

Abstract
Histone methyltransferases (HMTs) are enzymes that catalyze the methylation of lysine or arginine residues of histone proteins, a key post-translational modification (PTM). Aberrant expression or activity of these enzymes can lead to abnormal histone methylation of cancer-related genes and thus promote tumorigenesis. Histone methyltransferases have been implicated in chemotherapeutic resistance and immune stimulation, making these enzymes potential therapeutic targets of interest, and chemically targeting these proteins provides an avenue for novel drug development in cancer therapy. This review aims to discuss the evolution of chemical approaches that have emerged in the last five years to design probes targeting these enzymes, including inhibition through noncovalent inhibitors, covalent inhibitors, and targeted protein degradation through proteolysis targeting chimeras (PROTACs). This review also highlights how these compounds have been used to study the myriad of HMT functions in cancer progression and treatment response. The recent advancement of some of these drugs into human clinical investigation and even to regulatory approval highlights HMTs as a promising class of targets for chemical intervention and novel therapy development.
Graphical Abstract
Introduction
Within every cell, DNA is wrapped around an octamer of core proteins, deemed histones (H2A, H2B, H3, H4), to form nucleosomes that in turn, are wrapped into fibers that form tightly packed chromatin, which prevents DNA from becoming tangled and protects it from damage1. Strikingly, the extended tails of histones protruding from the nucleosomes are rich in positively-charged lysine and arginine residues that undergo a wide variety of post-translational covalent modifications, such as acetylation or methylation. These modifications impact chromatin compaction and can ultimately determine gene expression, genomic stability, and cell lineage development2-4. Histone methylation, in which enzymes called histone methyltransferases (HMTs) catalyze the transfer of one, two, or three methyl groups from the cofactor S-adenosyl-L-methionine (SAM) to the lysine or arginine residues5, produces modifications that are considered to be relatively stable and can be carried on through subsequent cell divisions, thus implicating them as critical players in both normal development and pathogenesis6.
In recent years, the importance of epigenetic modifications in cancer has become more widely recognized as an increasing number of tumorigenesis promoting phenotypes and pathways have been shown to be unrelated to mutational burden or chromosomal abnormalities. In particular, histone methylation has been implicated in not only metastatic outgrowth, but also in chemotherapeutic resistance and immune stimulation. Thus, there is an urgent need to develop compounds targeting these proteins to study their functions in cancer and for clinical use. HMTs, divided into two large subfamilies of histone lysine methyltransferases (HKMs) and histone arginine methyltransferases (HRMs), are highly specific and their activity has been implicated in both transcriptional activation and repression, depending on the methylation site (Figure 1A)1, 7. However, consistent with the interdependent nature of biology, the final effect of methylation marks on chromatin is determined by the interplay between multiple histone modifications (Figure 1B)8. Among lysine methyltransferases, Enhancer of Zeste Homolog 2 (EZH2), a catalytic subunit of the Polycomb Repressor Complex 2 (PRC2), has been widely reported to repress gene expression in a variety of cancer subtypes via H3K27me. EZH2 is associated with resistance to the chemotherapy drug Cisplatin and to targeted kinase inhibitors such as Gefitinib, and has been reported to negatively impact anti-tumor immunity9-13. Similarly, lysine methyltransferase KMT2A, also called Mixed Lineage Leukemia 1 (MLL1), has been associated with multiple cancer subtypes including leukemia, digestive cancers, prostate cancers, and breast cancers, and has been reported to contribute to oncogene induced senescence14-17. Within leukemia, specifically MLL rearranged (MLL-r) leukemia, MLL1 is cleaved and the N-terminus side of the protein then fuses to a number of identified partner proteins that interact with, and subsequently recruit, another lysine methyltransferase Disruptor of Telomeric Silencing 1 Like protein (DOT1L), another lysine methyltransferase. DOT1L catalyzes the mono-, di-, and tri-methylation of histone H3 lysine 79 (H3K79), which notably, is located in the H3 nucleosome core rather than on the tail18, 19. This hypermethylation of H3K79 results in aberrant transcriptional activation of leukemia promoting genes, thus contributing to tumor outgrowth and disease maintenance20. Another lysine methyltransferase of interest is the Nuclear Receptor-Binding SET Domain (NSD), which catalyzes the mono- and di-methylation of histone H3 lysine 36 (H3K36)21. The dysregulation and dysfunction of the 3 members of the NSD family (NSD1, NSD2, NSD3) are associated with several types of cancer22.
Figure 1: Histone methyltransferases in cancer and methods to target them.

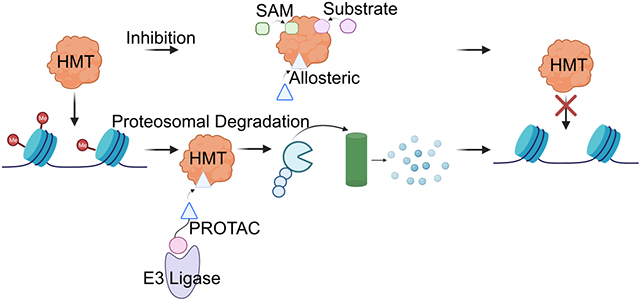
A. Phylogenetic tree of human histone methyltransferases, divided into histone lysine methyltransferases (HKM) and histone arginine methyltransferases (HRM)7. B. Dysregulation of histone methylation can lead to tumor initiation and cancer progression. C. Mechanism of lysine monomethylation using SAM cofactor. D. Methods of methyltransferase inhibition. E. Targeted protein degradation through PROTACs.
Within the arginine methyltransferase (HRM) subfamily, the protein arginine methyltransferases (PRMTs) methylate the arginine residues in histone proteins in a mono-, symmetric di-, or asymmetric di-methyl manner23. To date, nine PRMTs have been identified in mammals and all PRMTs share four conserved sequence motifs with a THW loop that comprises the SAM binding pocket24, 25. There have been concerted efforts to develop selective inhibitors against the various PRMT family members due to their increasingly important roles in various cancer types. Coactivator-Associated Arginine Methyltransferase I (CARM1, also known as PRMT4), PRMT5, and PRMT7 have emerged as increasingly promising therapeutic targets following reports that overexpression and dysregulation of these proteins promotes tumorigenesis in a range of solid and blood cancers26-30. Thus, aberrant expression or activity of any number of HMTs can disrupt transcriptional regulation of disease-related genes in normal tissue, including those functioning as oncogenes and tumor suppressors, and ultimately promote tumorigenesis31. The breadth of disease-contributing outcomes these modifications have makes these enzymes therapeutic targets of interest not only as monotherapies, but also in combination strategies to overcome drug resistance and promote immune responses. Of note, multiple inhibitors targeting EZH2, DOT1L, and multiple PRMT family members have been developed and, in some instances, have advanced to Phase I clinical trials. However, as we will discuss later in the Clinical Advancement section of this review, the lack of clinical response of some of these compounds and the discontinuation of these clinical trials suggest a need for more chemical strategies to target these proteins. While the reasons behind this lack of clinical benefit is still unknown, these compounds remain important and useful probes to study the catalytic functions of these enzymes in a variety of cancers. As a consequence, recent medicinal chemistry efforts have been devoted to further developing and improving good chemical probes for these HMTs.
Structurally, the catalytic process of HMTs involves two pockets, the SAM binding pocket and the substrate binding pocket, that bring SAM and the histone tail (substrate) into close proximity for methylation (Figure 1D). Inhibitor design for methyltransferases has been focused on targeting one of these two essential pockets by either mimicking the cofactors that can bind to this pocket, such as SAM, SAH, or the nucleoside 5’methylthioadenosine (MTA), competing with SAM for HMT binding, or blocking the histone tail from binding to the HMT (Figure 1D)32. Additionally, the increasing use, development, and popularity of high throughput screening assays, including that of virtual screenings, makes identification of different types of HMT targeting inhibitors, including allosteric HMT inhibitors, more accessible33, 34. Among these probes, both noncovalent inhibitors and covalent inhibitors have been designed and developed, all of which have been optimized to study the biological functions of the targeted protein and assess the translational potential. Together, computational chemistry and structural biology have become valuable tools in rational drug design. Molecular docking with solved crystal structures has been used to guide design and explain the structure activity relationship (SAR). Importantly, docking analyses and co-crystal structures are often used to build covalent inhibitors based upon noncovalent inhibitors by installing an electrophilic warhead at a position where it can reach and react with a functional group on the target residue, such as SH on Cystine35, 36. More recently, targeted protein degradation has become a novel chemical biology strategy gaining momentum in the field of cancer therapeutics. This degradation approach, specifically the development and use of proteolysis-targeted chimeras (PROTACs), can lead to near-complete loss of the target protein, providing a strategy that can be used to study protein function beyond their partial catalytic functional (Figure 1E)37.
The assessment and characterization of chemical probes has been furthered by advancements in the chemical biology field, including biochemical, biophysical, and cellular assay development. Among these assays, scintillation proximity assay (SPA) remains the gold standard enzymatic assay to evaluate small molecule potency and selectivity36, 38. Other biochemical assays, such as thermal shift assays and alpha assays have also been developed and are used to assess binding affinity and target engagement of the newly designed compound with the target protein in a high throughput manner39. In conjunction, these assays have been used to assess the competitive nature of the developed inhibitors with either SAM or the peptide substrate and provide insights into binding pocket interactions38, 40. To assess the on-target effect of these compounds, multiple cellular assays are used. Most commonly, western blotting of related methylation marks can reveal in vitro potency and selectivity while cell viability assays are used to assess the overall effect of targeted HMT inhibition or degradation32, 35, 40. Genetic knockout or knockdowns of the compound target are valuable methods in determining if the probe can phenocopy a complete loss of the protein of interest or if it can ameliorate the critical tumor-promoting function of the protein41. Using this combination of tools has resulted in the development of several HMT targeting compounds that have been used to further study the functions of various HMT family members in different cancers.
The development of these chemical probes has led to exciting studies elucidating the yet unknown roles and functions of various HMTs in cancer. Using these compounds in monotherapy studies has uncovered functional vulnerabilities used to design more thoughtful and intentional combinatorial therapeutic strategies. Although most of these compounds have not advanced to clinical application, and those that have advanced to patient use have shown only modest clinical responses, the fundamental studies carried out with these probes will reveal the true clinical potential of this family of compounds and can create a space in personalized medicine for HMT targeting in cancer therapy42-44.
Chemically Targeting HMTs
SAM Mimetic Noncovalent Inhibitors
SAM mimetics directly fit into the SAM binding pocket and compete the cofactor off to inhibit the catalytic function of HMTs45. Early generation SAM mimetic noncovalent inhibitors, specifically those targeting DOT1L, have piloted the advancement of HMT targeting chemical innovation through the combination of computational chemistry, biochemical assays, and molecular biology assays, followed by an optimized inhibitor advancing to clinical trial two years later. EPZ-4777, Epizyme’s first generation DOT1L inhibitor, has led the way for further development and improvement of chemical properties of DOT1L targeting inhibitors (Figure 2A). Specially, the co-crystal structure of EPZ-4777 with DOT1L was solved bound to the SAM binding pocket (Figure 2B)46. Interestingly, this structure showed that EPZ-4777 induced a conformational change of the DOT1L protein in order to accommodate the large and bulky t-butylphenyl group. Despite its selectivity, EPZ-4777 has generally poor pharmacokinetic properties due to its similarity toward SAM. Following structure-guided optimization, Epizyme developed EPZ-5676 (Figure 2A), which showed higher potency in cellular assays and improved pharmacokinetic properties47. However, clinical evaluation of this molecule resulted in only modest anti-tumoral effects in patients and drug resistance invariably occurred with long term treatment, highlighting a continued need for the development of DOT1L targeting inhibitors42 .
Figure 2: Noncovalent SAM Mimetic Inhibitors of DOT1L.

A. Structures of DOT1L SAM mimetic inhibitors EPZ-4777, EPZ-5676, and 3Dia2. B. Superposition of binding pockets of DOT1L displaying residues displaced upon EPZ-4777 binding. C. Structure of DOT1L SAM competitive inhibitor Compound 10. D. PDB:6TEL shows Compound 10 bound to DOT1L.
Recently, groups have set out to try to identify compounds that are both hydrophilic enough to bind to the pocket and hydrophobic to enter cells while having improved ADME properties in animal models. Specifically, replacing EPZ-4777’s tertiary amine with a carbon atom and introducing a secondary amine at the C5’ position was theorized to improve ADME properties by preventing N-dealkylase recognition and the use of an adenosinylcyclopentandiol in the main structure to avoid depurination led to the synthesis of 3Dia2 (Figure 2A)48. Concurrently, efforts to circumvent SAM-related pharmacokinetic limitations led to the recently reported DOT1L inhibitors Compound 10 and Compound 11 (Figure 2C, Table S1)43, 49. In a high throughput screening against DOT1L, the sole strong hit was found to bind in a SAM adjacent pocket and compete with SAM for binding in the vicinity49. Optimizations to this screening hit included the addition of a 2-pyrimidinylamino group to create a strong hydrogen bond with Asn241 and increase the compound’s binding affinity to the protein (Figure 2D). The addition of a fluorine group to the 3-chlorophenyl moiety improved the molecule’s pharmacokinetic profile. Several modifications were made to increase potency including the addition of a aminocarbonyl and a fluorine to improve stacking, the modification of the 3-fluoropyridyl group with a 2,2-difluorobenzo[d][1,3]dioxole-4-yl group to better align van der Waals contacts, and the replacement of the methoxy group with a piperazine to decrease logD. This compound was found to be orally bioavailable and showed efficacy in reducing leukemia in patient derived xenograft (PDX) models43.
Similarly, MTA has been shown to be a potent inhibitor of PRMT5, providing the starting point for the discovery of SAM mimetic inhibitors LLY-283 and JNJ-64619178 (Table S1)35. LLY-283, JNJ-64619178, and PF-06939999 are the first compounds to show potency, selectivity, and cellular activity against PRMT5 (Table S1)50, 51. While JNJ-64619178’s extension past the 5’ end of the ribose is reminiscent of EPZ-4777’s strategy of expanding the molecule into the SAM binding pocket, JNJ6461978 instead extends from the SAM binding pocket into the substrate arginine binding pocket in a pseudo-irreversible binding mode with increased residence time and extended pharmacodynamics effects, leading to more favorable pharmacokinetic and safety properties52.
Additionally, the increasing use, development, and popularity of high throughput screening assays, including that of virtual screenings, makes identification of potential SAM mimetic small molecules more accessible33, 34. Through a ligand-based pharmacophore screen, existing natural product inhibitor massonianoside B (MA) was identified as a potent and selective DOT1L SAM competitive inhibitor that can inhibit cellular H3K79 methylation and suppress MLL fusion target gene expression (Table S1)34. In 2020, the first PRMT7 specific SAM mimetic inhibitors, SGC8158 and its prodrug analog SGC3027, were derived from SGC0911, a hit in a scintillation proximity assay (SPA) screen against PRMT7 (Table S1)38.
The extremely hydrophilic nature of SAM combined with the high endogenous cellular levels of the molecule lends itself to chemical design challenges in SAM mimetic inhibitor development as probes must be polar enough to outcompete SAM in the binding pocket but hydrophobic enough to cross cell membranes53. Improper balance results in low cell permeability and low cellular activity, a phenomenon that has contributed to the slow acting and low acting in vitro and in vivo reputation of HMT inhibitors53. Despite these design obstacles, SAM mimetic molecules remain to be an attractive HMT inhibition approach as the lack of structural similarity between different SAM binding pockets allows for inhibitor selectivity45.
Substrate Competitive Noncovalent Inhibitors
An attractive alternative inhibitor development strategy for HMT is to identify small molecules that compete directly with the histone tail that binds to the HMT substrate binding site. Consistent with the approaches to designing SAM mimetic noncovalent inhibitors, modifications to existing substrate competitive noncovalent inhibitors have led to promising updates in HMT targeting chemical development. Early inhibitors of Nuclear Lysine Methyltransferase 1C (G9a), including UNC0638, utilized a central quinazoline moiety, but poor pharmacokinetic properties of these compounds prevented their advancement past in vitro pre-clinical studies (Figure 3A)54. Optimization of UNC0638 produced a peptide pocket binding G9a inhibitor EML741 via a scaffold hopping approach by replacing the quinazoline scaffold with a 1,4-benzodiazepine core (Figure 3A)32. The resultant inhibitor EML741 maintained the potency of other G9a inhibitors but exhibited heightened selectivity toward G9a, improved cellular and better blood brain barrier permeability, and the ability to reduce in vitro methylation of H3K9 (Figure 3B)32.
Figure 3: Substrate Competitive Noncovalent Inhibitors.

A. Structures of G9a inhibitors UNC0638 and EML741. B. PDB:6MBO shows EML741 bound to substrate pocket. C. Structure of PRMT4 inhibitor TP-064. D. PDB:5U4X shows TP-064 (orange) bound to the substrate pocket and SAH (green) bound to the SAM pocket in PRMT4. E. Structure of PRMT5 inhibitor EPZ015666. F. PDB4X61 shows SAM (red) and EPZ01566 (green) bound to PRMT5:MEP50 (orange).
High throughput chemical library screening was implemented to identify TP-064 as one of the first selective and potent PRMT4 binders (IC50 < 10nM, Figure 3C). Surface plasmon resonance (SPR) revealed that TP-064-PRMT4 binding requires the presence of SAM, and competition assays further confirmed it as a non-SAM mimetic. The crystal structure showed that TP-064 occupied the substrate binding site of PRMT4 and forms three crucial hydrogen bonds with Glu258, Met260, and His415 in PRMT4 (Figure 3D). Selectivity against PRMT4 was generated via pi stacking interactions with Phe163 and a hydrogen bond with Asn266, which is not observed in PRMT6 and PRMT3 (Figure 3D). In cellular assays, TP-064 successfully reduced methylation and inhibited cell proliferation in multiple myeloma cell lines40.
Another successful example of substrate competitive inhibitors of HMT is the PRMT5 inhibitor EPZ015666, which was developed via optimization of an HTS hit (Figure 3E)44. In this case, the initial hit identified through HTS showed modest inhibitory activity against PRMT5 and contain a scaffold that made unique cation-pi interactions with the protein’s substrate binding pocket (Figure 3F). Modifications to this hit included changes to the alkoxy phenyl ring to enhance potency. Multiple rounds of SAR, including introducing a less lipophilic aryl amide improved in vivo clearance rates and yielded EPZ015666, an orally bioavailable selective PRMT5 inhibitor that was shown to have anticancer effects in xenograft models44, 55. Substrate competitive inhibitors normally do not have to have optimal polarity to be effective compared to SAM competitive compounds discussed above53. However, as with SAM competitive inhibitors, substrate competitive inhibitors can also exhibit lower cellular activity as they still must outcompete the endogenous histone protein.
Allosteric Noncovalent Inhibitors
With the cellular activity challenges associated with SAM and substrate competitive HMT inhibitors, allosteric inhibition is an attractive alternative approach to modulating HMT function. Synthesis of allosteric HMT inhibitors has been less common than SAM mimetics and substrate binding noncovalent compounds because the design and development of these molecules is less intuitive and discovered compounds can be derived from existing inhibitors56. Using rational design based on inhibitors of other PRMTs, an initial PRMT4 inhibitor was identified and modified into a more potent PRMT4 binder. Co-crystal structure modeling confirmed the importance of the 1-amino-3-phenoxypropan-2-ol moiety shown to form 3 essential hydrogen bonds with PRMT4, as with TP-064 discussed above. Several rounds of SAR optimization improved biochemical and cellular potency and further structural refinements focused on improving the ADME properties created a selective and potent allosteric inhibitor, EZM2302, able to decrease methylation levels in vivo (Table S1)56.
In combating the design obstacles of creating these compounds, the availability of high throughput screening becomes increasingly useful. For example, the lysine methyltransferase absent, small, or homeotic-like 1 (ASH1L) has no reported small molecule inhibitors due to the structural challenge of circumventing the protein’s autoinhibitory loop to access its catalytic site57. Using NMR screening of SET domain targeting fragmented compounds, initial compound AS-5 was discovered to bind to ASH1L (Table S1). Structural optimization to fill the catalytic pocket of ASH1L resulted in a compound with more promising characterization but whose polarity prevented it from achieving high levels of cell permeability. Introducing a 1-(triflluoromethylsulfonyl) piperidine moiety at the yielded AS-99, whose co-crystal structure was shown to strongly bind to the SET domain near the autoinhibitory loop of ASH1L (Table S1)57. This loop exists only in four other H3K36 HMTs, making this compound a highly specific allosteric inhibitor.
Similarly, high throughput screening assays led to the discovery and development of two allosteric EED inhibitors, EED226 and A-395 (Table S1)58, 59. In both screens, modifications of hits guided by SAR study resulted in the reported compounds, which were both able to decrease H3K27 trimethylation marks in cellular assays and overall, demonstrated that small molecule binders of EED are an effective approach to disassemble the PRC2 complex, as observed with EZH2 inhibitors 58, 59. Without the pressure for a compound to fit into a substrate specific site on the target molecule of interest, allosteric inhibitors can offer higher specificity in inhibition and often, have been associated with decreased chances of off-target toxicities, a major concern in advancing chemical probes from in vitro studies to in vivo studies and clinical use60.
Covalent Inhibition of HMTs
Covalent inhibition strategies are an increasingly popular alternative for enzymatic inhibition as these probes form irreversible bonds for the duration of the half-life of the target protein and form a drug-protein complex that is not susceptible to classical equilibrium61. These inhibitors depend on a single target residue for much of their binding affinity and can have improved selectivity between proteins with high sequence homology, and potential solve the issue raised by SAM or substrate competitive effect in vitro and in vivo. While covalent inhibitors have been part of the drug design field since the late 19th century, their clinical use was limited until the last 30 years due to the toxicity associated with many covalent inhibitor metabolites and the off-target toxicities experienced by patients62. The development of covalent inhibitors with the right balance of reactivity and selectivity has been key to their increased success in the past few decades.
SAM Mimetic Covalent Inhibitors
The use of co-crystal structures is valuable in designing covalent inhibitors; visualizing and modeling hypothetical chemical interactions with a target cysteine residue in the protein of interest makes for more deliberate drug design and can improve the selectivity of the interaction. In the case of PRMT covalent inhibitor development, the co-crystal structure of PRMT5 with SAM competitive inhibitor MTA revealed a targetable small pocket near both the cysteine residue C449 and the MTA adenine ring that could potentially accommodate a covalent binding warhead35. Covalent inhibitors were developed by attaching electrophilic warheads, including acetals, hemiaminals, ketones, and aldehydes, to the adenine ring of the LLY-283 scaffold (Table S1). The covalent binding ability of the compounds was measured using a jump dilution assay and revealed that the reactive 5-membered cyclic hemiaminal Compound 9 covalently bound to the PRMT5/MEP50 complex rapidly and with an extremely slow off rate. The aldehyde Compound 10 also showed covalent binding. The co-crystal structure of Compound 10 with the PRMT5/MEP50 complex suggests that after nucleophilic attack by the cysteine residue, the resulting hydroxy group is then eliminated to form a vinyl-thio ether (Figure 4A). As aldehydes have rarely been used as covalent warheads, this novel mechanism of covalent inhibition warrants further investigation. Additionally, it is likely that Compound 9 is partially hydrolyzed to 10 under assay conditions, raising questions upon the identity of the active species in these experiments. While Compound 9 was able to inhibit methylation and reduce cell viability, it offers only a slight potency increase over LLY-283 and future optimization may produce more potent covalent PRMT5 inhibitors35.
Figure 4: Covalent Inhibitors.
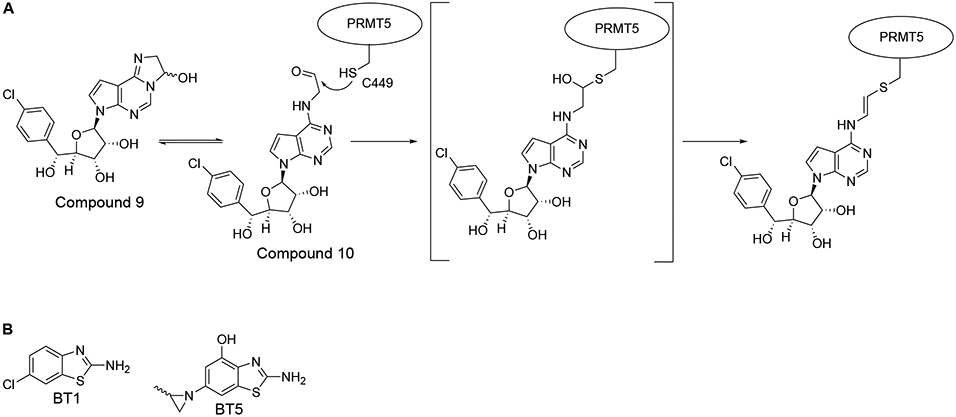
A. Proposed mechanism of covalent PRMT5 inhibitors. B. Structures of inhibitors BT1 and BT5.
Substrate Competitive Covalent Inhibitors
Similarly, the development of the first potent and cell-active covalent PRMT6 inhibitor and of a new covalent SETD8 inhibitor was started by using previously reported noncovalent inhibitors, MS203 and UNC0379 respectively, as base molecules (Table S1)63, 64. Protein sequence alignment and co-crystal structure modeling of MS023 and PRMT6 revealed Cys50, a cysteine residue unique to the PRMT6 substrate-binding site, as a target residue. An acrylamide warhead was attached to the MS023 scaffold in a position to react with Cys50, giving compound MS117 (Table S1). Kinetic studies to evaluate kinact/Ki confirmed irreversible binding and mass spectrometry analysis revealed that MS117 could efficiently form a single modified adduct with PRMT6. Scintillation proximity assays (SPA) confirmed potent inhibition against PRMT6 with an IC50 in the low nanomolar range, which was further confirmed by dose dependent decrease of H3R2 methylation levels in cellular studies36. However, MS117 showed only modest selectivity for PRMT6 over the other Type I PRMTs, showing that the covalent inhibition strategy requires further development for this target.
The substrate-competitive SETD8 inhibitor UNC0379 features a quinazoline core common to other potent methyltransferase inhibitors, but suffers from low in vitro potency. Adding an aminoethyl group to the inhibitor’s quinazoline core improved the potency of the molecule by forming a hydrogen bond with N339. A crystal structure of the improved compound with SETD8 revealed an opportunity to develop a covalent inhibitor that could bind to the surface exposed C311 cystine residue of SETD8. MS453 was synthesized by linking an electrophilic acrylamide warhead to the 4-position of the quinazoline core (Table S1). Mass spectrometry detected a single covalent adduct, confirming covalent binding and testing against a panel of over 30 other methyltransferases confirmed selectivity for SETD8. However, MS453, like its parent compound, suffered from poor membrane permeability which limited its in vivo efficacy65.
Allosteric Covalent Inhibitors
In instances where a promising tool compound does not exist, the use of high throughput chemical library screening can illuminate chemicals that were not initially used for HMT targeting. As with ASH1L discussed previously, the SET domain of NSD1 contains an autoinhibitory loop that has challenged inhibitor development, with previously reported compounds suffering from low affinity, low selectivity, or lack of cellular activity66. The first reported NSD1 covalent inhibitor was developed using a combination of NMR chemical fragment library screening, structural modeling, and chemical optimization assays67. NMR screening of a chemical fragment library against NSD1 showed that 6-chloro-1,3-benzothiazol-2-amine (BT1) (Figure 4B) bound to the SET domain. Examination of the binding analogs BT2 and BT3 revealed that the compounds bound near the SAM cofactor and induced significant rearrangement of the autoinhibitory loop (Table S1)67. The C2062 cysteine residue was also within the binding site of BT2, suggesting the potential for covalent inhibition67. Mass spectrometry confirmed that the introduction of a thiocyanate group caused covalent interactions with NSD1 and structural modeling showed a disulfide bond forming between the in-situ reduced thiol and the side chain of C2062. Further covalent warhead screening revealed that compound binding was sensitive to steric bulk, leading to the development of a methylaziridine warhead inhibitor, named BT5, that displayed time dependent binding to NSD1, characteristic of covalent inhibition (Figure 4B). BT5 showed selectivity against the NUP98/NSD1 fusion, strong growth inhibition, and cellular downregulation of H3K36 di-methylation in NUP98-NSD1 fused cells67.
While the number of noncovalent and covalent HMT inhibitors has grown significantly in recent years, many inhibitors, including some of the ones discussed above, showed weakened cellular activity in vitro or lack in vivo efficacy even with strong binding in biochemical assays. The transient nature of enzymatic inhibition gives diseased cells the opportunity to switch their survival mechanisms and upon repeated dosing of the compound, resistance often develops. As our knowledge of HMTs broadens, it is becoming increasing clear that most HMTs perform multiple functions, some of which may be independent of their canonical catalytic roles.
HMT Proteolysis Targeting Chimeras (PROTACs)
Targeted protein degradation, specifically PROTACS, have emerged as a solution for targeting multifunctional HMTs. PROTACs are bifunctional molecules consisting of a moiety that binds to the target protein, sometimes referred to as the bait moiety, and a moiety that binds an E3 ligase such as Von Hippel-Lindau (VHL) or Cereblon (CRBN)68. SAM competitive, substrate competitive, and allosteric binders have all been utilized as the bait moiety in PROTAC design. The molecule brings the target protein into proximity with the E3 ligase, which is then able to polyubiquitinate the target. The polyubiquitin group signals to the proteasome to degrade the target protein, thus removing all of the protein’s function in contrast to the loss of function of a single binding site. The full effect of these bifunctional molecules in treating cancer are still under evaluation relative to traditional inhibition.
SAM Mimetic PROTACs
Earlier this year, a series of EZH2 specific PROTACs was developed and reported by linking Cereblon binder 4-hydroxythalidomide to SAM mimetic EZH2 inhibitors GSK126 and EPZ6438, also known as Tazemetostat, which is approved in clinical trials for the treatment of patients with metastatic or locally advanced epithelioid sarcoma (Table S1)39, 69. Molecular docking of these compounds with the PRC2 complex revealed that both inhibitors contained solvent exposed fragments that could be used as a linking point for attachment of the CRBN binder without disrupting inhibitor binding to the complex and in-depth SAR studies were used to optimize linked length for both degraders. The GSK126 based degraders showed stronger degradation of EED and SUZ12 compared to EZH2 while the EPZ6438 based compounds degraded all complex components (Table S1). Thermal shift assays revealed that the EPZ6438 linked compound binds specifically to EZH2 and does not directly interact with SUZ12 or EED but this compound was able to increase the ubiquitination levels of all subunits, indicating that PROTAC binding to just a single subunit may be enough to bring the entire complex close to the E3 ligase to be destabilized. Additionally, complete degradation of EZH2 was shown to completely abolish its oncogenic function, including decreasing methylation, a feat that has not been achieved with any of the currently available EZH2 inhibitors39.
Substrate Competitive PROTACs
While there are multiple HMT inhibitors under clinical evaluation that can potently inhibit the protein’s methyltransferase activity, it has been reported that these catalytic inhibitors cannot phenocopy genetic knockdown of the protein, suggesting a role for an additional non-enzymatic function70. The first PRMT5 degrader was developed by linking substrate competitive inhibitor EPZ015666, discussed above, to a VHL ligand (Figure 3E, Table S1)71. Molecular docking analysis of the inhibitor in complex with PRMT5 revealed that the oxetane moiety of the inhibitor was solvent exposed and did not interact with other protein residues, making it a suitable position for attach an E3 ligase targeting ligand. SAR studies indicated that longer PEG linkers resulted in degraders that were able to reduce cellular PRMT5 protein levels to a larger extent. Following reports that the VHL binder VHL-2 produces more effective degraders, VHL-1 was replaced to synthesize MS4322, which was shown to be highly selective for PRMT5 in a global proteomics study and showed promising pharmacokinetic properties in mice (Table S1)71.
Complex Targeting PROTACs
Targeting individual subunits of protein complexes for degradation has been hypothesized to destabilize the entire complex, making these components targets of interest in the development of chemical degradaders39, 72. Attempted complex degradation of rearranged MLL1 fusion protein complexes through targeted degradation of the H3K4 presenter protein WDR5 resulted first in MS33, an OICR-9429 based PROTAC with a VHL ligand that exhibited dose dependent degradation of WDR5 in MLL-r leukemia cells (Table S1)73. The crystal structure of the WDR5-MS33-VCB ternary complex showed that the linker adopted an extended conformation that resulted in a decreased number of protein-protein interactions between WDR5 and VCB. A shorter linker, the additional of methyl and fluoro groups to the WDR5 binding head, and a methylated VHL binder created a significantly more potent degrader, MS67, that was able to induce degradation after 2 hours, decrease H3K4 methylation levels, and inhibit cell growth in several MLL-r AML cell lines (Table S1)41. Similarly, attempted interruption of the PRC2 complex through targeted degradation of the individual subunits resulted in the EED targeting PROTAC UNC6852, which was developed by linking the sulfone moiety of EED226, discussed above, to the VHL ligand VH032 using a 3-carbon linker (Table S1). This compound induced strong degradation of EED, EZH2, and SUZ12, and was able to decrease both cellular H3K27me3 levels and cell proliferation74.
Targeted protein degradation is an attractive alternative to traditional inhibition methods and degraders in preclinical stages of development have been reported to be more potent than their parent inhibitors. Despite promising preclinical reports, PROTACs for HMTs are still in very early stages of development and need to be further optimized. While there are few published loss of total function studies of any specific protein, with more PROTAC development, these molecules will help us understand how drug induced complete protein depletion can affect entire biological systems.
Clinical Advancement of HMT Targeting Compounds
Unsurprisingly, SAM mimetics are the main class of HMT targeting compounds that have advanced to the clinical trial stage. The first SAM mimetic to enter clinical testing was Epizyme’s EPZ-5676 (Pinometostat, Figure 2A) in 2012, reaching Phase II clinical trials for MLL-r leukemia treatment before being stopped due to a myriad of reasons, including metabolic instability, low response among patients, and drug resistance (NCT01684150, NCT02141828, NCT03701295)47, 75. Of the 51 adult MLL-r leukemia patients enrolled in NCT01684150 (Phase I), only 3 patients completed the study, with the remaining 48 patients withdrawing early due primarily to disease progression, lack of efficacy, and adverse events42. Overall, it was concluded that the levels of DOT1L inhibition and subsequent H3K79 methylation reduction attained by Pinomenostat monotherapy was not sufficient to acheive significant clinical benefit in most adult patients in this disease setting42. While Pinometostat fails to produce significant clinical benefit for regulatory approval, it continues to be an important HMT inhibitor and has been the basis for continued probe improvement, as discussed above. It has also been used as tool compounds to study the roles of DOT1L in different disease contexts and to study drug resistance mechanisms to determine treatment induced targetable vulnerabilities. To date, Pinomentostat continues to be in clinical trials, though as part of combination strategies rather than as a single agent treatment (NCT03701295, NCT03724084)76, 77.
In 2013, Tazemetostat, Epizyme’s SAM mimetic EZH2 inhibitor, was advanced to clinical testing in both solid and hematologic malignancies (NCT01897571, Table S1). While complete or partial response was only observed in 8 out of 21 patients enrolled in this Phase I study, the observation of a dose dependent reduction in H3K27 methylation marks in skin biopsies, the absence of Grade 3 and 4 adverse events, and the durability of response spurred further clinical investigation78. The Phase II arm of this trial revealed that Tazemetostat was more effective in EZH2 mutant tumors, with over half of the mutant subtype population exhibiting durable complete or partial response. Owing to the positive results of this trial, Tazemetostat was granted regulatory approval in 2020, first for patients with locally advanced or metastatic epithelioid sarcoma and then for adults with relapsed or refractory follicular lymphoma79. It continues to be clinically tested in other cancer settings as a monotherapy (NCT02601950, NCT04179864, NCT04204941), further suggesting that targeting EZH2 alone is sufficient for anti-proliferative tumor activity in humans80-82. This evidence is encouraging for PRC2 subunit targeting PROTAC development, as degraders have been shown to have the potential to be faster acting than traditional inhibitors, and further preclinical studies using PROTACs to study whole complex destabilization or loss in cancer may advance clinical compound development for EZH2 targeting probes. Similar to Pinometostat, Tazemetostat has been continued to be studied to uncover EZH2 resistance mechanisms and find opportunities for effective combination approaches. More recently, in 2016, allosteric EED inhibitor MAK683 enterred Phase I and II trials for hematologic malignancies and in 2018 and 2019, SAM mimetic PRMT5 inhibitors PF-06939999 and JNJ-64619178 enterred Phase I trials for solid tumors, though no results have been reported to date (Table S1)83-85.
Conclusions and Future Directions
HMTs have emerged as an attractive therapeutic target class in oncology. Small molecule inhibitors and degraders targeting HMTs have already exhibited promising potential for cancer therapeutics, as seen by the number of inhibitors advancing to clinical trial stages of development, and continued advancements in chemical biology approaches combined with increasing scientific excitement for the role of HMTs in disease states and as mediators of drug response will lead to further development and improvement of these compounds. In parallel, the advancement and increasing availability of high throughput compound screening platforms have largely expanded the array of potential HMT selective inhibitors that already exist. Additionally, computational chemistry approaches, including the use of crystal structures and molecular docking, has aided in more deliberate and intentional drug design choices.
Despite the exciting advancements in HMT targeting compound development, it is widely recognized that epigenetic modifier probes as a whole are at a large disadvantage in terms of time needed for compound effectiveness compared to kinase inhibitors and traditional chemotherapy. The window between drug exposure and cancer cell death allows for early genomic and enhancer remodeling, leading to resistant population outgrowth. Simultaneously, this might provide a larger therapeutic window with less toxicity toward normal tissue. Furthermore, with the rising use of functional genomics to define genes required for metastatic growth and resistant states, epigenetic targets essential for these pro-tumoral functions can be leveraged and combination or scheduled “priming” strategies can be developed to prolong cellular effectiveness of multiple drugs and consequently improve overall response and survival86, 87.
Supplementary Material
Acknowledgments
We sincerely thank the members of the Qi laboratory their valuable comments. We apologize to authors of studies which, for reasons of text space, could not be referenced within this work. Figures were created with Biorender.com and Inkscape. This work was supported in part by the National Cancer Institute (NCI; grants P01 CA142106, R01CA233601, CA222218, CA176745 to J.Q.).
Keywords
- Chromatin
genomic DNA and its associated regulatory proteins. Specifically, chromatin is made up of nucleosomes strung together by strands of DNA and can be modified by chromatin remodeling complexes such as BAF
- Post-Translational Modification (PMT)
the enzymatic modification of proteins after translation
- Histone Methylation
epigenetic modification in which methyl groups are transferred to amino acids of histone proteins. Depending on the location of the deposited methyl group, this reaction will increase or decrease gene transcription
- S-Adenosyl Methionine (SAM)
common co-substrate involved in methyl group transfers and is the methyl donor for nearly all methylation reactions in the body
- Targeted Protein Degradation
using a small molecule that binds to both a protein of interest (POI) and an E3 ligase to target a disease-causing protein for destruction
- High Throughput Screening
a drug discovery process that uses automated testing of a large number of chemical fragments or probes against a specific biological target to identify potential candidates for novel inhibitors
- Molecular Docking
a computational chemistry technique that uses modeling software such as Molecular Operating Environment (MOE) or PyMOL to predict the preferred orientation, affinity, and interaction of a ligand in the binding site of a protein
- Phenocopy
Within the drug discovery and development process, this refers to the ability of a chemical probe to induce the same change in phenotype as a genetic knockdown
Footnotes
Supplemental Information
The Supporting Information can be found online free of charge.
Supplementary Table 1 contains the structures of the chemical probes targeting HMTs discussed in this review separated into compound type (i.e. Noncovalent Inhibitors, Covalent Inhibitors, and PROTACs). These sections appear in the order they are discussed and references for all compounds with available structures are provided in this table.
Declaration of Interests
J.Q. is scientific co-founder and consultant for Epiphanes, member of the Scientific Advisory Board of Talus, and receives grant funding from Novartis. All other authors declare no financial conflicts of interest.
References:
- (1).Kouzarides T Chromatin modifications and their function. Cell 2007, 128 (4), 693–705. DOI: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- (2).Zagni C; Chiacchio U; Rescifina A Histone methyltransferase inhibitors: novel epigenetic agents for cancer treatment. Curr Med Chem 2013, 20 (2), 167–185. DOI: 10.2174/092986713804806667. [DOI] [PubMed] [Google Scholar]
- (3).Khorasanizadeh S The nucleosome: from genomic organization to genomic regulation. Cell 2004, 116 (2), 259–272. DOI: 10.1016/s0092-8674(04)00044-3. [DOI] [PubMed] [Google Scholar]
- (4).Sawan C; Herceg Z Histone modifications and cancer. Adv Genet 2010, 70, 57–85. DOI: 10.1016/B978-0-12-380866-0.60003-4. [DOI] [PubMed] [Google Scholar]
- (5).Bannister AJ; Schneider R; Kouzarides T Histone methylation: dynamic or static? Cell 2002, 109 (7), 801–806. DOI: 10.1016/s0092-8674(02)00798-5. [DOI] [PubMed] [Google Scholar]
- (6).Barski A; Cuddapah S; Cui K; Roh TY; Schones DE; Wang Z; Wei G; Chepelev I; Zhao K High-resolution profiling of histone methylations in the human genome. Cell 2007, 129 (4), 823–837. DOI: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- (7).Wu H; Min J; Lunin VV; Antoshenko T; Dombrovski L; Zeng H; Allali-Hassani A; Campagna-Slater V; Vedadi M; Arrowsmith CH; et al. Structural biology of human H3K9 methyltransferases. PLoS One 2010, 5 (1), e8570. DOI: 10.1371/journal.pone.0008570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lee JS; Smith E; Shilatifard A The language of histone crosstalk. Cell 2010, 142 (5), 682–685. DOI: 10.1016/j.cell.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).van Vlerken LE; Kiefer CM; Morehouse C; Li Y; Groves C; Wilson SD; Yao Y; Hollingsworth RE; Hurt EM EZH2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl Med 2013, 2 (1), 43–52. DOI: 10.5966/sctm.2012-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yin X; Yang S; Zhang M; Yue Y The role and prospect of JMJD3 in stem cells and cancer. Biomed Pharmacother 2019, 118, 109384. DOI: 10.1016/j.biopha.2019.109384. [DOI] [PubMed] [Google Scholar]
- (11).Liu X; Lu X; Zhen F; Jin S; Yu T; Zhu Q; Wang W; Xu K; Yao J; Guo R LINC00665 Induces Acquired Resistance to Gefitinib through Recruiting EZH2 and Activating PI3K/AKT Pathway in NSCLC. Mol Ther Nucleic Acids 2019, 16, 155–161. DOI: 10.1016/j.omtn.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sun J; Cai X; Yung MM; Zhou W; Li J; Zhang Y; Li Z; Liu SS; Cheung ANY; Ngan HYS; et al. miR-137 mediates the functional link between c-Myc and EZH2 that regulates cisplatin resistance in ovarian cancer. Oncogene 2019, 38 (4), 564–580. DOI: 10.1038/s41388-018-0459-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Peng D; Kryczek I; Nagarsheth N; Zhao L; Wei S; Wang W; Sun Y; Zhao E; Vatan L; Szeliga W; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527 (7577), 249–253. DOI: 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Malik R; Khan AP; Asangani IA; Cieslik M; Prensner JR; Wang X; Iyer MK; Jiang X; Borkin D; Escara-Wilke J; et al. Targeting the MLL complex in castration-resistant prostate cancer. Nat Med 2015, 21 (4), 344–352. DOI: 10.1038/nm.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Grinat J; Heuberger J; Vidal RO; Goveas N; Kosel F; Berenguer-Llergo A; Kranz A; Wulf-Goldenberg A; Behrens D; Melcher B; et al. The epigenetic regulator Mll1 is required for Wnt-driven intestinal tumorigenesis and cancer stemness. Nat Commun 2020, 11 (1), 6422. DOI: 10.1038/s41467-020-20222-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tate JG; Bamford S; Jubb HC; Sondka Z; Beare DM; Bindal N; Boutselakis H; Cole CG; Creatore C; Dawson E; et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 2019, 47 (D1), D941–D947. DOI: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Capell BC; Drake AM; Zhu J; Shah PP; Dou Z; Dorsey J; Simola DF; Donahue G; Sammons M; Rai TS; et al. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev 2016, 30 (3), 321–336. DOI: 10.1101/gad.271882.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).van Leeuwen F; Gafken PR; Gottschling DE Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 2002, 109 (6), 745–756. DOI: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- (19).Chen Y; Zhu WG Biological function and regulation of histone and non-histone lysine methylation in response to DNA damage. Acta Biochim Biophys Sin (Shanghai) 2016, 48 (7), 603–616. DOI: 10.1093/abbs/gmw050. [DOI] [PubMed] [Google Scholar]
- (20).Okada Y; Feng Q; Lin Y; Jiang Q; Li Y; Coffield VM; Su L; Xu G; Zhang Y hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121 (2), 167–178. DOI: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- (21).Bennett RL; Swaroop A; Troche C; Licht JD The Role of Nuclear Receptor-Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb Perspect Med 2017, 7 (6). DOI: 10.1101/cshperspect.a026708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wagner EJ; Carpenter PB Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 2012, 13 (2), 115–126. DOI: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bedford MT; Richard S Arginine methylation an emerging regulator of protein function. Mol Cell 2005, 18 (3), 263–272. DOI: 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- (24).Bedford MT; Clarke SG Protein arginine methylation in mammals: who, what, and why. Mol Cell 2009, 33 (1), 1–13. DOI: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Schapira M; Ferreira de Freitas R Structural biology and chemistry of protein arginine methyltransferases. Medchemcomm 2014, 5 (12), 1779–1788. DOI: 10.1039/c4md00269e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wysocka J; Allis CD; Coonrod S Histone arginine methylation and its dynamic regulation. Front Biosci 2006, 11, 344–355. DOI: 10.2741/1802. [DOI] [PubMed] [Google Scholar]
- (27).Tarighat SS; Santhanam R; Frankhouser D; Radomska HS; Lai H; Anghelina M; Wang H; Huang X; Alinari L; Walker A; et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia 2016, 30 (4), 789–799. DOI: 10.1038/leu.2015.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Tamiya H; Kim H; Klymenko O; Kim H; Feng Y; Zhang T; Han JY; Murao A; Snipas SJ; Jilaveanu L; et al. SHARPIN-mediated regulation of protein arginine methyltransferase 5 controls melanoma growth. J Clin Invest 2018, 128 (1), 517–530. DOI: 10.1172/JCI95410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gu Z; Gao S; Zhang F; Wang Z; Ma W; Davis RE; Wang Z Protein arginine methyltransferase 5 is essential for growth of lung cancer cells. Biochem J 2012, 446 (2), 235–241. DOI: 10.1042/BJ20120768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cheng D; He Z; Zheng L; Xie D; Dong S; Zhang P PRMT7 contributes to the metastasis phenotype in human non-small-cell lung cancer cells possibly through the interaction with HSPA5 and EEF2. Onco Targets Ther 2018, 11, 4869–4876. DOI: 10.2147/OTT.S166412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).McGrath J; Trojer P Targeting histone lysine methylation in cancer. Pharmacol Ther 2015, 150, 1–22. DOI: 10.1016/j.pharmthera.2015.01.002. [DOI] [PubMed] [Google Scholar]
- (32).Milite C; Feoli A; Horton JR; Rescigno D; Cipriano A; Pisapia V; Viviano M; Pepe G; Amendola G; Novellino E; et al. Discovery of a Novel Chemotype of Histone Lysine Methyltransferase EHMT1/2 (GLP/G9a) Inhibitors: Rational Design, Synthesis, Biological Evaluation, and Co-crystal Structure. J Med Chem 2019, 62 (5), 2666–2689. DOI: 10.1021/acs.jmedchem.8b02008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Chen C; Zhu H; Stauffer F; Caravatti G; Vollmer S; Machauer R; Holzer P; Mobitz H; Scheufler C; Klumpp M; et al. Discovery of Novel Dot1L Inhibitors through a Structure-Based Fragmentation Approach. ACS Med Chem Lett 2016, 7 (8), 735–740. DOI: 10.1021/acsmedchemlett.6b00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Chen J; Park HJ Computer-Aided Discovery of Massonianoside B as a Novel Selective DOT1L Inhibitor. ACS Chem Biol 2019, 14 (5), 873–881. DOI: 10.1021/acschembio.8b00933. [DOI] [PubMed] [Google Scholar]
- (35).Lin H; Wang M; Zhang YW; Tong S; Leal RA; Shetty R; Vaddi K; Luengo JI Discovery of Potent and Selective Covalent Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors. ACS Med Chem Lett 2019, 10 (7), 1033–1038. DOI: 10.1021/acsmedchemlett.9b00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Shen Y; Li F; Szewczyk MM; Halabelian L; Park KS; Chau I; Dong A; Zeng H; Chen H; Meng F; et al. Discovery of a First-in-Class Protein Arginine Methyltransferase 6 (PRMT6) Covalent Inhibitor. J Med Chem 2020, 63 (10), 5477–5487. DOI: 10.1021/acs.jmedchem.0c00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Burslem GM; Crews CM Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181 (1), 102–114. DOI: 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Szewczyk MM; Ishikawa Y; Organ S; Sakai N; Li F; Halabelian L; Ackloo S; Couzens AL; Eram M; Dilworth D; et al. Pharmacological inhibition of PRMT7 links arginine monomethylation to the cellular stress response. Nat Commun 2020, 11 (1), 2396. DOI: 10.1038/s41467-020-16271-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Liu Z; Hu X; Wang Q; Wu X; Zhang Q; Wei W; Su X; He H; Zhou S; Hu R; et al. Design and Synthesis of EZH2-Based PROTACs to Degrade the PRC2 Complex for Targeting the Noncatalytic Activity of EZH2. J Med Chem 2021, 64 (5), 2829–2848. DOI: 10.1021/acs.jmedchem.0c02234. [DOI] [PubMed] [Google Scholar]
- (40).Nakayama K; Szewczyk MM; Dela Sena C; Wu H; Dong A; Zeng H; Li F; de Freitas RF; Eram MS; Schapira M; et al. TP-064, a potent and selective small molecule inhibitor of PRMT4 for multiple myeloma. Oncotarget 2018, 9 (26), 18480–18493. DOI: 10.18632/oncotarget.24883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Yu X; Li D; Kottur J; Shen Y; Kim HS; Park KS; Tsai YH; Gong W; Wang J; Suzuki K; et al. A selective WDR5 degrader inhibits acute myeloid leukemia in patient-derived mouse models. Sci Transl Med 2021, 13 (613), eabj1578. DOI: 10.1126/scitranslmed.abj1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Stein EM; Garcia-Manero G; Rizzieri DA; Tibes R; Berdeja JG; Savona MR; Jongen-Lavrenic M; Altman JK; Thomson B; Blakemore SJ; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131 (24), 2661–2669. DOI: 10.1182/blood-2017-12-818948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Perner F; Gadrey JY; Xiong Y; Hatton C; Eschle BK; Weiss A; Stauffer F; Gaul C; Tiedt R; Perry JA; et al. Novel inhibitors of the histone methyltransferase DOT1L show potent antileukemic activity in patient-derived xenografts. Blood 2020, 136 (17), 1983–1988. DOI: 10.1182/blood.2020006113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Duncan KW; Rioux N; Boriack-Sjodin PA; Munchhof MJ; Reiter LA; Majer CR; Jin L; Johnston LD; Chan-Penebre E; Kuplast KG; et al. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med Chem Lett 2016, 7 (2), 162–166. DOI: 10.1021/acsmedchemlett.5b00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Campagna-Slater V; Mok MW; Nguyen KT; Feher M; Najmanovich R; Schapira M Structural chemistry of the histone methyltransferases cofactor binding site. J Chem Inf Model 2011, 51 (3), 612–623. DOI: 10.1021/ci100479z. [DOI] [PubMed] [Google Scholar]
- (46).Yu W; Chory EJ; Wernimont AK; Tempel W; Scopton A; Federation A; Marineau JJ; Qi J; Barsyte-Lovejoy D; Yi J; et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun 2012, 3, 1288. DOI: 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- (47).Daigle SR; Olhava EJ; Therkelsen CA; Basavapathruni A; Jin L; Boriack-Sjodin PA; Allain CJ; Klaus CR; Raimondi A; Scott MP; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122 (6), 1017–1025. DOI: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Bon C; Si Y; Pernak M; Barbachowska M; Levi-Acobas E; Cadet Daniel V; Jallet C; Ruzic D; Djokovic N; Djikic T; et al. Synthesis and Biological Activity of a Cytostatic Inhibitor of MLLr Leukemia Targeting the DOT1L Protein. Molecules 2021, 26 (17). DOI: 10.3390/molecules26175300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Stauffer F; Weiss A; Scheufler C; Mobitz H; Ragot C; Beyer KS; Calkins K; Guthy D; Kiffe M; Van Eerdenbrugh B; et al. New Potent DOT1L Inhibitors for in Vivo Evaluation in Mouse. ACS Med Chem Lett 2019, 10 (12), 1655–1660. DOI: 10.1021/acsmedchemlett.9b00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bonday ZQ; Cortez GS; Grogan MJ; Antonysamy S; Weichert K; Bocchinfuso WP; Li F; Kennedy S; Li B; Mader MM; et al. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med Chem Lett 2018, 9 (7), 612–617. DOI: 10.1021/acsmedchemlett.8b00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Brehmer D; Wu T; Mannens G; Beke L; Vinken P; Gaffney D; Sun W; Pande V; Thuring J-W; Millar H; et al. Abstract DDT02-04: A novel PRMT5 inhibitor with potent in vitro and in vivo activity in preclinical lung cancer models. Cancer Research 2017, 77 (13 Supplement), DDT02-04–DDT02-04. DOI: 10.1158/1538-7445.am2017-ddt02-04. [DOI] [Google Scholar]
- (52).Lin H; Luengo JI Nucleoside protein arginine methyltransferase 5 (PRMT5) inhibitors. Bioorg Med Chem Lett 2019, 29 (11), 1264–1269. DOI: 10.1016/j.bmcl.2019.03.042. [DOI] [PubMed] [Google Scholar]
- (53).Ferreira de Freitas R; Ivanochko D; Schapira M Methyltransferase Inhibitors: Competing with, or Exploiting the Bound Cofactor. Molecules 2019, 24 (24). DOI: 10.3390/molecules24244492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Kubicek S; O'Sullivan RJ; August EM; Hickey ER; Zhang Q; Teodoro ML; Rea S; Mechtler K; Kowalski JA; Homon CA; et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell 2007, 25 (3), 473–481. DOI: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- (55).Chan-Penebre E; Kuplast KG; Majer CR; Boriack-Sjodin PA; Wigle TJ; Johnston LD; Rioux N; Munchhof MJ; Jin L; Jacques SL; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 2015, 11 (6), 432–437. DOI: 10.1038/nchembio.1810. [DOI] [PubMed] [Google Scholar]
- (56).Drew AE; Moradei O; Jacques SL; Rioux N; Boriack-Sjodin AP; Allain C; Scott MP; Jin L; Raimondi A; Handler JL; et al. Identification of a CARM1 Inhibitor with Potent In Vitro and In Vivo Activity in Preclinical Models of Multiple Myeloma. Sci Rep 2017, 7 (1), 17993. DOI: 10.1038/s41598-017-18446-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Rogawski DS; Ndoj J; Cho HJ; Maillard I; Grembecka J; Cierpicki T Two Loops Undergoing Concerted Dynamics Regulate the Activity of the ASH1L Histone Methyltransferase. Biochemistry 2015, 54 (35), 5401–5413. DOI: 10.1021/acs.biochem.5b00697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Genta S; Pirosa MC; Stathis A BET and EZH2 Inhibitors: Novel Approaches for Targeting Cancer. Curr Oncol Rep 2019, 21 (2), 13. DOI: 10.1007/s11912-019-0762-x. [DOI] [PubMed] [Google Scholar]
- (59).He Y; Selvaraju S; Curtin ML; Jakob CG; Zhu H; Comess KM; Shaw B; The J; Lima-Fernandes E; Szewczyk MM; et al. The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat Chem Biol 2017, 13 (4), 389–395. DOI: 10.1038/nchembio.2306. [DOI] [PubMed] [Google Scholar]
- (60).Nussinov R; Tsai CJ Allostery in disease and in drug discovery. Cell 2013, 153 (2), 293–305. DOI: 10.1016/j.cell.2013.03.034. [DOI] [PubMed] [Google Scholar]
- (61).Powers JC; Asgian JL; Ekici OD; James KE Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev 2002, 102 (12), 4639–4750. DOI: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- (62).Ghosh AK; Samanta I; Mondal A; Liu WR Covalent Inhibition in Drug Discovery. ChemMedChem 2019, 14 (9), 889–906. DOI: 10.1002/cmdc.201900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Mitchell LH; Drew AE; Ribich SA; Rioux N; Swinger KK; Jacques SL; Lingaraj T; Boriack-Sjodin PA; Waters NJ; Wigle TJ; et al. Aryl Pyrazoles as Potent Inhibitors of Arginine Methyltransferases: Identification of the First PRMT6 Tool Compound. ACS Med Chem Lett 2015, 6 (6), 655–659. DOI: 10.1021/acsmedchemlett.5b00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Shen Y; Szewczyk MM; Eram MS; Smil D; Kaniskan HU; de Freitas RF; Senisterra G; Li F; Schapira M; Brown PJ; et al. Discovery of a Potent, Selective, and Cell-Active Dual Inhibitor of Protein Arginine Methyltransferase 4 and Protein Arginine Methyltransferase 6. J Med Chem 2016, 59 (19), 9124–9139. DOI: 10.1021/acs.jmedchem.6b01033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Butler KV; Ma A; Yu W; Li F; Tempel W; Babault N; Pittella-Silva F; Shao J; Wang J; Luo M; et al. Structure-Based Design of a Covalent Inhibitor of the SET Domain-Containing Protein 8 (SETD8) Lysine Methyltransferase. J Med Chem 2016, 59 (21), 9881–9889. DOI: 10.1021/acs.jmedchem.6b01244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Coussens NP; Kales SC; Henderson MJ; Lee OW; Horiuchi KY; Wang Y; Chen Q; Kuznetsova E; Wu J; Chakka S; et al. High-throughput screening with nucleosome substrate identifies small-molecule inhibitors of the human histone lysine methyltransferase NSD2. J Biol Chem 2018, 293 (35), 13750–13765. DOI: 10.1074/jbc.RA118.004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Huang H; Howard CA; Zari S; Cho HJ; Shukla S; Li H; Ndoj J; Gonzalez-Alonso P; Nikolaidis C; Abbott J; et al. Covalent inhibition of NSD1 histone methyltransferase. Nat Chem Biol 2020, 16 (12), 1403–1410. DOI: 10.1038/s41589-020-0626-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Sakamoto KM; Kim KB; Kumagai A; Mercurio F; Crews CM; Deshaies RJ Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 2001, 98 (15), 8554–8559. DOI: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Hoy SM Tazemetostat: First Approval. Drugs 2020, 80 (5), 513–521. DOI: 10.1007/s40265-020-01288-x. [DOI] [PubMed] [Google Scholar]
- (70).Sun X; Gao H; Yang Y; He M; Wu Y; Song Y; Tong Y; Rao Y PROTACs: great opportunities for academia and industry. Signal Transduct Target Ther 2019, 4, 64. DOI: 10.1038/s41392-019-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Shen Y; Gao G; Yu X; Kim H; Wang L; Xie L; Schwarz M; Chen X; Guccione E; Liu J; et al. Discovery of First-in-Class Protein Arginine Methyltransferase 5 (PRMT5) Degraders. J Med Chem 2020, 63 (17), 9977–9989. DOI: 10.1021/acs.jmedchem.0c01111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Hsu JH; Rasmusson T; Robinson J; Pachl F; Read J; Kawatkar S; DH OD; Bagal S; Code E; Rawlins P; et al. EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem Biol 2020, 27 (1), 41–46 e17. DOI: 10.1016/j.chembiol.2019.11.004. [DOI] [PubMed] [Google Scholar]
- (73).Lu K; Tao H; Si X; Chen Q The Histone H3 Lysine 4 Presenter WDR5 as an Oncogenic Protein and Novel Epigenetic Target in Cancer. Front Oncol 2018, 8, 502. DOI: 10.3389/fonc.2018.00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Potjewyd F; Turner AW; Beri J; Rectenwald JM; Norris-Drouin JL; Cholensky SH; Margolis DM; Pearce KH; Herring LE; James LI Degradation of Polycomb Repressive Complex 2 with an EED-Targeted Bivalent Chemical Degrader. Cell Chem Biol 2020, 27 (1), 47–56 e15. DOI: 10.1016/j.chembiol.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Shukla N; Wetmore C; O'Brien MM; Silverman LB; Brown P; Cooper TM; Thomson B; Blakemore SJ; Daigle S; Suttle B; et al. Final Report of Phase 1 Study of the DOT1L Inhibitor, Pinometostat (EPZ-5676), in Children with Relapsed or Refractory MLL-r Acute Leukemia. Blood 2016, 128 (22), 2780. DOI: 10.1182/blood.V128.22.2780.2780. [DOI] [Google Scholar]
- (76).Pinometostat and Azacitidine in Treating Patients With Relapsed, Refractory, or Newly Diagnosed Acute Myeloid Leukemia With 11q23 Rearrangement. 2018. https://clinicaltrials.gov/ct2/show/NCT03701295 (accessed.
- (77).Pinometostat With Standard Chemotherapy in Treating Patients With Newly Diagnosed Acute Myeloid Leukemia and MLL Gene Rearrangement. 2018. https://clinicaltrials.gov/ct2/show/NCT03724084 (accessed.
- (78).Italiano A; Soria JC; Toulmonde M; Michot JM; Lucchesi C; Varga A; Coindre JM; Blakemore SJ; Clawson A; Suttle B; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 2018, 19 (5), 649–659. DOI: 10.1016/S1470-2045(18)30145-1. [DOI] [PubMed] [Google Scholar]
- (79).Morschhauser F; Tilly H; Chaidos A; McKay P; Phillips T; Assouline S; Batlevi CL; Campbell P; Ribrag V; Damaj GL; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol 2020, 21 (11), 1433–1442. DOI: 10.1016/S1470-2045(20)30441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).A Phase II, Multicenter Study of the EZH2 Inhibitor Tazemetostat in Adult Subjects With INI1-Negative Tumors or Relapsed/Refractory Synovial Sarcoma. 2015. https://clinicaltrials.gov/ct2/show/NCT02601950 (accessed.
- (81).CELLO-1, Study of Tazemetostat With Enzalutamide or Abiraterone/Prednisone in Subjects With Castration Resistant Prostate Cancer Who Have Not Received Chemotherapy. 2019. https://clinicaltrials.gov/ct2/show/NCT04179864 (accessed.
- (82).Tazemetostat in Combination With Doxorubicin as Frontline Therapy for Advanced Epithelioid Sarcoma. 2019. https://clinicaltrials.gov/ct2/show/NCT04204941 (accessed.
- (83).Safety and Efficacy of MAK683 in Adult Patients With Advanced Malignancies. 2016. https://clinicaltrials.gov/ct2/show/NCT02900651 (accessed.
- (84).A Dose Escalation Study Of PF-06939999 In Participants With Advanced Or Metastatic Solid Tumors. 2019. https://clinicaltrials.gov/ct2/show/NCT03854227 (accessed.
- (85).A Study of JNJ-64619178, an Inhibitor of PRMT5 in Participants With Advanced Solid Tumors, NHL, and Lower Ris kMDS. 2018. https://clinicaltrials.gov/ct2/show/NCT03573310 (accessed.
- (86).Dafflon C; Craig VJ; Mereau H; Grasel J; Schacher Engstler B; Hoffman G; Nigsch F; Gaulis S; Barys L; Ito M; et al. Complementary activities of DOT1L and Menin inhibitors in MLL-rearranged leukemia. Leukemia 2017, 31 (6), 1269–1277. DOI: 10.1038/leu.2016.327. [DOI] [PubMed] [Google Scholar]
- (87).Kumar S; Zeng Z; Bagati A; Tay RE; Sanz LA; Hartono SR; Ito Y; Abderazzaq F; Hatchi E; Jiang P; et al. CARM1 Inhibition Enables Immunotherapy of Resistant Tumors by Dual Action on Tumor Cells and T Cells. Cancer Discov 2021, 11 (8), 2050–2071. DOI: 10.1158/2159-8290.CD-20-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.