

ABSTRACT
Women entering pregnancy with elevated body mass index (BMI) face greater risk of adverse outcomes during pregnancy, delivery, and for their offspring later in life, potentially via epigenetics. If epigenetic programming occurs early during in utero development, the differential marks should be detectable in multiple tissues despite the known unique epigenetic profile in each.
We used early-pregnancy BMI as reflection of maternal metabolic milieu exposure in peri-conception and early-pregnancy period. We analysed DNA methylation in paired cord blood and placenta samples among 437 newborns from Gen3G, a pre-birth prospective cohort of primarily European descent. We measured DNA methylation in both tissues across the genome in >720,000 CpG sites using the Illumina MethylationEPIC array. At each site, we used linear mixed models (LMMs) with an unstructured variance-covariance matrix to test for an association between maternal early-pregnancy BMI and DNA methylation in both tissues (modelled as M-values). We adjusted for tissue-specific covariates, offspring sex, gestational age at delivery, and maternal smoking and age.
Women had a mean (SD) BMI of 25.4 (5.7) kg/m2 measured at first trimester visit (mean=9.9 weeks). Early-pregnancy BMI was associated with differential DNA methylation levels in paired-tissue analyses at two sites: cg10593758 (β=0.0126, SE=0.0025; P=4.07e-7), annotated to CRHBP, and cg0762168 (β=−0.0094, SE=0.0018; P=2.78e-7), annotated to CCDC97.
Application of LMMs in DNA methylation data from distinct fetal-origin tissues allowed us to identify CpG sites at which early-pregnancy BMI may have an epigenetic ‘programming’ effect on overall fetus development. One site (CRHBP) may play a role in hypothalamic-pituitary-adrenal axis regulation.
KEYWORDS: DNA methylation, epigenome-wide association study, body mass index, pregnancy, placenta, cord blood, developmental programming
INTRODUCTION
The number of women entering pregnancy with an elevated body mass index (BMI) has been increasing in the United States in recent years. In 2019, 29% of women were entering pregnancy with a BMI >30 kg/m2 (obesity category) [1]. Prior research has found associations between high pre-pregnancy maternal BMI and numerous adverse outcomes during pregnancy, at delivery, and through the offspring’s life course [2,3]. Such adverse outcomes include both low and high birth weight (or macrosomia)[2], as well as increased obesity risk and lower cardiovascular health across the offspring’s lifespan [3–5].
With rising obesity rates in women who are of reproductive age [6], there is growing scientific interest in investigating the mechanisms that lead to and may mediate these adverse outcomes, especially in long-term offspring adverse cardiometabolic health. DNA methylation is a particular epigenetic mark that is highly dynamic during in utero development, and may be part of foetal programming that leads to these long-term risks in offspring. 7 Pre-pregnancy elevated BMI may impact subsequent foetal programming; and is also associated with early-pregnancy BMI, as prior studies have shown that very little, if any, weight gain occurs in the first trimester [8]. Therefore, we sought to investigate if DNA methylation in offspring may be associated with early-pregnancy maternal BMI and the related adverse metabolic milieu present during that critical period that likely program offspring to later adverse outcomes. Early-pregnancy is a very sensitive period for nutritional exposures and long-term programming of cardio-metabolic diseases. For example, in the landmark Dutch famine study, exposure to severe caloric restriction in the early pregnancy was specifically associated with heart disease and obesity, but also to specific changes in blood DNA methylation collected in middle-aged participants (about six decades after exposure) [9–13].
A common challenge in investigating epigenetic variations lies in the selection of tissue for investigation given that DNA methylation profiles are unique to each tissue and cell type [14,15]. Yet, if an environmental factor (e.g., maternal metabolic milieu related to elevated BMI) has a programming effect in early developmental processes (e.g., via DNA methylation), we hypothesized that the associations could be detectable in more than one tissue type. To test this hypothesis, we proposed a paired tissue approach using both cord blood and placenta collected at delivery. Cord blood and placenta are both vital links of the in utero environment and foetal development [14]. Despite the fact that tissue-specific epigenetic modifications may have important physiological implications at the tissue level and in the overall organism, if an early exposure impacts the overall foetus epigenetic programming, it should be detectable in more than one tissue [16]. Thus, analysing both tissues may reveal biological insight beyond what we could find from solely analysing DNA methylation in each alone. Using paired placenta and cord blood samples carefully collected in Gen3G, a pre-birth prospective cohort, we tested associations between maternal BMI measured in early-pregnancy and paired-tissue DNA methylation levels at over 720,000 individual CpG sites across the genome to test specific loci that may be part of the metabolic programming of future adverse cardiometabolic outcomes in the offspring.
RESEARCH DESIGN AND METHODS
Population
The Genetics of Glucose regulation in Gestation and Growth (Gen3G) Study is a prospective pre-birth cohort from Quebec, Canada. Study staff recruited women eighteen years of age or older in the first trimester of a singleton pregnancy to participate in the Gen3G study, provided there was no prior history of diabetes in medical records or at a first trimester screening. Detailed description of the Gen3G cohort has been published elsewhere [17]. All participants provided written consent prior to study enrolment according to the Declaration of Helsinki. All protocols were approved by the IRB at the Center Hospitalier Universitaire de Sherbrooke. For this study, we included individuals of European descent whose delivery occurred at >37 weeks of gestation and from whom we had collected both foetal placenta tissues and cord blood. We provided a study participants flow in Supplementary Figure 1.
Measurements
Study staff conducted three research visits over the course of pregnancy, one between 5–16 weeks of pregnancy (recruitment at first trimester), one between 24–30 weeks (to align with second trimester screening for gestational diabetes), and one at delivery. In the first visit, study staff measured anthropometric data and participants completed questionnaires on lifestyle, demographics, and medical history. Staff measured weight using a calibrated electronic scale and height using a wall stadiometer, and BMI was calculated as weight divided by squared height (kg/m2) during the first visit, which occurred at a mean (range) gestational age of 9.9 (4.1–16.4) weeks. Lifestyle questionnaires included questions on current and previous maternal smoking. We excluded women who had pre-existing diabetes based on medical history and blood testing at first trimester visit. At birth, study staff collected gestational age at delivery and sex of child from medical records.
Biological Specimens
Trained research staff collected foetal placenta tissue and cord blood samples at delivery. Staff collected biological samples according to established protocol, collecting a 1 cm3 foetal placental sample approximately 5 cm from umbilical cord insertion, as well as cord blood less than 30 min postpartum. Placental samples were stored in RNAlater (Qiagen) first at −4°C for 24 h and then at −80°C until DNA extraction occurred. Cord blood was treated with aprotinin to prevent protein degradation, EDTA as an anticoagulant, centrifuged, and subsequently stored at −80°C until DNA extraction.
Laboratory Procedures
Lab personnel performed DNA extraction on placenta and cord blood samples using the AllPrep DNA/RNA/Protein Mini Kit (Qiagen). DNA purity was assessed using Spectrophotometer (Ultrospec 2000 UV/Visible; Pharmacia Biotech) with an absorbance ratio set at 260–280 nm. We completed bisulphite conversion of 460 pairs of purified placenta and cord blood DNA samples. We measured epigenome-wide DNA methylation using the Infinium MethylationEPIC BeadChip (Illumina), an array which provides information on >850,000 CpG sites throughout the genome. We randomly allocated samples to position and plate, however, each of the ten total plates contained an equal number of cord blood vs. placenta samples.
Data QC
We imported data into R using the minfi package to perform sample and probe level quality control. We first excluded samples that failed based on lab QC (placenta n = 5, cord blood n = 3). Then we examined sex mismatch between predicted and recorded sex and excluded where there was a mismatch (placenta n = 1), as well as SNP mismatch between tissues that were labelled as coming from the same individual (placenta n = 6, cord blood n = 6). We also removed cord blood samples that further QC determined had been contaminated with maternal blood (n = 2), and samples that did not have a paired sample in the other tissue after the previously described sample exclusions (placenta n = 11, cord blood n = 12). We included 437 pairs of placenta and cord blood tissues in our final analyses for this current study (see Supplementary Figure 1).
Following division into separate datasets by tissue, we performed normalization using preprocessFunNorm() in the minfi package, which includes noob background correction, and used the RCP function from the ENmix package to correct for probe-type bias. We assessed the effectiveness of these methods after each step by checking the alignment of 10 duplicates from each tissue.
On the processed data set of beta-values, we computed a detection P-value for each probe in each sample and excluded probes that had a nonsignificant detection (P > 0.05) for 5% or more of the samples (placenta n = 1869 probes, cord blood n = 1754 probes). We then matched CpG sites by ID to ensure that we ran the analysis only on CpG sites that were present in the data from both tissues, which resulted in excluding additional probes (placenta n = 501, cord blood n = 616 probes). From the paired CpG sites, probes previously identified as cross reactive [18] were removed (n = 43,108). We also removed probes that are known to have a SNP at the single base extension (n = 5154), a SNP at the CpG (n = 5070) and probes that are found on the sex chromosomes (n = 17,737). We also removed probes with a SNP found underneath the probe (n = 71,218) leaving a total of 722,279 probes in our final analysis. Finally, we adjusted for sample plate using ComBat from the sva package, and logit transformed the β-values to M-values for statistical analysis [19].
Statistical Methods
For the 437 participants included in this analysis, we reported sample characteristics as a mean (with SD), or proportion (Table 1, Supplementary Table 1). Since the outcome, DNA methylation, is measured for both paired cord blood and placenta samples, we ran a linear mixed model with an unstructured variance–covariance matrix at each CpG site. We chose a model of the form:
Table 1.
Characteristics of Gen3G mother–child pairs included in analyses of DNA methylation in paired tissues (cord blood and placenta).
| Maternal characteristics | Mean (SD) or n (%) |
|---|---|
| N = 437 | |
| Gestational age at visit 1 (weeks) | 9.9 (2.4) |
| BMI (kg/m2) at visit 1 | 25.4 (5.7) |
| Maternal age (years) | 28.3 (4.2) |
| Gravidity (primigravid) | 145 (33.2%) |
| Maternal ethnicity (white)* | 434 (100%) |
| Smoke during pregnancy (yes) | 39 (8.9%) |
| Child characteristics | |
| Gestational age at delivery (weeks) | 39.6 (1.0) |
| Offspring sex (male) | 229 (52.4%) |
| Birth weight (grams) | 3,440.4 (422.6) |
SD = standard deviation, n = number of participants in category, N = number of cohort participants with available data for measure, BMI = body mass index. *N = 434 participants with available ethnicity data in this analysis.
where Yij represents the methylation values at a single CpG site, represents participant level covariates for participant i, and represents the tissue specific covariates for participant i in tissue j (either cord blood or placenta), and represents the random error term. At the participant level, we adjusted for sex of child, gestational age at delivery, maternal smoking (yes/no) and maternal age. At the tissue level, we performed cell-type adjustments using the Bakulski reference panel in cord blood [20] and the first 10 PCs estimated by the ReFACTor method as a proxy for placental cell types [21]. Bakulski cord blood cell type estimates are based on an algorithm previously shown to correctly estimate cell proportions in whole blood and were validated with actual counts of cord blood cell types [20]. The ReFACTor method uses sparse PCA on a subset of CpG sites to estimate cell types without requiring the need for actual cell counts while still accounting for cell type heterogeneity [21]. We chose ReFACTor as this method has been shown to properly account for false discoveries [22]. As the number of cell types in cord blood and the number of proxy cell types in placenta differed, we first obtained the residuals at each CpG site separately in the two tissues by regressing out the cell types in a linear regression of the form:
with M representing the vector of methylation M-values at a single CpG site, X the matrix of estimated or proxy cell types across all participants, β the estimated coefficients, and ε the residuals. We then took the residuals for each CpG site in the separate tissues and used them as our outcomes for the paired analysis. We considered a p < 1e-6 as suggestive signals in our paired-tissue analysis. At the identified CpG sites, we conducted analyses in each separate tissue to assess if the association was driven by only one tissue type, using the same covariates as in the paired analysis and the appropriate cell type adjustment for each tissue. In the single tissue analyses we performed a multiple linear regression of the methylation M-values and the covariates using robust linear regression. We conducted all analyses using R version 3.6.1 and Bioconductor version 3.10.
Comparison to Previously Published Meta-analysis of Pre-pregnancy BMI and Offspring Blood DNA Methylation at Birth and in Adolescence
We performed a lookup analyses for 22 CpG sites identified in a prior meta-analysis EWAS reported by Sharp et al. that showed association between maternal BMI and DNA methylation in cord blood (up to n = 7,523; significant associations across the genome P < 1.06 × 10−7 accounting for multiple testing using Bonferroni) and that showed persistent association with DNA methylation in blood cells collected in adolescence (up to n = 1,817). For the 20 CpG sites that were available in our Gen3G paired tissue dataset, we aligned the Sharp et al. meta-analysis results with our paired tissue results for each available CpG to compare direction of effect and observed p-values.
RESULTS
Characteristics of the Gen3G mother–child pairs included in the current analysis are presented in Table 1. The mean (SD) early-pregnancy BMI of women included in our study was 25.4 (5.7) kg/m [2] collected at a mean (range) gestational age of 9.9 (4.1–16.4) weeks. Mean (SD) maternal age was 28.3 (4.2) years, and 8.9% of women reported smoking during pregnancy. Mean BMI and age in our included sample were comparable to the rest of the Gen3G cohort that was not included, which had mean BMI and age values of 25.7 kg/m2 (5.9) and 28.3 years (4.5) as seen in Supplementary Table 1.
The linear mixed model regression approach yielded two CpG sites with a suggestive raw p-value (P < 1e-6). Epigenome wide results are presented in Figure 1. The two sites, cg07621682 (P = 4.1 ×10−7) and cg10593758 (P = 5.5 × 10−7) are annotated to, respectively, the gene body of CCDC97 (Coiled-Coil Domain Containing 97) and the first exon of CRHBP (Corticotropin-Releasing Hormone-Binding Protein) genes. For cg10593758 at CRHBP, greater early-pregnancy BMI was associated with higher DNA methylation levels, while for cg07621682 at CCDC97, greater early-pregnancy BMI was associated with lower DNA methylation in our paired-tissue analyses.
Figure 1.
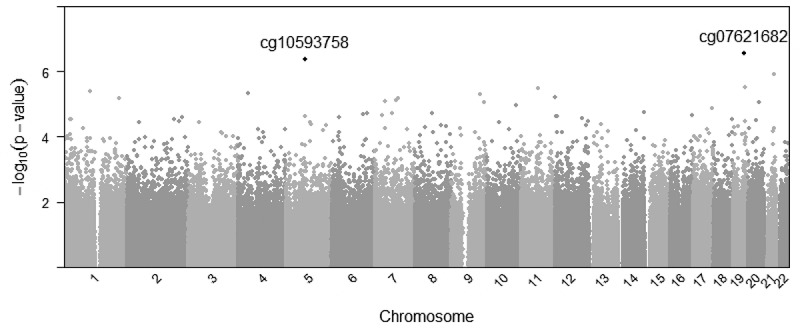
Manhattan plot representing associations between maternal body mass index and paired-tissue DNA methylation at 722,229 probes across the genome, CPG sites reaching p < 1e-6 are listed.
We presented in Table 2 characteristics of the two loci identified and paired-tissue results, along with the mean and standard deviation of normalized beta-values (estimated DNA methylation %) in individual tissues in Supplementary Table 2. These values confirmed that there is variability in the level of measured DNA methylation in both tissues at these specific CpG sites. We also presented the results of our regression analyses for associations between maternal BMI and DNA methylation in individual tissues at these two CpG sites (Supplementary Table 2), including the same covariates (maternal characteristics and cell composition adjustments) that were used in the paired-tissue EWAS. The associations observed from regression in individual tissues indicated that the directions of effect in each tissue were consistent with the paired-tissue analysis, and that the paired-tissue associations detected were not driven by a single tissue.
Table 2.
Associations between maternal BMI and DNA methylation (DNAm) in paired tissues (cord blood and placenta) and characteristics of identified CpGs.
| |
|
|
|
|
|
Maternal BMI association with newborn paired tissue DNAm* |
||
|---|---|---|---|---|---|---|---|---|
| Probe ID | Chr | Position | UCSC Gene Name | UCSC Ref Gene Group | Relation to CpG Island | β estimate (95% CI) | p-value | |
| cg07621682 | chr19 | 41,827,759 | CCDC97 | Body | Open Sea | −0.0093 | (−0.0129, −0.0057) | 4.09 × 10−7 |
| cg10593758 | chr5 | 76,248,743 | CRHBP | 5ʹ UTR; 1st exon | N. Shore | 0.0125 | (0.0077, 0.0174) | 5.50 ×10−7 |
We further examined our top hits from the paired and single individual tissue analyses in a 10kb region surrounding the CpG sites that reached our suggestive cut-off of p < 1e-6. As illustrated in Figure 2a, cg10593758 is located in exon 1 of CRHBP[23]. Within this region, we observed a clustering of probes around our top signal cg10593758 in all three analyses (paired-tissue, placenta, and cord blood). We also confirmed that cg10593758 is the most significant probe in that region in each respective tissue. We illustrated the regional associations for CCDC97 (Figure 2b), yet this locus had few probes available on the array within a 10 kb region of the top hit cg07621682.
Figure 2.
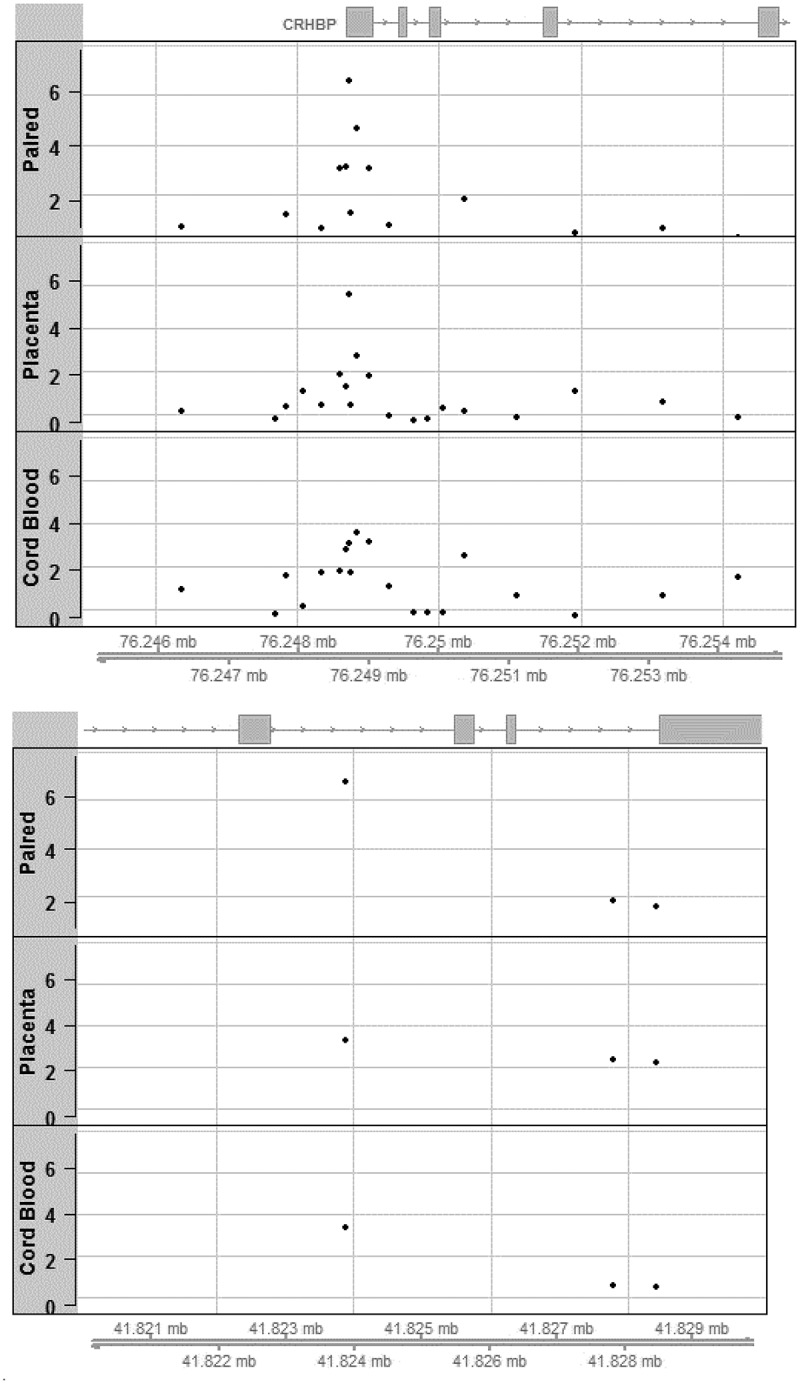
(a) Regional association plots aligned by position on chromosome 5 for locus at CRHBP around cg10593758 identified in the paired tissue analysis; levels of significance of associations are illustrated using negative log p-values (on Y-axis) for paired tissue (top) as well as in single tissue analyses (middle: placenta; bottom: cord blood). (b) Regional association plots aligned by position on chromosome 19 for locus at CCDC97 around cg07621682 identified in the paired tissue analysis; levels of significance of associations are illustrated using negative log p-values (on Y-axis) for paired tissue (top) as well as in single tissue analyses (middle: placenta; bottom: cord blood).
We performed look-up of previously reported CpG sites based on EWAS meta-analyses between maternal BMI and cord blood DNA methylation, focusing our attention on CpG sites where some persistence of association was observed in later life using blood DNA methylation from samples collected in adolescence (Sharp et al.) suggesting a ‘programming effect.’ Among the 22 CpG sites that Sharp et al. had identified, we had 20 CpG sites with data available for analyses in our paired-tissue DNA methylation dataset: we found similar direction of effect for association between maternal BMI and offspring DNA methylation at 15 CpG sites in our paired-tissue analyses among the 20 CpG available sites initially reported (see Supplementary Table 3). For example at cg16877087 (near RBMS1), Sharp et al. had reported that greater maternal BMI was associated with lower DNA methylation in cord blood (β = −0.0006; SE = 0.0001; Bonferroni adjusted P = 0.002) and in adolescent blood cells (β = −0.0003; SE = 0.0001; unadjusted P = 0.02), and in our analyses, we showed similarly that greater early pregnancy BMI was nominally associated with lower DNA methylation in our paired-tissue analyses (β = −0.0051; SE = 0.0023; P = 0.03).
DISCUSSION
Using a paired-tissue analytical approach leveraging placenta and cord blood samples collected in the same mother–child dyads, we found that higher maternal early-pregnancy BMI was associated with higher DNA methylation levels at cg10593758 (CRHBP) and with lower DNA methylation levels at cg07621682 (CCDC97) commonly in both tissues. Both loci are located within a DNAse hypersensitivity cluster based on ENCODE [23,24]. We investigated differentially methylated CpG sites associated with early-pregnancy maternal BMI with the hypothesis that if we find consistent signals in two different tissues at birth, this may indicate that multiple other tissues and cells experienced similar foetal epigenetic programming due to the same in utero metabolic environmental exposures and may be part of the mechanisms leading to long-term offspring adverse outcomes.
Very few pre-birth cohort studies have collected and compared DNA methylation levels in multiple tissues from the same newborn, thus most prior epigenetic association studies have investigated DNA methylation in a single tissue. Both cord blood and the placenta are commonly used in epigenetic studies of developmental origins of health and disease [14]. Cord blood is derived from the mesoderm germ cell layer during foetal development, and cord blood DNA methylation has been suspected to mediate environmental exposures, such as gestational weight gain, with offspring adverse outcomes [25]. The placenta forms from the trophectoderm in the first pregnancy stage [26], and is the initial foetal organ to form in offspring development [14]. It is located at the maternal-foetal interface, and develops during early embryogenesis in a unique epigenetic process. The organ’s main functions are to transfer nutrients towards the foetus, produce hormones to sustain pregnancy, as well as safeguard the foetus against infections while enhancing foetal growth [27]. Existing literature reports that pre-pregnancy obesity is associated with worsened placental function and increased global DNA methylation within the placenta[3]. For these reasons, both cord blood and placenta are strong tissues to investigate potential in utero programming. We know that DNA methylation is tissue-specific, yet it can be illuminating to understand methylation patterns across different tissue in relation to a common exposure (here high maternal BMI) to understand if variations in methylation in more than one tissue can potentially imply variations in all tissues [16]. Consistent associations in two tissues provide greater support that foetal epigenetic programming due to early-pregnancy maternal BMI may be present across multiple other tissues, as compared to when DNA methylation levels in single tissue has been studied in prior literature. By using the presented robust paired-tissue analytical approach, we have extended upon previous strategies.
Our findings at cg10593758 showed highly consistent associations across the two tissues and we found very similar associations at multiple probes in the 5ʹ untranslated region (UTR) of exon 123 of CRHBP, bringing confidence that we identified a signal of biological relevance. The corticotropin-releasing hormone binding protein (CRHBP) plays a part in the regulation of the canonical stress-responsive hypothalamic-pituitary-adrenal (HPA) axis, as well as the regulation of the neuronally-released corticotropin-releasing hormone (CRH) [28]. The HPA axis is a key component of stress adaptation and response [29]. When an individual experiences stress, the CRH hormone is released from the hypothalamus, which signals the pituitary gland to secrete the adrenocorticotropic hormone (ACTH) into the bloodstream, and then down to the adrenal glands to release cortisol, our main stress-response hormone [30]. CRHBP is expressed in the pituitary and brain in most mammals, as well as additionally in the placenta and liver in humans. CRHBP in the plasma binds plasma CRH, which then limits CRH receptor activation and the pituitary from releasing ACTH. Therefore, along with binding CRH that is released during pregnancy by the placenta, CRHBP is also thought to bind CRH that is released as part of the HPA axis in the stress response process [31]. Our study results indicate that higher early-pregnancy maternal BMI is associated with higher DNA methylation levels at CRHBP. Given that cg10593758 is located near the transcriptional start of CRHBP [23], the expected genomic regulatory process from higher DNA methylation near the promoter region would lead to lower expression levels of CRHBP and subsequent dysregulation of the buffering of CRH in the context of the stress response.
Previous studies have reported CRHBP genetic variation may be associated with humans having an increased risk of suicidal behaviour if they experience childhood trauma [32]. Reports have also shown that genetic variants in CRHBP are associated with the degree of effectiveness of SSRI antidepressants on individuals [28,33]. Additionally, prior epidemiological literature has revealed associations between maternal BMI and depression, anxiety, and emotional difficulties in offspring [34–36]. Future studies could investigate if the identified DNA methylation markers in CRHBP may be mediating the association between entering pregnancy with an elevated BMI and the risk of adverse mental health outcomes and adaptability to stressful events in offspring.
The coiled-coil domain-containing protein 97 (CCDC97) is the subject of limited literature that suggests the gene may be a susceptible locus for coronary heart disease risk [37], but additional investigations are needed. This region was poorly covered by the EPIC array, and the identified probe was not located into a known genomic regulatory region. Therefore, our finding must be cautiously interpreted.
We also performed look-up analyses leveraging findings of a prior large meta-analysis that had performed EWAS for associations between pre-pregnancy maternal BMI and DNA methylation levels in newborn blood from 19 independent cohorts (up to n = 7,523 mother–offspring participant pairs) [38]. Of the 20 sites that were genome-wide significant in the original cord blood EWAS and believed to be persistent in adolescence (up to n = 1,817 with blood DNA methylation), we found that early pregnancy BMI had similar direction of effect for association with DNA methylation in our Gen3G paired-tissue analyses at 15 CpG sites suggesting overall offspring epigenetic ‘programming.’ Of note, our finding at RBMS1 is of particular interest given the consistency of signal across time (in Sharp et al.) and across tissues (in our analyses). Genetic variants at RBMS1 have previously been associated with risk of type 2 diabetes [39–41], body mass index [42], and other adiposity phenotypes [42] in various populations, yet none of these variants are in close proximity to the CpG site identified. This suggests that both genetic and epigenetic variations at this locus may influence risk of metabolic diseases over the life course.
Strengths and Limitations
Our strengths include the use of a large number of cord blood and placenta samples. We also utilized the Illumina MethylationEPIC array, which is the most recent genome-wide array to investigate DNA methylation levels with coverage of over 850,000 CpG sites[43]. Our analyses utilized standardized phenotype information from our prospective cohort, as well as comprehensive data that allowed us to adjust for multiple confounders. Our analytic approach applied to examine the paired tissues jointly detected signals that would not have been detected in single tissue analyses with the same number of included individuals.
Our study also has limitations. Despite having access to two tissues, we still could not directly investigate tissues of interest (brain, liver, etc.) to support our hypothesis of overall foetal programming, as it is ethically not possible in human cohorts to have these tissues in healthy newborns. An additional limitation is that our cohort is comprised primarily of European women, so we may be unable to generalize our findings across ethnicities[27]. We also do not know if our DNA methylation findings have true biological functional consequences or are associated with longer term health outcomes in offspring. Further, Gen3G was part of the Sharp et al. meta-analysis, which may have resulted in some potential overfitting of lookups. However, the cohort contributed a smaller sample set of 170 infants (out of 9,340 mother–child pairs) using a previous dataset with cord blood DNA methylation using the Illumina 450k array, so the overlap is limited. Finally, we must be cautious in extending associations to maternal adiposity or other related metabolic factors, as BMI is a crude indicator of the maternal metabolic profile that may influence in utero environment [38].
Conclusion
DNA methylation alterations in cord blood or placenta tissue can broaden our understanding of mechanisms of foetal programming of offspring phenotypes leading to potential adverse outcomes over the life course. Our study found two CpG sites that were associated with early-pregnancy maternal BMI; and most notably, one located in a key regulatory region of CRHBP which plays a critical role in hypothalamic-pituitary-adrenal axis regulation and stress. Future studies are necessary to investigate potential functional downstream pathways of alteration of DNA methylation at this locus in key organs/tissues (e.g., brain) and whether identified DNA methylation markers are associated with later health outcomes in offspring life course.
Supplementary Material
Acknowledgments
We thank participants of the Gen3G cohort who contributed to this study, as well as clinical research nurses and research assistants for recruiting women and obtaining their informed consent. We also thank the CHUS biomedical laboratory for performing some of the assays used in this study. Finally, we thank the Harvard Medical School for providing the computational resources required to conduct the analyses.
Funding Statement
Work presented in this study was supported by an American Diabetes Association Pathways Award #1-15-ACE-26 (MFH); Gen3G has also been supported by Fonds de recherche du Québec en santé #20697 (MFH); Canadian Institute of Health Research #MOP 115071 (MFH), and Diabète Québec grants (PP and LB). NG was supported by a grant from the National Institute of Health (R01HD094150). SML work was supported by a grant from the NHLBI (K01HL125858). AC was supported by a grant from the National Institute of Environmental Health Sciences (R01ES031259). LB is a senior scholar from the Fond de la recherche du Québec en santé (FRQS);American Diabetes Association [1-15-ACE-26];Canadian Institutes of Health Research (CIHR)[115071];Diabète Québec;Diabète Québec;Fonds de Recherche du Québec - Santé;Fonds de Recherche du Québec - Santé [20697];National Institutes of Health (NIH)[R01HD094150];National Heart, Lung, and Blood Institute [K01HL125858].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Ethical disclosure
Institutional approval was obtained for Gen3G participants following the principles outlined in the Declaration of Helsinki. All women recruited in the study provided written informed consent prior to study enrollment.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Driscoll AK, Gregory E.. Increases in Prepregnancy Obesity: united States, 2016-2019. NCHS Data Brief. 2020;392:1–8. [PubMed] [Google Scholar]
- [2].Schummers L, Hutcheon JA, Bodnar LM, et al. Risk of adverse pregnancy outcomes by prepregnancy body mass index: a population-based study to inform prepregnancy weight loss counseling. Obstet Gynecol. 2015;125(1):133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shrestha D, Ouidir M, Workalemahu T, et al. Placental DNA methylation changes associated with maternal prepregnancy BMI and gestational weight gain. Int J Obesity. 2020;44. DOI: 10.1038/s41366-020-0546-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Maffeis C, Morandi A.. Effect of Maternal Obesity on Foetal Growth and Metabolic Health of the Offspring. Obes Facts. 2017;10(2):112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Martin CL, Jima D, Sharp GC, et al. Maternal pre-pregnancy obesity, offspring cord blood DNA methylation, and offspring cardiometabolic health in early childhood: an epigenome-wide association study. Epigenetics. 2019;14(4):325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Geurtsen M, Jaddoe V, Gaillard R, et al. Associations of maternal early-pregnancy blood glucose and insulin concentrations with DNA methylation in newborns. Clinical Epigenetics. 2020;12(1):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kadakia R, Zheng Y, Zhang Z, et al. Maternal pre-pregnancy BMI downregulates neonatal cord blood LEP methylation. Pediatr Obes. 2017;12(Suppl 1):57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fattah C, Farah N, Barry SC, et al. Maternal weight and body composition in the first trimester of pregnancy. Acta Obstet Gynecol Scand. 2010;89(7):952–955. [DOI] [PubMed] [Google Scholar]
- [9].Stein AD, Ravelli AC, Lumey LH. Famine, third-trimester pregnancy weight gain, and intrauterine growth: the Dutch Famine Birth Cohort Study. Hum Biol. 1995;67(1):135–150. [PubMed] [Google Scholar]
- [10].Zheng W, Huang W, Zhang Z, et al. Patterns of Gestational Weight Gain in Women with Overweight or Obesity and Risk of Large for Gestational Age. Obes Facts. 2019;12(4):407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gaillard R, Steegers EA, Franco OH, et al. Maternal weight gain in different periods of pregnancy and childhood cardio-metabolic outcomes. The Generation R Study. Int J Obesity. 2015;39(4):677–685. [DOI] [PubMed] [Google Scholar]
- [12].Hivert MF, Rifas-Shiman SL, Gillman MW, et al. Greater early and mid-pregnancy gestational weight gains are associated with excess adiposity in mid-childhood. Obesity. 2016;24(7):1546–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Godfrey KM, Reynolds RM, Prescott SL, et al. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017;5(1):53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen J, Li Q, Rialdi A, et al. Influences of Maternal Stress during Pregnancy on the Epi/genome: comparison of Placenta and Umbilical Cord Blood. Journal of depression & anxiety. 2014;3(2):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rakyan VK, Down TA, Thorne NP, et al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Res. 2008;18(9):1518–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Huang YT, Chu S, Loucks EB, et al. Epigenome-wide profiling of DNA methylation in paired samples of adipose tissue and blood. Epigenetics. 2016;11(3):227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Guillemette L, Allard C, Lacroix M, et al. Genetics of Glucose regulation in Gestation and Growth (Gen3G): a prospective prebirth cohort of mother-child pairs in Sherbrooke, Canada. BMJ Open. 2016;6(2):e010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pidsley R, Zotenko E, Peters TJ, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17(1):208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Du P, Zhang X, Huang -C-C, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11(1). DOI: 10.1186/1471-2105-11-587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bakulski KM, Feinberg JI, Andrews SV, et al. DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics. 2016;11(5):354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rahmani E, Zaitlen N, Baran Y, et al. Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies. Nat Methods. 2016;13(5):443–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Brägelmann J, Lorenzo Bermejo J. A comparative analysis of cell-type adjustment methods for epigenome-wide association studies based on simulated and real data sets. Brief Bioinform. 2019;20(6):2055–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Infinium MethylationEPICManifest Column Headings . (2014). Illumina. https://support.illumina.com/content/dam/illumina-support/documents/downloads/productfiles/methylationepic/infinium-methylationepic-manifest-column-headings.pdf
- [25].Kresovich JK, Zheng Y, Cardenas A, et al. Cord Blood DNA Methylation and Adiposity Measures in Early and Mid-childhood. Clin Epigenetics. 2017;9(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Turco MY, Moffett A. Development of the human placenta. Development. 2019;146(22):dev163428. [DOI] [PubMed] [Google Scholar]
- [27].Hivert M-F, Cardenas A, Allard C, et al. Interplay of Placental DNA Methylation and Maternal Insulin Sensitivity in Pregnancy. Diabetes. 2020;69(3):484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kalin NH. Corticotropin-Releasing Hormone Binding Protein: stress, Psychopathology, and Antidepressant Treatment Response. Am J Psychiatry. 2018;175(3):204–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Herman JP, McKlveen JM, Ghosal S, et al. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr Physiol. 2016;6(2):603–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Adinoff B, Iranmanesh A, Veldhuis J, et al. Disturbances of the stress response: the role of the HPA axis during alcohol withdrawal and abstinence. Alcohol health and research world. 1998;22(1):67–72. [PMC free article] [PubMed] [Google Scholar]
- [31].Ketchesin KD, Stinnett GS, Seasholtz AF. Corticotropin-releasing hormone-binding protein and stress: from invertebrates to humans. Stress. 2017;20(5):449–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Roy A, Hodgkinson CA, Deluca V, et al. Two HPA axis genes, CRHBP and FKBP5, interact with childhood trauma to increase the risk for suicidal behavior. J Psychiatr Res. 2012;46(1):72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].O’Connell CP, Goldstein-Piekarski AN, Nemeroff CB, et al. Antidepressant Outcomes Predicted by Genetic Variation in Corticotropin-Releasing Hormone Binding Protein. Am J Psychiatry. 2018;175(3):251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mina TH, Lahti M, Drake AJ, et al. Prenatal exposure to very severe maternal obesity is associated with adverse neuropsychiatric outcomes in children. Psychol Med. 2017;47(2):353–362. [DOI] [PubMed] [Google Scholar]
- [35].Robinson M, Zubrick SR, Pennell CE, et al. Pre-pregnancy maternal overweight and obesity increase the risk for affective disorders in offspring. J Dev Orig Health Dis. 2013;4(1):42–48. [DOI] [PubMed] [Google Scholar]
- [36].Rodriguez A. Maternal pre-pregnancy obesity and risk for inattention and negative emotionality in children. J Child Psychol Psychiatry. 2010;51(2):134–143. [DOI] [PubMed] [Google Scholar]
- [37].Miller CL, Pjanic M, Wang T, et al. Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat Commun. 2016;7:12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sharp GC, Salas LA, Monnereau C, et al. Maternal BMI at the start of pregnancy and offspring epigenome-wide DNA methylation: findings from the pregnancy and childhood epigenetics (PACE) consortium. Hum Mol Genet. 2017;26(20):4067–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Qi L, Cornelis MC, Kraft P, et al., Diabetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium . Genetic variants at 2q24 are associated with susceptibility to type 2 diabetes. Hum Mol Genet. 2010;19(13):2706–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kazakova EV, Chen M, Jamaspishvili E, et al. Association between RBMS1 gene rs7593730 and BCAR1 gene rs7202877 and Type 2 diabetes mellitus in the Chinese Han population. Acta Biochim Pol. 2018;65(3):377–382. [DOI] [PubMed] [Google Scholar]
- [41].Sajuthi SP, Sharma NK, Chou JW, et al. Mapping adipose and muscle tissue expression quantitative trait loci in African Americans to identify genes for type 2 diabetes and obesity. Hum Genet. 2016;135(8):869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pulit SL, Stoneman C, Morris AP, et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet. 2019;28(1):166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 2016;8(3):389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.