

ABSTRACT
Disruption of histone acetylation-mediated gene control is a critical step in Alzheimer’s Disease (AD), yet chromatin analysis of antagonistic histone acetyltransferases (HATs) and histone deacetylases (HDACs) causing these alterations remains uncharacterized. We report the first Tip60 HAT versus HDAC2 chromatin (ChIP-seq) and transcriptional (RNA-seq) profiling study in Drosophila melanogaster brains that model early human AD. We find Tip60 and HDAC2 predominantly recruited to identical neuronal genes. Moreover, AD brains exhibit robust genome-wide early alterations that include enhanced HDAC2 and reduced Tip60 binding and transcriptional dysregulation. Orthologous human genes to co-Tip60/HDAC2 D. melanogaster neural targets exhibit conserved disruption patterns in AD patient hippocampi. Notably, we discovered distinct transcription factor binding sites close or within Tip60/HDAC2 co-peaks in neuronal genes, implicating them in coenzyme recruitment. Increased Tip60 protects against transcriptional dysregulation and enhanced HDAC2 enrichment genome-wide. We advocate Tip60 HAT/HDAC2 mediated epigenetic neuronal gene disruption as a genome-wide initial causal event in AD.
KEYWORDS: Amyloid precursor protein, sequencing, histone acetylation, Alzheimer’s disease, KAT5
Introduction
Alzheimer’s Disease (AD) is a chronic neurodegenerative disorder affecting the elderly and is the most common cause of dementia. The disease is hallmarked by amyloid-β (Aβ) plaque accumulation, Tau-mediated neurofibrillary tangles, and neuronal cell death in the brain that is accompanied by debilitating cognitive deficits in AD patients that worsen as they age. The severity and speed of AD progression are dependent upon complex interactions between genetics, age, and environmental factors [1,2,3], all of which are orchestrated, at least in part, by epigenetic histone acetylation mediated gene control mechanisms. Indeed, decreased chromatin histone acetylation levels have been reported in the brains of animal models and human patients that have multiple types of neurodegenerative diseases that include AD [4,5]. These alterations have been shown to cause an epigenetic blockade of neuroplasticity gene transcription that contributes to cognitive impairment [6,7]. More recently, a compelling study using the brains of AD patients reported an age-associated genome-wide reduction of the histone acetylation H4K16 modification that is proposed to contribute to epigenetic gene alteration mediated neurodegeneration [8]. Despite these informative findings, to date, all AD-associated genome-wide epigenetic studies are limited to examining chromatin histone acetylation patterns and alterations already generated. Thus, little is known about the genome-wide distribution of the antagonizing histone acetyltransferase (HAT) and histone deacetylase (HDAC) enzymes that act to modify the neural epigenome by generating and erasing specific cognition-linked acetylation marks, respectively, and thus serve as the causative agents of memory-impairing histone acetylation alterations in AD.
Appropriate histone acetylation homoeostasis in the brain is maintained by HATs and HDACs that, in general, activate and repress neural gene expression profiles, respectively. Disruption of this finely tuned HAT and HDAC epigenetic balance causes transcriptional dysregulation that is a key step in AD aetiology [1,4,7,9]. In support of this concept, we and others have reported reduced HAT Tip60 (KAT5) [6] and enhanced HDAC2 [7] recruitment to a set of critical neuroplasticity genes in AD animal models and human patients that causes reduced histone acetylation at these gene loci with concomitant transcriptional repression. Nevertheless, whether similar alterations of Tip60 and HDAC2 chromatin distribution with concomitant transcriptional dysregulation are a genome-wide phenomenon that occurs as an early initial event in AD progression remains unknown.
Here we report the first genome-wide study profiling Tip60 HAT versus HDAC2 chromatin distribution and transcriptional dynamics in the brains of amyloid precursor protein (APP) Drosophila melanogaster larvae that effectively model early human AD neurodegeneration both epigenetically and pathologically. We find that Tip60 and HDAC2 are predominantly recruited to identical neuronal genes with enrichment peaking across entire gene bodies. Astoundingly, prior to Aβ deposition, AD larval brains exhibited robust genome-wide binding disruptions: enhanced HDAC2 and reduced Tip60 binding with concomitant transcriptional dysregulation. Orthologous human genes to co-Tip60/HDAC2 AD-associated neural targets identified in D. melanogaster exhibit conserved disruption patterns in the human AD hippocampus. Notably, we discovered eight transcription factors (TFs) binding close or within Tip60/HDAC2 co-peaks in neuronal genes, implicating them in coenzyme recruitment to these loci. Strikingly, increased Tip60 protects against transcriptional dysregulation and enhanced HDAC2 enrichment genome-wide. Based on these results, we advocate that Tip60 HAT/HDAC2 mediated epigenetic transcriptional dysregulation is a genome-wide initial causal event in the AD brain that can be reversed by restoring Tip60/HDAC2 balance.
Results
Tip60 protects against early and late transcriptome-wide alterations in the AD-associated neurodegenerative brain
Mild cognitive impairment (MCI) is a debilitating hallmark during the early pre-clinical stages of AD, yet the molecular events that trigger these impairments are unclear. We and others have shown that such preclinical AD pathologies in humans are conserved in the well-characterized AD-associated human APP D. melanogaster model that inducible and pan-neuronally express human APP [6,10]. Third-instar larvae that model early staged APP-induced neurodegeneration show deficits in cognitive ability and synaptic plasticity, axonal transport and outgrowth, and apoptotic neuronal cell death in the brain [6,11,12]. APP flies also display Aβ plaque accumulation in the aged adult fly eye via human conserved endogenous gamma [10] and beta-secretase cleavage pathways [13]. Thus, we first asked whether APP flies also display Aβ deposition in the fly brain and whether its deposition is associated with the early pre-clinical AD defects modelled during larval stages. We focused our studies on the mushroom body (MB) Kenyon cell region as we have shown that Tip60, robustly produced in the MB, is required for MB role in learning and memory and that MB morphology is disrupted in the aged seven-day-old APP fly brain [14]. Anti-Aβ immunofluorescence studies [15,16] revealed that APP expression in the D. melanogaster brain results in diffuse Aβ deposits that appear in the MB of seven-day-old flies (Figure 1(aii and bii)). These Aβ deposits are unobservable in an earlier AD stage modelled in third-instar larvae (Figure 1(ai and bi)). These results suggest that molecular mechanisms distinct from Aβ deposition trigger early AD pre-clinical impairments.
Figure 1.
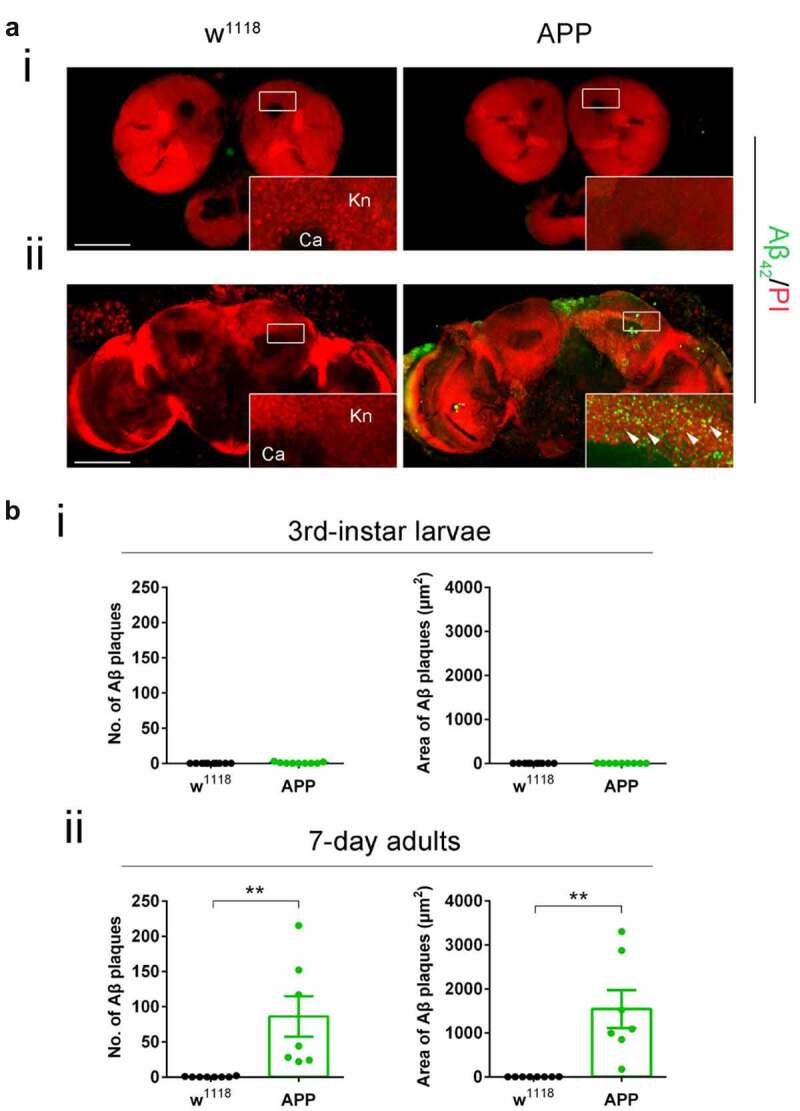
Diffuse amyloid deposits are abundant in the mushroom body (MB) in seven-day-old APP adults but not in third-instar APP larvae. (a) Representative images. Aβ deposits were stained with anti-Aβ42 antibody (green). Nuclei were stained with PI (red). The Kenyon (Kn) cell region (boxed) was zoomed in to display Kn cells and Aβ deposits. (i) Immunostaining of brains of third-instar larvae shows a negligible Aβ42 signal in APP flies compared to no Aβ42 signal in w1118 flies. (ii) Immunostaining of brains of seven-day-old adults shows evident Aβ deposits in APP flies compared to w1118 flies. Arrowheads indicate Aβ deposits. No Aβ42 signal was detected in the Calyx (Ca) region. Scale bar represents 100 μm. (b) Aβ deposits were quantified by both number and size. (i) Quantification of Aβ deposit numbers and areas in the third-instar larval brain Kn region. n = 9 ~ 10. (ii) Quantification of Aβ deposit numbers and area in the seven-day-old adult brain Kn region. n = 8 ~ 9. **p < 0.01; unpaired student’s t-test. All data are shown as mean ± SEM.
Gene expression [17,18] and genetic variation [3,19] studies in AD patients and animal models indicate that alteration in gene control contributes to disease pathology. Nevertheless, whether genome-wide gene expression alterations trigger MCI before Aβ deposition remains to be further elucidated as gene studies predominantly rely on aged AD brain samples. To address this question, we profiled genome-wide transcriptional changes during early neurodegeneration stages modelled in APP larval brains and later stages modelled in the aged seven-day-old APP fly heads. As we previously identified disruption of Tip60/HDAC2 mediated neuronal gene control as a potential early mechanism underlying neuronal deficits in APP flies [6], we also asked whether increasing Tip60 HAT activity would protect against potential genome-wide early and late-stage gene alterations.
For transcriptome analysis, RNA was isolated from the brains of staged third-instar larvae and from the heads of seven-day-old flies that were w1118 control flies or flies expressing either APP or APP; Tip60 under the control of the pan-neuronal elav-GAL4 driver. We used RNA-seq to quantify gene expression changes. Principal component analysis (PCA) analysis (S1A and S1B Figs) and hierarchically clustered heatmaps (Figure S1C and S1D) show homogeneity within replicates and variability between groups. Importantly, with increased Tip60 expression, as development proceeds from the third-instar larval stage (Figure S1A and S1C) to the seven-day-old adult stage (Figure S1B and S1D), the APP; Tip60 transcriptome becomes more similar to the w1118 transcriptome than to the APP transcriptome (Figure S1B, S1D and S1I). Further, tissue enrichment with the human orthologs of the top 2000 APP-induced gene alterations underscores the neural specificity in gene expression defects (Figure S1E and S1F). In adult fly heads that also exhibit Aβ deposits in the brain, we identified significant alterations in gene expression (APP vs. w1118: 1493 up/1641 down). Surprisingly, in the absence of Aβ deposits in the early APP larval stage of the brain, we observed greater changes in gene expression (APP vs. w1118: 1750 up/2261 down) when compared with adult APP fly heads (Figure 2(a) and S1 Table: S1-1 and S1-3). Consistent with our prior findings demonstrating Tip60 protection against AD defects modelled in APP flies, increased Tip60 expression led to notable protection in gene expression changes in both APP larval (APP; Tip60 vs. APP: 311 up/338 down) and adult heads (APP; Tip60 vs. APP: 1023 up/1280 down) (Figure 2(a) and S1 Table: S1-2 and S1-4). We next analysed the data with the goal of identifying Tip60 rescued genes and associated biological processes specifically reprogrammed by Tip60. To this end, we analysed the distribution and intersection between down- and up-regulated genes in APP vs. w1118 and APP; Tip60 vs. APP in larval (Figure 2(b)) and adult (Figure 2(c)) stages. In the APP larval brain, approximately 11% (458/4011) of gene changes (APP vs. w1118) are specifically protected against by increased Tip60 and are referred to here as ‘Tip60 reprogrammed genes.’ In APP adult heads, approximately 60% (1898/3134) of the APP-induced genes (APP vs. w1118) were identified as Tip60 reprogrammed genes. Thus, the number of Tip60 rescued genes is significantly greater in the adult stage than in the larval stage. Gene ontology (GO) analysis revealed that among the top 25 biological processes associated with the Tip60 reprogrammed genes identified in adult flies, axon and dendrite-related pathways were enriched (Figure 3(d) and S2 Table: S2-3), while cell-cycle regulation processes and RNA metabolic processes were enriched in the larval stage (Figure 3(a) and S2 Table: S2-2). Lipid metabolic pathways were enriched for Tip60 rescued genes in both adult and larval stages (Figure 3(b,c) and S2 Table: S2-4 and S2-1). In summary, our transcriptomic analysis reveals that Tip60 protects against genome-wide gene expression alterations important for neuronal function during early and late-stage AD-associated neurodegeneration with enhanced protection during later stages.
Figure 2.
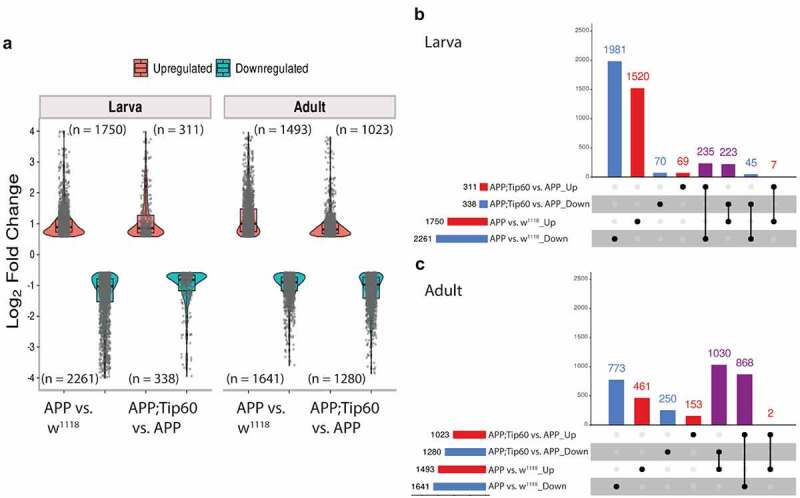
Tip60 protects against early (third-instar larval) and late (seven-day-old adult) transcriptomic deregulation in the APP AD-associated neurodegenerative brain. (a) Log2 fold changes of differentially expressed genes (padj ≤0.05 and log2FoldChange of ≤-0.583 and ≥0.583) determined by RNA-seq in the third-instar larval and adult heads in APP vs. w1118 and APP;Tip60 vs. APP. Changes were prominent in both third-instar larval and adult APP heads, while Tip60-induced changes initiated in the third-instar larval head and were prominent in the adult head: indicating the effect of Tip60 over time. (b and c) The upSet plot represents the distribution and intersection of down and up-regulated genes between APP vs. w1118 and APP;Tip60 vs. APP in third-instar larval (b) and adult (c) heads. Rows represent the number of genes in each comparison (APP vs. w1118 and APP;Tip60 vs. APP), and columns represent the number of genes per interaction. The red and blue bars represent the up and down-regulated genes, respectively. The black filled dots indicate the association between rows. The red and blue columns represent genes uniquely up-regulated and down-regulated genes, respectively, in given comparisons, while the purple columns represent Tip60 reprogrammed genes.
Figure 3.

Heatmaps depicting the relative expression pattern of genes misregulated in APP larval and adult heads and are rescued by Tip60. Representation of genes from the most representative biological processes in the top 25 pathways enriched from the rescue gene list. (a) Heatmap of genes representing the cell-cycle regulation processes and RNA metabolic processes in the third instar larval head. Heatmap of genes representing the lipid metabolic pathways in the (b) third-instar larval head and (c) the adult head. (d) Heatmap of genes representing the axon and dendrite related pathways in the adult head. Log-transformed gene expression values are displayed as colours ranging from red to blue, as shown in the key. Red represents an increase in gene expression, while blue represents a decrease in expression.
Increased Tip60 protects against enhanced repressor HDAC2 recruitment along the neuronal gene bodies during early AD neurodegeneration
We previously demonstrated a disruption in Tip60/HDAC2 balance in the larval APP brain exhibited by reduced Tip60 and increased HDAC2 protein levels that is alleviated by increasing Tip60 expression [6]. Consistent with these findings, our RNA-seq read counts here revealed reduced Tip60 and increased HDAC2 transcriptional levels in both larva and adult APP vs. w1118 comparison while additional Tip60 expression resulted in a decrease in HDAC2 levels in both larval and adult APP; Tip60 vs. APP comparison (Figure S1G). Thus, to further elucidate the role of Tip60 and HDAC2 in the early transcriptional dysregulation we observed in the larval brain prior to Aβ deposition, we profiled genome-wide enrichment of Tip60 and HDAC2 by ChIP-seq in larval heads obtained from w1118, APP, or APP; Tip60 genotypes (S3 Table). The peaks identified by ChIP-seq (Figure S2A and S2D) were first annotated to gain insight into their distribution over the genome (Figure S2B and S2E). Interestingly, approximately 60% of the peaks identified for both HDAC2 and Tip60 enrichment were along the gene body (exon and intron regions). A comparative analysis of the genes associated with these peaks between the genotypes (APP, APP; Tip60, w1118) revealed ~79% commonality for HDAC2 and ~88% commonality for Tip60 (Figure S2C and S2F). Strikingly, a comparative analysis of Tip60 and HDAC2 target genes in the w1118 control revealed that 83% of HDAC2 bound genes overlap with 97% of Tip60 bound genes (Figure S2G). Further, PCA analysis (Figure S3A and S3B) and hierarchically clustered heatmaps (Figure S3C and S3D) showed homogeneity within replicates and variability between groups in both HDAC2 and Tip60. These results suggest that similar genes were regulated in each genotype by Tip60 or HDAC2 and Tip60-induced enrichment in APP; Tip60 was more similar to control w1118 genotype, suggesting that increased Tip60 protects against these gene changes.
GO analysis of the genes bound by HDAC2 or Tip60 in the three genotypes reveals that the majority of biological pathways associated with HDAC2 and Tip60 bound genes within a genotype and across genotypes are similar. These results indicate that Tip60 and HDAC2 binding enrichment across genotypes are mostly associated with similar pathways (Figure S4 and S4 Table: S4-1 and S4-3 to S4-6). Interestingly, we found that 4133 HDAC2 target genes were unique to the control w1118 genotype as they were not found in the APP and APP; Tip60 genotypes. GO analysis reveals that these genes are primarily associated with cellular pathways regulating cell cycle (Figure S5 and S4 Table: S4-2). However, the gene ratio (ratio of the number of genes bound by HDAC2 or Tip60 to the number of genes related to the pathway) for these unique w1118 pathways was small and thus not identified in the top 20 pathways of overall HDAC2 bound genes in the w1118 genotype.
We next performed enrichment quantification of the identified Tip60 and HDAC2 ChIP-seq peaks in w1118 control, APP, and APP;Tip60 larval heads to determine whether their chromatin binding was altered in APP larval heads (APP vs. w1118) and whether increased Tip60 could protect against potential binding changes (APP;Tip60 vs. APP). Our findings revealed that in the APP larval heads, there were robust changes in binding enrichment for both HDAC2 (5400 peaks with increased binding and 6571 peaks with decreased binding) and Tip60 (1562 peaks with increased binding and 2023 peaks with decreased binding) (Figure 4(a) and S5 Table: S5-1 and S5-3). In addition, tissue enrichment of the top 2000 APP-induced peak enrichment unveils the HDAC2 and Tip60 neural specificity (Figure S3E and S3F). Increased Tip60 levels induced a significant reduction in HDAC2 binding (2718 peaks with increased binding and 8960 peaks with decreased binding) and minimal changes in Tip60 binding (4 peaks with increased binding and 1 peak with decreased binding) (Figure 4(a) and S5 Table: S5-2 and S5-4). This Tip60 mediated trend in reduced HDAC2 binding is evident by the change in the ratio of decreased binding to increased binding (5.5:4.5 in APP vs. w1118 to 7.7:2.3 in APP;Tip60 vs. APP): an increase in the number of peaks with decreased binding (6571 in APP vs. w1118 to 8960 in APP;Tip60 vs. APP) decrease in the number of peaks with increased binding (5400 in APP vs. w1118 to 2718 in APP;Tip60 vs. APP), and a decrease in the median of log2FoldChange with increase in HDAC2 binding. Further, we correlated the gene expression alterations (RNA-seq: APP vs. w1118) with HDAC2 or Tip60 binding changes (ChIP-seq: APP vs. w1118). We did not observe a strong correlation between overall gene expression and HDAC2 or Tip60 binding (Figure S6), suggesting that some gene alterations may represent downstream effects resulting from disruption of direct Tip60 and HDAC2 gene targets.
Figure 4.
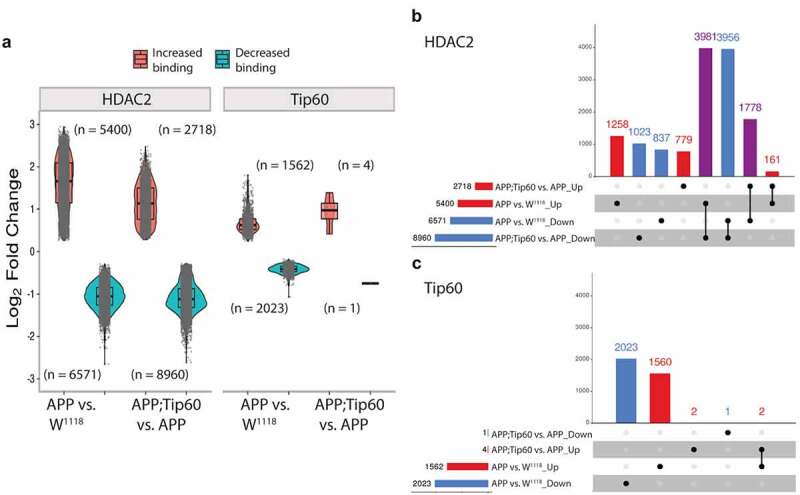
Increased Tip60 protects against enhanced HDAC2 enrichment in APP larval heads. (a) Log2 fold changes of differentially bound peaks (padj ≤0.05) of HDAC2 and Tip60 in APP vs. w1118 and APP;Tip60 vs. APP. APP-induced changes (APP vs. w1118) were prominent in both HDAC2 and Tip60 samples, while Tip60-induced changes (APP;Tip60 vs. APP) were prominent only in HDAC2 samples. (b and c) The upSet plot represents the distribution and intersection of differentially bound peaks between APP vs. w1118 and APP;Tip60 vs. APP from HDAC2 (b) and Tip60 (c) samples. Rows represent the number of peaks in each comparison (APP vs. w1118 and APP;Tip60 vs. APP), and columns represent the number of peaks per interaction. The red and blue bars represent the increased and decreased binding of HDAC2 or Tip60, respectively. The black filled dots indicate the association between rows. The red and blue columns represent peaks unique to a given comparison, while the purple columns represent the peaks rescued by Tip60 expression.
We next analysed the distribution and intersection of altered peaks in larval heads between genotypes APP vs. w1118 and APP;Tip60 vs. APP for HDAC2 (Figure 4(b)) and Tip60 (Figure 4(c)). Remarkably, for HDAC2 binding, approximately 48% (5759/11971) of the total number of peaks altered in APP larval head (APP vs. w1118) were restored by an increase in Tip60 levels (APP;Tip60 vs. APP). Thus, we refer to these peaks as ‘Tip60 reprogrammed HDAC2 peaks.’ The Tip60 reprogramming effect was primarily observed for HDAC2 binding, visualized with both profile plots (Figure 5(ei and Fi)) and heatmaps (Figure 5(eii and Fii)). As 60% of the identified Tip60 and HDAC2 peaks were enriched along the gene body, we visualized the ChIP-seq read densities of the significantly altered peak enrichment +/- 0.5 kilobase from the centre region of the gene body (Figures 5 and S7). In APP larval heads (Figure 5(a–d)), the increase in binding of HDAC2 (Figure 5(b)) and decrease in binding of Tip60 (Figure 5(c)) highly predominates over the decrease in binding of HDAC2 (Figure 5(a)) and increase in binding of Tip60 (Figure 5(d)). Further, increased Tip60 levels protected against alterations in the HDAC2 and Tip60 binding pattern in the APP larval heads (Figure S7A–D). Taken together, these results suggest that Tip60 exerts its neuroprotective action at least in part via protection against inappropriate enhancement of repressor HDAC2 genome-wide enrichment along neuronal gene bodies.
Figure 5.
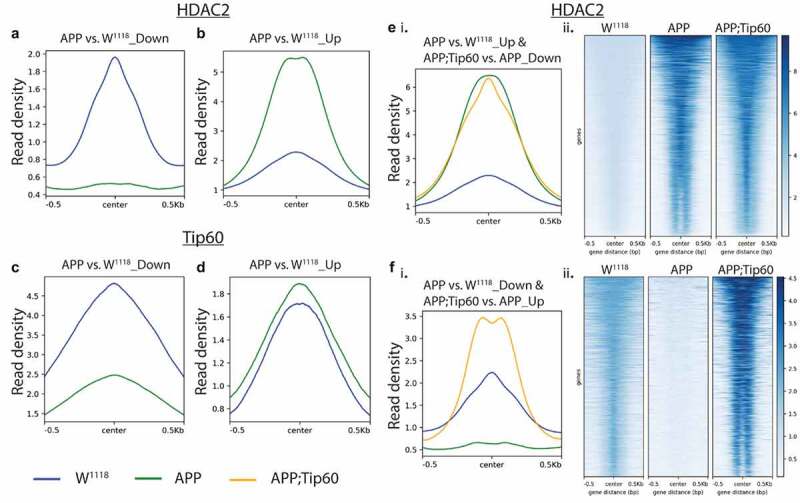
Tip60 expression protected against alterations in the HDAC2 binding pattern along the gene body in APP larval heads. (a and b) Profile plots representing decreased (a) and increased (b) binding of HDAC2 in APP larval heads. (c and d) Profile plots representing the decreased (c) and increased (d) binding of Tip60 in APP larval heads. Profile plots also represent the significant increase in HDAC2 binding (B) and decrease in Tip60 binding (C) in APP larval heads. (Ei. and Fi.) Profile plots representing the rescue effect (reversal in APP-induced binding pattern) of Tip60 expression on HDAC2 binding. (E ii. and F ii.) The corresponding heatmaps represent the Tip60 rescue effect. Sequencing data centred + /- 0.5 kilobase from the centre region of the gene body.
Tip60/HDAC2 co-regulated genes functionally modulate AD neurodegeneration in vivo and are conserved in the human AD brain
We observed that Tip60 and HDAC2 are recruited to genes in a binding enrichment pattern that is disrupted during early AD neurodegeneration, predominantly by enhanced HDAC2 binding over Tip60. To identify genes associated with Tip60 and HDAC2 that are misregulated under early AD-associated APP conditions, we compared all differentially expressed genes from our RNA-seq analysis (APP vs. w1118 and APP vs. APP;Tip60) with the protein-encoding genes bound by Tip60 in control w1118 larval heads and the protein-encoding genes bound by HDAC2 binding in APP larval heads (Figure S8A). Remarkably, this analysis revealed that 77% (or 3137 genes) of the total number of genes identified were identical direct target genes for both HDAC2 and Tip60. Together, these results indicate that Tip60 and HDAC2 co-regulate an identical set of genes and that this control is altered during early AD conditions at least in part by enhanced HDAC2 binding that may also displace Tip60 binding. Comparison of the top 20 biological processes, enriched in GO analysis, each for Tip60 and HDAC2 protein-encoding genes revealed that 17 of these biological processes are identical, further confirming that Tip60 and HDAC2 co-regulate overlapping biological processes (Figure S8B and S8C). These processes included axon guidance, associative learning, and neuron differentiation, underscoring the importance of the co-regulatory function of Tip60 and HDAC2 in neural function and cognition.
We next asked whether these genes are functionally involved in modifying AD-associated neurodegeneration, in vivo. To address this question in an unbiased fashion, we selected 50 genes from the top 20 enriched biological processes that were present in both Tip60 and HDAC2 GO analysis (Figure S8 and Table S6). To assess whether these 50 genes could functionally modulate AD neurodegeneration in vivo we used the well-characterized D. melanogaster neurodegenerative eye screen that enables us to assess the ability of a gene to functionally modulate AD-associated neurodegeneration. Due to the unavailability of GMR-APP and GMR-Aβ42 fly lines, we opted to use Gal4-GMR-tau transgenic flies (Bloomington stock #51367) to drive the expression of the mutant form of human tau V337M in all retinal cell types. This well-characterized transgenic fly lines induce expression of h-tauV337M in the retina causes a moderately rough eye phenotype at 25°C, characterized by a fused and disordered ommatidia with missing mechanosensory bristles [20]. We reasoned that because both tau and Aβ42 induced pathways are both associated with AD neurodegeneration and studies support that Aβ42 and tau neurodegenerative abnormalities are linked through common upstream drivers [21] our tau induced selection strategy would still serve as a robust in vivo functional triage system to identify AD associated genes. We next determined whether RNAi-mediated knockdown of the genes of interest was able to modify this Tau-induced phenotype by comparing the rough eye phenotype of the Gal4-GMR Tau control flies to the surface of the control Gal4-GMR Tau crossed with RNAi flies. We found that out of 50 genes we were able to obtain RNAi fly lines for only 14 genes showed either enhancement or suppression of the GMR Tau rough eye phenotype. These results are summarized in S7 Table. This is likely due either because the target gene was not robustly downregulated enough by RNAi to show an observable eye phenotype or because the gene was not involved in modifying tau induced eye neurodegeneration. Importantly, the positive outcomes we observed were as we would predict in that genes inappropriately downregulated in the APP larval brain enhanced the neurodegenerative phenotype when further down regulated by RNAi and genes inappropriately upregulated in the APP larval fly showed suppression of the eye neurodegenerative phenotype when down-regulated by RNAi. The functions of these 14 genes include diverse roles in neuronal function and neurodegenerative disease and are referred to here as ‘Tip60/HDAC2 AD genes’: Shroom, oc, nwk, nmo, Syn, Appl, Dop1R1, RhoGAP100F, NetB, flw, trx, Thor, Dl, and CG7275. The results of the GMR Tau eye screen functionally triaged our mass data sets from both ChIP and RNA sequencing to further streamline mechanistic analysis underlying Tip60 and HDAC2 co-regulation of genes functionally involved in early AD-associated neurodegeneration.
To gain further understanding into this new phenomenon of Tip60/HDAC2 co-regulation of target genes, we next wished to analyse Tip60 and HDAC2 binding at high resolution at their target gene loci. Because the number of Tip60/HDAC2 co-bound gene targets we identified was extensive (10,826 genes in total (Figure S2G), for high-resolution analysis and visualization of Tip60 and HDAC2 binding at specific coordinates within gene loci, we used Integrative Genomics Viewer (IGV) to focus exclusively on analysing the 14 genes selected based on their ability to modify neurodegeneration in the GMR-tau eye screen (Figures 6 and S9). These genes play critical roles in synaptic plasticity and neuronal developmental processes that include synaptic vesicle function (Syn and nwk), axonal outgrowth (NetB), neuronal signalling pathways (nmo, flw, Dop1R1, Dl, Appl, and RhoGAP100F), gene regulation (trx and Thor), actin filament formation and stabilization (Shroom), and neurodevelopment process (oc) [22]. Remarkably, our results revealed that Tip60 and HDAC2 not only both bind within each of these genes in all fly genotypes analysed (APP, APP;Tip60, w1118) but remarkably, at almost identical genomic coordinates, suggesting that Tip60 and HDAC2 are co-recruited to the same docking sites within gene loci (Figures 6 and S9). Furthermore, the same trend of inappropriately enhanced HDAC2 enrichment in APP vs. w1118 control that was protected against upon increased Tip60 levels (APP;Tip60 vs. APP) was observed at almost all of these genomic coordinates (S8 Table). Taken together, our results support a model by which Tip60 and HDAC2 co-regulate identical neuronal target genes via recruitment to overlapping binding sites within these gene bodies and that this regulation is disrupted in the AD brain.
Figure 6.
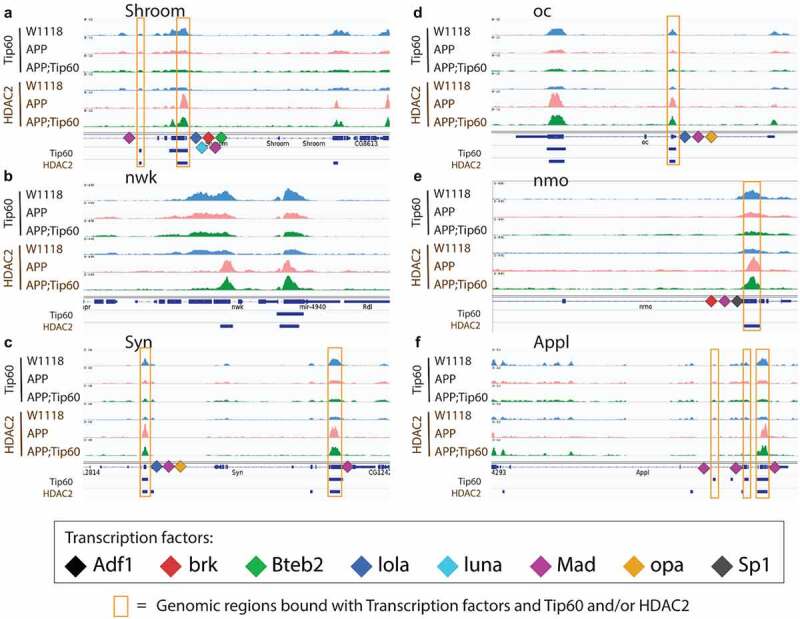
Tip60 and HDAC2 bind at similar genomic coordinates and co-regulate synaptic plasticity and neuronal developmental process-related genes. (a-f) The figure depicts the genome browser track view of high resolution Tip60 and HDAC2 peak analysis in three genotypes (w1118, APP, and APP;Tip60) at the following genes that were identified to functionally modify neurodegeneration in the GMR-tau eye screen: Shroom (a), nwk (b), Syn (c), oc (d), nmo (e), and Appl (f) loci. The orange box highlights the blue bars below the gene features panel depicting the location of genomic region peaks bound by both Tip60 and HDAC2. These co-Tip60/HDAC2 bound peaks also contain consensus binding motifs for the following transcription factors: Adf1, brk, Bteb2, lola, luna, Mad, opa, and Sp1).
Tip60 and HDAC2 both can interact with transcription factors (TFs) that aid in their gene recruitment and regulatory functions [23,24,25,26,27] that we speculate may be disrupted in early AD stages. Thus, we asked whether there are conserved TF motif binding sites within genes altered for both expression and Tip60 and HDAC2 binding during the early AD stages. To address this question, the genes selected for this analysis were triaged by comparing ChIP-seq and RNA-seq data sets for AD-associated alterations (APP vs. w1118) to select for down-regulated genes with reduced Tip60 and enhanced HDAC2 binding and up-regulated genes with enhanced Tip60 and reduced HDAC2 binding. Only those gene alterations that were protected against by increased Tip60 levels were selected for motif analysis. The selected genes were termed as up-regulated rescue (UpRegRes) list and a down-regulated rescue (DownRegRes) list (Figure S10). GO analysis of these genes revealed that the top biological processes were enriched for functions in learning and memory, axon guidance and extension, neurogenesis and neuron development, and gene silencing and chromatin modification (S9 Table), further underscoring the importance of Tip60 and HDAC2 in neuronal functions disrupted in AD. Motif enrichment analysis was performed to identify the TFs controlling the rescue genes’ transcription. HDAC2 and Tip60 bound coordinates altered by APP expression were also included for motif discovery (S10 Table). The analysis revealed eight TFs with known function in neuronal gene control (Figure 7(a)) and motif regions within the Tip60/HDAC2 AD genes (Figure 7(b)). Remarkably, many of these TFs are located either within or close to the identified Tip60 and HDAC2 co-peaks (Figures 6 and S9). Notably, 9 of the 14 Tip60/HDAC2 AD genes have Mad binding sites. Our results suggest that recruitment of Tip60 and HDAC2 by common TFs within gene bodies may be a general mechanism by which these chromatin regulators co-regulate neuronal gene expression.
Figure 7.
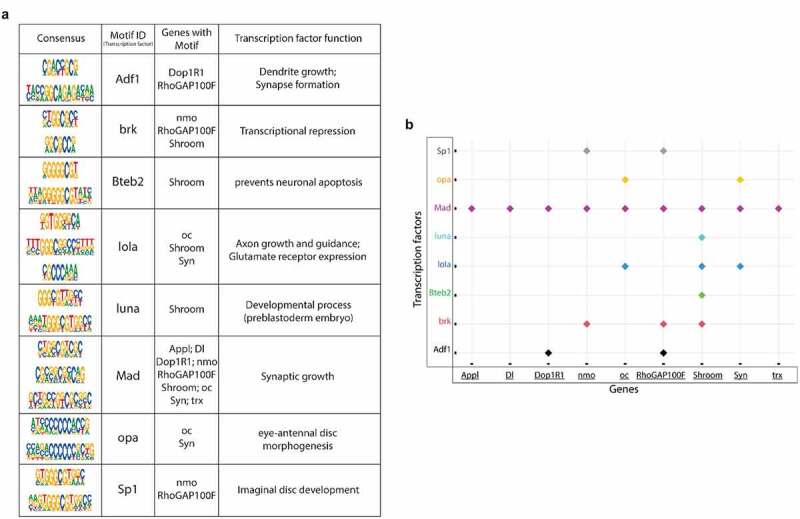
Transcription factor (TF) motifs significantly enriched within the rescue gene list and the associated Tip60/HDAC2 AD genes. TF motifs were identified using the MEME-Chip platform (CentriMo). (a) Consensus sequences and their corresponding TFs bound and the associated Tip60/HDAC2 AD genes. (b) Plot representing the association of Tip60/HDAC2 AD genes and the TFs.
Finally, we asked whether the Tip60/HDAC2 binding alterations and gene dysregulation we observed in APP larval heads were also reflected at the protein level. To address this, we used mass spectrometry (MS) analysis of proteins isolated from the larval brains of w1118, APP, and APP;Tip60 genotypes to identify significantly differentially regulated proteins [abs(FC) > 1.5 and q-value ≤0.1] with APP (APP vs. w1118) and Tip60 (APP;Tip60 vs. APP) expression (S11 Table). Analysis of the ~1100 most enriched proteins, identified by MS, revealed that 74 of these proteins were altered in their levels in the APP larval brain and 67 of these in Tip60 expressed brains (Figure S11). GO analysis revealed that these proteins regulate methylation [histone (Art1 and Art4) and mRNA (Art4)], axon guidance and transport (Dys), nucleocytoplasmic shuttling (Ntf-2), and glutamate (Galphas) and cholinergic (Dys) pathways (S12 Table). Comparison of proteomics and next-generation sequencing (RNA-seq and ChIP-seq) data reveal that 23% (17/74) of these altered proteins are directly encoded by Tip60/HDAC2 co-regulated genes misregulated in the APP larval brain while only 11% (7/67) are altered in the APP; Tip60 larval brain (Figures 8 and Table S13). These results suggest that early AD-associated alterations in epigenetic gene regulation persist to the protein level.
Figure 8.
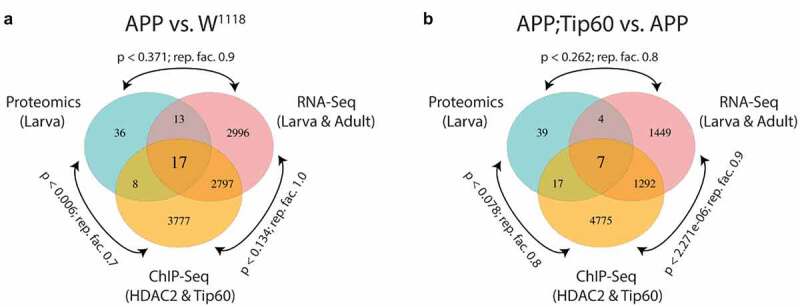
RNA-seq, ChIP-seq, and mass spectrometry data convey the integrative and independent gene expression regulation induced by APP and Tip60 expression. (a and b) Venn diagram of differentially regulated genes in the third instar larval and adult heads (RNA-seq), genes with differentially binding of Tip60 and HDAC2 in the third instar larval heads (ChIP-seq), and differentially regulated proteins in the third instar larval heads (mass spectrometry) from (a) APP vs. w1118 comparison and (b) APP;Tip60 vs. APP comparison. A representation factor (rep. fact.) >1 indicates more overlap than expected of two independent groups, a representation factor <1 indicates less overlap than expected, and a representation factor of 1 indicates that the two groups by the number of genes expected for independent groups of genes. A p value <0.05 is considered significant.
Tip60/HDAC2 co-regulation of neuronal genes is disrupted in hippocampus of AD patients
Our results thus far revealed that Tip60 and HDAC2 co-regulate neuronal target genes via recruitment to overlapping binding sites within gene bodies that is disrupted in the AD brain. To confirm human AD relevance for this new phenomenon, we next asked whether human orthologs for the 14 Tip60 and HDAC2 co-regulated direct target genes we identified from our D. melanogaster ChIP-seq and RNA-seq analysis (Figures 6 and S9) are co-targets of Tip60 and HDAC2 in the human hippocampus and if binding was compromised in AD patient hippocampus similar to what we observed in the APP fly. To address these questions, we performed ChIP analysis using chromatin prepared from age-matched human healthy control and AD hippocampus. We quantified enrichment of Tip60 and HDAC2 within gene bodies using real-time PCR. Remarkably, all 14 genes tested were found to be direct gene targets for both Tip60 and HDAC2 in the human hippocampus (Figure 9). Further, ChIP analysis using chromatin from AD patients revealed similar results to what we observed in APP larval heads in that Tip60 enrichment was significantly decreased at 12 of the 14 genes (Figure 9(a)) and HDAC enrichment was enhanced at 7 of the 14 genes tested (Figure 9(b)). Strikingly, five of these genes (FCHSD2, OTX1, SYDE1, PPP1CB, SHROOM2, DCAF13, and NLK) in the AD hipppocampus showed the same trend in Tip60/HDAC2 binding dysregulation that we identified in the APP larval brain exemplified by reduced Tip60 binding and enhanced HDAC2 binding within the same gene loci. Our findings reveal that Tip60/HDAC2 co-regulatory mechanisms underlying neuronal gene expression that are disrupted during early AD stages in the fly brain via reduced Tip60 binding and enhanced HDAC2 binding are conserved in the hippocampus of human AD patients.
Figure 9.
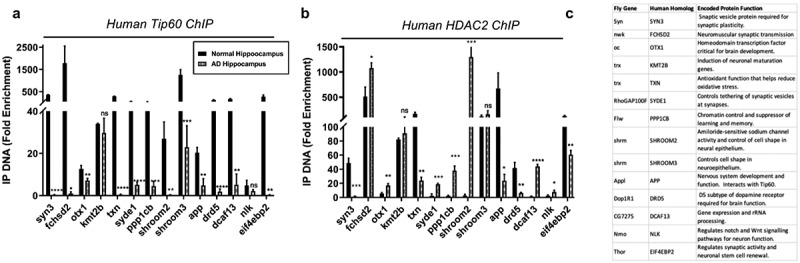
Human homologs of co-Tip60/HDAC2 D. melanogaster neural gene targets exhibit conserved Tip60 and HDAC2 binding patterns in normal versus AD patient hippocampi. Chromatin was isolated from healthy control and AD hippocampus (n = 3 brains per condition). Histograms represent ChIP enrichment using the EZ-Magna ChIP Kit (Millipore, MA, USA) with antibodies to (a) Tip60 and (b) HDAC2. For each ChIP experiment a control reaction containing mouse IgG polyclonal antibodies was performed simultaneously. Real-time PCR was performed on DNA purified from each of the ChIP reactions using primer pairs specific for each gene loci. Fold enrichment of the respective genes was calculated relative to the IgG control using the standard curve method as described by the EZ-Magna ChIP Kit manual. Statistical significance was calculated using unpaired Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars indicate SEM. (See S12 Table for primer sequences) (c) Table depicting D. melanogaster and human homolog gene names and conserved gene functions.
Discussion
Here we report the first genome-wide HAT versus HDAC profiling study assessing epigenetic alterations initiated during early stages of AD-associated neurodegeneration modelled in the D. melanogaster APP larval brain. A key finding from our analysis revealed that both Tip60 and HDAC2 binding is not exclusively restricted to promoter regions but are also enriched predominantly along the gene bodies, suggesting these enzymes may act to both initiate and then maintain gene regulatory control in a poised state [28,29,30]. Additionally, since gene-body bound TFs have also been shown to regulate RNA splicing by binding to pre-mRNAs to recruit HATs that increase histone acetylation to facilitate RNA Polymerase elongation and exon exclusion or HDACs that reduce histone acetylation to slow RNA Polymerase elongation and exon inclusion, Tip60 and HDAC2 might also function in this capacity [29,31]. Furthermore, we observed robust alterations in binding enrichment for both HDAC2 and Tip60 in the AD larval brain well before Aβ deposition and lethality, indicating that chromatin remodelling changes are an initial event in neurodegenerative progression and not a consequence. Notably, our analysis showed a predominant increase in binding of HDAC2 (Figure 5(b)) and a decrease in binding of Tip60 (Figure 5(c)) within central gene bodies of their target loci. These findings extend prior studies showing enhanced HDAC2 recruitment to a focused subset of synaptic genes in AD fly [6] and mouse models [7] by revealing for the first time that an increase in HDAC2 binding is a broad genome-wide AD-associated phenomenon that occurs significantly within gene bodies resulting in their dysregulation. A similar complimentary trend in a marked reduction in genome-wide H4K16 acetylation in the human AD brain [32], which notably is the preferential acetylation target for Tip60, has recently been reported. Thus, our results indicate that some histone acetylation changes [9,32,33] functionally contributing to AD may be initiated at the level of altered Tip60 and HDAC2 antagonistic enzyme recruitment within the central gene body regions.
Another significant finding originating from our work is that Tip60 and HDAC2 co-regulate a similar set of genes that function in cognition linked neural processes disrupted early in AD progression. Comparison of transcriptionally altered genes enriched with HDAC2 in APP larval heads and transcriptionally altered genes enriched with Tip60 in the w1118 larval heads revealed that, remarkably, 77% of these genes are identical and misregulated in the AD fly brain (Figure S8A). Further, GO analysis of Tip60 versus HDAC2 target genes revealed that 17 of the top 20 most enriched biological processes identified for each enzyme also overlapped and included functions like axonal guidance, associative learning, and neuron differentiation: underscoring their importance in cognitive function and relevance to AD (Figure S8B and S8C). Thus, while other groups have proposed that HAT and HDAC enzymatic activities may both be present in close proximity to each other on gene regulatory regions [34,35] we are the first to report co-docking of Tip60/HDAC2 on chromatin targets that mediates a co-regulatory function for neural genes at a genome-wide level. Finally, we find that almost one-fourth of the proteins altered in the APP larval brain (17/74) are encoded by dysregulated Tip60/HDAC2 co-target genes (Figure 8(a)), indicating that such early AD-associated Tip60/HDAC2 epigenetic alterations persists at the RNA and the protein level.
How might Tip60 and HDAC2 be co-recruited to similar genomic loci within neural genes? It is well-documented that both HATs and HDACs interact with the same TF that facilitates their recruitment to gene loci to promote chromatin remodelling and transcriptional control. For instance, NF-kB interacts with and is acetylated by p300/CBP and deacetylated by HDAC1/HDAC2 to increase and decrease target gene expression, respectively [36]. However, whether HATs and HDACs can bind simultaneously to the same gene by being recruited by either different TFs in close proximity within a given gene locus or by the same TF remains to be elucidated. Here, in our motif enrichment analysis of Tip60 and HDAC2 ChIP-seq peaks, we identify eight TFs with known function in neuronal gene control (Figure 7) that are located either within or in proximity to the Tip60 and HDAC2 co-peaks we identified within AD-associated neural gene loci (Figures 6 and S9). These findings indicate that these TFs are involved in the co-recruitment of Tip60 and HDAC2 to common gene regulatory regions. Most notably, Mad binds to 9 of the 14 AD-associated genes we analysed and, remarkably, is present at the identical coordinates within the Appl (β amyloid protein precursor-like) gene within co-Tip60 and HDAC2 peaks (Figure 6). Accordingly, in prior studies, Mad has been shown to interact with both Tip60 and HDAC2 in other systems to activate and repress gene expression, respectively [24,37]. Our results support a model by which Mad, along with other TFs within a given gene body, serve as docking sites for recruitment of both HDAC2 and Tip60 either separately and within proximity to one another or simultaneously, thus keeping genes poised for rapid activation or repression. We speculate that these scenarios are not mutually exclusive of one another and, importantly, may explain the rapid histone acetylation changes within activity-dependent neural genes that drive their swiftly fluctuating transcriptional responses [34,38,39]. Intriguingly, some of the TFs we identify have been previously implicated in AD. For example, Sp1 dysregulation identified in the AD frontal cortex has been proposed to alter its regulation of APP and Tau target genes [40], while human SMAD (human ortholog of fly Mad) activity is also reduced in the AD brain, causing dysregulation of downstream signalling pathway mediated gene expression [41].
In the present study, a pivotal discovery with clinical relevance is that increased Tip60 levels protect against altered HDAC2 binding and gene disruption in larval AD brains. Essentially, such Tip60 mediated neuroprotection against epigenetic gene dysregulation is a genome-wide phenomenon as evidenced by our observation that 5400 genes display inappropriately enhanced HDAC2 binding and that increased Tip60 protects against such increases for 74% (3981/5400) of these affected genes in the AD larval brain (Figure 4(b)). Interestingly, we observed such inappropriate enhanced HDAC2 binding significantly in the gene body’s central region (Figure 5). Further, high-resolution mapping of Tip60 and HDAC2 peaks within AD-associated neuronal genes reveal that enhanced HDAC2 and reduced Tip60 binding in the APP larval head occurs within several Tip60/HDAC2 co-docking sites, with such inappropriate enhanced HDAC2 enrichment reduced with increased Tip60 levels (Figures 6, Figures 5) and S9). Similar trends in altered Tip60/HDAC2 co-regulation of human orthologs of these genes were observed in the human AD hippocampus (Figure 9), highlighting human relevance and the remarkable conservation in Tip60/HDAC2 epigenetic mechanisms between AD flies and human patients. Together, our findings support a model (Figure 10) that increased HDAC2 in the AD larval and human brain [7] displaces genome-wide Tip60 recruitment within gene bodies that may be initiated at co-Tip60/HDAC2 docking sites, causing harmful changes in gene expression that persist and worsen during disease progression. Tip60 may mediate its neuroprotective role in epigenetic gene control by either reducing HDAC2 levels, a phenomenon which we previously demonstrated to occur at the transcriptional level [6] and/or by displacing inappropriately enhanced HDAC2 binding levels to restore Tip60 mediated gene regulation.
Figure 10.
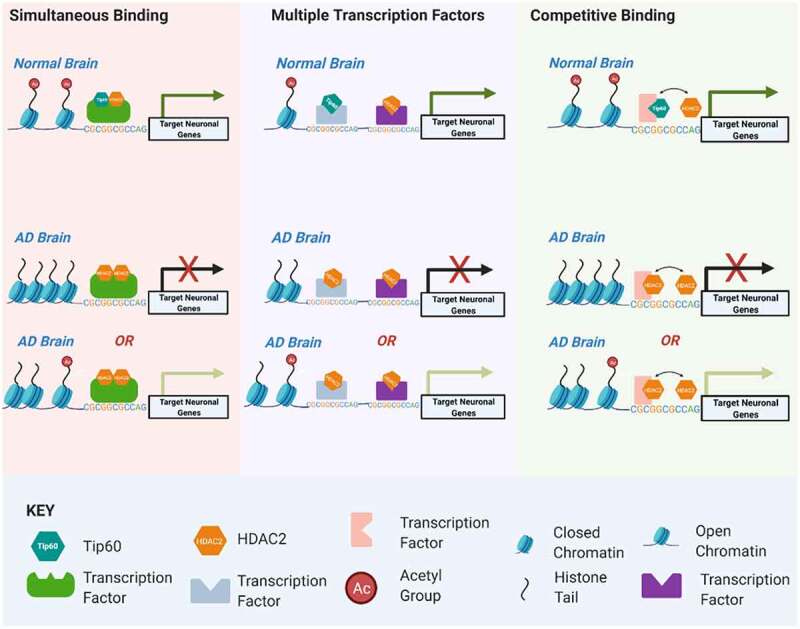
Model for Tip60 and HDAC2 co-mediated neuronal gene control. Our results support a model by which transcription factors (TFs) within a given neuronal gene body serve as docking sites for recruitment of both HDAC2 and Tip60 either simultaneously to the same TF, separately to multiple TFs within close proximity to one another or competitively to a given TF. We speculate these scenarios are not mutually exclusive of one another and may explain the rapid histone acetylation changes within activity-dependent neural genes that drive their swiftly fluctuating transcriptional responses. Early disruption of Tip60/HDAC homoeostasis in the AD brain promotes enhanced HDAC2 recruitment over Tip60 that may be initiated at co-Tip60/HDAC2 docking sites and causes inappropriate up or down regulation of target genes. We speculate these deleterious changes in gene expression persist and worsen during AD progression.
Our study proposes a mechanism involving aberrant Tip60 and HDAC2 co-recruitment to genes genome-wide to explain how histone acetylation changes are initiated in AD, providing informative directions for chromatin-mediated therapeutic avenues. For example, HDAC inhibitors (HDACi) lack target specificity and act to increase global acetylation [12,42,43], reducing their applicability as safe cognition promoting therapeutics, thus promoting exploration into more specific HAT activators that can potentially reset AD associated site-specific histone acetylation disruption. Our findings underscore this concept by showing that HDAC2 has a reduced gene target specificity compared with Tip60, as evidenced by the far more HDAC2 genome-wide target genes altered in the APP larval brain (Figure 4(a)) when compared to Tip60. Nevertheless, increased Tip60 specifically protects against altered HDAC2 binding at most genes in the APP larval heads (Figure 4(b)) and at many of the co-Tip60/HDAC2 docking sites with TF binding motifs (Figures 6, Figure 7 and S9), highlighting the relevance for Tip60 and/or Tip60/HDAC2 interacting TFs as more specific therapeutic targets. Further, these Tip60/HDAC2 binding alterations at specific gene loci are present before Aβ accumulation is detectable, supporting these sites as potentially valuable early AD biomarker ‘hot spots’ that are easy to track. Recently, we reported that disruption of Tip60 and HDAC2 balance in the brain is a common event in other neurodegenerative diseases modelled in D. melanogaster: Huntington’s disease, amyotrophic lateral sclerosis, and Parkinson’s disease [44]. Further studies may reveal a therapeutic potential for targeting Tip60 in these disorders as well. Together, our findings warrant future epigenetic therapeutic studies intended to restore Tip60 mediated histone acetylation homoeostasis for earlier and more selective treatment for AD and potentially other neurodegenerative disorders.
Materials and methods
Fly stocks
Fly strains and crosses. All fly lines were raised under standard conditions at 22°C on standard yeasted Drosophila media (Applied Scientific Jazz Mix Drosophila Food; Thermo Fisher Scientific, MA, USA). The pan-neuronal driver elav C155 and the transgenic UAS lines carrying human APP 695 isoform (UAS-APP) were obtained from Bloomington Drosophila Stock. Generation and characterization of APP and Tip60 expression levels for double-transgenic UAS APP;Tip60WT fly lines used for these studies are described in [45] and show equivalent APP levels between APP and APP;Tip60WT genotypes and increased Tip60 exogenous expression for double transgenic APP;Tip60 fly lines. For all experiments presented here, transgenic expression levels for APP and Tip60 in each of these UAS fly lines were revalidated using a quality control qPCR strategy [45,46] with RNA extracted from the same pooled larval or adult heads samples used for the ChIP-Seq, RNA-Seq and proteomics experiments to ensure samples showed equivalent APP expression levels between APP and APP;Tip60WT genotypes and increased Tip60 levels in APP;Tip60WT genotype. GMR-Gal4 driver was used to drive the expression of the mutant form of human tau V337M in all retinal cell types and was obtained from Bloomington Drosophila Stocks (stock #51367). Bloomington Stock numbers for all RNAi fly lines used for the GMR-Gal4 neurodegenerative eye screen are listed in Table S7. The w1118 line served as the genetic background control. All experimental crosses were performed at a normal physiological temperature of 25°C with 12 hour light/dark cycles.
Immunofluorescence, imaging, and quantification
For anti-Aβ42 immunofluorescence samples were prepared as described in Zhang et al. (2020). Briefly, larval or adult brains were dissected in PBS, fixed in fixation buffer containing 0.7% paraformaldehyde and 0.9% lysine for 1 h at room temperature, washed three times in PBS containing 0.5% Triton X-100 (PBST) for 15 min each time at room temperature, and blocked for 1 h at room temperature in PBST containing 5% normal goat serum, and incubated with primary anti-Aβ42 (1:100, #05-831-I, Millipore, MA, USA) antibody in blocking solution overnight at 4°C. Samples were washed three times in PBST for 15 min each time at room temperature and incubated with goat anti-mouse Alexa Fluor 488 (1:300, #A28175, Invitrogen, CA, USA) and propidium iodide (PI, a final concentration of 1.5 μM) for 2 h at room temperature. After washing three times in PBST for 15 min each time, samples were mounted in VECTASHIELD antifade mounting media (Vector Laboratories, CA, USA).
For imaging, samples were analysed as described in Zhang et al. (2020). Confocal microscopy was performed using a ZEISS microscope (LSM 700, ZEISS, NY, USA). The optical intervals were 5.94 μm z-sections for 100× magnifications and 0.79 μm z-sections for 200× magnifications. The optical intervals were determined by the optimized pinhole diameters which are 33.3 μm at 1 Airy Unit (AU) for 100× magnification and 25.1 μm at 1 AU for 200× magnification. Consecutive z-stacks through the entire Kn were used for quantification. Consecutive subsets of the z-stacks approximately at the level of centre Kn were used for the final projection and display. The quantification of Aβ plaques and apoptosis in different genotypes was measured under 200× magnification using Image J software.
For the neurodegenerative eye screen, flies were anaesthetised with Anesthifly and attached to glass slides with gorilla tape. Fly eyes were imaged at 50x magnification using a Zeiss Stemi 2000-C microscope and a Leica MC 190 HD camera with Leica Application Suite (LAS v4.12). Enhancers/suppressors were identified by visualizing changes in ommatidia pattern and eye shape. Enhancers were classified as genes that decreased the uniformity of ommatidia and caused indentations in eye shape. Suppressors were classified as genes that increased the uniformity of ommatidia and evenness of eye shape. All experiments were blinded to prevent bias.
RNA isolation
Total RNA was isolated from third-instar larval brains or seven-day-old adult heads using the QIAGEN RNeasy Mini Kit (#74106, QIAGEN, MD, USA) following the manufacturer’s protocol. The quality, quantity, and purity of RNA were determined using a Nanodrop spectrophotometer (Thermo Fisher Scientific, MA, USA) and 2100 Bioanalyzer (Agilent Technologies, CA, USA). RNA samples with an RNA integrity number (RIN) ≥8.0 were used for sequencing.
RNA-seq library preparation, sequencing, and analysis
One hundred ng of total RNA was used to prepare libraries using TruSeq Stranded Total RNA kit (Illumina, CA, USA) according to the manufacturer’s instructions. The final libraries at the concentration of 4 nM were sequenced on NextSeq 500 platform (Illumina, CA, USA) using 75 bp paired-end sequencing. Raw FASTQ sequencing reads were aligned to the D. melanogaster genome (Ensembl version BDGP6) using RNA-Seq by Expectation-Maximization (RSEM) [47]. Total read counts were obtained using RSEM’s calculate-expression function. Principal component analysis (PCA) and heatmap clustering (Euclidean distance) were performed to cluster the samples and identify the batch effects and sample heterogeneity. All the plots were constructed using R/Bioconductor.
Differential gene expression analysis
Differential gene expression between any two genotypes was tested using the DESeq2: a statistical tool that employs shrinkage estimates to compute fold changes [48]. Raw RNA-seq read counts from biological replicates of each genotype were used as the input for DESeq2. For both larval and adult data, all three genotypes (w1118, APP, and APP;Tip60) were analysed together using a single model matrix, and the desired pairwise comparisons were then extracted. Only genes that displayed log2FoldChange of ≤-0.583 and ≥0.583 in their expression levels, with an adjusted p-value ≤0.05, were used for the UpSet plot [49] and gene ontology (GO) analysis (FlyEnrichr) [50]. Among the ontologies in GO analysis, GO Biological Process GeneRIF was included in our downstream analysis. Heatmaps were generated using the ComplexHeatmap package [51]. The TissueEnrich package is used to calculate enrichment of tissue-specific genes in a set of input genes [52].
Chromatin immunoprecipitation (ChIP)
Chromatin was extracted and sheared from ~200 third-instar larval heads per replicate. To obtain larval heads, the first one-third of the larvae (anterior head region) were isolated. Remaining fat bodies were carefully dissected and discarded. All larval heads were inspected visually to ensure that the entire CNS was intact. Using the GAL4-inducible system to target gene expression exclusively in the nervous system of the larvae ensures virtually no variability in gene expression in the samples used. For IPs, we used truChIP Chromatin Shearing Kit (Covaris Inc., MA, USA) following the manufacturer’s instructions. Briefly, protein-DNA cross-links were made at RT for 5 min with 1% formaldehyde, and the tissue was pulverized using the CryoPrep (Covaris Inc., MA, USA). Cells were lysed and nuclei were prepared using Covaris lysis buffer. Sonication of DNA was performed using a Covaris E220 Ultrasonicator for 15 min. The sheared chromatin was immunoprecipitated using the EZ-Magna ChIP A Chromatin Immunoprecipitation Kit (Millipore, MA, USA) following the manufacturer’s instructions. Sheering quality and chromatin quantity was determined using Agilent Bioanalyzer DNA 1000 kit (Agilent Technologies, CA, USA). Briefly, ChIP was performed with 30 μg of sheared chromatin using anti-Rpd3 (ab1767, Abcam, MA, USA), anti-Tip60 (ab23886, Abcam, MA, USA). The eluted material from the immunoprecipitation along with an input sample was then purified using a QIAquick PCR purification kit (QIAGEN, MD, USA).
ChIP-seq library preparation, sequencing, and analysis
ChIP-seq libraries were prepared from the ChIP-enriched DNA samples using the Accel-NGS 2SPlus DNA Library Kit (Swift Biosciences, MI, USA), following the 350 base pair insert guide of the protocol. After library preparation, all libraries were normalized and sequenced using the standard Illumina loading protocol on the Illumina NextSeq 500 Sequencer (Illumina, CA, USA). Sequence read fragments were aligned to the D. melanogaster BDGP6 genome using the BWA-MEM aligner [53]. Samtools was used to filter the resulting alignments to remove reads with mapping quality below q30 and any remaining duplicate reads, and then to merge replicated BAM files for each factor and condition [54]. Peak calling was performed on the reads that passed filters for each replicate in addition to the merged alignments using macs2 with default settings [55]. The resulting peaks were annotated for genomic features using the HOMER annotatePeaks.pl tool [56]. Replicated peak calls were used to estimate the irreproducibility discovery rate (IDR) and create consensus peak sets with IDR ≤0.05 [57]. Regions of interest were defined by intersecting the consensus peak sets with Ensembl BDGP6.22 annotation release 98. The featureCounts tool from the subread software package was used to generate read counts for each region of interest [58]. PCA and heatmap clustering (Euclidean distance) were performed to cluster the samples and identify the batch effects and sample heterogeneity. All the plots were constructed using R/Bioconductor.
Differential binding analysis
Differential binding of peaks (region of interests) between any two genotypes was tested using the DESeq2 [48]. Raw read counts, for each region of interest, from biological replicates of each genotype were used as the input for DESeq2. For Tip60 and HDAC2 samples, all three genotypes (w1118, APP, and APP;Tip60) were analysed together using a single model matrix, and the desired pairwise comparisons were then extracted. Peaks with an adjusted p-value ≤0.05 were used for the UpSet plot analysis [49]. Genes associated with these peaks were further used for GO analysis (FlyEnrichr) [50]. Among the ontologies in GO analysis, GO Biological Process GeneRIF was included in our downstream analysis.
Visualization of ChIP-seq data
The merged BAM files for each genotype were converted to BPM normalized BigWig files using bamCompare. ComputerMatrix was used to calculate scores per genome regions (Differentially bound regions from DESeq2) and prepared an intermediate file that can be used with plotHeatmap and plotProfiles [59]. The reference point for the plotting was the centre of the region with a window of +/- 0.5 kilobase. For ChIP-Seq track generation, BigWig files were used with Integrated Genomics Viewer (IGV_Linux_2.8.6) [60]. The BED files used for IGV contain genomic coordinates of the significantly enriched peaks of genes resulted from the eye-screen (S6 Table).
Motif enrichment analysis
We performed DNA motif enrichment analysis, central motif enrichment analysis or CentriMo [61], to detect the positional enrichment of previously characterized TF binding motifs in the Tip60 and HDAC2 bound sequences (Figure S10). The Combined Drosophila Databases (TF motifs) provided in the web version of the CentriMo were used as the input for CentriMo. The default options were used for the analysis, and the statistical significance of discovered motifs was estimated using P values and E-values derived from a one-tailed binomial test (S8 Table).
Protein isolation, identification, and analysis
Protein was extracted from dissected third-instar larval brains of three genotypes (w1118, APP, and APP;Tip60) and was sent to Bioproximity LLC for proteomic profiling. Samples were subjected to enzymatic digestion with sequencing-grade trypsin. The digested peptides were cleaned-up by solid-phase extraction (SPE) protocol. Each digestion mixture was analysed by UPLC-MS/MS (Ultra performance liquid chromatography-tandem mass spectrometer). The LC was performed on an Easy-nLC 1200 system (Thermo Fisher Scientific, MA, USA) fitted with a heated, 25 cm Easy-Spray column. The LC was interfaced to a quadrupole-Orbitrap mass spectrometer (Q-Exactive HF-X, Thermo Fisher Scientific, MA, USA). TMGF (Mascot Generic Format) files were searched using X!Tandem and Open Mass Spectrometry Search Algorithm (OMSSA). Protein intensity values were calculated using OpenMS to measure the area under the curve of identified peptides. The Perseus software platform was used for protein quantification, cross-comparisons between genotypes, and multiple-hypothesis testing (Benjamini–Hochberg FDR: t-test p-value adjusted to account for multiple testing) [62]. Proteins with q < 0.05 and |FC| > 1.5, determined as significantly changed proteins, were used for downstream analysis. Protein–protein interaction networks among the significantly changed proteins were visualized using STRING on the Cytoscape platform (Cytoscape_v3.7.2) [63]. Functional enrichment analysis was performed using FlyEnrichr [50] and GO Biological Process GeneRIF was included in our downstream analysis.
ChIP-qPCR (human)
For all human studies, human hippocampal samples were obtained from the National Disease Research Interchange, with informed consent by all donors. The control brains included three males with an age range of 70–85 years. The AD brains were from one male and two females with an age range of 73–87 years.
Chromatin was extracted and sheared from ~120 mg human hippocampus using truChIP Chromatin Shearing Kit (Covaris Inc., MA, USA) following the manufacturer’s instructions. Briefly, protein-DNA crosslinks were made at RT for 5 min with 1% formaldehyde and the tissue were pulverized using the CryoPrep (Covaris Inc., MA, USA). Cells were lysed and nuclei were prepared using Covaris lysis buffer. Sonication of DNA was performed using a Covaris E220 Ultrasonicator for 13 min. The sheared chromatin was immunoprecipitated using the EZ-Magna ChIP Chromatin Immunoprecipitation Kit (Millipore, MA, USA) following the manufacturer’s instructions. Briefly, ChIP was performed with 50ug of sheared chromatin using anti-Tip60 (ab23886, Abcam, MA, USA), anti-HDAC2 (ab12169, Abcam, MA, USA), and Normal Mouse IgG Polyclonal Antibody control (Millipore, MA, USA). Eluted material from the immunoprecipitation was purified using a QIAquick PCR purification kit (QIAGEN, MD, USA) and used directly for real-time PCR.
qRT-PCRs were performed in a 20 uL reaction volume containing cDNA, 1 M Power SYBR Green PCR Master Mix (Applied Biosystems, CA, USA), and 10 M forward and reverse primers (S14 Table). Primer sets were designed by NCBI/Primer-BLAST (www.ncbi.nlm.nih.gov/tools/primer-blast/). RT-qPCR was performed using an ABI 7500 Real-Time PCR system (Applied Biosystems, CA, USA) following the manufacturer’s instructions. Fold enrichment for all the respective genes was calculated relative to the non-specific Mouse IgG Polyclonal Antibody control.
Statistical analysis
Statistical analysis of RNA-seq, ChIP-seq, and mass spectrometry (MS) data differences between the two groups were considered statistically significant with q < 0.05 (FDR < 0.05, controlled by Benjamini–Hochberg). For the ChIP-seq analysis, sample sizes were w1118 = 3; APP = 2; APP;Tip60 = 2. For RNA-seq analysis, the sample size for third-instar larva was w1118 = 2; APP = 2; APP;Tip60 = 2 and for seven-day-old adult flies was w1118 = 3; APP = 3; APP;Tip60 = 3. For the MS analysis, sample sizes were w1118 = 3; APP = 2; APP;Tip60 = 2.
The statistical significance of the overlap between two groups of genes (Venn diagrams) was calculated and represented by the representation factor and the associated hypergeometric probability (http://nemates.org/MA/progs/overlap_stats.html). The number of genes in the genome is considered as 13,500 (based on the number of genes obtained from RNA-seq data). A representation factor >1 indicates more overlap than expected of two independent groups, a representation factor <1 indicates less overlap than expected, and a representation factor of 1 indicates that the two groups by the number of genes expected for independent groups of genes. A p value <0.05 is considered significant. For correlation analysis, a value of r greater than 0.7 is considered a strong correlation. Anything between 0.5 and 0.7 is a moderate correlation, and anything less than 0.4 is considered a weak or no correlation. A p value <0.05 is considered significant.
Model figure is created using BioRender.com
Supplementary Material
Acknowledgments
Research reported in this publication utilized the MetaOmics Shared Resource at Sidney Kimmel Cancer Center at Jefferson Health. We thank Adam Ertel and Gaurav Kumar for their help with the initial bioinformatics analysis. We are grateful for the human subjects and their families for participation in this study.
Funding Statement
This work was supported by the National Institutes of Neurological Disorders and Stroke of the NIH under the Award Number [R01HD057939] to Dr. Felice Elefant;National Institute of Neurological Disorders and Stroke
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Sanchez-Mut JV, Graff J.. Epigenetic alterations in Alzheimer’s disease. Front Behav Neurosci. 2015;9:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Masters CL, Bateman R, Blennow K, et al. Alzheimer’s disease. Nat Rev Dis Primers. 2015;1(1):15056. [DOI] [PubMed] [Google Scholar]
- [3].Karch CM, Cruchaga C, Goate AM.. Alzheimer’s disease genetics: from the bench to the clinic. Neuron. 2014;83(1):11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Saha RN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006;13(4):539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Berson A, Nativio R, Berger SL, et al. Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 2018;41(9):587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Panikker P, Xu, SJ, Zang, H, et al. Restoring Tip60 HAT/HDAC2 balance in the neurodegenerative brain relieves epigenetic transcriptional repression and reinstates cognition. J Neurosci. 2018;38(19):4569–4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Graff J, Rei D, Guan J-S, et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483(7388):222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nativio R, Donahue G, Berson A, et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat Neurosci. 2018;21(4):497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lu X, Wang L, Yu C, et al. Histone acetylation modifiers in the pathogenesis of Alzheimer’s disease. Front Cell Neurosci. 2015;9(226):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fossgreen A, Bruckner B, Czech C, et al. Transgenic drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blistered-wing phenotype. Proc Natl Acad Sci U S A. 1998;96(23):13703–13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pirooznia SK, Sarthi J, Johnson AA, et al. Tip60 HAT activity mediates APP induced lethality and apoptotic cell death in the CNS of a drosophila Alzheimer’s disease model. PLoS One. 2012;7(7):e41776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Johnson AA, Sarthi J, Pirooznia SK, et al. Increasing Tip60 HAT levels rescues axonal transport defects and associated behavioral phenotypes in a drosophila Alzheimer’s disease model. J Neurosci. 2013;33(17):7535–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Greeve I, Kretzschmar D, Tschape JA, et al. Age-dependent neurodegenertion and Alzheimer-amyloid plaque formation in transgenic drosophila. J Neurosci. 2004;24(16):3899–38906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xu S, Wilf R, Menon T, et al. Epigenetic control of learning and memory in drosophila by Tip60 HAT action. Genetics. 2014;198(4):1571–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Iijima K, Liu H-P, Chiang A-S, et al. Dissecting the pathological effects of human Abeta40 and Abeta42 in drosophila: a potential model for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101(17):6623–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Iijima K, Chiang H-C, Hearn SA, et al. Abeta42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS One. 2008;3(2):e1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Grothe MJ, Sepulcre J, Gonzalez-Escamilla G, et al. Molecular properties underlying regional vulnerability to Alzheimer’s disease pathology. Brain. 2018;141(9):2755–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Patel H, Dobson RJB, Newhouse SJ. A meta-analysis of Alzheimer’s disease brain transcriptomic data. J Alzheimers Dis. 2019;68(4):1635–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kunkle BW, Grenier-Boley B, Sims, R, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Blard O, Feuillette S, Bou J, et al. Cytoskeleton proteins are modulators of mutant tau-induced neurodegeneration in Drosophila. Hum Mol Genet. 2007;16(5):555–566. [DOI] [PubMed] [Google Scholar]
- [21].Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron. 2008;60(4):534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Larkin A, Marygold SJ, Antonazzo G, et al. FlyBase: updates to the Drosophila melanogaster knowledge base. Nucleic Acids Res. 2020;49(D1):D899–D907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tea JS, Chihara T, Luo L. Histone deacetylase Rpd3 regulates olfactory projection neuron dendrite targeting via the transcription factor Prospero. J Neurosci. 2010;30(29):9939–9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Frank SR, Parisi T, Taubert S, et al. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 2003;4(6):575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hlubek F, Lohberg C, Meiler J, et al. Tip60 is a cell-type-specific transcriptional regulator. J Biochem. 2001;129(4):635–641. [DOI] [PubMed] [Google Scholar]
- [26].Aghdassi A, Sendler M, Guenther A, et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. 2012;61(3):439–448. [DOI] [PubMed] [Google Scholar]
- [27].Yang WM, Inouye C, Zeng Y, et al. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc Natl Acad Sci U S A. 1996;93(23):12845–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang Z, Zang C, Cui K, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138(5):1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Greer CB, Tanaka Y, Kim YJ, et al. Histone deacetylases positively regulate transcription through the elongation machinery. Cell Rep. 2015;13(7):1444–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang L, Zhang F, Rode S, et al. Ethylene induces combinatorial effects of histone H3 acetylation in gene expression in Arabidopsis. BMC Genomics. 2017;18(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rambout X, Dequiedt F, Maquat L. Beyond transcription: roles of transcription factors in pre-mRNA splicing. Chem Rev. 2018;118(8):4339–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nativio R, Donahue G, Berson A, et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat Neurosci. 2018;21(4):497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Stilling RM, Fischer A. The role of histone acetylation in age-associated memory impairment and Alzheimer’s disease. Neurobiol Learn Mem. 2011;96(1):19–26. [DOI] [PubMed] [Google Scholar]
- [34].Peserico A, Simone C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J Biomed Biotechnol. 2011;2011:371832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yamagoe S, Kanno T, Kanno Y, et al. Interaction of histone acetylases and deacetylases in vivo. Mol Cell Biol. 2003;23(3):1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen L-F, Greene WC. Shaping the nuclear action of NF-κB. Nat Rev Mol Cell Biol. 2004;5(5):392–401. [DOI] [PubMed] [Google Scholar]
- [37].Laherty CD, Yang W-M, Sun J-M, et al. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell. 1997;89(3):349–356. [DOI] [PubMed] [Google Scholar]
- [38].Karnay AM, Elefant F. Chapter 14 - Drosophila epigenetics. In: Tollefsbol TO, editor. Handbook of epigenetics. Second ed. Academic Press; 2017. p. 205–229. [Google Scholar]
- [39].Katan-Khaykovich Y, Struhl K. Dynamics of global histone acetylation and deacetylation in vivo: rapid restoration of normal histone acetylation status upon removal of activators and repressors. Genes Dev. 2002;16(6):743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Citron BA, Dennis JS, Zeitlin RS, et al. Transcription factor Sp1 dysregulation in Alzheimer’s disease. J Neurosci Res. 2008;86(11):2499–2504. [DOI] [PubMed] [Google Scholar]
- [41].Ueberham U, Rohn S, Ueberham E, et al. Pin1 promotes degradation of Smad proteins and their interaction with phosphorylated tau in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2014;40(7):815–832. [DOI] [PubMed] [Google Scholar]
- [42].Fischer A, Sananbenesi F, Mungenast A, et al. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci. 2010;31(12):605–617. [DOI] [PubMed] [Google Scholar]
- [43].Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1):32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Beaver M, Bhatnagar A, Panikker P, et al. Disruption of Tip60 HAT mediated neural histone acetylation homeostasis is an early common event in neurodegenerative diseases. Sci Rep. 2020;10(1):18265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pirooznia SK, Chiu K, Chan MT, et al. Epigenetic regulation of axonal growth of Drosophila Pacemaker cells by histone acetyltransferase Tip60 controls sleep. Genetics. 2012;192(4):1327–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lorbeck M, Pirooznia K, Sarthi J, et al. Microarray analysis uncovers a role for Tip60 in nervous system function and general metabolism. PloS One. 2011;6(4):e18412–e18412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Li B, Dewey CNJBB. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome Li B, Dewey CNJBB. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Love MI, Huber W, Anders SJGB. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. BMC Genome Biology. 2014;15(12):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Conway JR, Lex A, Gehlenborg NJB. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics. 2017;33(18):2938–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kuleshov MV, Jones, MR, Rouillard, AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acid Research. 2016;44(W1):W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gu Z, Eils R, Schlesner MJB. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–2849. [DOI] [PubMed] [Google Scholar]
- [52].Jain A, Tuteja GJB. TissueEnrich: tissue-specific gene enrichment analysis. Bioinformatics. 2019;35(11):1966–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li HJAPA. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Quantitative Biology, Genomics, 2013;1:1–3. [Google Scholar]
- [54].Li H, Handsaker, B, Wysoker, A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhang Y, Liu, T., Meyer, CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biology. 2008;9(9):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Heinz S, Benner, C., Spann, N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular Cell. 2010;38(4):576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li Q, Brown, JB, Huang, H, et al. Measuring reproducibility of high-throughput experiments. Bioinformatics. 2011;5(3):1752–1779. [Google Scholar]
- [58].Liao Y, Smyth GK, Shi WJB. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Nucleic Acid Research.2014;30(7):923–930. [DOI] [PubMed] [Google Scholar]
- [59].Ramírez F, et al. deepTools2: a next generation web server for deep-sequencing data analysis. 2016;44(W1):W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Robinson JT, Thorvaldsdottir, H, Winckler, W, et al. Integrative genomics viewer. Nature Biotechnology. 2011;29(1):24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Bailey TL, Machanick PJNAR. Inferring direct DNA binding from ChIP-seq. 2012;40(17):e128–e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tyanova S, Temu, T, Sinitcyn, P, et al. The Perseus computational platform for comprehensive analysis of (prote) omics data. Nature Methods. 2016;13(9):731. [DOI] [PubMed] [Google Scholar]
- [63].Shannon P, Markiel, A, Ozier, O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13(11):2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.