

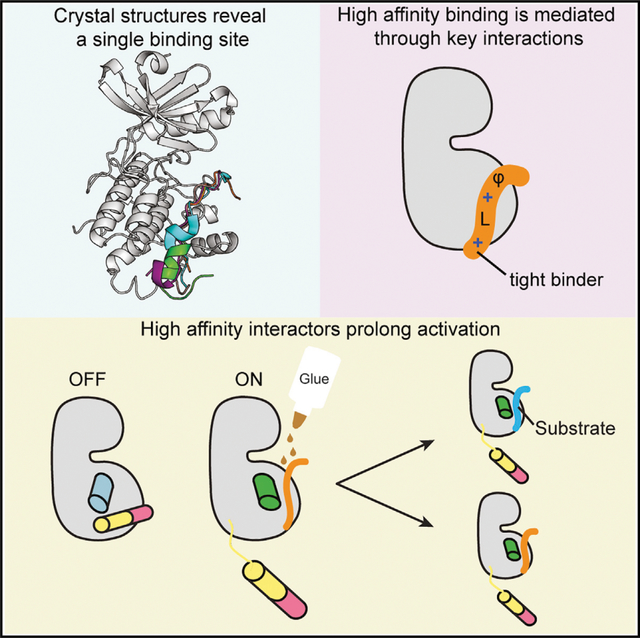
SUMMARY
Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a signaling protein required for long-term memory. When activated by Ca2+/CaM, it sustains activity even after the Ca2+ dissipates. In addition to the well-known autophosphorylation-mediated mechanism, interaction with specific binding partners also persistently activates CaMKII. A long-standing model invokes two distinct S and T sites. If an interactor binds at the T-site, then it will preclude autoinhibition and allow substrates to be phosphorylated at the S site. Here, we specifically test this model with X-ray crystallography, molecular dynamics simulations, and biochemistry. Our data are inconsistent with this model. Co-crystal structures of four different activators or substrates show that they all bind to a single continuous site across the kinase domain. We propose a mechanistic model where persistent CaMKII activity is facilitated by high-affinity binding partners that kinetically compete with autoinhibition by the regulatory segment to allow substrate phosphorylation.
In brief
Özden et al. report high-resolution structures of CaMKII bound to peptides from proteins that are crucial for memory. All interactors dock onto the same single binding site using conserved interactions. A model is proposed for how high-affinity interactors prolong CaMKII activity by competing with autoinhibition.
Graphical Abstract
INTRODUCTION
Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a central signaling protein that controls cellular functions such as synaptic plasticity, cytoskeletal regulation, cell growth and division, gene transcription, and ion channel modulation (Lisman et al., 2012). CaMKII biology has been an active focus of research especially because of its crucial role in long-term potentiation (LTP), which is the basis for long-term memory (Malenka et al., 1989; Sloutsky and Stratton, 2020). CaMKII is highly abundant in the forebrain postsynaptic density (PSD) fraction, where it makes up to 2% of the total protein (Erondu and Kennedy, 1985). CaMKII is a multisubunit complex made up of 12–14 subunits and is oligomerized by the hub domain (Figure 1A). Each CaMKII subunit contains a Ser/Thr kinase domain, autoinhibitory/regulatory segment, variable linker region, and hub domain (Figure 1B). CaMKII is governed by many molecular interactions, but structural details of these interactions are lacking (Figure 1C; Lynch, 2004).
Figure 1. CaMKII architecture and the interaction partners at excitatory synapses.
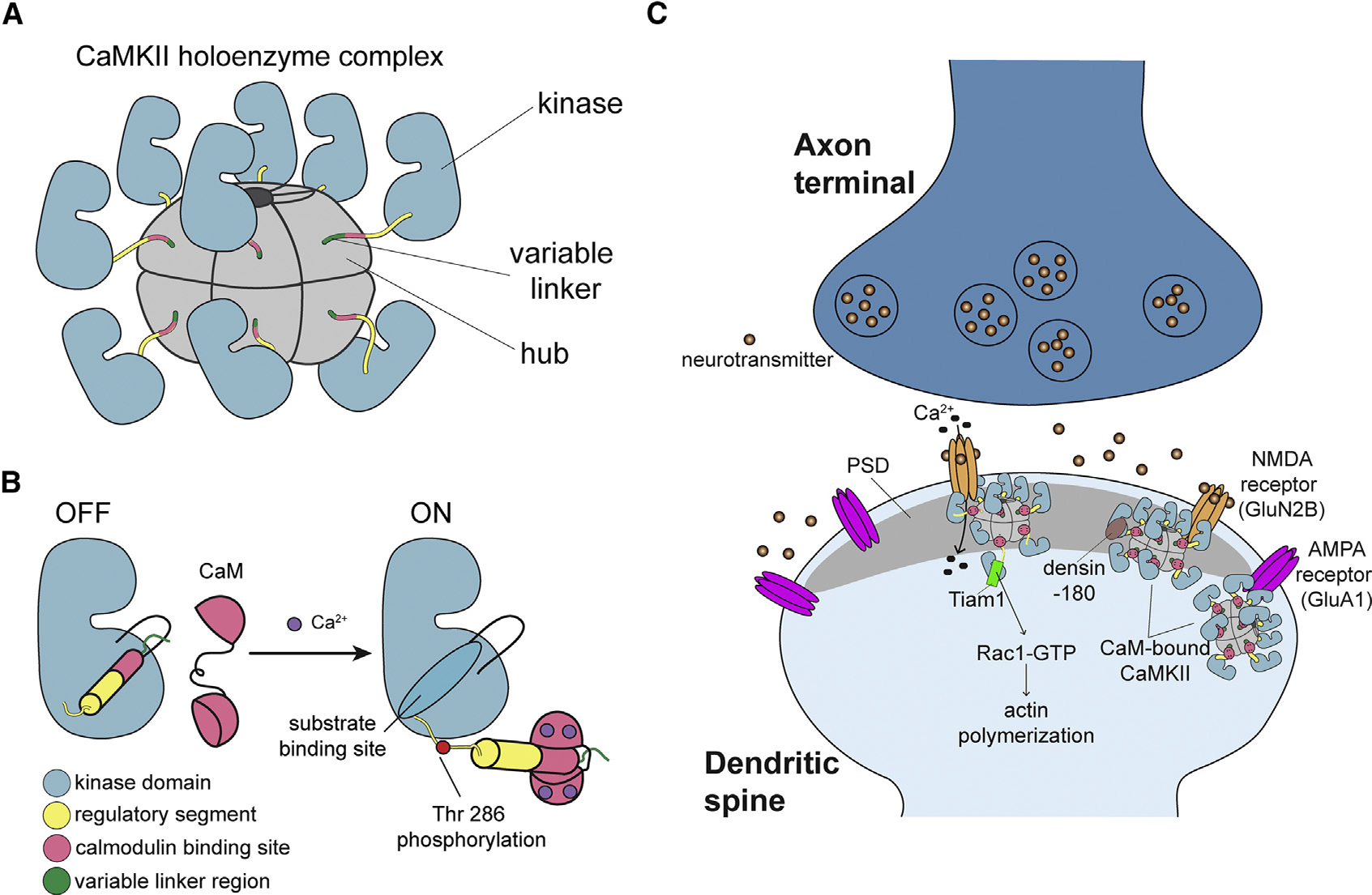
(A) The architecture of a dodecameric CaMKII holoenzyme.
(B) Ca2+/CaM binding activates CaMKII by competitively binding the regulatory segment, thereby freeing the substrate binding site. Active CaMKII autophosphorylates at Thr 286.
(C) CaMKII interactions at the excitatory postsynaptic structure, mostly in the postsynaptic density (PSD), of the dendritic spine.
Ca2+/calmodulin (CaM) activates CaMKII by binding to the regulatory segment, thereby freeing the substrate binding site. This well-understood mechanism triggers trans-autophosphorylation of T286, which renders CaMKII constitutively active until T286 is dephosphorylated. CaMKII has also been shown to maintain activity in the absence of Ca2+/CaM and T286 phosphorylation by another mechanism that invokes binding partners (Bayer et al., 2001, 2006; Ishida and Fujisawa, 1995; Saneyoshi et al., 2019), for which we lack a clear mechanism. There are three binding partners (NMDA [N-methyl-D-aspartate] receptor, Tiam1, and Drosophila EAG [ether-à-go-go] [dEAG]) that have been shown to be activators of CaMKII after the Ca2+ stimulus dissipates, with or without T286 phosphorylation (Bayer et al., 2001, 2006; Saneyoshi et al., 2019; Sun et al., 2004). The GluN2B subunit of the NMDA receptor is a known substrate of CaMKII (Strack and Colbran, 1998; Strack et al., 2000), and so far, it is the best-studied CaMKII activator (Omkumar et al., 1996). Ca2+/CaM-activated CaMKII has been shown to form a persistent complex with GluN2B, which locks CaMKII in an active conformation, as long as binding persists (Bayer et al., 2001; Strack et al., 2000). Persistent binding of CaMKII to GluN2B and the resultant activation have been explained by a hypothetical model (Bayer et al., 2006). In this model, GluN2B first binds to Ca2+/CaM-activated CaMKII close to the active site (S site), presenting Ser 1303 for phosphorylation. Then, GluN2B dissociates from CaMKII and re-binds at the base of the CaMKII C lobe (T site), freeing the S site to bind and phosphorylate other substrates. This model has been widely accepted in the field, but to date, there is no structural data supporting it.
Another known activator is Tiam1, a Rac guanine-nucleotide exchange factor (RacGEF), and it is phosphorylated at multiple sites by CaMKII. The carboxy tail of Tiam1 also forms a stable complex with CaMKII using a pseudosubstrate sequence (alanine at the phosphorylation site) (Saneyoshi et al., 2019). The third activator is dEAG, a potassium channel in Drosophila (Sun et al., 2004). Tiam1 and dEAG behave similarly to GluN2B in that binding maintains CaMKII activation in the absence of Ca2+/CaM (Saneyoshi et al., 2019; Sun et al., 2004). A recent structure shows that dEAG binds over an extensive surface on the CaMKII kinase domain (Castro-Rodrigues et al., 2018). On the other hand, densin-180 (LRRC7) is a postsynaptic scaffolding protein. It also forms a stable complex with CaMKII through a pseudosubstrate sequence (isoleucine at the phosphorylation site), but unlike GluN2B or Tiam1, it inhibits kinase activity selectively (Jiao et al., 2011; Walikonis et al., 2001). Finally, the AMPA receptor subunit GluA1 is a CaMKII substrate but not a known activator (Barria et al., 1997a; Mammen et al., 1997). GluA1 phosphorylation by CaMKII is important for synaptic plasticity (Barria et al., 1997b; Derkach et al., 1999; Diering and Huganir, 2018).
In the current study, we solved co-crystal structures of the CaMKII kinase domain bound to peptides from GluN2B, Tiam1, densin-180, and GluA1. Using these structures as starting points, we compared molecular dynamics (MD) simulations of the kinase domain in complex with GluN2B, Tiam1, and a previously solved structure with CaMKII bound to an inhibitor, CaMKIIN (Chao et al., 2010). Combining this structural information with the biophysical and biochemical measurements allowed us to clarify important interactions that drive binding and propose a working model for maintaining CaMKII activity in the absence of Ca2+/CaM.
RESULTS
The studies outlined below include co-crystallization of the CaMKII kinase domain bound to peptides from the following CaMKII binding partners: GluN2B (residues 1,289–1,310), GluA1 (residues 818–837), densin-180 (residues 797–818), and Tiam1 (residues 1,541–1,559). We also include CaMKIIN in these studies, a known endogenous inhibitor of CaMKII (Chang et al., 2001; Wang et al., 2008; Zhang et al., 2001). Unless otherwise specified, all experiments were conducted in the background of an inactivating CaMKII mutation (D135N). This mutation effectively disables kinase activity by removing the catalytic base while preserving the structural integrity of the kinase. Indeed, an overlay of the wild-type (WT) kinase domain with the D135N kinase domain structure shows very few deviations (root-mean-square deviation [RMSD] = 0.309).
In addition to analysis of the crystal structures, we also performed MD simulations of the kinase domain in complex with extended versions of peptides to assess any contributions from the terminal ends that would not be observed in the crystal structures. All simulations were performed in the NPT (isothermal-isobaric) ensemble at 300 K. One production simulation was performed for the kinase:GluN2B and kinase:Tiam1 complexes, producing trajectories of 2.19 μs and 2.05 μs, respectively. Two independent simulations were performed for the kinase:CaMKIIN complex. These were 0.46 μs and 1.84 μs in length for an aggregate of 2.3 μs. RMSD and root-mean-square fluctuation (RMSF) for the kinase domain and the peptide were calculated over the course of the trajectories (Figures S1A–S1C).
The CaMKII kinase domain has a single binding site
We first used isothermal titration calorimetry (ITC) to measure the binding affinity of peptides to the CaMKII kinase domain (D135N). We showed that four of five peptides bound to the kinase with high affinity, whereas GluA1 bound with significantly lower affinity (Figures 2A–2E). The Kd values for single-site binding ranged from 39 nM to more than 55 μM (GluN2B Kd = 107 ± 47 nM, CaMKIIN Kd = 39 ± 24 nM, densin-180 Kd = 585 ± 114 nM, Tiam1 Kd = 1.1 ± 0.1 μM, GluA1 Kd >55 μM) (Table S4). From these data, it appears that GluA1 is the outlier, whereas the other partners bind with relatively high affinity.
Figure 2. There is a single binding site on the CaMKII kinase domain.
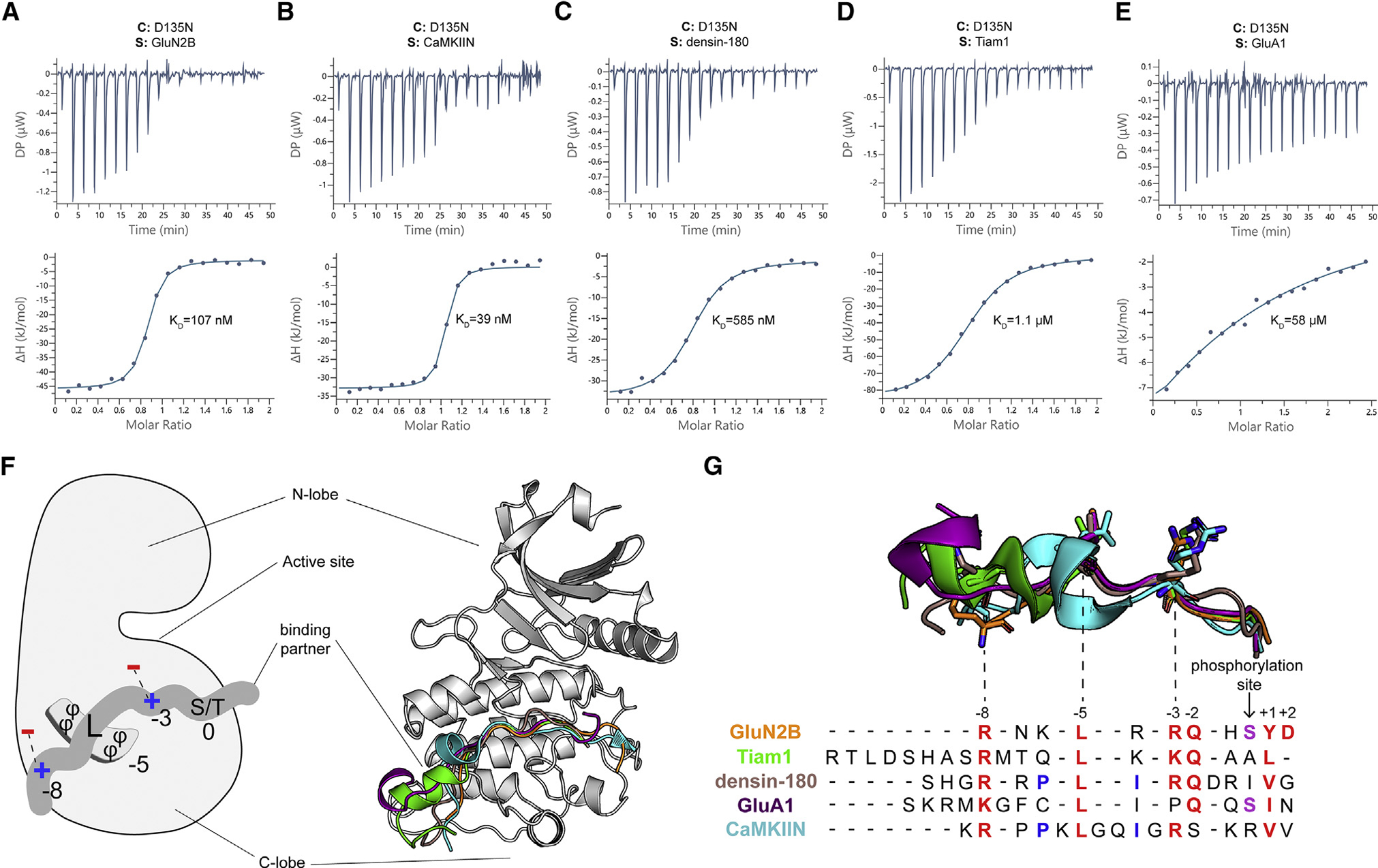
(A–E) Representative ITC measurements of D135N kinase domain binding to peptide partners. Contents of the cell (C) and syringe (S) are indicated. The mean Kd value from two independent measurements is labeled.
(F) Left: schematic of the interactions with binding partners. The phi symbol indicates hydrophobic residues. Right: overlay of five cocrystal structures, peptides shown as cartoon in corresponding colors seen in key to the right.
(G) The sequence alignment of CaMKII binding partners. Binding partner position numbering is based on the prototypical GluN2B substrate with the phosphorylation site set to zero. Aligned peptide structures are shown above for reference. Conserved residues are colored red. Phosphorylatable residues at the phosphorylation site are colored purple. Residues involved in a docking event with W214 are colored blue.
We then determined 12 total crystal structures of the CaMKII kinase domain (residues 7–274, lacking the regulatory domain) bound to one of four peptides: GluN2B, GluA1, densin-180, and Tiam1 (Figures 2F and S2A–S2D). We resolved 14–19 residues in each peptide. All structures have resolutions in the range of 1.85–3.1 Å (Tables S1–S3). We solved eight structures of the kinase domain bound to the GluN2B peptide (WT and phosphomimetic S1303D) with different ligands in the nucleotide binding pocket (Tables S1 and S2). We also solved structures of the kinase domain bound to peptides of Tiam1, densin-180, and GluA1, with ATP bound or empty nucleotide binding pockets (Table S3). In a structure with ATP and S1303D GluN2B bound (PDB: 7KL1), there is strong density between the aspartate side chain and gamma phosphate group, indicating formation of a covalent bond similar to what would be expected for transfer of the gamma phosphate to the carboxylate (Figure S2E).
Across all structures, we observed that peptides interacted with the same surface on the kinase domain. The overall fold of the kinase domain is similar in all structures. So far, predicting CaMKII interaction partners has been difficult because of conformational heterogeneity. Our structures revealed that several peptides have helical turns, which shifted the register of conserved interactions. In the CaMKIIN structure, there is an internal short helical turn (Chao et al., 2010). Tiam1 and GluA1 peptides also adopted short helical turns, but they were shifted to the proximal (N-terminal) end compared with CaMKIIN. GluN2B and densin-180 peptides bound in completely extended conformations with no helical turns. We now provide an updated sequence alignment based on our structural observations (numbering is based on the prototypical GluN2B substrate with the phosphorylation site set to zero, and the inserted residues are not counted) (Figure 2G). The two peptides that are substrates (GluN2B and GluA1) have the phosphosite facing the nucleotide binding pocket so that both peptides are docked at the active site, ready to be phosphorylated. The critical residues of the binding partner mediating this interaction are conserved at positions +1, −2, −3, −5, and −8, which are discussed in detail below (Figure 3).
Figure 3. Conserved binding motifs on the catalytic domain surface.
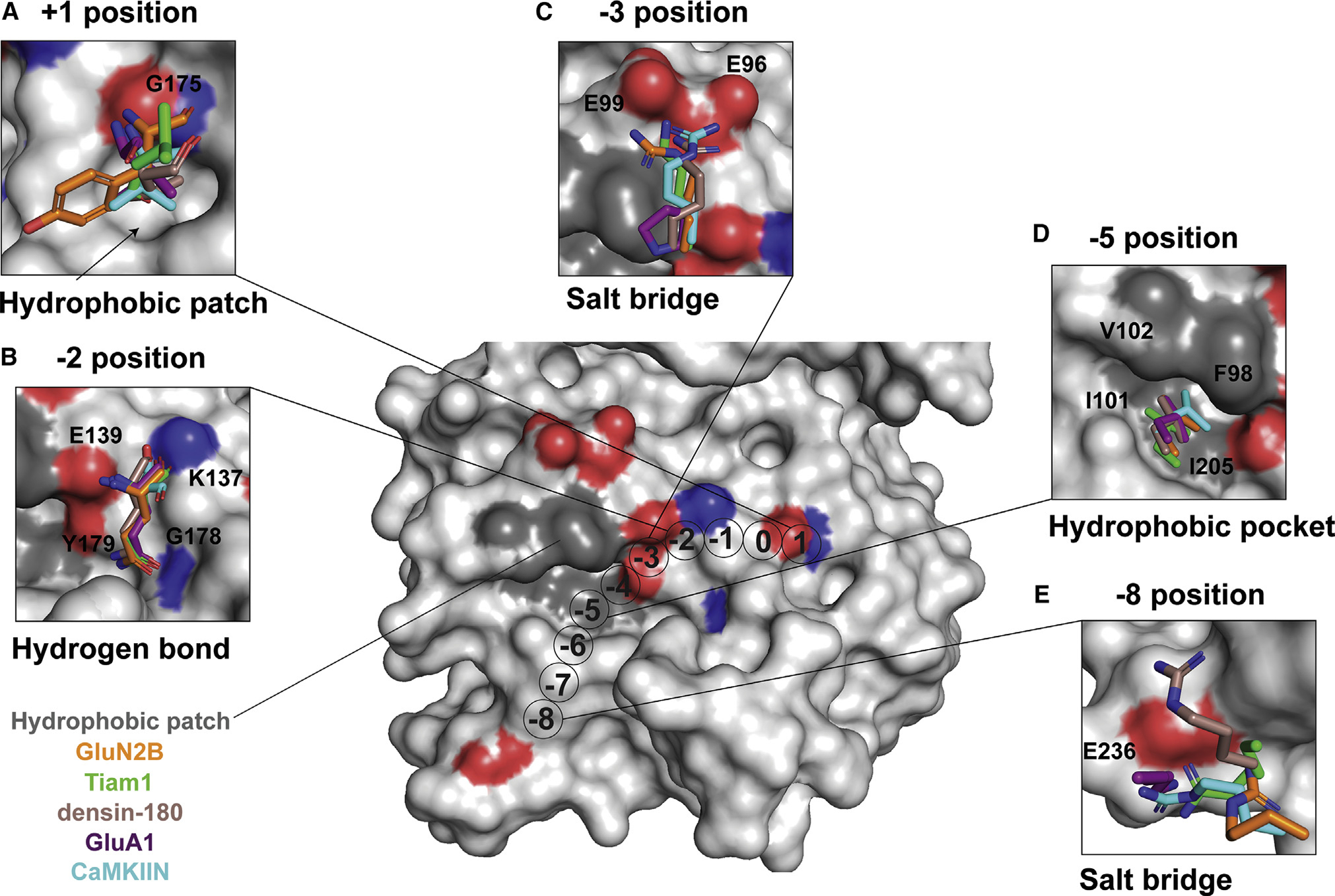
Shown is a surface representation of the CaMKII kinase domain, highlighting residues that mediate interactions with binding partners. The color code matches previous figures (GluN2B, orange; Tiam1, green; densin-180, brown; GluA1, purple; CaMKIIN, cyan).
(A) At the +1 position, a hydrophobic patch (arrow) is formed by F173, P177, L185, and Y222. GluA1, CaMKIIN, and densin-180 have Val or Ile at the +1 position, which are buried in this hydrophobic groove. Backbone atoms of the +1 residue hydrogen bond with the backbone of G175 (highlighted red and blue on the structure).
(B) At the −2 position, all interactors have a glutamine except for CaMKIIN, which has a serine. The glutamine side-chain amide oxygen forms a hydrogen bond with the backbone of G178, and the amino group interacts with the side chain of Y179. The backbone carbonyl interacts with the side chain of K137, and the backbone amino group interacts with E139.
(C) At the −3 position, lysine or arginine interacts with E96 and E99. The basic residues of interaction partners are positioned 2.4–4.2 Å between E96 and E99. GluA1 has a proline at the −3 position, which is flipped away from E96/99.
(D) At the −5 position, a conserved leucine across all interactors nestles into a hydrophobic pocket formed by F98, I101, V102, and I205.
(E) Lysine or arginine at the −8 position forms a salt bridge with E236.
Interactions driving binding to the CaMKII kinase domain
A hydrophobic residue at the +1 position
Our alignment is consistent with previous studies showing that a small hydrophobic residue is preferred at the +1 position (Castro-Rodrigues et al., 2018; Ishida et al., 1998; Songyang et al., 1996; Stokoe et al., 1993; White et al., 1998; Figure 2G). Our structures revealed that a hydrophobic patch adjacent to G175 in the kinase domain forms a docking site for small- and medium-sized hydrophobic residues (valine, isoleucine, and leucine) (Figure 3A). In GluN2B, tyrosine is larger and polar, so the side chain is not buried in the hydrophobic pocket, unlike the other binding partners. In all structures, backbone atoms of the +1 position form hydrogen bonds with the backbone of G175.
In the Tiam1 crystal structure, the +1 leucine does not interact with this groove. We hypothesized that this is because it is the C-terminal residue on the peptide used for crystallization. To test this, we extended the Tiam1 sequence and performed MD simulations, which showed that the +1 leucine does interact with the hydrophobic patch. In the simulations, we observed that the +1 leucine and the +4 isoleucine interact strongly over the course of the trajectory with an average distance of 3.2 Å (Figure S3A). We compared this with simulations of GluN2B and CaMKIIN, which also show a relatively strong interaction between the +1 residue and the hydrophobic patch (Figures S3B and S3C). In GluN2B and Tiam1, the +1 residue fluctuates in and out of the pocket, as shown by the larger distribution of distance from the pocket. The +4 residue of GluN2B also strongly interacts with the pocket, similar to Tiam1, whereas the CaMKIIN +2 residue interacts less.
Ionic interactions at the −2 position
Glutamine is commonly found at the −2 position of CaMKII interactors (Castro-Rodrigues et al., 2018; Ishida et al., 1998; Songyang et al., 1996; White et al., 1998). Indeed, all four peptides in our structures have glutamine at −2, allowing us to resolve the important interactions mediated by the backbone and the side chain at this position. There is extensive hydrogen bonding between the −2 glutamine and G178, Y179, K137, and E139 (Figure 3B). CaMKIIN has a serine at the −2 position, and the side chain is flipped relative to glutamine, which enables tighter interactions with E139 and K137 side chains (Chao et al., 2010). This observation is consistent with a previous study showing that CaMKIIN binding to CaMKII was reduced when this serine was mutated to alanine (Coultrap and Bayer, 2011).
A conserved salt bridge at the −3 position
Like many kinases, CaMKII prefers a basic residue at the −3 position (Rust and Thompson, 2011). In the crystal structures, all interactors except for the low-affinity binder GluA1 have a basic residue at the −3 position (arginine or lysine) that forms salt bridges with two glutamic acid residues (E96, E99) located on the αD helix of the kinase domain (Figures 3C and 4A; Castro-Rodrigues et al., 2018; Chao et al., 2010; Rellos et al., 2010; Yang and Schulman, 1999). These salt bridges are not formed with GluA1 because there is a proline at the −3 position. In the GluA1 structure, the side chain of E96 is flipped away from the peptide compared with the other four structures, where it is oriented toward the binding partner (Figure 4A, inset).
Figure 4. Electrostatic interactions with a basic residue at the −3 position facilitate high-affinity binding.
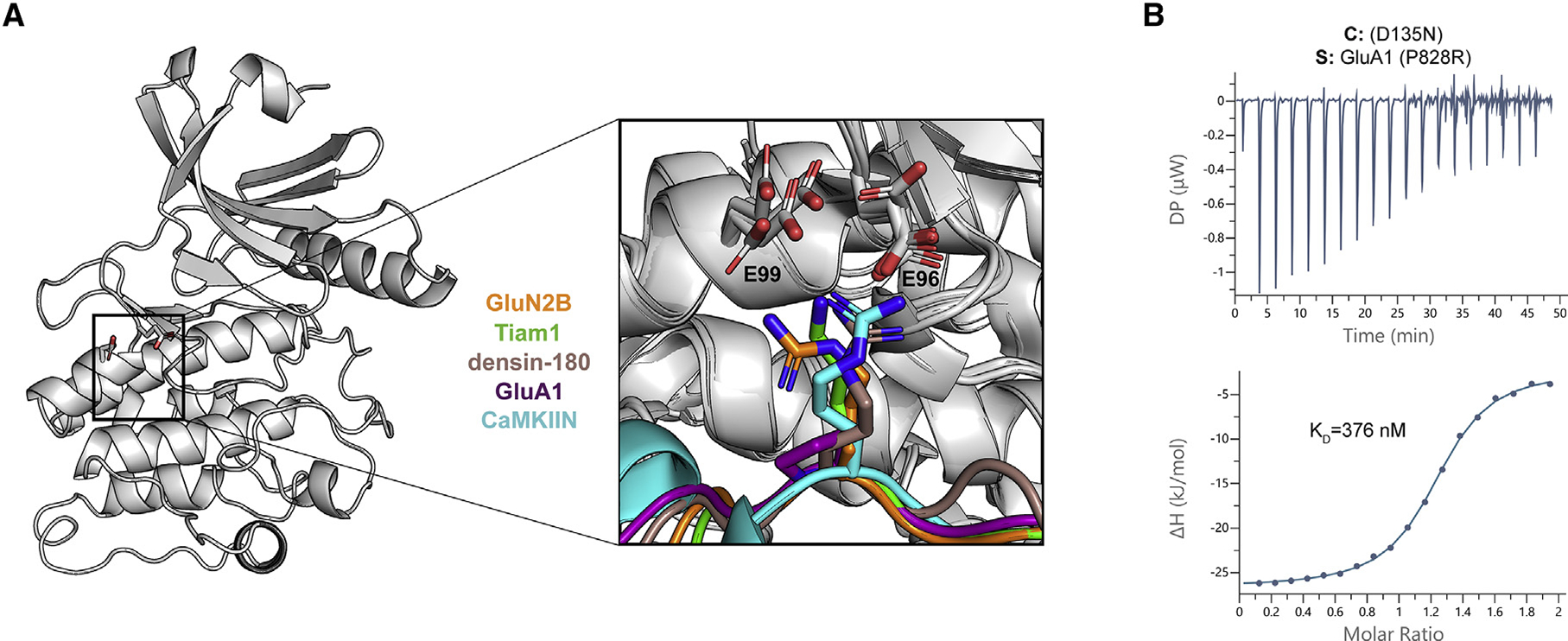
(A) CaMKII kinase domain shown as a cartoon; the E96 and E99 residues are shown as sticks. A magnified view of all five co-crystal structures is overlaid to highlight the basic residue at the −3 position (except for GluA1 in purple) interacting with the two glutamic acids in the kinase domain.
(B) ITC data for the D135N CaMKII kinase domain (cell) and GluA1 with P828R mutation (syringe). The mean Kd value is from two independent measurements.
We hypothesized that the reason for GluA1’s lower affinity was the lack of these salt bridges. To test this, we measured binding of a mutant GluA1 peptide where the −3 proline was changed to an arginine (GluA1 P828R). This mutation increased binding more than 140-fold to Kd = 376 nM (Figure 4B). To further validate this, we created single and double charge reversal mutations (E→K) and compared the behavior of GluA1 P828R with a high-affinity interactor, GluN2B. Single mutants (E96K or E99K) decreased the affinity to GluA1 P828R and GluN2B by 7- to 65-fold, whereas the double mutant (E96/99K) decreased affinity by 75- to 140-fold (Figure S4; Table S4). These data indicate the importance of these salt bridges for determining binding affinity.
We also wanted to investigate the role of E96/99 in ATP binding. E96 is well conserved across kinases and known to be important for ATP binding (Rellos et al., 2010). Almost 50% of kinases have an aspartate or glutamate at this position (E96), which might explain the basophilic kinase substrate preference (Vulpetti and Bosotti, 2004). In structures where ATP/Mg2+ is bound (GluN2B, Tiam1), the side chain of E96 interacts with the hydroxyl groups of ribose in ATP (Figure S5A). In MD simulations with CaMKIIN, interactions with ATP were very different. We observed a unique interaction at the −3 position, which resulted in a conformation that distorted ATP binding (Figure S5B). MD trajectories predict that CaMKIIN binding is facilitated by E96 in the presence of ATP, whereas E99 plays essentially no role. We performed ITC measurements, comparing binding of CaMKIIN to the kinase domain with charge reversal mutations (E96K, E99K) in the presence of an unhydrolyzable ATP analog (AMP-PNP [Adenylyl-imidodiphosphate]). Consistent with the prediction from the simulation, the affinity of E99K was only 2-fold weakened, whereas that of E96K was weakened by 10-fold (Figures S4 and S5C–S5F; Table S4). Further efforts are needed to interrogate the role of ATP coordination in CaMKIIN inhibition. E99 is not as well-conserved, even across other CaM kinases (Figures S6A–S6C), indicating that it may be more important for substrate specificity and potentially compensate if E96 is mutated (Zha et al., 2012).
Hydrophobic interaction at the −5 position
All CaMKII interactors studied here have a conserved leucine at the −5 position, which fits into a hydrophobic pocket on the kinase domain comprised of F98, I101, V102, and I205 (Figures 3D and 5A). In these structures, leucine is 3.3–4.5 Å from the four hydrophobic residues, indicating a tight interaction, as found by measuring the distance between the closest carbon atoms in each residue. In the CaMKIIN structure, a turn motif, facilitated by two glycines, orients leucine at the −8 position into this pocket to make it nominally at the −5 position (Chao et al., 2010). In simulations of GluN2B, Tiam1, and CaMKIIN, this interaction also demonstrates high structural integrity with a pairwise RMSD below 2 Å (Figure 5B).
Figure 5. Hydrophobic interactions mediate binding.
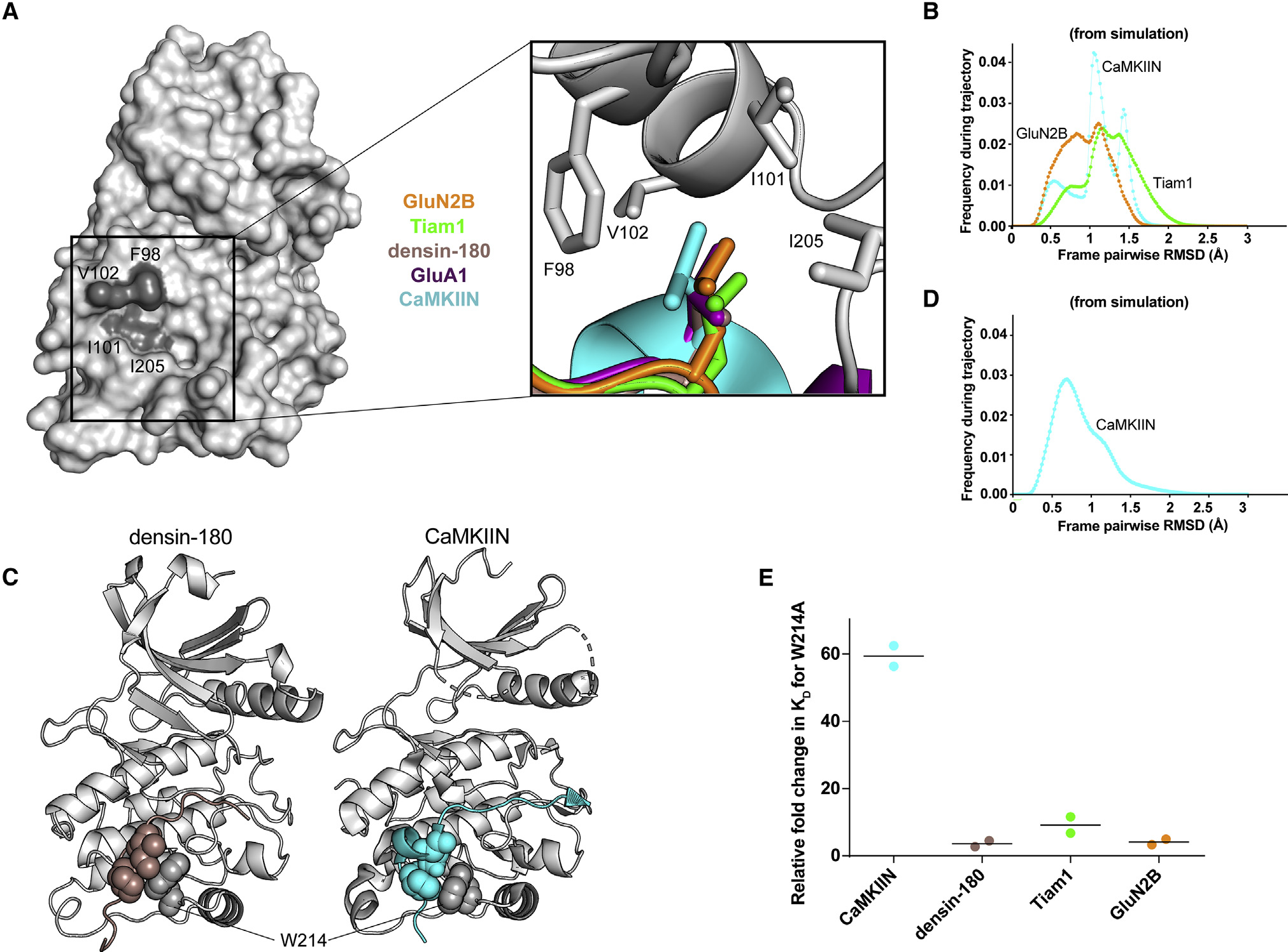
(A) Surface representation of the CaMKII kinase domain, with residues forming the hydrophobic pocket labeled (dark gray). Inset: overlay of leucine residues from all co-crystal structures bound in the hydrophobic pocket.
(B) Histograms of RMSD from MD simulations between every pair of trajectory frames for F98, I101, V102, and I205 with −5 peptide leucine.
(C) Crystal structures with sphere representation of the isoleucine and proline or leucine residues of densin-180 (brown) and CaMKIIN (cyan) docking onto W214 (gray) of the kinase domain.
(D) RMSD histogram from MD simulations for W214 interacting with isoleucine and proline of CaMKIIN.
(E) Kd values were extracted from ITC data, and relative fold changes were calculated by dividing the observed Kd from the mutant (W214A) by the D135N kinase domain. Individual data points are shown, and the line indicates the average.
Docking site mediated by W214
In the densin-180 and CaMKIIN structures, the side chains of proline and isoleucine (highlighted blue in Figure 2G) pack against W214 on the kinase domain (Figure 5C). For densin-180, the guanidino group of R808 at the −7 position hydrogen bonds with the backbone carbonyl group of W214 and the side chain of Q224. In simulations of CaMKIIN, the pairwise RMSD calculated for W214 interacts with isoleucine and proline below 1.5 Å, indicating a strong and persistent interaction over the course of the trajectory (Figure 5D).
We measured binding to a kinase domain mutated at this tryptophan (W214A) to discern its contribution to these interactions. The W214A mutation significantly disrupted CaMKIIN binding by lowering the affinity 60-fold, whereas the effects on the other binding partners were not as severe (4- to 10-fold decreased affinity) (Figures 5E and S7A–S7D; Table S4). CaMKIIN docking onto W214 likely stabilizes its helical motif, which properly orients the −5 leucine into the hydrophobic pocket. When W214 is mutated, the −5 leucine interaction is unlikely to be maintained in CaMKIIN, which results in disrupted binding. Densin-180 does not have a helical motif, which may explain why the W214A mutant does not have a drastic effect on binding. CaMKIIN has been characterized as a CaMKII-specific inhibitor despite the fact that CaMKII has high similarity to the CaMK-family kinases. This specificity is likely driven by the presence of this tryptophan residue because the other family members have tyrosine or methionine at this position (Figure S6A).
Salt bridge at the −8 position
We observed an electrostatic interaction between a conserved basic residue at the −8 position (arginine or lysine) and a glutamate (E236) at the base of the C lobe (Figures 3E and 6A). This has not been described previously, likely because this position is quite far from the phosphorylation site (~30 Å), and substrate alignments have not been accurate enough to highlight the conservation at this position. We tested the effect of mutating E236 to lysine (E236K) on binding affinity. The E236K mutation completely abolished Tiam1 binding (Figures 6B and S7E; Table S4). For densin-180, CaMKIIN, and GluN2B, E236K binding affinity was reduced ~17-fold (Kd = 10 ± 7 μM), ~21-fold (Kd = 825 ± 6 nM), and ~35-fold (Kd = 3.5 ± 1 μM), respectively (Figures 6B and S7F–S7H; Table S4). A previous study noted that E236K did not significantly affect GluN2B binding in biochemical assays (Bayer et al., 2006); however, in that measurement, low-micromolar binding would not be discernable from high-nanomolar binding as in our ITC measurements.
Figure 6. Electrostatic interaction with E236 facilitates binding.
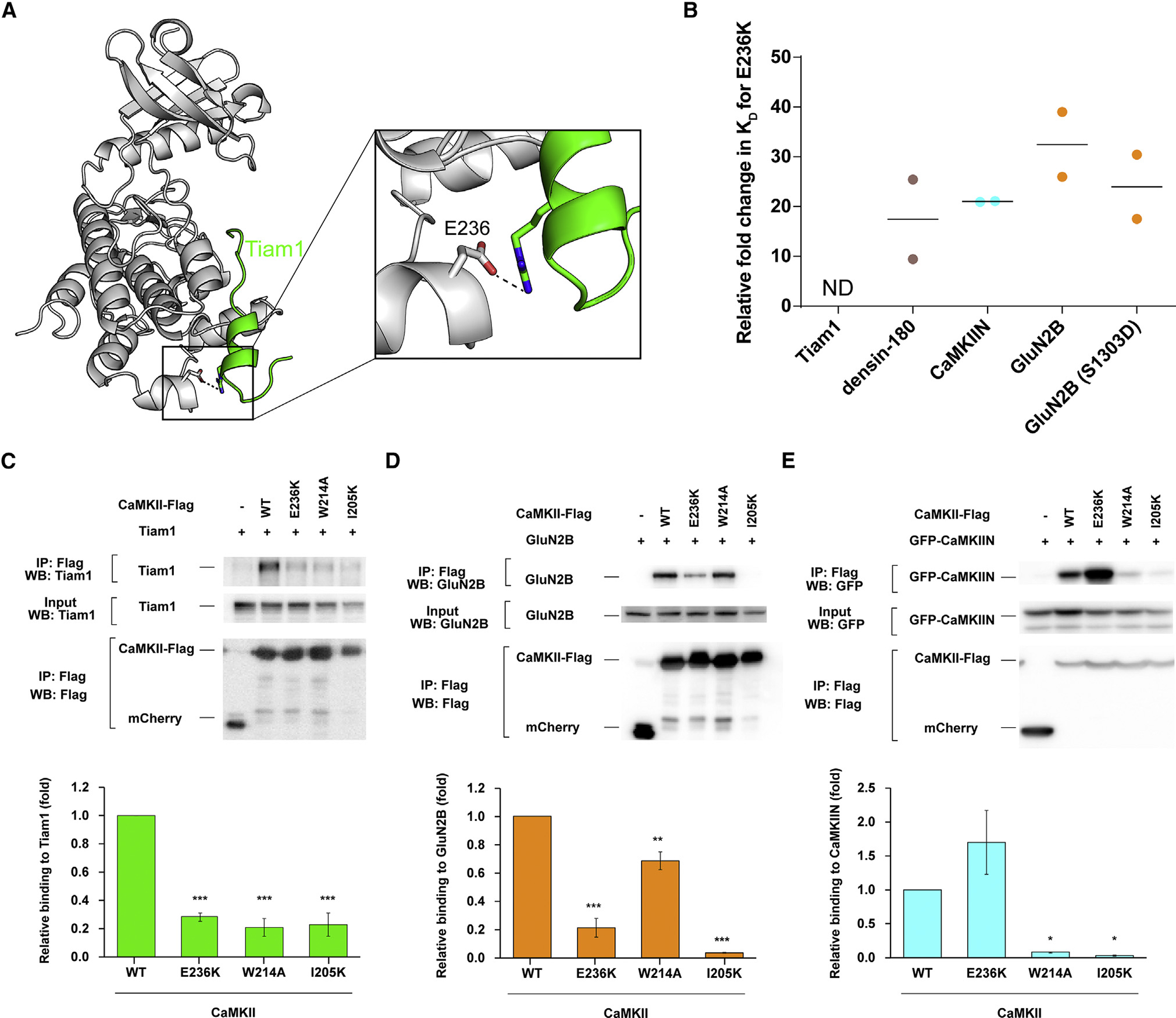
(A) View of the interaction between CaMKII E236 (gray) and Tiam1 R1549 (green).
(B) Kd values were extracted from ITC data, and relative fold changes were calculated by dividing the observed Kd from the mutant (E236K) by the D135N kinase domain. Individual data points are shown, and the line indicates the average.
(C–E) Effects of CaMKII mutations (E236K, W214A, and I205K) on interactions with Tiam1, GluN2B, and CaMKIIN. HEK293T cells were co-transfected with FLAG-tagged CaMKII variants and Tiam1-mGFP (modified GFP), GluN2B, or EGFP-CaMKIIN. Cell lysates were immunoprecipitated with FLAG antibody, and samples were immunoblotted with Tiam1, GluN2B, GFP, and FLAG antibodies. Representative blots are shown in the top panels. Quantification of the co-immunoprecipitation from 3 or 4 independent experiments is shown in a graph of (C) Tiam1 (n = 3), (D) GluN2B (n = 3), and (E) CaMKIIN (n = 4). Error bars indicate standard error of the means. The amount of co-immunoprecipitated Tiam1, GluN2B, or CaMKIIN was normalized by the amount in cell lysate and immunoprecipitated CaMKII. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with WT CaMKII; one-way ANOVA with Shaffer’s post hoc test comparisons. WT, wild type; IP, immunoprecipitation.
We wondered whether substrate phosphorylation would alter this interaction that is so far from the active site. To test this, we compared the affinity between the E236K kinase domain and WT GluN2B and the phosphomimetic GluN2B (S1303D, S→D at position 0). There was very little effect on S1303D GluN2B binding to D135N (Kd = 493 ± 47 nM) (Figure S8A), whereas affinity for the E236K mutant was reduced ~24-fold (Kd = 12 ± 4.5 μM) (Figures 6B and S8B; Table S4). This indicates that E236K significantly weakens binding to S1303D GluN2B to approximately the same extent as the non-phosphorylated peptide.
Generalizing CaMKII binding partners
We mined the curated database of CaMKII phosphorylation sites (n = 418) for consistencies with our updated alignment and found additional similarities (Figure S6D; http://www.phosphosite.org; Hornbeck et al., 2015). Hydrophobic residues (I, V, L, M, and F) are found at +1, Q is found at −2, and R is overwhelmingly found at −3 but also at −2 and −4. Smaller hydrophobic residues (L, V, I, and M) are found at −5 and −6. Generally, the minus direction disfavors P, D, and E. In contrast, acidic residues D and E are highly favored at +2. GluN2B has D at the +2 position, and we note an interaction with K56 in several structures, which was also observed in the dEAG-bound structure (PDB: 5H9B; Castro-Rodrigues et al., 2018). S, which may serve as a second phosphorylation site, is strongly disfavored at nearly every position. The Phosphosite Plus database only allows analysis out to the +7/−7 positions, so we could not compare residues at the −8 position and beyond. The actual −8 position may be farther than anticipated from alignments because of secondary structural elements. Overall, these features are consistent with important residues identified in our structures.
Interrogating CaMKII interactions with full-length binding partners
In order to assess the importance of the residues on CaMKII identified in the crystallographic analysis, we used a pull-down assay to assess the effect of CaMKII mutations on interactions with full-length binding partners. We co-transfected HEK293 cells with FLAG-tagged full-length CaMKIIα and full-length Tiam1, GluN2B, or GFP-CaMKIIN. Binding was assessed by Western blot after immunoprecipitation with an anti-FLAG antibody. Immunoprecipitation showed a remarkable loss of binding for Tiam1 and GluN2B when E236 is mutated, similar to our ITC data (Figures 6C and 6D). E236K CaMKII resulted in better binding to CaMKIIN compared with WT CaMKII in HEK293 cells (Figure 6E). Our ITC results showed tight binding between the E236K CaMKII kinase domain and CaMKIIN (825 nM), and it is known that the E236K mutation disrupts regulatory segment binding (Yang and Schulman, 1999), facilitating more CaMKIIN binding to E236K compared with the WT because the binding site is more available. The far salt bridge interaction mediated by E236 facilitates high-affinity binding to CaMKII and may also be critical for CaMKII specificity because it is not conserved across other CaMK-family kinases.
The immunoprecipitation experiments also corroborate our peptide binding data that show the importance of hydrophobic interactions. Indeed, mutating the hydrophobic pocket at the −5 position (I205K) showed significant loss of CaMKII binding to Tiam1, GluN2B, and CaMKIIN (Figures 6C–6E). The W214A mutation also disrupted binding to Tiam1, GluN2B, and CaMKIIN. The pull-down assay revealed a significant effect on Tiam1 binding to W214A, which is in agreement with ~9-fold lower affinity from ITC measurements (Figure 6C). The W214A mutation slightly weakened the interaction with full-length GluN2B, and we observed 4-fold lower affinity in our ITC measurement using GluN2B peptide (Figure 6D). Consistent with our ITC data, CaMKIIN binding to W214A was remarkably weakened compared with the WT (Figure 6E). CaMKIIN is a high-affinity inhibitor of CaMKII, and its specificity likely arises from this W214 docking site, which is not present in other CaMK-family members (Figures S6A and S6C). In this way, W214 might serve as a docking site for CaMKII-specific interactions. A previous study suggested that the pantothenate segment of coenzyme A packs against W214 of CaMKII to mediate this interaction (McCoy et al., 2013).
The αD helix rotates outward to accommodate binding partners
CaMKII is unique among protein kinases in that it does not require phosphorylation in the activation loop. In fact, CaMKII structures have highlighted that the autoinhibited conformation of CaMKII actually resembles many other kinases’ active conformations (Nolen et al., 2004; Rosenberg et al., 2005). Upon activation, the αD helix in CaMKII undergoes a 45° rotation outward (Pellicena and Schulman, 2014; Rellos et al., 2010; Rosenberg et al., 2005). In the autoinhibited conformation, the regulatory segment stabilizes the αD helix in the inward conformation (Rosenberg et al., 2005).
We then compared the structures of autoinhibited kinase (PDB: 2VZ6, residues 13–300; Rellos et al., 2010), the uninhibited kinase (PDB: 6VZK; Torres-Ocampo et al., 2020), and the kinase bound to GluN2B. It is clear from this comparison that the αD helix rotates outward to accommodate GluN2B binding, just as it does when the regulatory segment is removed (Figure 7A). This is also true for Tiam1, GluA1, densin-180, and CaMKIIN (Chao et al., 2010). The rotation of the αD helix effectively presents the hydrophobic patch in a competent position for interacting with the binding partner. The αD helix houses three of the four residues that make up the hydrophobic pocket for the −5 leucine to dock in, which is likely a large contribution to an observed gain in kinase domain stability upon substrate binding (Ishida and Fujisawa, 1995) (Figure S9A), similar to what we have observed previously with regulatory segment binding, which is mediated by L290 in the regulatory segment (Torres-Ocampo et al., 2020). We measured the stability of all kinase domain mutants bound to GluN2B and showed that the melting temperatures are stabilized compared with the kinase domain alone (Tm = 33°C) (Figure S9B). Notably, the E96/99K mutant is only stabilized by 1°C, indicating that these mutations destabilize the kinase and worsen the binding affinity to GluN2B.
Figure 7. GluN2B binding interferes with autoinhibition.
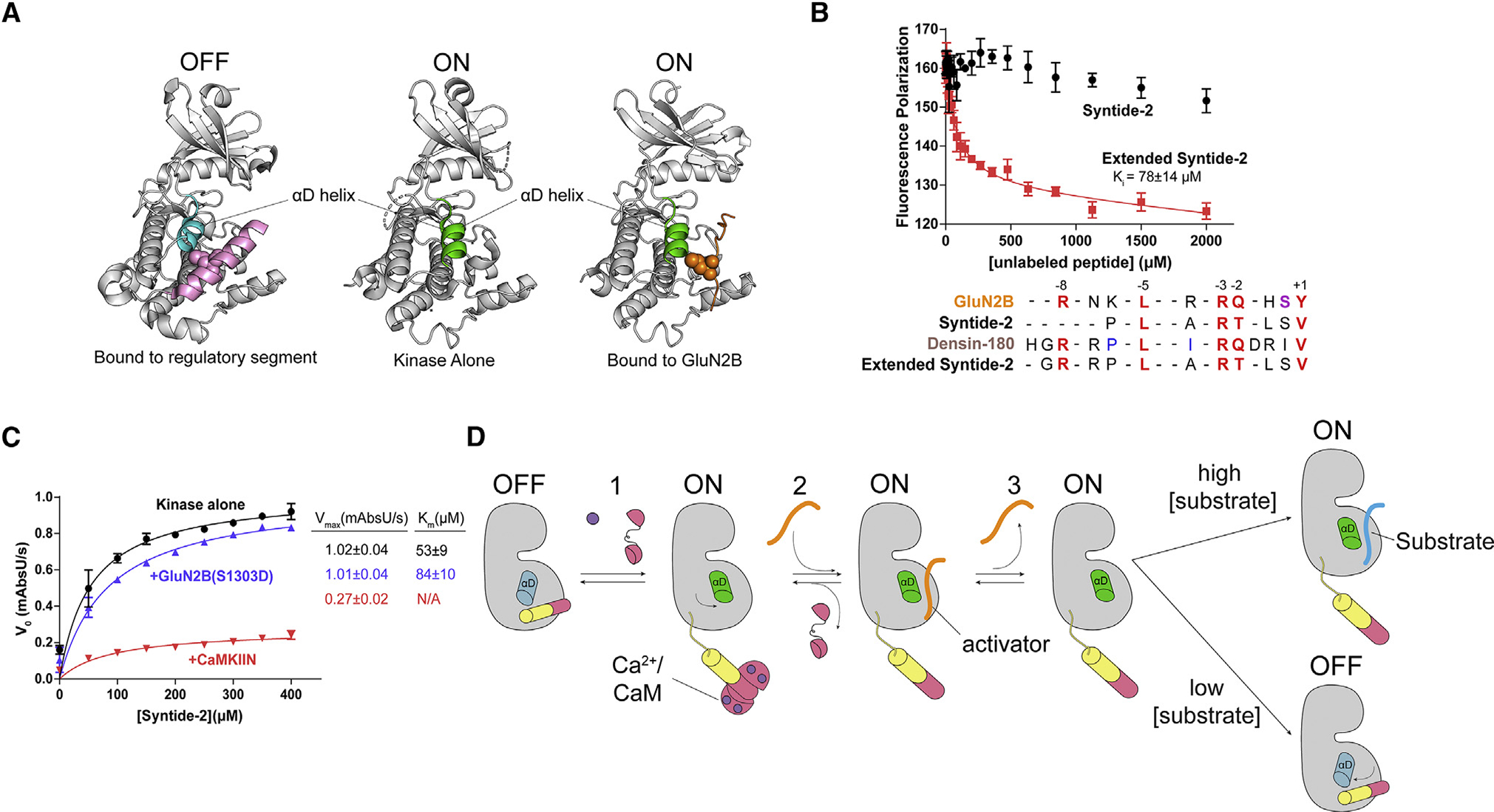
(A) Comparison of the αD helix between crystal structures of the autoinhibited with regulatory segment (pink) bound (left; PDB: 2VZ6), uninhibited kinase with no regulatory segment (center; PDB: 6VZK), and active kinase with GluN2B (orange) bound (right; PDB: 7UIS). The αD helix goes from rotated inward in the off-state (blue) to rotated outward 45° in the on state (green).
(B) Competition assay against GluN2B using Syntide-2 (black) and the extended version of Syntide-2 (red). Error bars indicate standard deviation from triplicate at each data point. Sequence alignment of GluN2B, Syntide-2, densin-180, and the extended Syntide-2 is shown below the graph. For clarity, the alignments start at the +1 position (C-terminal end) and contain the −8 basic residue (N-terminal end). Full sequences used are listed in the STAR Methods.
(C) Coupled kinase assay results with kinase alone and in the presence of GluN2B and CaMKIIN. The significance of the change in Km values is significant when using unpaired t test with Welch’s correction (p = 0.0208).
(D) Proposed model of maintaining CaMKII activity by binding to a high-affinity activator. CaMKII binds Ca2+/CaM and is activated, and the αD helix rotates out (Rxn 1). A high-affinity activator binds to the substrate binding site (Rxn 2). When the Ca2+ signal dissipates, CaM dissociates from CaMKII, but the activator remains bound, competing with the regulatory segment. The activator dissociates, but the αD helix remains in the active conformation (Rxn 3). In high [substrate], the substrate will bind and be phosphorylated. In low [substrate], the activator will rebind or the αD helix will rotate back in to accommodate regulatory segment binding.
Testing the S and T site binding model
Because our structures revealed that all interactors bound to the same single site on the kinase domain, we wanted to directly test the idea of the 2-site (S and T site) binding model (Bayer et al., 2006). Previous studies have shown that GluN2B peptide could not be competed off by Syntide-2 (the synthetic CaMKII substrate), even at low millimolar concentrations, which we also observed (Figure 7B; Strack et al., 2000). As noted previously (Strack et al., 2000), a sequence alignment of Syntide-2 compared with other substrates reveals that the Syntide-2 peptide is shorter at the N terminus and is therefore missing the −8 position basic residue necessary for mediating the far salt bridge. To test whether this is the reason why Syntide-2 could not compete with GluN2B, we created an extended version of Syntide-2 by adding three residues to the N terminus, mimicking densin-180 (Figure 7B, inset). The extended version of Syntide-2 did compete off GluN2B (Ki ~ 78 ± 14 μM) (Figure 7B). This result shows that Syntide-2 and GluN2B occupy the same binding site, indicating that there are not two separate binding sites on the kinase domain. When we tried to measure affinity for Syntide-2 peptide using ITC, we did not see any appreciable binding despite using protein/peptide concentrations high enough to characterize an interaction with a Kd of several hundred micromolar (Figure S10). When we tested the extended version of Syntide-2 in ITC, we obtained a Kd of 2.6 ± 0.9 μM (Figure S10), providing further evidence that this far salt bridge at the −8 position facilitates tight binding.
A conundrum thus arises. If there are not two distinct binding sites, then how is CaMKII activity maintained by specific binding partners like GluN2B? The mode of activation must invoke the phosphorylated form of GluN2B because this is observed under activating conditions. In a canonical kinase reaction, the kinase first binds substrate and ATP, then transphosphorylation occurs, and finally the substrate is released and ADP is replaced by another ATP molecule. We measured the affinity of the phosphomimetic GluN2B peptide (S1303D) (Kd = 493 ± 47 nM) and observed only a ~5-fold reduction in affinity compared to WT GluN2B (Kd = 107 ± 46 nM) (Figures S8A and S2B). In the presence of AMP-PNP, there is also tight binding of S1303D GluN2B (Kd = 277 ± 30) (Figure S8C). We were also able to crystallize the kinase domain bound to GluN2B S1303D, which showed the same conformation as bound to WT GluN2B (backbone RMSD, 0.266 Å) (Figure S9C). We hypothesized that phosphoGluN2B would act as a competitive inhibitor for Syntide-2 because of this tight binding. We performed kinase assays using Syntide-2 as a substrate in the presence of saturating GluN2B S1303D. As a control, we also measured activity against Syntide-2 in the presence of CaMKIIN. Indeed, high levels of CaMKIIN completely inhibit Syntide-2 phosphorylation, whereas GluN2B acts as a competitive inhibitor, exemplified by comparable Vmax values but an increased Km value (Figure 7C). These data indicate that GluN2B and Syntide-2 bind to the same site.
A proposed model for persistent CaMKII activity
Based on our data, we propose a mechanistic model to explain the observations of prolonged CaMKII activity in the presence of specific binding partners. Let us consider the mode of activation by GluN2B as the activator, shown in Figure 7D. As described above, upon Ca2+/CaM binding and release of autoinhibition, the kinase domain undergoes a conformational change where the αD helix rotates out and the kinase is in the “on” state (Figure 7A). After dissociation of GluN2B, a conformational change in the αD helix is required for the regulatory segment to re-bind but not for a new substrate to bind. For this reason, we hypothesize that, under conditions of high substrate concentrations, substrate binding will be faster than regulatory segment re-binding. Under conditions where the activator concentration is high, activator rebinding will be favorable because this high-affinity binder will have a relatively high on rate and low off rate.
DISCUSSION
CaMKII recognizes a broad range of interaction partners, but the structural details driving these interactions had not been fully elucidated. We addressed this by solving co-crystal structures of four binding partners bound to the CaMKII kinase domain, which allowed us to highlight the interactions mediating binding. The minimum consensus sequence for CaMKII targets is R-X-X-S/T (Kennelly and Krebs, 1991; Pearson and Kemp, 1991); however, our results have expanded this understanding. If we incorporate our structural observations, then the full consensus sequence is R/K-X-X-ϕ-X-R/K-X-X-S/T-ϕ. We postulate that, if all positions are satisfied, as shown here, then the binding affinity will be high nanomolar to low micromolar. Interaction partners lacking 1–2 of these positions will have poorer affinities but may still be viable interactors because a typical kinase substrate has an affinity of several hundred micromolar (Endicott et al., 2012). For example, GluA1 lacks a basic residue at the −3 position, which yields an affinity of more than 55 μM. The biological ramifications of the weak interaction between CaMKII and GluA1 will need to be further investigated where localization of CaMKII to this receptor by another factor or avidity effects of holoenzyme localization may be driving GluA1 phosphorylation in vivo (Mao et al., 2014). Our observation might explain the molecular basis of a recent study that reported lower levels of S831 phosphorylation in the hippocampus (Hosokawa et al., 2015). We report GluA1 as a low-affinity substrate, which would explain why densin-180 was interpreted as a selective inhibitor because GluA1 would not be strong enough to compete with densin-180 binding (Jiao et al., 2011). Given the large size and scaffolding role of densin-180 (180 kDa) compared with CaMKIIN, the densin-180 interaction might be more important for CaMKII localization than widespread inhibition.
There has been a lack of mechanistic clarity in understanding how certain CaMKII interactors lead to prolonged activation in the absence of calcium. In 2001, without a crystal structure to guide an understanding, a set of extraordinary data revealed that GluN2B binding yielded autonomous CaMKII activity (Bayer et al., 2001). This work came on the heels of an extensive mutagenesis study mapping how the regulatory domain interacts with the surface of the kinase (Yang and Schulman, 1999). One of the findings from this work was that the mechanism of autoinhibition is distinct from substrate binding, especially when residues far from the active site were tested. To orient thinking about these interactions, residues close to the active site were termed the S site (for substrate binding), and residues far from the active site were termed the T site (for Thr286 binding) (Pellicena and Schulman, 2014; Schulman, 2004). In 2006, these reference points were implemented into a 2-site binding model to explain this autonomous activity with GluN2B binding (Bayer et al., 2006).
We were well poised to systematically test the S and T site model using biochemistry and structural biology. In the described model, F98 is part of the S site, and I205 is part of the T site (Bayer et al., 2006). Conversely, we find that all interactors, including activators, bind to the CaMKII kinase domain using a continuous single site across the surface of the kinase domain (Castro-Rodrigues et al., 2018; Chao et al., 2010). In these structures, F98 and I205 (along with I101 and V102) make up a single hydrophobic pocket that houses the −5 leucine residue, first described as “docking site B” (Figures 3D and 5A) by Chao et al. (2010). So, F98 and I205 are performing the same role instead of comprising separate sites. A large body of evidence for the S and T site model has been based on experiments that compare a short synthetic substrate, Syntide-2 (which lacks a basic residue at the −8 position), with another substrate, AC3 (which contains a −8 basic residue). Syntide-2 has been deemed an S site binder, whereas AC3 binds to the S and T site. This distinction stemmed from data showing that, if CaMKII is bound to Syntide-2 or AC3 and then GluN2B is added, then GluN2B binds CaMKII-Syntide-2 but not CaMKII-AC3 (Bayer et al., 2006). These data were interpreted so that GluN2B was able to bind to the T site, whereas Syntide-2 remained bound at the S site. Based on our data, an updated interpretation is that GluN2B competes off Syntide-2 because GluN2B binds CaMKII with more than 1,000-fold higher affinity.
CaMKII has high affinity (submicromolar to low micromolar) for the activator peptides GluN2B, Tiam1, and dEAG (Castro-Rodrigues et al., 2018). This is somewhat unique because typically these interactions are in the Kd range of 200 μM (Endicott et al., 2012). We postulate that these high-affinity interactors allow the interaction partners to kinetically compete with regulatory segment rebinding by stabilizing the active conformation with the αD helix rotated outward. Under conditions where substrate concentration is high, CaMKII will phosphorylate substrate because the kinase is in the active conformation and is in the proper conformation to interact with substrates. The data presented here provide clarity regarding the interactions between CaMKII and its binding partners, which will be crucial for guiding biological experiments to assess the downstream effects of specific interactions. Future experiments will necessarily invoke the complexities of the CaMKII holoenzyme structure. CaMKII-activator binding provides a mechanism to maintain CaMKII activity in the absence of Ca2+. It is intriguing to consider other enzymes that might be modulated in a similar way by high-affinity binding partners as a mechanism for prolonged activation in the absence of other stimuli.
Limitations of the study
A limitation of our study is that, for most experiments performed here, we used a monomeric version of the CaMKII kinase domain, but in cells, CaMKII oligomerizes into higher-order complexes. The holoenzyme structure will likely impact how CaMKII interacts with binding partners. Another limitation is that we have not measured the rate constants for substrate binding compared with regulatory segment binding, which would inform the model we have proposed here.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Margaret Stratton (mstratton@umass.edu).
Materials availability
All unique/stable reagents generated in this study will be made available on request but we may require a completed Materials Transfer Agreement.
Data and code availability
Atomic structures have been deposited in the PDB, listed in the Star Methods. Molecular dynamics simulation files are deposited in Mendeley and can be accessed using https://doi.org/10.17632/9m3bg3h5rs.1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
METHOD DETAILS
Cloning
All CaMKIIα variants were expressed with an N-terminal His-SUMO tag in a pET vector. For mammalian expression, CaMKIIα was expressed with a C-terminal Flag tag in pDEST12.2 vector [9]. Tiam1-mGFP was subcloned in a pCAGGS [9], GluN2B was in pGW-CMV (Kim et al., 1996), CaMKIIN-GFP was in pEGFP vector [25], and mCherry-Flag was in pDEST12.2 vector [9]. All point mutations were created using site-directed mutagenesis.
Protein expression and purification
WT CaMKIIα kinase domain (residues 7–274) was co-expressed with λ phosphatase in E. coli BL21(DE3). Inactive constructs (D135N) and CaMKIIN2 (Rattus norvegicus) were expressed without λ phosphatase. The cells were induced at 18°C with 1 mM isopropyl β-a-D-1-thiogalactopyranoside (IPTG) and grown overnight. Following to ~16 h incubation, cell pellets were resuspended in Buffer A (25 mM Tris, pH 8.5, 50 mM KCl, 40 mM imidazole, 10% glycerol) and commercially available protease inhibitors (0.5 mM benzamidine, 0.2 mM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF), 0.1 mg/mL trypsin inhibitor, 0.005 mM leupeptin, 1 μg/mL pepstatin), 1 μg/mL DNase/50 mM MgCl2 were added, then lysed. All following purification steps were performed using an ÄKTA pure chromatography system at 4°C. Filtered cell lysate was loaded onto a 5 mL His Trap FF Ni Sepharose column (GE Research), and eluted with 50% Buffer B (25 mM Tris-HCl pH 8.5, 150 mM KCl, 1 M imidazole, 10% glycerol). The protein was desalted from excess imidazole using a HiPrep 26/10 Desalting column using Buffer C (25 mM Tris-HCl pH 8.5, 40 mM KCl, 40 mM imidazole, 2 mM tris(2-carboxyethyl)phosphine (TCEP), 10% glycerol). His SUMO tags were cleaved with Ulp1 protease overnight at 4°C. Cleaved His SUMO tags were separated by a subtractive NiNTA step prior to an anion exchange step. Proteins were eluted from HiTrap Q-FF with a KCl gradient. Eluted proteins were concentrated and further purified in gel filtration buffer (25 mM Tris-HCl pH 8.0, 150 mM KCl, 1 mM TCEP, 10% glycerol) using Superdex 75 10/300 GL size exclusion column. Fractions (>95% purity) were pooled, concentrated, aliquoted and flash frozen in liquid nitrogen, and stored at −80°C until needed.
We use rat full-length CaMKIIN2 protein in our ITC measurements, coupled-kinase assays and cellular immunoprecipitation experiments. The CaMKII-bound CaMKIIN co-crystal structure (PDB:3KL8) was obtained by using a peptide derived from rat CaMKIIN1. In our MD simulations, the full length human CaMKIIN1 amino acid sequence was used. Amino acid sequences of each construct are listed below:CaMKIIN2 (Rattus norvegicus): MSEILPYGEDKMGRFGADPEGSDLSFSCRLQDTNSFFAGNQAKRPPKLGQIGRAKRVVIEDDRIDDVLKGMGEKPPSGV.
CaMKIIN1 peptide (Rattus norvegicus): KRPPKLGQIGRSKRVVIA CaMKIIN1 (Homo sapiens): MSEVLPYGDEKLSPYGDGGDVGQIFSCRLQDTNNFFGAGQNKRPPKLGQIGRSKRVVIEDDRIDDVLKNMTDKAPPGV.
Peptide synthesis
Peptides used for co-crystallization were synthesized and amidated at the C-terminus (Genscript, RIKEN Research Resource Center). Peptide used for fluorescence polarization assays were synthesized with an additional N-terminal 5-FAM.
Full length peptide sequences are as follows:
Human GluA1 (residues 818–837): SKRMKGFCLIPQQSINEAIR [Numbering for GluA1 is for mature peptide, excluding 18 amino acid long signal peptide, following convention in the field.].
Human GluN2B (residues 1289–1310): KAQKKNRNKLRRQHSYDTFVDL
Human GluN2B(S1303D) (residues 1289–1310): KAQKKNRNKLRRQHDYDTFVDL.
Human densin-180 (residues 797–818): SKSRSTSSHGRRPLIRQDRIVG
Mouse Tiam1 (residues 1541–1559): RTLDSHASRMTQLKKQAAL.
Syntide-2: PLARTLSVAGLPGKK Extended Syntide-2: GRRPLARTLSVAGLPGKK.
Full length peptide sequence for fluorescence polarization assays are as follows:
Human GluN2B(S1303D) (residues 1289–1310): KAQKKNRNKLRRQHDYDTFVDL.
Crystallization and X-Ray data collection
Initial crystallization screening was done using the sitting vapor diffusion method at 4°C with commercially available screening kits. If needed, initial hits were optimized by the hanging vapor diffusion method. Final conditions for crystal formation and cryo solutions are listed below, all grown at 4°C. The peptide-to-protein ratio was kept at 3:1 throughout the co-crystallization attempts. ATP-bound crystal structures were obtained by adding 0.2 μL of reservoir solution containing 10 mM ATP/40 mM MgCl2 to crystal drops. For crystals that had methyl 6-O-(N-heptylcarbamoyl)-α-D-glucopyranoside (hecameg), a detergent in the crystallization condition (a detergent), the crystals were moved to a new drop without Hecameg, and containing 1 mM ATP/4 mM MgCl2. Diffraction data were collected from crystals flash-frozen in liquid nitrogen at a wavelength of 1.5418 Å using a Rigaku MicroMax-007 HF X-ray source, which was coupled to a Rigaku VariMax HF optic system (UMass Amherst). The X-ray data was collected at 100 K. Crystallization and cryo conditions are listed below: 6X5G: 0.1 M Bicine pH 9, 10% PEG 20K, 2% 1,4-Dioxane (Cryoprotectant: 20% glycerol)6X5Q: 0.1 M Tris pH 8, 28% PEG 4K (Cryoprotectant: 20% glycerol) 7UIQ: 0.1 M HEPES pH 7, 16% PEG 6K, 0.1% Triton X-114 (Cryoprotectant: 20% ethylene glycol)7UJQ: 0.1 M Bis-tris methane pH 6.2, 0.1 M ammonium sulfate, 25% PEG 3350, 19mM Hecameg (Cryoprotectant: 20% glycerol) 7UJP: 0.1 M Bis-tris methane pH 6.2, 0.1 M ammonium sulfate, 25% PEG 3350, 19mM Hecameg (Cryoprotectant: 20% glycerol)7UJR: 0.1 M Bis-tris propane pH 6.2, 0.1 M ammonium sulfate, 20% PEG 3350, 5% ethanol (Cryoprotectant: 20% ethylene glycol)7UJS: 0.1 M Bis-tris propane pH 6.6, 0.1 M ammonium sulfate, 20% PEG 3350 (Cryoprotectant: 20% ethylene glycol)7UIR: 0.1 M HEPES pH 7, 16% PEG 6K, 0.1% Triton X-114 (Cryoprotectant: 20% ethylene glycol) 7UJT: 0.1 M Bis-tris propane pH 6.5, 0.2 M Sodium chloride, 25% PEG 3350 (Cryoprotectant: 20% glycerol). 7KL0: 0.1 M Bis-tris methane pH 6.5, 0.1 M Ammonium sulfate, 20% PEG 6000, 19mM Hecameg (Cryoprotectant: 20% ethylene glycol)7KL1: 0.1 M Bis-tris methane pH 6.5, 0.1 M Ammonium sulfate, 20% PEG 6000, 19mM Hecameg (Cryoprotectant: 20% ethylene glycol)7UIS: 0.1 M Bis-tris propane pH 6.5, 0.2 M Sodium chloride, 25% PEG 3350 (Cryoprotectant: 20% glycerol).
Data processing and structure determination
Datasets were integrated, merged, and scaled using HKL-2000. The structures were solved by molecular replacement (MR) with Phaser using the coordinates of CaMKIIα kinase domain (PDB ID: 6VZK, 100% amino acid sequence identity) as a search model (McCoy et al., 2007). Peptides were built into electron density using Coot and refinement was performed with REFMAC5 (Emsley et al., 2010; Murshudov et al., 1997; Winn et al., 2011).
Isothermal titration calorimetry
ITC data were obtained using a MicroCal PEAQ-ITC automated calorimeter (Malvern Panalytical, Westborough, MA). Before each titration, interaction partners were dissolved in gel filtration buffer and final buffer conditions of CaMKII kinase domain and interaction partners were matched. GluN2B peptides and CaMKIIN protein contain a single tyrosine residue, which enabled accurate concentration determination. The other peptides lack aromatic residues, and these required crude weight to estimate concentration. Titrations were performed with different peptides as titrant into the cell containing D135N or other mutant CaMKII kinase domains (concentrations are indicated in Table S4). All titrations were performed using the standard 19 injection method, modified to run at 20°C. The standard 19 injection method in the PEAQ ITC automated control software (v1.40) is one injection of 0.4 μL followed by 18 2 μL injections with a spacing between injections of 150s, a stir speed of 750 rpm, and the reference power set to 10. Data were analyzed using PEAQ ITC analysis software (v1.40) using the one-site fitting model, and to eliminate integration of some baseline noise, some integration markers were set manually to obtain better fit.
Fluorescence polarization assay
The competition assay was conducted at 1 μM of CaMKII kinase domain (25 mM Tris pH 7.5, 150 mM KCl, 1 mM TCEP, 10% glycerol) with 60 nM fluorescein-labeled GluN2B peptide (dissolved in 25 mM Tris pH 7.5, 150 mM KCl, 0.02% Tween, 0.02% Triton) and then titrated increasing concentrations of unlabeled peptide (25 mM Tris pH 7.5, 150 mM KCl, 1 mM TCEP, 10% glycerol) at varying concentrations in Corning low volume 384-well black flat bottom plates. The final volume in each well was 20 μL. The fluorescence polarization was measured using a Synergy H1 hybrid plate reader (Biotek) with a filter of 485/20 nm excitation and 528/20 nm emission. Data were fit using a one-site binding equation in GraphPad PRISM version 6.0.
Differential scanning calorimetry
All protein samples were diluted to 0.5 mg/mL in DSC buffer (25 mM Tris pH 8,150 mM KCl, 1 mM TCEP, 10% glycerol). DSC measurements were performed on a MicroCal Automated PEAQ-DSC instrument (Malvern Panalytical, Westborough, MA). Unless otherwise indicated, after a 5 min pre-scan equilibration step, samples were scanned from 10–120°C at a scan rate of 90°C/h with no feedback. Data were analyzed using MicroCal PEAQ-DSC software, and baseline-subtracted data were fit to a non-two-state fitting model to obtain apparent Tm values.
Molecular dynamics simulations
Trajectories were run for 1.2 μs–3.7 μs aggregate time for each complex. All simulations are available for download from Mendeley: https://doi.org/10.17632/9m3bg3h5rs.1. Starting conformations were taken from co-crystal structures of CaMKIIα kinase domain in complex with peptides derived from GluN2B (PDB:7UJP) and Tiam1 (PDB:7UIR). The sequences used for interaction partners in the simulations are as follows: Tiam1 (1513–1581, adding 28 N-terminal residues and 22 C-terminal residues) as well as GluN2B (1263–1328, adding 32 N-terminal residues and 20 C-terminal residues), and the full-length CaMKIIN protein (adding 41 N-terminal residues and 22 C-terminal residues). For CaMKIIN, Coot (Winn et al., 2011) was used to superimpose the structure of a peptide derived from rat CaMKIIN in complex with the kinase domain from C. elegans CaMKII (PDB:3KL8) with the structure of GluN2B in complex with the kinase domain of CaMKIIα bound to a Mg2+ ion and an ATP molecule (PDB:7UJP). The CaMKIIN peptide and the CaMKIIα kinase domain were then merged into a single structure. Crystallographically unresolved atoms were added, peptides were extended in both N-terminal and C-terminal directions using PDBFixer, part of the OpenMM package (Eastman et al., 2013).
All simulations were performed with Gromacs 2020.2 (Huang et al., 2017; Pall et al., 2015) and the CHARMM36m force field (Huang et al., 2017). Protein complexes were solvated with TIP3P water (Jorgensen et al., 1983) in dodecahedral boxes that extended 1 nm past the protein in all dimensions. Na+ ions were added in sufficient quantity to charge neutralize each system. Systems were energy-minimized by steepest descent for 50,000 steps using a 0.01 nm step size until the maximum force exerted on any atom fell below 1000 kJ/mol/nm. Solvent was equilibrated for 1 ns at constant temperature and volume (NVT ensemble) and another 2 ns at constant temperature and pressure (NPT ensemble) with a positional restraint applied to protein heavy atoms. An additional 10 ns of unrestrained dynamics were simulated prior to collection of production data in order to allow the polypeptide chains to relax from their crystallographic conformations. Periodic boundary conditions were used in all dimensions, bonds were constrained with the LINCS algorithm (Hess, 2008), virtual sites (v-sites) were used to remove the fastest degrees of freedom to facilitate a 4 fs time step (Feenstra et al., 1999), particle mesh Ewald (PME) was used to treat electrostatic interactions (Essmann et al., 1995), the v-rescale thermostat (Bussi et al., 2007) with a 0.1 ps coupling time constant was used to maintain the temperature at 300 K, and a cut-off distance of 1.2 nm for neighbor list, Coulomb interactions, and Van der Waals interactions was used throughout system preparation and production simulations. The Parrinello–Rahman barostat (Parrinello and Rahman, 1981) with a 2 ps coupling time constant was used to maintain a pressure of 1 bar during NPT equilibration and production simulations. V-site parameters for ATP were determined using MkVsites (Larsson et al., 2020). GPU-accelerated simulations were performed on two hardware architectures: 1) 2× Intel Xeon Silver 4110 cpu (8 Cores), 2× nVidia Tesla V100 gpu; 2) 2× Intel Xeon Gold 6140 cpu (18 Cores), 2× nVidia Tesla V100 gpu. In order to obtain comparable aggregate lengths of production simulations for the three simulated systems, two independent simulations of the kinase:CaMKIIN complex were carried out.
To calculate RMSD and RMSF during the simulations, trajectories were aligned about the Cα of the kinase domain. RMSD was calculated with VMD (Humphrey et al., 1996). RMSF was calculated with ProDy (Bakan et al., 2011) as interfaced with VMD. RMSD and RMSF of the peptide were calculated with the kinases aligned to determine how the residues of the peptide fluctuated with respect to the kinase.
One production simulation was performed for each of the wild type kinase:GluN2B and wild type kinase:Tiam1 complexes, producing trajectories of 2.19 μs and 2.05 μs, respectively. Two independent simulations were performed for the wild type kinase:CaMKIIN. The CaMKIIN trajectories were 0.46 μs and 1.84 μs in length, for an aggregate of 2.3 μs. The GluN2B trajectories were 0.68 μs and 0.57 μs in length, for an aggregate of 1.25 μs. Trajectories were analyzed using MDTraj (McGibbon et al., 2015). Hydrophobic contact distance between two residues was defined as the shortest distance between side chain heavy atoms. Pairwise RMSD between frames for specific hydrophobic interactions (kinase residues F98, I101, V102, I205, and peptide residue in −5 position adjusted from sequence alignment; kinase residue W214, Ile at −5, Pro at −10 in CaMKIIN peptide, see Figure 2B) was calculated based on side chain heavy atoms for the residues involved in the interaction: 5 residues for the first interaction, 3 residues for the second.
Coupled kinase assays
Kinase activity was monitored using a Synergy H1 microplate reader (Biotek) as previously described (Barker et al., 1995; Chao et al., 2010). The assay was conducted in 50 mM Tris, 150 mM KCl, 10 mM MgCl2, 2 mM ATP, 1 mM Phosphoenolpyruvate (Alfa Aesar), 0.2 mM Nicotinamide adenine dinucleotide (Sigma), 10 units/mL Pyruvate kinase (Sigma), 30 units/mL Lactate dehydrogenase (Millipore Sigma), varying concentrations of Syntide-2 (Lifetein). The final CaMKII kinase domain concentration was 5 nM. 500 nM CaMKIIN protein or 8.14 μM NMDAR(S1303D) peptide was preincubated with CaMKII kinase domain before adding to the reaction mix of corresponding experiments. The reactions were started by the addition of ATP to the reaction mix and the absorbance was measured at 340 nM at 30°C at 10 and 15 s intervals for 10 min. The rate was obtained by calculating the maximum observed slope of each reaction. Data were fit using the Michaelis-Menten equation in GraphPad PRISM version 6.01.
Immunoprecipitation
HEK293T cell lysates were prepared in lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM Na3VO4, 10 mM NaF, 1 mM β-glycerophosphate, 1 × phosphatase inhibitor cocktail (Nacalai, Kyoto, Japan), 1× cOmplete tablet (Roche, Basel, Switzerland) and centrifuged at 14,000 rpm for 10 min at 4°C. The supernatant was subjected to immunoprecipitation using 20 μL of the anti-Flag antibody-beads (Sigma) for 2–4 h at 4°C. Beads were washed with 1 mL of lysis buffer for three times. Bound proteins were eluted with SDS-PAGE sample buffer and subjected to western blotting.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical testing was performed using GraphPad Prism software. The number of replicates and details of statistical tests used for immunoblotting and kinetics assays are indicated in the figure or table legends. Software used to analyze molecular dynamics simulations statistics are listed in the Method details section.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| GFP Polyclonal Antibody | Invitrogen | CAT# A-11122; RRID:AB_221569 |
| Tiam1 Antibody (C-16) | Santa Cruz Biotechnology | CAT# sc-872; RRID:AB_2240410 |
| Flag-M2 | Sigma-Aldrich | CAT# F1804; RRID:AB_262044 |
| Anti-GluN2B Antibody | Millipore | CAT# 06-600, RRID:AB_310193 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| BL21(DE3) Competent cells | New England Biolabs | CAT# C2527I |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| IPTG | GoldBio | CAT# I2481C100; CAS# 367-93-1 |
| TCEP | GoldBio | CAT# TCEP25; CAS# 51805-45-9 |
| Pepstatin | Sigma-Aldrich | CAT# 11359053001; CAS# 26305-03-3 |
| AEBSF | Sigma-Aldrich | CAT# 76307; CAS# 30827-99-7 |
| Leupeptin | Alfa Aesar | CAT# J61188-MC; CAS# 103476-89-7 |
| Benzamidine | Sigma-Aldrich | CAT# 12072-10G; CAS# 618-39-3 |
| DNase I | Sigma-Aldrich | CAT# 10104159001; CAS# 9003-98-9 |
| Aprotinin | Sigma-Aldrich | CAT# 10981532001; CAS# 9087-70-1 |
| Tween 20 | Acros Organics | CAT# 23336-2500; CAS# 9005-64-5 |
| Triton X-100 | Acros Organics | CAT# 21568-2500; CAS# 9002-93-1 |
| AMPPNP | Tocris | CAT# 6086; CAS# 72957-42-7 |
| ATP | Sigma-Aldrich | CAT# A3377-10G; CAS# 34369-07-8 |
| Syntide-2 | LifeTein | CAT# 187809 |
| L-Lactate Dehydrogenase | Sigma-Aldrich | CAT# 10127876001; CAS# 9001-60-9 |
| Pyruvate Kinase | Sigma-Aldrich | CAT# 10128163001; CAS# 9001-59-6 |
| Phosphoenolpyruvate | Alfa Aesar | CAT# B20358-14; CAS# 4265-07-0 |
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | CAT# 11873580001 |
|
| ||
| Deposited data | ||
|
| ||
| WT kinase in complex with GluN2B and ADP | This study | PDB:7UJS |
| WT kinase in complex with GluN2B | This study | PDB:7UJR |
| D135N kinase in complex with GluN2B and ATP | This study | PDB:7UJP |
| D135N kinase in complex with GluN2B and Hecameg | This study | PDB:7UPQ |
| D135N kinase in complex with phosphomimetic GluN2B and ATP | This study | PDB:7KL1 |
| D135N kinase in complex with phosphomimetic GluN2B and ATP | This study | PDB:7UJT |
| D135N kinase in complex with phosphomimetic GluN2B | This study | PDB:7UIS |
| D135N kinase in complex with phosphomimetic GluN2B and Hecameg | This study | PDB:7KL0 |
| D135N kinase in complex with Tiam1 and ATP | This study | PDB:7UIR |
| D135N kinase in complex with Tiam1 | This study | PDB:7UIQ |
| D135N kinase in complex with densin-180 | This study | PDB:6X5G |
| D135N kinase in complex with GluA1 | This study | PDB:6X5Q |
| MD simulations | This study | https://doi.org/10.17632/9m3bg3h5rs.1 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T | ATCC | CAT# CRL-3216 |
|
| ||
| Recombinant DNA | ||
|
| ||
| CaMKII expressin plasmids in pET | This study | N/A |
| Λ Phosphatase | This study | N/A |
| ULP1 protease | This study | N/A |
| CaMKII with a C-terminal FLAG tag in pDEST12.2 | (Saneyoshi et al., 2019) | N/A |
| Tiam1-mGFP in pCAGGS | (Saneyoshi et al., 2019) | N/A |
| GluN2B in pGW-CMV | (Kim et al., 1996) | N/A |
| CaMKIIN-GFP in pEGFP | (Chang et al., 2001) | N/A |
| mCherry-Flag in pDEST12.2 | (Saneyoshi et al., 2019) | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism 6 | Dotmatics | https://www.graphpad.com/dl/96314/10B92408/ |
| Pymol | Schrödinger | https://pymol.org/2/ |
| HKL-2000 | HKL Research, Inc | https://hkl-xray.com/ |
| Coot | (Emsley et al., 2010) | https://www.ccp4.ac.uk/download/#os=windows |
| REFMAC5 | (Murshudov et al., 1997) | https://www.ccp4.ac.uk/download/#os=windows |
| Phaser | (McCoy et al., 2007) | https://www.ccp4.ac.uk/download/#os=windows |
| MicroCal PEAQ-ITC Analysis Software | Malvern Panalytical | https://www.malvernpanalytical.com/en/support/software-download/ |
| MicroCal PEAQ-DSC Software | Malvern Panalytical | https://www.malvernpanalytical.com/en/support/software-download/ |
| PDBFixer | (Eastman et al., 2013) | https://anaconda.org/conda-forge/pdbfixer |
| Gromacs 2020.2 | (Pall et al., 2015) (Huang et al., 2017) | https://anaconda.org/conda-forge/pdbfixer |
| VMD | (Humphrey et al., 1996) | https://www.ks.uiuc.edu/Development/Download/download.cgi?PackageName=VMD |
| ProDy | (Bakan et al., 2011) | http://prody.csb.pitt.edu/downloads/ |
| MDTraj | (McGibbon et al., 2015) | https://www.mdtraj.org/1.9.8.dev0/index.html |
Highlights.
CaMKII kinase domain binds all interactors through a single continuous binding site
A salt bridge interaction far from active site increases binding affinity
αD helix changes conformation between autoinhibited and active “on” states
High-affinity binders may prolong activation by maintaining the “on” state
ACKNOWLEDGMENTS
We thank Roger Colbran, Howard Schulman, Eric Strieter, and Peter Chien for helpful discussions. We thank Josh Pajak for help with MD simulations. This work was supported by funds from NIGMS R01123157 (to M.S.); Umass Amherst Biophysical Core Facility RRID:SCR_019063, SPIRITS 2019 of Kyoto University; Grants-in-Aid for Scientific Research JP18H04733 and JP18H 05434 from the MEXT; CREST JPMJCR20E4 from JST, Japan; and HFSP research grant (RGP0020/2019) (to Y.H.).
Footnotes
DECLARATION OF INTERESTS
Y.H. received research funds from Fujitsu Laboratories and Dwango.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111064.
REFERENCES
- Bakan A, Meireles LM, and Bahar I (2011). ProDy: protein dynamics inferred from theory and experiments. Bioinformatics 27, 1575–1577. 10.1093/bioinformatics/btr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker SC, Kassel DB, Weigl D, Huang X, Luther MA, and Knight WB (1995). Characterization of pp60c-src tyrosine kinase activities using a continuous assay: autoactivation of the enzyme is an intermolecular autophosphorylation process. Biochemistry 34, 14843–14851. 10.1021/bi00045a027. [DOI] [PubMed] [Google Scholar]
- Barria A, Derkach V, and Soderling T (1997a). Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. J. Biol. Chem. 272, 32727–32730. 10.1074/jbc.272.52.32727. [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, and Soderling TR (1997b). Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 276, 2042–2045. 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, and Schulman H (2001). Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411, 801–805. 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- Bayer KU, LeBel E, McDonald GL, O’Leary H, Schulman H, and De Koninck P (2006). Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J. Neurosci. 26, 1164–1174. 10.1523/jneurosci.3116-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G, Donadio D, and Parrinello M (2007). Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101. 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Castro-Rodrigues AF, Zhao Y, Fonseca F, Gabant G, Cadene M, Robertson GA, and Morais-Cabral JH (2018). The interaction between the Drosophila EAG potassium channel and the protein kinase CaMKII involves an extensive interface at the active site of the kinase. J. Mol. Biol. 430, 5029–5049. 10.1016/j.jmb.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, and Soderling TR (2001). Calcium/calmodulin-dependent protein kinase II inhibitor protein: localization of isoforms in rat brain. Neuroscience 102, 767–777. 10.1016/s0306-4522(00)00520-0. [DOI] [PubMed] [Google Scholar]
- Chao LH, Pellicena P, Deindl S, Barclay LA, Schulman H, and Kuriyan J (2010). Intersubunit capture of regulatory segments is a component of cooperative CaMKII activation. Nat. Struct. Mol. Biol. 17, 264–272. 10.1038/nsmb.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, and Bayer KU (2011). Improving a natural CaMKII inhibitor by random and rational design. PLoS One 6, e25245. 10.1371/journal.pone.0025245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach V, Barria A, and Soderling TR (1999). Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc. Natl. Acad. Sci. USA 96, 3269–3274. 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diering GH, and Huganir RL (2018). The AMPA receptor code of synaptic plasticity. Neuron 100, 314–329. 10.1016/j.neuron.2018.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman P, Friedrichs MS, Chodera JD, Radmer RJ, Bruns CM, Ku JP, Beauchamp KA, Lane TJ, Wang LP, Shukla D, et al. (2013). OpenMM 4: a reusable, extensible, hardware independent library for high performance molecular simulation. J. Chem. Theor. Comput. 9, 461–469. 10.1021/ct300857j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott JA, Noble ME, and Johnson LN (2012). The structural basis for control of eukaryotic protein kinases. Annu. Rev. Biochem. 81, 587–613. 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- Erondu NE, and Kennedy MB (1985). Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. J. Neurosci. 5, 3270–3277. 10.1523/jneurosci.05-12-03270.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, and Pedersen LG (1995). A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Feenstra KA, Hess B, and Berendsen HJC (1999). Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 20, 786–798. . [DOI] [PubMed] [Google Scholar]
- Hess B (2008). P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theor. Comput. 4, 116–122. 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520. 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa T, Mitsushima D, Kaneko R, and Hayashi Y (2015). Stoichiometry and phosphoisotypes of hippocampal AMPA-type glutamate receptor phosphorylation. Neuron 85, 60–67. 10.1016/j.neuron.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, and MacKerell AD Jr. (2017). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Ishida A, and Fujisawa H (1995). Stabilization of calmodulin-dependent protein-kinase-ii through the autoinhibitory domain. J. Biol. Chem. 270, 2163–2170. 10.1074/jbc.270.5.2163. [DOI] [PubMed] [Google Scholar]
- Ishida A, Shigeri Y, Tatsu Y, Uegaki K, Kameshita I, Okuno S, Kitani T, Yumoto N, and Fujisawa H (1998). Critical amino acid residues of AIP, a highly specific inhibitory peptide of calmodulin-dependent protein kinase II. FEBS Lett. 427, 115–118. 10.1016/s0014-5793(98)00405-0. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Jalan-Sakrikar N, Robison AJ, Baucum AJ 2nd, Bass MA, and Colbran RJ (2011). Characterization of a central Ca2+/Calmodulin-dependent protein kinase IIα/β binding domain in densin that selectively modulates glutamate receptor subunit phosphorylation. J. Biol. Chem. 286, 24806–24818. 10.1074/jbc.m110.216010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Kennelly PJ, and Krebs EG (1991). Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J. Biol. Chem. 266, 15555–15558. 10.1016/s0021-9258(18)98436-x. [DOI] [PubMed] [Google Scholar]
- Kim E, Cho KO, Rothschild A, and Sheng M (1996). Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins. Neuron 17, 103–113. 10.1016/s0896-6273(00)80284-6. [DOI] [PubMed] [Google Scholar]
- Larsson P, Kneiszl RC, and Marklund EG (2020). MkVsites: a tool for creating GROMACS virtual sites parameters to increase performance in all-atom molecular dynamics simulations. J. Comput. Chem. 41, 1564–1569. 10.1002/jcc.26198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, and Raghavachari S (2012). Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 13, 169–182. 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA (2004). Long-term potentiation and memory. Physiol. Rev. 84, 87–136. 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, and Waxham MN (1989). An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature 340, 554–557. 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- Mammen AL, Kameyama K, Roche KW, and Huganir RL (1997). Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J. Biol. Chem. 272, 32528–32533. 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- Mao LM, Jin DZ, Xue B, Chu XP, and Wang JQ (2014). Phosphorylation and regulation of glutamate receptors by CaMKII. Sheng Li Xue Bao 66, 365–372. [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. 10.1107/s0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy F, Darbandi R, Lee HC, Bharatham K, Moldoveanu T, Grace CR, Dodd K, Lin W, Chen SI, Tangallapally RP, et al. (2013). Metabolic activation of CaMKII by coenzyme A. Mol. Cell 52, 325–339. 10.1016/j.molcel.2013.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández C, Hernandez CX, Schwantes CR, Wang LP, Lane TJ, and Pande VS (2015). MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 109, 1528–1532. 10.1016/j.bpj.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, and Dodson EJ (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255. 10.1107/s0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nolen B, Taylor S, and Ghosh G (2004). Regulation of protein kinases. Mol. Cell 15, 661–675. 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, and Kennedy MB (1996). Identification of a phosphorylation site for calcium/calmodulindependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 271, 31670–31678. 10.1074/jbc.271.49.31670. [DOI] [PubMed] [Google Scholar]
- Pall S, Abraham MJ, Kutzner C, Hess B, and Lindahl E (2015). Tackling exascale software challenges in molecular dynamics simulations with GROMACS. Lect. Notes Comput. Sci. 8759, 3–27. [Google Scholar]
- Parrinello M, and Rahman A (1981). Polymorphic transitions in single-crystals - a new molecular-dynamics method. J. Appl. Phys. 52, 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Pearson RB, and Kemp BE (1991). Protein kinase phosphorylation site sequences and consensus specificity motifs: tabulations. Methods Enzymol. 200, 62–81. 10.1016/0076-6879(91)00127-i. [DOI] [PubMed] [Google Scholar]
- Pellicena P, and Schulman H (2014). CaMKII inhibitors: from research tools to therapeutic agents. Front. Pharmacol. 5, 21. 10.3389/fphar.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rellos P, Pike ACW, Niesen FH, Salah E, Lee WH, von Delft F, and Knapp S (2010). Structure of the CaMKIId/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation. PLoS Biol. 8, e1000426. 10.1371/journal.pbio.1000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg OS, Deindl S, Sung RJ, Nairn AC, and Kuriyan J (2005). Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell 123, 849–860. 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Rust HL, and Thompson PR (2011). Kinase consensus sequences: a breeding ground for crosstalk. ACS Chem. Biol. 6, 881–892. 10.1021/cb200171d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneyoshi T, Matsuno H, Suzuki A, Murakoshi H, Hedrick NG, Agnello E, O’Connell R, Stratton MM, Yasuda R, and Hayashi Y (2019). Reciprocal activation within a kinase-effector complex underlying persistence of structural LTP. Neuron 102, 1199–1210.e6. 10.1016/j.neuron.2019.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman H (2004). Activity-dependent regulation of calcium/calmodulin-dependent protein kinase II localization. J. Neurosci. 24, 8399–8403. 10.1523/jneurosci.3606-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloutsky R, and Stratton MM (2020). Functional implications of CaMKII alternative splicing. Eur. J. Neurosci. 54, 6780–6794. 10.1111/ejn.14761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z, Lu KP, Kwon YT, Tsai LH, Filhol O, Cochet C, Brickey DA, Soderling TR, Bartleson C, Graves DJ, et al. (1996). A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol. Cell Biol. 16, 6486–6493. 10.1128/mcb.16.11.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokoe D, Caudwell B, Cohen PTW, and Cohen P (1993). The substrate specificity and structure of mitogen-activated protein (MAP) kinase-activated protein kinase-2. Biochem. J. 296, 843–849. 10.1042/bj2960843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, and Colbran RJ (1998). Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl- D-aspartate receptor. J. Biol. Chem. 273, 20689–20692. 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB, and Colbran RJ (2000). Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 275, 23798–23806. 10.1074/jbc.m001471200. [DOI] [PubMed] [Google Scholar]
- Sun XX, Hodge JJ, Zhou Y, Nguyen M, and Griffith LC (2004). The eag potassium channel binds and locally activates calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 279, 10206–10214. 10.1074/jbc.m310728200. [DOI] [PubMed] [Google Scholar]
- Torres-Ocampo AP, Ozden C, Hommer A, Gardella A, Lapinskas E, Samkutty A, Esposito E, Garman SC, and Stratton MM (2020). Characterization of CaMKII alpha holoenzyme stability. Protein Sci. 29, 1524–1534. 10.1002/pro.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulpetti A, and Bosotti R (2004). Sequence and structural analysis of kinase ATP pocket residues. Farmaco 59, 759–765. 10.1016/j.farmac.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Walikonis RS, Oguni A, Khorosheva EM, Jeng CJ, Asuncion FJ, and Kennedy MB (2001). Densin-180 forms a ternary complex with the (alpha)-subunit of Ca2+/calmodulin-dependent protein kinase II and (alpha)-actinin. J. Neurosci. 21, 423–433. 10.1523/jneurosci.21-02-00423.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Li N, Liu X, Zheng Y, and Cao X (2008). A novel endogenous human CaMKII inhibitory protein suppresses tumor growth by inducing cell cycle arrest via p27 stabilization. J. Biol. Chem. 283, 11565–11574. 10.1074/jbc.m800436200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- White RR, Kwon YG, Taing M, Lawrence DS, and Edelman AM (1998). Definition of optimal substrate recognition motifs of Ca2+-calmodulin-dependent protein kinases IV and II reveals shared and distinctive features. J. Biol. Chem. 273, 3166–3172. 10.1074/jbc.273.6.3166. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242. 10.1107/s0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, and Schulman H (1999). Structural examination of autoregulation of multifunctional calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 274, 26199–26208. 10.1074/jbc.274.37.26199. [DOI] [PubMed] [Google Scholar]
- Zha M, Zhong C, Ou Y, Han L, Wang J, and Ding J (2012). Crystal structures of human CaMKIα reveal insights into the regulation mechanism of CaMKI. PLoS One 7, e44828. 10.1371/journal.pone.0044828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li N, Yu J, Zhang W, and Cao X (2001). Molecular cloning and characterization of a novel calcium/calmodulin-dependent protein kinase II inhibitor from human dendritic cells. Biochem. Biophys. Res. Commun. 285, 229–234. 10.1006/bbrc.2001.5175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic structures have been deposited in the PDB, listed in the Star Methods. Molecular dynamics simulation files are deposited in Mendeley and can be accessed using https://doi.org/10.17632/9m3bg3h5rs.1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.