

Abstract
Cell-free DNA (cfDNA) in the circulating blood plasma of patients with cancer contains tumour-derived DNA sequences that can serve as biomarkers for guiding therapy, for the monitoring of drug resistance, and for the early detection of cancers. However, the analysis of cfDNA for clinical diagnostic applications remains challenging because of the low concentrations of cfDNA, and because cfDNA is fragmented into short lengths and is susceptible to chemical damage. Barcodes of unique molecular identifiers have been implemented to overcome the intrinsic errors of next-generation sequencing, which is the prevailing method for highly multiplexed cfDNA analysis. However, a number of methodological and pre-analytical factors limit the clinical sensitivity of the cfDNA-based detection of cancers from liquid biopsies. In this Review, we describe the state-of-the-art technologies for cfDNA analysis, with emphasis on multiplexing strategies, and discuss outstanding biological and technical challenges that, if addressed, would substantially improve cancer diagnostics and patient care.
Dying cells release their DNA into blood plasma, where it is fragmented by nucleases into cell-free DNA (cfDNA). cfDNA consists of short (≈160 nt) double-stranded DNA fragments that are continuously cleared from the bloodstream1–3 (the half-life of cfDNA is 5–150 min). cfDNA is therefore a ‘snapshot’ of the dying cells throughout the whole body, and it can be used to detect a broad and diverse set of diagnostic biomarkers for a variety of diseases. In particular, cfDNA has gained traction for cancer diagnostics over the past 5 years4–13, partly because of the high cost and complexity often associated with radiological examinations and tissue biopsies.
Circulating tumour DNA (ctDNA) is the subset of cfDNA molecules that are tumour-derived. Because ctDNA is analysed from a sample of cfDNA, the terms ‘cfDNA diagnostics’ and ‘ctDNA diagnostics’ are often used interchangeably. Tumour-specific mutations can be used to distinguish ctDNA from healthy cfDNA, and the fraction of cfDNA molecules at a particular genomic locus that bears a mutation is the variant allele fraction (VAF). The primary technical challenge of cfDNA-based diagnostics is the accurate detection and quantitation of mutation VAFs.
Diagnosing cancer on the basis of cfDNA is challenging because of the short length of cfDNA molecules, the low concentration of cfDNA in plasma, the high sequence similarity between cancer-derived and healthy human DNA, and the large number of markers that must be simultaneously analysed to achieve high clinical sensitivity. These challenges render older technologies for nucleic acid testing (in particular, methods based on the quantitative polymerase chain reaction (qPCR) for the identification of infectious pathogens) inadequate for cfDNA. Despite the innovations in high-throughput sequencing instruments, library-preparation methods and bioinformatics pipelines reported in the past 15 years, cfDNA diagnostics have yet to see widespread introduction in clinical settings, in part because no single dominant technology reliably, simply and affordably addresses all these challenges. In this Review, we discuss the current methodological limitations of cfDNA analysis for cancer diagnostics.
cfDNA diagnostics in cancer care
The United States Food and Drug Administration (FDA) defines an in vitro diagnostic as a test done on samples, such as blood or tissue, that have been taken from the human body. Within this broad definition, there are three definitions of diagnostics that are typically considered in the context of cancer: non-FDA-approved analytic tests based on patient-derived biospecimens that provide information on the presence, characteristics or evolution of the patient’s disease14; tests approved or cleared by the FDA via 510(k) (https://www.fda.gov/medical-devices/device-approvals-denials-and-clearances/510k-clearances), de novo (https://www.fda.gov/medical-devices/premarket-submissions/de-novo-classification-request) or pre-market approval pathways (https://www.fda.gov/medical-devices/premarket-submissions/premarket-approval-pma); and pathology tests for the definitive identification and classification of a malignancy. Here we use the first (and broadest) definition of a diagnostic test, which includes tests for screening, prognosis, therapy selection and post-treatment monitoring. Many of these tests have not received FDA approval or clearance; nonetheless, they are informative for cancer care and are recommended by guidelines (such as those of the National Comprehensive Cancer Network). The typical use of nucleic acid tests can be divided into three main stages: pre-analytical steps that result in a purified DNA sample, the analysis of the DNA sample and the clinical interpretation of the results. Using non-small-cell lung cancer (NSCLC) as a model disease, Fig. 1 describes some cfDNA tests and how they would fit into the clinical diagnostic workup-and-treatment workflow.
Fig. 1 |. cfDNA tests in clinical diagnostics for NSCLC, and treatment workflow.
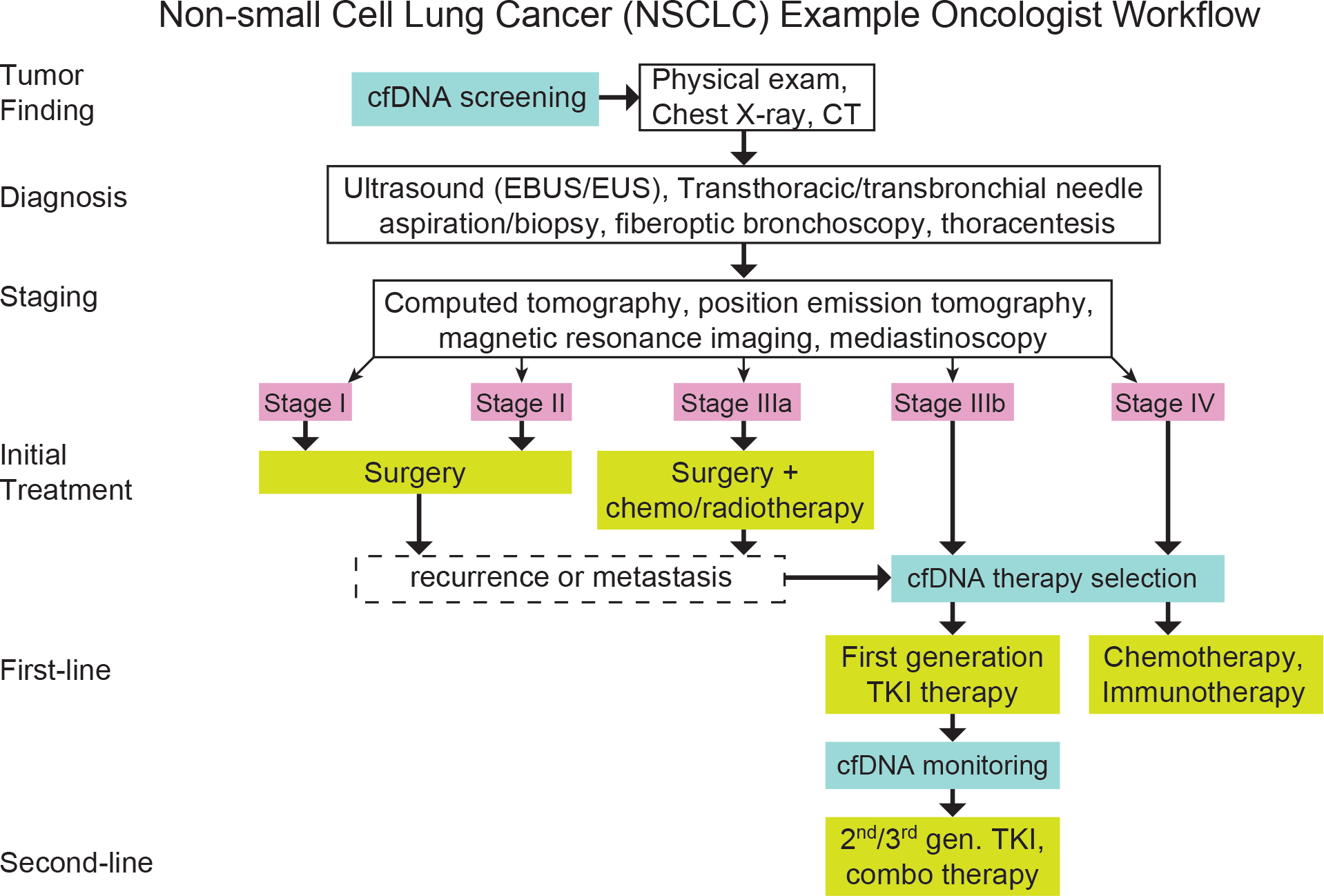
cfDNA screening can be used in combination with a physical exam, a chest X-ray and a CT scan. Approximately 70% of patients with NSCLC are diagnosed at a late stage (stages III or IV) following overt clinical symptoms. The most common use of cfDNA analysis is in therapy selection for patients at stages IIIb or IV. The technologies of cfDNA analysis can be used for post-treatment monitoring, including the detection of recurrence and de novo resistance mutations. TKI, tyrosine kinase inhibitor.
Today, approximately 70% of patients with NSCLC are diagnosed at late stages of the disease (stages III and IV) following overt clinical symptoms. Only 30% of patients are diagnosed at stage I or II, often following incidental findings or screening exams based on chest computed tomography (CT) scans15. Because patient outcomes are worse when NSCLC is detected at later stages (despite advances in targeted therapies and immunotherapies16–18), major initiatives in the United States19,20 and across the world21–24 are aiming to improve and expand early screening efforts.
Currently, the most common use of cfDNA analysis is in therapy selection for patients at stages IIIb and IV. For example, tests for mutations in the epidermal growth factor receptor (EGFR) stratify patients based on the likelihood of response to targeted therapies such as erlotinib23,25,26 or osimertinib24,27. There are more than 100 cfDNA diagnostic tests in clinical trials in the United States (https://clinicaltrials.gov/), and several commercial cfDNA-based laboratory-developed tests (LDTs) are being routinely ordered by oncologists28,29. In addition to specific mutation markers for resistance or sensitivity to targeted therapeutics such as tyrosine kinase inhibitors (TKIs), tumour-specific DNA can also be analysed more broadly for overall tumour mutational burden (TMB) that positively correlates with the efficacy of immunotherapies, such as inhibitors of programmed death-1 (PD-1; ref. 30), programmed death ligand-1 (PD-L1; ref. 31) or cytotoxic T-lymphocyte-associated protein 4 (CTLA4; ref. 32). The approximation of TMB from cfDNA analysis (known as blood tumour mutation burden, bTMB)33 is an important recent use case of cfDNA-based immunotherapy guidance. Similarly, genome-wide microsatellite instability correlates with immunotherapy effectiveness34,35, and represents another set of promising markers for cfDNA-based therapy guidance.
As technologies for cfDNA analysis advance36–41, they are also being considered for the assessment of post-treatment monitoring, including the detection of mutations associated with recurrence or de novo drug resistance, which may inform the modification of therapy regimens, especially if they involve combination therapies41–44. Recurrence monitoring has been applied in research settings for a number of different cancer types (including breast cancer45,46, colorectal cancer47,48 and lung cancer49). We expect that cfDNA-based cancer-monitoring tests will soon become commercially available.
The possibility of early cancer screening via cfDNA diagnostics has been widely discussed11,50,51,55. In a limited number of cancer types in which high-risk individuals can be identified by age, lifestyle habits or geographic locations, cancer screening via cfDNA is becoming a reality (this is the case for colorectal cancer52,53 and nasopharyngeal cancer53,54). However, the recent discovery of a notable presence of cancer-associated mutations in healthy individuals56,57 suggests that it will be challenging to develop diagnostic tests with high sensitivity and specificity for early pan-cancer detection in asymptomatic populations. These challenges are exacerbated by the cost and dangers associated with applying diagnostic workups to healthy individuals.
Cytosine methylation in cfDNA is being explored as a marker for the detection of early cancers. As with TMB for mutations, global methylome profiling does not consider individual methylation markers at specific genomic loci; rather, genome-wide hypomethylation has been suggested as a universal marker for cancer58. On the one hand, because genome-wide methylation does not inform about the location of the tumour, and because epigenetic features are less conserved across cell divisions, clinical actionability is limited to the early detection of cancer. On the other hand, approaches relying on targeted bisulfite sequencing59–61 are more suitable for the detection of specific cancer types. In fact, combined with bioinformatics including haplotype phasing62, methylation markers on cfDNA can be used to identify the tissue of origin63, and may represent a substantial advantage for early detection applications.
There are numerous clinical trials that employ mutational analysis of cfDNA. Owing to the long timeframes required for prospective clinical trials, many ongoing trials using cfDNA as a stratification marker (such as NCT02418234 for non-small-cell lung cancer, NCT00730158 for colorectal cancer, and NCT01349959 for breast cancer) are based on low-plex PCR methods. With the maturation of next-generation sequencing (NGS) technology, multiple clinical trials relying on NGS of specific gene panels using cfDNA samples have been initiated by companies (NCT02889978 by Grail, NCT03477474 by Guardant Health, and NCT02620527 by Foundation Medicine).
Pre-analytical factors and limitations
Pre-analytical factors describe the biological variables and handling protocols of the sample. And because different protocols impact the quality, quantity or characteristics of the DNA sample to be analysed (Fig. 2a), they can have an especially outsized impact on the overall accuracy of the tests.
Fig. 2 |. Pre-analytical factors impacting the accuracy of cfDNA analysis.
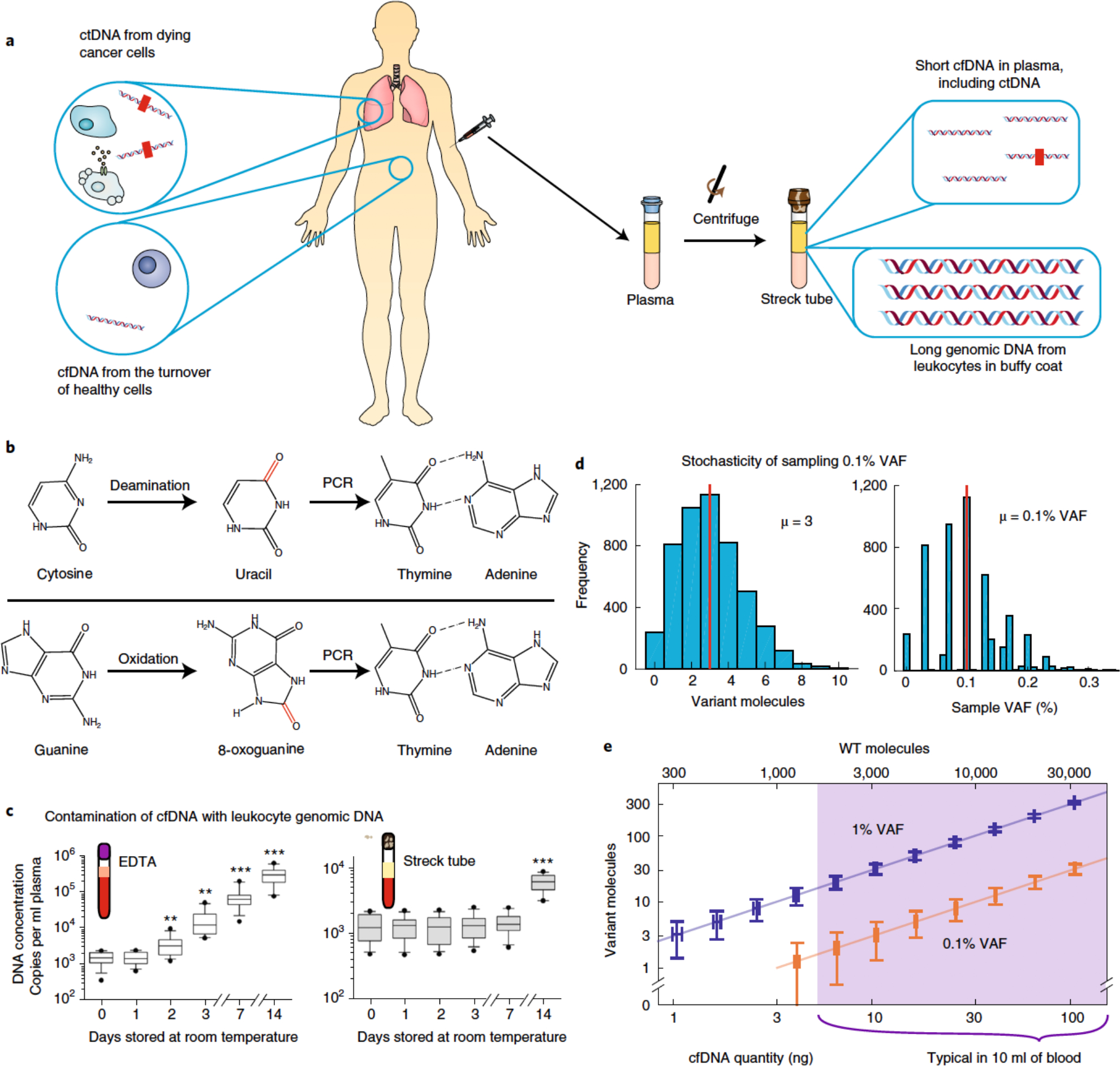
a, Whereas the buffy-coat layer of blood is rich in genomic DNA from peripheral mononuclear blood cells, blood plasma contains relatively low quantities of extracellular DNA (that is, cfDNA). cfDNA results from dying cells in the entire body, including healthy cells (depicted in white) and tumour cells (shown in brown) dying from apoptosis, necrosis and immune cytotoxicity. Thus, only a small fraction of cfDNA comprises tumour-derived ctDNA. The red rectangles within the ctDNA strands denote tumour-specific mutations. b, cfDNA in blood may be damaged during sample collection, transport and storage, resulting in modified nucleosides that are incorrectly recognized by DNA polymerases during PCR amplification. This leads to amplicon DNA sequences with variants that may be interpreted as cancer-specific mutations. The schematics show cytosine deamination and guanine oxidation, the two most commonly observed types of DNA damage. c, Contamination of cfDNA with genomic DNA from leukocytes. Except for blood cancers, genomic DNA from leukocytes will not contain cancer-specific ctDNA. Thus, contamination of cfDNA with leukocyte genomic DNA will dilute the fraction of cfDNA that contains useful information, rendering the mutation analysis of downstream DNA more difficult and the quantitation of VAF less accurate. EDTA, ethylenediaminetetraacetic acid. d, Poisson distribution of tumour-mutation molecules and VAF in a blood sample. An adult human has roughly 5 l of blood in circulation, and sampling 10 ml of blood for cfDNA analysis introduces variations in VAF owing to small-number statistics. Assuming a ‘ground truth’ of 0.1% cancer-mutation VAF in the entire 5 l of blood supply and a 10 ng sample of cfDNA in a 4 ml plasma sample, the number of cancer-mutation molecules present will range between 0 and 10, corresponding to an observed VAF range of 0–0.3% for any given DNA locus. No technology improvements can transcend this sampling variation; only the use of larger-volume blood samples can mitigate this VAF-irreproducibility challenge. The Matlab code used to generate these results is provided as Supplementary Information. e, Visualization of molecule-number variations owing to cfDNA sampling. The vertical and horizontal error bars show the analytically calculated standard-deviation values for different cfDNA input quantities and mutation VAFs. WT, wild-type. Panel c adapted from ref. 71.
Many biological variables that impact the quantity and characteristics of cfDNA are difficult to fully control. For example, cfDNA is partially cleared through the urine, so an individual who has recently imbibed a large amount of fluids may have lower concentrations of cfDNA. Also, because cfDNA is derived from all dying cells in the body, an individual’s physical health state, which is influenced by exercise64 and by any bacterial or viral infection65–67, will affect cfDNA concentrations. Moreover, the quantity of tumour-derived ctDNA molecules in cfDNA depends on the mass of the tumours and their proximity and accessibility to the circulatory system.
In contrast to biological variables, sample-collection and sample-handling protocols could, in principle, be fully controlled to maximize the reproducibility of results from two aliquots of the same blood sample. Ideally, fresh venous blood samples should be immediately centrifuged to separate the plasma from red blood cells and the buffy coat (this step is typically done twice, to minimize contamination from the buffy coat). Subsequently, the cfDNA should be immediately extracted from the plasma, followed by downstream analysis by digital PCR (dPCR) or by NGS. In practice however, there are often unavoidable delays associated with the transport, aliquoting and storage of the blood sample. These have two main risks: chemical damage of the cfDNA (Fig. 2b) and contamination of cfDNA by leukocyte genomic DNA (Fig. 2c). DNA can in fact undergo hydrolysis, deamination and oxidative damage both in vivo in the body and in vitro in the blood collection tube68,69 (more than 20 types of damage have been identified70). The products of these undesired chemical reactions are non-canonical nucleosides that can be spuriously recognized as mutations, and these processes are suspected to impact cfDNA analysis accuracy. Although there are some commercial kits (such as the New England Biolab’s preCR) that claim to repair such damage, the general consensus is that damage repair is imperfect and results in many false-positive variant calls. A highly specific and high-yield method for reversing DNA damage could greatly improve the ultimate limits of cfDNA-based diagnostics.
In collection tubes, leukocytes in the blood slowly die and release their genomic DNA into the plasma layer. This genomic DNA contributes to an increased background of wild-type DNA, and reduces the effective mutation VAF, rendering the detection of rare cancer mutations more difficult. Whole blood can be stored for 1–7 days at room temperature before incurring an obvious increase in the quantity of plasma DNA owing to leukocyte genomic DNA71. Some blood collection tubes increase the stability of leukocytes in collected blood samples67,71. Methods for improving the stability of leukocytes in blood, or for differentiating cfDNA from leukocyte genomic DNA, would improve the accuracy and reproducibility of cfDNA diagnostics.
The low quantity of cfDNA in blood and the low VAFs of cancer mutations mean that Poisson sampling statistics can reduce the reproducibility of the profiling of mutation VAFs (Fig. 2d,e). This limitation implies that the detection and quantitation of low-VAF mutations can suffer from irreproducibility and lowered clinical sensitivity, regardless of the downstream analysis technology. The only way to overcome this limitation is through the use of larger quantities of cfDNA; for this reason, many commercial LDTs for cfDNA require 2 tubes of 10 ml blood as input, which allows near 100% clinical sensitivity at 1% VAF, and over 90% sensitivity at 0.1% VAF. An adult human can lose up to 14% (roughly 700 ml; ref. 72) of their total blood supply (approximately 5 l) without experiencing substantial adverse effects. However, for some patients with cancer, it is practically difficult to collect more than about 50 ml of blood for cfDNA analysis.
Because a substantial fraction of cfDNA is filtered by the kidneys into the urine, this biofluid is another source of cfDNA molecules73. Adults usually pass between 800 ml and 2 l of urine per day, all of which could potentially be collected and used for extracting cfDNA without any adverse effects. However, the high-yield purification and concentration of cfDNA from large volumes of urine is technically challenging. Also, although urine restricts contamination from genomic DNA from cellular debris, cfDNA molecules in urine are substantially shorter than cfDNA molecules in blood74, which presents unique challenges for cfDNA extraction yields75,76 and for PCR amplification. Biochemical and physical methods that reliably and affordably purify and analyse cfDNA at scale from urine could revolutionize cfDNA-based cancer diagnostics.
Low-plex approaches to cfDNA analysis
Traditional nucleic acid tests (such as those used for the detection of HIV and other viral infections, and of methicillin-resistant Staphylococcus aureus (MRSA) and other pathogens) use low-plex instruments and assays that detect a small number of target DNA sequences. Typically, these assays are ran using qPCR77; other FDA-approved detection assays involve chemiluminescence78,79, isothermal DNA amplification80 or transcription-mediated amplification81. In these tests, there is typically a single binary decision (such as whether anti-retroviral therapy should be used) that the nucleic acid test is meant to inform. However, cancer is a complex disease with many different biological pathways and many different treatment options. For example, there are dozens of FDA-approved targeted therapies and immunotherapies for NSCLC (https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm279174.htm); furthermore, many drugs can be used in combination to maximize a therapeutic effect (https://clinicaltrials.gov/)28. Consequently, more information is typically required than simply the presence or absence of a tumour. The presence of specific DNA mutations can not only inform the therapy regimens most likely to be effective for a patient, but also provide snapshots of tumour responses to the treatment and the potential emergence of drug-resistant tumours. The My Cancer Genome database (https://www.mycancergenome.org/) lists hundreds of mutations with known effects on cancer treatment, and other databases (such as the Catalog Of Somatic Mutations In Cancer (https://cancer.sanger.ac.uk/cosmic) and cBioPortal (http://www.cbioportal.org/)) compile more than 100,000 mutations that have been observed in patients with cancer.
Because cfDNA exists in plasma in very low quantities (about 2.5 ng of cfDNA per ml of plasma in healthy individuals, and about 10 ng of cfDNA per ml of plasma in patients with cancer), repeated low-plex testing on different sample aliquots is not practical. Consequently, low-plex tests such as those based on qPCR typically target one or a few specific mutations to guide the use of a single drug. For example, the presence of the EGFR-T790M mutation in a patient with lung cancer confers resistance to erlotinib82, informs the use of osimertinib83 and can alternatively be used for the detection of recurrence in erlotinib-resistant tumours.
Currently, dPCR is the most used method for the low-plex analysis of cfDNA. By performing end-point PCR on 20,000 individual reaction droplets, Bio-Rad’s digital droplet PCR (ddPCR) allows for the accurate detection and quantitation of known DNA mutations without separate calibration reactions84. Compared with commercial qPCR assays (such as the amplification refractory mutation system85), ddPCR assays achieve a better VAF limit of detection (by roughly 20-fold; 0.05% versus 1%) as well as a more accurate quantitation of VAF. However, the high cost of ddPCR instruments (list price, roughly $100,000) and the low number of installed ddPCR instruments are a challenge for the widespread adoption of ddPCR-based cfDNA assays, especially when compared with NGS instruments with similar cost and much higher multiplexing (Fig. 3). Because ddPCR is currently only capable of analysing one potential mutation per reaction, an unreasonably high quantity of cfDNA samples would be needed to profile many different mutations.
Fig. 3 |. NgS for cfDNA analysis.
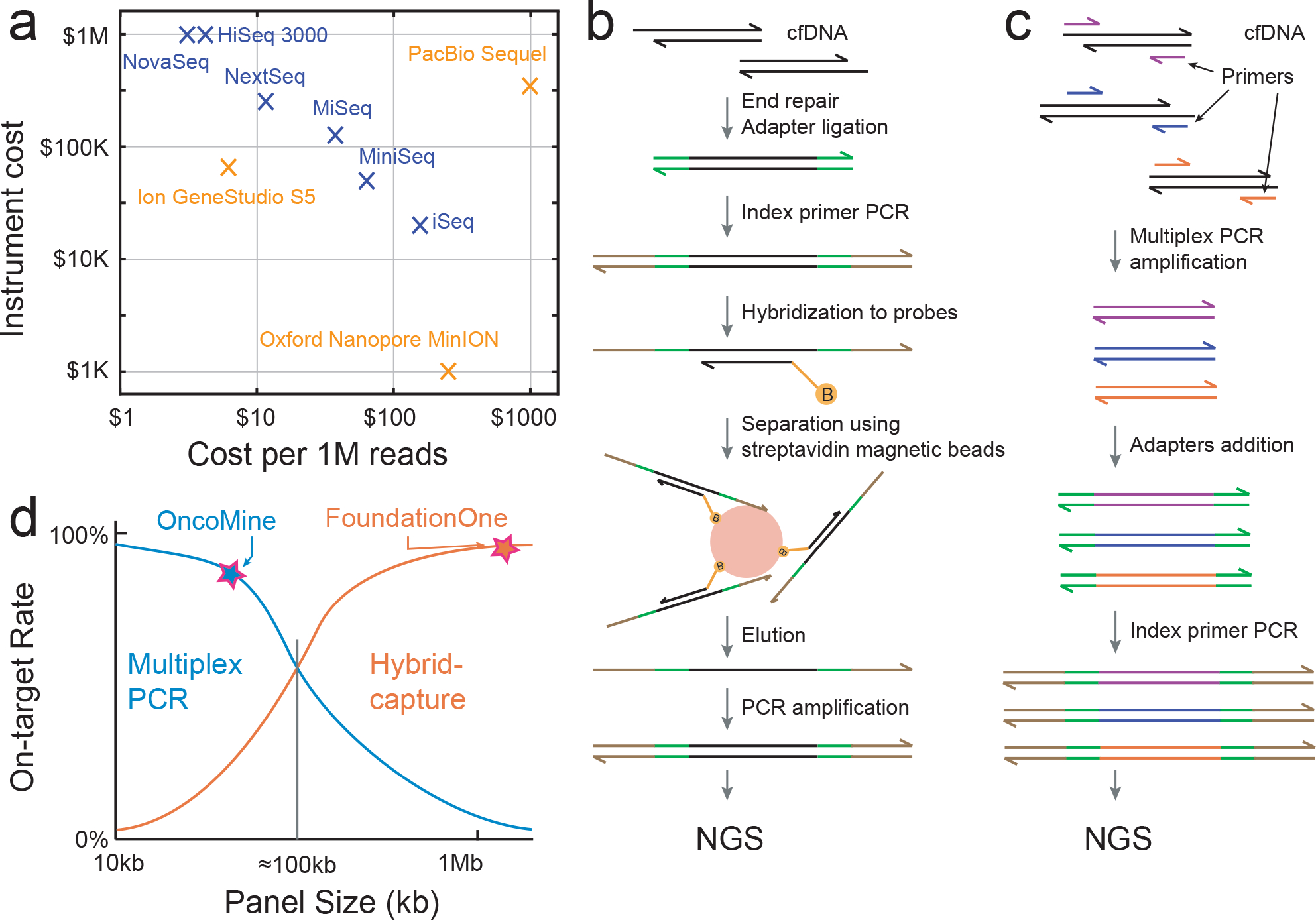
a, Current sequencing platforms and their cost. b, Example of NGS library-preparation workflow for target enrichment via ligation and hybrid capture. B (orange circle) represents a biotin label. c, Example of NGS library-preparation workflow for target enrichment via multiplex PCR. d, Comparison of hybrid-capture and multiplex-PCR target enrichment in terms of on-target rate and panel size. The approximate panel sizes and on-target rates of the commercial panels Oncomine (multiplex PCR) and Guardant360 (hybrid capture) are shown.
There are other methods for the low-plex detection of mutations with low VAF in cfDNA. These include electrochemistry86,87, isothermal amplification with CRISPR (clustered regularly interspaced short palindromic repeats)88, nanoparticles87 and single-molecule fluorescence89. These emerging technologies have primarily focused on improving analytic sensitivity, by either requiring lower quantities of input cfDNA or by detecting lower-mutation VAFs. More efforts should be devoted towards the massive scale-up of the multiplexing capabilities of these technologies, to render them suitable for the broader analysis of cfDNA for cancer diagnostics.
At the border between low-plex technologies and massively multiplexed technologies, the MassARRAY system90 (Agena) allows for many different primers to be added to the same reaction. A potential single-nucleotide extension is performed, and the potential extension products are then simultaneously analysed via mass spectrometry. The most recent assays claim a limit of detection of 0.1% mutation VAF and the detection of up to 40 different known mutations from a single sample91. This performance puts the MassARRAY system at roughly the needed multiplexing capacity for the actionable detection of mutations for the diagnosis of individual cancer types. Also, the low marginal cost of sample testing and the rapid workflow make the system an attractive alternative to NGS methods. As with digital PCR, the major adoption barrier for MassARRAY is the high up-front cost of the instrument ($250,000).
NGS methods for cfDNA analysis
In NGS, DNA molecules in a solution that bear pre-defined adapter sequences are randomly sampled, and the NGS instrument provides the sequences of the sampled molecules from the 5′ end up to a defined length limit (the read length). For the popular NGS instruments and sequencing kits commercialized by the company Illumina, the read length varies between 75 and 300 nt, and the number of reads (sampled molecules) varies between 4 million and 10 billion. Other major NGS platforms include those of Ion Torrent, Oxford Nanopore and Pacific Biosciences (Fig. 3a).
NGS offers orders-of-magnitude higher multiplexing capabilities than other technologies for the analysis of nucleic acids, and is thus the dominant approach for cfDNA analysis. Because cancer mutations follow a long-tailed distribution, a very large number of genetic loci should be simultaneously observed to ensure high clinical sensitivity92. Also, the cost of performing NGS has been dropping exponentially (halving roughly every 18 months93).
Illumina NGS systems are commonly used for cfDNA analysis because of the high accuracy and the low marginal cost of NGS reads (Fig. 3a). In India and other countries, Ion Torrent NGS instruments have gained higher market penetration than those of Illumina owing to the lower up-front cost of the instrument. The third-generation NGS platforms (from Oxford Nanopore and Pacific Biosciences)94,95 are not currently competitive for cfDNA analysis because the primary advantage of these platforms are long-read lengths of more than 10,000 nt (cfDNA is about 160 nt long).
If all the NGS reads are deployed to randomly sample all cfDNA molecules in a plasma sample, then the fraction of all reads that corresponds to useful information about cancer-related genes will be very small. The human genome is more than 3 × 109 nt long, and current knowledge in cancer biology is limited to roughly 1,000 possibly cancer-related genes96, each with about 4,000 nt of protein-coding sequence, corresponding to a total of 4 × 106 nt. Hence, simplistically, roughly 99.9% of the NGS reads would be wasted on portions of the human genome with little information that is cancer-relevant, resulting in grossly increased NGS costs. Target enrichment is the process by which the composition of the cfDNA library is adjusted to increase the relative concentrations of DNA sequences corresponding to the genomic loci of interest. The two most popular methods for target enrichment today are ligation and hybrid capture, and multiplex PCR.
In a typical ligation-and-hybrid-capture workflow (Fig. 3b), cfDNA first undergoes end-repair to produce flush ends with a single 3′ A tail. Adapters are then ligated to both ends of the duplex. Index primers are further appended via PCR using primers against the universal adapter sequences (this step also serves to pre-amplify the cfDNA). Subsequently, the amplicons are denatured and then hybridized to biotinylated probe sequences. Streptavidin-coated magnetic beads are then used to capture the biotinylated probes and any cfDNA amplicons bound to the probes (other cfDNA amplicons are removed via washing). The probes correspond to the genes or loci of interest; consequently, the captured cfDNA amplicons will be enriched in these genes or loci. However, owing to the non-specific binding of cfDNA amplicons to the probes or the magnetic beads, enrichment is imperfect. Hybrid-capture probes are available commercially (from Twist Biosciences, Integrated DNA Technologies, Nimblegen and Agilent). NGS panels for cfDNA that rely on hybrid capture are also available (from FoundationACT32, Guardant360 (ref. 33) and Roche Avenio97).
Target enrichment via multiplexed PCR allows for different genes or loci to be simultaneously amplified using different PCR primer sequences98,99, and adapters and indexes are appended afterwards using PCR or ligation (Fig. 3c). With the large number of PCR primers present, some amount of primer dimers and non-specific amplicons from other regions of the genome are likely to form. A majority of these undesired molecules can be removed by size-selection steps in the NGS library-preparation process (for example, by using Agencourt AMPureXP100) to remove primer dimers and non-specific amplicons with grossly different lengths than the expected amplicons. Because the amplicon concentration of the loci of interest doubles with every PCR cycle, the fold-enrichment can be much higher than that of hybrid capture. For example, with 20 PCR cycles, the loci of interest are enriched up to 106-fold, whereas it is difficult even with optimized hybrid-capture protocols to ensure that non-specific binding is less than 1 part in 104. Thus, multiplexed PCR is generally able to achieve higher on-target rates than hybrid capture, especially for smaller NGS panels (the on-target rate of an NGS library is the fraction of all reads that correspond to the genes or loci of interest). Additionally, performing multiplexed PCR is generally less complicated than performing hybrid capture, as it has shorter total turnaround and hands-on times. Commercial NGS panels from Thermo Fisher Oncomine and Paragon CleanPlex use multiplexed PCR for target enrichment101,102.
On-target rates of hybrid-capture methods are generally high for panels larger than 100 kilobases (kb), but low for smaller panels. In contrast, on-target rates of multiplex PCR methods are generally high for panels smaller than 10 kb, but low for larger panels. In general, ligation and hybrid capture is preferred for large NGS panels covering over 100 kb, and multiplex PCR is preferred for small panels covering less than 10 kb (Fig. 3d).
Both methods for the preparation of NGS libraries face the same three main limitations: PCR amplification and NGS read errors that result in false-positive variant calls; imperfect representation of the original cfDNA molecules in the NGS library, resulting in false negatives; and sequencing non-uniformity that either reduces mutation sensitivity or substantially increases costs (Fig. 4).
Fig. 4 |. Primary limitations of NgS-based cfDNA analysis.
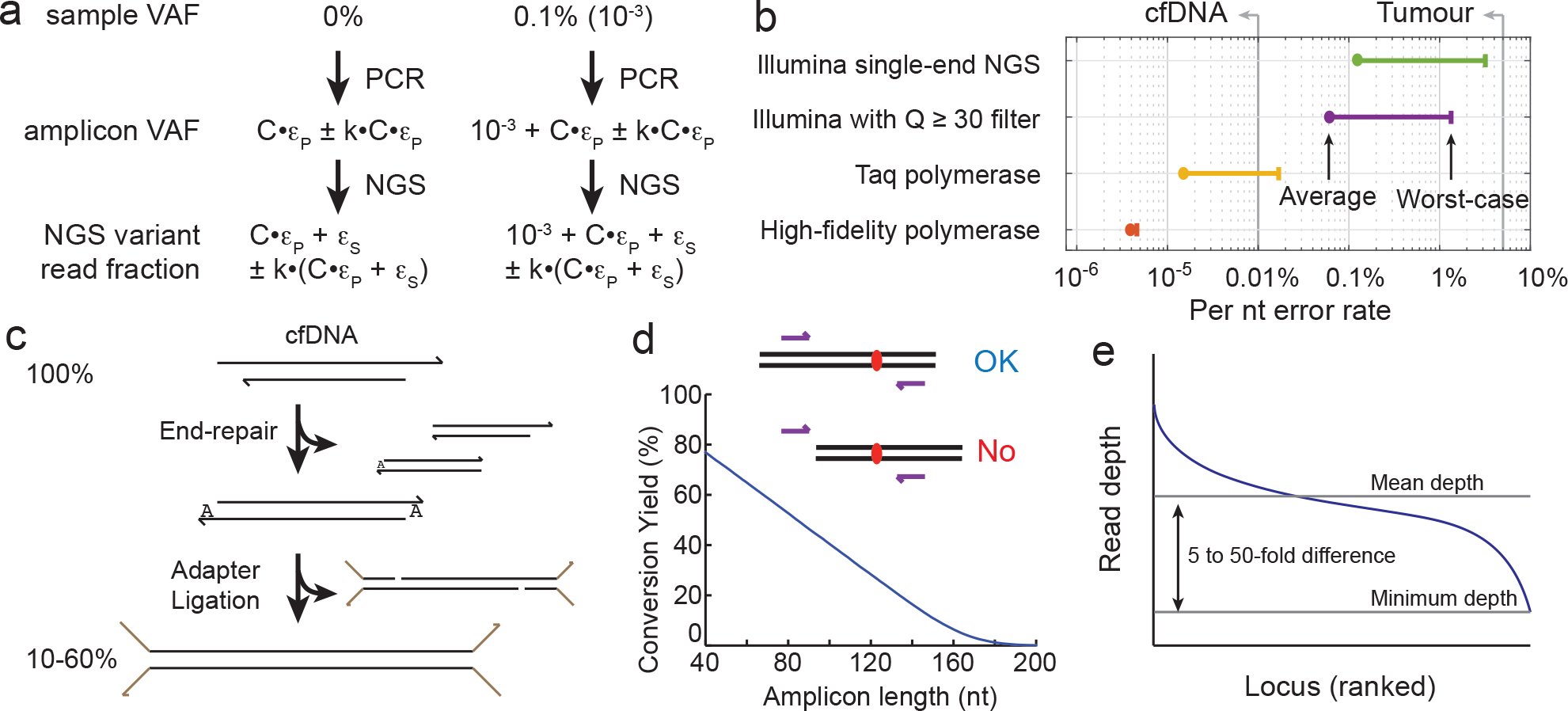
a, Misincorporation errors by the DNA polymerase and intrinsic errors of NGS constrain the limit of detection of mutations. The limit of detection is defined as the lowest VAF of a mutation that can be confidently distinguished from a pure WT sample (which would correspond to a 0% VAF). The PCR misincorporation rate (ϵP), the number of PCR cycles (C) and the NGS intrinsic error rate (ϵS) all increase the fraction of NGS reads that correspond to variant sequences for a 0% VAF sample. Owing to variations in the error rates, which depend on experimental protocol minutiae, the actual fraction of NGS reads corresponding to variants will vary from run to run. Consequently, the combined error rate (C × ϵP + ϵS) should be much (2-fold or more) lower than the limit of detection. k is a sequence-dependent constant. b, Typical error rates for PCR amplification and NGS147–152. All error rates exhibit some sequence bias. The plot shows average error rates and worst-case error rates. Single-pass NGS intrinsic errors are lowest for Illumina platforms. Average sequencing error rates for single-pass sequencing by Ion Torrent, Pacific Biosciences and Oxford Nanopore range from 1% to 20%. The DNA-polymerase error rates shown are per extension; through the course of a PCR reaction, the misincorporation rate is multiplied by the number of cycles. High-fidelity polymerases refer to enzymes with 3′ > 5′ proofreading capabilities. The three high-fidelity polymerases most frequently used for NGS are Phusion, NEB Q5 and KAPA HiFi. The vertical lines labelled ‘cfDNA’ and ‘Tumour’ indicate the currently achievable limit of detection for VAF based on NGS. c, Imperfect end-repair and ligation efficiencies limit the conversion yield of cfDNA for ligation and hybrid-capture protocols. The conversion yield is the fraction of the original cfDNA molecules represented as amplicons in the NGS library. For ligation and hybrid-capture workflows, conversion yield is primarily bottlenecked by ligation efficiency, and secondarily by DNA extraction and DNA end-repair. The conversion yields listed in the literature are typically high-end estimates, because there are different ways to estimate the total quantity of input cfDNA, and they can differ by a factor of 2 or more. d, cfDNA breakpoints limit the conversion yield of cfDNA for multiplex PCR protocols. Long amplicons have a high probability of not being able to amplify the original cfDNA molecule of interest, owing to the original molecule not spanning the nucleotides of the desired amplicon. e, Non-uniformity increases the total NGS reads needed to ensure the minimum depth needed for achieving a defined limit of detection. The mean NGS read depth can be calculated as the total NGS reads multiplied by the on-target rate and divided by the amplicons or loci; however, the minimum depth can be a factor of 5 to 50 lower than the mean depth. Because sequencing depth limits analytical sensitivity to low-VAF mutations, some mutations will have worse (higher) VAF limits of detection than others. The Matlab code used to generate these results is available as Supplementary Information.
Errors in PCR amplification and in NGS can result in NGS reads containing variant sequences even when the sample is purely wild-type (that is, 0% in VAF). Theoretically, if the error rate was absolutely reproducible from procedure to procedure, then the actual mutation VAF of an unknown sample could be mathematically computed by subtracting the expected false-variant reads owing to errors. For example, if 2% of the reads from an NGS library contain a particular mutation, and the aggregated errors result in a reproducible 1% of NGS reads being that variant, then one can infer the true sample VAF as 2% – 1% = 1%. In practice, however, there are run-to-run variations owing to different enzyme lots and slight differences in experimental temperatures, times and concentrations; thus, the aggregated error rate is not perfectly reproducible. Furthermore, the error rate is not identical for all sequences; it can vary depending on the exact nucleotide being sequenced and its neighbouring sequences. Hence, it is difficult to standardize NGS panels to confidently claim the detection of mutations below about 1% in VAF, even when using high-fidelity polymerases to reduce PCR errors and the filtering of Phred quality score to Q ≥ 30 (which indicates a mean error rate of 0.1% per nucleotide, based on manufacturer specifications) to reduce the intrinsic errors of NGS (Fig. 4a,b). Moreover, because of the large number of nucleotides sequenced and sequence biases in error rate, variant calls at ≤1% cannot be reliably made even with Q ≥ 30 filters.
The conversion yield—the fraction of original cfDNA molecules that are represented in the final NGS library—depends on whether the library-preparation method used is ligation and hybrid capture or multiplex PCR. For example, a 10 ml blood sample could contain a single-digit number of copies of DNA molecules with tumour-specific mutations; hence, protocols with low conversion yields could end up losing all the mutant DNA molecules and report a false negative. Therefore, conversion yield is an important determinant of the clinical sensitivity of cfDNA assays. Panel developers are incentivized to report optimistic numbers, based on the lowest reasonable estimate of the number of original cfDNA molecules present at a particular genomic locus. Standard DNA-quantitation methods based on the fluorescence of an intercalating dye (Qubit), the absorbance of DNA at 260 nm (measured by Nanodrop) and ddPCR can differ by more than 2-fold, depending on the DNA-size distribution, the presence and concentration of single-stranded nucleic acids, DNA sequence, fragmentation patterns, solution buffer and chemical impurities. Thus, conversion-yield metrics today are estimates based on imperfect underlying measurements of cfDNA quantity.
For ligation and hybrid capture, the conversion yield is primarily limited by imperfect end-repair and ligation efficiencies (Fig. 4c). Because both ends of a cfDNA fragment must be ligated to adapters for the cfDNA to be amplified in subsequent steps, imperfect ligation yields have a quadratic effect on the conversion yield. In some specific library-preparation protocols (such as DuplexSeq103) that rely on ligation to both strands of the same cfDNA fragment for further error correction, all four ligation reactions must be complete for the molecule to be represented. Reported conversion yields for ligation and hybrid capture vary between 10% and 60% (refs. 103–105).
For multiplex PCR, the conversion yield is primarily limited by the fraction of cfDNA molecules that cannot be amplified because the molecules do not span the length of the amplicon (Fig. 4d). Assuming that the typical length of cfDNA is 160 nt (refs. 106,107), the theoretical conversion yield of multiplex PCR can be calculated on the basis of the length of the amplicon: a 100 nt amplicon would exhibit a conversion yield of approximately (160–100)/160 = 37.5%, and conversion yield would drop precipitously for longer amplicons. Furthermore, recent studies suggest the existence of a population of very short ctDNA molecules in blood108,109 that had not been systematically characterized owing to limitations in DNA extraction and NGS-library preparation. Multiplex PCR analysis of this short-ctDNA population would probably lead to very low yields. Exosomal DNA also exists in blood plasma and contains cancer-specific DNA mutations110,111. The longer lengths of exosomal DNA (more than 2,500 nt) render these fragments relatively easy to amplify by PCR.
In ligation and hybrid capture, and multiplex PCR protocols, some loci or amplicons are sequenced to much higher depth than others (Fig. 4e), owing to sequence-based hybridization kinetics that impact the amplification yield of PCR and the efficiency of hybrid capture. The rate constants of DNA-hybridization kinetics can vary by more than three orders of magnitude for primers or probes of the same length at the same temperature and buffer conditions112. The concentrations of different PCR primers or hybridization probes can be adjusted to counteract the differences in kinetics between targets (for example, by increasing the concentrations of primers or probes with slow kinetics) to make the NGS read depths more uniform; however, the adjustment is imperfect, and for commercial NGS panels there is typically a 5- to 50-fold gap between the mean and minimum sequencing depths.
Unlike sequencing error and conversion yield, sequencing non-uniformity increases the cost of achieving a desired sensitivity rather than setting hard limits on sensitivity. For example, sequencing to 200× depth is typically sufficient to make confident mutation calls at 5% VAF; thus, sequencing to 1,000× mean depth is sufficient to ensure a 5% VAF limit of detection for all loci in an NGS panel with a 5-fold gap between mean and minimum depths. Nonetheless, for cfDNA analysis where typical commercial panels (such as Guardant360) perform NGS reads per sample costing over $1,000 (against a list price of $6,000), reducing cost through improved depth uniformity remains a priority.
Technologies using unique molecular identifiers
In the cfDNA of patients with cancer, the VAF of cancer-specific mutations can vary between 0.01% and 10%, depending on disease stage and patient-specific disease characteristics4. Mutations in cfDNA may have low VAFs because the disease is at an early stage and the tumour mass is small, or because subclonal mutations are present in only a subset of the tumour cells. Subclonal mutations are especially important for therapy selection, because rare subclones with resistance mutations can lead to rapid treatment failure owing to subclone expansion under therapy42. Achieving a limit of detection of mutation VAFs of 0.1% or lower is thus crucial in clinical settings.
Unique molecular identifiers (UMIs) are currently the most popular method for overcoming the PCR and NGS errors, to reliably detect and quantitate mutations at ≤0.1% VAFs40,113–115. The NanoSeq method can achieve error rates of less than 10−9 errors per base pair (bp)116. SaferSeqS can detect VAFs as low as 1 in 100,000 DNA template molecules with a background mutation rate of <5 × 10−7 mutants per bp (ref. 117). The key idea of UMIs is to attach a unique DNA sequence to each original molecule of DNA in the cfDNA sample (Fig. 5a). When the UMI is subsequently PCR-amplified and sequenced, all NGS reads with the same UMI sequence can be interpreted as being derived from the same original DNA sequence. Spurious mutation reads generated from PCR or NGS errors are likely to be a minority of the reads within the family of reads with the same UMI sequence (Fig. 5b); instead, true mutations will generate families of reads in which all or a majority of the reads have the mutation (Fig. 5c). The bioinformatic interpretation of NGS data with UMIs starts with grouping different NGS reads into ‘UMI family’ reads on a DNA locus with an identical UMI sequence. Subsequently, a ‘vote’ is taken for each UMI family, with the identified dominant or majority sequence being accepted as the true sequence of the original DNA molecule. Using UMIs, many PCR and NGS errors can be corrected, and the limit of detection of mutation VAFs can be brought below the PCR and NGS error rates. UMIs can be applied in both ligation and hybrid capture, and multiplex PCR protocols (although it is more difficult for the latter when the plex number is high).
Fig. 5 |. Methods for the accurate detection of mutations with ≤1% in VAF.
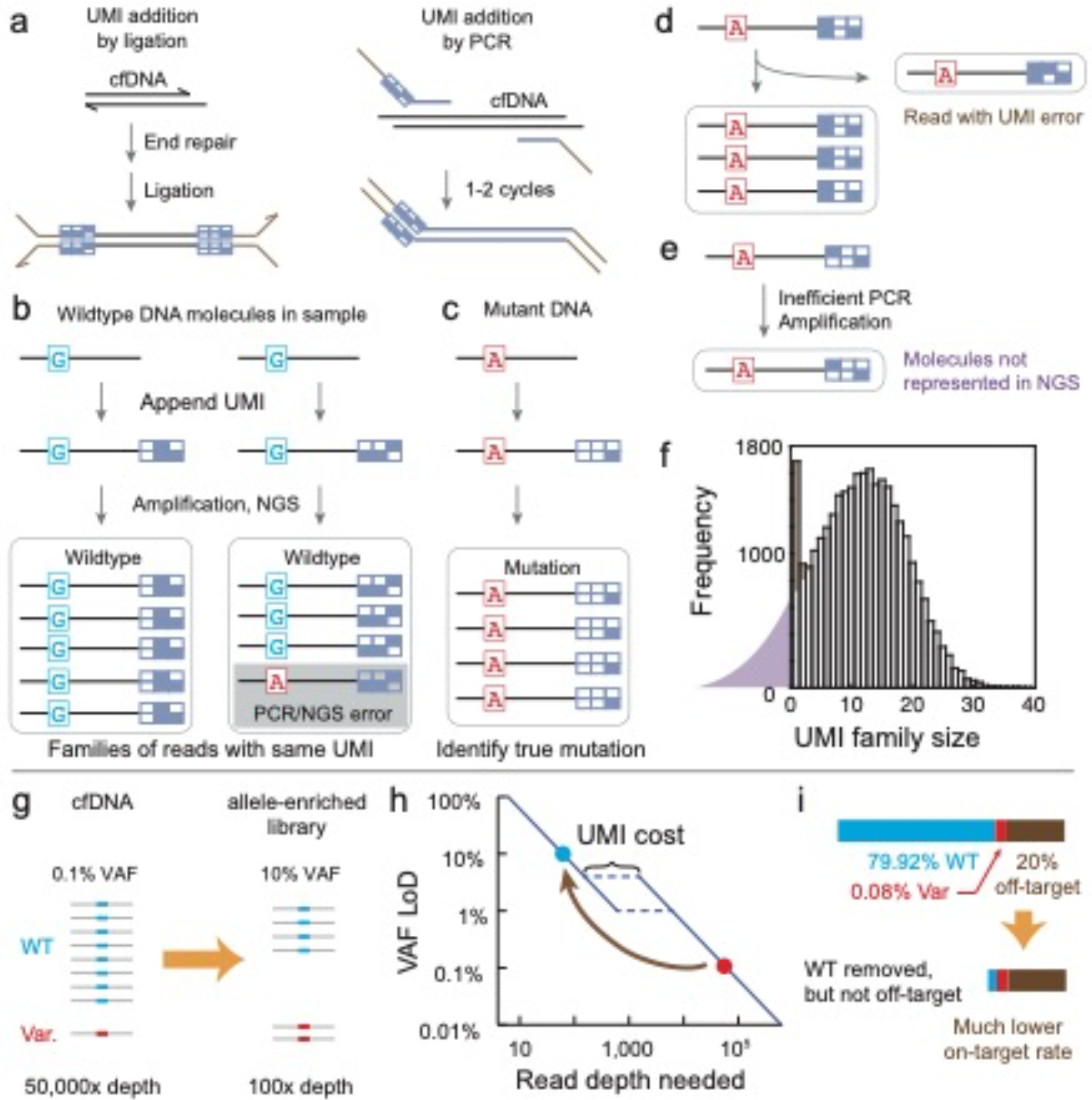
a, Unique molecular identifiers (UMIs) are used to overcome the limits of detection imposed by the misincorporation errors of PCR and the intrinsic errors of NGS. UMIs are unique sequences that are attached to each original cfDNA molecule (here shown as a 2D barcode). UMIs are typically random degenerate sequences such as ‘NNNNNN’, but can also comprise specific designed DNA sequences with error-correction or error-detection properties. UMIs can be incorporated into ligation and hybrid-capture protocols or into multiplex PCR protocols. Adapter sequences are shown in brown. b, UMIs correct most PCR and NGS errors. In the absence of UMIs, the NGS results showing 1 mutant A (red) read and 8 WT G (cyan) reads may suggest that the A mutation has a VAF of 11%. By sorting different reads by UMIs, one can bioinformatically determine that all 9 reads were derived from 2 original DNA molecules in the sample, which are both likely to be WT in sequence. However, there is a small chance that the right group of reads was actually derived from an early stage PCR error. c, True mutations will probably be represented by a family of reads with the same UMI that predominantly corresponds to the same mutation sequence. d, The UMI strategy is imperfect in error correction because PCR or NGS errors can occur within the UMI barcode sequence. The reads bearing UMI errors cannot be easily distinguished from a true family of UMIs corresponding to reads derived from another original cfDNA molecule. Thus, both the number of mutants and WT molecules may be overestimated. e, Different UMI sequences can have considerable and unpredictable impact on the efficiency of PCR amplification, resulting in some molecules being poorly amplified and thus not well represented in the NGS library. This can result in an effectively lower conversion yield than that without the use of UMIs, and can also lead to false negatives. f, Typical distribution of UMI family sizes for an NGS library; these results are from a 3 Mb ligation-and-hybrid-capture panel. The median UMI family size, which roughly follows a normal distribution, is approximately 13. The distribution of UMI family sizes suggests that a fraction (≈20%) of UMI families are not represented in the library (purple region), owing to the UMI amplification bias described in e. The number of UMI families with size 1 and 2 is unusually high; the excess families (brown region) are probably UMI errors (described in d). Because it is not possible to distinguish which of the UMI families of sizes 1 and 2 are UMI errors, a typical bioinformatic workflow will ignore all UMI families with sizes less than 3, resulting in an even greater loss of effective conversion yield. g, Allele enrichment seeks to increase the representation of the NGS library by the variant alleles (that is, cancer mutations). This is typically accomplished either through the selective removal of WT alleles via probe hybridization118 or enzymatic degradation120, or through the selective PCR amplification of variant alleles124,127. h, NGS read depth required for different VAF sensitivities. There is a nonlinear increase in depth required between 1% and 5% in VAF sensitivity, owing to the overhead required for UMIs to suppress the intrinsic error of NGS. Thus, enriching variant VAFs from 0.1% to 10% can, in principle, reduce the required NGS reads by more than 500-fold. LoD, limit of detection. i, The reads savings provided by allele enrichment are typically bottlenecked by on-target rates. By depleting the majority of WT reads in an NGS library, the relative fraction of off-target reads (that is, non-specific amplification of primer dimers at other genomic loci) becomes much higher in an allele-enriched library. For an original library with on-target fraction at a reasonable 80%, the NGS reads saving would be limited to a factor of 5, even if all on-target WT molecules or reads are perfectly removed. Thus, NGS libraries need to be close to 100% in on-target rates to fully realize the potential of allele enrichment.
Because UMIs can only function effectively to correct PCR and NGS errors when the UMI family size is large enough to allow for a majority vote, UMIs increase the required sequencing depth and cost by at least a factor of 5. Furthermore, unlike standard NGS, the amount of input cfDNA needs to be carefully controlled when UMIs are used. For example, when a DNA sample is sequenced to a mean 30,000× depth for a particular gene panel, the average UMI family size will be 10 if the input amount is 10 ng of cfDNA, but will only be 2 if the input amount is 50 ng of cfDNA.
The bioinformatic interpretation of UMIs is also somewhat challenging because the UMI sequences themselves could have PCR or NGS errors (Fig. 5d), which are difficult to distinguish from sequences with small UMI family sizes, owing to poor PCR-amplification efficiency (Fig. 5e). The typical bioinformatic workflow ignores all sequence information from UMI families with fewer than either 5 or 3 reads, which effectively mitigates detection and quantitation inaccuracies due to UMI errors, but also discards information from a large number of original cfDNA molecules, effectively reducing the conversion yield. An average UMI family size of 12 will thus result in a roughly 30% drop in effective conversion yield (Fig. 5f). The use of smaller UMI family sizes would greatly increase the number of original molecules whose information is discarded.
Technologies for allele enrichment
Strategies for allele enrichment refer to library-preparation methods that seek to detect low-VAF mutations by increasing the VAFs upstream of sequencing (Fig. 5g). For example, a cfDNA sample that contains a 0.1% VAF of a particular mutation may generate a library that has a 10% VAF for the same mutation; the latter is simple to detect and quantitate even with low-depth sequencing. Thus, in contrast to the UMI strategy, which increases sequencing costs by more than 10-fold, allele-enrichment methods can decrease the sequencing cost while achieving better limits of detection (Fig. 5h).
Allele enrichment can be achieved either through the removal of wild-type alleles or through the selective amplification of variant alleles. For example, oscillatory electrophoresis can amplify the mobility differences of DNA molecules differing by even a single nucleotide, and allows for effective removal of wild-type sequences118,119. Recently, wild-type-specific probes and double-strand specific nucleases have been used to selectively degrade wild-type DNA molecules120, likewise improving VAF by depleting wild-type alleles. The selective enrichment of variant alleles is typically achieved through PCR methods in which the wild-type DNA sequences are prevented from being PCR-amplified. Examples of this approach include blocker PCR121, locked-nucleic-acid and peptide-nucleic-acid clamp PCR122,123, ICE-COLD PCR124–126 and blocker displacement amplification127.
Allele-enrichment technologies are not currently broadly applied for three reasons. First, allele-enrichment methods have generally not been demonstrated to perform robustly at high-multiplex conditions. Multiple allele-enrichment methods have been shown to work for fewer than 20-plex primers or probes121–124,127, but even the smallest cfDNA NGS panels today are at least 50-plex (and many panels are over 1,000-plex). Second, the VAF fold-enrichment needs to be stable and reproducible for accurate sample-VAF quantitation. If the VAF of a particular mutation is always increased 100-fold through allele enrichment, then one can infer a 0.1% mutation VAF on the basis of an observed 10% VAF in the NGS library; however, if the VAF fold-enrichment varies between 50 and 200 across different runs, then VAF quantitation becomes much less accurate. Third, allele-enrichment technologies typically struggle with on-target rates (the fraction of NGS reads that map to the gene loci of interest, regardless of whether it is wild-type or a mutant). This is because while wild-type DNA sequences are removed, off-target reads such as those from primer dimers and those from the non-specific amplification of other portions of the genome are not (Fig. 5i); hence, high-performance allele-enrichment technologies are primarily bottlenecked by off-target reads (with respect to potential savings in NGS costs).
Inaccessible cfDNA markers
The types of cancer markers in DNA can be grouped into four classes: mutations, gene fusions, copy-number variations (CNVs; including loss of heterozygosity) and aneuploidy128. In previous sections, we have primarily discussed methods for the detection of mutations (including point substitutions and small insertions or deletions ≤50 nt). This is because mutations exhibit qualitatively different sequences at defined coding positions within genes, and hence are the easiest to detect in cfDNA.
Fusions and mutations are similar in that they result in qualitatively different sequences, but differ in that the unique cancer-specific sequence can reside at many different DNA loci129,130 (Fig. 6c). For example, the breakpoint for a ROS1 gene fusion in NSCLC can occur at any of the roughly 140,000 nt in the 44 introns of the ROS1 gene. Considering that a typical NGS panel for a cfDNA mutation is only about 100 kb, the detection of fusions at the cfDNA level is technically possible; however, the full coverage of the intron regions for high clinical sensitivity is economically non-viable when applied to many potential fusions. Some commercial panels detect fusions in cfDNA by focusing on intron loci that have been shown to have higher chances of being a fusion breakpoint; however, these sacrifice clinical sensitivity. For these reasons, fusions are typically detected from mature mRNA, in which the introns are spliced out and the number of possible fusion sequences is limited131,132.
Fig. 6 |. CNVs and gene fusions are challenging biomarkers for cfDNA analysis.
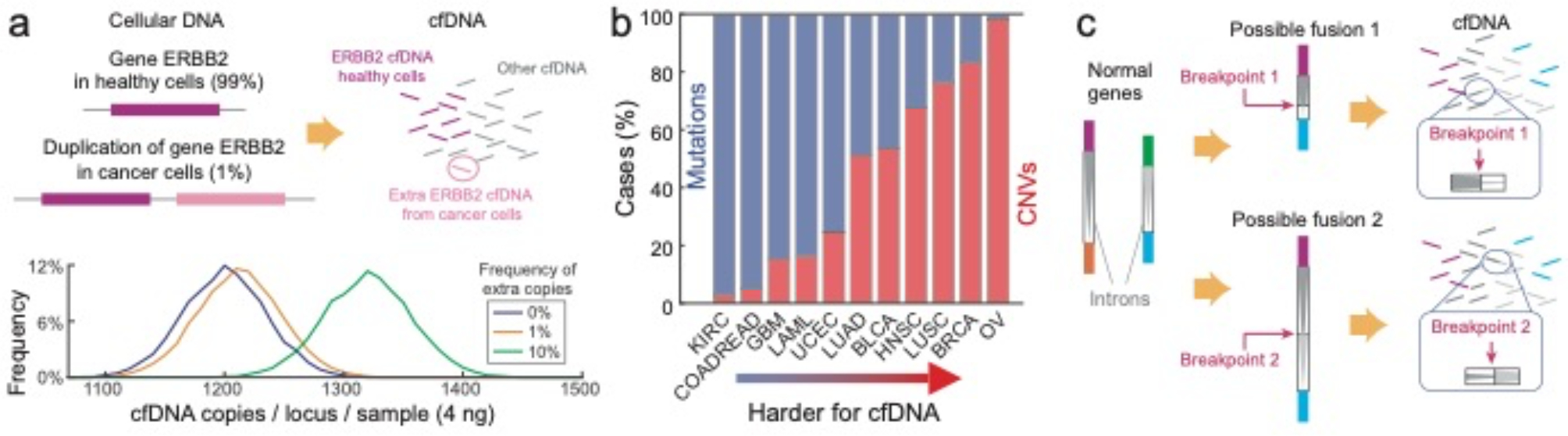
a, Because the fraction of all cfDNA that is tumour-derived is frequently under 1%, the stochasticity of molecular sampling renders CNVs difficult to distinguish from regular samples. The plot shows the expected distribution of the number of molecules present per locus in a 4 ng sample of cfDNA for 0%, 1% and 10% VAFs (based on 5,000 stochastic simulations). Even for a sample with 10% VAF, the molecular count overlaps substantially with the 0%-VAF sample, resulting in imperfect clinical sensitivity and specificity for a CNV call based on a single locus. The Matlab code used to generate these results is available as Supplementary Information. b, The distribution of mutation versus CNV markers varies drastically by cancer type; for example, ovarian cancer bears almost exclusively CNV markers128. Thus, there is a pressing unmet need to reliably quantitate CNVs in cfDNA. KIRC, kidney renal clear cell carcinoma; COADREAD, colon adenocarcinoma and rectum adenocarcinoma; GBM, glioblastoma multiforme; LAML, acute myeloid leukaemia; UCEC, uterine corpus endometrial carcinoma; LUAD, lung adenocarcinoma; BLCA, bladder urothelial carcinoma; HNSC, head and neck squamous cell carcinoma; LUSC, lung squamous cell carcinoma; BRCA, breast invasive carcinoma; OV, ovarian carcinoma. c, Gene fusions153 are difficult to detect in cfDNA because the fusion breakpoints can occur at any of a very large number of intron positions. Because of the long lengths of introns and the variable nature of fusion components, a very large (>200 kb) intron panel would be needed for high clinical sensitivity for a single fusion type (such as EML4-ALK). In contrast, fusions are more easily detected in RNA using tissue biopsies, because exon splicing results in a much smaller number of well-defined mature mRNA sequences.
Because the ends of duplicated regions often reside in repetitive DNA sequences133–135, CNVs do not typically contain any unique sequences. Therefore, rather than searching for the presence of a unique DNA sequence, CNV profiling requires accurate quantitation of the potentially duplicated gene relative to other genes. However, because the fraction of cfDNA that is tumour-derived can be 1% or lower, the stoichiometric excess of DNA corresponding to the CNV gene is very small (Fig. 6a,b). The small stoichiometric excess is often obfuscated by the Poisson-distribution nature of sampling cfDNA: a typical sample containing 10 ng of cfDNA corresponds to about 3,000 haploid-genome equivalents, so the number of DNA molecules at each locus will follow a distribution with a standard deviation of √3,000 ≈ 55, corresponding to an excess of nearly 2%. This challenge is partially mitigated by the fact that genes are long, so multiple distinct non-overlapping cfDNA species are available for each gene. However, technical difficulties in appending UMIs compound the limitations arising from Poisson statistics, and current commercial cfDNA assays exhibit a CNV limit of detection of roughly 20% in VAF32,33, which implies low clinical sensitivity.
Aneuploidy is similar to CNV in that typically there are no unique sequences that serve as distinctive cancer markers. However, aneuploidy is easier to detect than CNVs, owing to the vastly greater number of loci available for statistical comparisons. For example, a gene including introns may be up to 50 kb long, but even the shortest chromosome (chromosome 22) is about 50 Mb long; this 1,000-fold difference can result in a 30-fold lower coefficient of variation owing to Poisson sampling. For this reason, in non-invasive prenatal diagnostics for Down’s syndrome136, aneuploidy in cfDNA is routinely detected at 4% in VAF. Although aneuploidy has been observed in cancer137, it is not currently considered clinically actionable; consequently, most commercial cancer cfDNA panels do not include assays for it.
Accuracy requirements in cancer screening
There is strong enthusiasm regarding the possible use of cfDNA markers for the early detection of cancers in asymptomatic individuals138,139. For example, the company GRAIL has raised more than US$1.4 billion in funding over the past 3 years to develop cfDNA technologies and to run clinical trials for early cancer detection54. In this section, we discuss the biological, statistical and social challenges associated with the screening of populations for the early detection of cancers.
One key biological challenge of cfDNA-based cancer screening is that a large fraction of healthy individuals will have low levels of cancer-associated DNA sequences in cfDNA. These may arise from clonal haematopoiesis140–142, somatic mutations or somatic mosaicism, and could lead to false-positive results. Another central challenge is that a large fraction of individuals with early stage cancer will have undetectable cancer-specific mutations in cfDNA. This may be because the tumours have poor access to the circulatory system or because pathogenic mutations from cancer pathways are not currently understood, and could lead to false negatives. Because of these biological challenges, it is not possible for any cancer-screening test to achieve 100% specificity and 100% sensitivity (Fig. 7a).
Fig. 7 |. Accuracy requirements for the screening of early cancers via cfDNA analysis.
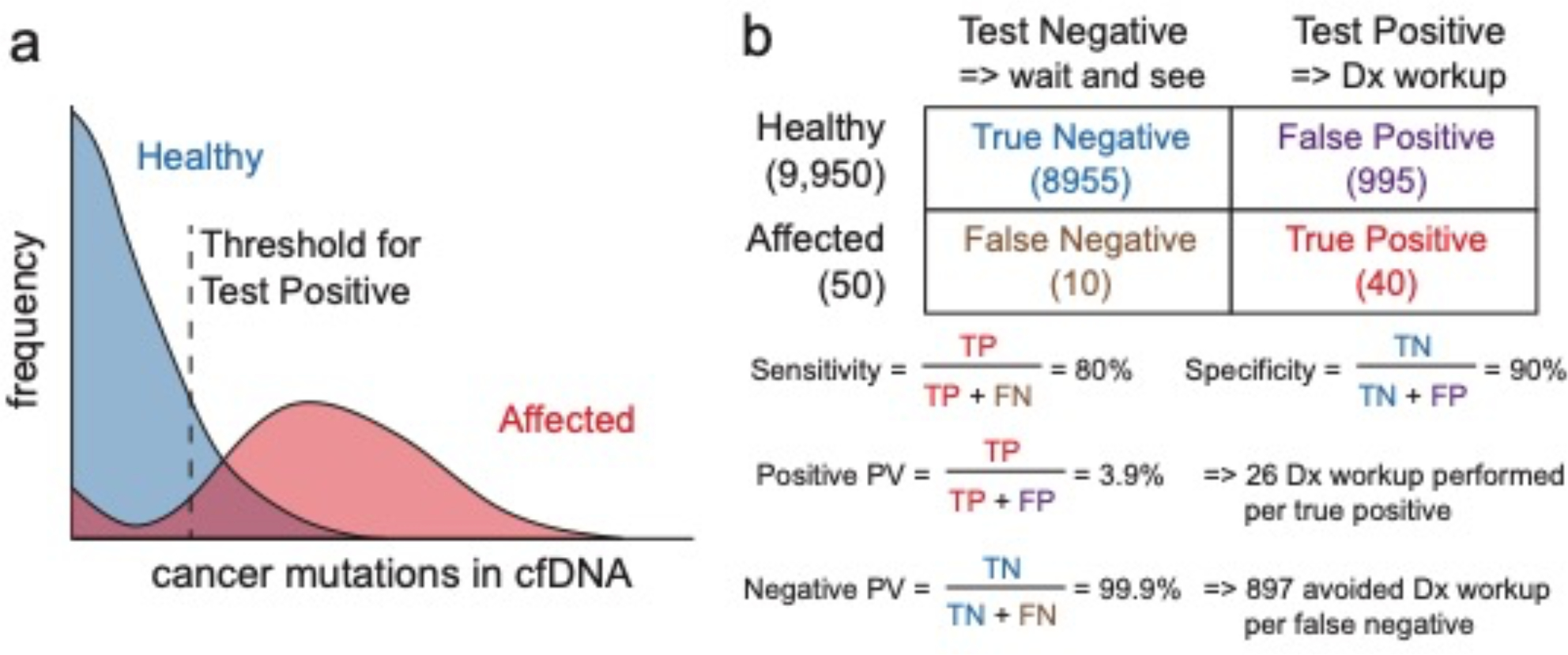
a, Hypothetical distribution of cancer mutations in cfDNA for healthy and affected individuals. The test will report a positive when the observed mutations and VAF combinations exceed some threshold, and will have both false positives (healthy individuals above the threshold) and false negatives (affected individuals below the threshold). The threshold determines the trade-off between clinical sensitivity and specificity. b, Hypothetical test results for a test with 80% sensitivity and 90% specificity, for a tested population of 10,000 where 0.5% have early stage cancer. TP, true positive; TN, true negative; FP, false positive; FN, false negative.
For a hypothetical test with 80% sensitivity and 90% specificity (Fig. 7b) used to test 10,000 samples in which 0.5% of the population have early stage cancer, because the prior probability of early stage cancer is low (0.5%), the posterior probability of an individual with a positive test result having early stage cancer is a modest 3.9% (this is the test’s positive predictive value). In early cancer screening, the typical next step for individuals who test positive is to undergo a diagnostic workup that includes endoscopy, X-rays, CT scans and possibly biopsies (Fig. 1). A 3.9% positive predictive value therefore means that 25 unnecessary diagnostic workups would be performed for each patient with early stage cancer. Taking into account the potential medical harm and the economic costs of the diagnostic workup, this may not be an acceptable trade-off.
Therefore, the realization of early cancer screening via cfDNA analysis will require that the specificity and sensitivity of the test be very close to 100%, that substantial improvements are made to the safety and costs of diagnostic workups to confirm any positive findings, or that methods for ‘enriching’ the tested population, for example via selection by age or by history of family cancers, are employed. As a point of comparison, the FDA-approved assay Cologuard for the early detection of colorectal cancer in stool has a clinical sensitivity of 94% and a clinical specificity of 87% (ref. 53), and it is recommended for individuals aged 50 years or older. Also, the follow-up colonoscopy exam for individuals with a positive Cologuard test is considered relatively safe and inexpensive. Other recent advances in early cancer screening include the detection of Epstein–Barr viral DNA for nasopharyngeal cancer56, and the use of combinations of protein and cfDNA markers for the detection of resectable tumours57. Global hypomethylation62 and specific promoter hypermethylation66 are promising markers for the early screening of multiple cancer types. However, depending on the specific cancer type, the lower disease incidence and higher medical risk of diagnostic workups (in particular, for gliomas and other brain cancers, and for pancreatic cancer) render early cancer detection a far more difficult problem to consider from societal and economical viewpoints.
Outlook
More than 70 years after its discovery138, cfDNA is starting to impact cancer care; numerous clinical trials are in progress in North America, Europe and Asia to assess its diagnostic utility. However, many technical challenges and opportunities remain for cfDNA diagnostics to have sufficiently high clinical sensitivity for its widespread use in cancer monitoring. Technologies for analysing cfDNA can broadly be split into rapid low-plex methods, and expensive and slow high-plex NGS-based methods. Most cfDNA diagnostic applications require information from multiple markers to achieve high clinical sensitivity and to inform treatment strategy, making NGS-based methods the preferred choice. Simultaneously, biological, statistical, clinical, physical, chemical and economical constraints mean that only a small portion of all potentially available information is currently accessible by using commercial cfDNA panels, and that opportunities are rife for improving the state-of-the-art through scientific innovation (Table 1). Minimally invasive cancer-diagnostic methods hold potential because common tissue-sampling techniques (such as tumour biopsies) and medical-imaging techniques that require exposure to ionizing radiation are limited to high-risk individuals and to individuals with already identified lesions. In contrast, diagnostics based on liquid biopsies are suitable for repeat sampling and can potentially be used for early cancer detection or screening. Earlier detection and continuous monitoring could help stratify individuals (Fig. 7). Identifying a set of biomarkers in cfDNA with sufficient specificity and sensitivity for the early detection of cancer may be challenging if the analysis is limited to DNA mutations. Hence, in addition to gene fusions and CNVs, which are currently challenging to detect in cfDNA, other sources of biomarkers (such as cell-free RNA143, exosomes144, methylation patterns63,64,67, protein levels54 and circulating tumour cells145,146) may be included in biomarker panels for the detection of early cancers, especially if applied to asymptomatic individuals. The multiplexed detection of analytes holds potential for the improvement of the sensitivity of cancer detection and for clinical utility. However, this will require further technical advances in sample preparation and in the methods of analysis.
TABLE I:
Current methods and challenges in cfDNA analysis
| Challenge | Solution | Comments |
|---|---|---|
|
| ||
| DNA damage | DNA repair | Need high-yield method to reverse DNA oxidation and deamination |
| cfDNA sampling stochasticity | Larger blood volumes | Concern for patient health |
| cfDNA sampling stochasticity | Urine cfDNA | Need for extracting cfDNA from large volumes; process short cfDNA |
|
| ||
| Detecting mutations with ≤1% VAF | Digital PCR | Single-plex, only known mutations |
| Detecting mutations with ≤1% VAF | Mass spectrometry | Medium-plex, only known mutations |
| Detecting mutations with ≤1% VAF | NGS with UMIs | Expensive and low conversion yield |
| Detecting mutations with ≤1% VAF | NGS with allele enrichment | Low-plex, inaccurate quantitation, and low on-target rates |
|
| ||
| High conversion yield from cfDNA | N/A | Need high-yield end-repair and ligation |
| NGS depth uniformity | Primer/probe conc. tuning | labor-intensive and imperfect uniformity |
|
| ||
| Detecting fusions in cfDNA | Very large NGS panel | Very expensive because introns are long |
| Detecting CNVs in cfDNA | NGS, ddPCR | No current solution for detection ≤5% VAF |
|
| ||
| Rapid cfDNA diagnostics | Nanopore sequencing | High error rates and expensive reads |
| Affordable cfDNA diagnostics | N/A | Current cfDNA NGS panels have list price over $4,000 |
Supplementary Material
Acknowledgements
We thank S. Skates for useful discussions regarding diagnostic economics and outcomes. L.N.K. was supported by NIH grant P01CA163222. A.A.P. was supported by NIH grants R01CA197486 and U01CA233364. D.Y.Z. was supported by NIH grants R01CA203964 and U01CA233364, and by CPRIT grant RP180147.
Footnotes
Competing interests
P.S., L.R.W. and A.A.P. are consultants for NuProbe USA. A.A.P. is a consultant for Binary Genomics and has equity ownership in the company. D.Y.Z. is a co-founder of, and holds significant equity in, NuProbe Global, Torus Biosystems and Pana Bio.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41551-021-00837-3.
Peer review information Nature Biomedical Engineering thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Lo YD et al. Rapid clearance of fetal DNA from maternal plasma. Am. J. Hum. Genet. 64, 218–224 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diehl F et al. Circulating mutant DNA to assess tumour dynamics. Nat. Med. 14, 985–990 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jahr S et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 61, 1659–1665 (2001). [PubMed] [Google Scholar]
- 4.Wan JC et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat. Rev. Cancer 17, 223–238 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Diaz LA Jr & Bardelli, A. Liquid biopsies: genotyping circulating tumour DNA. J. Clin. Oncol. 32, 579–586 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thierry AR et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumour DNA. Nat. Med. 20, 430–435 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Alix-Panabieres C & Pantel K Clinical applications of circulating tumour cells and circulating tumour DNA as liquid biopsy. Cancer Discov. 6, 479–491 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Mok T et al. Detection and dynamic changes of EGFR mutations from circulating tumour DNA as a predictor of survival outcomes in NSCLC patients treated with first-line intercalated erlotinib and chemotherapy. Clin. Cancer Res. 21, 3196–3203 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Hao TB et al. Circulating cell-free DNA in serum as a biomarker for diagnosis and prognostic prediction of colorectal cancer. Br. J. Cancer 111, 1482–1489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Azad AA et al. Androgen receptor gene aberrations in circulating cell-free DNA: biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin. Cancer Res. 21, 2315–2324 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Lebofsky R et al. Circulating tumour DNA as a non-invasive substitute to metastasis biopsy for tumour genotyping and personalized medicine in a prospective trial across all tumour types. Mol. Oncol. 9, 783–790 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwarzenbach H, Hoon DSB & Pantel K Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 11, 426–437 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Goyal L et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 7, 252–263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only: Guidance for Industry and Food and Drug Administration Staff (US FDA, 2013); https://www.fda.gov/regulatory-information/search-fda-guidance-documents/distribution-vitro-diagnostic-productslabeled-research-use-only-or-investigational-use-only [Google Scholar]
- 15.Dietel M et al. Diagnostic procedures for non-small-cell lung cancer (NSCLC): recommendations of the European Expert Group. Thorax 71, 177–184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herbst RS et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–1550 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Vansteenkiste J et al. Final results of a multi-center, double-blind, randomized, placebo-controlled phase II study to assess the efficacy of MAGE-A3 immunotherapeutic as adjuvant therapy in stage IB/II non-small cell lung cancer (NSCLC). J. Clin. Oncol. 25, 7554 (2007). [Google Scholar]
- 18.Gettinger SN et al. Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J. Clin. Oncol. 33, 2004–2012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burstein HJ et al. Clinical cancer advances 2017: annual report on progress against cancer from the American Society of Clinical Oncology. J. Clin. Oncol. 35, 1341–1367 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Wender R et al. American Cancer Society lung cancer screening guidelines. CA Cancer J. Clin. 63, 107–117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baldwin DR et al. UK Lung Screen (UKLS) nodule management protocol: modelling of a single screen randomised controlled trial of low-dose CT screening for lung cancer. Thorax 66, 308–313 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Field JK et al. International association for the study of lung cancer computed tomography screening workshop 2011 report. J. Thorac. Oncol. 7, 10–19 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Spigel DR et al. Final efficacy results from OAM4558g, a randomized phase II study evaluating MetMAb or placebo in combination with erlotinib in advanced NSCLC. J. Clin. Oncol. 29, 7505 (2011). [Google Scholar]
- 24.Mok TS et al. Osimertinib or platinum-pemetrexed in EGFR T790M positive lung cancer. N. Engl. J. Med. 376, 629–640 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou C et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer. Lancet Oncol. 12, 735–742 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Gatzemeier U et al. Results of a phase III trial of erlotinib (OSI-774) combined with cisplatin and gemcitabine (GC) chemotherapy in advanced non-small-cell lung cancer (NSCLC). J. Clin. Oncol. 22, 7010 (2004). [Google Scholar]
- 27.Oxnard GR et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non-small-cell lung cancer. J. Clin. Oncol. 34, 3375–3382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark TA et al. Analytical validation of a hybrid capture-based next-generation sequencing clinical assay for genomic profiling of cell-free circulating tumour DNA. The. J. Mol. Diagnostics 20, 686–702 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanman RB et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumour DNA. PLoS ONE 10, e0140712 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paz-Ares L et al. plus chemotherapy for squamous non–small-cell lung cancer. N. Engl. J. Med. 379, 2040–2051 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Fabrizio D et al. A blood-based next-generation sequencing assay to determine tumour mutational burden (bTMB) is associated with benefit to an anti-PD-L1 inhibitor, atezolizumab. Cancer Res. 78, 5706–5706 (2018).30115693 [Google Scholar]
- 32.Hellmann MD et al. (2018). Nivolumab plus ipilimumab in lung cancer with a high tumour mutational burden. N. Engl. J. Med. 378, 2093–2104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gandara DR et al. Blood-based tumour mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 24, 1441–1448 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Le DT et al. PD-1 blockade in tumours with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le DT et al. Mismatch repair deficiency predicts response of solid tumours to PD-1 blockade. Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rolfo C et al. Liquid biopsies in lung cancer: the new ambrosia of researchers. Biochim. Biophys. Acta 1846, 539–546 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Xiong L et al. Dynamics of EGFR mutations in plasma recapitulates the clinical response to EGFR-TKIs in NSCLC patients. Oncotarget 8, 63846–63856 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romano G et al. A preexisting rare PIK3CAE545K subpopulation confers clinical resistance to MEK plus CDK4/6 inhibition in NRAS melanoma and is dependent on S6K1 signaling. Cancer Discov. 8, 556–567 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldberg SB et al. Early assessment of lung cancer immunotherapy response via circulating tumour DNA. Clin. Cancer Res. 24, 1872–1880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Narayan A et al. Ultrasensitive measurement of hotspot mutations in tumour DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res. 72, 3492–3498 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thress KS et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small-cell lung cancer harboring EGFR T790M. Nat. Med. 21, 560–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kwong LN et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat. Med. 18, 1503–1510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Z et al. Lung adenocarcinoma harboring EGFR T790M and in trans C797S responds to combination therapy of first-and third-generation EGFR TKIs and shifts allelic configuration at resistance. J. Thorac. Oncol. 12, 1723–1727 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Corcoran RB & Chabner BA Application of cell-free DNA analysis to cancer treatment. N. Engl. J. Med. 379, 1754–1765 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Garcia-Murillas I et al. Mutation tracking in circulating tumour DNA predicts relapse in early breast cancer. Sci. Transl. Med. 7, 302ra133 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Dawson SJ et al. Analysis of circulating tumour DNA to monitor metastatic breast cancer. N. Engl. J. Med. 368, 1199–1209 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Reinert T et al. Analysis of plasma cell-free DNA by ultradeep sequencing in patients with stages I to III colorectal cancer. JAMA Oncol. 5, 1124–1131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tie J et al. Circulating tumour DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 8, 346ra92 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abbosh C, Birkbak NJ & Swanton C Early stage NSCLC—challenges to implementing ctDNA-based screening and MRD detection. Nat. Rev. Clin. Oncol. 15, 577–586 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Oxnard GR et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin. Cancer Res. 20, 1698–1705 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phallen J et al. Direct detection of early-stage cancers using circulating tumour DNA. Sci. Transl. Med. 9, eaan2415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Imperiale TF et al. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 370, 1287–1297 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Chan KA et al. Analysis of plasma Epstein-Barr virus DNA to screen for nasopharyngeal cancer. N. Engl. J. Med. 377, 513–522 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Cohen JD et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359, 926–930 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lecomte T et al. Detection of free circulating tumour associated DNA in plasma of colorectal cancer patients and its association with prognosis. N. Engl. J. Med. 370, 1287–1297 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Hu Y et al. False-positive plasma genotyping due to clonal hematopoiesis. Clin. Cancer Res. 24, 4437–4443 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Heitzer E, Haque IS, Roberts CE & Speicher MR Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 20, 71–88 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Sina AAI et al. Epigenetically reprogrammed methylation landscape drives the DNA self-assembly and serves as a universal cancer biomarker. Nat. Commun. 9, 4915 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lehmann-Werman R et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl Acad. Sci. USA 113, E1826–E1834 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deng J et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat. Biotechnol. 27, 353–360 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu RH et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat. Mater. 16, 1155–1161 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Teschendorff AE & Relton CL Statistical and integrative system-level analysis of DNA methylation data. Nat. Rev. Genet. 19, 129–147 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Guo S et al. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumour tissue-of-origin mapping from plasma DNA. Nat. Genet. 49, 635–642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tug S et al. Exercise-induced increases in cell-free DNA in human plasma originate predominantly from cells of the haematopoietic lineage. Exerc. Immunol. Rev. 21, 164–173 (2015). [PubMed] [Google Scholar]
- 65.Moreira VG, Prieto B, Rodr.ıguez JSM & Alvarez FV Usefulness of cell-free plasma DNA, procalcitonin and C-reactive protein as markers of infection in febrile patients. Ann. Clin. Biochem. 47, 253–258 (2010). [DOI] [PubMed] [Google Scholar]
- 66.Burnham P et al. Urinary cell-free DNA is a versatile analyte for monitoring infections of the urinary tract. Nat. Commun. 9, 2412 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Siljan WW et al. Circulating cell-free DNA is elevated in community acquired bacterial pneumonia and predicts short-term outcome. J. Infect. 73, 383–386 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Lindahl T Instability and decay of the primary structure of DNA. Nature 362, 709–715 (1993). [DOI] [PubMed] [Google Scholar]
- 69.Richter C, Park JW & Ames BN Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl Acad. Sci. USA 85, 6465–6467 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cooke MS, Evans MD, Dizdaroglu M & Lunec J Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 17, 1195–1214 (2003). [DOI] [PubMed] [Google Scholar]
- 71.Norton SE, Lechner JM, Williams T & Fernando MR A stabilizing reagent prevents cell-free DNA contamination by cellular DNA in plasma during blood sample storage and shipping as determined by digital PCR. Clin. Biochem. 46, 1561–1565 (2013). [DOI] [PubMed] [Google Scholar]
- 72.Manning JE in Emergency Medicine: A Comprehensive Study Guide (ed. Tintinalli JE) 227 (McGraw-Hill, 2004). [Google Scholar]
- 73.Botezatu I et al. Genetic analysis of DNA excreted in urine: a new approach for detecting specific genomic DNA sequences from cells dying in an organism. Clin. Chem. 46, 1078–1084 (2000). [PubMed] [Google Scholar]
- 74.Koide K et al. Fragmentation of cell-free fetal DNA in plasma and urine of pregnant women. Prenat. Diagnosis 25, 604–607 (2005). [DOI] [PubMed] [Google Scholar]
- 75.Tani M & Beck S Epigenome-wide association studies for cancer biomarker discovery in circulating cell-free DNA: technical advances and challenges. Curr. Opin. Genet. Dev. 42, 48–55 (2017). [DOI] [PubMed] [Google Scholar]
- 76.Reckamp KL et al. A highly sensitive and quantitative test platform for detection of NSCLC EGFR mutations in urine and plasma. J. Thorac. Oncol. 11, 1690–1700 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Swanson P et al. Performance of the automated Abbott RealTime HIV-1 assay on a genetically diverse panel of specimens from London: comparison to VERSANT HIV-1 RNA 3.0, AMPLICOR HIV-1 MONITOR v1. 5, and LCx R○ HIV RNA quantitative assays. J. Virol. Methods 137, 184–192 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Castle PE Performance of carcinogenic human papillomavirus (HPV) testing and HPV16 or HPV18 genotyping for cervical cancer screening of women aged 25 years and older: a subanalysis of the ATHENA study. Lancet Oncol. 12, 880–890 (2011). [DOI] [PubMed] [Google Scholar]
- 79.Sandri MT et al. Comparison of the Digene HC2 assay and the Roche AMPLICOR human papillomavirus (HPV) test for detection of high-risk HPV genotypes in cervical samples. J. Clin. Microbiol. 44, 2141–2146 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Misawa Y et al. Application of loop-mediated isothermal amplification technique to rapid and direct detection of methicillin-resistant Staphylococcus aureus (MRSA) in blood cultures. J. Infect. Chemother. 13, 134–140 (2007). [DOI] [PubMed] [Google Scholar]
- 81.Ethridge SF et al. Performance of the Aptima HIV-1 RNA qualitative assay with 16- and 32-member specimen pools. J. Clin. Microbiol. 48, 3343–3345 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pao W et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2, e73 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mok TS et al. Osimertinib or platinum–pemetrexed in EGFR T790M–positive lung cancer. N. Engl. J. Med. 376, 629–640 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hindson BJ et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 83, 8604–8610 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Newton CR et al. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 17, 2503–2516 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Das J et al. An electrochemical clamp assay for direct, rapid analysis of circulating nucleic acids in serum. Nat. Chem. 7, 569–575 (2015). [DOI] [PubMed] [Google Scholar]
- 87.Lin M et al. Electrochemical detection of nucleic acids, proteins, small molecules and cells using a DNA-nanostructure-based universal biosensing platform. Nat. Protoc. 11, 1244–1263 (2016). [DOI] [PubMed] [Google Scholar]
- 88.Gootenberg JS et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dressman D, Yan H, Traverso G, Kinzler KW & Vogelstein B Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl Acad. Sci. USA 100, 8817–8822 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Khoo C et al. Molecular methods for somatic mutation testing in lung adenocarcinoma: EGFR and beyond. Transl. Lung Cancer Res. 4, 126–141 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ragoussis J Genotyping technologies for genetic research. Annu. Rev. Genomics Hum. Genet. 10, 117–133 (2009). [DOI] [PubMed] [Google Scholar]
- 92.Murtaza M et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497, 108–112 (2013). [DOI] [PubMed] [Google Scholar]
- 93.Wetterstrand KA DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP) (National Human Genome Research Institute, accessed 19 July 2018); www.genome.gov/sequencingcostsdata [Google Scholar]
- 94.Laver T et al. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 3, 1–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Carneiro MO et al. Pacific biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genomics 13, 375 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Taylor AD, Micheel CM, Anderson IA, Levy MA & Lovly CM The path (way) less traveled: a pathway-oriented approach to providing information about precision cancer medicine on My Cancer Genome. Transl. Oncol. 9, 163–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Diehn M et al. Early prediction of clinical outcomes in resected stage II and III colorectal cancer (CRC) through deep sequencing of circulating tumour DNA (ctDNA). J. Clin. Oncol. 35, 3591 (2017).28892431 [Google Scholar]
- 98.Couraud S et al. Non-invasive diagnosis of actionable mutations by deep sequencing of circulating-free DNA in non-small-cell lung cancer: findings from BioCAST/IFCT-1002. Clin. Cancer Res. 20, 4613–4624 (2014). [DOI] [PubMed] [Google Scholar]
- 99.Song P et al. Selective multiplexed enrichment for the detection and quantitation of low-fraction DNA variants via low depth sequencing. Nat. Biomed. Eng. 5, 690–701 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Meyer M & Kircher M Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, pdb.prot5448 (2010). [DOI] [PubMed] [Google Scholar]
- 101.Hovelson DH et al. Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumours. Neoplasia 17, 385–399 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dupuis JR et al. HiMAP: robust phylogenomics from highly multiplexed amplicon sequencing. Mol. Ecol. Resour. 18, 1000–1019 (2018). [DOI] [PubMed] [Google Scholar]
- 103.Schmitt MW et al. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl Acad. Sci. USA 109, 14508–14513 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Blakely CM et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 49, 1693–1704 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kinde I et al. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl Acad. Sci. USA 108, 9530–9535 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lo YD et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2, 61ra91 (2010). [DOI] [PubMed] [Google Scholar]
- 107.Thierry AR et al. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 38, 6159–6175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Snyder MW, Kircher M, Hill AJ, Daza RM & Shendure J Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 164, 57–68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mouliere F et al. Enhanced detection of circulating tumour DNA by fragment size analysis. Sci. Transl. Med. 10, 466 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Thakur BK et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 24, 766–769 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Balaj L et al. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2, 180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang JX et al. Predicting DNA hybridization kinetics from sequence. Nat. Chem. 10, 91–98 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Newman AM et al. Integrated digital error suppression for improved detection of circulating tumour DNA. Nat. Biotechnol. 34, 547–555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kou R et al. Benefits and challenges with applying unique molecular identifiers in next generation sequencing to detect low frequency mutations. PLoS ONE 11, e0146638 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jabara CB, Jones CD, Roach J, Anderson JA & Swanstrom R Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc. Natl Acad. Sci. USA 108, 20166–20171 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Abascal F et al. Somatic mutation landscapes at single-molecule resolution. Nature 593, 405–410 (2021). [DOI] [PubMed] [Google Scholar]
- 117.Cohen JD et al. Detection of low-frequency DNA variants by targeted sequencing of the Watson and Crick strands. Nat. Biotechnol. 39, 1220–1227 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pel J et al. Nonlinear electrophoretic response yields a unique parameter for separation of biomolecules. Proc. Natl Acad. Sci. USA 106, 14796–14801 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kidess E et al. Mutation profiling of tumour DNA from plasma and tumour tissue of colorectal cancer patients with a novel, high-sensitivity multiplexed mutation detection platform. Oncotarget 6, 2549–2561 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Song C & Liu et al. Elimination of unaltered DNA in mixed clinical samples via nuclease-assisted minor-allele enrichment. Nucleic Acids Res. 44, e146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Seyama T et al. A novel blocker-PCR method for detection of rare mutant alleles in the presence of an excess amount of normal DNA. Nucleic Acids Res. 20, 2493–2496 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Arcila M, Lau C, Nafa K & Ladanyi M Detection of KRAS and BRAF mutations in colorectal carcinoma: roles for high-sensitivity locked nucleic acid-PCR sequencing and broad-spectrum mass spectrometry genotyping. J. Mol. Diagnostics 13, 64–73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Orum H et al. Single base pair mutation analysis by PNA directed PCR clamping. Nucleic Acids Res. 21, 5332–5336 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Milbury CA, Li J & Makrigiorgos GM Ice-COLD-PCR enables rapid amplification and robust enrichment for low-abundance unknown DNA mutations. Nucleic Acids Res. 39, e2 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li J et al. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat. Med. 14, 579–584 (2008). [DOI] [PubMed] [Google Scholar]
- 126.Zuo Z et al. Application of COLD-PCR for improved detection of KRAS mutations in clinical samples. Mod. Pathol. 22, 1023–1031 (2009). [DOI] [PubMed] [Google Scholar]
- 127.Wu LR et al. Multiplexed enrichment of rare DNA variants via sequence-selective and temperature-robust amplification. Nat. Biomed. Eng. 1, 714–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ciriello G et al. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 45, 1127–1133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mertens F, Johansson B, Fioretos T & Mitelman F The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 15, 371–381 (2015). [DOI] [PubMed] [Google Scholar]
- 130.Wang Q et al. Application of next generation sequencing to human gene fusion detection: computational tools, features and perspectives. Brief. Bioinform. 14, 506–519 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Maher CA et al. Transcriptome sequencing to detect gene fusions in cancer. Nature 458, 97–101 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhao Q et al. Transcriptome-guided characterization of genomic rearrangements in a breast cancer cell line. Proc. Natl Acad. Sci. USA 106, 1886–1891 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zack TI et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Whale AS et al. Comparison of microfluidic digital PCR and conventional quantitative PCR for measuring copy number variation. Nucleic Acids Res. 40, e82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pinkel D et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 20, 207–211 (1998). [DOI] [PubMed] [Google Scholar]
- 136.Chiu RW et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: large scale validity study. Brit. Med. J. 342, c7401 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kops GJ, Weaver BA & Cleveland DW On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 5, 773–785 (2004). [DOI] [PubMed] [Google Scholar]
- 138.Mandel P & Metais P Les acides nucleiques du plasma sanguin chez la homme. C. R. Seances Soc. Biol. Fil. 142, 241–243 (1948). [PubMed] [Google Scholar]
- 139.Bettegowda C et al. Detection of circulating tumour DNA in early- and late-stage human malignancies. Sci. Transl. Med. 6, 224ra224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Aravanis AM, Lee M & Klausner RD Next-generation sequencing of circulating tumour DNA for early cancer detection. Cell 168, 571–574 (2017). [DOI] [PubMed] [Google Scholar]
- 141.Razavi P et al. Cell-free DNA (cfDNA) mutations from clonal hematopoiesis: implications for interpretation of liquid biopsy tests. J. Clin. Oncol. 35, 11526 (2017). [Google Scholar]
- 142.Genovese G et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schwarzenbach H, Nishida N, Calin GA & Pantel K Clinical relevance of circulating cell-free microRNAs in cancer. Nat. Rev. Clin. Oncol. 11, 145–156 (2014). [DOI] [PubMed] [Google Scholar]
- 144.Azmi AS, Bao B & Sarkar FH Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev. 32, 623–642 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.De Mattos-Arruda L et al. Circulating tumour cells and cell-free DNA as tools for managing breast cancer. Nat. Rev. Clin. Oncol. 10, 377–389 (2013). [DOI] [PubMed] [Google Scholar]
- 146.de Bono JS et al. Circulating tumour cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 14, 6302–6309 (2008). [DOI] [PubMed] [Google Scholar]
- 147.Potapov V & Ong JL Examining sources of error in PCR by single-molecule sequencing. PLoS ONE 12, e0181128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schirmer M et al. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 43, e37 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schirmer M, Damore R, Ijaz UZ, Hall N & Quince C Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC Bioinformatics 17, 125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Minoche AE, Dohm JC & Himmelbauer H Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biol. 12, R112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jain M et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Quail MA et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 13, 341 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Leary RJ et al. Development of personalized tumour biomarkers using massively parallel sequencing. Sci. Transl. Med. 2, 20ra14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.