

ABSTRACT
Gastrointestinal dysfunction is a common symptom of acute mountain sickness (AMS). The gut microbiota and γδ T cells play critical roles in intestinal disease. However, the mechanistic link between the microbiota and γδ T cells in hypoxia-induced intestinal injury remains unclear. Here, we show that hypoxia-induced intestinal damage was significantly alleviated after microbiota depletion with antibiotics. Hypoxia modulated gut microbiota composition by promoting antimicrobial peptides angiogenin-4 secretions. The abundance of Clostridium in the gut of mice after hypoxia significantly decreased, while the abundance of Desulfovibrio significantly increased. Furthermore, Desulfovibrio-derived phosphatidylethanolamine and phosphatidylcholine promoted γδ T cell activation. In CD1d-deficient mice, the levels of intraepithelial IL-17A and γδ T cells and intestinal damage were significantly decreased compared with those in wild-type mice under hypoxia. Mechanistically, phospholipid metabolites from Desulfovibrio are presented by intestinal epithelial CD1d to induce the proliferation of IL-17A-producing γδ T cells, which aggravates intestinal injury. Gut microbiota-derived metabolites promote hypoxia-induced intestinal injury via CD1d-dependent γδ T cells, suggesting that phospholipid metabolites and γδ T cells can be targets for AMS therapy.
KEYWORDS: Gut microbiota, hypoxia, intestinal injury, metabolites, γδ T cells
Introduction
Acute mountain sickness (AMS) is a potentially fatal disease closely related to acute hypoxia after rapid ascent to plateaus above 2500 meters.1,2 It is the leading cause of hypoxia-related disease unique to plateau areas, with high-altitude cerebral edema (HACE) and high-altitude pulmonary edema (HAPE) being the most serious presentations of the disease.2 Despite the recent development of preventive and therapeutic applications targeting basal oxyhemoglobin saturation for human AMS patients,3–5 the overall effect of treatment for the disease is still not ideal. In addition to the usual presentation occurring with headache, chest tightness, palpitation, panting and other related conditions, AMS also presents as intestinal symptoms such as nausea, vomiting, and diarrhea.6,7 However, the detailed mechanisms responsible for hypoxia-induced intestinal dysfunction have not been clearly defined. Importantly, as the largest mucosal tissue among body organs, the intestine is continually exposed to antigens and metabolites from the diet and microbiota.8,9 Intestinal flora imbalance and immune dysfunction are closely related to various diseases, such as inflammatory bowel disease (IBD), colorectal cancer, primary biliary cholangitis (PBC), immune-mediated liver injury.10–12 Recent studies with 16S rRNA sequencing analysis indicate that the diversity and composition of intestinal commensal microbiota in individuals from the plains are different from those in individuals from plateau areas.13 Thus, the relationship between host immune response to the gut microbiota and intestinal injury in AMS is of particular interest.
The intestine contains a large number of innate immune cells, with a large proportion of γδ T cells in intraepithelial lymphocytes (IELs).14 γδ T cells are believed to protect against the invasion of foreign pathogens and perform epithelial barrier surveillance under physiological conditions.14 However, many studies have also shown that γδ T cells are responsible for driving the inflammatory response, which participates in the pathogenesis of infectious diseases and tumors.15,16 Because of the dual properties of innate and adaptive immunity, γδ T cells can recognize specific antigens and rapidly secrete proinflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-17A, and interferon (IFN)-γ17 The upregulated deSUMOylase SENP7 in intestinal epithelial cells contributes to the expansion of TNF-α-producing and IFN-γ-producing γδ T cells during IBD.16 Moreover, regulatory T (Treg) cells can maintain intestinal homeostasis by inhibiting IL-17A-producing γδ T cells.18 Although the epithelial layer of the intestine contains a high proportion of γδ T cells involved in various intestinal diseases, the role of intraepithelial γδ T cells in the acute hypoxia processes and their relationship with the gut microbiota are still unclear.
Specifically, γδ T cells can be recognized and activated by lipid antigens presented by CD1d, a nonpolymorphic major histocompatibility class (MHC) I-like molecule.19 CD1d molecules are mainly expressed on the surface of antigen-presenting cells, hepatocytes and intestinal epithelial cells.20–23 Sulfatide, a lipid antigen, in the blood can be presented to a specific subset of γδ T cells through CD1d on the surface of antigen-presenting cells.24 Exogenous lipid antigens such as phosphatidylethanolamine (PE) and phosphatidylglycerol (PG) in the liver can also be presented to γδ T cells or natural killer T (NKT) cells through CD1d expressed by hepatocytes to promote activation.22,25–27 Moreover, γδ T cells in the mouse intestine can respond to lipid antigens presented by CD1d, including PE, PG and phosphatidylcholine (PC).23 Since the intestines constantly encounter lipid antigens derived from the commensal microbiota and acute hypoxia can significantly change the gut microbiota composition and structure, we speculate that lipid antigens derived from the intestinal microbiota may be related to the host immune response after acute hypoxia.
Here, we studied the host-microbiota interactions in hypoxia-induced intestinal injury. Our findings reveal a novel mechanism by which lipid antigens from intestinal microbiota, especially Desulfovibrio, are presented to γδ T cells through CD1d expressed on small intestinal epithelial cells, which promotes the production of IL-17A and exacerbates hypoxia-induced intestinal injury. These findings imply that modifying the intestinal flora during hypoxia might be a promising method for the prevention and treatment of AMS.
Results
Gut microbiota alteration during hypoxia aggravates intestinal damage
We established an acute mountain sickness model using rearing conditions with 5.0% oxygen concentration for 48 h. To investigate the role of the intestinal microbiota in the occurrence and development of AMS, we first observed phenotypic changes in lung and intestinal tissues between hypoxic (5.0% oxygen concentration) and normoxic (20.9% oxygen concentration) conditions after oral gavage with an antibiotic cocktail (4Abx) or sterile water (Figure 1a). As shown in Figure 1, mice treated with antibiotics were significantly protected against intestinal injury and pulmonary edema by hypoxia. To characterize the degree of acute altitude sickness, the histological images and water content of lung tissue were observed, showing that the degree of pulmonary edema in the mice treated with antibiotics was attenuated compared with sterile water-fed controls (Figure 1b,c). Compared with mice fed sterile water, antibiotic-fed mice exhibited a mild disease process with significant reduction in intestinal villi shortening, inflammatory cell infiltration and intestinal wall thinning (Figure 1d). Furthermore, a tendency toward increased expression of tight junction proteins such as claudin-1, ZO-1 in antibiotic-fed mice during hypoxia (Figure 1e,f). We also assessed intestinal permeability using the permeability marker FITC-dextran. The results showed that serum FITC-dextran concentrations were significantly reduced in antibiotic-treated mice during hypoxia compared with sterile water-fed controls (Figure 1g). Meanwhile, serum LPS levels were significantly reduced in hypoxic mice treated with antibiotics (Figure 1h). Besides, a measurable decreased in intestine apoptosis (Figure 1i) were observed in antibiotic-fed mice compared with sterile water-fed controls. Corresponding with their intestinal barrier function, the pro-inflammatory factors IL-6 and IL-17A are significantly increased under hypoxic conditions, which can be partially suppressed by combined antibiotic treatment (Figure 1j). The above results indicate that the elimination of commensal bacteria can delay the progression of AMS.
Figure 1.
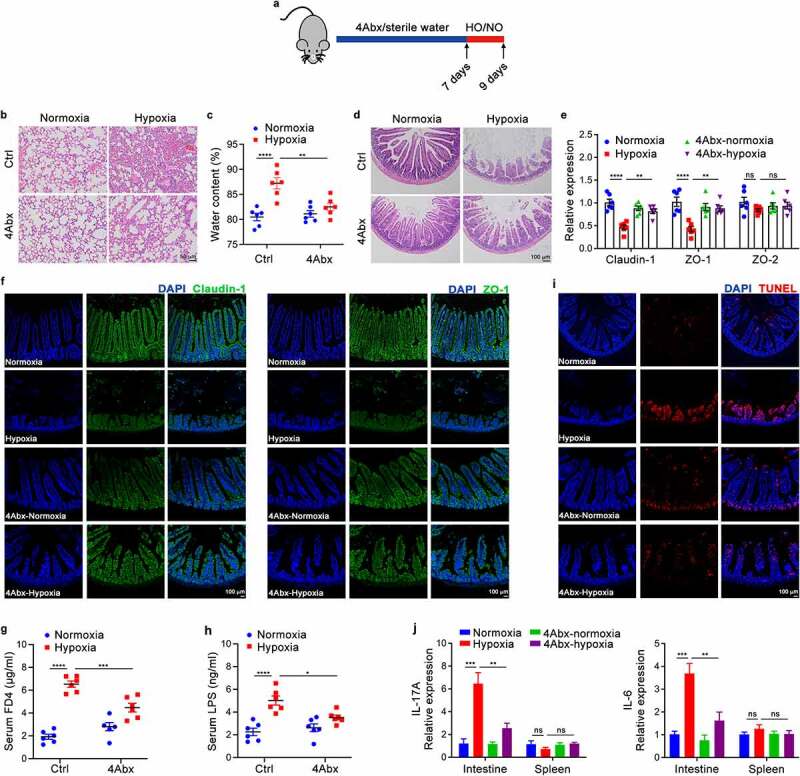
Depletion of the Gut Microbiome Attenuates Intestinal Damage and Pulmonary Edema Caused by Hypoxia. (a) A schematic diagram of SPF mice was administered combined antibiotics (4Abx) or sterile water (Ctrl) by oral gavage for 7 days followed by hypoxia (5.0% oxygen concentration) or normoxia (20.9% oxygen concentration) for 48 hours (n = 3–6). (b) Histopathological analysis of lung tissues from the different groups; representative pictures of H&E staining are shown. Scale bars, 50 µm. (c) The whole lung water content is measured using the (wet-dry)/wet weight ratio. (d) Histopathological analysis of small intestine tissues from the different groups; representative pictures of H&E staining are shown. Scale bars, 100 µm. (e) Real-time qPCR analysis of claudin-1, ZO-1 and ZO-2 gene expression in the intestinal epithelium tissues. (f) Immunofluorescence staining of claudin-1 and ZO-1 was performed to examine intestinal barrier function; representative images are shown. Green: claudin-1 or ZO-1; blue: DAPI. Scale bars, 100 µm. (g) Serum FITC-dextran concentrations in mice after gavage administration. (h) ELISA analysis of LPS concentration in the serum of mice. (i) Immunofluorescence staining of TUNEL was performed to examine apoptotic cells; representative images are shown. Red: TUNEL‐positive nuclei; blue: DAPI. Scale bars, 100 µm. (j) Real-time qPCR analysis of IL-6 and IL-17A gene expression in the intestinal epithelium tissues. The data are representative of three independent experiments and shown by the mean ± SEM. *p <0.05, **p <0.01, ***p <0.001, ****p <0.0001 by one-way ANOVA test with Tukey’s posttest (c, e, g, h, j). HO, hypoxia; NO, normoxia; Ctrl, control; FD4, FITC-dextran 4; LPS, lipopolysaccharide; ns, no significance.
To determine whether alteration of the commensal microbiota is responsible for the disease process, we also examined phenotypic changes in mice fed fecal microbiota from hypoxic and normoxic mice (Figure S1a). Mice transplanted with fecal bacteria from hypoxic mice displayed significantly worse intestinal damage than their counterparts transplanted with fecal bacteria from normoxic mice (Figure S1b). Similarly, a decrease in tight junction proteins (Figure S1c,d) and a significant increase in intestinal cell apoptosis (Figure S1e) and the levels of proinflammatory factors (Figure S1f) were observed in mice transplanted with hypoxic mouse feces. Together with the findings in mice treated with the antibiotic cocktail or fecal bacteria, these data indicate that commensal bacteria have profound effects in promoting AMS development.
Hypoxia significantly reduces the abundance of Clostridium XIVa and increases Desulfovibrio in the intestine
To further explore the role of the gut microbiota in hypoxia-induced intestinal injury, we performed 16S rRNA gene sequencing to analyze the difference in gut microbial community composition between hypoxic and normoxic mice. At the phylum level, the abundance of Firmicutes was significantly increased in mice exposed to acute hypoxia, accompanied by a decrease in the abundance of Bacteroidetes and Proteobacteria (Figure 2a). At the genus level, Desulfovibrio and Alloprevotella were more abundant in the hypoxic group than in the control group, while the abundance of Clostridium was decreased (Figure 2a). Unweighted principal component analysis (PCA) and principal coordinates analysis (PCoA) also indicated a noticeable difference between hypoxic and normoxic mice (Figure 2b). Furthermore, volcano plot analysis depicted differences in genera, including 5 upregulated and 9 downregulated genera (Figure 2c). It has been previously shown that Clostridium XIVa (C. XIVa) suppresses intestinal inflammation by inducing an IL-10-producing Treg cell population.28 Moreover, as the most abundant sulfate-reducing bacteria in the intestine, Desulfovibrio promotes the occurrence and development of IBD through the production of hydrogen sulfide and acetate.29 Based on these findings combined with our 16S rRNA gene sequencing analysis results, we identified a bacterial taxon, Desulfovibrio, that was significantly overrepresented in hypoxic mouse intestines, while another taxon, C. XIVa, was enriched in healthy intestines (Figure 2c-e and Figure S2a). In addition, a similar trend was also observed in linear discriminant analysis effect size (LEfSe) analysis (Figure S2b).
Figure 2.
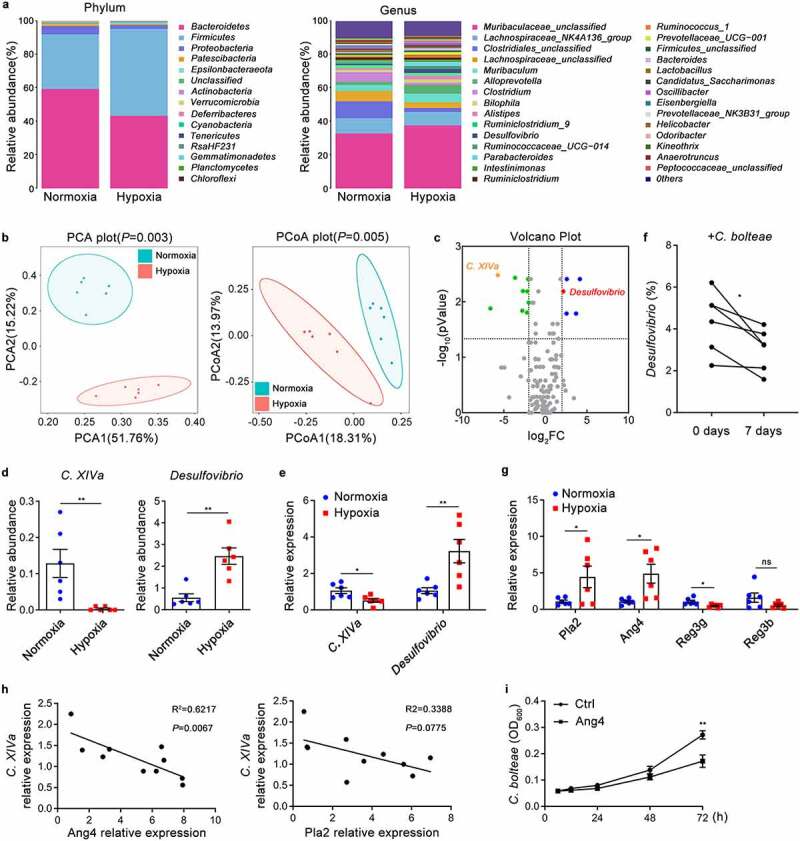
Hypoxia Contributes to the Alteration of Gut Microbiota and Associated with Antimicrobial Peptides. (a-d) 16S rRNA analysis was performed on the cecal contents from SPF mice under hypoxia (5.0% oxygen concentration) or normoxia (20.9% oxygen concentration) for 48 hours (n = 6). (a) The relative abundance of intestinal microbiota at the phylum level and genus level. (b) Principal component analysis (PCA) and Principal co-ordinates analysis (PCoA) analysis based on the relative abundance of operational taxonomic units (97% similarity). (c) Volcano plot of differentially expressed genera. (d) The relative abundance of C. XIVa and Desulfovibrio in the cecal contents of normoxic and hypoxic mice was analyzed by 16S rRNA gene sequencing. (e) The relative abundance of C. XIVa and Desulfovibrio in the cecal contents of normoxic and hypoxic mice was quantified by Real-time qPCR analysis (n = 6). (f) Real-time qPCR analysis of Desulfovibrio in SPF mice after C. bolteae administered orally for 7 days (n = 6). (g) Real-time qPCR analysis of antimicrobial peptide Pla2, Ang4, Reg3g and Reg3b gene expression in the intestinal epithelium tissues (n = 6). (h) Correlation analysis between the relative content of C. XIVa and the relative expression of Ang4 or Pla2 in the intestine of SPF mice (n = 8). (i) Spectrophotometry of C. bolteae growth at different time points following recombinant Ang4 (5 µg/ml each) stimulation (n = 3). The data are representative of three independent experiments and shown by the mean ± SEM. *p <0.05, **p <0.01 by unpaired Student’s t-test (d, e, g, i), paired Student’s t-test (f) or linear regression analysis (h). ns, no significance.
Hypoxia modulates gut microbiota composition by promoting angiogenin-4 secretion
Notably, a significant inverse correlation between the abundances of the genera Desulfovibrio and C. XIVa (including Clostridium bolteae) in the intestine was observed according to 16S rRNA sequencing analysis (Figure S2c). To further confirm the relationship, Clostridium bolteae was given to mice by daily oral gavage for a period of 7 days. The data demonstrated that the relative abundance of Desulfovibrio in the intestines of mice receiving gavage with Clostridium bolteae was significantly reduced, suggesting that C. XIVa may inhibit the growth of Desulfovibrio (Figure 2f). Next, we explored how hypoxia inhibits the growth of C. XIVa. Since the commensal microbiota is known to be directly regulated by antibacterial peptides (AMPs),30,31 we first measured the expression of AMPs in the intestinal epithelial layer. The expression of mRNA encoding phospholipase A2 (PlA2) and angiogenin-4 (Ang4) was upregulated while the expression of regenerating islet-derived 3 gamma (Reg3g) and regenerating islet-derived 3 beta (Reg3b) was downregulated in the intestinal epithelial layer of hypoxic mice (Figure 2g). The expression of Ang4, but not Pla2, was negatively associated with the relative abundance of C. XIVa (Figure 2h), and Ang4 significantly inhibited the growth of Clostridium bolteae (Figure 2i). These results indicated that the increased Ang4 in the intestines during hypoxia inhibits the growth of Clostridium XIVa, which weakens the inhibitory effect on Desulfovibrio, resulting in an increase in the abundance of Desulfovibrio.
γδ T cells-derived IL-17A promotes hypoxia-induced intestinal injury
IL-17 can not only promote the differentiation and migration of granulocytes by inducing cytokines and chemokines and mediating tumor-associated inflammation,15 but also participate in protective immunity against infection by regulating the cell-mediated immune response.32 Previous studies have shown that the intestinal microbiota can induce the expression of IL-17A; however, its role in hypoxia-induced intestinal injury has not been clearly identified. We have mentioned that IL-17A production is dramatically increased in the small intestine of hypoxic mice (Figure 1j). Moreover, IL-17A-deficient mice exhibited a significant reduction in intestinal damage (Figure 3a) and increased tight junction proteins under hypoxic conditions compared with WT mice (Figure 3b). Meanwhile, serum FD4 and LPS levels were significantly reduced in IL-17A-deficient mice compared with WT mice during hypoxia (Figure 3c-d). Taken together, these data suggest that IL-17A might play a major role in hypoxia-mediated intestinal injury. To further explore the immune mechanisms mediated by the gut microbiota during hypoxia-induced intestinal injury, we analyzed IL-17A-producing immune cells in the small intestine epithelial layer from hypoxic and normoxic mice by flow cytometry. Our data showed a notable increase in the abundance of γδ T cells in the intestines of hypoxic mice, but there was no difference in the proportion of T helper 17 (Th17), natural killer (NK), and NKT cells (Figure 3e). In addition, γδ T cells were dramatically increased in only the small intestine epithelial layer of mice after acute hypoxia exposure but not in other immune organs (Figure S3a). Moreover, γδ T cells in the small intestine epithelial layer of hypoxic mice showed elevated T cell activation markers, including CD44 and CD69, compared with normoxic mice (Figure 3f). Expanded and activated γδ T cells showed the maximum capacity for IL-17A production (Figure 3g). Compared with those in normoxic mice, a higher proportion of γδ T cells in the intestinal epithelium of hypoxic mice were positive for RORγt, a crucial transcription factor for IL-17A secretion by γδ T cells (Figure 3h). On the other hand, the production of IFN-γ by γδ T cells was not significantly different between hypoxic and normoxic mice, nor was the expression of the transcription factor T-bet (Figure 3g and Figure S3b). Notably, the proportions of CD4+ T, CD8+ T, NKT and NK cells among the IL-17-positive cells did not change significantly after hypoxia. γδ T cells were the highest producers of IL-17A in the small intestine of hypoxic mice but not normoxic mice (Figure 3i). To assess the functional importance of γδ T cells in intestinal barrier disruption and promotion of injury, mice were injected with monoclonal antibodies against γδ T cells prior to exposure to hypoxia. The results demonstrated that the anti-γ/δ TCR antibody effectively inhibited γδ T cells in the intestine (Figure S3c) and significantly suppressed the progression of intestinal damage (Figure S3d-f). Collectively, these data suggest that γδ T cells, the major producers of IL-17A during acute hypoxia, accumulate in the intestinal epithelial layer to aggravate hypoxia-induced intestinal injury.
Figure 3.
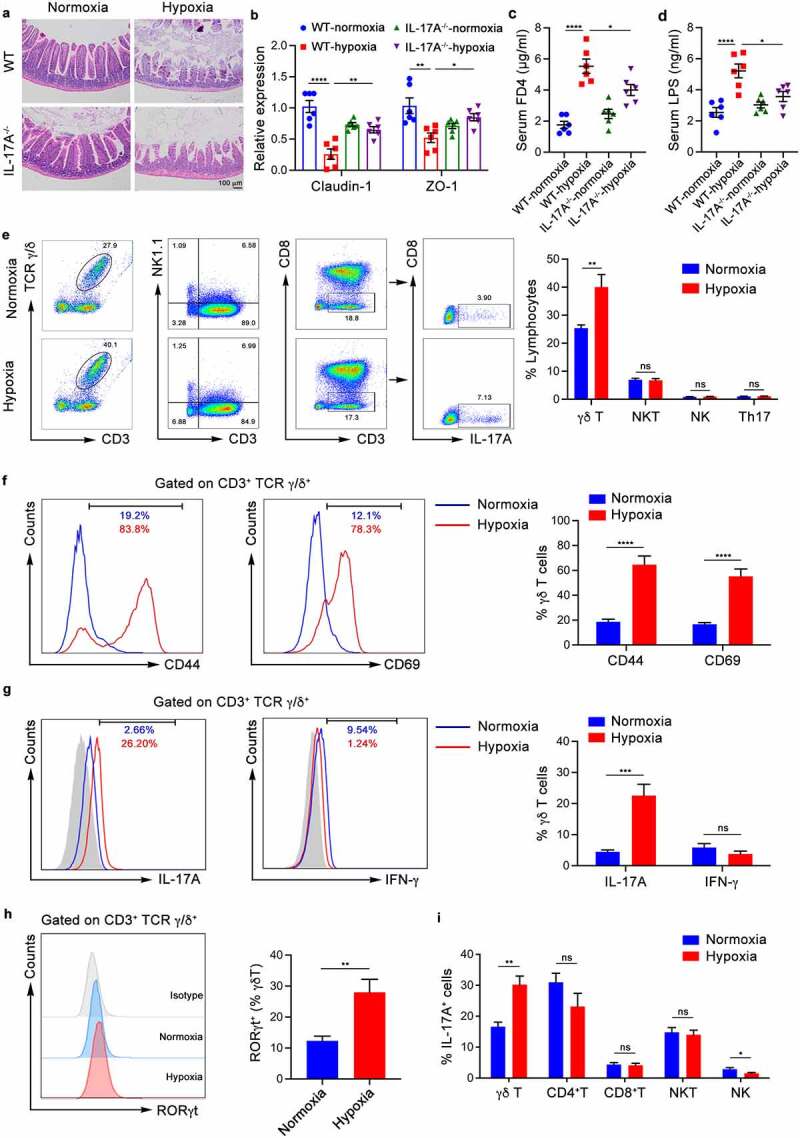
Proliferation and Activation of γδ Τ Cells in the Small Intestine Epithelium and Production of IL-17A Promote Hypoxia-induced Intestinal Injury. (a-d) WT mice and IL-17A-deficient (IL-17A−/−) mice were housed in hypoxia (5.0% oxygen concentration) or normoxia (20.9% oxygen concentration) for 48 hours (n = 6). (a) Histopathological analysis of small intestine tissues from the different groups; representative pictures of H&E staining are shown. Scale bars, 100 µm. (b) Real-time qPCR analysis of claudin-1, ZO-1 and gene expression in the intestinal epithelium tissues. (c) Serum FITC-dextran concentrations in mice after gavage administration. (d) ELISA analysis of LPS concentration in the serum of mice. (e-i) WT mice were exposed to hypoxia (5.0% oxygen concentration) or normoxia (20.9% oxygen concentration) for 48 hours (n = 6). (e) The frequency of γδ T, NK, NKT and Th17 cells from the epithelial layer of the small intestine were identified by flow cytometry. Representative scatter plot and quantitative data are shown. (f) CD44 and CD69 expression in γδ T cells from the epithelial layer of the small intestine of normoxic mice (blue) and hypoxic mice (red) was analyzed. Representative images and quantitative histograms are shown. (g) IL-17A and IFN-γ expression in γδ T cells from the epithelial layer of the small intestine of normoxia mice (blue) and hypoxia mice (red) was analyzed. Representative images and quantitative histograms are shown. (h) RORγt expression in γδ T cells from the epithelial layer of the small intestine of normoxia mice (blue) and hypoxia mice (red) was analyzed. Representative images and quantitative histograms are shown. (i) The proportion of γδ T, CD4+T, CD8+T, NKT and NK cells in the IL-17A-positive cells of the small intestine epithelial layer was analyzed by flow cytometry. The data are representative of three independent experiments and shown by the mean ± SEM. *p <0.05, **p <0.01, ***p <0.001, ****p <0.0001 by one-way ANOVA test with Tukey’s posttest (b-d) or unpaired Student’s t-test (e-i). FD4, FITC-dextran 4; LPS, lipopolysaccharide; ns, no significance.
γδ T cells are activated by Desulfovibrio to produce IL-17A
We further studied the genera mentioned in Figure 2 and found that the abundance of Desulfovibrio was significantly positively correlated with the proportion of γδ Τ cells and IL-17A production, and the opposite effect was observed for C. XIVa (Figure 4a,b). To explore whether the microbiota might affect the proliferation of γδ Τ cells and IL-17A production, combined antibiotics were used to eliminate the intestinal bacteria of mice as before. The antibiotic-treated mice had a sharp decrease in the proportion of γδ Τ cells and IL-17A production (Figure 4c,d). Furthermore, mice gavaged with Desulfovibrio piger (D. piger) showed a higher proportion of γδ Τ cells and more IL-17A production than PBS-treated control mice (Figure 4e,f and Figure S3g). Simultaneously, there was a significant increase in the proportion of γδ T cells IL-17A after stimulation with D. piger. (Figure 4g,h). These results indicate that Desulfovibrio in the intestines promote the proliferation of γδ T cells and the secretion of IL-17A during acute hypoxia, thereby aggravating hypoxia-induced intestinal injury.
Figure 4.
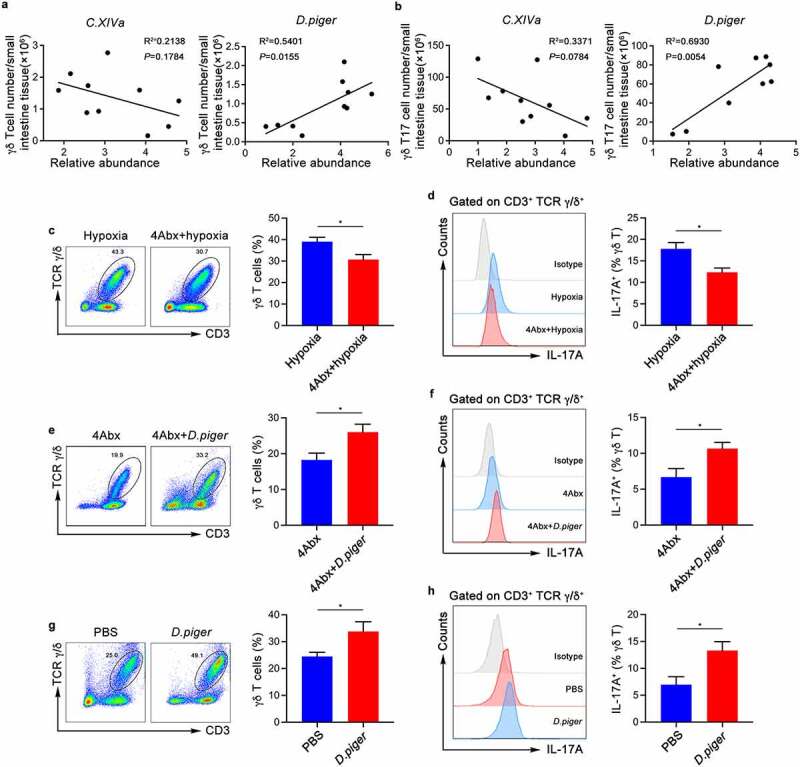
Commensal Microbiota Induces γδ Τ Cells Proliferation and IL-17A Production. (a) Correlation between the relative abundance of C. XIVa or Desulfovibrio and the numbers of intestinal intraepithelial γδ T cells in SPF wild-type mice (n = 10). (b) Correlation between the relative abundance of C. XIVa or Desulfovibrio and the numbers of intestinal intraepithelial γδ T17 cells in SPF wild-type mice (n = 10). (c-d) The frequency of γδ T cells (c) from the small intestinal epithelial layer of mice treated with sterile water or 4Abx and IL-17A production (d) were determined by flow cytometry. Representative pictures and quantitative data are show (n = 6). (e-f) 4Abx-treated mice were given hypoxia (5.0% oxygen concentration) for 48 hours after intragastric administration of D. piger or PBS. Representative pictures and quantitative data are show (n = 6). (g-h) Total cells of the small intestine epithelial layer were co-cultured with D. piger for 3 days. the frequency of γδ T cells and IL-17A production were determined by flow cytometry. Representative pictures and quantitative data are show (n = 6). The data are representative of three independent experiments and shown by the mean ± SEM. *p <0.05 by linear regression analysis (a and b) or unpaired Student’s t-test (c-h).
Desulfovibrio activate intestinal intraepithelial γδ T cells in a CD1d-dependent manner
γδ T cells can be activated by the microbiota in different ways, including direct stimulation by bacteria and recognition of lipid antigens from bacteria presented by CD1d molecules.22 To explore the mechanism by which Desulfovibrio promote the proliferation and activation of γδ T cells in the intestinal epithelial layer, we first constructed an AMS model with CD1d-deficient mice and observed the phenotypic changes. The results demonstrated that the CD1d-deficient mice were partially resistant to the intestinal damage and intestinal barrier breakdown caused by hypoxia (Figure 5a,b). Meanwhile, serum FD4 and LPS levels were significantly reduced in CD1d-deficient mice compared with WT mice during hypoxia (Figure 5c-d). Consistently, the CD1d-deficient mice had a decreased proportion of γδ T cells and reduced IL-17Α production compared to wild-type mice under hypoxic conditions (Figure 5e,f). In addition, in the intestinal epithelial layer of CD1d-deficient mice, the proportion of γδ T cells in IL-17A-producing cells was also downregulated (Figure 5g). This finding indicated that CD1d might participate in the proliferation of γδ T cells. However, we further found that there was no difference in the proportion of CD1d-positive cells in CD45-negative cells, representing epithelial cells, between hypoxic mice and normoxic mice, and the same was true in CD45-positive cells, representing intraepithelial leukocytes (Figure 5h). Notably, the proportion of CD45-negative cells in CD1d-expressing cells was higher than that of CD45-positive cells regardless of the oxygen concentration (Figure 5i). Furthermore, the corresponding cell numbers also exhibited the same trend, suggesting that CD1d expressed in small intestinal epithelial cells may play an irreplaceable role in hypoxia-induced intestinal injury (Figure 5j). Next, we confirmed that total cells derived from the small intestine epithelium of wild-type mice, but not CD1d-deficient mice, showed a significant increase in the proportion of γδ T cells after stimulation with D. piger (Figure 5k). In summary, these results indicate that Desulfovibrio mediate the expansion and activation of γδ T cells in the small intestine epithelial layer mainly through CD1d signaling.
Figure 5.
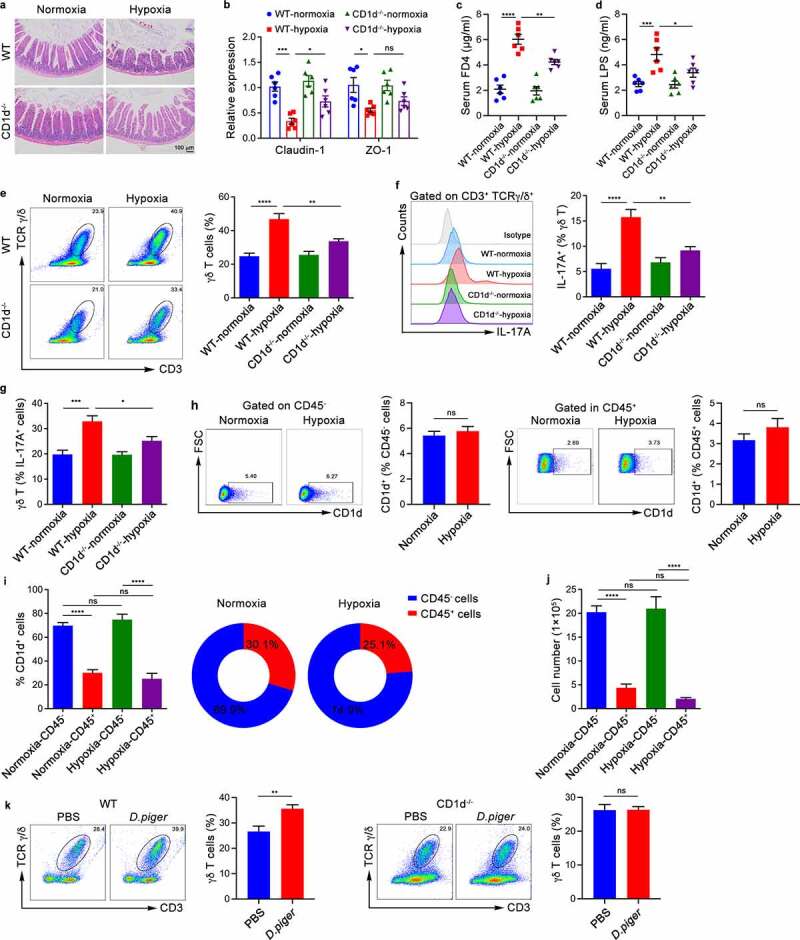
Commensal Microbiota Activate Intestinal Intraepithelial γδ T Cells in a CD1d-dependent Manner. (a-g) WT mice and CD1d-deficient (CD1d−/−) mice were exposed to hypoxia (5.0% oxygen concentration) or normoxia (20.9% oxygen concentration) for 48 hours (n = 6). (a) Histopathological analysis of small intestine tissues from the different groups; representative pictures of H&E staining are shown. Scale bars, 100 µm. (b) Real-time qPCR analysis of claudin-1, ZO-1 and mRNA expression in the intestinal epithelium tissues. (c) Serum FITC-dextran concentrations in mice after gavage administration. (d) ELISA analysis of LPS concentration in the serum of mice. (e) The frequency of γδ T cells from the epithelial layer of the small intestine were identified by flow cytometry. Representative scatter plot and quantitative data are shown. (f) IL-17A expression in γδ T cells from the epithelial layer of the small intestine were analyzed. Representative histograms and quantitative data are shown. (g) The proportion of γδ T cells in the IL-17A-positive cells of the small intestine epithelial layer was measured by flow cytometry. (h-j) WT mice were exposed to hypoxia (5.0% oxygen concentration) or normoxia (20.9% oxygen concentration) for 48 hours (n = 6). (h) CD1d expression in the CD45-negative or CD45-positive cells of the small intestine epithelial layer was measured by flow cytometry. Representative scatter plot and quantitative data are show (n = 6 per group). (i) The proportion of CD45-positive or CD45-negative cells in the CD1d-positive cells of the small intestine epithelial layer was measured by flow cytometry. (j) The number of CD45-positive or CD45-negative cells expressing CD1d in the epithelial layer of the small intestine was measured by flow cytometry. (k) Total cells derived from the small intestine epithelial layer of WT or CD1d-/- mice were stimulated with D. piger for 3 days, the frequency of γδ T cells and IL-17A production were determined by flow cytometry. Representative scatter plot and quantitative data are show (n = 6). The data are representative of three independent experiments and shown by the mean ± SEM. *p <0.05, **p <0.01, ***p <0.001, ****p <0.0001 by one-way ANOVA test with Tukey’s posttest (b-g, i and j) or unpaired Student’s t-test (h, k). FD4, FITC-dextran 4; LPS, lipopolysaccharide; ns, no significance.
The phospholipid antigens PE and PC derived from Desulfovibrio are responsible for the expansion and activation of γδ T cells
To explore the key factors influencing the expansion and activation of intraepithelial γδ T cells under hypoxic conditions, we further detected CD1d-associated lipid antigens in the intestine through lipid metabolomics analysis. PCA and partial least squares discriminant analysis (PLS-DA) revealed that there were remarkable differences in lipid metabolites between the hypoxic and normoxic groups (Figure 6a). Heatmap analysis also illustrated the same phenomenon (Figure 6b). Lipid antigens derived from bacteria, including PE and PC, are known to induce the proliferation and activation of γδT-17 cells through CD1d molecules.22 Therefore, we compared PE and PC expression in the cecal contents of hypoxic and normoxic mice using metabolomics. The results showed that PE and PC expression and their corresponding signals were significantly increased in the gut of hypoxic mice (Figure 6c–eand Fig. S4a,b). Furthermore, we used enzyme-linked immunosorbent assay (ELISA) to confirm that the expression of PE and PC was indeed significantly increased in the gut of hypoxic mice compared with normoxic mice (Figure 6f). After administration of exogenous lipid antigen PE or PC, intestinal intraepithelial γδ T cell frequency and IL-17A production were partially recovered in Abx-treated wild-type mice but not in Abx-treated CD1d-deficient mice (Figure 6g,h). Notably, consistent with the changes under hypoxic conditions, the expression of the cytokines IL-6 and IL-17A in the epithelial layer increased significantly after intraperitoneal injection of PE or PC, indicating that PE and PC can be presented by CD1d molecules to induce proinflammatory responses and aggravate intestinal damage (Figure 6i). In an in vitro coculture system, γδ T cells stimulated by CD1d tetramers loaded with PE or PC showed significant proliferation (Figure 6j).
Figure 6.
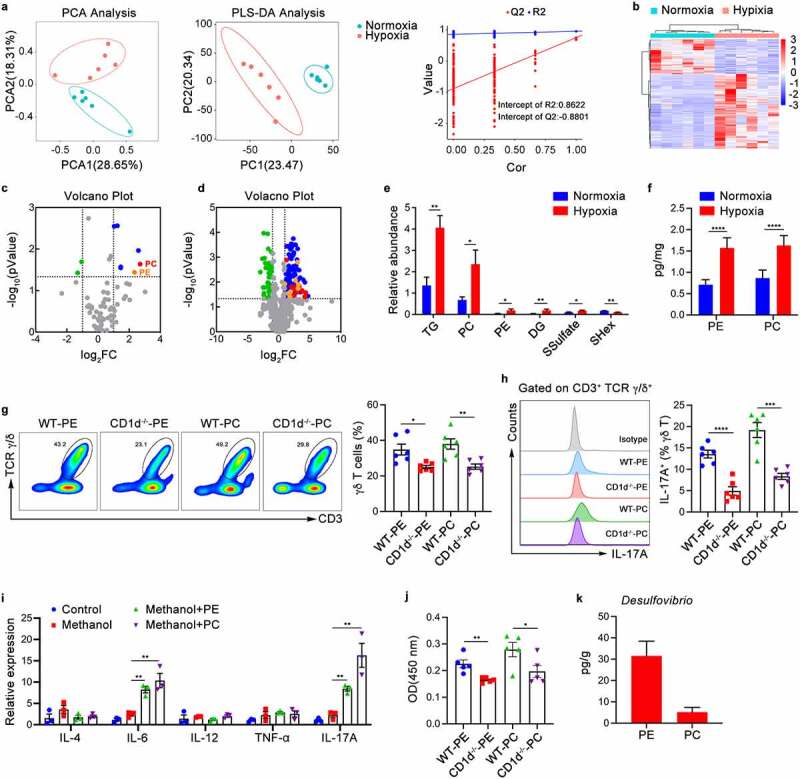
Lipid Antigens PE or PC Derived from Desulfovibrio Are Responsible for the Expansion and Activation of γδ T Cells. (a-e) Lipid metabolomics was performed on the cecal contents from SPF mice under hypoxia (5.0% oxygen concentration) or normoxia (20.9% oxygen concentration) for 48 hours (n = 5–6). (a) PCA and partial least squares (PLS) analysis illustrates the differences in lipid metabolites. (b) The relative abundance of significant lipid metabolites depicted by the heatmap. (c) Volcano plot of the total amount of differentially lipid metabolites. (d) Volcano plot of differentially lipid metabolites. (e) The relative abundance of significant lipid metabolites (top 6) in the cecal contents of normoxic and hypoxic mice was analyzed. (f) Phosphatidylethanolamine (PE) and phosphatidylcholine (PC) in the cecal contents of hypoxic and normoxic mice as examined by ELISA (n = 6). (g-i) 4Abx-treated WT or CD1d−/− mice were given hypoxia (5.0% oxygen concentration) or normoxia (20.9% oxygen concentration) for 48 hours after intraperitoneal injection of lipid antigen PE or PC. Representative pictures and quantitative data are show (n = 3–6). (g-h) The frequency of γδ T cells (g) and IL-17A production (h) were determined by flow cytometry. Representative pictures and quantitative data are show. (i) Real-time qPCR analysis of multiple cytokines gene expression in the intestinal epithelium tissues. (j) Purified γδ T cells were co-cultured with PE or PC-loaded intestinal epithelial cells (IECs) for 3 days. the proliferation of γδ T cells were determined by CCK8 assay (n = 6). (k) The PE or PC content of D. piger were examined by ELISA (n = 3). The data are representative of three independent experiments and shown by the mean ± SEM. *p <0.05, **p <0.01, ***p <0.001, ****p <0.0001 by unpaired Student’s t-test (e-h, j, k) or one-way ANOVA test with Tukey’s posttest (i). PE, Phosphatidylethanolamine; PC, phosphatidylcholine.
Next, we depicted the relationship between the gut microbiota and lipid antigens through combined 16S rRNA gene sequencing and lipid metabolomics analysis, which showed that Desulfovibrio is significantly positively correlated with the lipid antigens PE or PC (Figure S4c). To further confirm whether the increase in Desulfovibrio abundance under hypoxia provides more PE or PC for γδ T activation, we used D. piger, the most abundant Desulfovibrio species, as an example and detected the lipid metabolite content of D. piger. The ELISA results showed that D. piger contains high levels of the lipid antigens PE and PC (Figure 6k). Collectively, these data indicate that under hypoxic conditions, the increase in Desulfovibrio in the intestine provides more PE and PC that can be presented by CD1d, promoting the proliferation of γδ T cells and the production of IL-17A and thereby exacerbating hypoxia-induced intestinal damage.
Discussion
Altitude sickness is a common high-altitude clinical syndrome after exposure to low-pressure and low-oxygen environments and is especially common when individuals who typically reside in plains areas quickly enter plateau areas over 2500 meters above sea level.2 Shortness of breath and central nervous system symptoms such as headache and dizziness are the main symptoms of acute altitude sickness, which can lead to life-threatening high altitude pulmonary edema and high altitude cerebral edema if left untreated.1 However, affected individuals can also experience gastrointestinal symptoms such as nausea, vomiting and diarrhea after entering plateau areas, which may further worsen dyspnea, dizziness and headache. Although increasing evidence indicates that intestinal mucosal injury is the main cause of gastrointestinal dysfunction, the pathogenesis of gastrointestinal reactions related to high altitude is still unclear.
As mucosal tissue exposed to large amounts of microorganisms is colonized by a variety of bacterial communities under normal physiological conditions, damage to the intestinal mucosa may be related to an imbalance of the flora.33,34 The intestinal commensal microbiota is closely associated with the pathogenesis of multiple disorders, such as nervous system diseases and respiratory diseases.35,36 Previous studies have shown that Alzheimer’s disease is positively correlated with Shigella or trimethylamine oxide in the intestine.37,38 In addition, coronavirus disease 2019 is related to the impaired synthesis of bacterial metabolites such as short-chain fatty acids or L-isoleucine.39 Furthermore, the development of pulmonary edema and cerebral edema may be influenced not only directly by hypoxic stress-related reactions but also indirectly through systemic immune regulation mediated by the distal gut microbiota. Our findings demonstrate an important role of gut microorganisms in AMS through antibiotic gavage and fecal bacterial transplantation experiments. The results indicated that intestinal damage and pulmonary edema were significantly reduced in mice with antibiotic cocktail treatment before hypoxia exposure, and intestinal tight junction proteins, such as claudin-1 and ZO-1, which are markers of intestinal barrier integrity, were increased. In addition, the levels of IL-6 and IL-17A changed in line with disease severity. IL-6 can promote the differentiation of naive T cells into Th17 cells and is also considered to contribute to high-altitude pulmonary edema.40–42 Our results reveal that IL-17A is functionally important for the progression and severity of acute mountain sickness. Moreover, we demonstrated that intragastric administration of bacterial populations isolated from cecal contents of mice exposed to hypoxia for 48 hours significantly aggravated intestinal damage. Thus, these findings show a critical link between the gut microbiota and hypoxia-induced intestinal damage.
Despite increasing evidence linking the commensal microbiota to hypoxia-related diseases,43–45 the published studies have mainly focused on oxidative stress and active alveolar reabsorption.46,47 In contrast, what happens to the intestinal flora after acute hypoxia and how the intestinal flora can regulate the balance of intestinal mucosal protection and injury by regulating immune cells are still unclear. Our data support that the composition of the intestinal flora changes significantly after hypoxia, specifically showing a decrease in C. XIVa and an increase in Desulfovibrio. Overcolonization of C. XIVa in the intestine was found to be responsible for the expansion of Treg cells, which suggests that this species can inhibit inflammation and protect the integrity of the intestinal barrier.28 In contrast, Desulfovibrio have been proven to disrupt the intestinal barrier because they can reduce sulfate to produce hydrogen sulfide.29 Furthermore, our study revealed that Ang4, an antibacterial peptide produced by intestinal epithelial cells, is increased under hypoxia, inhibits C. XIVa, and affecting the growth of Desulfovibrio. The abundance of Desulfovibrio was increased, which aggravated the intestinal damage induced by hypoxia.
γδ T cells are believed to protect against the invasion of foreign pathogens and protect the integrity of the mucosal barrier under physiological conditions.48 However, many studies have also revealed that γδ T cells are responsible for driving the inflammatory response, participating in the pathogenesis of infectious diseases and tumors.15,16 Our study revealed a crucial role of small intestinal intraepithelial γδ T cells in promoting hypoxia-induced intestinal injury upon activation by intestinal commensal bacteria. We observed a significant increase in the proportion of intraepithelial γδ T cells in the hypoxic group compared with the control group, and intraepithelial γδ T cells are the main source of IL-17A due to upregulation of the nuclear transcription factor RORγt. Not only did the abundance of γδ T cells increase, but these cells also showed elevated surface expression of CD44 and CD69 in the hypoxic group compared to the control group. Interestingly, either loss of IL-17A or elimination of γδ T cells under hypoxia partially restored the integrity of the small intestinal epithelium, indicating that the mice had the ability to resist hypoxia-induced intestinal injury. Previous studies have provided strong evidence of a link between the expansion of γδ T cells and the amounts of local bacteria.22 Our data support that intestinal bacterium, especially Desulfovibrio, can stimulate the proliferation of intraepithelial γδ T cells and the production of IL-17A in an AMS model, suggesting that the intestinal microbiota may contribute to hypoxia-induced intestinal damage. Taken together, our data indicate that the amplification of γδ T cell responses through intestinal bacterial stimulation leads to the development of hypoxia-induced intestinal injury.
γδ T cell activation can be directly stimulated not only by classical antigens but also by lipid antigens presented by CD1d, a nonpolymorphic MHC class I-like molecule.19 CD1d molecules are mainly expressed on the surface of antigen-presenting cells, hepatocytes and intestinal epithelial cells.20–23 Lipid antigens derived from bacteria, such as PE, PC and PG, can be presented to γδ T cells through CD1d molecules, thereby promoting proliferation, activation and the production of cytokines such as IL-17A.22,23 Our research shows that the proportion of intraepithelial γδ T cells and IL-17A production under hypoxic conditions in the small intestine of CD1d-deficient mice are reduced compared with those in wild-type mice, with CD1d-deficient mice showing an ability to partly resist hypoxia-induced intestinal damage. A variety of cells in the intestinal epithelial layer, including epithelial cells, macrophages and dendritic cells (DCs), can express CD1d, but the most numerous cells are epithelial cells. Consistently, γδ T cells cultured in vitro cannot be activated or proliferate without intestinal epithelial cells, despite the addition of bacterial antigens. The above result indicates that CD1d expressed on small intestinal epithelial cells plays an essential role in triggering acute inflammation during hypoxia-induced intestinal injury. Nonetheless, our data show that the abundance of CD1d expressed on small intestinal epithelial cells does not change based on oxygen concentration. Therefore, nontargeted lipid metabolomics analysis was used to detect lipid antigens in feces that can be presented by CD1d to activate γδ T cells. The results indicated that lipid antigens such as PE and PC, and not CD1d, are responsible for the excessive activation of γδ T cells in the intestinal epithelial layer after hypoxia. To further determine whether the increase in Desulfovibrio abundance under hypoxia provides more PE and PC, we used D. piger, the most abundant Desulfovibrio species, as an example and detected its lipid metabolite content. The results show that Desulfovibrio contain high levels of the lipid antigens PE and PC, indicating that Desulfovibrio are an important factor that drives hypoxia-induced intestinal damage.
Our work shows that the increased Ang4 in the intestines during hypoxia inhibits the growth of C. XIVa, thereby weakening the inhibitory effect on Desulfovibrio, resulting in an increase in their abundance. Desulfovibrio can provide PE and PC, which are mainly presented by CD1d expressed on small intestinal epithelial cells. The lipid antigens generated under hypoxic conditions are presented to intraepithelial γδ T cells, prompting them to predominantly produce IL-17A and aggravating hypoxia-induced intestinal damage. Taken together, these findings suggest that disrupting the microbiota-induced activation of γδ T cells during hypoxia might be a promising method for the prevention and treatment of AMS.
Materials and methods
Mice
C57BL/6 J mice were purchased from the Shanghai SLAC Laboratory Animal Co.,Ltd; CD1d-deficient and IL-17A-deficient mice were obtained from the Jackson Laboratory. All the above mice were kept under the specific pathogen-free conditions and the study was approved by Experimental Animal Ethics Committee of the First Affiliated Hospital, Zhejiang University (No. 2021–0019-1).
Mouse treatment
six- to eight-week-old male mice were subjected to hypoxia (5.0% O2-95.0% N2) in an automatic animal hypoxic device (Maworde GC-A01, China). Then descending to a plain altitude (20.9% O2-79.1% N2) at a constant velocity after 48 hours of hypoxia. For bacterial clearance experiments, mice were intragastrically administered a mixture of ampicillin (1 g/L, Sigma, US), neomycin trisulfate (1 g/L, Sigma, US), metronidazole (1 g/L, Sigma, US), and vancomycin (500 mg/L, Sigma, US) (4Abx) for 7 days before hypoxia. For the fecal bacterial transplantation, wild-type SPF mice were gavaged with antibiotic cocktail for 7 days, followed by gut microbiota from hypoxic and normoxic mice for 7 days. For lipid antigen recovery experiments, mice were injected intraperitoneally with 20 mg PC or PE 6 times every 2 days and experiments were performed after the last injection. For bacterial suspension gavage, mice treated with antibiotics were given 1*109 cfu Clostridium bolteae or Desulfovibrio for 7 days.
Histology
Intestines and lungs were harvested and fixed in 10% neutral buffered formalin for 24 hours at room temperature. The tissue was then rinsed with 70% ethanol, graded dehydrated. Sample were immersed in paraffin, embedded, sectioned and stained with H&E.
Water content
lungs were harvested and the wet weights were measured. The whole lung was dried at 65°C for 3 days to obtain the dry weight. The whole lung water content is measured using the (wet-dry)/wet weight ratio.
Intestinal permeability
Intestinal permeability was performed using the FITC-conjugated dextran 4kDa (FD4) method. Mice were gavaged with PBS containing 600 mg/kg body weight of FITC-dextran (Sigma, US). Four hours after gavage, 100 µL serum was collected, and the fluorescence intensity was detected at an excitation wavelength of 485 nm using a multi-plate reader (SpectraMax M5/M5e, US). Serum FITC-dextran concentration was calculated from a standard curve of serially diluted FITC-dextran.
ELISA
After the bacterial solution was centrifuged, the bacterial pellet was washed 2–3 times with PBS, and the cells were lysed by sonication at 300 w, 10s/10s, and 20 minutes to obtain a suspension. cecal contents were grounded with methanol to obtain a homogenate. The PE and PC levels in the feces homogenate and bacterial suspension were generally measured using the ELISA kits according to the manufacturer’s instructions. Serum LPS levels were measured using an ELISA kit (Cusabio, China) according to the manufacturer’s instructions.
Quantitative RT-PCR
Total RNA was extracted from small intestine tissue using TRIzol reagent (Invitrogen, US) following manufacturer’s instruction. cDNA was synthesized with the HiScript II 1st Strand cDNA Synthesis Kit (Vazyme, China). Relative gene expression was analyzed according to the instructions of the SYBR Premix Ex Taq kit ChamQ Universal SYBR qPCR Master Mix (Vazyme, China). The relative expression of mRNA for each gene was normalized to that of β-actin. For bacterial identification, genomic DNA of fecal flora was isolated using the QIAamp DNA Stool Mini Kit (QIAGEN, US) and qPCR was performed; the relative abundance of each bacterium was normalized to the amounts of total bacteria (16S). All primer were synthesized by Sangon (Shanghai, China). The primers were listed in Table 1.
Table 1.
Primer sequences.
| Gene | 5’-3’ | 3’-5’ |
|---|---|---|
| Claudin-1 | TGCCCCAGTGGAAGATTTACT | CTTTGCGAAACGCAGGACAT |
| ZO-1 | GCCGCTAAGAGCACAGCAA | GCCCTCCTTTTAACACATCAGA |
| ZO-2 | ATGGGAGCAGTACACCGTGA | GCTGAACGGCAAACGAATGG |
| Bacterial 16S rRNA | AGAGTTTGATCMTGGCTCAG | CTGCTGCCTYCCGTA |
| Clostridium XIVa 16S rRNA | AAATGACGGTACCTGACTAA | CTTTGAGTTTCATTCTTGCGAA |
| Desulfovibrio16S rRNA | CCGTAGATATCTGGAGGAACATCAG | ACATCTAGCATCCATCGTTTACAGC |
| IL-17A | TCAGCGTGTCCAAACACTGAG | CGCCAAGGGAGTTAAAGACTT |
| IL-6 | CTGCAAGAGACTTCCATCCAG | AGTGGTATAGACAGGTCTGTTGG |
| IL-4 | GGTCTCAACCCCCAGCTAGT | GCCGATGATCTCTCTCAAGTGAT |
| IL-12 | CAATCACGCTACCTCCTCTTTT | CAGCAGTGCAGGAATAATGTTTC |
| TNF-α | CAGGCGGTGCCTATGTCTC | CGATCACCCCGAAGTTCAGTAG |
| Pla2 | CTATGCCTTCTATGGATGCCAC | CAGCCGTTTCTGACAGGAGT |
| Ang4 | GGTTGTGATTCCTCCAACTCTG | CTGAAGTTTTCTCCATAAGGGCT |
| Reg3b | CTCTCCTGCCTGATGCTCTT | GTAGGAGCCATAAGCCTGGG |
| Reg3g | TCAGGTGCAAGGTGAAGTTG | GGCCACTGTTACCACTGCTT |
Immunofluorescence staining
The paraffin sections of small intestine tissues were dewaxed, dehydrated with graded alcohol, and then added with 0.01 mol/L sodium citrate buffer (pH 6.0) for antigen retrieval. Add 10% BSA to the samples and block for 30 minutes at room temperature, then remove the supernatant. Primary antibodies (anti-mouse claudin-1, anti-mouse ZO-1; Abcam) at 1:50 dilution was added dropwise to the samples, incubated overnight at 4°C, and washed 3 times with PBS after returning to room temperature. Next, samples were incubated with 1:100 diluted secondary antibody (FITC-conjugated anti-rabbit; Abcam) for 30 min at room temperature and washed 3 times with PBS. Finally, incubate with 5 mL of DAPI staining solution for 10 min and washed 3 times with PBS. Stained sections were imaged using an FV3000 confocal microscope (Olympus, Japan) and analyzed using Olympus confocal software FV31S-SW.
Tunel staining
TUNEL staining was performed using TMR Tunel Cell Apoptosis Detection Kit (Servicebio, China). The intestines were washed with PBS and fixed by immersion in 10% neutral buffered formalin for 24 h. Treat with 0.1% Triton X-100 for 5 minutes, then incubate with TdT enzyme solution for 1 hour at 37°C. Tunel-positive cells were imaged using Olympus confocal software FV31S-SW.
16S sequencing analysis
16S sequencing analysis was carried out at Lc-Bio Technologies Co.,Ltd. Total RNA was isolated from cecal contents using the QIAamp DNA Stool Mini Kit (QIAGEN, US) according to the manufacturer’s instructions. The primers used were 341 F (5’-CCTACGGGNGGCWGCAG-3’) and 805 R (5’-GACTACHVGGGTATTCTAATCC-3’) for PCR amplification of the variable region of 16S rDNA (V3+ V4). Evaluation of PCR amplification products by 2% agarose gel electrophoresis and recovery of target gene fragments using AxyPrep PCR Clean-up Kit (Axygen, US) according to manufacturers’ instructions. Libraries on purified PCR products were quantified using the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, US) on the Qbit Fluorometric Quantitation System. The QIIME2 process was used to analyze alpha diversity and beta diversity and the pictures were drawn with the R (v3.5.2) package.
The antimicrobial testing assays
Recombinant Ang4 (Novoprotein, China) (5 µg/ml) was added to Reinforced Clostridium Medium (RCM) containing Clostridium bolteae (JCM 12243) and incubated at 37°C under anaerobic conditions for 6, 12, 24, 48 and 72 hours. Bacterial growth was then measured by absorbance at 600 nm using a NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific, US).
Cell preparation
Mice were euthanized, and intestines, Peyer’s patches, spleen, and thymus were collected. Single-cell suspensions were prepared as previously described with minor modifications.22 Briefly, spleens, peyer’s patches, and thymi were ground and passed through a 200-mesh steel mesh. The spleen requires an additional step of lysis of red blood cells. The small intestine was cut into small pieces with scissors, digested with Hank’s buffer containing 1 mM EDTA for 25 min at 37°C, and passed through a 200-mesh steel mesh. siIEL and IECs were obtained by gradient centrifugation with 40% and 60% percoll.
Flow cytometry
For surface staining, add an appropriate amount of fluorescently labeled primary antibody to the cell suspension after blocking and incubate at 4°C for 30 minutes. For intracellular staining, cells were stimulated for 5 hours with 50 ng ml−1 PMA (Sigma, US), 1 mg ml−1 ionomycin (Sigma, US), and 10 mg ml−1 monensin (BD Biosciences, US), followed by surface staining. Cells were then fixed and permeabilized according to the instructions of BD Cytofix/Cytoperm Fixation/Permeabilization Solution kit (BD Biosciences, US), and finally incubated with an appropriate amount of fluorescently labeled intracellular antibodies at 4°C for 30 minutes. All samples were collected on a FACSCanto™ II flow cytometer (BD Biosciences, US) and analyzed using FlowJo software (Tree Star). Transcription factor staining was performed based on the True-Nuclear™ Transcription Factor Buffer Set (Biolegend, US). The antibodies were listed in Table 2.
Table 2.
Antibodies details.
| Antibodies | Source | Identifier |
|---|---|---|
| Αnti-mouse CD45-PerCP | ΒioLegend | Clone: 30-F11; Cat#:103130; RRID:AB_893339 |
| Αnti-mouse CD3-PE/Cyanine7 | ΒioLegend | Clone: 17A2; Cat#:100220; RRID:AB_1732057 |
| Αnti-mouse CD3-FITC | ΒioLegend | Clone: 17A2; Cat#:100204; RRID:AB_312661 |
| Αnti-mouse TCR γ/δ-APC | ΒioLegend | Clone: GL3; Cat#:118116; RRID:AB_1731813 |
| Αnti-mouse TCR γ/δ-PE | ΒioLegend | Clone: UC7-13D5; Cat#:107508; RRID:AB_345266 |
| Αnti-mouse TCR γ/δ-AF488 | ΒioLegend | Clone: GL3; Cat#:118128; RRID:AB_2562771 |
| Αnti-mouse NK1.1-APC | ΒioLegend | Clone: PK136; Cat#:108710; RRID:AB_313397 |
| Αnti-mouse CD4-PE | ΒioLegend | Clone: GK1.5; Cat#:100408; RRID:AB_312693 |
| Αnti-mouse CD4-FITC | ΒioLegend | Clone: GK1.5; Cat#:100406; RRID:AB_312691 |
| Αnti-mouse CD8-Pacific Blue | ΒioLegend | Clone: 53–6.7; Cat#:100725; RRID:AB_493425 |
| Αnti-mouse CD8-FITC | ΒioLegend | Clone: 53–6.7; Cat#:100706; RRID:AB_312745 |
| Αnti-mouse CD25-APC | ΒioLegend | Clone: 3C7; Cat#:101910; RRID:AB_2280288 |
| Αnti-mouse IL-17A-PE | ΒioLegend | Clone: TC11-18H10.1; Cat#:506904; RRID:AB_315464 |
| Αnti-mouse CD44-FITC | BD PharMingen | Clone: IM7; Cat#:553133; RRID:AB_2076224 |
| Αnti-mouse CD69-PE | ΒioLegend | Clone: H1.2F3; Cat#:104508; RRID:AB_313111 |
| Αnti-mouse IFN-γ-PE | ΒioLegend | Clone: XMG1.2; Cat#:505810; RRID:AB_315404 |
| Αnti-mouse RORγt-BV421 | BD Horizon | Clone: Q31-378; Cat#:562894; RRID:AB_2687545 |
| Αnti-mouse T-bet-PE | ΒioLegend | Clone: 4B10; Cat#:644810; RRID:AB_2200542 |
| Αnti-mouse FoxP3-PE | ΒioLegend | Clone: MF-14; Cat#:126404; RRID:AB_1089117 |
| Αnti-mouse CD1d-PE | ΒioLegend | Clone: 1B1; Cat#:123509; RRID:AB_1236547 |
| PE Mouse IgG1, κ Isotype Control | BD PharMingen | Clone: MOPC-21; Cat#:556650; RRID:AB_396514 |
| BV421 Mouse IgG2a, κ Isotype Control | BD Horizon | Clone: MOPC-173; Cat#:563464; RRID:AB_2869495 |
| PE Rat IgG1, κ Isotype Control | BD PharMingen | Clone: R3-34; Cat#:553925; RRID:AB_395140 |
In vitro bacterial stimulation in vitro
Mice were euthanized and intestinal intraepithelial lymphocytes were harvested. After the cells were plated in 12-well plates, inactivated bacteria were added to stimulate for 72 hours.
Antibody neutralization
For antibody-mediated depletion experiments, animals are injected intraperitoneally (i.p.) every 2–3 days. Inject monoclonal antibody (200 mg/mouse) against γδ-TCR (UC7-13D5, BioXCell, China) and the same concentration of monoclonal rat IgG as the control (BioXCell, China).
Metabolite extraction and MS analysis
Untargeted lipid metabolomes detected in cecal contents was carried out at Lc-Bio Technologies Co.,Ltd. Metabolite-containing supernatant was obtained after centrifugation at 4,000 g for 20 min from 20 µL of feces in 50% methanol. Mix 10 µL of each sample’s extract as a quality control sample. The obtained sample was detected by LC-MS system according to the machine instructions, and lipid metabolites in mouse feces were measured using high-resolution tandem mass spectrometer TripleTOF5600plus (SCIEX, UK).
In vitro co-culture
The small intestinal epithelial cells (5 × 105 per well) were plated overnight in 12-well plates and the lipid antigens PE or PC (2.5 μg ml−1) were added to culture for 24 hours.22 Then, IEC cells are irradiated and co-cultured with sorted γδ T cells (1 × 105 per well) for 3 days.
In vitro cell proliferation
The Cell Counting Kit-8 (CCK-8) assay was performed to measure the proliferation of γδ T cells stimulated by CD1d-antigen in vitro. Add 10 µL of CCK8 solution to the cells to be assayed in a 96-well plate and continue to incubate at 37°C for 30 minutes. Absorbance at 450 nm was detected with a microplate reader.
Statistics
Data are presented as mean ± SEM and GraphPad Prism (GraphPad Software) was used to analyze the data of each group. The values of each group were tested by Shapira-Wilkinson normal distribution, P >0.05. Student’s t-test, one-way ANOVA, and linear regression analysis were used to determine statistically significant differences. P <0.05 was considered significant. *P <0.05, **P <0.01, ***P <0.001, ****P <0.0001, ns represents not significant.
Supplementary Material
Acknowledgments
Not applicable.
Funding Statement
This work was supported by National Key Research and Development Program of China (2021YFA1301100, 2018YFC2000500, 2021YFA1301101), the Key Research & Development Plan of Zhejiang Province (2019C04005), Research Project of Jinan Microecological Biomedicine Shandong Laboratory (JNL2022012B), Fundamental Research Funds for the Central Universities (2022ZFJH003).
Disclosure statement
The authors report no conflict of interest.
Author details
Yuyu Li, Doc., Ph.D., No. 79 Qingchun Road, Shangcheng District, Hangzhou City, China, Email: liyuyu2019@zju.edu.cn, Tel: +86 13868083426
Yuchong Wang, Doc., Ph.D., No. 79 Qingchun Road, Shangcheng District, Hangzhou City, China, Email: 21918243@zju.edu.cn, Tel: +86 17816860886
Fan Shi, Mr, M.Sc., No. 79 Qingchun Road, Shangcheng District, Hangzhou City, China, Email: 22018103@zju.edu.cn, Tel: +86 15267752807
Xujun Zhang, Doc., Ph.D., No. 79 Qingchun Road, Shangcheng District, Hangzhou City, China, Email: 290516@zju.edu.cn, Tel: +86 15167118621
Yongting Zhang, Doc., Ph.D., No. 79 Qingchun Road, Shangcheng District, Hangzhou City, China, Email: 11918232@zju.edu.cn, Tel: +86 15250959699
Kefan Bi, Doc., Ph.D., No. 79 Qingchun Road, Shangcheng District, Hangzhou City, China, Email: bdgxbkf@zju.edu.cn, Tel: +86 13806529347
Xuequn Chen, Prof., Ph.D., No. 866 Yuhangtang Road, Xihu District, Hangzhou City, China, Email: chewyg@zju.edu.cn, Tel: +86 (0571)-88208182
Lanjuan Li, Prof., MD; Ph.D., No. 79 Qingchun Road, Shangcheng District, Hangzhou City, China, Email: ljli@zju.edu.cn, Tel: +86 (0571)-87236458
Hongyan Diao, Prof., MD; Ph.D., No. 79 Qingchun Road, Shangcheng District, Hangzhou City, China, Email: diaohy@zju.edu.cn, Tel: +86 18658160678
Author contributions
H. Diao and L.L. designed and supervised the study. Y.Li, Y.W., X.Z., Y.Z. and K.B. performed the experiments and data analysis. Y.Li, H.D., L.L., and X.C. contributed to writing and discussing the manuscript.
Data availability statement
The data that support the findings of this study are openly available in NCBI, GEO: GSE199645 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE199645).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2022.2096994.
References
- 1.Wilson R. Acute high-altitude illness in mountaineers and problems of rescue. Ann Intern Med. 1973;78(3):421–20. doi: 10.7326/0003-4819-78-3-421. [DOI] [PubMed] [Google Scholar]
- 2.Basnyat B, Murdoch DR. High-altitude illness. Lancet. 2003;361(9373):1967–1974. doi: 10.1016/S0140-6736(03)13591-X. [DOI] [PubMed] [Google Scholar]
- 3.Williamson J, Oakeshott P, Dallimore J. Altitude sickness and Acetazolamide. BMJ. 2018;361:k2153. doi: 10.1136/bmj.k2153. [DOI] [PubMed] [Google Scholar]
- 4.Davis C, Hackett P. Advances in the prevention and treatment of high altitude illness. Emerg Med Clin North Am. 2017;35:241–260. doi: 10.1016/j.emc.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Simancas-Racines D, Arevalo-Rodriguez I, Osorio D, Franco JV, Xu Y, Hidalgo R. Interventions for treating acute high altitude illness. Cochrane Database Syst Rev. 2018;6:CD009567. doi: 10.1002/14651858.CD009567.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meier D, Collet TH, Locatelli I, Cornuz J, Kayser B, Simel DL, Sartori C. Does this patient have acute mountain sickness?: the rational clinical examination systematic review. JAMA. 2017;318:1810–1819. doi: 10.1001/jama.2017.16192. [DOI] [PubMed] [Google Scholar]
- 7.Roach RC, Hackett PH, Oelz O, Bartsch P, Luks AM, MacInnis MJ, Baillie JK. The 2018 lake louise acute mountain sickness score. High Alt Med Biol. 2018;19:4–6. doi: 10.1089/ham.2017.0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. 2012;489:231–241. doi: 10.1038/nature11551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma X, Zhou Z, Zhang X, Fan M, Hong Y, Feng Y, Dong Q, Diao H, Wang G. Sodium butyrate modulates gut microbiota and immune response in colorectal cancer liver metastatic mice. Cell Biol Toxicol. 2020;36(5):509–515. doi: 10.1007/s10565-020-09518-4. [DOI] [PubMed] [Google Scholar]
- 11.Hou X, Yang Y, Chen J, Jia H, Zeng P, Lv L, Lu Y, Liu X, Diao H. Tcrβ repertoire of memory t cell reveals potential role for Escherichia coli in the pathogenesis of primary biliary cholangitis. Liver Int. 2019;39:956–966. doi: 10.1111/liv.14066. [DOI] [PubMed] [Google Scholar]
- 12.Chen J, Wei Y, He J, Cui G, Zhu Y, Lu C, Ding Y, Xue R, Bai L, Uede T, et al. Natural killer t cells play a necessary role in modulating of immune-mediated liver injury by gut microbiota. Sci Rep. 2014;4:7259. doi: 10.1038/srep07259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu LL, Ma ZJ, Ren M, Wei YM, Liao YH, Shen YL, Fan SM, Li L, Wu QX, Gao ZS, et al. Distinct features of gut microbiota in high-altitude Tibetan and middle-altitude han hypertensive patients. Cardiol Res Pract. 2020;2020:1957843. doi: 10.1155/2020/1957843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nielsen MM, Witherden DA, Havran WL. γδ t cells in homeostasis and host defence of epithelial barrier tissues. Nat Rev Immunol. 2017;17(12):733–745. doi: 10.1038/nri.2017.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, Ameh S, Sandel D, Liang XS, Mazzilli S, et al. Commensal microbiota promote lung cancer development via γδ t cells. Cell. 2019;176(5):998–1013 e1016. doi: 10.1016/j.cell.2018.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suhail A, Rizvi ZA, Mujagond P, Ali SA, Gaur P, Singh M, Ahuja V, Awasthi A, Srikanth CV. Desumoylase senp7-mediated epithelial signaling triggers intestinal inflammation via expansion of gamma-delta t cells. Cell Rep. 2019;29(11):3522–3538 e3527. doi: 10.1016/j.celrep.2019.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonneville M, O’Brien RL, Born WK. γδ t cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol. 2010;10(7):467–478. doi: 10.1038/nri2781. [DOI] [PubMed] [Google Scholar]
- 18.Park SG, Mathur R, Long M, Hosh N, Hao L, Hayden MS, Ghosh S. T regulatory cells maintain intestinal homeostasis by suppressing γδ t cells. Immunity. 2010;33(5):791–803. doi: 10.1016/j.immuni.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luoma AM, Castro CD, Adams EJ. γδ t cell surveillance via cd1 molecules. Trends Immunol. 2014;35(12):613–621. doi: 10.1016/j.it.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colgan SP, Hershberg RM, Furuta GT, Blumberg RS. Ligation of intestinal epithelial cd1d induces bioactive il-10: critical role of the cytoplasmic tail in autocrine signaling. Proc Natl Acad Sci U S A. 1999;96(24):13938–13943. doi: 10.1073/pnas.96.24.13938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dieude M, Striegl H, Tyznik AJ, Wang J, Behar SM, Piccirillo CA, Levine JS, Zajonc DM, Rauch J. Cardiolipin binds to cd1d and stimulates cd1d-restricted γδ t cells in the normal murine repertoire. J Immunol. 2011;186(8):4771–4781. doi: 10.4049/jimmunol.1000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li F, Hao X, Chen Y, Bai L, Gao X, Lian Z, Wei H, Sun R, Tian Z. The microbiota maintain homeostasis of liver-resident γδ t-17 cells in a lipid antigen/cd1d-dependent manner. Nat Commun. 2017;7:13839. doi: 10.1038/ncomms13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russano AM, Bassotti G, Agea E, Bistoni O, Mazzocchi A, Morelli A, Porcelli SA, Spinozzi F. Cd1-restricted recognition of exogenous and self-lipid antigens by duodenal γδ+ t lymphocytes. J Immunol. 2007;178:3620–3626. doi: 10.4049/jimmunol.178.6.3620. [DOI] [PubMed] [Google Scholar]
- 24.Bai L, Picard D, Anderson B, Chaudhary V, Luoma A, Jabri B, Adams EJ, Savage PB, Bendelac A. The majority of cd1d-sulfatide-specific t cells in human blood use a semiinvariant vdelta1 tcr. Eur J Immunol. 2012;42:2505–2510. doi: 10.1002/eji.201242531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeissig S, Murata K, Sweet L, Publicover J, Hu Z, Kaser A, Bosse E, Iqbal J, Hussain MM, Balschun K, et al. Hepatitis b virus-induced lipid alterations contribute to natural killer t cell-dependent protective immunity. Nat Med. 2012;18:1060–1068. doi: 10.1038/nm.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yanagisawa K, Yue S, van der Vliet HJ, Wang R, Alatrakchi N, Golden-Mason L, Schuppan D, Koziel MJ, Rosen HR, Exley MA. Ex vivo analysis of resident hepatic pro-inflammatory cd1d-reactive t cells and hepatocyte surface cd1d expression in hepatitis c. J Viral Hepat. 2013;20:556–565. doi: 10.1111/jvh.12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diao H, Kon S, Iwabuchi K, Kimura C, Morimoto J, Ito D, Segawa T, Maeda M, Hamuro J, Nakayama T, et al. Osteopontin as a mediator of nkt cell function in t cell-mediated liver diseases. Immunity. 2004;21:539–550. doi: 10.1016/j.immuni.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 28.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. Treg induction by a rationally selected mixture of clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 29.Kushkevych I, Dordevic D, Vitezova M. Possible synergy effect of hydrogen sulfide and acetate produced by sulfate-reducing bacteria on inflammatory bowel disease development. J Adv Res. 2021;27:71–78. doi: 10.1016/j.jare.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mergaert P, Kikuchi Y, Shigenobu S, Nowack ECM. Metabolic integration of bacterial endosymbionts through antimicrobial peptides. Trends Microbiol. 2017;25:703–712. doi: 10.1016/j.tim.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 31.Chung LK. Pros: antimicrobial defense in the gastrointestinal tract. Semin Cell Dev Biol. 2019;88:129–137. doi: 10.1016/j.semcdb.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGeachy MJ, Cua DJ, Gaffen SL. The il-17 family of cytokines in health and disease. Immunity. 2019;50:892–906. doi: 10.1016/j.immuni.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wells JM, Rossi O, Meijerink M, Baarlen P. Epithelial crosstalk at the microbiota-mucosal interface. Proc Natl Acad Sci U S A. 2011;108:4607–4614. doi: 10.1073/pnas.1000092107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bi K, Zhang X, Chen W, Diao H. MicroRNAs regulate intestinal immunity and gut microbiota for gastrointestinal health: a comprehensive review. Genes (Basel). 2020;11:1075. doi: 10.3390/genes11091075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren Z, Wang H, Cui G, Lu H, Wang L, Luo H, Chen X, Ren H, Sun R, Liu W, et al. Alterations in the human oral and gut microbiomes and lipidomics in covid-19. Gut. 2021;70:1253–1265. doi: 10.1136/gutjnl-2020-323826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gu S, Chen Y, Wu Z, Chen Y, Gao H, Lv L, Guo F, Zhang X, Luo R, Huang C, et al. Alterations of the gut microbiota in patients with coronavirus disease 2019 or h1n1 influenza. Clin Infect Dis. 2020;71:2669–2678. doi: 10.1093/cid/ciaa709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao Q, Wang Y, Wang X, Fu S, Zhang X, Wang RT, Zhang X. Decreased levels of circulating trimethylamine n-oxide alleviate cognitive and pathological deterioration in transgenic mice: a potential therapeutic approach for Alzheimer’s disease. Aging (Albany NY). 2019;11:8642–8663. doi: 10.18632/aging.102352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mancuso C, Santangelo R. Alzheimer’s disease and gut microbiota modifications: the long way between preclinical studies and clinical evidence. Pharmacol Res. 2018;129:329–336. doi: 10.1016/j.phrs.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 39.Zhang F, Wan Y, Zuo T, Yeoh YK, Liu Q, Zhang L, Zhan H, Lu W, Xu W, Lui GCY, et al. Prolonged impairment of short-chain fatty acid and l-isoleucine biosynthesis in gut microbiome in patients with covid-19. Gastroenterology. 2021. doi: 10.1053/j.gastro.2021.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kimura A, Kishimoto T. Il-6: regulator of treg/th17 balance. Eur J Immunol. 2010;40:1830–1835. doi: 10.1002/eji.201040391. [DOI] [PubMed] [Google Scholar]
- 41.Lee JY, Hall JA, Kroehling L, Wu L, Najar T, Nguyen HH, Lin WY, Yeung ST, Silva HM, Li D, et al. Serum amyloid a proteins induce pathogenic th17 cells and promote inflammatory disease. Cell. 2020;183:2036–2039. doi: 10.1016/j.cell.2020.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mazzeo RS. Altitude, exercise and immune function. Exerc Immunol Rev. 2005;11:6–16. [PubMed] [Google Scholar]
- 43.Olson CA, Iniguez AJ, Yang GE, Fang P, Pronovost GN, Jameson KG, Rendon TK, Paramo J, Barlow JT, Ismagilov RF, et al. Alterations in the gut microbiota contribute to cognitive impairment induced by the ketogenic diet and hypoxia. Cell Host Microbe. 2021;29:1378–1392 e1376. doi: 10.1016/j.chom.2021.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khalyfa A, Ericsson A, Qiao Z, Almendros I, Farre R, Gozal D. Circulating exosomes and gut microbiome induced insulin resistance in mice exposed to intermittent hypoxia: effects of physical activity. EBioMedicine. 2021;64:103208. doi: 10.1016/j.ebiom.2021.103208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lucking EF, O’Connor KM, Strain CR, Fouhy F, Bastiaanssen TFS, Burns DP, Golubeva AV, Stanton C, Clarke G, Cryan JF, et al. Chronic intermittent hypoxia disrupts cardiorespiratory homeostasis and gut microbiota composition in adult male Guinea-pigs. EBioMedicine. 2018;38:191–205. doi: 10.1016/j.ebiom.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baloglu E, Nonnenmacher G, Seleninova A, Berg L, Velineni K, Ermis-Kaya E, Mairbaurl H. The role of hypoxia-induced modulation of alveolar epithelial na(+)- transport in hypoxemia at high altitude. Pulm Circ. 2020;10:50–58. doi: 10.1177/2045894020936662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaur P, Prasad S, Kumar B, Sharma SK, Vats P. High-altitude hypoxia induced reactive oxygen species generation, signaling, and mitigation approaches. Int J Biometeorol. 2021;65:601–615. doi: 10.1007/s00484-020-02037-1. [DOI] [PubMed] [Google Scholar]
- 48.Wang X, Lin X, Zheng Z, Lu B, Wang J, Tan AH, Zhao M, Loh JT, Ng SW, Chen Q, et al. Host-derived lipids orchestrate pulmonary γδ t cell response to provide early protection against influenza virus infection. Nat Commun. 2021;12:1914. doi: 10.1038/s41467-021-22242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are openly available in NCBI, GEO: GSE199645 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE199645).