

Abstract
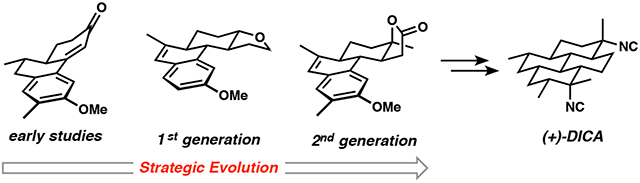
A full account of the development of a concise and highly stereoselective synthesis of (+)-7,20-diisocyanoadociane (DICA)—a structurally complex isocyanoditerpene with potent antiplasmodial activity—is described. The strategy that evolved relies on the rapid construction of unsaturated tricyclic precursors designed to undergo stereocontrolled Birch reductions and a subsequent “bay ring” formation to generate the isocycloamphilectane core. This report is divided into three sections: (1) a description of the initial strategy and the results that focused our efforts on a single route to the DICA core, (2) a discussion of the precise choreography needed to enable a 1st-generation formal synthesis of (±)-DICA, and (3) the execution of a 13-step 2nd-generation synthesis of (+)-DICA that builds on important lessons learned in the 1st-generation effort.
Graphical Abstract
Introduction.
7,20-Diisocyanoadociane (DICA, 1, Figure 1) has become the flagship member of the isocyanoterpene (ICT) family of secondary metabolites, perhaps owing to the combination of its early isolation, its beautiful fusion of four chair cyclohexanes, and its potent antiplasmodial activity. Among the large number of ICTs, which are unusual for the presence of isonitrile functional groups, the cyclo- and isocycloamphilectane sub-families distinguish themselves by the fused tetracyclic architectures shown in Figure 1. This group of ICTs possesses stereochemically complex, sparsely functionalized, largely saturated pyrene cores that present significant synthetic challenges.1 The individuals in this group of secondary metabolites are clearly related by cationic rearrangements and eliminations that occur during their biosynthesis.2 Although ICTs in general have antiplasmodial activity, DICA is among the most active. It is also curiously abundant in the marine sponge from which it is isolated; it makes up about 2% of the dry weight of collected material.3 Nonetheless, the advent of an efficient synthesis—one that is generalizable to many of the tetracyclic members of the family and perhaps other ICTs—might permit a deeper foray into structure/activity relationships, as well as the generation of chemical probes for interrogation of the as-yet unconfirmed mechanisms of antiplasmodial action.
Figure 1.

Representative tetracyclic ICTs. Values in brackets are IC50 values against Plasmodium falciparum strain Dd2.
Multiple groups have expended significant efforts toward the synthesis of DICA, which poses notable synthetic challenges in its axial and equatorially disposed tertiary isonitriles as well as the all-trans pyrene framework.4 The complete saturation in the DICA scaffold provides no intrinsic, functional-group-related inspiration for the generation of the core structure; only the isonitriles are present to stimulate possible disconnections. Three tactics have been used for isonitrile installation in syntheses of DICA (Scheme 1A). Corey and Magriotis5 made strategic use of a 7,20-diketone (5), which was converted to a bis-axial diol by nucleophilic methylation. Activation as the corresponding trifluoroacetates permitted TiCl4-facilitated displacement with TMSCN, generating all four stereoisomers of DICA; the desired isomer could be isolated by HPLC. An alternative approach involved the stereoretentive bis-Curtius rearrangement of diacid substrate 6 by Fairweather and Mander,6 also previously used by Schindeler and Piers in their synthesis of related ICT 4 (Figure 1). In a critical development, the Shenvi group discovered a Sc(OTf)3-mediated invertive displacement of stereochemically defined tertiary trifluoroacetates.7 The isonitrile stereocontrol problem was thereby reduced to the stereoselective synthesis of tertiary carbinol precursors of type 7.8 The simultaneous introduction of both isonitriles in the Shenvi synthesis of DICA demonstrated that both the axial and equatorial tertiary carbinols could be converted to isonitriles with inversion of configuration (7 to 1). Naturally, the choice of isonitrile precursors—tert-alkyl amines or tertiary alcohols—directly influences the strategy by which the carbocyclic structure is constructed.
Scheme 1.

Synthetic approaches to the completion of DICA, including tactics for introduction of the key isonitrile functional groups and strategies for the synthesis of the tetracyclic architecture of DICA
Two main types of approaches have been used to assemble the perhydropyrene scaffold of DICA (Scheme 1B). The cyclohexane-based carbocyclic core immediately brings to mind the Diels–Alder reaction as a key bond-forming strategy. The groups of Corey,5 Miyaoka9 and Shenvi8 each applied double Diels–Alder strategies. The former two approaches were strategically similar and involved two intramolecular cycloadditions separated by several functional group manipulations, ultimately leading to tetracyclic intermediates 8 and 9. Corey and Magriotis showcased the utility of ketones as both an enabling functional group for architecture building and precursors to the isonitriles. Shenvi’s approach featured the novel use of a Danishefsky’s-diene-like dendralene for rapid buildup of complexity; tricyclic intermediate 10 is fashioned in only 6 steps in the longest linear sequence from commercially available materials. This Diels–Alder strategy was designed to unite two simple starting materials and to differentiate the C7 and C20 future isonitrile-bearing carbons for targeted application of their invertive displacement method. Alternatively, the groups of Mander,6 Vanderwal10 and Thomson11 did not apply cycloaddition chemistry, but rather traversed through unique partially saturated phenanthrene intermediates; each synthesis featured a distinct “bay-ring” cyclization to complete the tetracyclic core.
While the DICA syntheses from the groups of Corey, Mander, and Thomson were single efforts, Miyaoka and co-workers used a similar Diels–Alder strategy and the Shenvi group applied their sequenced dendralene cycloaddition approach toward kalihinol and amphilectane members of the ICTs. Our own work on ICTs also converged on a common strategic theme that we continue to use in the context of several different polycyclic natural products of this family.12 In this article, we provide a full account of the project’s evolution from our earliest ideas, through our first-generation formal synthesis,10a to our second-generation,10b highly optimized, 13-step stereocontrolled synthesis of (+)-DICA.
Results and Discussion.
A. Early Route Scouting: Development of a Birch Reduction Approach
While the lack of functional handles in DICA could be considered a challenge because it limits the options for strategic disconnections without the retrosynthetic addition of superfluous functionality, it could equally be viewed as a motivator for creativity. At the time of project inception, only the Corey and Mander groups had produced viable end-games. We elected to target Corey’s dione because the challenging installation of two tertiary carboxylic acids—one axial and one equatorial, as in the Mander synthesis6—seemed destined to make for a longer synthesis. We therefore initially focused on the evaluation of new and potentially efficient ways to construct the hydrocarbon framework rather than developing a novel isonitrile installation strategy. A main consideration during early idea generation was the near symmetry of DICA, which in several cases provided us with two complementary options to implement a single overall strategy (see below).
Although ultimately unsuccessful, our earliest plans were part of an important thought lineage that contributed to an eventual successful approach to DICA. First, inspired by Swaminathan’s reported anionic oxy-Cope/transannular Michael addition cascade of spirocyclic enones to perhydrophenalenedione products,13 we designed 14 as the cascade precursor to 15 (Scheme 2A). Unfortunately, the stereochemical outcome of the cascade was unexpectedly unsuitable for the purposes of a synthesis of DICA.14 In the course of these studies, we contributed a stereochemical correction to the prior work, gained an improved mechanistic understanding of the cascade itself, and ultimately took advantage of the approach in a recent formal synthesis of the pseudopterosins.15 Despite the detour, we were unperturbed and continued to target perhydrophenalenedione 15 as a versatile intermediate. The next design consisted of a Diels–Alder/aldol condensation idea as formulated in 16. This Diels–Alder approach was fraught with poor reactivity and unreliable reactions and was consequently abandoned. In retrospect, Shenvi’s approach that was disclosed after our initial efforts better applied the Diels–Alder reaction to the synthesis of DICA.
Scheme 2.

Early strategies targeting tricyclic intermediates.
With a few discouraging approaches behind us, we embarked on a route relying on another classic six-membered ring forming reaction: the Robinson annulation.16 We envisioned closing the bay ring last, starting from perhydrophenanthrenes of type 18 or 20 (Scheme 2B). Although we initially intended to target 18 and install the last two carbons independently using the corresponding C7 and C20 ketone enolate functions, elaboration of 17 to 18 was unsuccessful. An alternate idea involved installation of these carbon atoms early; thus dione 19 was generated by reductive allylation of an enedione precursor.17 These early efforts were promising, because three of five stereocenters could be equilibrated to deliver 19 in an all-trans stereochemical configuration, obviating the need for kinetically controlled stereochemical induction. However, conversion to 20 via Robinson or related annulations was not successful. Although these specific approaches never led to tetracyclic DICA precursors, we continued to strategize around intermediates related to 18 and 20. Given that the major challenge up to that point revolved around annulation of a third ring, we pursued a new strategy of rapidly assembling a tricyclic hydrophenanthrene framework.
One of the most powerful means of polycyclic ring construction involves stitching together aromatic rings.18 In addition, given the known proclivity for dissolving metal reductions of both fused aromatic rings19 and enones20 to afford thermodynamically preferred trans-ring junctions (Scheme 3A), we were attracted to a strategy centered on stereocontrolled reduction of an aromatic precursor. The strategy rests on rapid access to an unsaturated aromatic intermediate, followed by saturation and a bay ring cyclization to introduce the fourth ring. While global reduction of a substituted phenanthrene such as 21 (Scheme 3B) was intellectually appealing, we opted to instead target hydrophenanthrenone 23 (Scheme 3C), which seemed logical both due to its ready accessibility from tetralones and the precedent for reduction under dissolving metal conditions.21
Scheme 3.

Honing in on an aromatic ring reduction strategy toward DICA.
The pursuit of a Birch reduction approach began with a Robinson annulation of known tetralone 2522 and methyl vinyl ketone (MVK, Scheme 4A). Phenanthrenone 26, lacking the methyl group found at the C15 position of DICA, was targeted to limit the number of possible diastereomers produced and to focus solely on the stereochemical outcome of the key Birch reduction. Robinson annulation of tetralone 25 with MVK smoothly delivered phenanthrenone 26, albeit as a 1:1 mixture of diastereomers. Addition of lithium metal to 26 in liquid ammonia, methanol and THF at −40 °C generated reduced products with spectral data indicating the desired reactivity, although lacking stereoselectivity. In search of a highly selective strategy, we turned our efforts elsewhere.23 At this point, we chose to exploit the near symmetry of DICA, and the cyclohexenone and anisole moieties of 28 were interchanged, pointing to new intermediate 29 (Scheme 4B). With the two stereogenic centers now possessing a vicinal relationship, we hoped that the diastereoselectivity of the Robinson annulation would improve, as precedent suggested.24
Scheme 4.

The full reduction of a hydrophenanthrenone and two possible isomeric intermediates drawn from the near symmetry of DICA.
The Robinson annulation of tetralone 30 and MVK indeed afforded a single diastereomer of hydrophenanthrenone 31 (Scheme 5). In this case, the methyl group was included on C15 because an aromatic starting material was commercially available and it would not cause any stereochemical complications in this setting. Exposure of 31 to lithium in ammonia selectively reduced the enone to cyclohexanone 32, but with the undesired cis-ring fusion, while the aromatic ring remained intact. Substituting tert-butanol with isopropanol or ethanol did not result in aromatic ring reduction and still provided the cis-isomer as the major product. Higher temperature dissolving metal reductions of the Benkeser type25 were evaluated and lithium in ethylenediamine26 was found to reduce the aromatic ring of 31 with 3:1 diastereoselectivity favoring the undesired cis-ring junction (33);27 however, hydrolysis and isomerization of the methoxycyclohexadiene could not be accomplished. The use of lithium or sodium in HMPA28 caused decomposition, while calcium29 did not effect reduction of 31 at all. Reduction with magnesium in methanol led to an equimolar mixture of ring junction diastereomers, as determined after reoxidation of the C7 alcohol to ketone; the arene was not reduced. Although hydrogenation of 31 did deliver predominantly the desired C4/C13 trans-stereochemistry, it also naturally left the arene untouched, which would necessitate subsequent reduction steps. Overall, we were interested in a more streamlined reaction that could reveal the all-trans network salient to DICA in a single step. As a result of these complications, we turned to a route in which the trans-stereochemistry of the ring junction was introduced earlier.
Scheme 5.

Hydrophenanthrenone synthesis and reduction attempts
A silver lining from these efforts is the further support for the unusual cis-selective reduction behavior of conjugated hydrophenanthrenones of type 29 under dissolving metal conditions (as opposed to the high trans-selectivity observed with “regular” ring-fused cyclohexenones). As the literature is mixed with regards to stereochemical outcomes, we suggest that care be taken when using precedent to assign the stereochemistry of products arising from these reactions.21
B. Key Aromatic Intermediate Bearing a Trans-Ring Fusion
Conceding that we might be unable to effect a one-step reduction of 31 with the desired stereochemical control to yield an advanced tricyclic intermediate, we envisioned a new key aromatic intermediate. As the key challenges of operating on 31 were enone reduction to set the trans-ring fusion and recalcitrant reduction of the aromatic, the new key intermediate instead featured a pre-installed trans-ring fusion and lack of the methyl group on C15, which should make reduction more facile (Scheme 6). Our new intermediary target 35 consisted of a tricyclic system embedded with a dihydronaphthalene, which we predicted to be amenable to reduction under Birch conditions.
Scheme 6.

New key aromatic intermediate with preinstalled trans-ring fusion.
Cyclohexenone 36 (Scheme 7), whose synthesis was optimized such that it could be procured in a robust three-step, two-distillation process,10a served as a substrate for trans-selective conjugate addition.30 Cuprate addition to 36 was most effectively executed with CuI•2LiCl as prescribed by Reetz.31 Ozonolysis and aldehyde/aromatic cyclodehydration was implemented to generate the advanced aromatic intermediate 39.32 Reduction with lithium in ammonia and methanol followed by treatment with acidic methanol furnished the expected enone 40 bearing an all-trans hydrocarbon core as anticipated.
Scheme 7.

Successful synthesis and Birch reduction of anisole 35
The next objective was to reduce the enone of 39, thereby setting the C12-stereogenic center, and then to perform an enolate alkylation with a group destined to produce the fourth ring of the DICA scaffold (Scheme 8A). Treatment of enone 39 with lithium in ammonia followed by allyl iodide generated allylated product 40 along with undesired side products resulting from diallylation and enolate protonation (likely resulting from enolate equilibration). The kinetic product had the allyl group in an equatorial position, in agreement with the strong literature precedence for equatorial allylation of octalones under typical Birch reduction/alkylation conditions.33 The C11, allyl-bearing stereogenic center required careful handling to avoid epimerization to the thermodynamically preferred axial isomer 41, which could be purposefully accessed by treatment with base. This unusual phenomenon was readily explained by relief of a syn-pentane interaction that is present with the allyl group equatorial (Scheme 8B). Interestingly, the drive for 40 to relieve non-bonded interactions is manifested in its reactivity; nucleophilic methylation of 40 generated equatorial alcohol 43 by formal axial approach of the methyl nucleophile. This behavior is in stark contrast to the typical equatorial methyl addition seen in cyclohexanone electrophiles and likely arises reactivity through potential low-energy twist-boat conformation 42, allowing for nucleophile addition from the more accessible bottom face.34 Although other issues (see below) ultimately led us to abandon this approach, we suspected that this interesting and potentially useful formal axial methylation would prove more problematic with the required C15 methyl group in place, as in the transformation of 44 to 45 (Scheme 8B).
Scheme 8.

Fourth ring closure proves difficult, and some interesting configurational preferences in tricyclic intermediates. Note: although drawn in a three-dimensional perspective, the three-dimensional depictions of compounds do not necessarily represent their lowest energy conformations, as they likely exist largely in twist-boat conformations as shown in part B.
Conversion of the alkene in 40 to an aldehyde for bay-ring cyclization proved to be surprisingly difficult (Scheme 8A). At the hydroxyketone oxidation state, lactol formation and decomposition occurred upon attempted oxidative cleavage (40 to 46). Surprisingly, alkene oxidation of the corresponding diketone 47 also met with failure; in both of these cases, it is possible that C11 epimerization under the reaction conditions is one of the contributors to the formation of many different products. Because the reductive allylation of 39 bearing a free alcohol was already poor-yielding, likely due to competitive proton transfer, the decision was made to protect the alcohol. Enolate methylation of TBS ether 49 followed by reductive allylation consistently afforded 51 in reasonable yields and diastereoselectivity. Unfortunately, a variety of alkene oxidation reactions on 51—including ozonolysis, Johnson–Lemieux oxidative cleavage, and epoxidation (not shown)—were not successful; overoxidation as well as reactivity of the C20 carbonyl appeared to be the major issues. Attempts at protecting the C20 carbonyl also failed to produce satisfactory results. Although we were successful in generating 52 via radical alkene nitration,35 our attempts to use this intermediate in ring closures were fruitless. At this time, the long step-count drove us to reevaluate this strategy for late-stage installation of the two-carbon unit.
C. A First-Generation Synthesis of (±)-DICA
Our early work cemented the dihydronaphthalene moiety as a viable unsaturated precursor that could be stereoselectively reduced to more advanced saturated intermediates that appeared promising for a synthesis of DICA. Our problems with the late-stage installation and manipulation of the two-carbon unit designed as a precursor to the fourth ring led us to consider its early introduction via an enone tandem vicinal difunctionalization approach (Scheme 9).36 In this way, a 4-substituted cyclohexene of type 56 might undergo conjugate arylation and subsequent enolate trapping with a two-carbon electrophile. The resident stereogenic center and the known preference for trans-selective enone vicinal difunctionalization promised a high degree of relative stereocontrol among C4, C13, and C8. The critical dihydronaphthalene substructure could be constructed by the same type of Friedel–Crafts-type cyclodehydration as used previously, only in the more highly functionalized case of aldehydes of type 55.
Scheme 9.

Change in strategy to permit early installation of two-carbon unit.
The key enone tandem vicinal difunctionalization was initially executed by modifying the workup of the cuprate conjugate addition used previously (Scheme 8) to retain the enoxysilane (see 36 to 57, Scheme 10A). Purification of 57 and treatment with methyllithium revealed the corresponding lithium enolate that was trapped with ethyl bromoacetate to deliver 58. This early intermediate contains all of the carbon atoms required for the perhydropyrene framework of DICA. An attempt to maintain the electrophilic ester for future bay-ring cyclization during the dihydronaphthalene installation led to partial C8 epimerization (see 59). To remove the liability of the C8 stereocenter the ketone of 58 was reduced, as was the ester, to give 60. Oxidative cleavage of the alkene provided aldehyde 61 in impure form, which could be moved forward through a successful cyclodehydration reaction; however, the desired dihydronaphthalene formation was accompanied by concomitant cyclization of the 1,4-diol to the corresponding tetrahydrofuran, generating 12. The same reactivity was observed when the primary alcohol was protected (see 62 to 12). Although double cyclodehydration to forge both the dihydronaphthalene and the THF ring looked attractive in terms of step count, the ozonolysis of precursor 62 produced a complex mixture of products that did not deliver 12 cleanly. The decision was made to intentionally trigger tetrahydrofuran formation by treating diol 60 with tosyl chloride. Ozonolysis of 63 delivered the aldehyde and several overoxidation products, the mixture of which was used directly for cyclodehydration to give 12. Birch reduction of 12 delivered 64 exclusively with the desired trans-stereochemistry. The route from 36 to 64 was streamlined by rendering the tandem vicinal difunctionalization into a single pot sequence and telescoping that crude reaction mixture into the LiAlH4 reduction (Scheme 10B). Replacement of the ozonolysis with a dihydroxylation and diol cleavage sequence avoided THF oxidation and thus improved material throughput in the preparation of 12. The cleaner material thus produced engaged in a higher-yielding Birch reduction, permitting access to 64 in multi-gram quantities.
Scheme 10.

Dihydronaphthalene assembly and Birch reduction
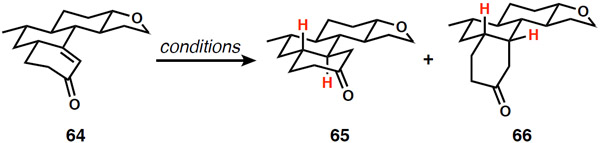
With the two-carbon unit for eventual cyclization of the bay-ring already in place (albeit in the unusual form of a THF ring), the enone of 64 required only reduction to introduce the C12 stereogenic center. Based on our previous experience (Scheme 8) in combination with the strong literature precedence for related reductions giving the desired trans-ring fusion,20 the reduction of 64 under dissolving metal conditions was evaluated (Table 1). Surprisingly, the major product using typical conditions presented a cis-ring fusion (entry 1). Other alkali metals at colder temperatures improved the outcome to 1:1 dr (entries 2 and 3). Addition of a bulky alcohol led to preferential formation of the cis-diastereomer (entry 4). Interestingly, this change in selectivity supports precedent that t-BuOH acts as the proton donor, leading to a possible steric effect during the protonation step. In any case, no dissolving metal conditions were found that tipped the selectivity toward the desired stereochemical outcome. Hydride reagents known to effect conjugate reductions led to greater selectivity for the cis-ring fusion (entries 5 and 6).37 Fortunately, hydrogenation using heterogeneous catalysts reliably installed the required trans-stereochemistry in reasonable yields (entries 7–10). The highest selectivities were observed with Rh/C (entry 9), although overreduction under the high required pressure necessitated an alcohol oxidation step to reinstall the C20 ketone.
Table 1.
Stereochemical control in enone reduction and introduction of the C12 stereogenic center
| |||
|---|---|---|---|
| Entry | Conditions | Yielda | 65:66 (trans:cis) |
| 1 | Li, NH3, THF, −40 °C | 64% | 1:2 |
| 2 | Na, NH3, THF, −78 °C | 85% | 1:1 |
| 3 | K, NH3, THF, −78 °C | 82% | 1:1 |
| 4 | K, t-BuOH, NH3, THF, −78 °C | 80% | 1:3 |
| 5 | Karstedt’s catb, Et3SiHc | 92% | 1:5 |
| 6 | t-BuCu, DIBAL-H, HMPA, THF | 86% | 1:>20 |
| 7 | H2, Pd/C, EtOAc | 94% | 6:1 |
| 8 | H2, Rh/alumina, EtOAcd | 93% | 8:1 |
| 9 | H2, Rh/C, EtOAcd | 93% | 15:1 |
| 10 | H2, Pt/C, EtOAc | trace | nd |
| 11 | H2, Ru/C, EtOAc | trace | 10:1 |
Yield of purified material after column chromatography
Karstedt’s Pt hydrosilylation catalyst
followed by TBAF, THF to convert enoxysilane to ketone
followed by PCC, Celite, CH2Cl2 to reoxidize some carbinol.
With installation of the C12 stereocenter complete, all that remained was a ‘simple’ THF ring-opening followed by enolate-mediated bay-ring closure. The most direct means of accomplishing this feat would be to treat 65 with a Lewis acid to encourage nucleophilic attack by a halide at the ethereal methylene position.38 Several conditions that were effective on 2-methyltetrahydrofuran were evaluated on 65, but without success, owing to undesired reactivity with the ketone (Scheme 11); no compound of type 67 was ever observed. Because we had seen inadvertent THF oxidation in 63 earlier (Scheme 10), we pursued C─H oxidations in parallel to nucleophilic ring opening. Treatment of 65 with CrO3 in acetic acid39 revealed a path forward via 68 and, though optimization did not lead to improved yields, provided enough material to evaluate downstream steps. The next objective was to convert the carbonyl of the lactone into an electrophilic moiety capable of cyclizing with an enol or enolate derived from the C20 ketone. Ketal protection of 68 followed by hydride reduction revealed the original diol functionality (69). Selective masking of the secondary alcohol was needed to convert the primary alcohol into an electrophilic center. Silyl protection of the primary alcohol of 69 followed by attempted benzylation of the secondary carbinol surprisingly yielded 70, likely due to facile silyl group transfer between the alcohols prior to kinetically preferred benzylation of the primary alcohol. The sequential alcohol protection strategy was altered, and the primary and secondary alcohols protected as a trityl ether and a benzyl ether, respectively, affording 72. Mild hydrolytic conditions revealed keto-alcohol 73, which was oxidized and cyclized via acid-catalyzed aldol condensation, thus generating the final ring on sub-milligram scale (see 74).
Scheme 11.

First successful bay-ring cyclization en route to DICA
With the general path laid out to advance tetrahydrofuran 65 to tetracyclic enone 74, a more efficient route was sought. At first, an improved C─H oxidation to either the lactol or lactone was targeted (Scheme 12A). A variety of Cr(VI)-based oxidations were screened, but the most reliable oxidation reaction in this series was the initially applied CrO3 in aqueous acetic acid. Lactol 75 was a major product from the oxidation with RuO4,40 TFDO,41 KMnO442 and others, with in situ-generated chromyl peroxide (CrO3-n-Bu4NIO4)43 emerging as the preferred option. Unfortunately, owing to difficulties in handling the lactol, we were unable to process it cleanly to tetracyclic products. Naturally, this outcome was disappointing because all that would have been required to access Corey’s dione was enolate alkylation of an activated form of the primary alcohol that was part of the lactol. Alternatively, further oxidation to the diketoaldehyde (not shown) might have permitted bay-ring-closure via aldol condensation; unfortunately, that type of oxidation was not successful. As a result, we opted to make use of lactone 68 in the final stages of our approach. Since hydrogenation of the alkene in 64 also resulted in some ketone reduction, the crude product was telescoped directly into the CrO3 C─H oxidation reaction to effect both oxidations concurrently giving 68 in an acceptable two-step yield (Scheme 12B). Most of the loss of material occurred during the oxidation step.
Scheme 12.

C─H oxidation as a means to process the 1,4-diol protected as a THF ring
From lactone 68, the sequence to Corey’s dione was lengthy but straightforward, and frequent telescoping of crude material improved the overall efficiency (Scheme 13). In a sequence that served to differentiate the three oxygenated carbon atoms to permit clean closure of the last ring, lactone 68 was converted to 73 over five steps in 75% overall isolated yield. Oxidation to the aldehyde set up for an efficient acid-catalyzed aldol condensation, affording enone 74. The methyl group at C15 was installed via the enolate, yielding a diastereomeric mixture of product 76. Hydrogenation of the alkene and hydrogenolysis of the benzyl group was accomplished in the same step. Partial undesired reduction of the C2 carbonyl was rendered inconsequential due to the necessary subsequent oxidation of the C7 secondary alcohol. From the newly-formed diketone, base-mediated equilibration of C15 finally produced (±)-Corey’s dione (5).
Scheme 13.

Intercepting (±)-Corey’s dione completes a formal synthesis of racemic DICA
A racemic formal synthesis was thereby completed via a rather lengthy—24 steps from commercially available materials—sequence, but featured numerous attractive characteristics that seemed likely to form the foundation of a particularly concise 2nd-generation synthesis. At this point we inspected our route to Corey’s dione, extracted lessons learned, and evaluated the issues that needed addressing to apply the overarching design to a short, complete synthesis of enantioenriched DICA. Overall, the strategy of rapidly creating a tricyclic styrene and leveraging stereocontrolled reductions successfully delivered DICA’s all-trans perhydropyrene framework, and the high degree of stereochemical control and the apparent robustness of most steps were major sources of enthusiasm. With a few minor changes to the key aromatic intermediate and some reorchestration of steps, many unproductive operations could be obviated, with a much shorter total synthesis as a very likely consequence.
D. A Short Stereocontrolled Second-Generation Synthesis of (+)-DICA
Having established a viable synthetic design for the tetracyclic architecture of DICA, the following issues needed to be addressed in a 2nd-generation synthesis: (1) the establishment of a stereocontrolled endgame for isonitrile introduction; (2) the careful editing of the perhydropyrene-building events to minimize unproductive steps; and (3) the identification of a source of asymmetry to permit an enantiocontrolled synthesis. The successful incorporation of these relatively small number of changes transformed a ~25-step formal synthesis of racemic material with a stereorandom endgame into a highly stereocontrolled, enantioselective total synthesis completed in half the number of steps.
As discussed previously, the strategy for introduction of DICA’s two isonitrile-bearing stereogenic centers acutely impacts the selection of intermediates. An important development that eased the burden of selecting an isonitrile installation strategy was Shenvi’s successful implementation of the stereoinvertive isonitrile introduction7b to simultaneously access the axial and equatorial tertiary isonitriles of DICA,8 which was reported after our first-generation synthesis. This end-game reduced the isonitrile problem to the stereoselective formation of a penultimate intermediate bearing stereochemically defined C7 and C20 tertiary carbinols (Shenvi’s diol, 7). A few critical structural changes to the 1st-generation’s key aromatic intermediate (see 12 to 77) enabled our much-improved 2nd-generation synthesis (Scheme 14).
Scheme 14.

Formal synthesis analysis and redesign to an improved 2nd-generation approach to DICA.
A first priority was to establish an enantioenriched cyclohexanone starting material onto which we could project the established strategy to ultimately yield an asymmetric synthesis of DICA. Unfortunately, in spite of our previous successes with asymmetric organocatalytic Robinson annulations,12,44 we struggled to access cyclohexanone 80 in highly enantioenriched form (Scheme 15A). The planned organocatalytic Michael addition between aldehyde 78 and MVK was successful in providing ketoaldehyde 79 in ~80% ee according to analysis of a derivative. However, after much experimentation with established45 and other methods, we were unable to find conditions for aldol condensation that were efficient without compromising the enantiopurity of the product (best case 66% ee, 61% yield10a). In search of synthetic equivalents to 80, we surmised that its lower homolog, 1,1-disubstituted alkene 82, could be used as an effective surrogate to deliver the same aldehyde intermediate 81 (Scheme 15B). This idea took on critical importance when we realized that 82, known as dehydrocryptone, can be made easily from the inexpensive monoterpene perillaldehyde (Scheme 15C).46 Process improvements of the known procedure of Razdan and co-workers46b enabled the preparation of up to 30 grams of 82 from (−)-perillaldehyde in just three steps and a single distillation.
Scheme 15.

Conception of a chiral pool approach to an asymmetric synthesis of DICA
To confirm the utility of dehydrocryptone as a starting material, we used it first to converge upon tetracyclic THF (−)-12 in enantiopure form (Scheme 16A). Without optimization, the same sequence of conjugate arylation/enolate trapping and reduction gave diol 84, which was cyclized to THF 85 under standard conditions. Whereas previously we had generated a C2-aldehyde by oxidative cleavage, here epoxidation followed by acid treatment directly delivered (−)-12.47 The aldehyde forged by Meinwald rearrangement of the intermediate epoxide (isolated in crude form) could not be observed, as it underwent spontaneous Friedel–Crafts-type cyclodehydration. Formally, this two-pot transformation accomplished a dehydrogenative coupling between two sp2 carbon atoms. Further, it completed our proof-of-principle for the strategic use of dehydrocryptone—and ultimately perillaldehyde—as a chiral pool starting material for DICA synthesis.
Scheme 16.

Dehydrocryptone enables access to key intermediates in enantioenriched form
The next key improvement involved the early installation of the C7 methyl group (Scheme 16B), which would result in the desired tertiary axial carbinol protected in lactone form (see, for example, 77 in Scheme 14). This change serves to differentiate C7 and C20, such that the stereocontrolled isonitrile introduction might be easily applied downstream. The key question was whether or not the lactone derived from the tertiary carbinol would be stable to the acidic conditions necessary for cyclodehydration to the dihydronaphthalene ring system. Rather than immediate hydride reduction of the intermediate from tandem vicinal difunctionalization of 82, the ketoester product 86 was instead isolated. Methylmagnesium bromide addition to the ketone functionality proceeded with near perfect equatorial delivery and the formed alkoxide spontaneously closed onto the pendant ester to cleanly provide lactone 87. Attempts at a one-pot conjugate addition/enolate trapping/nucleophile-triggered lactone formation sequence did provide product 87 in low yields; however, this sequence proved difficult to optimize owing to the large quantity of metal salts and additives already present in the reaction mixture prior to the addition of nucleophilic methylating agents. The crude lactone product was subjected to the two-step formal dehydrogenative C─C coupling sequence described above, yielding 88 without any apparent decomposition of the potentially sensitive tertiary lactone.
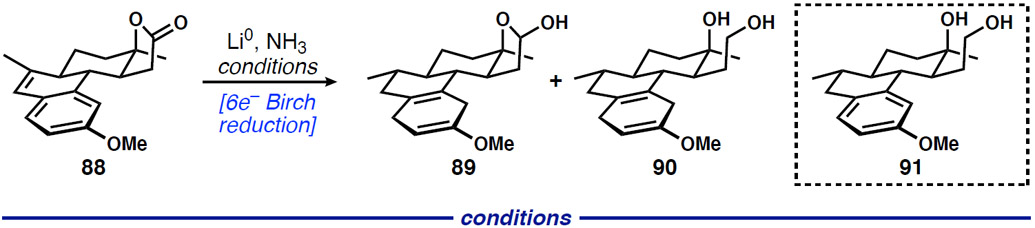
With the tetracyclic product in hand, we began efforts to realize a 6-electron Birch reduction of 88, in which the styrene, the lactone, and the electron-rich aromatic ring are each reduced to lactol/cyclohexadiene 89. The challenge inherent in this proposed transformation lies in the preservation of the lactol (anion) function, because overreduction to the diol (90) leads to the attendant need for multi-step differentiation of oxygenated carbon atoms similar to our 1st-generation approach. Maintaining the lactol provides that requisite differentiation, which is critical for shortening the end-game. Initial evaluation of the Birch reduction conditions employed in our 1st-generation synthesis (Scheme 10) showed rapid and complete formation of the lactone-derived diol prior to aromatic ring reduction (91). Attempts to accomplish full reduction to 90 were unsuccessful; the aromatic ring was only reduced in ~40% of the material after several hours under these conditions (Table 2, entry 1). We observed poor solubility of 88 prior to the addition of lithium, so we increased the ratio of THF to NH3 from 1:4 to 1:2, which led to increased solubility and consumption of the aromatic ring to ~75% (entry 2). Attempts to further increase the ratio of THF to NH3 led to biphasic reaction mixtures. Unexpectedly, replacement of methanol as a proton source with t-butanol completely suppressed lactol reduction with reasonable conversion of the arene. Although much of the mass balance had not undergone aromatic ring reduction (entry 3), this finding was critical to our 2nd-generation goals. At this stage, we do not understand the origins of the beneficial outcome provided by t-butanol. Next, we focused on improving reduction of the aromatic ring. Limited product solubility consistently plagued this reaction and was likely the cause for unreliable reactivity; thus, we investigated inverse order of addition. To our delight, adding 88 as a solution in THF to a mixture of ammonia, t-butanol, and lithium resulted in a homogeneous reaction mixture and 89% consumption of the aromatic ring (entry 4). A quick analysis of ethereal co-solvents showed that 1,2-dimethoxyethane led to complete aromatic ring consumption (entry 6).
Table 2.
Optimization of the 6-electron Birch reduction of 88 with avoidance of formation of diol 90
| ||||||
|---|---|---|---|---|---|---|
| Entn | H+ source | Co-solvent | Co-solvent: NH3 ratio |
Prodoct | Inverse addition |
% Arene redutcion |
| 1 | MeOH | THF | 1:4 | 90 | no | 40% |
| 2 | MeOH | THF | 1:2 | 90 | no | 75% |
| 3 | t-BuOH | THF | 1:5 | 89 | no | 46% |
| 4 | t-BuOH | THF | 1:1 | 89 | yes | 89% |
| 5 | t-BuOH | dioxane | 1:3 | 89 | yes | 88% |
| 6 | t-BuOH | DME | 1:3 | 89 | yes | >95% |
| 7 | s-BuOH | DME | 1:3 | 89 | yes | 86% |
 |
(1) |
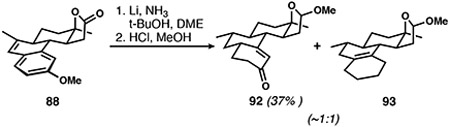
As alluded to above, what initially appeared to be a clean reduction led to the isolation of unexpectedly low quantities of enone 92 (Eq. 1) generated by hydrolysis of the initial Birch reduction product. After a thorough investigation of the crude product mixtures, we found that cyclohexene 93 was formed in equimolar quantities along with desired cyclohexenone 92. Cyclohexene products have frequently been reported as unintended over-reduced side products of certain anisole derivatives.48 Armed with the literature knowledge that reaction conditions often have little impact on attenuating their formation, we set out to evaluate reductions of the C15-methyl derivative in the hope that a higher-yielding reduction might be attained.
The revised route began with identical conditions as used previously for tandem vicinal difunctionalization of enone 82, except for the use of the aryl Grignard reagent incorporating the methyl group at C15 to give 94 (Scheme 17). Subsequent Grignard addition/lactonization occurred in essentially quantitative yield, delivering 95. We also utilized this opportunity to reevaluate the conditions used for styrene formation via the formal dehydrogenative coupling process that consists of alkene epoxidation followed by Meinwald rearrangement/cyclodehydration. Initially, we aimed to effect epoxidation with a peracid, whose by-product carboxylic acid would ideally be powerful enough to induce rearrangement and ring closure. A significant effort was undertaken to use m-CPBA for this proposed cascade because we had already determined that it was a viable epoxidizing reagent in this context. Perhaps unsurprisingly, we found that the corresponding m-chlorobenzoic acid was not a potent enough acid to cause the desired transformation, even when high-boiling solvents (benzene or toluene at reflux) were used to permit thermal instigation of epoxide rearrangement. Turning to the peroxy acid of a stronger carboxylic acid, the use of trifluoroperacetic acid (TFPAA) did indeed lead to styrene 77 in a single step. Unfortunately, we were unable to optimize this reaction to yield more than ~25% of the desired product. TFPAA is known to oxidize electron-rich arenes,49 which appeared to be the major problem with this particular reaction protocol. In several instances of incomplete reactions under protic conditions, we observed intermediates that were tentatively characterized as diastereomeric aldehydes that would result from Meinwald rearrangement, prior to cyclodehydration. Of course, Lewis acids are also competent effectors of Meinwald rearrangements.50 In our case, the rearrangement would directly produce a Lewis-acid-activated aldehyde poised for attack from the proximal arene. To further streamline this cascade reaction, we opted to use DMDO as a traceless oxidizing agent. As a result, only removal of acetone after oxygen transfer was required prior to addition of the appropriate Lewis acid. Redissolution of the crude epoxide followed by treatment with an appropriate Lewis acid allowed for the rapid screening of conditions. Several classes of Lewis acids were investigated, including aluminum-based MABR [methylaluminum bis(4-bromo-2,6-di-tert-butylphenoxide)], as well as those based on indium, bismuth and boron.51 Optimized conditions used an excess of boron trifluoride etherate at 0 °C, providing the desired styrene 77 in 76% yield on milligram scale, which translated to 84% yield on a gram scale.
Scheme 17.

Synthesis of tetracyclic lactone 77 bearing the methyl group at C15, with optimization of a one-pot epoxidation/Meinwald rearrangement/cyclodehydration
Unfortunately, our previously optimized Birch reduction conditions used for the reduction of 88 (Table 2) were ineffective, because the C15-methylated Birch precursor 77 (Scheme 18) was insoluble in DME and could not be added to the reaction mixture as a solution. A short screen identified 1,4-dioxane as a solvent capable of dissolving 77; however, extremely slow reactivity led to exclusive ring-opening to undesired diol 96 and minimal aromatic reduction. We found that use of s-butanol, previously identified as the least sterically-hindered proton source that did not cause lactol opening and reduction, significantly improved the reaction outcome. Following hydrolysis, enone 98 was isolated in 70% yield. Under these conditions, the lactol was converted to the corresponding acetal (2:1 dr) and the C15 stereogenic center was introduced as a mixture of diastereomers. These mixtures were expected to be of no consequence in downstream chemistry. Furthermore, the over-reduced cyclohexene byproduct was not observed, indicating a return to the expected Birch reduction outcome. Hydrogenation of enone 98 under conditions used in the first-generation synthesis (H2, Rh/C, EtOAc) was prone to over-reduction. While alcohol formation during reduction was not a problem in the 1st-generation synthesis because the next step was a CrO3 oxidation, in the present situation, the oxidation would necessitate an extra step before aldol-based ring-closure. Performing the reduction of 98 in methanol with Rh/alumina avoided over-reduction of the ketone and consistently delivered 99 in essentially quantitative yield. The addition of NaHCO3 during hydrogenation equilibrated the C15 methyl group to exclusively the equatorial position; however, we found it more convenient to effect this epimerization later in the synthesis because the forthcoming aldol condensation reaction reintroduced a thermodynamic mixture of C15 epimers.
Scheme 18.

Efficient Birch reduction with C15 methyl group and hydrogenation of the resulting cyclohexenone
At this point, lactol ether ring opening and aldol condensation remained for the formation of the tetracyclic scaffold of DICA. Because liberation of the aldehyde from the cyclic mixed acetal must precede ring closure, we were restricted to acidic or buffered (amine + acid) conditions. Application of the acidic conditions used in our 1st-generation synthesis was ineffective, but we identified enamine catalysis as a promising method for formation of 100 (Table 3). Product formation was first observed using pyrrolidine, water, and acetic acid (entry 1); however, significant decomposition also occurred. Gradual improvement resulted from replacing acetic acid with oxalic acid (entry 2) and changing amines (entries 3, 4, and 5). Even though it is not obvious from examination of the yields in entries 4 and 5, DL-proline provided the cleanest reactivity. Use of a large excess of DL-proline led to substantially improved efficiency (entry 6) and to a reliable reaction able produce material in synthetically useful quantities of isolated, pure material (Eq. 2).
Table 3.
Optimization of aldol condensation to close the fourth ring of DICA
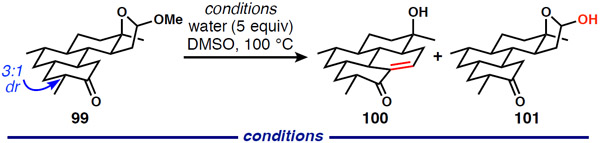
| ||||
|---|---|---|---|---|
| Entry | Amine (equiv.) |
Acid (equiv.) |
NMR yield 100 |
NMR yield 101 |
| 1 | pyrrolidine (5) | AcOH (113) | 26% | 10% |
| 2 | pyrrolidine (5) | oxalic acid (20) | 34% | 10% |
| 3 | piperidine (5) | oxalic acid (20) | 11% | 27% |
| 4 | morpholine (5) | oxalic acid (20) | 32% | 18% |
| 5 | DL-proline (5) | oxalic acid (20) | 30% | 20% |
| 6 | DL-proline (25) | oxalic acid (20) | 63% | 4% |
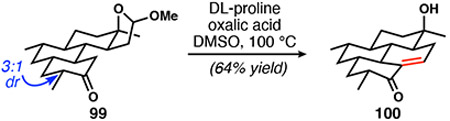 |
(2) |
Although enone 100 remained a diastereomeric mixture at C15, which presumably corresponded to the thermodynamic ratio under the forcing aldol conditions, subsequent hydrogenation under basic conditions proved highly selective and equilibrated both α-stereocenters to the all-equatorial configurations (Eq. 3). Based on the results of hydrogenations in the absence of base, C11 was formed with high kinetic selectivity, but the inclusion of base ensured complete epimerization of C15 to afford diastereomerically pure ketoalcohol 102, a compound prepared by Shenvi and co-workers en route to DICA.
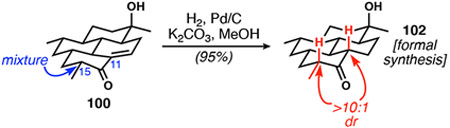 |
(3) |
With the hydrocarbon framework complete, the lone challenge remaining was selective axial methylation at C20 as a precursor to isonitrile introduction. Shenvi had previously converted ketoalcohol 102 to 7 (“Shenvi’s diol”) by a two-step Peterson alkenylation followed by oxymercuration/reduction. It was clear that a dramatic improvement would result from a direct axial methylation to install the C20 equatorial tertiary carbinol.52 Such a task is no small feat as the propensity for nucleophilic methylation reagents to add in an equatorial sense to substituted cyclohexanones is well-documented34a,53 and there are few methods shown to reverse this selectivity. Shenvi had already noted the ineffectiveness of Yamamoto’s bulky aluminum reagents54 to deliver an axial methyl group to 102, and this result was confirmed in our experiments. We therefore embarked on a screen of organometallic methylating agents (Scheme 19A), wherein the application of MeLi under standard conditions provided, as expected, predominantly equatorial delivery. Initial attempts to utilize Me3Al as an alternative, more electrophilic surrogate of Yamamoto’s reagent did increase the amount of desired axially-methylated product, albeit only modestly (entry 2). Lower temperatures with the Trapp solvent mixture (entry 3) resulted in near exclusive equatorial methylation. The application of dimethylzinc in a non-ethereal solvent (entry 4) appeared promising, with essentially no equatorial methylation product observed; however, this reaction was plagued by apparent elimination of the C7 tertiary carbinol (tentative assignment of structure 104, although it could never be purified to homogeneity), which could not be optimized against. Our breakthrough (entry 5) was inspired by Ashby and coworkers, who extensively studied the stereochemistry of trialkylaluminum additions to ketones and described the ability to control for axial addition by stoichiometry and solvent selection.55 The reported preferred axial addition to 4-tert-butylcyclohexanone with excess Me3Al also proved effective with our complex ketoalcohol substrate 102. Gratifyingly, we found that treatment of 102 with excess Me3Al in toluene at cryogenic temperatures and warming to ambient temperature provided methyl addition in ca. 2–2.5:1 dr and with 63% isolated yield of the desired diastereomer 7. Although these conditions produce 7 in comparable yield to Shenvi’s alkenylation/oxymercuration sequence (60%), our one step procedure is preferable due to the ease of setup, avoidance of stoichiometric mercury, and trivial separation of diastereomers. Moreover, our findings illustrate that a great deal has yet to be uncovered in the realm of axial carbonyl alkylations, and opens the door to further improvement of this important reaction type.
Scheme 19.

Endgame features an unusual axial nucleophilic methylation of ketone 102
All that remained to complete the synthesis of DICA (1) was the stereoinvertive bis-isocyanation of diol 7 (Scheme 19B). The conditions developed and applied by Shenvi worked exactly as reported; thus, treatment of the bis-trifluoroacetate of diol 7 with trimethylsilyl cyanide and scandium triflate furnished 1 in 59% yield as a 5:1 mixture of DICA to other diastereomers. To date, approximately 30 mg DICA have been prepared to support ongoing biological and physicochemical studies, and we are encouraged that this short route will permit access as needed for further studies of this fascinating antimalarial natural product.
Conclusion.
Sometimes, small changes to a synthesis design can have seemingly outsized effects on the overall outcome. That was surely the case as we adapted key lessons of our lengthy 1st-generation formal synthesis of DICA to a strategy that became a concise 2nd-generation enantiospecific total synthesis (Scheme 20). We first identified a chiral pool starting point in (−)-dihydrocryptone (82), which is readily available from perrilaldehyde. From this precursor, introduction of the tertiary carbinol at C7 early in the synthesis differentiated C7 and C20. This tactic led to the retention of a higher oxidation state at C10 in the form of lactone 95, which we hoped could be reduced to the aldehyde oxidation state (lactol ether, see 99) for bay-ring closure via aldol chemistry. This single change obviated the need to oxidize the THF ring in 65, and to spend several steps differentiating the oxygenated carbon atoms in late-stage intermediates. Finally, as a result of the early introduction of the C7 carbinol, access to Shenvi’s diol (7)—the precursor for stereoinvertive isonitrile introduction—would require only the stereoselective formation of the C20 equatorial tertiary carbinol via an admittedly non-trivial axial ketone methylation, which was solved using Ashby’s protocol with trialkylaluminums. We initially considered versions of this 2nd-generation approach that either did or did not include the methyl group at C15 (see 95). In the end, the early introduction of this methyl group also proved critical, because in addition to saving a late-stage enolate methylation step, its presence dramatically increased the efficiency of the key 6-electron Birch reduction. To really hone in on the key changes, it is helpful to compare key aromatic intermediates 12 and 77 from the two approaches; the early introduction of two methyl groups and the higher oxidation state in 77 permitted us to cut the length of the synthesis in half. Put another way, these seemingly small changes resulted in a 2nd-generation synthesis in which every step is productive, generating either new C─C, C─N, or C─O bonds, and/or new stereogenic centers, and ultimately leads to a particularly rapid buildup of the complexity of the antiplasmodial isocyanoterpene, 7,20-diisocyanoadociane.
Scheme 20.

Summary of improvements that led to 2nd-generation total synthesis of DICA
Experimental Part.
The following compounds were reported previously, and the experimental protocols for their preparation and full characterization data can be found in the references indicated: 15,8,10b, 55,10a, 78,10b, 1210a, 2522, 3610a, 5910a, 64-6610a, 6810a, 73-7610a, 7710b, 78-8010a, 8210a,46b, 84-8510a, 9410b, 9510b, 98-9910b, 1008,10b, 102-10310b. Some procedures below involve compounds S2, S3, and S4, whose preparations can be found in the Supporting Information.
General
All reactions were carried out under an argon atmosphere with dry solvents under anhydrous conditions, unless otherwise noted. Argon balloons were the sole inert atmosphere used. Reactions run at an ambient temperature of 20–25 °C are designated as room temperature. Reactions that were performed open to air utilized solvent dispensed from a wash bottle or solvent bottle, and no precautions were taken to exclude water. Reactions that were performed open to air utilized solvent dispensed from a wash bottle or solvent bottle, and no precautions were taken to exclude water. Yields refer to chromatographically and spectroscopically homogeneous materials, unless otherwise stated. Dry tetrahydrofuran (THF), diethyl ether (Et2O), dichloromethane (CH2Cl2), toluene, benzene (C6H6) and methanol (MeOH) were obtained by passing commercially available formulations through activated alumina columns. tert-Butyl alcohol (t-BuOH), N,N-diisopropylamine (i-Pr2NH), triethylamine (NEt3), pyridine (py), hexamethylphosphoramide (HMPA), benzyl chloride (BnCl) and trimethylsilyl chloride (TMSCl) were purified by distillation from CaH2. Ethyl bromoacetate was purified by washing three times with 2M aq. Na2CO3, twice with brine, drying over MgSO4, filtering and distillation. Allyl iodide was purified by distillation and stored over copper beads at −20 °C. Copper(I) iodide (CuI) was purified by Soxlet extractor with THF then drying the solid under high vacuum. Methyl vinyl ketone (MVK) was purified by distillation. Alkyllithium reagents were titrated using 2,6-di-(tert-butyl)-4-methylphenol (BHT) as the sacrificial proton source and fluorene as an indicator in THF or using diphenylacetic acid in THF. Grignard reagents were titrated using salicylaldehyde phenylhydrazone in THF.
Thin layer chromatography was performed on 0.25 mm EMD glass-backed TLC plates impregnated or Merck silica gel 60 F254 TLC plates with a fluorescent dye and visualized with UV light and KMnO4 in K2CO3/NaOH/water, p-anisaldehyde in ethanol/aqueous H2SO4/AcOH, cerium ammonium molybdate in aqueous H2SO4, or phosphomolybdic acid in ethanol and heat as a developing agent. Forced flow (flash) chromatography was performed on EMD Silica 60, mesh 0.04-0.063 silica gel. NMR spectra were recorded on Bruker 500 MHz or 600 MHz instrument, obtained at 298 K unless otherwise noted and calibrated to residual undeuterated solvent as an internal reference. Chemical shifts are reported in ppm with the following abbreviations to explain multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, quin = quintuplet, sext = sextet, sep = septet, bs = broad signal, m = multiplet. All coupling constants are apparent J values measured at the indicated field strengths and reported in Hertz (Hz). FT-IR spectra were recorded on a Perkin-Elmer spectrum RX1 or Varian 640-IR spectrometer. High-resolution mass spectra (HRMS) were recorded on a Waters LCT Premier spectrometer using ESI-TOF (electrospray ionization-time of flight) or Waters GCT Premier spectrometer using GC-CI, as indicated. Melting points were measured on a MEL-TEMP II capillary apparatus and stand uncorrected.
Tetracycle 12 from 60:
see reference 10a.
Tetracycle 12 from 62:
To a 5 mL round bottom flask containing S3 (7 mg, 0.017 mmol) was added a solution of TsOH•H2O (2 mg, 0.011 mmol, predried with 2 mL toluene over a Hickmann still). The reaction was refluxed over a Hickmann still for 2 hours. The reaction was cooled, diluted with sat. aq. NaHCO3, EtOAc and the layers separated. The organic layer was washed with brine, dried over MgSO4, filtered and all volatiles removed in vacuo. The crude oil was purified by column chromatography (SiO2, 5:1 hexanes/EtOAc) to afford 12 (3 mg, 66%, single diastereomer) as a colorless oil.
Tetracycle 12 from 63:
To a 10 mL round bottom flask containing S4 (63 mg, 0.218 mmol) was added a solution of TsOH•H2O (2 mg, 0.011 mmol, predried with 2 mL toluene over a Hickmann still). The reaction was heated to reflux in an oil bath under a Hickmann still for 2 hours. The reaction was cooled, diluted with sat. aq. NaHCO3, EtOAc and the layers separated. The organic layer was washed with brine, dried over MgSO4, filtered and all volatiles removed in vacuo. The crude oil was purified by column chromatography (SiO2, 5:1 hexanes/EtOAc) to afford 12 (31 mg, 52%, single diastereomer) as a colorless oil.
Phenanthrenone 26:
A 25 mL round bottom flask containing 25 (201 mg, 1.06 mmol) and ethyl formate (0.15 mL, 1.86 mmol) in THF (5 mL) was cooled in an ice bath and treated with 1 M t-BuOK/THF (1.3 mL, 1.3 mmol). The ice bath was removed upon complete addition and stirring continued for 1 hour. The reaction was treated with 6 M HCl (2 mL) and water (8 mL), then extracted CH2Cl2 (3 x 5 mL). The organic layers were combined, washed with half sat. brine (10 mL), dried over MgSO4, filtered and all volatiles removed in vacuo. The crude mixture in a 10 mL round bottom flask was stirred with MVK (0.30 mL, 3.60 mmol) and powdered NaOH (7 mg, 0.18 mmol) [exotherm]. After 20 minutes the reaction was diluted with sat. NH4Cl (5 mL) and extracted twice with CH2Cl2 (2 x 5 mL). The organic layers were combined, dried over MgSO4, filtered and all volatiles removed in vacuo. A 25 mL round bottom flask containing the crude material in THF (4 mL) was treated with NaOH (210 mg, 5.25 mmol) in water (4 mL) at 0 °C. The ice bath was immediately removed and stirring continued for 36 hours. The reaction was quenched with half sat. NH4Cl (10 mL) and extracted CH2Cl2 (2 x 10 mL). The organic layers were combined, washed with brine (5 mL), dried over MgSO4, filtered and all volatiles were removed. The residue was purified by column chromatography (SiO2, 5:1 hexanes/EtOAc) to afford 26 (195 mg, 74% over 3 steps, 1:1 dr) as a colorless oil. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 7.33 (d, J = 8.7 Hz, 1H), 7.25 (d, J = 2.6 Hz, 1H), 7.21 (d, J = 2.6 Hz, 1H), 7.13 (d, J = 8.5 Hz, 1H), 6.98 (dd, J = 8.7, 2.7 Hz, 1H), 6.95 (dd, J = 8.5, 2.6 Hz, 1H), 6.61 (s, 2H), 3.82 (s, 3H), 3.81 (s, 3H), 3.09 (qt, J = 7.2, 3.6 Hz, 1H), 2.97 (dquintet, J = 12.0, 6.0 Hz, 1H), 2.92-2.85 (m, 1H), 2.73-2.66 (m, 1H), 2.62-2.54 (m, 2H), 2.53-2.42 (m, 2H), 2.23-2.12 (m, 3H), 2.04 (dt, J = 12.9, 4.2 Hz, 1H), 1.84-1.75 (m, 4H), 1.40-1.38 (m, 2H), 1.37 (d, J = 6.8 Hz, 3H), 1.32 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3, at 77 ppm) δ 200.4, 200.3, 159.1, 158.3, 158.0, 157.9, 137.4, 137.0, 132.1, 131.3, 130.5, 128.1, 120.6, 120.3, 118.5, 118.2, 108.7, 108.3, 55.3 (2 C), 40.2, 37.5, 37.2, 36.9, 36.8, 32.4, 32.1, 31.0, 30.4, 30.1, 22.7, 21.1; IR (thin film) 2927, 2856, 1660, 1611, 1589, 1493 cm−1; HRMS (ESI) calculated for C16H18O2 [M+Na]+ 265.1205 found 265.1210.
Tetralone 30:
A 100 mL round bottom flask containing DME (24 mL) and NaH (0.970 g of a 60% in mineral oil, 24.3 mmol) was cooled in an ice bath and dimethylmalonate (2.7 mL, 23.6 mmol) added dropwise. After the reaction was warmed to room temperature S2 (3.00 g, 11.6 mmol) was added in one portion, the flask fitted with a reflux condenser and the reaction placed in an oil bath at 110 °C. After 20 hours, the reaction was cooled, the volatiles stripped in vacuo and the solid mass dissolved in two-thirds sat. NH4Cl (60 mL) and EtOAc (60 mL). The aqueous layer was washed with additional EtOAc (20 mL). The combined organic phases were washed with brine (20 mL), dried over MgSO4, filtered all volatiles removed in vacuo. The crude material was used without further purification.
The crude diester was placed into a 250 mL round bottom flask, diluted with 1:1:1 THF/MeOH/water (60 mL) and treated with powdered NaOH (4.64 g, 116 mmol). The flask was placed into an oil bath at 70 °C. After 1.5 hours, the reaction was cooled, the volatiles stripped in vacuo and crude material taken up in water (120 mL). The aqueous solution was washed with Et2O (2 x 20 mL), then conc. HCl (40 mL) was added and the aqueous phase extracted with Et2O (2 x 60 mL). The organic layers were combined, washed with brine (20 mL), dried over MgSO4, filtered and all volatiles removed in vacuo. The crude solid was used without further purification.
The crude diacid in a 100 mL round bottom flask was dissolved in diglyme (35 mL) and placed in an oil bath at 170 °C for 1 hour. The reaction was cooled and all volatiles were removed in vacuo. The obtained oil was used without further purification.
The crude acid in a 100 mL round bottom flask was treated with polyphosphoric acid (38 g, prepared from 25 g H3PO4 and 15 g P2O5) and heated at 90 °C in an oil bath for 3 hours. The external heating was removed and the reaction mixture was treated slowly with ice (150 g), then 5 M NaOH (20 mL) and the resulting aqueous solution extracted with CH2Cl2 (2 x 60 mL). The organic extracts were combined, washed with brine (20 mL), dried over MgSO4, filtered, and the volatiles were removed in vacuo. The crude solid was dissolved in hexanes (20 mL), filtered over Celite, concentrated to approximately half volume and crystallized in a freezer overnight. The solvent was removed via cannula, the crystals washed with hexanes (2 x 2 mL) and the remaining solvent removed in vacuo to afford 30 (1.44 g, 60% over 4 steps) as a white solid (mp = 68–69 °C). 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 7.44 (s, 1H), 7.01 (s, 1H), 3.87 (s, 3H), 2.88 (dd, J = 15.9, 3.6 Hz, 1H), 2.70 (dd, J = 13.1, 1.5 Hz, 1H), 2.59 (dd, J = 15.9, 10.3 Hz, 1H), 2.33-2.26 (m, 2H), 2.26-2.24 (m, 3H), 1.13 (d, J = 6.1 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 198.5, 156.6, 136.4, 133.9, 130.9, 130.9, 106.6, 55.5, 46.9, 37.2, 30.9, 21.4, 16.6; IR (thin film) 2951, 1678, 1510, 1496, 1302, 1210 cm−1; HRMS (ESI) calculated for C13H16O2 [M+H]+ 205.1228 found 205.1221.
Phenanthrenone 31:
A 50 mL round bottom flask containing NaH (304 mg of a 60% in mineral oil, 7.60 mmol) in Et2O (15 mL) was cooled in an ice bath and treated sequentially with ethyl formate (0.95 mL, 11.8 mmol), 30 (602 mg, 2.95 mmol) and ethanol (0.05 mL, 0.86 mmol). Stirring was continued at 0 °C for 20 minutes before the bath was removed. After an additional 1 hour and 40 minutes, 0.25 M NaOH (40 mL) was added, the layers separated and the aqueous phase extracted with Et2O (2 x 10 mL). The aqueous layer was acidified with 6 M HCl (10 mL) and extracted with Et2O (40 mL and 10 mL). The organic extracts were combined, washed with brine (10 mL), dried over MgSO4, filtered and all volatiles removed in vacuo to afford crude product that was used without further purification.
The crude vinylogous acid in a 100 mL round bottom flask was dissolved in CH2Cl2 (3 mL) and NEt3 (3 mL, 21.5 mmol) at room temperature and treated with MVK (0.75 mL, 9.00 mmol). After 13 hours of reaction at room temperature, all volatiles were removed in vacuo to afford crude product that was used without further purification.
The crude solid was dissolved in THF (8 mL), cooled to 0°C and treated by dropwise addition with KOH (980 mg, 17.5 mmol dissolved in 8 mL water). After complete addition, the cold bath was removed and the reaction was stirred for 5 days. The volatiles were removed in vacuo and the remaining mixture was partitioned between sat. NH4Cl (10 mL) and EtOAc (30 mL), and the layers were separated. The aqueous layer was extracted with EtOAc (10 mL). The combined organic extracts were washed with brine (10 mL), dried over MgSO4, filtered, and all volatiles were removed in vacuo. The crude material was resubjected to the same conditions as described above for an additional 4 days. The same work up procedure was repeated. Crystallization from CH2Cl2/hexanes afforded 31 (501 mg, 66%, single diastereomer) as an off-white crystalline solid (mp = 188–190 °C). 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 7.15 (s, 1H), 6.94 (s, 1H), 6.64 (s, 1H), 3.83 (s, 3H), 2.79 (dd, J = 16.3, 4.1 Hz, 1H), 2.63-2.54 (m, 2H), 2.45-2.37 (m, 2H), 2.28-2.25 (m, 1H), 2.22 (s, 3H), 1.74-1.61 (m, 2H), 1.16 (d, J = 6.5 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 200.3, 158.8, 156.4, 132.4, 131.2, 130.9, 129.5, 119.8, 105.1, 55.3, 43.3, 38.3, 37.2, 35.3, 27.0, 19.5, 16.2; IR (thin film) 2949, 2913, 1654, 1205 cm−1; HRMS (ESI) calculated for C17H20O2 [M+H]+ 257.1541 found 257.1529.
Ketone 32:
A 10 mL round bottom flask was charged with excess lithium metal and ammomia at −40 °C, then 31 (10 mg, 0.039 mmol) and t-BuOH (58 mg, 0.78 mmol) in THF (0.5 mL) was added dropwise. After 30 min another portion of t-BuOH (58 mg, 0.78 mmol) in THF (0.2 mL) was added. The reaction was quenched with solid NH4Cl, warmed to room temperature, diluted with water and extracted with EtOAc. The organic extract was dried over MgSO4, filtered and all volatiles were removed. The crude solid was purified by column chromatography (SiO2, 8:1 hexanes/EtOAc) to afford 32 (8.0 mg, 80%, 8:1 dr) as a white wax. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) major δ 6.87 (s, 1H), 6.50 (s, 1H), 3.79 (s, 3H), 3.20 (td, J = 8.6, 4.3 Hz, 1H), 3.09 (ddd, J = 14.1, 3.7, 2.1 Hz, 3H), 2.86 (dd, J = 16.3, 5.1 Hz, 1H), 2.57-2.55 (m, 2H), 2.47-2.19 (m, 5H), 2.19 (s, 3H), 1.86 (ddq, J = 14.1, 9.2, 4.6 Hz, 2H), 1.14 (d, J = 6.5 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) major δ 211.5, 156.0, 137.5, 130.9, 127.0, 125.0, 109.0, 55.3, 47.6, 42.4, 39.0, 37.3, 29.7, 27.3, 26.3, 19.2, 15.8; IR (thin film) 2919, 2853, 1710 cm−1; HRMS (ESI) calculated for C17H22O2 [M+NH4]+ 276.1964 found 276.1967.
Cyclohexadienes 33:
To a 25 mL round bottom flask was added 31 (20 mg, 0.080 mmol), t-BuOH (0.38 mL, 4.0 mmol), ethylenediamine (0.8 mL), and THF (0.8 mL). A glass stir bar was added and the reaction mixture was stirred vigorously at room temperature. Lithium metal (25 mg, 3.6 mmol) was added over the course of 2 hours. After stirring overnight. the mixture was diluted with THF (5 mL) and cannula transferred into a stirring mixture of water (10 mL) and Et2O (15 mL). The layers were separated and the aqueous phase was extracted with Et2O. The organic extracts were combined, washed with brine, dried over MgSO4, filtered and all the volatiles were removed in vacuo to afford crude 33 (3:1 dr). The relative configurations of the isomers of 33 were ascertained by conversion of the mixture to 32 and 34 by oxidation. This compound was not purified any further.
Dihydronaphthalene 35:
A 100 mL round bottom flask containing 38 (2.36 g, 9.07 mmol), TsOH•H2O (86 mg, 0.45 mmol) and toluene (70 mL) was heated to reflux in an oil bath under a Dean–Stark trap. After 2 hours the reaction was cooled, diluted with Et2O (100 mL), washed with sat. aq. NaHCO3 (20 mL), brine (20 mL), dried over MgSO4, filtered and all of the volatiles were removed in vacuo. The crude material was purified by column chromatography (SiO2, 6:1 hexanes/EtOAc) to afford 35 (1.78 g, 81%) as a white solid that was crystallized from Et2O to afford a single cubic crystal (mp = 132–134 °C). 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 6.97 (d, J = 7.9 Hz, 1H), 6.73 (d, J = 7.5 Hz, 1H), 6.72-6.67 (m, 1H), 6.29 (s, 1H), 3.80 (s, 3H), 3.04 (d, J = 13.6 Hz, 1H), 2.87 (t, J = 12.7 Hz, 1H), 2.58-2.39 (m, 5H), 1.92 (s, 3H), 1.63 (t, J = 11.4 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 210.5, 158.5, 138.1, 136.0, 127.7, 126.3, 124.1, 110.8, 110.1, 55.2, 43.8, 42.2, 40.9, 40.7, 28.7, 20.6; IR (thin film) 2918, 2849, 1713, 1606, 1571, 1242, 1159 cm−1; HRMS (ESI) calculated for C16H18O2 [M+Na]+ 265.1205 found 265.1210.
Ketone 37:
3-Methoxyphenylmagnesium bromide was prepared by addition of 3-bromoanisole (3.0 mL, 23.7 mmol) in THF (3.0 mL) to magnesium metal (0.865 g, 35.6 mmol) in THF (12 mL) activated by dibromoethane, at such a rate as to maintain reflux. To a 250 mL round bottom flask charged with 36 (2.00 g, 13.3 mmol), CuI (0.255 g, 0.133 mmol), LiCl (0.125 g, 2.95 mmol), and TMSCl (1.9 mL, 15.0 mmol) in THF (80 mL) cooled to 0 °C was added 3-methoxyphenylmagnesium bromide (13.5 mL of a 1.2 M solution, 16.2 mmol) dropwise over 30 minutes. The reaction was stirred for an additional hour and quenched at 0 °C by addition of 1 M HCl (50 mL) and warmed to room temperature. The mixture was extracted with Et2O (50 mL and 25 mL). The organic extracts were washed with 3 M HCl (40 mL), water (20 mL), 3:1 sat. aq. NH4Cl/5 M NaOH (40 mL), brine (20 mL), dried over MgSO4, filtered and all of the volatiles were removed in vacuo. The crude oil was purified by column chromatography (SiO2, 10:1 hexanes/EtOAc) to afford 37 (2.67 g, 78%, 1:1 dr) as a colorless oil. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 7.28-7.25 (m, 2H), 6.79 (td, J = 15.7, 7.6 Hz, 6H), 5.81 (ddd, J = 17.0, 10.7, 6.1 Hz, 1H), 5.67 (dt, J = 17.8, 8.9 Hz, 1H), 5.00 (d, J = 10.5 Hz, 2H), 4.93 (d, J = 17.3 Hz, 1H), 4.82 (d, J = 17.1 Hz, 1H), 3.82 (s, 6H), 2.88 (td, J = 20.4, 11.3 Hz, 2H), 2.59-2.41 (m, 8H), 2.22-2.09 (m, 4H), 2.05-1.99 (m, 2H), 1.55 (quintetd, J = 12.9, 4.9 Hz, 2H), 1.01 (d, J = 7.0 Hz, 3H), 0.84 (d, J = 6.9 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 210.69, 210.66, 159.9, 159.8, 145.0, 144.8, 143.3, 138.8, 129.9, 129.8, 119.8, 119.5, 115.7, 113.6, 113.5, 113.4, 111.5, 111.5, 55.2, 49.7, 49.5, 48.2, 47.7, 46.4, 45.7, 41.1, 41.0, 37.2, 36.2, 25.3, 25.1, 18.9, 11.5; IR (thin film) 2959, 1714, 1600, 1487, 1263, 1046 cm−1; HRMS (ESI) calculated for C17H22O2 [M+Na]+ 281.1518 found 281.1511.
Ketoaldehyde 38:
A 250 mL round bottom flask containing 37 (2.67 g, 10.3 mmol) in CH2Cl2 (100 mL) was treated with ozone at −78 °C. After a persistent blue color, the excess ozone was discharged by bubbling oxygen through the solution until the color disappeared, and PPh3 (3.22 g, 12.2 mmol) was added. The reaction was allowed to warm to room temperature over several hours. All of the volatiles were removed and the residue purified by column chromatography (SiO2, 3:1 hexanes/EtOAc) to afford 38 (2.40 g, 88%, 1:1 dr) as a colorless oil. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 9.59 (s, 1H), 9.47 (s, 1H), 7.28 (q, J = 8.0 Hz, 2H), 6.79 (td, J = 17.7, 7.7 Hz, 6H), 3.79 (d, J = 8.0 Hz, 6H), 3.12 (q, J = 9.8 Hz, 1H), 2.88 (td, J = 11.5, 5.4 Hz, 1H), 2.73-2.67 (m, 1H), 2.64-2.46 (m, 8H), 2.41-2.36 (m, 2H), 2.25 (qd, J = 7.2, 2.5 Hz, 1H), 2.20-2.14 (m, 1H), 1.87-1.67 (m, 2H), 1.60 (dtd, J = 16.7, 11.3, 5.0 Hz, 1H), 1.11 (d, J = 7.0 Hz, 3H), 1.01 (d, J = 7.3 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 209.50, 209.47, 205.5, 203.2, 160.1, 160.0, 143.72, 143.69, 130.3, 130.1, 119.6, 119.3, 113.6, 113.3, 112.2, 112.0, 55.2, 49.4, 49.2, 47.4, 47.2, 47.0, 46.7, 44.8, 41.0, 41.1, 40.2, 27.9, 26.2, 11.2, 7.1; IR (thin film) 2943, 2719, 1716, 1600, 1488, 1263, 1046 cm−1; HRMS (ESI) calculated for C16H20O3 [M+Na]+ 283.1310 found 283.1307.
Enone 39:
A 1 L 3-neck round bottom flask fitted with a low-temperature thermometer was charged with a glass stir bar, 35 (1.77 g, 7.30 mmol) in THF (75 mL), ammonia (230 mL), and MeOH (15 mL, 370 mmol), and the mixture was cooled to −60 °C. Lithium metal (2.50 g, 260 mmol) was added slowly in small pieces at −40 °C. After the blue color discharged, solid NH4Cl (20 g, 374 mmol) was added and the ammonia was allowed to evaporate overnight. The white residue taken up in water (50 mL) and extracted with EtOAc (100 mL, 50 mL and 25 mL). The organic extracts were combined, washed with brine (20 mL), dried over MgSO4, filtered and all of the volatiles were removed in vacuo. To a 300 mL flask containing the crude material was added MeOH (75 mL) followed by 6 M HCl (7.5 mL). After 8 hours, sat. aq. NaHCO3 (100 mL) was added and after 1 hour of stirring ~60 mL of the volatiles were removed in vacuo. The remaining solution was extracted with EtOAc (75 mL, 50 mL and 2 x 25 mL). All organic extracts were combined and were washed with brine (30 mL); the aqueous wash was back extracted with EtOAc (25 mL). The combined organics were dried over MgSO4, filtered and all of the volatiles were removed in vacuo. The crude material was purified by column chromatography (SiO2, 1:1→1:3 hexanes/EtOAc) to afford 39 (1.20 g, 70%) as a white solid that was recrystallized from Et2O to afford colorless prisms (mp = 139–140 °C). 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 5.80 (s, 1H), 3.64 (tt, J = 10.3, 4.8 Hz, 1H), 2.40-2.24 (m, 4H), 2.15-2.02 (m, 4H), 1.89-1.85 (m, 2H), 1.66-1.58 (m, 1H), 1.38 (dddt, J = 12.5, 9.4, 6.3, 3.1 Hz, 1H), 1.26-1.09 (m, 3H), 1.07-0.98 (m, 1H), 0.92 (d, J = 9.6 Hz, 3H), 0.78 (qd, J = 10.9, 3.1 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 200.6, 169.0, 121.0, 70.5, 48.9, 45.2, 43.4, 38.1, 37.4, 36.8, 35.5, 34.9, 29.3, 28.5, 19.3; IR (thin film) 3392, 2926, 2857, 1658, 1057 cm−1; HRMS (ESI) calculated for C15H22O2 [M+Na]+ 257.1518 found 257.1521.
Allylated Ketone 40:
To a 25 mL round bottom flask containing a glass stir bar and lithium metal (7.7 mg, 1.1 mmol) was condensed ammonia (7 mL) and THF (1.5 mL) was added. At −50 °C, 39 (76 mg, 0.32 mmol) in THF (1.5 mL) was added. The reaction was cooled to −78 °C then treated with isoprene (0.1 mL) and stirred until all of the lithium was discharged. To the resulting white slurry was added allyl iodide (69 mg, 0.41 mmol) at −78 °C. The reaction was stirred for 2.5 hours before sat. aqueous NH4Cl (3 mL) was added slowly and the mixture was warmed to room temperature. The mixture was partitioned between water (5 mL) and EtOAc (15 mL). The organic layer was washed with brine (5 mL), dried over MgSO4, filtered and all of the volatiles were removed in vacuo. The crude material was purified by column chromatography (SiO2, 4:1→1:2 hexanes/EtOAc) to afford 40 (38 mg, 43%, 4:1 dr) as a white wax. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 5.71-5.60 (m, 1H), 5.12-4.97 (m, 2H), 3.54 (t, J = 10.4 Hz, 1H), 2.48 (td, J = 15.7, 8.2 Hz, 2H), 2.32-2.22 (m, 3H), 2.11 (d, J = 9.7 Hz, 1H), 2.02-1.96 (m, 2H), 1.75-1.59 (m, 4H), 1.32-1.11 (m, 5H), 0.99-0.75 (m, 6H), 0.61 (q, J = 10.4 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) major δ 216.0, 134.9, 117.2, 70,7, 53.2, 50.9, 48.0, 46.6, 42.6, 39.9, 39.8, 39.4, 37.2, 36.5, 35.5, 28.8, 28.3, 20.0, minor δ 214.4, 134.9, 116.6, 70.6, 51.3, 50.2, 47.3, 42.4, 40.8, 38.1, 38.0, 36.3, 35.4, 34.4, 34.2, 30.4, 28.4, 19.9; IR (thin film) 3388, 2923, 2858, 1702, 1444, 1051, 1023 cm−1; HRMS (ESI) calculated for C18H28O2 [M+Na]+ 299.1987 found 299.1994.
Diol 43:
CeCl3 (45 mg, 0.18 mmol) was added to a 1-dram vial and was dried under vacuum at 160 °C for 5 hours. The solid was cooled under argon and THF (0.6 mL) was added, and the resulting mixture was stirred for 30 minutes before being cooled in an ice bath. The mixture was treated with MeMgCl (0.07 mL of a 2.7 M solution in THF, 0.19 mmol). After 30 minutes at 0 °C, a solution of 40 (10 mg, 0.036 mmol) in THF (0.3 mL) was added dropwise at 0°C. The ice bath was removed after 10 minutes. After 5 hours, the reaction was cooled to 0°C and quenched with 3 M HCl (1 mL), then extracted Et2O (3 x). The organic extracts were combined, washed with sat. aq. NaHCO3, dried over MgSO4, filtered and all of the volatiles were removed in vacuo. The crude material was purified by column chromatography (SiO2, 3:1→1:1 hexanes/EtOAc) to provide 43 (6.5 mg, 62%, single diastereomer) as a white solid. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 6.03-5.95 (m, 1H), 5.09-5.02 (m, 2H), 3.50-3.46 (m, 1H), 2.71-2.66 (m, 1H), 2.34-2.32 (m, 1H), 2.23-2.18 (m, 1H), 2.06-1.95 (m, 2H), 1.65 (dt, J = 13.2, 3.6 Hz, 1H), 1.55 (dt, J = 12.9, 3.3 Hz, 1H), 1.50-1.44 (m, 1H), 1.41-1.30 (m, 4H), 1.27 (t, J = 7.1 Hz, 1H), 1.22 (s, 3H), 1.21-1.02 (m, 6H), 0.90 (t, J = 5.7 Hz, 3H), 0.95-0.83 (m, 2H), 0.70 (t, J = 9.4 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 140.5, 115.1, 73.6, 71.6, 49.1, 48.0, 47.4, 44.2, 42.6, 42.1, 39.4, 37.4, 35.1, 34.8, 30.8, 29.3, 28.4, 20.4; IR (thin film) 3389, 3322, 2922, 1638, 1403, 1045 cm−1; HRMS (ESI) calculated for C19H32O2 [M+NH4]+ 310.2746 found 310.2743.
Dione 47:
To a 1-dram vial of 40 (14 mg, 0.051 mmol, 3:1 dr) and celite (60 mg) in CH2Cl2 (0.8 mL) was added PCC (30 mg, 0.14 mmol) at room temperature. After 16 hours, the mixture was diluted with Et2O, passed through a silica gel plug and concentrated. The crude material was purified by column chromatography (SiO2, 4:1 hexanes/EtOAc) to provide 47 (12 mg, 85%, 3:1 dr) as a white solid. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 5.70-5.57 (m, 1H), 5.07-4.98 (m, 2H), 2.56-2.21 (m, 10H), 2.05-1.89 (m, 1H), 1.81-1.72 (m, 2H), 1.68-1.58 (m, 1H), 1.41-1.32 (m, 1H), 1.30-1.17 (m, 3H), 1.10 (q, J = 10.6 Hz, 1H), 1.03-0.82 (m, 4H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) major δ 214.7, 210.7, 134.6, 117.6, 52.7, 51.6, 48.7, 47.6, 45.5, 42.3, 41.0, 39.8, 39.7, 37.2, 36.3, 29.9, 28.6, 20.0, minor δ 213.7, 211.0, 134.3, 116.9, 51.2, 51.1, 46.9, 44.0, 43.1, 42.1, 41.0, 37.9, 36.1, 34.3, 33.9, 30.2, 30.1, 19.9; IR (thin film) 2960, 2916, 2859, 1713 cm−1; HRMS (ESI) calculated for C18H26O2 [M+Na]+ 297.1830 found 297.1836.
TBS ether 49:
To 100 mL round bottom flask containing 39 (1.20 g, 5.12 mmol), imidazole (0.525 g, 7.71) and DMAP (60 mg, 0.49 mmol) in CH2Cl2 (25 mL) was added TBSCl (0.950 g, 6.30 mmol) at room temperature. After 15 hours, half sat. aq. NaHCO3 (100 mL) was added and the mixture was extracted with EtOAc (100 mL and 50 mL). The organic extracts were combined, washed with brine (20 mL), dried over MgSO4, filtered, and all of the volatiles were removed in vacuo. The crude material was purified by column chromatography (SiO2, 6:1 hexanes/EtOAc) to provide 49 (1.67 g, 93%) as a white solid which was recrystallized from pentane to afford colorless prisms (mp = 110–112 °C). 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 5.84 (s, 1H), 3.61 (tt, J = 10.8, 4.3 Hz, 1H), 2.42-2.26 (m, 3H), 2.16-2.10 (m, 1H), 2.08-2.00 (m, 2H), 1.94-1.82 (m, 3H), 1.68-1.62 (m, 1H), 1.42-1.34 (m, 1H), 1.32-1.21 (m, 2H), 1.14 (q, J = 12.5 Hz, 1H), 1.01 (qd, J = 12.5, 3.2 Hz, 1H), 0.95 (d, J = 6.5 Hz, 3H), 0.89 (s, 9H), 0.78 (qd, J = 10.9, 3.0 Hz, 1H), 0.06 (d, J = 1.8 Hz, 6H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 200.4, 168.9, 121.1, 71.3, 48.9, 45.3, 43.5, 38.1, 37.7, 36.9, 35.6, 35.4, 29.5, 28.5, 25.9, 19.2, 18.2, −4.6; IR (thin film) 2929, 2857, 1677, 1092 cm−1; HRMS (ESI) calculated for C21H36O2Si [M+Na]+ 371.2382 found 371.2388.
Enone 50:
LDA was prepared in a 50 mL round bottom flask by addition of n-BuLi (1.90 mL of a 2.7 M solution in hexanes, 5.13 mmol) to diisopropylamine (0.81 mL, 5.78 mmol) in THF (17 mL) at 0 °C. To the stirring LDA solution at −78 °C was added 49 (1.55 g, 4.45 mmol) dissolved in THF (9 mL). After 10 minutes, HMPA (1.0 mL, 5.75 mmol) was added neat followed by iodomethane (0.83 mL, 13.3 mmol). The cold bath was removed after an additional 10 minutes and the reaction stirred for 50 minutes before half sat. aq. NH4Cl (20 mL) was added. The solution was extracted with EtOAc (50 mL and 25 mL). The organic extracts were combined, washed with brine (20 mL), dried over MgSO4, filtered, and all of the volatiles were removed in vacuo. The crude mixture was purified by column chromatography (SiO2, 10:1 hexanes/EtOAc) to afford 50 (1.51 g, 93%, 13:1 dr) as a white wax. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 5.75 (s, 1H), 3.63 (tt, J = 10.8, 4.4 Hz, 1H), 2.47-2.40 (m, 1H), 2.35 (dq, J = 11.6, 5.5 Hz, 1H), 2.05 (dq, J = 13.3, 3.6 Hz, 1H), 2.00-1.83 (m, 5H), 1.77 (dt, J = 13.7, 4.7 Hz, 1H), 1.44-1.23 (m, 4H), 1.11 (d, J = 6.9 Hz, 3H), 1.06-0.98 (m, 1H), 0.95 (d, J = 6.3 Hz, 3H), 0.89 (s, 9H), 0.74 (qd, J = 10.6, 3.1 Hz, 1H), 0.06 (d, J = 1.8 Hz, 6H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 202.9, 168.1, 119.5, 71.2, 49.9, 45.7, 42.8, 37.8, 37.6, 37.4, 37.2, 36.0, 35.5, 29.6, 25.9, 19.2, 18.2, 15.4, −4.6; IR (thin film) 2928, 2856, 1675, 1627, 1249, 1092, 863, 835, 775 cm−1; HRMS (ESI) calculated for C22H38O2Si [M+H]+ 363.2719 found 363.2715.
Allylated Ketone 51:
To a 25 mL round bottom flask containing a glass stir bar and lithium metal (4.0 mg, 0.58 mmol) was condensed ammonia (7 mL) and THF added (4 mL). At −78 °C, 50 (85 mg, 0.23 mmol) and t-BuOH (17 mg, 0.23 mmol) dissolved in THF (3 mL) was added. After stirring for 10 minutes, isoprene (0.1 mL) was added and the mixture was stirred until all lithium was discharged. To the white slurry was added allyl iodide (0.045 mL, 0.50 mmol) at −78 °C. The reaction was stirred for 2.5 hours before sat. aq. NH4Cl (3 mL) was added slowly and the mixture was warmed to room temperature. The mixture was partitioned between water (5 mL) and EtOAc (15 mL). The organic phase was washed with brine (5 mL), dried over MgSO4, filtered, and all of the volatiles were removed in vacuo. The crude material was purified by column chromatography (SiO2, 30:1 hexanes/EtOAc) to afford 51 (54 mg, 56%, >10:1 dr) as a colorless oil. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 5.68 (ddt, J = 16.9, 10.0, 7.0 Hz, 1H), 5.01 (d, J = 7.0 Hz, 1H), 4.99 (s, 1H), 3.49 (tt, J = 10.1, 4.8 Hz, 1H), 2.64 (dqd, J = 10.3, 6.9, 3.4 Hz, 1H), 2.44 (dt, J = 13.3, 6.3 Hz, 1H), 2.31-2.25 (m, 1H), 2.19 (dt, J = 10.1, 4.9 Hz, 1H), 2.02-1.88 (m, 4H), 1.64-1.57 (m, 2H), 1.39 (dt, J = 13.5, 3.3 Hz, 1H), 1.28-1.21 (m, 1H), 1.16-1.06 (m, 3H), 1.04 (d, J = 6.9 Hz, 3H), 0.89 (s, 9H), 0.97-0.77 (m, 6H), 0.56 (qd, J = 10.5, 2.5 Hz, 1H), 0.06 (s, 6H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 217.9, 135.1, 116.7, 71.6, 54.4, 51.9, 47.7, 47.1, 42.7, 40.1, 40.0, 38.5, 37.7, 37.5, 36.5, 35.8, 28.4, 25.9, 19.9, 18.3, 16.3, −4.48, −4.55; IR (thin film) 2927, 2857, 1709, 1461, 1249, 1089, 835, 775 cm−1; HRMS (ESI) calculated for C25H44O2 [M+Na]+ 427.3008 found 427.3014.
Nitroalkene 52:
A 1-dram vial containing 51 (30 mg, 0.0741 mmol) and 4Å MS (32 mg) in DCE (0.5 mL) was stirred for 15 minutes before AgNO2 (116 mg, 0.754 mmol) and TEMPO (20 mg, 0.128 mmol) was added. The flask was sealed and heated at 70 °C in an aluminum heating block for 30 hours. The flask was cooled, filtered over Celite with EtOAc, then concentrated in vacuo. The crude material was purified by column chromatography (SiO2, 10:1 hexanes/EtOAc) to afford 52 (23 mg, 69%, >20:1 E/Z) as a light red oil. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 7.15 (dt, J = 13.5, 7.0 Hz, 1H), 6.98 (d, J = 13.5 Hz, 1H), 3.51 (d, J = 0.4 Hz, 1H), 2.65-2.60 (m, 2H), 2.48 (dt, J = 15.9, 8.4 Hz, 1H), 2.30-2.28 (m, 1H), 1.66 (t, J = 13.9 Hz, 2H), 1.52-1.43 (m, 3H), 1.09 (d, J = 6.5 Hz, 3H), 1.31-0.78 (m, 12H), 0.91 (s, 9H), 0.62-0.58 (m, 1H), 0.07 (s, 6H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 215.8, 140.1, 138.3, 71.0, 52.3, 51.5, 47.2, 46.9, 42.3, 39.9, 38.0, 37.7, 36.9, 36.2, 35.4, 33.1, 28.0, 25.6, 19.6, 17.9, 16.0, −4.7, −4.9; IR (thin film) 2928, 2857, 1706, 1527, 1461, 1349, 1089, 860, 835, 775 cm−1; HRMS (ESI) calculated for C25H43NO4Si [M+Na]+ 472.2859 found 472.2862.
Enoxysilane 57:
3-Methoxyphenylmagnesium bromide was prepared by addition of 3-bromoanisole (1.9 mL, 15.0 mmol) in THF (1.9 mL) to magnesium metal (440 mg, 18.1 mmol) in THF (7.5 mL) activated by dibromoethane, at such a rate as to maintain a temperature of 40–50 °C. A 50 mL round bottom flask was charged with THF (17 mL, rigorously degassed by purging with argon under sonication) and CuI•2LiCl (0.80 mL of a 0.5 M solution in THF, 0.40 mmol). At 0 °C, 36 (602 mg, 4.01 mmol) in THF (6 mL) and TMSCl (0.55 mL, 4.3 mmol) was added, followed by the dropwise addition of 3-methoxyphenylmagnesium bromide (3.4 mL of a 1.41 M solution, 4.8 mmol) over the course of 5 minutes. Stirring at 0 °C was continued for 1.5 hours before NEt3 (2 mL) was added and the reaction was poured into a rapidly stiring solution of sat. aq. NH4Cl (25 mL), ice (25 g), Et2O (25 mL), and NEt3 (2 mL) cooled in an ice bath. After all salts had dissolved, additional Et2O (25 mL) was added, the layers were separated and the organic phase was washed with half sat. aq. NaHCO3 (20 mL), brine (15 mL), dried over MgSO4, filtered, and all of the volatiles were removed in vacuo. The crude oil was flushed through a pad of Florisil®, pretreated with hexanes and 1% NEt3, with hexanes 1% NEt3 to afford 57 (830 mg, 62%, >10:1 conjugate addition dr) as a colorless oil. 1H NMR (500 MHz, C6D6 at 7.15 ppm) δ 7.16-7.12 (m, 1H), 7.01 (dt, J = 6.7, 1.9 Hz, 1H), 6.93 (d, J = 7.6 Hz, 0.5H), 6.89 (d, J = 7.6 Hz, 0.5H), 6.71-6.68 (m, 1H), 5.70-5.62 (m, 1H), 5.03-4.88 (m, 3H), 3.41 (dq, J = 8.6, 2.6 Hz, 1H), 3.37 (s, 1.5H), 3.36 (s, 1.5 H), 2.35-2.28 (m, 1H), 2.19-2.13 (m, 2H), 1.69-1.58 (m, 2H), 1.51 (ddt, J = 11.4, 8.3, 3.1 Hz, 1H), 1.41-1.32 (m, 1H), 0.88 (d, J = 6.9 Hz, 1.5H), 0.86 (d, J = 6.9 Hz, 1.5H), 0.17 (s, 4.5H), 0.16 (s, 4.5H); 13C{1H} NMR (126 MHz, C6D6 at 128 ppm) mixture δ 160.4, 151.6, 151.3, 148.50, 148.46, 141.4, 141.1, 129.6, 129.5, 121.2, 121.1, 115.0, 114.8, 114.6, 113.5, 111.8, 111.7, 108.2, 107.5, 54.7, 47.2, 46.1, 45.4, 44.7, 38.1, 37.3, 30.2, 29.6, 22.5, 21.8, 19.5, 14.2, 0.4; IR (thin film) 2958, 2873, 2835, 1665, 1251, 1184, 845 cm−1; HRMS (ESI) calculated for desilylation only, C17H22O2 [M+Na]+ 281.1518 found 281.1523.
Ketoester 58:
To 25 mL round bottom flask containing enoxysilane 57 (819 mg, 2.48 mmol) in THF (10 mL) was added MeLi (1.9 mL of a 1.40 M solution in Et2O, 2.6 mmol) at −78 °C. After 30 minutes the flask was placed into an ice bath and stirring was continued for 30 minutes. The reaction was recooled to −78 °C and treated with HMPA (0.90 mL, 5.2 mmol) and stirred until homogeneous. After the addition of ethyl bromoacetate (0.70 mL, 6.3 mmol), the reaction was kept at −78 °C for 30 minutes. The cold bath was removed. After 1 hour the reaction was poured into half sat. aq. NH4Cl (30 mL) and extracted with EtOAc (30 mL and 10 mL). The organic extracts were combined, washed with brine (10 mL), dried over MgSO4, filtered, and all of the volatiles were removed in vacuo. The crude oil was purified by column chromatography (SiO2, 5:1 hexanes/EtOAc) to afford 58 (768 mg, 90%, >10:1 alkylation dr) as a white wax. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 7.28-7.24 (m, 1H), 6.80-6.72 (m, 3H), 5.78 (ddd, J = 17.2, 10.6, 5.8 Hz, 0.5H), 5.65 (ddd, J = 17.2, 10.3, 8.0 Hz, 0.5H), 5.01-4.96 (m, 1H), 4.89 (dt, J = 17.3, 1.6 Hz, 0.5H), 4.84-4.80 (m, 0.5H), 4.06-3.94 (m, 2H), 3.81 (s, 3H), 3.09 (dddd, J = 16.7, 12.5, 8.9, 3.8 Hz, 1H), 2.63 (t, J = 11.7 Hz, 0.5H), 2.60-2.50 (m, 2.5H), 2.43 (ddd, J = 16.5, 9.4, 6.9 Hz, 1H), 2.22-2.00 (m, 3H), 1.92 (ddd, J = 20.2, 16.7, 3.5 Hz, 1H), 1.54 (ddd, J = 12.6, 7.8, 5.0 Hz, 1H), 1.20-1.16 (m, 3H), 0.98 (d, J = 7.0 Hz, 1.5H), 0.81 (d, J = 6.9 Hz, 1.5H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) mixture δ 210.02, 209.97, 172.7, 172.7, 159.97, 159.85, 143.3, 143.0, 142.8, 138.5, 130.0, 129.8, 115.9, 113.5, 111.90, 111.88, 60.31, 60.29, 55.19, 55.18, 53.6, 53.0, 52.3, 52.5, 47.4, 46.7, 41.2, 41.0, 37.4, 36.4, 32.45, 32.44, 26.0, 25.7, 19.0, 14.1, 11.0; IR (thin film) 3076, 2962, 2938, 2836, 1731, 1715, 1599, 1584, 1262, 1195, 1156, 1041 cm−1; HRMS (ESI) calculated for C21H28O4 [M+Na]+ 367.1885 found 367.1866.
Dihydronaphthalene 59:
A 100 mL round bottom flask containing 58 (30 mg, 0.087 mmol) dissolved in CH2Cl2 (2 mL) was cooled to −78 °C and treated with a stream of ozone. After the solution became blue, the solution was sparged with oxygen until the color dissipated. A single portion of AcOH (0.2 mL), Zn powder (30 mg, 0.46 mmol) and AgNO3 (1 mg, 0.0059 mmol) was added. After stirring at −78 °C for 2 hours, the bath was removed and stirring was continued for 2 hours. The reduction was determined complete by TLC and the solution was filtered over Celite, washing with CH2Cl2. The filtrate was washed with water, sat. aq. NaHCO3, brine, dried over MgSO4, filtered, and all of the volatiles were removed in vacuo. A round bottom flask containing this crude aldehyde dissolved in toluene was treated with a catalytic amount of TsOH•H2O and heated to 140 °C in an oil bath under a Dean–Stark trap. After 2 hours the reaction was cooled, diluted with Et2O, washed with half sat. aq. NaHCO3, and brine. The aqueous phases were combined and back extracted with Et2O. All organic extracts were combined, dried over MgSO4, filtered, and all of the volatiles were removed in vacuo to afford crude product 59 as a 1.6:1 mixture of diastereomers, which was not purified to homogeneity because it was deemed of no value to the synthesis effort.
Diol 60:
A 100 mL 3-neck round bottom flask fitted with a thermometer was charged with Et2O (15 mL), cooled in an ice bath and LiAlH4 (260 mg, 6.85 mmol) was added. After 10 minutes, 58 (768 mg, 2.23 mmol) in Et2O (7 mL) was added over the course of 30 minutes, ensuring the internal temperature remained below 4 °C. The ice bath was removed. After 3 hours, EtOAc (0.25 mL) was added at 0 °C followed by water (0.25 mL), 5 M NaOH (0.25 mL), water (0.75 mL), and Na2SO4 (0.50 g). After 30 minutes of vigorous stirring, the contents were filtered over Celite® and the filter cake was washed with Et2O (150 mL). The filtrate was concentrated and the residue purified by column chromatography (SiO2, 1:2 hexanes/EtOAc) to afford 60 (624 mg, 91%, 4:1 dr) as a viscous colorless oil. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) mixture of diastereomers and slow rotation δ 7.22 (bs, 1H), 6.76-6.69 (m, 3H), 5.83-5.67 (m, 1H), 4.99 (dd, J = 10.3, 1.4 Hz, 0.5), 4.91-4.89 (m, 0.5), 4.85-4.79 (m, 1H), 3.81 (s, 3H), 3.59-3.54 (m, 1H), 3.45-3.37 (m, 1H), 3.31-3.23 (m, 1H), 2.27-2.20 (m, 1H), 2.12 (ddq, J = 12.6, 8.5, 4.1 Hz, 1H), 1.93-1.82 (m, 2H), 1.71-1.40 (m, 5H), 1.19 (quintet, J = 12.7, 2.7 Hz, 1H), 0.89 (d, J = 6.8 Hz, 0.2H), 0.88 (d, J = 7.0 Hz, 1.3H), 0.86 (d, J = 6.9 Hz, 0.2H), 0.80 (d, J = 6.9 Hz, 1.3H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) mixture or diastereomers and slow rotation δ 145.0, 144.8, 144.1, 139.6, 115.1, 112.7, 74.8, 74.6, 62.2, 62.1, 55.1, 55.1, 49.91, 49.87, 48.1, 47.4, 37.6, 36.6, 35.05, 35.00, 34.7, 34.5, 23.3, 23.2, 19.0, 11.1; IR (thin film) 3304, 3077, 2997, 2932, 2882, 1599, 1584, 1487, 1260, 1046 cm−1; HRMS (ESI) calculated for C19H28O3 [M+Na]+ 327.1936 found 327.1933.
TBS ether 62:
To a 1-dram vial containing 60 (15 mg, 0.050 mmol) in CH2Cl2 (1 mL) was added DMAP (7 mg, 0.057 mmol) and NEt3 (0.05 mL, 0.36 mmol). After the addition of TBSCl (19 mg, 0.13 mmol), the reaction stirred for 3 days. The reaction was quenched with sat. aq. NaHCO3 and extracted thrice with EtOAc. The organic extracts were combined and washed with brine, dried over MgSO4, filtered, and all of the volatiles were removed in vacuo. The crude oil was purified by column chromatography (10:1 hexanes/EtOAc) to afford 62 (18 mg, 86%) as a colorless oil. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 7.21 (bs, 1H), 6.81-6.61 (m, 3H), 5.75-5.68 (m, 1H), 4.98 (dd, J = 10.3, 2.0 Hz, 0.5H), 4.89 (dt, J = 10.5, 1.6 Hz, 0.5H), 4.84-4.78 (m, 1H), 4.66 (bs, 1H), 3.80 (s, 3H), 3.60-3.54 (m, 1H), 3.34 (dtd, J = 14.7, 10.1, 4.4 Hz, 1H), 3.28-3.20 (m, 1H), 2.25-2.12 (m, 2H), 1.93-1.80 (m, 1H), 1.74-1.50 (m, 4H), 1.45-1.34 (m, 2H), 1.29-1.12 (m, 1H), 0.88 (d, J = 1.6 Hz, 10.5H), 0.80 (d, J = 6.9 Hz, 1.5H), 0.043 (s, 1.5H), 0.038 (s, 1.5H), 0.035 (s, 1.5H), 0.031 (s, 1.5H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 145.4, 145.1, 144.2, 139.9, 115.0, 112.6, 74.3, 74.2, 63.1, 55.12, 55.11, 50.60, 50.54, 48.3, 47.6, 37.8, 36.7, 36.6, 35.0, 34.9, 34.4, 34.3, 25.9, 23.4, 23.2, 19.0, 18.2, 11.2, −5.5, −5.6; IR (thin film) 3416, 3077, 2953, 2929, 2858, 2882, 1599, 1584, 1257, 1080, 1048, 836, 777 cm−1; HRMS (ESI) calculated for C25H42O3Si [M+Na]+ 441.2801 found 441.2784.
Ether 63:
To a 25 mL round bottom flask containing 60 (510 mg, 1.68 mmol) in pyridine (8 mL) was added p-toluenesulfonyl chloride (820 mg, 4.30 mmol) at room temperature. The flask was placed in a 40 °C oil bath. After 15 hours, the reaction was cooled to room temperature and quenched with sat. aq. NaHCO3 (10 mL), water (5 mL), and the resulting mixture was extracted with EtOAc (30 mL). The organic extract was washed with 6 M HCl (3 x 10 mL) followed by water (5 mL). The acidic aqueous washings were combined, back extracted with EtOAc (10 mL) and all of the organic phases were combined. The organic solution was washed with sat. aq. NaHCO3, (10 mL), brine, dried over MgSO4, filtered and all volatiles removed in vacuo. The crude oil was purified by column chromatography (SiO2, 10:1 hexanes/EtOAc) to afford 63 (358 mg, 74%, 11:1 dr) as a colorless oil. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) major δ 7.24 (t, J = 7.8 Hz, 0.5H), 7.23 (t, J = 7.9 Hz, 0.5H), 6.79-6.72 (m, 3H), 5.80-5.70 (m, 1H), 4.99 (d, J = 10.5 Hz, ), 4.92 (d, J = 10.5 Hz, 1H), 4.84 (dd, J = 17.2, 11.2 Hz, 1H), 3.88 (qd, J = 9.1, 2.6 Hz, 1H), 3.84-3.79 (m, 1H), 3.82 (s, 3H), 3.16 (ddq, J = 16.4, 10.6, 5.5 Hz, 1H), 2.31 (dt, J = 15.5, 10.9 Hz, 1H), 2.21 (ddq, J = 12.0, 8.2, 3.9 Hz, 1H), 2.12-2.04 (m, 1H), 1.97 (dq, J = 13.7, 3.5 Hz, 1H), 1.83-1.40 (m, 7H), 1.32-1.20 (m, 1H), 0.95 (d, J = 7.0 Hz, 1.4H), 0.91 (d, J = 7.0 Hz, 0.2H), 0.84 (d, J = 7.0 Hz, 1.4H), 0.79 (d, J = 6.9 Hz, 0.2H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) major δ 144.8, 144.6, 144.1, 139.8, 129.5, 129.4, 115.0, 112.7, 110.9, 82.8, 82.6, 67.1, 67.1, 55.1, 52.1, 52.0, 50.3, 49.9, 48.0, 47.4, 37.3, 36.2, 30.6, 30.5, 30.0, 30.0, 23.51, 23.45, 19.1, 11.6; IR (thin film) 3073, 2934, 2871, 1600, 1583, 1261, 1047, 911, 778, 701 cm−1; HRMS could not be obtained.
Diol 69 via lactone 68:
To a 1-dram vial containing 65 (30 mg, 0.111 mmol) in AcOH (0.3 mL) was added CrO3 (51 mg, 0.51 mmol) in 9:1 AcOH/water (0.8 mL) open to air. After 23 hours, i-PrOH (0.10 mL) was added and stirring was continued for 20 minutes. The solution was poured into a stirred mixture of ice cold water (7 mL) and Et2O (5 mL), and the solution was treated with 5 M NaOH (3 mL) at 0 °C and extracted with Et2O (5 mL). The organic extracts were combined, washed with water (2 x 2 mL) and sat. aq. NaHCO3 (2 mL). The aqueous washings were combined and back extracted with Et2O (3 mL). All organic extracts were combined, washed with brine (2 mL), dried over MgSO4, filtered, and all of the volatiles were removed in vacuo to afford crude lactone 68 which was used without further purification. Crude C─H oxidation product 68 was dissolved in benzene (2.5 mL) in a 10 mL round bottom flask. Ethylene glycol (0.03 mL, 0.54 mmol) and TsOH•H2O (2 mg, 0.011 mmol) were added. The solution was heated to reflux in an oil bath under a Hickman Still. After 2 hours the reaction was cooled, diluted with EtOAc (5 mL) and washed with sat. aq. NaHCO3 (2 mL) and brine (2 mL). The organic phase was dried over MgSO4, filtered, and all of the volatiles were removed in vacuo. The crude lactone dissolved in THF (1.0 mL) was added to LiAlH4 (16 mg, 0.42 mmol) in THF (1.0 mL) at 0 °C. The flask was immediately removed from the cold bath and stirred for 10 minutes before being heated to 40 °C in an oil bath for 1 hour. After cooling to 0 °C, EtOAc (0.05 mL), water (0.02 mL), 5 M NaOH (0.02 mL), water (0.06 mL), and Na2SO4 were carefully added sequentially. The solution was filtered over Celite and washed with Et2O (~40 mL until no more product eluted as determined by TLC analysis) and all of the volatiles were removed in vacuo. The crude material was purified by column chromatography (SiO2, EtOAc) to afford 69 (15 mg, 42% over 3 steps, >10:1 dr) as a white semi-solid. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 3.94 (s, 4H), 3.81-3.62 (m, 2H), 3.47-3.31 (m, 2H), 2.05-1.46 (m, 10H), 1.35-1.10 (m, 7H), 0.92-0.81 (m, 7H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 109.4, 76.2, 64.3, 64.1, 62.3, 50.1, 47.9, 47.7, 45.6, 43.3, 41.6, 40.7, 39.4, 37.6, 34.2, 32.1, 31.4, 26.0, 20.2.
Benzyl Alcohol 70:
To a 1-dram vial containing 69 (11 mg,0.034 mmol) was added imidazole (5.5 mg, 0.081 mmol), DMAP (2.5 mg, 0.020 mmol) and CH2Cl2 (0.5 mL). The mixture was cooled to 0 °C and TBSCl (7.0 mg,0.046 mmol) was added and the cooling bath was removed. After 10 hours, sat. aq. NaHCO3 was added and the mixture was extracted with EtOAc (3 x). The organic extracts were combined, washed with sat. aq. brine, dried over MgSO4, filtered, and all of the volatiles were removed in vacuo. The crude material and TBAI (2 mg, 0.0054 mmol) in a 1-dram vial were dissolved in THF (0.4 mL) and cooled to 0 °C. The mixture was treated with KHMDS (0.2 mL of a 0.5 M solution in toluene, 0.10 mmol) and benzyl chloride (0.02 mL, 0.17 mmol). The cold bath was immediately removed. After 1 hour the reaction was recooled to 0 °C and quenched with half sat. aq. NaHCO3 and extracted with EtOAc (3 x). The organic extracts were combined, washed with sat. aq. brine, dried over MgSO4, filtered, and all of the volatiles were removed. The crude material was purified by column chromatography (SiO2, 20:1 hexanes/EtOAc) to afford 70 (8.5 mg, 47%) as a colorless oil. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 7.34-7.26 (m, 5H), 4.53-4.41 (m, 2H), 3.93-3.87 (m, 2H), 3.81-3.76 (m, 1H), 3.75-3.64 (m, 2H), 3.55-3.48 (m, 2H), 2.11 (d, J = 12.0 Hz, 1H), 2.04-2.02 (m, 1H), 1.92-1.86 (m, 1H), 1.74-1.35 (m, 10H), 1.32-0.95 (m, 5H), 0.95-0.77 (m, 13H), 0.62-0.50 (m, 1H), 0.04 (s, 3H), 0.03 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 138.6, 128.3, 127.81, 127.77, 127.44, 124.41, 109.5, 73.2, 71.8, 69.3, 64.0, 63.9, 48.6, 47.3, 42.9, 42.0, 41.8, 41.7, 38.6, 38.4, 35.0, 34.8, 31.3, 26.4, 25.9, 24.2, 20.0, 17.9, −4.6, −4.7.
Trityl ether 71:
A 5 mL round bottom flask was charged with 69 (15 mg, 0.046 mmol), DMAP (6.8 mg, 0.056 mmol), NEt3 (0.03 mL, 0.22 mmol) and triphenylmethyl chloride (18 mg, 0.064 mmol) in DCE (1 mL). After stirring the reaction at 85 °C for 3 hours the solution was cooled, diluted with sat. aq. NaHCO3 and extracted with EtOAc (3 x). The organic extracts were combined, washed with sat. aq. brine, dried over MgSO4, filtered, and all of the volatiles were removed in vacuo. The crude material was purified by column chromatography (SiO2, 5:1 hexanes/EtOAc) to afford 71 (15 mg, 58%) as a colorless oil. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 7.44 (d, J = 7.5 Hz, 6H), 7.31 (t, J = 7.6 Hz, 6H), 7.23 (t, J = 7.3 Hz, 3H), 3.85-3.79 (m, 2H), 3.72-3.67 (m, 1H), 3.63-3.58 (m, 1H), 3.47-3.43 (m, 1H), 3.30-3.24 (m, 1H), 3.13 (dt, J = 9.1, 6.1 Hz, 1H), 1.92-1.80 (m, 4H), 1.69-1.46 (m, 7H), 1.39-0.98 (m, 9H), 0.87 (d, J = 6.4 Hz, 3H), 0.91-0.78 (m, 3H), 0.70 (t, J = 8.9 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 171.1, 143.9, 128.7, 128.6, 127.8, 127.0, 109.4, 87.4, 77.3, 77.0 76.8, 74.0, 64.2, 64.0, 63.3, 60.4, 49.5, 47.5, 45.7, 44.4, 43.0, 41.6, 40.0, 37.9, 36.0, 34.5, 31.4, 29.3, 25.1, 21.1, 20.1, 14.2.
Benzyl ether 72:
A 1-dram vial was charged with oil free KH (5 mg , ~0.12 mmol) and THF (0.2 mL) was added. At 0 °C, a solution of TBAI (1 mg, 0.0026 mmol and alcohol 71 (15 mg, 0.026 mmol) was added via THF (total of 0.5 mL). After the addition of benzyl chloride (1 drop), the reaction was heated to 50 °C in an oil bath for 2 hours. The reaction was cooled, then treated with water and extracted with EtOAc (3 x). The organic extracts were combined, washed with sat. aq. brine, dried over MgSO4, filtered, and all of the volatiles were removed in vacuo. The crude material was purified by column chromatography (SiO2, 15:1 hexanes/EtOAc) to afford 72 (11 mg, 64%) as a colorless oil. 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 7.47 (d, J = 7.4 Hz, 5H), 7.34-7.22 (m, 15H), 4.44 (d, J = 12.3 Hz, 1H), 4.40 (d, J = 12.3 Hz, 1H), 3.87-3.81 (m, 2H), 3.66-3.64 (m, 1H), 3.29 (bs, 1H), 3.10 (t, J = 6.0 Hz, 2H), 2.19 (d, J = 10.8 Hz, 1H), 1.98-1.84 (m, 3H), 1.78-1.55 (m, 7H), 1.46-1.37 (m, 2H), 1.36-1.23 (m, 6H), 1.13-1.03 (m, 2H), 0.99-0.86 (m, 3H), 0.91 (d, J = 6.3 Hz, 3H) 0.70-0.65 (m, 1H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 144.4, 139.4, 128.7, 128.2, 127.71, 127.68, 127.65, 127.2, 127.0, 126.8, 109.5, 78.0, 69.6, 64.3, 64.0, 62.2, 60.4, 48.4, 47.3, 42.7, 42.0, 41.5, 39.5, 38.7, 38.1, 35.1, 34.8, 31.2, 24.1, 22.7, 21.1, 19.9, 14.2.
Ketoester 86:
A flame-dried round bottom flask was equipped with a stir bar and magnesium powder was added. The flask was evacuated, filled with argon, evacuated, and flame dried with stirring. Upon cooling to room temperature, the flask was filled with argon, and THF (14 mL) was added. The suspension was treated with dibromoethane (1 drop) under sonication and heating. With vigorous stirring, a solution of 3-bromoanisole (3.5 mL, 27.64 mmol) in THF (3.5 mL) was added slowly over 20 minutes to the magnesium suspension, maintaining an internal temperature of 55–60 °C. Following complete addition, the reaction mixture was stirred for 30 minutes, and transferred via cannula to a flame-dried flask under argon. A 0.25 M solution of CuI•2LiCl was prepared: LiCl (381.6 mg, 9.00 mmol) was added to a 50 mL round bottom flask with stir bar, evacuating, and flame drying while stirring. Upon cooling to room temperature, the flask was removed from vacuum and CuI (856.8 mg, 4.50 mmol) was added as a solid. The mixture was evacuated and filled with argon. To the solid mixture was added anhydrous THF (18 mL), and the suspension was stirred vigorously until dissolved. To afford the desired 0.25 M solution of CuI•2LiCl. To a three-neck flame-dried round bottom flask under argon equipped with an internal thermometer was added of THF (18 mL) followed by 3-methoxyphenylmagnesium bromide (6.8 mL of a 1.20 M solution, 8.128 mmol). The solution was cooled to −78 °C and CuI•2LiCl (2.7 mL of a 0.25 M solution, 0.661 mmol) was added dropwise over 5 minutes. After stirring at −78 °C for 10 minutes, a solution of enone 82 (900 mg, 6.608 mmol) in THF (5.4 mL) was added dropwise over 15 minutes. Following addition, the resulting yellow reaction mixture was stirred for 2 hours at −78 °C. The cooling bath was removed and the reaction mixture was stirred for 1 hour while warming to room temperature. The reaction mixture was then cooled to −78 °C, HMPA (2.34 mL, 13.4 mmol) was added over 2 minutes, and the suspension was stirred vigorously for 1 hour at −78 °C. Ethyl bromoacetate (2.70 mL, 24.4 mmol) was added dropwise over 5 minutes. After stirring for 10 minutes at −78 °C, the bath was removed and the reaction mixture was stirred for 24 hours at room temperature. Upon completion, the reaction mixture was poured into half saturated ammonium chloride (100 mL). The phases were separated, and the aqueous phase was extracted with EtOAc (2 x 100 mL). The combined organic extracts were washed with brine (2 x 50 mL), dried with MgSO4, filtered through cotton, and concentrated in vacuo to a yellow oil. The crude product mixture was purified by chromatography (SiO2, 5–15% EtOAc in hexanes) to afford 86 (1.13g, 52%) as a colorless oil. The functionality on 86 experiences restricted rotation, and two 13C peaks are missing: 1H NMR (600 MHz, CDCl3 at 7.27 ppm) δ 7.20 (t, J = 7.4 Hz, 1H), 6.73 (t, J = 6.6 Hz, 2H), 6.66 (s, 1H), 4.58 (d, J = 14.0 Hz, 2H), 4.06–3.95 (m, 2H), 3.78 (s, 3H), 3.16–3.08 (m, 1H), 2.85 (td, J = 11.7, 2.6 Hz, 1H), 2.71 (t, J = 11.7 Hz, 1H), 2.67 (td, J = 13.6, 5.9 Hz, 1H), 2.56 (d, J = 13.5 Hz, 1H), 2.44 (dd, J = 16.7, 9.1 Hz, 1H), 2.15–2.07 (m, 1H), 1.96 (dd, J = 16.8, 3.2 Hz, 1H), 1.88 (ddd, J = 26.0, 13.5, 3.7 Hz, 1H), 1.49 (s, 3H), 1.18 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 209.6, 172.6, 159.6, 145.7, 142.8, 129.5, 112.8, 111.7, 60.3, 55.2, 53.5, 52.3, 51.3, 41.3, 32.3, 32.2, 19.5, 14.1; IR (film) 2930, 1714, 1153 cm−1; HRMS (ESI) calculated for C20H26O4 [M + Na]+ 353.1729 found 353.1728.
Dihydronaphthalene 88 via lactone 87:
To a flame-dried round bottom flask with a stir bar under argon was added ketone 86 (2.50 g, 7.57 mmol) followed by THF (37.8 mL). The reaction mixture was cooled to 0 °C, and methylmagnesium bromide (3.26 mL of a 2.55 M solution, 8.32 mmol) was added dropwise over 10 minutes. The reaction mixture was stirred for 2 hours at 0 °C, the bath was removed, and the reaction mixture was stirred at room temperature for 1 hour. Upon completion, the reaction mixture was diluted with Et2O (100 mL) and poured into saturated, aqueous NH4Cl (100 mL). The phases were separated, and the aqueous phase was extracted with Et2O (2 x 100 mL). The combined organic extracts were dried over MgSO4, filtered through cotton, and concentrated in vacuo to provide crude lactone 87 as a viscous oil. The crude mixture was used in the next step without further purification.
The crude lactone 87 was dissolved in CH2Cl2 (126 mL) in a round bottom flask. Solid NaHCO3 (8.26 g, 98.4 mmol) was added, followed by batch-wise addition of m-CPBA (3.73 g, 70% w/w, 15.1 mmol) over 10 minutes. The reaction mixture was capped with outlet needle and stirred for 16 hours. Upon completion, sodium thiosulfate (100 mL) was added, and the biphasic mixture was stirred vigorously for 1 hour. The mixture was extracted with EtOAc (3 x 100 mL), and the combined extracts were washed sequentially with saturated aqueous NaHCO3 (100 mL) and brine (100 mL). The organic extracts were dried with MgSO4, filtered through cotton, and concentrated in vacuo to a colorless foam. The crude epoxide was used in the next step without purification. To a round bottom flask with a stir bar was attached a Dean–Stark apparatus with condenser. Benzene (150 mL) and TsOH•H2O (173 mg, 0.91 mmol) were added. The solution was heated to reflux in an oil bath for 30 minutes to remove water, and then cooled to 50 °C. A solution of crude epoxide in benzene (10 mL) was added dropwise over 5 minutes. The reaction mixture was heated to reflux in an oil bath for 1.5 hours, then cooled to room temperature and poured into saturated aqueous NaHCO3 (300 mL). The biphasic mixture was extracted with Et2O (100 mL and 200 mL, sequentially), and the combined organic extracts were washed with brine (100 mL). The organic extracts were dried with MgSO4, filtered through cotton, and concentrated in vacuo to a brown solid. The crude product mixture was dry loaded onto silica gel and purified by silica gel chromatography (SiO2, 10–20% EtOAc in hexanes) to afford 88 (1.15g, 51% over 3 steps) as an off-white solid: 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 6.97 (d, J = 8.2 Hz, 1H), 6.72 (dd, J = 8.2, 2.3 Hz, 1H), 6.56 (s, 1H), 6.27 (s, 1H), 3.81 (s, 3H), 3.25 (dd, J = 17.4, 7.0 Hz, 1H), 2.54 (dd, J = 10.7, 7.0 Hz, 1H), 2.40–2.28 (m, 2H), 2.06–1.92 (m, 2H), 1.88 (s, 3H), 1.55–1.50 (m, 2H), 1.47 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 176.2, 158.2, 137.9, 137.1, 128.9, 126.2, 123.6, 111.5, 109.8, 84.8, 55.4, 44.3, 42.4, 39.6, 38.7, 34.8, 27.8, 23.5, 20.4; IR (film) 2926, 1754, 1606, 1494, 1224, 933, 728 cm−1; HRMS (ESI) calculated for C19H22O3 [M + Na]+ 321.1467 found 321.1461.
Enone 92:
A three-neck round bottom flask with glass stir bar, internal thermometer, cold finger condenser, and septum was cooled to −78 °C. Ammonia (34 mL) was condensed into the reaction flask, and lithium (311 mg, 45.1 mmol) was added over 2 minutes. After stirring for 10 minutes at −78 °C, t-BuOH (6.0 mL, 63 mmol) and DME (6.0 mL) were added. The reaction mixture was warmed to reflux, and a solution of 88 (269 mg, 0.90 mmol) in DME (5.0 mL) was added dropwise over 2 minutes. The reaction mixture was warmed to reflux until colorless. t-BuOH (4.3 mL, 45 mmol) was added, followed by lithium (311 mg, 45.1 mmol). The reaction mixture was warmed to reflux until colorless. Upon completion, solid NH4Cl (5.8 g, 108 mmol) was added, and the reaction mixture was warmed to room temperature over 3 hours to allow the ammonia to evaporate. H2O (30 mL) was added and the biphasic mixture was extracted with EtOAc (3 x 20 mL), and the combined organic extracts were washed with brine (2 x 10 mL). The organic extracts were dried with MgSO4, filtered through cotton, and concentrated in vacuo to a colorless oil. The crude acetal was dissolved in methanol (11.8 mL) in a large vial, and 6.0 M aqueous HCl (1.07 mL) was added. The reaction mixture was capped and stirred overnight. The reaction mixture was poured into saturated aqueous NaHCO3 (20 mL), and the mixture was extracted with EtOAc (3 x 20 mL). The combined organic extracts were washed with brine (10 mL), then dried with MgSO4, filtered through cotton, and concentrated in vacuo to a colorless oil. The crude mixture was purified by chromatography (SiO2, 15–20% EtOAc in hexanes) to afford a 3:1 mixture of diastereomers of 92 (100.4 mg, 37% over 2 steps) as a colorless oil: 1H NMR (500 MHz, CDCl3 at 7.27 ppm) δ 5.79–5.75 (m, 1H), 5.01 (major, dd, J = 5.9, 5.1 Hz, 0.75H), 4.98 (minor, d, J = 6.2 Hz, 0.25H), 3.36 (major, s, 2.25H), 3.32 (minor, s, 0.75H), 2.55 (minor, quintet, J = 6.8 Hz, 0.25H), 2.44–2.25 (m, 4H), 2.22– 2.10 (m, 1H), 2.09–2.00 (contains major, m,1.75H), 1.94–1.78 (m, 3H), 1.73–1.60 (m, 2H), 1.47–1.14 (m, 8H), 0.97 (minor, d, J = 6.4 Hz, 0.75H), 0.96 (major, d, J = 6.5 Hz, 2.25H); 13C{1H} NMR (126 MHz, CDCl3 at 77 ppm) δ 199.9, 167.8, 121.7, 121.6, 105.2, 104.6, 83.1, 81.8, 55.5, 55.1, 50.4, 49.1, 48.8, 48.7, 43.6, 43.4, 43.2, 41.3, 40.1, 38.8, 37.3, 36.9, 35.1, 35.0, 34.8, 34.7, 29.4, 28.9, 28.4, 28.1, 26.2, 25.9, 19.23, 19.20; IR (film) 2927, 1672, 1100, 1032 cm−1; HRMS (ESI) calculated for C19H28O3 [M + Na]+ 327.1936 found 327.1931.
Supplementary Material
ACKNOWLEDGMENTS
Over the course of this project, funding was provided by the NSF (CHE-1262040) and the NIH (AI-138139). P.C.R. was funded in part by a UC Irvine Regents’ Dissertation Fellowship, and A.S.K. was supported by an NSF Graduate Fellowship.
Footnotes
Supporting Information. Experimental protocols and characterization data for intermediates not explicitly shown in the body of the paper, as well as NMR spectral data for all new compounds.
REFERENCES
- 1.Schnermann MJ; Shenvi RA Syntheses and biological studies of marine terpenoids derived from inorganic cyanide. Nat. Prod. Rep 2015, 32, 543–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garson MJ; Simpson JS Marine isocyanides and related natural products—structure, biosynthesis and ecology. Nat. Prod. Rep 2004, 21, 164–179. [DOI] [PubMed] [Google Scholar]
- 3.Baker JT; Wells JR; Oberhansli WE; Hawes GB A new diisocyanide of novel ring structure from a sponge. J. Am. Chem. Soc 1976, 98, 4010–4012. [Google Scholar]
- 4.Aside from DICA, the only other perhydropyrene ICT that has succumbed to synthesis is 8-isocyanocycloamphilec-10-ene 4, the unnatural enantiomer of which was made by Schindeler and Piers: Schindeler TW Ph.D. Dissertation, University of British Columbia, 1998. [Google Scholar]
- 5.Corey EJ; Magriotis PA Total synthesis and absolute configuration of 7,20-diisocyanoadociane. J. Am. Chem. Soc 1987, 109, 287–289. [Google Scholar]
- 6.(a) Fairweather KA; Mander LN A Formal Total Synthesis of the Marine Diterpenoid Diisocyanoadociane. Org. Lett 2006, 8, 3395–3398. [DOI] [PubMed] [Google Scholar]; (b) Fairweather KA; Crabtree SR; Mander LN “A Formal Total Synthesis of the Marine Diterpenoid, Diisocyanoadociane” In Strategies and Tactics in Organic Synthesis, Harmata M Ed.; Elsevier Academic Press: Oxford, 2008, Vol. 7, pp 35–58 [Google Scholar]; (c) Mander LN Exploitation of Aryl Synthons in the Synthesis of Polycyclic Natural Products. Synlett 1991, 134–144. [Google Scholar]
- 7.(a) Pronin SV; Shenvi RA Synthesis of a Potent Antimalarial Amphilectene. J. Am. Chem. Soc 2012, 134, 19604–19606 [DOI] [PubMed] [Google Scholar]; (b) Pronin SV; Reiher CA; Shenvi RA Stereoinversion of tertiary alcohols to tertiary-alkyl isonitriles and amines. Nature 2013, 501, 195–199. [DOI] [PubMed] [Google Scholar]
- 8.Lu H-H; Pronin SV; Antonova-Koch Y; Meister S; Winzeler EA; Shenvi RA Synthesis of (+)-7,20-Diisocyanoadociane and Liver-Stage Antiplasmodial Activity of the Isocyanoterpene Class. J. Am. Chem. Soc 2016, 138, 7268–7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyaoka H; Okubo Y; Muroi M; Mitome H; Kawashima E A Formal Synthesis of Antimalarial Diterpenoid 7,20-Diisocyanoadociane. Chem. Lett 2011, 40, 246–247. [Google Scholar]
- 10.(a) Roosen PC; Vanderwal CD A Formal Enantiospecific Synthesis of 7,20-Diisocyanoadociane. Angew. Chem. Int. Ed 2016, 55, 7180–7183 [DOI] [PubMed] [Google Scholar]; (b) Karns AS; Ellis BD; Roosen PC; Chahine Z; Le Roch KG; Vanderwal CD Synthesis of the Antiplasmodial Isocyanoterpene 7,20-Diisocyanoadociane. Angew. Chem. Int. Ed 2019, 58, 13749–13752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson EE; Thomson RJ A Strategy for the Convergent and Stereoselective Assembly of Polycyclic Molecules. J. Am. Chem. Soc 2018, 140, 1956. [DOI] [PubMed] [Google Scholar]
- 12.(a) Daub ME; Prudhomme J; Le Roche KG; Vanderwal CD Synthesis and Potent Antimalarial Activity of Kalihinol A. J. Am. Chem. Soc 2015, 137, 4912–4915 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Daub ME; Prudhomme J; Ben Mamoun C; Le Roche KG; Vanderwal CD Antimalarial Properties of Simplified Kalihinol Analogues. ACS Med. Chem. Lett 2017, 8, 355–360 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Daub ME; Roosen PC; Vanderwal CD General Approaches to Structurally Diverse Isocyanoditerpenes. J. Org. Chem 2017, 82, 4533–4541. [DOI] [PubMed] [Google Scholar]
- 13.Rao CSS; Kumar G; Rajagopalan K; Swaminathan S Base catalysed rearrangement of a spiro oxy-cope system and related studies: A new entry into perhydrophenalene and perhydroacenaphthylene systems. Tetrahedron 1982, 38, 2195–2199. [Google Scholar]
- 14.Roosen PC; Vanderwal CD Investigations into an Anionic Oxy-Cope/Transannular Conjugate Addition Approach to 7,20-Diisocyanoadociane. Org. Lett 2014, 16, 4368–4371. [DOI] [PubMed] [Google Scholar]
- 15.Ramella V; Roosen PC; Vanderwal CD Concise Formal Synthesis of the Pseudopterosins via Anionic Oxy-Cope/Transannular Michael Addition Cascade. Org. Lett 2020, 22, 2883–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gawley RE The Robinson Annelation and Related Reactions. Synthesis 1976, 777–794. [Google Scholar]
- 17.(a) Stork G; Logusch EW Reductive alkylation of enediones. J. Am. Chem. Soc 1980, 102, 1218–1219 [Google Scholar]; (b) Stork G; Logusch EW Reductive alkylation of enediones. 2. Synthesis of corticosteroids. J. Am. Chem. Soc 1980, 102, 1219–1220. [Google Scholar]
- 18.(a) Mander LN Exploitation of Aryl Synthons in the Synthesis of Polycyclic Natural Products. Synlett 1991, 134–144 [Google Scholar]; (b) Parr BT; Economou C; Herzon SB A concise synthesis of (+)-batzelladine B from simple pyrrole-based starting materials. Nature 2015, 525, 507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Hook JM; Mander LN Recent developments in the Birch reduction of aromatic compounds: applications to the synthesis of natural products. Nat. Prod. Rep 1986, 3, 35–85 [Google Scholar]; (b) Rabideau PW; Marcinow Z The Birch Reduction of Aromatic Compounds. Org. React 1992, 42, 1–334. [Google Scholar]
- 20.(a) Caine D Reduction and Related Reactions of α,β-Unsaturated Carbonyl Compounds with Metals in Liquid Ammonia. Org. React 1976, 23, 1 [Google Scholar]; (b) Keinan E; Greenspoon N “Reduction of α,β-unsaturated carbonyl compounds” in The Chemistry of Enones; Patai S; Rappoport Z, Ed.; John Wiley & Sons, Ltd, 1989; pp 923–1022 [Google Scholar]; (c) Huffman JE “Reduction of C=X to CHXH by Dissolving Metals and Related Methods” In Comprehensive Organic Synthesis; Trost BM; Fleming I, Ed.; Elsevier Science Ltd., 1991; pp 107–127. [Google Scholar]
- 21.(a) The literature on stereochemistry of phenanthrenone reduction is mixed with both cis-ring junction outcomes:Bansal RC; Browne CE; Eisenbraun EJ; Thomson JS Synthesis of cis-1,2,3,4,4a,9,10,10a-octahydrophenanthrene. J. Org. Chem 1988, 52, 452–455 [Google Scholar]; (b) Chatterjee A; Chaudhuri SRR; Chatterjee SK Stereospecific cis-reduction of hexahydro-3-oxophenanthrene derivatives. Total synthesis of a new class of ring-C aromatic steroid. J. Chem. Soc., Chem. Commun 1982, 24–25; and trans-ring junction outcomes [Google Scholar]; (c) Gesson J-P; Jacquesy J-C; Jacquesy R Cyclisation of diarylalkanes in the superacidic media SbF5-HF: Synthesis of spiroketones and phenanthrenones. Nouv. J. Chim 1977, 1, 511 [Google Scholar]; (d) Robinson MJT Steroids and related compounds—I: Synthesis of 1:2:3:4:9:10:11“β”: 12“α”-octahydro-7-methoxy-2“α”:8:11“β”-trimethyl-3-oxophenanthrene. Tetrahedron 1957, 1, 49–66. [Google Scholar]
- 22.Nadeau E; Ventura DL; Brekan JA; Davies HM Controlling Factors for C─H Functionalization versus Cyclopropanation of Dihydronaphthalenes. J. Org. Chem 2010, 75, 1927–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dissolving metal reductions of ketones typically provide equatorial alcohols with high selectivity. Similarly, we observe the four diastereomers to be differentiated not at the carbinol but only at the ring fusion.
- 24.White JD; Hrnciar P; Stappenbeck F Asymmetric Total Synthesis of (+)-Codeine via Intramolecular Carbenoid Insertion. J. Org. Chem 1999, 64, 7871–7884. [Google Scholar]
- 25.(a) Benkeser RA; Sgihotri RK; Burbous ML; Kaiser EM; Mallan JM; Ryan PW Highly Selective Lithium—Amine Reducing Systems. The Selective Reduction of Aromatic Compounds by Lithium in Mixed Amine Solvents. J. Org. Chem 1964, 29, 1313–1316 [Google Scholar]; (b) Kaiser EM A Comparison of Methods Using Lithium/Amine and Birch Reduction Systems. Synthesis 1972, 391–415. [Google Scholar]
- 26.Garst ME; Dolby LJ; Esfandiari S; Fedoruk NA; Chamberlain NC; Avey AA Reductions with Lithium in Low Molecular Weight Amines and Ethylenediamine. J. Org. Chem 2000, 65, 7098–7104. [DOI] [PubMed] [Google Scholar]
- 27.Li, t-BuOH, EDA, THF reduced only the alkene of the enone. The carbonyl remained intact. [Google Scholar]
- 28.Kotlarek W Alkali metal: Hexamethylphosphoramide reduction. Part V. Reduction of some mono- and dialkylanisoles by lithium in HMPA. Pol. J. Chem 1983, 57, 809–815. [Google Scholar]
- 29.Benkeser RA; Laugal J; Rappa A A safe and convenient new procedure for reducing aromatic compounds to birch-type products. Tetrahedron Lett. 1984, 25, 2089–2092. [Google Scholar]
- 30.(a) Posner GH Conjugate Addition Reactions of Organocopper Reagents. Org. React 1972, 19, 1–113. [Google Scholar]; (b) Blumenkopf TA; Heathcock CH Stereochemistry of the Sakurai reaction. Additions to cyclohexenones and cycloheptenones. J. Am. Chem. Soc 1983, 105, 2354–2358. [Google Scholar]
- 31.Reetz MT; Kindler A The Kharasch reaction revisited: CuX3Li2-catalyzed conjugate addition reactions of Grignard reagents. J. Organomet. Chem 1995, 502, C5–C7. [Google Scholar]
- 32.Ichiki M; Tanimoto H; Miwa S; Saito R; Sato T; Chida N Synthesis of (−)-Morphine: Application of Sequential Claisen/Claisen Rearrangement of an Allylic Vicinal Diol. Chem. Eur. J 2013, 19, 264–269. [DOI] [PubMed] [Google Scholar]
- 33.(a) Nemoto H; Fujita S; Nagai M; Fukumoto K; Kametani T A novel strategy for the stereoselective total synthesis of C-17 spiro steroids. Total synthesis of 19-norcanrenone - a formal total synthesis of 19-norspironolactone. J. Am. Chem. Soc 1988, 110, 2931–2938 [Google Scholar]; (b) Li H-S; Rampersaud AA; Archer RA; Pawlak JM; Beavers LS; Schimdt RJ; Kauffman RF; Bensch WR; Bumol TF; Apelgren LD; Eacho PI; Perry DN; McClure DB; Gadski RA Synthesis and Biological Evaluation of a New Series of Sterols as Potential Hypocholesterolemic Agents. J. Med. Chem 1995, 38, 277–288 [DOI] [PubMed] [Google Scholar]; (c) Bielska AA; Ory DA; Covey DF Synthesis of the enantiomer of the oxysterol-antagonist LY295427. Steroids 2011, 76, 986–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.(a) Ashby EC; Laemmle JT Stereochemistry of organometallic compound addition to ketones. Chem. Rev 1975, 75, 521–546. [Google Scholar]; (b) Greeves N; Lyford L; Pease JE Ligand effects on diastereoselective addition of organocerium reagents to aldehydes and cyclic ketones. Tetrahedron Lett. 1994, 35, 285–288 [Google Scholar]; (c) Alcaraz C; Groth U Ligand Effects in Diastereoselective Additions of Organocerium Reagents to Carbonyl Substrates. Angew. Chem. Int. Ed 1997, 36, 2480–2482. [Google Scholar]
- 35.Maity S; Manna S; Rana S; Naveen T; Mallick A; Maita D Efficient and Stereoselective Nitration of Mono- and Disubstituted Olefins with AgNO2 and TEMPO. J. Am. Chem. Soc 2013, 135, 3355–3358. [DOI] [PubMed] [Google Scholar]
- 36.(a) Touré BB; Hall DG Natural Product Synthesis Using Multicomponent Reaction Strategies. Chem. Rev 2009, 109, 4439–4486 [DOI] [PubMed] [Google Scholar]; (b) Noyori R; Suzuki M Prostaglandin Syntheses by Three-Component Coupling. New Synthetic Methods (49). Angew. Chem. Int. Ed 1984, 23, 847–876 [Google Scholar]; (c) Chapdelaine MJ; Hulce M Tandem Vicinal Difunctionalization: β-Addition to α,β-Unsaturated Carbonyl Substrates Followed by α-Functionalization. Org. React 1990, 38, 225–653; [Google Scholar]; (d) Dowling MS; Vanderwal CD Ring-Closing Metathesis of Allylsilanes/Electrophilic Desilylation To Prepare exo-Methylidenecycloalkanes. Short Syntheses of Teucladiol and Poitediol. J. Am. Chem. Soc 2009, 131, 15090–15091 [DOI] [PubMed] [Google Scholar]; (e) Dowling MS; Vanderwal CD Ring-Closing Metathesis of Allylsilanes As a Flexible Strategy toward Cyclic Terpenes. Short Syntheses of Teucladiol, Isoteucladiol, Poitediol, and Dactylol and an Attempted Synthesis of Caryophyllene. J. Org. Chem 2010, 75, 6908–6922. [DOI] [PubMed] [Google Scholar]
- 37.(a) Johnson CR; Raheja RK Hydrosilylation of enones: platinum divinyltetramethyldisiloxane complex in the preparation of triisopropylsilyl and triphenylsilyl enol ethers. J. Org. Chem 1994, 59, 2287–2288 [Google Scholar]; (b) Tsuda T; Hayashi T; Satomi H; Kawamoto T; Saegusa T Methylcopper(I)-catalyzed selective conjugate reduction of α,β-unsaturated carbonyl compounds by di-iso-butylaluminum hydride in the presence of hexamethylphosphoric triamide. J. Org. Chem 1986, 51, 537–540 [Google Scholar]; (c) Daniewski AR; Kiegiel, Stereoselective Conjugate Reduction of (+)-(7aS)-5,6,7,7a-Tetrahydro-7a-Methyl-1,5-Indandione. Synth. Commun 1988, 18, 115–118. [Google Scholar]
- 38.(a) Krüerke U Halogen-austausch an chlorsilanen und die tetrahydrofuran-spaltung durch brom- und jodosilane. Chem. Ber 1962, 95, 174–182. [Google Scholar]; (b) Karger MH; Mazur Y Mixed sulfonic-carboxylic anhydrides. II. Reactions with aliphatic ethers and amines. J. Org. Chem 1971, 36, 532–540. [Google Scholar]; (c) Beard CD; Baum K; Grakauskas V Synthesis of some novel trifluoromethanesulfonates and their reactions with alcohols. J. Org. Chem 1973, 38, 3673–3677. [Google Scholar]; (d) Forbus TR Jr; Taylor SL; Martin JC Reactions of the readily accessible electrophile, trifluoroacetyl triflate: A very reactive agent for trifluoroacetylations at oxygen, nitrogen, carbon, or halogen centers. J. Org. Chem 1987, 52, 4156–4159. [Google Scholar]; (e) Ohshita J; Iwata A; Kanetani F; Kunai A; Yamamoto Y; Matui C Ring-opening iodo- and bromosilation of cyclic ethers by treatment with iodo- and bromotrialkylsilane equivalents. J. Org. Chem 1999, 64, 8024–8026. [Google Scholar]; (f) Iqbal J; Srivastava RR Cobalt(II)chloride catalyzed cleavage of ethers with acyl halides: Scope and mechanism. Tetrahedron 1991, 47, 3155–3170. [Google Scholar]; (g) Node M; Kajimoto T; Nishide K; Fujita E; Fuji K Regioselective ring opening of unsymmetrical cyclic ethers wit the AlCl3-NaI-acetonitrile system: Application to hydroxylation of ent-kaurene. Tetrahedron Lett. 1984, 25, 219–222. [Google Scholar]
- 39.Wettstein A; Mischer K Zur konstitution des cafesterols. Helv. Chim. Acta 1942, 25, 718–731. [Google Scholar]
- 40.(a) Courtney JL "Ruthenium Tetroxide Oxidations" In Organic Syntheses by Oxidation with Metal Compounds; Mijs WJ; de Jonge CRHI, Ed.; Plenum Press: New York, 1986; pp 445–468. [Google Scholar]; (b) Lee JS; Cao H; Fuchs PL Ruthenium-catalyzed mild C─H oxyfunctionalization of cyclic steroidal ethers. J. Org. Chem 2007, 72, 5820–5823. [DOI] [PubMed] [Google Scholar]
- 41.Curci R; D’Accolti L; Florentino M; Fusco C; Adam W; González-Nuñez ME; Mello R Oxidation of acetals, an orthoester, and ethers by dioxiranes through alpha-CH insertion. Tetrahedron Lett. 1992, 33, 4225–4228. [Google Scholar]
- 42.Zhao D; Lee DG Heterogeneous permanganate oxidations. Part 6: Selective oxidation of arenes. Synthesis 1994, 915–916. [Google Scholar]
- 43.Lee S; Fuchs PL An efficient C─H oxidation protocol for the alpha-hydroxylation of cyclic steroidal ethers. Org. Lett 2004, 6, 1437–1440. [DOI] [PubMed] [Google Scholar]
- 44.White AM; Dao K; Vrubliauskas D; Konst ZA; Pierens GK; Mándi A; Andrews KT; Skinner-Adams TS; Clarke ME; Narbutas PT; Sim DC-M; Cheney KL; Kurtán T; Garson MJ; Vanderwal. C. D. Catalyst-controlled stereoselective synthesis secures the structure of the antimalarial isocyanoterpene pustulosaisonitrile-1. J. Org. Chem 2017, 82, 13313–13323. [DOI] [PubMed] [Google Scholar]
- 45.(a) Chen K; Baran PS Total synthesis of eudesmane terpenes by site-selective C─H oxidations. Nature 2009, 459, 824–828 [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC; Sarlah D; Shaw DM Total synthesis and revised structure of biyouyanagin A. Angew. Chem. Int. Ed 2007, 46, 4708–4711 [DOI] [PubMed] [Google Scholar]; (c) Wang J; Ma A; Ma D Organcatalytic Michael addition of aldehydes to gamma-keto-alpha,beta-unsaturated esters. An efficient entry to versatile chiral building blocks. Org. Lett 2008, 10, 5425–5428 [DOI] [PubMed] [Google Scholar]; (d) Houjeiry T; Poe SL; McQuade DT Synthesis of optically active 4-substituted 2-cyclohexenones. Org. Lett 2012, 14, 4394–4397. [DOI] [PubMed] [Google Scholar]
- 46.(a) Stevens RV; Albizati KF Synthetic approach to the amphilectane diterpenes: The use of nitriles as terminators of carbocation-olefin cyclizations. J. Org. Chem 1985, 50, 632–640 [Google Scholar]; (b) Siegel C; Gorton PM; Uliss DB; Handrick GR; Dalzell HC; Razdan RK Synthesis of racemic and ptically active delta9-tetrahydrocannabinol (THC) metabolites. J. Org. Chem 1991, 56, 6865–6872 [Google Scholar]; (c) Money T; Wong MKC in Stud. Nat. Prod. Chem, Ed. Atta ur R, Elsevier; 1995, 16, 123–288. [Google Scholar]
- 47.(a) This type of reactivity (Meinwald rearrangement of epoxide with in situ cyclodehydration) is known from the work of Kumar, though they generated the epoxide from aldehyde precursors via sulfonium ylide additions:Kumar S A convenient and general synthesis of cata- and peri-condensed polycyclic aromatic hydrocarbons with a Fjord-region. Synthesis 2001, 841–844. [Google Scholar]; (b) For an application in natural product synthesis, see:Liao X; Stanley LM; Hartwig JF Enantioselective total synthesis of (−)-taiwaniaquinone H and (−)-taiwaniaquinol B by iridium-catalyzed borylation and palladium-catalyzed asymmetric-alpha-arylation. J. Am. Chem. Soc 2011, 133, 2088–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.(a) Wilds AL; Nelson NA A superior method for reducing phenol ethers to dihydro derivatives and unsaturated ketones. J. Am. Chem. Soc 1953, 75, 5360–5365 [Google Scholar]; (b) Krapcho AP; Bothner-By AA J. Am. Chem. Soc 1959, 81, 3658–3666 [Google Scholar]; (c) Bible RH; Burtner RR Derivatives of podocarpic acid. IV. Reduction of the aromatic ring. J. Org. Chem 1961, 26, 1174–1180 [Google Scholar]; (d) Dryden HL; Webber GM; Burtner RR; Cella JA The use of sodium metal in the Birch reduction of aromatic compounds. J. Org. Chem 1961, 26, 3237–3245 [Google Scholar]; (e) Chinn LJ; Dryden HL Studies in the total synthesis of steroids and their analogs. I. Synthesis of 19-nortestosterone. J. Org. Chem 1961, 26, 3904–3910 [Google Scholar]; (f) Bennett CR; Cambie RC Chemistry of the podocarpaceae. XI. Reductions of totarol. Tetrahedron 1966, 22, 2845–2860 [Google Scholar]; (g) Cherkasov AN; Ponomarev AM; Pivnitskii KK Reduction of estrogen methyl ethers by alkali metals in ethers. J. Org. Chem USSR; 1971, 7, 955–961 [Google Scholar]; (h) Ponomarev AM; Cherkasov AN; Pivnitskii KK 1,2-Addition during the reduction of aromatic compounds with alkali metals in ethers. J. Org. Chem USSR; 1971, 7, 1477–1479 [Google Scholar]; (i) Hancock WS; Mander LN; Massy-Westropp RA The rearrangement of some epoxy podocarpanoic acids. Aust. J. Chem 1977, 30, 1093–1107 [Google Scholar]; (j) Chatterjee A; Hazra BG Total synthesis of ring-c aromatic 18-nor steroid. Tetrahedron 1980, 36, 2513–2519 [Google Scholar]; (k) Collins DJ; Jacobs HA Steric and stereoelectronic effects in the hydrogenolysis and Birch reduction of some hindered tertiary-benzylic carbinols. Aust. J. Chem 1987, 40, 1989–2004 [Google Scholar]; (l) Cambie RC; Mezler MR; Rutledge PS; Woodgate PD Organoiron complexes of podocarpic acid derivatives. J. Organomet. Chem 1990, 398, 117–131 [Google Scholar]; (m) Lukács A; Szabó L; Hazai L; Szántay C; Mák M; Gorka A; Szántay C Seven-membered ring formation through Grewe-cyclization. Tetrahedron 2001, 57, 5843–5850 [Google Scholar]; (n) Nagase H; Imaide S; Hirayama S; Nemoto T; Fujii H Essential structure of opioid k receptor agonist nalfurafine for binding to the k receptor 2: Synthesis of decahydro(iminoethano)phenanthrene derivatives and their pharmacologies. Bioorg. Med. Chem. Lett 2012, 22, 5071–5074. [DOI] [PubMed] [Google Scholar]
- 49.Liotta R; Hoff WS Trifluoroperacetic acid. Oxidation of aromatic rings. J. Org. Chem 1980, 45, 2887–2890. [Google Scholar]
- 50.Rickborn B, Acid-catalyzed Rearrangements of Epoxides. Elsevier Science Ltd.: 1991; Vol. 3. [Google Scholar]
- 51.(a) Maruoka K; Murase N; Bureau R; Ooi T; Yamamoto H Lewis acid-promoted selective rearrangement of trisubstituted epoxides to aldehydes or ketones. Tetrahedron 1994, 50, 3663–3672 [Google Scholar]; (b) Ranu BC; Jana U Indium(III) chloride-promoted rearrangement of epoxides: a selective synthesis of substituted benzylic aldehydes and ketones. J. Org. Chem 1998, 63, 8212–8216. [Google Scholar]
- 52.We were able to replace the two-step Peterson with a salt-free Wittig methylenation of 104 (85% yield), but this adjustment still requires a subsequent oxymercuration/reduction step to access DICA. See SI for details.
- 53.(a) Cahiez G; Duplais C; Buendia J Chemistry of organomanganese(II) compounds. Chem. Rev 2009, 109, 1434–1476 [DOI] [PubMed] [Google Scholar]; (b) Reetz MT (1982) “Organotitanium reagents in organic synthesis a simple means to adjust reactivity and selectivity of carbanions.” In: Synthetic and Structural Problems. Topics in Current Chemistry, vol 106. Springer, Berlin, Heidelberg. pp 1–52. [Google Scholar]
- 54.(a) Yamamoto H; Maruoka K Organoaluminum reagents for selective reactions. Pure & Appl. Chem 1988, 760, 21–26 [Google Scholar]; (b) Yamamoto H; Saito S Designer Lewis acid catalysts for selective organic synthesis. Pure & Appl. Chem 1999, 71, 239–245. [Google Scholar]
- 55.Ashby EC; Roling PV; Yu SH, Stereoselective alkylation reactions. I. Organomagnesium and organoaluminum addition to 4-tert-butylcyclohexanone. Unusual stereoselectivity involving trimethylaluminum alkylation in benzene. J. Org. Chem 1972, 37, 1918–1925. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.