying false variants and false regulatory relationships (6). Relatedly, artifacts such as an apparent imbalance between insertions and deletions (indels) have been attributed to systematic misassemblies in GRCh38 (15–17). Overall, these errors and omissions in GRCh38 introduce biases in genomic analyses, particularly in centromeres, satellites, and other complex regions.
Another major concern regards the influence of the reference genome on analysis of variation across large cohorts for population and clinical genomics. Several studies, such as the 1000 Genomes Project (1KGP) (18) and gnomAD (6), have provided information about the extent of genetic diversity within and between human populations. Many analyses of Mendelian and complex diseases use these catalogs of single-nucleotide variants (SNVs), small indels, and structural variants (SVs) to rank and prioritize potential causal variants on the basis of allele frequencies (AFs) and other evidence (19–21). When evaluating these resources, the overall quality and representativeness of the human reference genome should be considered. Any gaps or errors in the sequence could obscure variation and its contribution to human phenotypes and disease.
In addition to omissions such as centromeric sequences or acrocentric chromosome arms, the current reference genome possesses other errors and biases, including within genes of known medical relevance (22, 23). Moreover, GRCh38 was assembled from multiple donors with clone-based sequencing, which creates an excess of artificial haplotype structures that can subtly bias analyses (1, 24). Over the years, there have been attempts to replace certain rare alleles with more common alleles, but hundreds of thousands of artificial haplotypes and rare alleles remain to this day (3, 25, 26). Increasing the continuity, quality, and representativeness of the reference genome is therefore crucial for improving genetic diagnosis, as well as for understanding the complex relationship between genetic and phenotypic variation.
The Telomere-to-Telomere (T2T) CHM13 genome addresses many of the limitations of the current reference (27). Specifically, the T2T-CHM13v1.0 assembly adds nearly 200 Mbp of sequence and resolves errors present in GRCh38. Here, we demonstrate the impact of the T2T-CHM13 reference on variant discovery and genotyping in a globally diverse cohort. This includes all 3202 samples from the recently expanded 1KGP sequenced with short reads (28) along with 17 samples from diverse populations sequenced with long reads (8, 27, 29). Our analysis reveals more than 2 million variants within previously unresolved regions of the genome, genome-wide improvements in SV discovery, and enhancement in variant calling accuracy across 622 medically relevant genes. In summary, our work demonstrates universal improvements in read mapping and variant calling, thereby broadening the horizon for future genomic studies.
Structural comparisons of GRCh38 and T2T-CHM13
Introducing the T2T-CHM13 genome
The T2T-CHM13 reference genome was primarily assembled from Pacific Biosciences (PacBio) High Fidelity (HiFi) reads augmented with Oxford Nanopore Technology (ONT) reads to close gaps and resolve complex repeats (27). The resulting T2T-CHM13v1.0 assembly was subsequently validated and polished, with a consensus accuracy estimated to be between Phred Q67 and Q73 (27, 30) and with only three minor known structural defects detected (30). The assembly is highly contiguous, with only five unresolved regions from the most highly repetitive ribosomal DNA (rDNA) arrays, representing only 9.9 Mbp of sequence out of >3.0 Gbp of fully resolved sequence. The version 1.0 assembly adds or revises 229 Mbp of sequence compared to GRCh38; these are “nonsyntenic” regions of the T2T-CHM13 assembly that do not linearly align to GRCh38 over a 1-Mbp interval. Furthermore, 189 Mbp of sequence are not covered by any primary alignments from GRCh38 and are resolved in the T2T-CHM13 assembly. Figure 1A is a summary diagram of the syntenic/nonsyntenic regions and their associated annotations for chromosomes 1 and 21 (figs. S1 to S4 give details for all chromosomes). Note that the subsequent T2T-CHM13v1.1 assembly (27) further resolves the rDNA regions using model sequences for some array elements, although for this study we analyze the v1.0 assembly, which does not contain these representations.
Fig. 1. Genomic comparisons of human assemblies GRCh38 and T2T-CHM13.
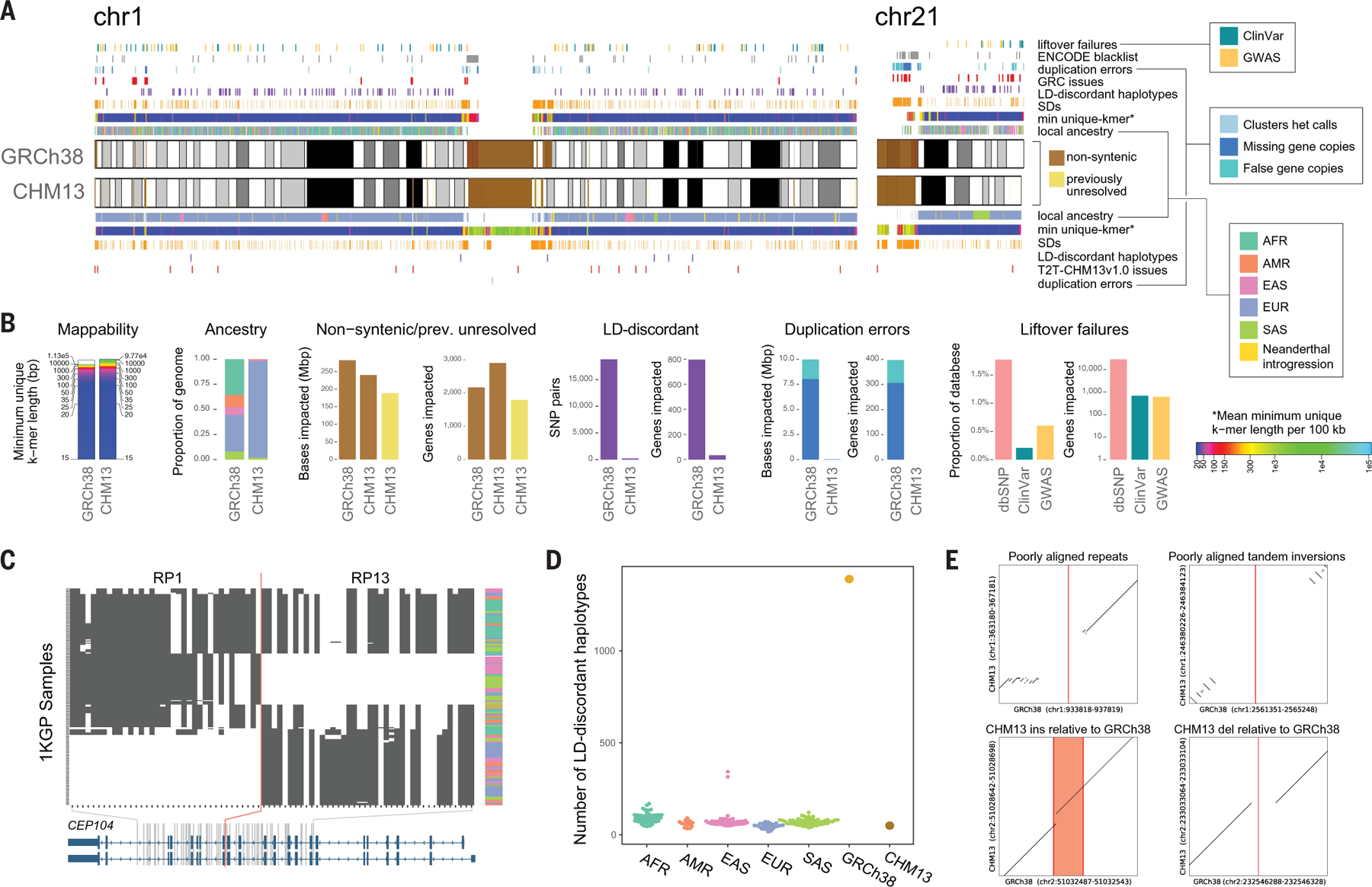
(A) Overview of annotations available for GRCh38 and T2T-CHM13 chromosomes 1 and 21 with colors indicated in legends, which are also used in (B) to (D). Colors for mean minimum (min) unique k-mers are defined in the legend with indicated asterisk. Cytobands are pictured as gray bands with red bands representing centromeric regions within ideograms. Complete annotations of all chromosomes can be found in figs. S1 to S4. Local ancestry is denoted using 1KGP superpopulation abbreviations (AFR, African; AMR, admixed American; EAS, East Asian; EUR, European; SAS, South Asian). (B) Summary of the number of bases and/or genes annotated by different features for the assemblies with colors indicated in the legends shown in (A). Note, dbSNP liftover failures (pink) are not annotated in (A). (C) Example of a clone boundary (red line) where GRCh38 possesses a combination of alleles that segregate in negative LD within the 1KGP sample (which we term as an “LD-discordant haplotype”). SNPs are depicted in columns; phased 1KGP samples are depicted in rows. White indicates reference allele genotype; black indicates alternative allele genotypes. Superpopulation ancestry of each sample is indicated in the rightmost column with colors indicated in local ancestry legend shown in (A). CEP104 splice isoforms (blue) are depicted at the bottom. (D) Tally of such LD-discordant haplotypes in a selection of 1KGP individuals, colored by population, as well as GRCh38 and T2T-CHM13. (E) Examples of variants that cannot be lifted over to T2T-CHM13 because of structural differences between the genomes. The position of the reference allele in GRCh38 is shown in red.
The bulk of the nonsyntenic sequence within T2T-CHM13 comprises centromeric satellites (190 Mbp) (31) and copies of segmental duplications (SDs; 218 Mbp) (32). These sequences could prove challenging for variant analysis, especially for variants identified using short-read sequencing. However, relative to GRCh38, we report an overall increase in unique sequence, defined as k-length strings (k-mers) found only once in the genome (e.g., 14.9 Mbp of added unique sequence when considering 50-mers, 23.5 Mbp for 100-mers, and 39.5 Mbp for 300-mers). These sequences delineate regions of confident mapping for short paired-end reads or longer reads, including those in previously unrepresented portions of the genome (Fig. 1B and figs. S5 and S6).
More than 106 Mbp of sequence absent from GRCh38 was identified in T2T-CHM13 within highly repetitive regions that require reads of more than 300 bp to uniquely map. Concomitantly, T2T-CHM13 possesses fewer exactly duplicated sequences (≥5 kbp) shared across chromosomes (excluding sequence pairs within centromeres) than GRCh38 (figs. S7 and S8). Specifically, GRCh38 possesses 28 large shared interchromosomal sequences, primarily consisting of pairs of subtelomeric sequences, with an additional 42 pairs involving at least one unplaced contig. All of these identical sequence pairs, except for one between two subtelomeres, are nonidentical in T2T-CHM13, as small but important differences between repetitive elements have now been resolved (27, 33).
T2T-CHM13 accurately represents the haplotype structure of human genomes
The human reference genome serves as the standard to which other genomes are compared, and is typically perceived as a haploid representation of an arbitrary genome from the population (25). In contrast with T2T-CHM13, which derives from a single homozygous complete hydatidiform mole, the Human Genome Project constructed the current reference genome via the tiling of sequences obtained from bacterial artificial chromosomes (BACs) and other clones with lengths ranging from ~50 to >250 kbp (24), which derived from multiple donor individuals. GRCh38 and its predecessors thus comprise mosaics of many haplotypes, albeit with a single library (RP11) contributing the majority (24).
To further characterize this aspect of GRCh38 and its implications for population studies, we performed local ancestry inference for both GRCh38 and T2T-CHM13 through comparison to haplotypes from the 1KGP (34) (Fig. 1A and figs. S2 and S9). Continental superpopulation-level ancestry was inferred for 72.9% of GRCh38 clones on the basis of majority votes of nearest-neighbor haplotypes. For the remaining 27.1% of clones, no single superpopulation achieved a majority of nearest neighbors, and ancestry thus remained ambiguous. This ambiguity occurred primarily for short clones with few informative SNPs (fig. S10), but also for some longer clones with potential admixed ancestry.
In accordance with Green et al. (24), we inferred that library RP11, which constitutes 72.6% of the genome, is derived from an individual of admixed African-American ancestry, with 56.0% and 28.1% of its component clones assigned to African and European local ancestries, respectively. The second most abundant library, CTD (5.5% of the genome), consists of clones of predominantly (86.3%) East Asian local ancestries, and the remaining libraries are derived from individuals of predominantly European ancestries. In contrast, T2T-CHM13 exhibits European ancestries nearly genome-wide (fig. S11). In addition, GRCh38 and T2T-CHM13 harbor 26.7 Mbp and 51.0 Mbp, respectively, of putative Neanderthal-introgressed sequences that originated from ancient inter-breeding between the two hominin groups ~60,000 years ago (24). The excess of introgressed sequence in T2T-CHM13, even when restricting to the genomic intervals of GRCh38 clones with confident ancestry assignments, is consistent with its greater proportion of non-African ancestry.
We hypothesized that the mosaic nature of GRCh38 would generate abnormal haplotype structures at the boundaries of clones used for its construction, producing combinations of alleles that are rare or absent from the human population. Indeed, some previous patches of the reference genome sought to correct abnormal haplotype structures wherever noticed because of their impacts on genes of clinical importance (e.g., ABO and SLC39A4) (3). Such artificial haplotypes would mimic rare recombinant haplotypes private to any given sample, but at an abundance and genomic scale unrepresentative of any living human. To test this hypothesis, we identified pairs of common [minor allele frequency (MAF) > 10%] SNP alleles always observed on the same haplotype [i.e., segregate in perfect (R2 = 1) linkage disequilibrium (LD)] in the 2504 unrelated individuals of the 1KGP and queried the allelic states of these SNPs in both GRCh38 and T2T-CHM13 (34).
In accordance with our expectations, we identified numerous haplotype transitions in GRCh38 absent from the 1KGP samples, with 18,813 pairs of LD-discordant SNP alleles (i.e., in perfect negative LD) distributed in 1390 narrow nonoverlapping clusters (median length = 3703 bp) throughout the genome (Fig. 1C). Such rare haplotype transitions are comparatively scarce in T2T-CHM13, with only 209 pairs of common high-LD SNPs (50 nonoverlapping clusters) possessing allelic combinations absent from the 1KGP sample (Fig. 1D). Using a leave-one-out analysis, we confirmed that T2T-CHM13 possesses a similar number of LD-discordant haplotypes as phased “haploid” samples from 1KGP, whereas GRCh38 vastly exceeds this range (Fig. 1E). By intersecting the GRCh38 results with the tiling path of BAC clones, we found that 88.9% (16,733 of 18,813) of discordant SNP pairs straddle the documented boundaries of adjacent clones (fig. S12). Of these, 45.9% (7686 of 16,733) of the clone pairs derived from different BAC libraries, whereas the remainder likely largely reflects random sampling of distinct homologous chromosomes from the same donor individual. Thus, our analysis suggests that T2T-CHM13 accurately reflects haplotype patterns observed in contemporary human populations, whereas GRCh38 does not.
T2T-CHM13 corrects genomic collapsed duplications and falsely duplicated regions
Genome assemblies often suffer from errors in complex genomic regions such as SDs. In the case of GRCh38, targeted sequencing of BAC clones has been performed to fix many such loci (3, 16, 35–38), but problems persist. To systematically identify errors in GRCh38 that could produce spurious variant calls, we leveraged the fact that T2T-CHM13 is an effectively haploid cell line that should produce only homozygous variants when its sequence is aligned to GRCh38. Thus, any apparent heterozygous variant can be attributed to mutations accrued in the cell line, sequencing errors, or read mapping errors. In the last case, assembly errors or copy number polymorphism of SDs produce contiguous stretches of heterozygous variants (39), which confound the accurate detection of paralog-specific variants (PSVs). By mapping PacBio HiFi reads from the CHM13 cell line (27) as well as Illumina-like simulated reads (150 bp) obtained from the T2T-CHM13 reference to GRCh38, we identified 368,574 heterozygous SNVs within the autosomes and chromosome X, of which 56,413 (15.3%) were shared between datasets. This evidence shows that each technology is distinctively informative as a result of differences in mappability (fig. S13 and table S1).
To home in on variants deriving from collapsed duplications, we delineated “clusters” of heterozygous calls (34) and identified 908 putative problematic regions (541 supported by both technologies) comprising 20.8 Mbp (Fig. 1 and fig. S13). Many of these loci intersected SD-associated regions (668/908; 73.6%) and centromere-associated regions (542/908; 59.7%) (31) as well as known GRCh38 issues (341/908; 37.55%). Variants flagged as excessively heterozygous in the population by gnomAD (6) were significantly enriched in these regions (10,000 permutations, empirical P = 1 × 10−4), representing 23.6% (87,005/368,574) of our discovered CHM13 heterozygous variants, which suggests that these spurious variants arise in genome screens and represent false positives (Fig. 1A and figs. S1 and S3).
We next “lifted over” (i.e., converted the coordinates of) 821 of these 908 putative problematic regions to the T2T-CHM13 assembly and used human copy number estimates [n = 268 individuals from the Simons Genome Diversity Project (SGDP)] (32, 40) to conservatively identify 203 loci (8.04 Mbp) evidencing missing copies in GRCh38 (fig. S14). These regions have an impact on 308 gene features, with 14 of the total 48 protein-coding genes fully contained within a problematic region, indicating that complete gene homologs are hidden from GRCh38-based population analyses of variation. Examples include DUSP22,a gene involved in immune regulation (41), as well as KMT2C, a gene implicated in Kleefstra syndrome 2 (42) (fig. S15). Additionally, we identified 30 SNPs within problematic regions with known phenotype associations from the GWAS Catalog (43). Finally, we evaluated the status of these regions in the T2T-CHM13 reference by following a similar approach to obtain 9193 heterozygous variants clustered in 11 regions—none of which overlapped GRCh38 problematic regions (table S2). As a result, we are now able to call variants in these 48 previously inaccessible protein-coding genes. We did identify one putative collapsed duplication in T2T-CHM13, based on the presence of a heterozygous variant cluster and reduced copy number in T2T-CHM13, localized to an rDNA array corrected in the most recent version of T2T-CHM13v1.1 (27).
Conversely, the T2T-CHM13 reference also corrects regions falsely portrayed as duplicated in GRCh38. Specifically, we identified 12 regions affecting 1.2 Mbp and 74 genes (including 22 protein-coding genes) with duplications private to GRCh38 and not found in T2T-CHM13 or the 268 genomes from SGDP (40) (fig. S14 and table S3). In contrast, only five regions affecting 160 kbp have duplications in T2T-CHM13 that are not in GRCh38 or the SGDP, which suggests that genuine rare variation cannot explain the excess of private duplications in GRCh38. Indeed, upon inspecting the CHM13 data, we deemed that these five loci are true duplications with support from mapped HiFi reads (30).
The five largest duplications in GRCh38, affecting 15 protein-coding genes on the q-arm of chromosome 21, involve BAC clones with sequence misplaced between gaps on the heterochromatic p-arm of the same chromosome. The Genome Reference Consortium (GRC)—an international team of researchers that has maintained and improved the reference genome and related resources since its initial publication— determined that admixture mapping incorrectly localized these five clones to the acrocentric short arm and therefore should not have been added to GRCh38 (34). Of the seven false duplications outside chromosome 21, two occur in short contigs between gaps, two occur adjacent to a gap, two occur on unlocalized “random” contigs, and one occurs as a tandem duplication (table S4). We provide an exhaustive list of falsely duplicated gene pairs corrected in T2T-CHM13 (table S5). Thus, T2T-CHM13 authoritatively corrects many false duplications, improving variant calling for short- and long-read technologies, including in medically relevant genes.
Liftover of clinically relevant and trait-associated variation from GRCh38 to T2T-CHM13
In transitioning to a different reference genome, it is imperative to document the locations of known genetic variation of biological and clinical relevance with respect to the updated coordinate system. To this end, we sought to lift over 802,674 unique variants in the ClinVar database and 736,178,420 variants from the NCBI dbSNP database (including 151,876 NHGRI-EBI GWAS Catalog variants) from the GRCh38 reference to the T2T-CHM13 reference. Liftover was successful for 800,942 (99.8%) ClinVar variants, 723,117,125 (98.2%) NCBI dbSNP variants, and 150,962 (99.4%) GWAS Catalog SNPs (table S6). We provide these lifted-over datasets as a resource for the scientific community within the UCSC Genome Browser and the NHGRI AnVIL, along with lists of all variants that failed liftover and the associated reasons (Fig. 1, A and B, and figs. S1 and S4). Critically, this resource includes 138,319 of 138,927 (99.6%) ClinVar variants annotated as “pathogenic” or “likely pathogenic.”
Of the 1732 ClinVar variants that failed to lift over, 1186 overlap documented insertions or deletions that distinguish the GRCh38 and T2T-CHM13 assemblies. The remaining 546 variants (<0.1% of all variants) lie within regions of poor alignment between the GRCh38 and T2T-CHM13 assemblies (Fig. 1E). The modes of liftover failure for variants in dbSNP and the GWAS Catalog follow similar distributions (table S6). In all, these annotated variants offer a resource to enable researchers to interpret genetic results using the T2T-CHM13 assembly.
T2T-CHM13 improves analysis of global genetic diversity based on 3202 short-read samples from the 1KGP dataset
T2T-CHM13 improves short-read mapping across populations
To investigate how the T2T-CHM13 assembly affects short-read variant calling, we realigned and reprocessed all 3202 samples from the 1KGP cohort (28) using the NHGRI AnVIL Platform (44) (figs. S16 and S17). In this collection, each sample is sequenced to at least 30× coverage with paired-end Illumina sequencing, with samples from 26 diverse populations across five major continental superpopulations (fig. S18). Although most samples are unrelated, the expanded collection includes 602 complete trios that we used to estimate the rate of false variants below on the basis of discordance with Mendelian expectations. We matched the analysis pipeline for GRCh38 (28) as closely as possible so that any major differences would be attributable to the reference genome rather than technical differences in the analysis software (34).
On average, BWA-MEM (45) maps an additional 7.4 × 106 (0.97%) of properly paired reads to T2T-CHM13 compared to GRCh38, even when considering the alternative (ALT) and decoy sequences used in the original analysis (fig. S19). Interestingly, even though more reads align to T2T-CHM13, the subsequent per-read mismatch rate is 20 to 25% lower across all continental populations. African samples continue to present the highest mismatch rate (Fig. 2A), as the observed mismatch rate includes both genuine sequencing errors, which are largely consistent across all samples, and any true biological differences between the read and the reference genome, which vary substantially according to the ancestry of the sample. Relatedly, T2T-CHM13 improved other mapping characteristics, including reducing the number of misoriented read pairs (Fig. 2A). Finally, by considering the alignment coverage across 500-bp bins across the respective genomes, we observed improvement in coverage uniformity within every sample’s genome when using T2T-CHM13 rather than GRCh38. For example, within gene regions, we noted a factor of 4 decrease in the standard deviation of the coverage (Fig. 2A) and similar improvements in other types of genomic regions among all population groups (fig. S20). Overall, these improvements in error rates, mapping characteristics, and coverage uniformity demonstrate the superiority of T2T-CHM13 as a reference genome for short-read alignment across all populations.
Fig. 2. Improvements to short-read mapping and variant calling.
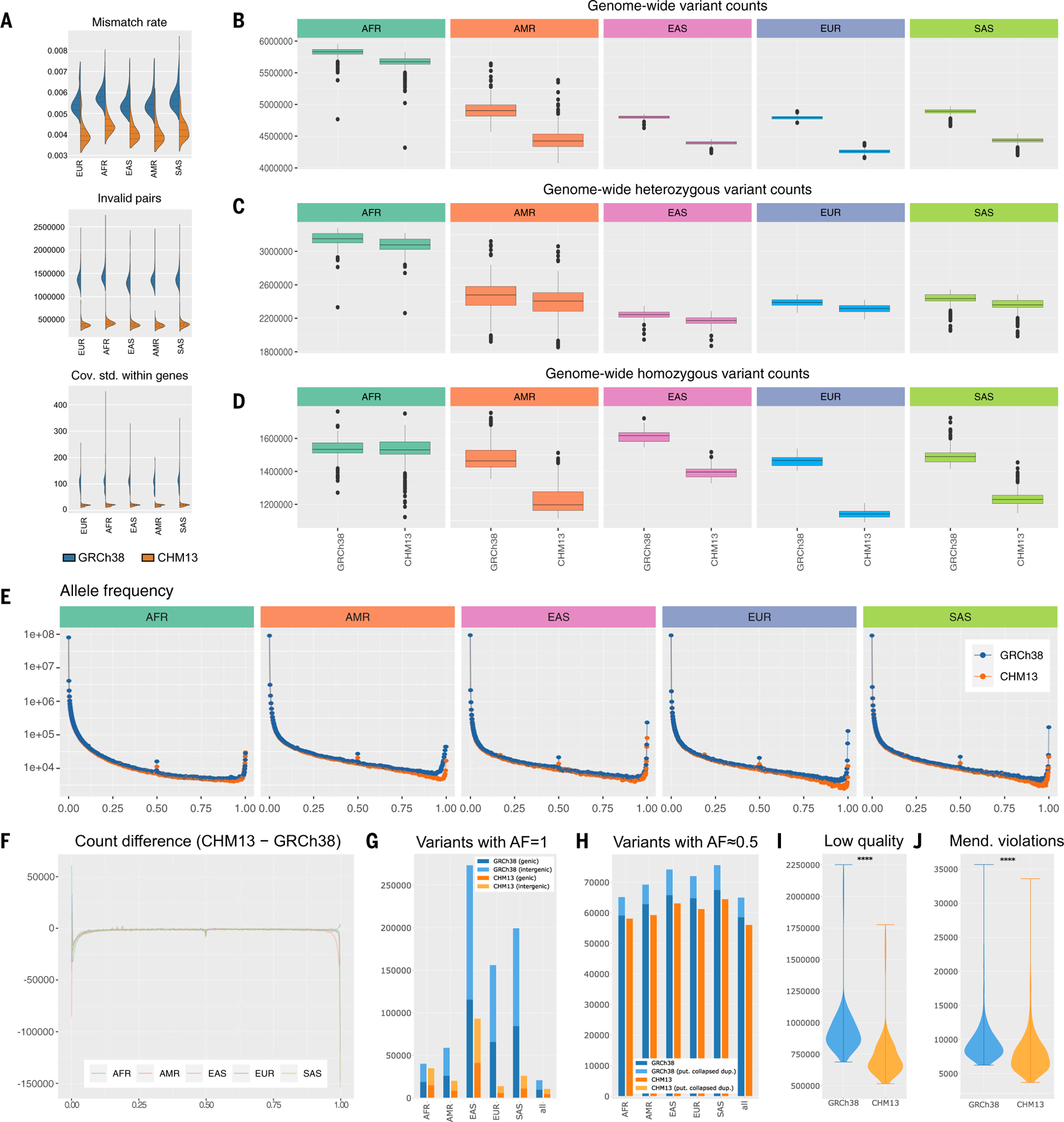
(A) Summary of alignment characteristics aligning to T2T-CHM13 instead of GRCh38. (B) Boxplot of overall number of variants found in each person across superpopulations, with colors indicated in Fig. 1A legend. (C) Boxplot of the number of heterozygous variants found in each person across superpopulations. (D) Boxplot of the number of homozygous variants found in each person across superpopulations. (E) AF distribution of each superpopulation relative to T2T-CHM13 and GRCh38. (F) Change in AF distribution. (G) Number of variants with AF equal to 100%, both within protein-coding genes and without. (H) Number of variants with AF equal to 50%, both within putative collapsed duplications and without. (I) Violin plot of the number of low-quality variants found when aligning to GRCh38 and T2T-CHM13. (J) Violin plot of the number of Mendelian violations found when aligning to GRCh38 and T2T-CHM13.
T2T-CHM13 improves variant calling across populations
From these alignments, we next generated SNV and small indel variant calls with the GATK Haplotype Caller, which uses a joint genotyping approach to optimize accuracy across large populations (46). Again, we matched the pipeline used in the prior 1KGP study, albeit with updated versions of some analysis tools, to minimize software discrepancies and attribute differences to changes in the reference genome. Across all samples, we identified 126,591,489 high-quality (“PASS”) variants relative to T2T-CHM13 (per-sample mean, 4,717,525; median, 4,419,802) compared to 125,484,020 variants relative to GRCh38 (per-sample mean, 5,101,897; median, 4,867,871), additionally noting a decrease in the number of called variants per individual genome (Fig. 2B and fig. S21). We performed all subsequent analyses using these high-quality variants, as the PASS filter successfully removed spurious variants (fig. S22), particularly in complex regions (fig. S23).
As with the improvement to the per-read mismatch rate, we attribute the reduction in the number of per-sample variant calls to improvements in the number of rare alleles, consensus errors, and structural errors in T2T-CHM13. This conclusion is supported by the observation that the number of heterozygous variants per sample is more similar (Fig. 2C and fig. S24) across reference genomes in contrast to homozygous variants (Fig. 2D and fig. S24). This discrepancy is especially pronounced in non-African samples, which have on average 200,000 to 300,000 more homozygous variants relative to GRCh38 than to T2T-CHM13, likely because ~70% of the GRCh38 sequence comes from an individual with African-American ancestry, and African populations are enriched for rare and private variants (18).
Further investigating this relationship, we computed the AFs of variants from unrelated samples from each of the five continental superpopulations (Fig. 2E). Although the distributions were nearly equivalent over most of the AF spectrum, we observed substantial differences for rare alleles (AF < 0.05); intermediate-frequency alleles, including errors where nearly all individuals are heterozygous (AF ≈ 0.5); and fixed or nearly fixed alleles (AF ≈ 0.95). The most prominent difference in AF distributions affected fixed or nearly fixed alleles in each assembly, for which all non-African superpopulations showed an excess of ~150,000 variants in GRCh38, whereas the African super-population showed an excess of 2364 variants in T2T-CHM13 (Fig. 2F). This observation was driven by a decrease in the number of completely fixed variants (100% AF) relative to GRCh38 (Fig. 2G). Such variants represent positions where the reference genome itself is the only sample observed to possess the corresponding allele. These alleles arise either because of genuine private variants in one of the GRCh38 donors, or from errors in the reference genome itself, and result in 100% of other individuals possessing two copies of the alternative allele. As a result, these “variants” will not be reported at all if the same reads are mapped to a different genome that does not have these private alleles. Interestingly, the number of such private “singleton” variants in T2T-CHM13 lies squarely within the observed range of singleton counts among 1KGP samples, adjusting for the difference in ploidy (fig. S25). In addition to the lower rate of private variation relative to GRCh38, T2T-CHM13 possesses fewer ultra-rare variants, effectively reducing the number of “nearly fixed” alleles in population data such as the 1KGP.
Finally, the reduction in AF ≈ 0.5 variants is largely explained by the corrections to collapsed SDs (table S1), as these regions are highly enriched for heterozygous PSVs in nearly all individuals. Such erroneous heterozygous variants are caused by the false pileup of reads from the duplicated regions to a single location (Fig. 2H). Collectively, the decrease in variants with AF = 1 and AF ≈ 0.5 largely explains the decrease in the overall number of variants observed per sample and across the entire population for T2T-CHM13.
Informed by these results, we considered the feasibility of calling variants using the T2T-CHM13 reference and then lifting over the results to GRCh38 for further analyses. Using a liftover tool to transform a variant call set for a single sample into a call set with respect to GRCh38 requires special handling to account for variants for which the two references have different alleles. Specifically, if one of the reference alleles is not present in the sample, it will be necessary to genotype the site against the T2T-CHM13 reference. Although this issue is less of a concern for large datasets such as the 1KGP, even these large samples will contain a small number of variants that become invisible when switching reference genomes (fig. S26). In addition, differences in variant representation, especially in regions of low complexity, may cause lifted variant sets to differ from those called against the target reference.
Reduction of Mendelian-discordant variants
As further quality control for the variant calls, we performed a Mendelian concordance analysis using the 602 trios represented in the 1KGP cohort. We observed a statistically significant decrease in both the number of low-quality variants [median, 890,701 (GRCh38) versus 682,609 (T2T-CHM13); P = 4.943 × 10−96, Wilcoxon signed-rank test] (Fig. 2I) and the number of Mendelian-discordant variants (i.e., variants found in children but not their parents, or homozygous parental variants not observed in their children) [median, 8879 (GRCh38) versus 7484 (T2T-CHM13); P = 7.346 × 10−96, Wilcoxon signed-rank test] (Fig. 2J) when aligned to T2T-CHM13 as compared to GRCh38. In addition to providing an estimate of the error rate for variant calls in this call set, this improvement has broad implications for clinical genetics analyses of de novo or somatic mutations, which have been implicated as causes of autism spectrum disorders (47) and many forms of cancer (48).
T2T-CHM13 improves SV analysis of 17 diverse long-read samples
T2T-CHM13 improves long-read mapping across populations
Next, we investigated the effects of using T2T-CHM13 as a reference genome for alignment and large SV calling from both PacBio HiFi and ONT long reads. To this end, we aligned reads and called SVs in 17 samples of diverse ancestries from the Human Pangenome Reference Consortium (HPRC+) (27) and the Genome in a Bottle Consortium (GIAB) (29), including two trios (table S7). All of these samples had HiFi data available, and 14 had also been sequenced with ONT (Fig. 3A), with mean read lengths of 18.1 kbp and 21.9 kbp and read N50 values of 18.3 kbp and 44.9 kbp, respectively (fig. S27).
Fig. 3. Improvements to long-read alignment and SV calling in CHM13.

(A) The coverage, ancestry, and sequencing platforms available for the 17 samples sequenced with long reads (headers: AFR, African; AMR, Admixed American; ASH, Ashkenazi; EAS, East Asian; SAS, South Asian; populations: ACB, African Caribbean in Barbados; ASH, Ashkenazi; CHS, Southern Han Chinese; CLM, Colombian in Medellin, Colombia; GWD, Gambian in Western Division, The Gambia; KHV, Kinh in Ho Chi Minh City, Vietnam; MSL, Mende in Sierra Leone; PJL, Punjabi in Lahore, Pakistan; PUR, Puerto Rican in Puerto Rico). (B) The genome-wide mapping error rate and the standard deviation of the coverage for T2T-CHM13 (orange) and GRCh38 (blue). The standard deviation was computed across each 500-bp bin of the genome. (C) The allele frequency of SVs derived from HiFi data in T2T-CHM13 and GRCh38 among the 17-sample cohort. The red arrows indicate fixed (100% frequency) variants. (D) The balance of insertions (INS) vs. deletion (DEL) calls in the 17-sample cohort in T2T-CHM13 and GRCh38. Variants in T2T-CHM13 are stratified by whether or not they intersect regions which are nonsyntenic with GRCh38. (E) The SV calls in T2T-CHM13 for two trios: a trio of Ashkenazi ancestry [child HG002, and parents HG003 (46XY), and HG004 (46XX)], and a trio of Han Chinese ancestry [child HG005, and parents HG006 (46XY) and HG007 (46XX)]. The red arrows indicate child-only, or candidate de novo, variants (DEL, Deletion; DUP, Duplication; INS, Insertion; INV, Inversion; TRA, Translocation). (F) The density of SVs called from HiFi data in the 17-sample cohort across T2T-CHM13. (G) Alignments of HiFi reads in the HG002 trio to T2T-CHM13 showing a deletion spanning an exon of the transcript AC134980.2 viewed using the Integrative Genomic Viewer (IGV). Pink horizontal rectangles indicate reads aligned to the forward strand; blue horizontal rectangles indicate reads aligned to the reverse strand. Thin black lines indicate split-read alignments. Small vertical rectangles indicate SNVs (H) Alignments of HiFi reads in the HG002 trio to the same region of GRCh38 as shown in (G), showing much poorer mapping to GRCh38 than to T2T-CHM13, viewed using IGV with colors same as (G).
In line with our short-read results, aligning long reads to T2T-CHM13 versus GRCh38 did not substantially change the number of reads mapped with either Winnowmap (49) or minimap2 (50) because most of the previously unresolved sequence in T2T-CHM13 represents additional copies of SDs or satellite repeats already partially represented in GRCh38 (fig. S28). However, aligning to T2T-CHM13 reduced the observed mismatch rate per mapped read by 5% to 40% across the four combinations of sequencing technologies and aligners because GRCh38 has more rare alleles. T2T-CHM13 also corrects structural errors in GRCh38 and is a complete assembly of the genome, which facilitates accurate alignment, similar to what we observed for short reads (Fig. 3B). Relatedly, we found that previously reported African-specific (51) and Icelandic-specific (52) sequences at least 1 kbp in length align with substantially greater identity and completeness to T2T-CHM13 than to GRCh38 (34) (fig. S29 and S30).
To study coverage uniformity, we next measured the average coverage across each 500-bp bin on a per-sample basis and computed the standard deviation of the coverage. Across all aligners and technologies, the median standard deviation of the per-bin coverage was reduced by more than a factor of 3, indicating more stable mapping to T2T-CHM13 (Fig. 3B). This difference in coverage uniformity was pronounced in satellite repeats and other regions of GRCh38 that are nonsyntenic with T2T-CHM13 (fig. S31 and S32). This coverage uniformity will broadly improve variant calling and other long read–based analyses.
T2T-CHM13 improves the balance of apparent indels
We next used our optimized SV-calling pipeline, including Sniffles (53), Iris, and Jasmine (54), to call SVs in all 17 samples (figs. S33 and S34) and consolidate them into a cohort-level call set in each reference from HiFi data. From these results, we observed a reduction from 5147 to 2260 SVs that are homozygous in all 17 individuals when calling variants relative to T2T-CHM13 instead of GRCh38 (Fig. 3C). Previous studies (16, 17) have noted the excess of such SV calls when using GRCh38 as a reference and attributed them to structural errors. Here, we found that using a complete and accurate reference genome naturally reduces the number of such variants. In addition, the number of indels was more balanced when calling against T2T-CHM13, whereas GRCh38 exhibited a bias toward insertions caused by missing or incomplete sequence (Fig. 3D), such as incorrectly collapsed tandem repeats (16). With respect to T2T-CHM13, we observed a small bias toward deletions, which likely results from the challenges in calling insertions with mapping-based methods and in representing SVs within repeats, as this difference is especially prominent in highly repetitive regions such as centromeres and satellite repeats (fig. S35). The variants we observed relative to T2T-CHM13 are enriched in the centromeres and subtelomeric sequences, likely because of a combination of repetitive sequence and greater recombination rates (17). We observed similar trends among SVs unique to single samples (fig. S36).
We also observed similar improvements in the insertion/deletion balance for large SVs (>500 bp) detected by Bionano optical mapping data in HG002 against the T2T-CHM13 reference, with an increase in deletions (1199 versus 1379) and a decrease in insertions (2771 versus 1431) with GRCh38 and T2T-CHM13, respectively (fig. S37). Using the T2T-CHM13 reference for Bionano optical mapping also improved SV calling around gaps in GRCh38 that are closed in T2T-CHM13 (fig. S38), which suggests that T2T-CHM13 offers improved indel balance relative to GRCh38 across multiple SV-calling methods.
T2T-CHM13 facilitates the discovery of de novo SVs
To investigate the impacts of the T2T-CHM13 reference on our ability to accurately detect de novo variants, we called SVs in both of our trio datasets using a combination of HiFi and ONT data and identified SVs only present in the child of the trio and supported by both technologies—approximately 40 variants per trio (Fig. 3E). Manual inspection revealed a few variants in each trio that were strongly supported with consistent coverage and alignment breakpoints, whereas the other candidates exhibited less reliable alignments, as noted in previous reports (54). In HG002, we detected six strongly supported candidate de novo SVs that had been previously reported (29, 54). In HG005, we detected a 1571-bp deletion at chr17:49401990 in T2T-CHM13 that was supported as a candidate de novo SV relative to both T2T-CHM13 and GRCh38 (fig. S39). This demonstrates the ability of T2T-CHM13 to be used as a reference genome for de novo SV analysis.
T2T-CHM13 enables the discovery of additional SVs within previously unresolved sequences
The improved accuracy and completeness of the T2T-CHM13 genome make it easier to resolve complex genomic regions. Within nonsyntenic regions, we identified a total of 27,055 SVs (Fig. 3D), the majority of which were deletions (15,998) and insertions (10,912). Of these SVs, 22,362 (82.7%; 8903 insertions, 13,334 deletions) overlap previously unresolved sequences in T2T-CHM13, whereas the remaining SVs are now accessible because of the accuracy of the T2T-CHM13 reference. The AF and size distributions for these variants mirror the characteristics of the syntenic regions, with rare variants (fig. S40) and smaller (30 to 50 bp) indels (fig. S41) being the most abundant. However, we also note some nonsyntenic regions with few or zero SVs identified. Many of these regions lie at the interiors of p-arms of acrocentric centromeres, which are gaps in T2T-CHM13v1.0 that have been resolved in later versions of the assembly; however, we also noticed depletions of SVs in a few other highly repetitive regions, such as the resolved human satellite array on chromosome 9 (Fig. 3F). We largely attribute the reduction in variant density to the low mappability of these complex and repetitive regions. Future improvements in read lengths and alignment algorithms are needed to further resolve such loci.
Within syntenic regions, we also noted improvements to alignment and variant calling accuracy, including the identification of variant calls not previously observed within homologous regions of GRCh38. For example, in T2T-CHM13, we observed a deletion in all of the samples of the HG002 trio in an exon of the olfactory receptor gene AC134980.2 (Fig. 3G), whereas the reads from those samples largely failed to align to the corresponding region of GRCh38 (Fig. 3H). Meanwhile, reads from African samples (fig. S42) aligned to both references at this locus. The difference in alignment among different samples is likely because the region is highly polymorphic for copy number variation; GRCh38 contains a reasonable representation of that region for the tested African samples, whereas the homologous region in T2T-CHM13 more closely resembles European samples (fig. S43). This highlights the need for T2T reference genomes for as many diverse individuals as possible to account for common haplotype diversity.
Variation within previously unresolved regions of the genome
T2T-CHM13 enables variant calling in previously unresolved and corrected regions of the genome
The T2T-CHM13 genome contains 229 Mbp of sequence that is nonsyntenic to GRCh38, which intersects 207 protein-coding genes (Fig. 4A and Table 1). Within these regions, we report 3,692,439 PASS variants across all 1KGP samples from short reads (Fig. 4B and Table 1). Comparing variants called in a subset of 14 HPRC+ samples with Illumina, HiFi, and ONT data, we found that 73 to 78% of the Illumina-discovered SNVs are concordant with variants identified with PacBio HiFi long-read data using the PEPPER-Margin-DeepVariant algorithm (51,306 to 74,122 matching SNVs and genotypes per sample) (55). Long reads discovered more than 10 times as many SNVs per sample as short reads in these regions, with 447,742 to 615,085 (41 to 43%) SNVs matching between HiFi and ONT with PEPPER-Margin-Deep-Variant. In nonsyntenic regions, 97% of the SNVs called by HiFi fell in centromeric regions of CHM13, so we stratified concordance by type of satellite repeat within the centromere. We found that nonsatellites in centric transition regions and monomeric satellites had higher concordance between HiFi and ONT, with >99% concordance in a few regions, but some as low as 50%. Human satellite (HSAT) regions, which pose some of the greatest challenges for read mapping and harbor abundant structural variation, exhibit the lowest rates of concordance between the platforms.
Fig. 4. Characterization of variants within regions of the genome resolved by T2T-CHM13.
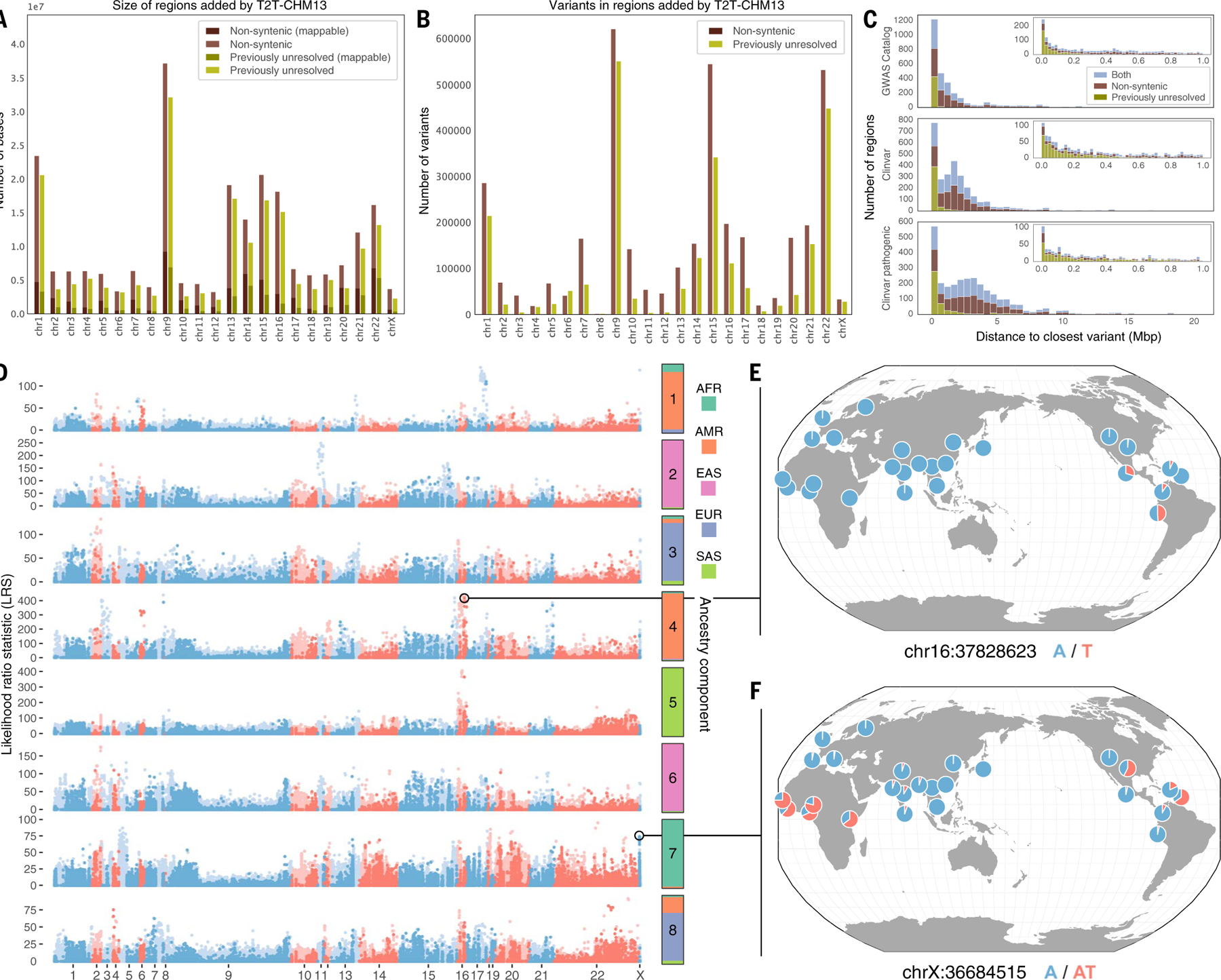
(A) Number of bases added in nonsyntenic and previously unresolved regions by chromosome, along with how many variants for each respective region are mappable (have contiguous unique 100mers). (B) Number of variants in nonsyntenic and previously unresolved regions by chromosome. (C) Distance from each previously unresolved–only, nonsyntenic-only, or overlapping region to the closest Clinvar or GWAS Catalog variant. Insets are zoomed to 1 Mbp. (D) Scan for variants in nonsyntenic (light blue and red) and previously unresolved (dark blue and red) regions that exhibit extreme patterns of allele frequency differentiation. Allele frequency outliers were identified for each of eight ancestry components, colored by the superpopulation membership of the corresponding 1KGP samples. Large values of the likelihood ratio statistic (LRS) denote variants for which AF differences in the corresponding ancestry component exceeds that of a null model based on genome-wide covariances in allele frequencies. (E and F) Population-specific allele frequencies of two highly differentiated variants in previously unresolved regions.
Table 1.
Overview of nonsyntenic and previously unresolved regions and their respective variant counts.
| Nonsyntenic | Previously unresolved | |
|---|---|---|
| Summary | ||
| Total span (bp) (excluding Ns) | 240,044,315 (228,569,315) | 189,036,735 (177,561,735) |
| Unique span (100mers) | 65,471,195 | 40,205,401 |
| Protein-coding genes | 207 | 207 |
| Entire region | ||
| 1KGP SNVs + indels (within genes) | 3,692,439 (138,829) | 2,370,384 (52,567) |
| Short-read SNVs per sample | 65,931 to 101,161 | 35,506 to 56,489 |
| Long-read SNVs per sample | 1,178,371 to 1,467,243 | 957,629 to 1,197,463 |
| Short-read SNVs confirmed by long reads | 73 to 78% | 64 to 69% |
| Long-read SNVs identified in short reads | 4 to 5% | 3% |
| SNVs concordant between long reads | 41 to 43% | 38 to 40% |
| High-confidence regions (excluding coverage abnormalities) | ||
| High-confidence region bases | 13,683,528 | 2,987,935 |
| Short-read SNVs confirmed by long reads in high-confidence regions | 95 to 96% | 84 to 88% |
| Long-read SNVs identified in short reads in high-confidence regions | 60 to 63% | 39 to 46% |
| SNVs concordant between long reads in high-confidence regions | 91 to 95% | 81 to 90% |
We further defined conservative high-confidence regions by excluding regions with abnormal coverage in any long-read sample (i.e., coverage outside of 1.5× the interquartile range). This effectively excludes difficult-to-map regions with excessively repetitive alignments as well as copy number–variable regions. After excluding abnormal coverage from nonsyntenic regions, 14 Mbp remained, and SNVs from HiFi and ONT long reads were 91 to 95% concordant (21,835 to 28,237 variants). We found 95 to 96% (14,575 to 18,949) of short-read SNVs in HiFi long-read calls, although 37 to 40% of HiFi SNVs were still missing from the short-read calls as a result of poorer mappability of the short reads (table S8). Although many nonsyntenic regions will require further method development [e.g., pangenome references (56)] to achieve accurate variant calls, the concordance of long- and short-read calls for tens of thousands of variants highlights previously unresolved sequences that are immediately accessible to both technologies.
Because these broadly defined nonsyntenic regions include inversions and other structural changes between GRCh38 and T2T-CHM13 that do not necessarily alter many of the variants contained within, we also considered a narrower class of “previously unresolved” sequences, representing segments of the T2T-CHM13 genome that do not align to GRCh38 with Winnowmap (49). Within these previously unresolved sequences, which span a total of 189 Mbp (Fig. 4A, Table 1, and fig. S44), we report a total of 2,370,384 PASS variants in 1KGP samples based on short reads, intersecting 207 protein-coding genes (Fig. 4B, Table 1, and fig. S45). We note that this set of 207 genes is distinct from the 207 genes that intersected with the nonsyntenic regions, and these two sets together comprise 329 unique genes. Because these previously unresolved sequences are enriched for highly repetitive sequences, concordance is slightly lower, such that 64 to 69% of the SNVs in each sample match variants found in PacBio HiFi long-read data from the same samples (24,371 to 36,501 matching SNVs and genotypes per sample), and 339,783 to 473,074 (38 to 40%) of SNVs match between HiFi and ONT. When removing difficult-to-map and copy number–variable regions as above, 3 Mbp of high-confidence regions remained. Within high-confidence regions, 84 to 88% of short-read SNVs in each sample matched variants found in each sample’s PacBio HiFi long-read data (2938 to 3811 matching SNVs and genotypes per sample), and 5544 to 8298 (81 to 90%) of SNVs matched between HiFi and ONT (table S8). Although these previously unresolved regions are more challenging than nonsyntenic regions, thousands of variants can still be called concordantly with short and long reads.
We noted homology between GRCh38 collapsed duplications and many T2T-CHM13 nonsyntenic and/or previously unresolved regions (137 regions comprising 6.8 Mbp), indicating that the T2T-CHM13 assembly corrects these sequences through the deconvolution of nearly identical repeats. Comparing total variants identified in the 1KGP dataset, we observed a significant decrease in variant densities of 41 protein-coding genes intersecting with GRCh38 collapsed duplications in T2T-CHM13 (mean, 27 variants per kbp) compared with GRCh38 (mean, 46 variants per kbp; P = 6.906 × 10−8, Wilcoxon signed-rank test) (fig. S46). Besides differences in local ancestries between the references, these higher variant densities in GRCh38 in part represent PSVs or misassigned alleles from missing paralogs (57). Conversely, 1KGP variants were significantly increased in 32 protein-coding genes contained within GRCh38 false duplications using the T2T-CHM13 reference genome (mean values of 48 variants per kbp in T2T-CHM13 versus 12 variants per kbp in GRCh38; P = 4.657 × 10–10, Wilcoxon signed-rank test).
To assess whether these corrected complex regions in T2T-CHM13 accurately reveal variation, we evaluated the concordance of variants generated from short-read Illumina and PacBio HiFi sequencing datasets of two trios from the GIAB consortium and the Personal Genome Project (58) and observed similar recall for Illumina data in T2T-CHM13 (20.1 to 28.3%) and GRCh38 (21.5 to 25.4%), but with improved precision in the variants identified (98.1 to 99.7% in T2T-CHM13 versus 64.3 to 67.3% in GRCh38) in a subset of the GRCh38 collapsed duplications (copy number < 10; ~910 kbp) (table S9). Corrected false duplications (1.2 Mbp) exhibited improved recall for Illumina data by a factor of 50 relative to HiFi in T2T-CHM13 (57.4 to 68.3%) versus GRCh38 (1.1 to 1.8%), as well as improved precision in T2T-CHM13 (98.5 to 99.3%) versus GRCh38 (76.5 to 95.8%) (table S9). These improvements show that variants can be discovered and genotyped in regions corrected by the T2T-CHM13 assembly.
Phenotypic associations and evolutionary signatures within nonsyntenic
T2T-CHM13 regions
Sequences in the T2T-CHM13 assembly that are nonsyntenic with GRCh38 offer opportunities for future genetic studies. Several such loci lie in close proximity to variation that has been implicated in complex phenotypes or disease, supporting their potential biomedical importance. These include eight loci occurring within 10 kbp of GWAS hits and 19 loci within 10 kbp of ClinVar pathogenic variants (Fig. 4C). In addition, 113 of 22,474 GWAS hits (representing 0.5% of all variants in the studies we tested) segregated in LD (R2 ≥ 0.5) with variants in nonsyntenic regions, thereby expanding the catalog of potential causal variants for these GWAS phenotypes (43) (fig. S47 and table S10).
Using short read–based genotypes generated from the 1KGP cohort, we also searched for variants within nonsyntenic regions that exhibit large differences in AF between populations— a signature that can reflect historical positive selection or demographic forces shaping these previously inaccessible regions of the genome. To study these signatures, we applied Ohana (59), a method that models individuals as possessing ancestry from k components and tests for ancestry component–specific frequency outliers. Focusing on continental-scale patterns (k = 8; fig. S48), we identified 5154 unique SNVs and indels across all ancestry components that exhibited strong deviation from genome-wide patterns of AF (99.9th percentile of distribution for each ancestry component; Fig. 4D). These included 814 variants over-lapping with annotated genes and 195 variants that intersected annotated exons.
We first focused on the 3038 highly differentiated nonsyntenic variants that lift over from T2T-CHM13 to GRCh38. These successful liftovers allowed us to make direct comparisons to selection results, generated with identical methods, using 1KGP phase 3 data aligned to GRCh38 (fig. S49) (34, 60). For 41.3% of the lifted-over variants, we found GRCh38 variants within a 2-kbp window that possessed similar or higher likelihood ratio statistics for the same ancestry component, indicating that these loci were possible to identify in scans of GRCh38 (fig. S50). The remaining 58.7% of lifted-over variants may represent regions of the genome where differences in the T2T-CHM13 and 1KGP phase 3 variant calling or filtering procedures lead to discrepancies in AFs between these two datasets. They may also indicate regions whose more accurate representation in T2T-CHM13 improves variant calling enough to resolve previously unknown signatures of AF differentiation (fig. S51). We then investigated the 943 variants that could not be lifted over from T2T-CHM13 to GRCh38 and were located in both previously unresolved sequences and regions deemed mappable from unique 100-mer analysis. Some of these variants overlap with genes, including several annotated with RNA transcripts in regions not present in the GRCh38 assembly (Fig. 4D and table S11).
We highlight two loci that exhibit some of the strongest allele frequency differentiation observed across ancestry components. The first locus, located in a centromeric alpha satellite on chromosome 16, contains variants that reach intermediate allele frequency in the ancestry component corresponding to the Peruvian in Lima, Peru (PEL) and other admixed American populations of 1KGP [AFs, 0.49 in PEL; 0.20 in CLM (Colombian in Medellin, Colombia) and MXL (Mexican ancestry in Los Angeles, California); absent or nearly absent elsewhere; Fig. 4E and figs. S52 and S53]. Variants at the second locus, located in a previously unresolved T2T-CHM13 sequence on the X chromosome that contains a multi-kbp imperfect AT tandem repeat, exhibit high AFs in the ancestry component corresponding to African populations of 1KGP and low AFs in other populations (AFs, 0.67 in African populations and 0.014 in European populations; Fig. 4F and figs. S54 and S55). The variant at this locus with the strongest signature of frequency differentiation also lies within 10 kbp of two pseudogenes, MOB1AP2–201 (MOB kinase activator 1A pseudogene 2) and BX842568.1–201 (ferritin, heavy polypeptide-like 17 pseudogene).
We note that as a consequence of the repetitive nature of the sequences in which they reside, many of the loci that we highlight here remain challenging to genotype with short reads, and individual variant calls remain uncertain. Nonetheless, patterns of AF differentiation across populations are relatively robust to such challenges and can still serve as proxies for more complex SVs whose sequences cannot be resolved by short reads alone. The presence of population-specific signatures at these loci highlights the potential for T2T-CHM13 to reveal evolutionary signals in previously unresolved regions of the genome.
Impact of T2T-CHM13 on clinical genomics
Variants of potential clinical relevance in T2T-CHM13
A deleterious variant in a reference genome can mislead the interpretation of a clinical variant identified in a patient because it may not be flagged as such using standard analysis tools. The GRCh38 reference genome is known to contain such variants that likely affect gene expression, protein structure, or protein function (25), although systematic efforts have sought to identify and remove these alleles (3). To determine the existence and location of loss-of-function variants in T2T-CHM13, we aligned the assembly to GRCh38 using dipcall (61) to identify and functionally annotate nucleotide differences (62) (Fig. 5A). This analysis identified 210 putative loss-of-function variants (defined as variants that affect protein-coding regions and predicted splice sites) affecting 189 genes, 31 of which are clinically relevant (23). These results are in line with work showing that the average diploid human genome contains ~450 putative loss-of-function variants affecting ~200 to 300 genes when low-coverage Illumina sequencing is applied (before stringent filtering) (63).
Fig. 5. T2T-CHM13 improves clinical genomics variant calling.

(A) Numbers of potential loss-of-function mutations in the T2T-CHM13 reference. (B) The counts of medically relevant genes affected by genomic features and variation in GRCh38 (blue) and T2T-CHM13 (orange) are depicted as bar plots on logarithmic scale. Light blue indicates genes affected in GRCh38 where homologous genes were not identified in T2T-CHM13 due to inability to lift over, with counts included in parentheses. (C) An example erroneous GRCh38 complex SV corrected in T2T-CHM13 affecting TNNT3 and LINC01150, displayed by sequence comparison using miropeats (88) with homologous regions colored in green and blue, respectively. HG002 PacBio HiFi data are displayed showing read coverages and mappings from IGV, with allele fractions of variant sites colored (red, T; green, A; blue, C; black, G) within histograms of read depth (0 to 50). (D and E) Snapshots of regions using IGV and UCSC Genome Browser representing a collapsed duplication in GRCh38 corrected in T2T-CHM13 affecting KCNJ18 (D) and a false duplication in GRCh38 affecting most of KCNE1 (E). SDs depicted on top are colored by sequence similarity to paralog (gray, 90 to 98%; orange, >99%). Read mappings and variants from HG002 Illumina, PacBio HiFi, and ONT (mappings only), with homozygous (light blue) and heterozygous (dark blue) variants depicted as dashes. Colors within histograms of read depth (0–120) are the same as described in (C). Copy number estimates, displayed as colors indicated in legends, across k-merized versions of the GRCh38 and T2T-CHM13 references as well as representative examples of the SGDP individuals. (F) An example CDS region of KCNJ18 (highlighted as a red box in D), with amino acids colored in alternating shades of blue and potential start codons (methionines) labeled in green using the UCSC Genome Browser codon-coloring scheme. Alignments of KCNJ18 (blue), KCNJ12 (orange), and KCNJ17 (pink) along with allele counts of variants in each gene identified on GRCh38 and T2T-CHM13 are shown as bar plots (to approximate scale per variant), with examples 1 to 7 described in table S14. (G) Schematic depicts a benchmark for 269 challenging medically relevant genes for HG002. The number of variant-calling errors from three sequencing technologies on each reference is plotted.
Of these 210 variants, 158 have been identified in at least one individual from the 1KGP, with most variants relatively common in human populations (median AF of 0.47), suggesting that they are functionally tolerated. The remaining variants not found in 1KGP individuals comprise larger indels, which are more difficult to identify with 1KGP Illumina data, as well as alleles that are rare or unique to CHM13. We curated the 10 variants affecting medically relevant genes and found seven that likely derived from duplicate paralogs: a 100-bp insertion also found in long reads of HG002, a stop gain in a final exon in one gnomAD sample, and an insertion in a homopolymer in a variable-number tandem repeat in CEL, which may be an error in the assembly. Understanding that the T2T-CHM13 assembly represents a human genome harboring potentially functional or rare variants that in turn would affect the ability to call variants at those sites, we have made available the full list of putative loss-of-function variants to aid in the interpretation of sequencing results (table S12).
T2T-CHM13 improves variant calling for medically relevant genes
We sought to understand how the transition from GRCh38 to the T2T-CHM13 reference might have an impact on variants identified in a previously compiled (23) set of 4964 medically relevant genes residing on human autosomes and chromosome X (representing 4924 genes in T2T-CHM13 via liftover; table S13). Of these genes, 28 mapped to previously unresolved and/or nonsyntenic regions of T2T-CHM13. We found more than twice as many medically relevant genes affected by rare or erroneous structural alleles on GRCh38 (n = 756 including 14 with no T2T-CHM13 lift-over) compared to T2T-CHM13 (n = 306) (Fig. 5B), of which 622 genes appear corrected in T2T-CHM13. This includes 116 genes falling in regions previously flagged as erroneous in GRCh38 by the GRC. The majority (82%) of affected clinically relevant genes in GRCh38 overlap SVs that exist in all 13 HiFi-sequenced individuals, likely representing rare alleles or errors in the reference (see above), including 13 of the 14 genes with no T2T-CHM13 liftover.
One example of a resolved gene structure involves TNNT3, which encodes Troponin T3, fast skeletal type, and is implicated in forms of arthrogryposis (64). When calling SVs with respect to GRCh38, TNNT3 was previously postulated to be affected by a complex structural rearrangement in all individuals, consisting of a 24-kbp inversion and 22-kbp upstream deletion, which also ablates LINC01150 (Fig. 5C). The GRC determined that a problem existed with the GRCh38 reference in this region (GRC issue HG-28). Analysis of this region in T2T-CHM13 instead shows a complex rearrangement with the 22-kbp region upstream of TNNT3 inversely transposed in the T2T-CHM13 assembly to the proximal side of the gene. Besides potentially affecting interpretations of gene regulation, this structural correction of the reference places TNNT3 >20 kbp closer to its genetically linked partner TNNI2 (65). Other genes have variable number tandem repeats (VNTRs) that are collapsed in GRCh38, such as one expanded by 17 kbp in most individuals in the medically relevant gene GPI. MUC3A was also flagged with a whole-gene amplification in all individuals, which we identified as residing within a falsely collapsed SD in GRCh38, further evidencing that finding (Fig. 1A).
Seventeen medically relevant genes reside within erroneous duplicated and putative collapsed regions in GRCh38 (tables S1 and S3), including KCNE1 (false duplication) and KCNJ18 (collapsed duplication) (Fig. 5, D and E). For these genes, we show that a significant skew in total variant density occurs in GRCh38 (58 variants per kbp for eight genes in collapsed duplications and 21 variants per kbp for seven genes in false duplications; P = 5.684 × 10–3 and 6.195 × 10–4, respectively, Mann-Whitney U test) versus the rest of the 4909 medically relevant gene set (40 variants per kbp) that largely disappears in T2T-CHM13 (40 variants per kbp in collapsed duplications and 47 variants per kbp in false duplications versus 41 variants per kbp for the remaining gene set; P = 0.8778 and 0.0219, respectively) (fig. S45). Examining KCNE1, we found that coverage is much lower than normal on GRCh38 for short and long reads and that most variants are missed because many reads incorrectly map to a likely false duplication (KCNE1B on the p-arm of chromosome 21). The k-mer–based copy number of this region in all 266 SGDP genomes supports the T2T-CHM13 copy number as well as its lack of duplication in GRCh37 (23). As for KCNJ18, which resides within a GRCh38 collapsed duplication at chromosome 17p11.2 (66), we found increased coverage and variants within HG002 using short- and long-read sequences in GRCh38 relative to T2T-CHM13.
To verify whether the additional variants identified using GRCh38 are false heterozygous calls from PSVs derived from missing duplicate paralogs, we compared the distributions of MAFs across the 49-kbp SD. We observed a shift in SNV proportions, with a relative decrease in intermediate-frequency alleles and a relative increase in rare alleles for KCNJ18 and KCNJ12 (another collapsed duplication residing distally at chromosome 17p11.2) in T2T-CHM13 compared with GRCh38 (P = 8.885 × 10–2 and 3.102 × 10–2, respectively; Mann-Whitney U test) (fig. S56). We matched the homologous positions of discovered alternative alleles in GRCh38 and T2T-CHM13 across the three paralogs—including the previously missing paralog located in a centromere-associated region on chromosome 17p KCNJ17, denoted KCNJ18–1 in T2T-CHM13—and observed that even true variants (i.e., non-PSVs) had discordant allele counts in KCNJ18 and KCNJ12 between the two references (Fig. 5F and table S14). Considering that rare variants of KCNJ18 contribute to muscle channelopathy-thyrotoxic periodic paralysis (66), including nine “pathogenic” or “likely pathogenic” variants in ClinVar, increased sensitivity to discover variants in patients using T2T-CHM13 would have a sub-stantial clinical impact. In summary, the improved representation of this gene and other collapsed duplications in T2T-CHM13 not only eliminates false positives but also improves detection and genotyping of true variants.
Clinical gene benchmark demonstrates that T2T-CHM13 reduces errors across technologies
Finally, to determine how the T2T-CHM13 genome improved the ability to assay variation broadly, we used a curated diploid assembly to develop a benchmark for 269 challenging medically relevant genes in GIAB Ashkenazi son HG002 (23), with comparable benchmark regions on GRCh38 and T2T-CHM13. We tested three short- and long-read variant call sets against this benchmark: Illumina-BWAMEM-GATK, HiFi-PEPPER-DeepVariant, and ONT-PEPPER-DeepVariant. Counts of both false positives and false negatives substantially decreased for all three call sets when using T2T-CHM13 as a reference instead of GRCh38 (Fig. 5G and table S15). The number of false positives for HiFi decreased by a factor of 12 in these genes, primarily because of the addition of missing sequences similar to KMT2C (fig. S15) and removal of false duplications of CBS, CRYAA, H19, and KCNE1 (Fig. 5G). As demonstrated above, T2T-CHM13 better represents these genes and others for a diverse set of individuals, so performance should be higher across diverse ancestries. Furthermore, the number of true positives decreased by a much smaller fraction than the errors (~14%); this is due to a reduction of true homozygous variants caused by T2T-CHM13 possessing fewer ultra-rare and private alleles (Fig. 2G). This benchmarking demonstrates concrete performance gains in specific medically relevant genes resulting from the highly accurate assembly of a single genome.
Discussion
Difficult regions of the human reference genome, ranging from collapsed duplications to missing sequences, have remained unresolved for decades. The assumptions that most genomic analyses make about the correctness of the reference genome have contributed to spurious clinical findings and mistaken disease associations (67–70). Here, we identify variation in difficult-to-resolve regions and show that the T2T-CHM13 reference genome universally improves genomic analyses for all populations by correcting major structural defects and adding sequences that were absent from GRCh38. In particular, we show that the T2T-CHM13 assembly (i) revealed millions of additional variants and the existence of additional copies of medically relevant genes (e.g., KCNJ17) within the 240 Mbp and 189 Mbp of nonsyntenic and previously unresolved sequence, respectively; (ii) eliminated tens of thousands of spurious variants and incorrect genotypes per sample, including within medically relevant genes (e.g., KCNJ18) by expanding 203 loci (8.04 Mbp) that were collapsed in GRCh38; (iii) improved genotyping by eliminating 12 loci (1.2 Mbp) that were duplicated in GRCh38; and (iv) yielded more comprehensive SV calling genome-wide, with an improved insertion/deletion balance, by correcting collapsed tandem repeats. Overall, the T2T-CHM13 assembly reduced false positive and false negative SNVs from short and long reads by as much as a factor of 12 in challenging, medically relevant genes. The T2T-CHM13 reference also accurately represents the haplotype structure of human genomes, eliminating 1390 artificial recombinant haplotypes in GRCh38 that occurred as artifacts of BAC clone boundaries. These improvements will broadly enable future discoveries and refine analyses across all of human genetics and genomics.
Given these advances, we advocate for a rapid transition to the T2T-CHM13 genome as a reference. Although we appreciate that transitioning institutional databases, pipelines, and clinical knowledge from GRCh38 to T2T-CHM13 will require substantial bioinformatics and clinical effort, we provide several resources to advance this goal. On a practical level, improvements to large genomic regions, such as entire p-arms of the acrocentric chromosomes, and the discovery of clinically relevant genes and disease-causing variants justify the labor and cost required to incorporate T2T-CHM13 into basic science and clinical genomic studies. On a technical level, T2T-CHM13 simplifies genome analysis and interpretation because it consists of 23 complete linear sequences and is free of “patch,” unplaced, or unlocalized sequences. Many of the corrections introduced by T2T-CHM13 were previously noted and addressed by the GRC as “fix patches,” but few studies use these existing resources. The reduced contig set of T2T-CHM13 also facilitates interpretation and is directly compatible with the most commonly used analysis tools. To promote this transition, we provide variant calls and several other annotations for the T2T-CHM13 genome within the UCSC Genome Browser and the NHGRI AnVIL as a resource for the human genomics and medical communities.
Finally, our work underscores the need for additional T2T genomes. Most urgently, the CHM13 genome lacks a Y chromosome, so our analysis relied on the incomplete representation of chromosome Y from GRCh38. A T2T representation of the Y chromosome should further improve mapping and variant analysis, especially with respect to variants on the Y chromosome itself. Furthermore, many of the previously unresolved regions in T2T-CHM13 are present in all human genomes and enable variant calling with traditional methods from short and/or long reads. However, many previously unresolved regions identified in the T2T-CHM13 genome exhibit substantial variation within and between populations, including satellite DNA (31) and SDs that are polymorphic in copy number and structure (32). Relatedly, the T2T-CHM13 reference provides a basis for calling millions of variants that were previously hidden, but many of these variants are challenging to resolve accurately with current sequencing technologies and analysis algorithms. Robust variant calling in these regions will require many hundreds or thousands of diverse haplotype-resolved T2T assemblies to construct a pangenome reference, such as the effort now underway by the Human Pangenome Reference Consortium (56). These assemblies will then motivate further development of methods for discovering, representing, comparing, and interpreting complex variation, as well as benchmarks to evaluate their respective performances (71, 72).
Through our detailed assessment of variant calling across global population samples, our study showcases T2T-CHM13 as a preeminent reference for human genetics. The annotation resources provided herein will help facilitate this transition, expanding knowledge of human genetic diversity by revealing hidden functional variation.
Methods summary
Haplotype structure
We examined the impact of the fact that GRCh38 comprises a mosaic of clones derived from multiple donor individuals on its haplotype structure. To this end, we searched for “LD-discordant” SNP pairs, defined as common (>10% MAF) SNPs that segregate in perfect LD (R2 = 1) in the 1KGP sample, but for which GRCh38 possesses a pair of alleles that are never observed together on a single phased haplotype among 1KGP samples (i.e., alleles in perfect negative LD). We then compared these results to the same analysis applied to each 1KGP sample using a leave-one-out strategy.
Duplication errors
We flagged putatively collapsed duplications as regions >5 kbp containing clusters of heterozygous variants identified from two CHM13 datasets [simulated Illumina-like reads from T2T-CHM13 reference v1.0 including the GRCh38 Y chromosome and PacBio HiFi reads (73)] mapping against GRCh38 and T2T-CHM13 references. False duplications were identified as regions, converted to T2T-CHM13 coordinates, with median read-depth copy numbers (32) lower in kmerized GRCh38 compared to kmerized T2T-CHM13 and 88% of SGDP individuals. Alternatively, false duplications were identified as regions >3 kbp with copy numbers greater in kmerized GRCh38 compared to kmerized T2T-CHM13 and 99% of SGDP individuals using a genome-wide sliding-window approach.
Liftover of resources from GRCh38 to T2T-CHM13
Using the GATK release 4.1.9 (74) LiftoverVcf (Picard) tool, we lifted dbSNP build 154 (75), the March 8, 2021 release of Clinvar (76), and GWAS Catalog v1.0 (43) from the GRCh38 assembly to the T2T-CHM13 assembly. Initial liftover was done with default LiftoverVcf parameters. A secondary round of liftover was performed to recover variants with swapped reference and alternative alleles between GRCh38 and T2T-CHM13. We cataloged variants that failed to lift over because they overlap an indel that distinguishes T2T-CHM13 and GRCh38 based on results from dipcall.
Short-read variant calling
To evaluate short-read small-variant calling between GRCh38 and T2T-CHM13, we used the NHGRI AnVIL (44) to align all 3,202 1KGP samples to CHM13 with BWA-MEM (45) and performed variant calling with GATK HaplotypeCaller (77) using a workflow modeled on the one developed by the New York Genome Center (NYGC) for 1KGP analysis performed on GRCh38 (28). As in the NYGC analysis, we recalibrated the variant calls with GATK VariantRecalibrator. We analyzed coverage statistics using samtools and AF using bedtools. To identify Mendelian-discordant variants, we used GATK VariantEval.
Long-read variant calling
To compare long-read mapping and large SV calling between T2T-CHM13 and GRCh38, we aligned HiFi and ONT data from 17 samples of diverse ancestry to each reference with both Winnowmap (49) and minimap2 (50) and called SVs with Sniffles (53). Variant calls were refined with Iris, and HiFi-derived calls from both aligners were merged with Jasmine (54); the resulting sets of 124,566 SVs in GRCh38 and 141,193 SVs in T2T-CHM13 to compute AFs and other cohort-level statistics. In addition, we constructed trio-level callsets for two trios—the HG002 and HG005 trios from the GIAB Consortium—to compare Mendelian discordance rates between the two references.
Concordance of variants analysis across sequencing type
To evaluate the variant calls in nonsyntenic regions, we derived concordance between variant calls generated with HiFi, ONT, and Illumina reads. For each sample, we used bcftools to filter the non-PASS variants, indels, and nonautosomal variants from each callset. We then used hap.py (78) to derive the precision, recall, and F1-score between each variant callset to determine how many variants are common between each pair of sets.
AF differentiation of nonsyntenic variants
Using short read–based variant calls within T2T-CHM13 nonsyntenic regions, we searched for variants with signatures of extreme AF differentiation across human populations. We performed this analysis with Ohana (59), a method that infers admixture components for each sample and quantifies frequency variation among the components. For outlier nonsyntenic variants with extreme patterns of AF differentiation, we used liftover to compare our results to previous results generated with 1KGP phase 3 data aligned to GRCh38 (60).
T2T-CHM13 dipcall and Variant Effect Predictor (VEP)
VEP (62) (version 102.0) was used to annotate variants generated by dipcall (61) when aligning the T2T-CHM13 reference genome (chm13_ v1.0_plus38Y.fa) to the GRCh38 reference genome (hg38.no_alt.fa). VCF files were annotated without the –filter_common and –canonical flags. CADD (79) v1.6 and raw SpliceAI (80) scores were added using both the CADD and SpliceAI plugins. Variants were filtered based on predicted HIGH functional impact.
HG002 medically relevant genes benchmark
To evaluate variant call accuracy when using T2T-CHM13 vs. GRCh38 as a reference, we developed equivalent small variant benchmarks for GIAB sample HG002 in 269 challenging, medically relevant genes. Methods were adapted from a companion manuscript that describes a curated benchmark for these genes created by using variants generated by dipcall (61) when aligning a trio-based hifiasm assembly to GRCh37 and GRCh38 (23).
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Zody, B. Grüning, H. Li, S. Langley, C. Langley, S.-H. Kim, G. Van der Auwera, V. Schneider, S. Salzberg, B. Langmead, A. Battle, and several of their lab members for helpful discussions. Certain commercial equipment, instruments, or materials are identified to specify adequate experimental conditions or reported results. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the equipment, instruments, or materials identified are necessarily the best available for the purpose. This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov) and the Maryland Advanced Research Computing Center (www.marcc.jhu.edu/).
Funding:
Supported by NIH grants R35GM133747 (R.C.M.), DP2MH119424 (M.Y.D.), R01HG006677 (A.S.), R01HG010485 (B.P.), U41HG010972 (B.P.), U01HG010961 (B.P., U24HG011853 (B.P.), OT2OD026682 (B.P.), UM1HG008898 (F.J.S.), 1R01HG011274–01 (K.H.M.), 1R21HG010548–01 (K.H.M.), 1U01HG010971 (K.H.M.), U24HG010263 (M.C.S.), U24HG006620 (M.C.S.), U01CA253481 (M.C.S.), R24DK106766–01A1 (M.C.S.), and R00HG009532 (R.M.L.); NSF grants DBI-1627442, IOS-1732253, and IOS-1758800 (M.C.S.); the Intramural Research Program of the National Human Genome Research Institute (P.A., A.R., S.K., A.M.P.); the Intramural Program of the National Institute of Standards and Technology (N.D.O., J.M., J.W., J.M.Z.); the Intramural Research Program of the National Library of Medicine (C.X.); the Mark Foundation for Cancer Research (19-033-ASP) (M.C.S.); and a Fulbright fellowship (D.C.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Competing interests: C.S.C. is an employee of DNAnexus. J.L. is a former employee and shareholder of Bionano Genomics. S.A. is an employee of Oxford Nanopore Technologies. F.J.S. has received travel funds and spoken at PacBio and Oxford Nanopore Technologies events. K.H.M., S.K., and D.E.M. have received travel funds to speak at symposia organized by Oxford Nanopore Technologies. K.H.M. is a SAB member of Centaura Inc.
Data and materials availability: Sequencing data: 1KGP short-read datasets available within the ENA under projects PRJEB31736 and PRJEB36890. Long-read data accessions presented in table S7. Variant calling workflows and downstream analysis scripts: Mappability analysis, https://github.com/msauria/T2T_MUK_Analysis and published in Zenodo (81). Local ancestry and genome masks, https://github.com/mccoy-lab/t2t-variants and published on Zenodo (82). Collapsed duplications and clinical genomics, https://github.com/mydennislab/t2t-variants and published on Zenodo (83). Short-read variant calling, https://github.com/schatzlab/t2t-variants and published on Zenodo (84). Jasmine v1.1.0 for long-read SV comparison, https://github. com/mkirsche/Jasmine and published on Zenodo (85). Iris v1.0.4 for insertion SV polishing, https://github.com/mkirsche/Iris and published on Zenodo (86). Aligned reads, variant calls, and other summarizations available within the NHGRI AnVIL Platform, along with notebooks and workflows for computing the analysis, https://anvil.terra.bio/#workspaces/anvil-datastorage/AnVIL_T2T. UCSC assembly hub browser with variant calls and genome annotations, http://genome.ucsc.edu/cgi-bin/hgTracks?genome=t2t-chm13-v1.0&hub Url=http://t2t.gi.ucsc.edu/chm13/hub/hub.txt. Mirror of short- and long-read variant calls, https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=T2T/CHM13/assemblies/variants/. Genome in a Bottle Challenging Medically-Relevant Gene Benchmarks, https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/AshkenazimTrio/HG002_NA24385_son/CMRG_v1.00/ and published at NCBI (87). Stratification bed files for collapsed duplications, false duplications, and local ancestry in GRCh38 are available for stratifying GIAB/GA4GH benchmarking results at https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/genome-stratifications/.
REFERENCES AND NOTES
- 1.International Human Genome Sequencing Consortium, Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). doi: 10.1038/35057062; [DOI] [PubMed] [Google Scholar]
- 2.International Human Genome Sequencing Consortium, Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004). doi: 10.1038/nature03001; [DOI] [PubMed] [Google Scholar]
- 3.Schneider VA et al. , Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27, 849–864 (2017). doi: 10.1101/gr.213611.116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stephens ZD et al. , Big Data: Astronomical or Genomical? PLOS Biol 13, e1002195 (2015). doi: 10.1371/journal.pbio.1002195; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sudmant PH et al. , An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015). doi: 10.1038/nature15394; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karczewski KJ et al. , The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). doi: 10.1038/s41586-020-2308-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seo J-S et al. , De novo assembly and phasing of a Korean human genome. Nature 538, 243–247 (2016). doi: 10.1038/nature20098; [DOI] [PubMed] [Google Scholar]
- 8.Shafin K et al. , Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol 38, 1044–1053 (2020). doi: 10.1038/s41587-020-0503-6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Navarro Gonzalez J et al. , The UCSC Genome Browser database: 2021 update. Nucleic Acids Res 49, D1046–D1057 (2021). doi: 10.1093/nar/gkaa1070; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore JE et al. , Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). doi: 10.1038/s41586-020-2493-4; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Consortium GTEx, The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020). doi: 10.1126/science.aaz1776; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taliun D et al. , Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021). doi: 10.1038/s41586-021-03205-y; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miga KH et al. , Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res 24, 697–707 (2014). doi: 10.1101/gr.159624.113; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Church DM et al. , Extending reference assembly models. Genome Biol 16, 13 (2015). doi: 10.1186/s13059-015-0587-3; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaisson MJP et al. , Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun 10, 1784 (2019). doi: 10.1038/s41467-018-08148-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaisson MJP et al. , Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611 (2015). doi: 10.1038/nature13907; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Audano PA et al. , Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 176, 663–675.e19 (2019). doi: 10.1016/j.cell.2018.12.019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.1000 Genomes Project Consortium, A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yandell M et al. , A probabilistic disease-gene finder for personal genomes. Genome Res 21, 1529–1542 (2011). doi: 10.1101/gr.123158.111; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kircher M et al. , A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315 (2014). doi: 10.1038/ng.2892; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gulko B, Hubisz MJ, Gronau I, Siepel A, A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat. Genet 47, 276–283 (2015). doi: 10.1038/ng.3196; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller CA et al. , Failure to detect mutations in U2AF1 due to changes in the GRCh38 reference sequence. J. Mol. Diagn 24, 219–223 (2022). doi: 10.1016/j.jmoldx.2021.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner J et al. , Curated variation benchmarks for challenging medically relevant autosomal genes. Nat. Biotechnol 1–9, (2022). doi: 10.1038/s41587-021-01158-1 [DOI] [PMC free article] [PubMed]
- 24.Green RE et al. , A draft sequence of the Neandertal genome. Science 328, 710–722 (2010). doi: 10.1126/science.1188021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ballouz S, Dobin A, Gillis JA, Is it time to change the reference genome? Genome Biol 20, 159 (2019). doi: 10.1186/s13059-019-1774-4; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zerbino DR, Frankish A, Flicek P, Progress, Challenges, and Surprises in Annotating the Human Genome. Annu. Rev. Genomics Hum. Genet 21, 55–79 (2020). doi: 10.1146/annurev-genom-121119-083418; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nurk S et al. , The complete sequence of a human genome. Science 376, 44–53 (2022). doi: 10.1126/science.abj6987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Byrska-Bishop M et al. , High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. bioRxiv 430068 (2021). doi: 10.1101/2021.02.06.430068 [DOI] [PMC free article] [PubMed]
- 29.Zook JM et al. , A robust benchmark for detection of germline large deletions and insertions. Nat. Biotechnol 38, 1347–1355 (2020). doi: 10.1038/s41587-020-0538-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mc Cartney AM et al. , Chasing perfection: Validation and polishing strategies for telomere-to-telomere genome assemblies. Nat. Methods 10.1038/s41592-022-01440-3 (2022). doi: 10.1038/s41592-022-01440-3 [DOI] [PMC free article] [PubMed]
- 31.Altemose N et al. , Complete genomic and epigenetic maps of human centromeres. Science 376, eabl4178 (2022). doi: 10.1126/science.abl4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vollger MR et al. , Segmental duplications and their variation in a complete human genome. Science 376, eabj6965 (2022). doi: 10.1126/science.abj6965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoyt SJ et al. , From telomere to telomere: The transcriptional and epigenetic state of human repeat elements. Science 376, eabk3112 (2022). doi: 10.1126/science.abk3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. See supplementary materials.
- 35.Steinberg KM et al. , Single haplotype assembly of the human genome from a hydatidiform mole. Genome Res 24, 2066–2076 (2014). doi: 10.1101/gr.180893.114; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huddleston J et al. , Reconstructing complex regions of genomes using long-read sequencing technology. Genome Res 24, 688–696 (2014). doi: 10.1101/gr.168450.113; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dennis MY et al. , The evolution and population diversity of human-specific segmental duplications. Nat. Ecol. Evol 1, 69 (2017). doi: 10.1038/s41559-016-0069; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Bleness M et al. , Finished sequence and assembly of the DUF1220-rich 1q21 region using a haploid human genome. BMC Genomics 15, 387 (2014). doi: 10.1186/1471-2164-15-387; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng H, Concepcion GT, Feng X, Zhang H, Li H, Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021). doi: 10.1038/s41592-020-01056-5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mallick S et al. , The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201–206 (2016). doi: 10.1038/nature18964; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J-P et al. , The phosphatase JKAP/DUSP22 inhibits T-cell receptor signalling and autoimmunity by inactivating Lck. Nat. Commun 5, 3618 (2014). doi: 10.1038/ncomms4618; [DOI] [PubMed] [Google Scholar]
- 42.OMIM Entry #617768, KLEEFSTRA SYNDROME 2; KLEFS2; www.omim.org/entry/617768.
- 43.Buniello A et al. , The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012 (2019). doi: 10.1093/nar/gky1120; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schatz MC et al. , Inverting the model of genomics data sharing with the NHGRI Genomic Data Science Analysis, Visualization, and Informatics Lab-space. Cell Genomics 2, 100085 (2022). doi: 10.1016/j.xgen.2021.100085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 1303.3997 (2013).
- 46.Poplin R et al. , Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 201178 (2021). doi: 10.1101/201178 [DOI]
- 47.Iossifov I et al. , The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). doi: 10.1038/nature13908; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alexandrov LB et al. , Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). doi: 10.1038/nature12477; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jain C et al. , Weighted minimizer sampling improves long read mapping. Bioinformatics 36 (suppl. 1), i111–i118 (2020). doi: 10.1093/bioinformatics/btaa435; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). doi: 10.1093/bioinformatics/bty191; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sherman RM et al. , Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet 51, 30–35 (2019). doi: 10.1038/s41588-018-0273-y; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beyter D et al. , Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat. Genet 53, 779–786 (2021). doi: 10.1038/s41588-021-00865-4; [DOI] [PubMed] [Google Scholar]
- 53.Sedlazeck FJ et al. , Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468 (2018). doi: 10.1038/s41592-018-0001-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kirsche M et al. , Jasmine: Population-scale structural variant comparison and analysis. bioRxiv 445886 (2021). doi: 10.1101/2021.05.27.445886 [DOI] [PMC free article] [PubMed]
- 55.Shafin K et al. , Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. Nat. Methods 18, 1322–1332 (2021). doi: 10.1038/s41592-021-01299-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miga KH, Wang T, The Need for a Human Pangenome Reference Sequence. Annu. Rev. Genomics Hum. Genet 22, 81–102 (2021). doi: 10.1146/annurev-genom-120120-081921; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hartasánchez DA, Brasó-Vives M, Heredia-Genestar JM, Pybus M, Navarro A, Effect of Collapsed Duplications on Diversity Estimates: What to Expect. Genome Biol. Evol 10, 2899–2905 (2018). doi: 10.1093/gbe/evy223; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ball MP et al. , A public resource facilitating clinical use of genomes. Proc. Natl. Acad. Sci. U.S.A 109, 11920–11927 (2012). doi: 10.1073/pnas.1201904109; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng JY, Stern AJ, Racimo F, Nielsen R, Detecting selection in multiple populations by modeling ancestral admixture components. Mol. Biol. Evol 39, msab294 (2022). doi: 10.1093/molbev/msab294; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yan SM et al. , Local adaptation and archaic introgression shape global diversity at human structural variant loci. eLife 10, e67615 (2021). doi: 10.7554/eLife.67615; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H et al. , A synthetic-diploid benchmark for accurate variant-calling evaluation. Nat. Methods 15, 595–597 (2018). doi: 10.1038/s41592-018-0054-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McLaren W et al. , The Ensembl Variant Effect Predictor. Genome Biol 17, 122 (2016). doi: 10.1186/s13059-016-0974-4; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.MacArthur DG et al. , A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823–828 (2012). doi: 10.1126/science.1215040; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sung SS et al. , Mutations in TNNT3 cause multiple congenital contractures: A second locus for distal arthrogryposis type 2B. Am. J. Hum. Genet 73, 212–214 (2003). doi: 10.1086/376418; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sheng J-J, Jin J-P, TNNI1, TNNI2 and TNNI3: Evolution, regulation, and protein structure-function relationships. Gene 576, 385–394 (2016). doi: 10.1016/j.gene.2015.10.052; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ryan DP et al. , Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell 140, 88–98 (2010). doi: 10.1016/j.cell.2009.12.024; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khalilipour N et al. , Familial Esophageal Squamous Cell Carcinoma with damaging rare/germline mutations in KCNJ12/KCNJ18 and GPRIN2 genes. Cancer Genet 221, 46–52 (2018). doi: 10.1016/j.cancergen.2017.11.011; [DOI] [PubMed] [Google Scholar]
- 68.Munchel S et al. , Targeted or whole genome sequencing of formalin fixed tissue samples: Potential applications in cancer genomics. Oncotarget 6, 25943–25961 (2015). doi: 10.18632/oncotarget.4671; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gürünlüoğlu K et al. , Whole exome sequencing analysis for mutations in isolated type III biliary atresia patients. Clin. Exp. Hepatol 6, 347–353 (2020). doi: 10.5114/ceh.2020.102156; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lalrohlui F, Zohmingthanga J, Hruaii V, Vanlallawma A, Senthil Kumar N, Whole exome sequencing identifies the novel putative gene variants related with type 2 diabetes in Mizo population, northeast India. Gene 769, 145229 (2021). doi: 10.1016/j.gene.2020.145229; [DOI] [PubMed] [Google Scholar]
- 71.Eizenga JM et al. , Pangenome Graphs. Annu. Rev. Genomics Hum. Genet 21, 139–162 (2020). doi: 10.1146/annurev-genom-120219-080406; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pritt J, Chen N-C, Langmead B, FORGe: Prioritizing variants for graph genomes. Genome Biol 19, 220 (2018). doi: 10.1186/s13059-018-1595-x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vollger MR et al. , Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. Ann. Hum. Genet 84, 125–140 (2020). doi: 10.1111/ahg.12364; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van der Auwera GA, O’Connor BD, Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media Inc., 2020); https://play.google.com/store/books/details?id=vsXaDwAAQBAJ. [Google Scholar]
- 75.Sherry ST, Ward M, Sirotkin K, dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res 9, 677–679 (1999). doi: 10.1101/gr.9.8.677; [DOI] [PubMed] [Google Scholar]
- 76.Landrum MJ et al. , ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46, D1062–D1067 (2018). doi: 10.1093/nar/gkx1153; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Van der Auwera GA et al. , From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krusche P et al. , Best practices for benchmarking germline small-variant calls in human genomes. Nat. Biotechnol 37, 555–560 (2019). doi: 10.1038/s41587-019-0054-x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rentzsch P, Schubach M, Shendure J, Kircher M, CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med 13, 31 (2021). doi: 10.1186/s13073-021-00835-9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jaganathan K et al. , Predicting Splicing from Primary Sequence with Deep Learning. Cell 176, 535–548.e24 (2019). doi: 10.1016/j.cell.2018.12.015; [DOI] [PubMed] [Google Scholar]
- 81.Sauria M, msauria/T2T_MUK_Analysis: T2T_resubmission (2021); https://zenodo.org/record/5596590.
- 82.McCoy R, Taylor D, Yan S, mccoy-lab/t2t-variants: First release (2021); https://zenodo.org/record/5591054.
- 83.Soto DC, mydennislab/t2t-variants: T2T-variants (2021); https://zenodo.org/record/5595398.
- 84.Schatz M, Zarate S, Aganezov S, schatzlab/t2t-variants: T2TVariants1.0 (2021); https://zenodo.org/record/5598342.
- 85.Kirsche M, Jasmine: Population-scale structural variant merging (2021); https://zenodo.org/record/5586905. [DOI] [PMC free article] [PubMed]
- 86.Kirsche M, Iris: Structural variant breakpoint and sequence refinement (2021); https://zenodo.org/record/5586965.
- 87.Wagner J, Olson ND, McDaniel J, Zook JM, Challenging medically-relevant genes benchmark set (NIST Public Data Repository, 2021) doi: 10.18434/MDS2-2475 [DOI]
- 88.Parsons JD, Miropeats: Graphical DNA sequence comparisons. Comput. Appl. Biosci 11, 615–619 (1995). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.