

Abstract
Stroke kills or disables approximately 15 million people worldwide each year. It is the leading cause of brain injury, resulting in persistent neurological deficits and profound physical handicaps. In spite of over 100 clinical trials, stroke treatment modalities are limited in applicability and efficacy, and therefore, identification of new therapeutic modalities is required to combat this growing problem. Poststroke oxidative damage and lactic acidosis are widely-recognized forms of brain ischemia/reperfusion injury. However, treatments directed at these injury mechanisms have not been effective. In this review, we offer a novel approach combining these well-established damage mechanisms with new insights into brain glucose handling. Specifically, emerging evidence of brain gluconeogenesis provides a missing link for understanding oxidative injury and lactate toxicity after ischemia. Therefore, dysfunctional gluconeogenesis may substantially contribute to oxidative and lactate damage. We further review that hypothermia initiated early in ischemia and before reperfusion may ameliorate gluconeogenic dysfunction and subsequently provide an important mechanism of hypothermic protection. We will focus on the efficacy of pharmacologically assisted hypothermia and suggest a combination that minimizes side effects. Together, this study will advance our knowledge of basic mechanisms of ischemic damage and apply this knowledge to develop new therapeutic strategies that are desperately needed in the clinical treatment of stroke.
Keywords: Gluconeogenesis, ischemic stroke, neuroprotection, PCK, pharmacological hypothermia
Introduction
Stroke claims approximately 15 million victims worldwide each year and is the leading cause of persistent neurological deficits and profound physical handicap.[1] Many injury mechanisms have been identified in the post-stroke brain. However, they were unsuccessfully treated in clinical trials. Current clinical treatments are limited to tissue plasminogen activator or surgery (e.g., thrombectomy) which is beneficial to only a proportion of stroke victims in a limited time window. Therefore, the identification of new therapeutic modalities is critical to improve clinical outcomes in stroke. Here, we address a new approach to oxidative and acidosis injury, and study how pharmacological hypothermia (PH) may combat these injuries and establish a potential neuroprotection strategy for stroke treatment.
Oxidative stress, the overproduction of reactive oxygen species (ROS), is a fundamental damage mechanism after stroke which increases brain edema, hemorrhagic transformation, cell death, and infarct volume.[2] The shift to glycolysis during ischemia and subsequent acidosis enhances ROS production.[3] Thus, aside from cellular energetics, glucose handling in the ischemic brain is intimately linked to cellular damage mechanisms. However, recent studies suggest this is not the whole story, showing gluconeogenic activity also occurs in the brain, especially after ischemic stroke.[4] Dysfunctional gluconeogenesis during stroke has the potential to enhance acidosis and ROS damage. Due to lack of ATP during ischemia, gluconeogenesis would be stunted, leading to accumulation of phosphoenolpyruvate (PEP) catalyzed by phosphoenolpyruvate carboxykinase (PCK). In turn, PEP is a source for lactic acidosis and thus ROS production, rather than the formation of glucose. Hypoxia-inducible factors (HIFs) are transcription factors that respond to decreases in cellular oxygen[5] and regulate transcription of gluconeogenic enzymes. A second transcription factor, X-box binding protein 1 (XBP-1s), functions with HIF-1α to control gluconeogenesis.[6] XBP-1s regulate gluconeogenic genes such as PCK[7,8,9] in combination with forkhead box O (FoxO) transcription factor. Under hypoxic and ischemic conditions, HIF1-α is activated and endoplasmic reticulum (ER) stress leads to increased XBP-1s. However, HIF-α has been shown to generate incomplete gluconeogenesis during hypoxia. The roles of HIF-α, XBP-1s, and FoxO have been studied in relation to gluconeogenesis during stroke and may act as the upstream regulators leading to dysfunctional gluconeogenesis and contributing to ROS, acidosis, and cell death.[10]
The clinical treatment of stroke is highly limited and new therapeutic modalities are urgently needed. During a stroke, focal ischemia ceases normal brain metabolism, and many physiologic adaptations and pathophysiological processes come into play. The central problem is impaired oxidative phosphorylation in mitochondria, the major source of brain ATP, due to oxygen and glucose deprivation. Within minutes after stroke, a shift from aerobic to anaerobic metabolism occurs in ischemic tissue to compensate for the lack of ATP. However, compared to aerobic metabolism, anaerobic metabolism of glucose is not efficient (2 vs. 36–38 ATP/glucose). Hyperglycolysis also produces lactic acid, leading to oxidative injury. The neuronal adaptation to induce hyperglycolysis, paradoxically, increases brain injury.[3,11,12] However, clinical trials targeting hyperglycolysis or ROS alone were ineffective, suggesting a blind spot in our understanding. The role of cerebral gluconeogenesis may provide a “missing link” in the mechanisms of acidosis and ROS damage. We wish to determine the role of gluconeogenesis in focal ischemia and reperfusion (I/R) injury, which is expected to provide new therapeutic targets.
Therapeutic hypothermia (TH) has long been considered a promising neuroprotective treatment after ischemic stroke.[13,14] Clinical trials have proved that hypothermia in ischemic patients is safely inducible, but they have not observed clear amelioration on brain injury thus far, albeit they did not fulfill the criteria of an early induction of hypothermia, delayed by the priority of reperfusion.[13] Clinic limitations in the induction of physical hypothermia include delays in cooling initiation and the onset of target temperature.[15] The late start necessitates a prolonged hypothermia duration, which requires labor-intensive medical and nursing efforts, and causes secondary complications.[16] The benefits of inter-ischemia hypothermia have been shown following myocardial infarction (MI).[17,18] Studies have reported that hypothermia before reperfusion can reduce infarction, prevent the death of myocardial cells, and improve patient outcome.[17,18,19,20,21] Considering the benefits of inter-ischemia hypothermia in MI, a more clinically manageable approach to overcome the obstacles for the application of hypothermia in ischemic stroke is highly desirable. PH offers an alternative means of hypothermia induction not subject to limitations of physical cooling. Eight classes of hypothermia-inducing drugs in animal models were studied[22] and DHC, a TRPV1 agonist, showed the greatest effect as a PH agent.[22,23,24] Unfortunately, the effective doses of DHC were toxic and induced complications. Thus, DHC by itself cannot act as a PH agent.[22] However, we and others showed that DHC at a low, non-toxic, dose acted synergistically with the phenothiazine class of drugs to induce a highly effective PH[25] with decreased brain glucose metabolism[26,27] and reduced stroke injury.[28] Here, we introduce the pharmacological combination of DHC and phenothiazines to induce PH and provide a novel therapeutic strategy that operates on brain glucose metabolism. Specifically, PH which is relatively easy to induce, targets brain gluconeogenesis, and thus decreases post-stroke acidosis and oxidative injury and improves functional recovery.
Gluconeogenesis, Limiting Enzymes, and Ischemic Stroke
Gluconeogenesis is a metabolic pathway that generates glucose from noncarbohydrate carbon substrates including amino acids and triglycerides. Gluconeogenesis is one of the several important mechanisms used by humans and other mammals to maintain glucose levels.[4] It is commonly believed that gluconeogenesis is present only in the liver, kidney, intestine, and muscle.[29] Although these tissues have gluconeogenic activity much greater than the brain, due to the application of modern molecular methods, there is now unambiguous evidence that gluconeogenesis also occurs in the brain.[4]
Gluconeogenesis is a multistep metabolic process that generates glucose from pyruvate or a related three-carbon compound (such as lactate). Gluconeogenesis consists of a series of eleven enzyme-catalyzed reactions. It begins in mitochondria, oxaloacetate (OAA) is decarboxylated and then phosphorylated to form PEP by the enzyme PCK with a GTP. The next steps are glycolysis in reverse. Fructose1,6-bisphosphatase converts fructose 1,6-bisphosphate to fructose 6-phosphate (F6P), using one ATP. Glucose-6-phosphate (G6P) is formed from F6P. The final reaction of gluconeogenesis is the formation of glucose from G6P by hydrolysis with glucose-6-phosphatase (G6PC), which requires another ATP. PCK, an enzyme in the lyase family, converts OAA into PEP and carbon dioxide through the cytosolic (PCK1) or mitochondrial (PCK2) isoforms.[4]
In ischemia, when mitochondrial oxidative phosphorylation and ATP production are disrupted, anaerobic glycolysis becomes the primary source of ATP. Anaerobic glycolysis alone cannot produce sufficient ATP to maintain brain functioning, instead lactate production leads to acidosis and ROS. It is expected that gluconeogenesis would increase after ischemia to provide additional substrate for energy production. However, the status of gluconeogenesis after ischemia is simply unknown. If there is increased gluconeogenesis, then this pathway may not function correctly because of lack of ATP. This dysfunctional (or stunted) gluconeogenesis will lead to excess PEP activity, in turn augmenting lactic acidosis since the formation of glucose will be interrupted by lack of ATP [Figure 1]. We must note that gluconeogenesis is anabolic, increasing the amount of glucose, while oxidative phosphorylation is catabolic and decreases the amount of glucose. Although they are coupled, it is technically difficult to determine the percent of glucose due to either pathway. In our recent study, we measured energy-dependent enzymes, PCKs, substrates, and products of gluconeogenesis to determine the status of gluconeogenesis.[30] All glucose handling pathways, including glycolysis and gluconeogenesis, are ancillary relative to oxidative phosphorylation, and are disrupted by ischemia. Therefore, ancillary pathways like gluconeogenesis greatly increase in relative importance after stroke when oxidative phosphorylation is inactive. Post-ischemic gluconeogenesis then may lead to a significant proportion of lactate and ROS, as our preliminary data suggest, which makes the relevance of gluconeogenesis to stroke injury highly novel. Our recent study demonstrated a key role of PCK (s) and its transcriptional regulation by HIF-XBP-1-FoxO in cerebral gluconeogenesis in stroke.[10,30]
Figure 1.
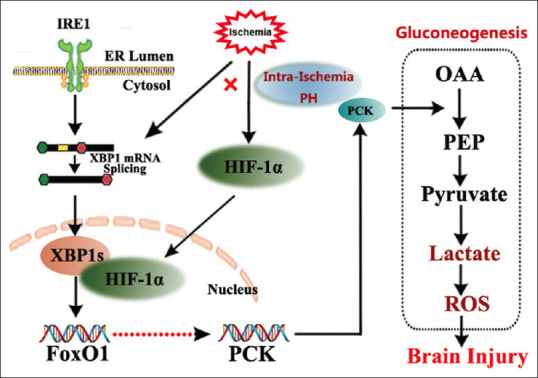
Regulation of PH on gluconeogenesis through HIF-1α/XBP-1/FoxO1. Gluconeogenesis is a multistep metabolic process that generates glucose from pyruvate or a related three-carbon compound (lactate, alanine). Conversion of pyruvate to PEP via oxaloacetate, catalyzed by pyruvate carboxylase and PEP carboxy kinase is one of the irreversible steps in the gluconeogenic pathway. HIF-1α regulation and XBP-1-FoxO Signal play the key roles in gluconeogenic activity through PCKs. HIF: Hypoxia-inducible factor, XBP-1: X-box binding protein 1, FoxO: Forkhead box O, PEP: Phosphoenolpyruvate, PH: Pharmacological hypothermia, ROS: Reactive oxygen species, OAA: Oxaloacetate, PCK: Phosphoenolpyruvate carboxykinase, ER: Endoplasmic reticulum
Hypoxia-Inducible Factor-1α, X-box Binding Protein-1s, and Forkhead Box O Regulation of Gluconeogenic Activity in Stroke
Stroke and endoplasmic reticulum stress
The normal ER function of protein synthesis is disrupted when a cell experiences stress conditions, such as starvation, redox imbalance, and metabolic failure, then the ER switches to a stress state, in which unfolded or misfolded proteins in the ER are accumulated due to an imbalance of ER homeostasis.[31,32,33,34] ER stress is an essential step in the progression of brain I/R injury,[35] which involves protein synthesis inhibition and selective activation of stress gene expression for cells to regulate signaling pathways for cell damage, repair, or death. ER stress induces cleavage of the mRNA coding the transcription factor called XBP-1 and the removal of a 26-bp intron from the full-length XBP-1 mRNA. The ligation of the two mRNA fragments creates a translational frameshift, which leads to the translation of processed XBP-1s,[8] which acts as a nuclear transcription factor. Furthermore, XBP-1s interact with FoxO transcription factor and directs it toward proteasome-mediated degradation. FoxO subfamily has emerged as a shared component among pathways regulating diverse cellular functions, such as differentiation, metabolism, proliferation, and survival.[36,37,38] In particular, FoxO1 acts as a cardinal regulator of whole-body energy homeostasis, including glucose output, adipocyte and muscle differentiation, and feeding behavior in the brain.[36,37]
X-box binding protein-1 interaction with hypoxia-inducible factor-1α
XBP-1 drives a transcriptional complex with HIF-1α that regulates the expression of HIF-1α target genes [Figure 1], [39,40] including direct binding to the promoters of genes coding gluconeogenic enzymes. XBP-1 transcriptional activity is induced following neonatal hypoxia-ischemia with HIF-1α overexpression. There is evidence of IRE-1α cleavage of XBP-1 after stroke, but the study of its target genes have not been conducted. Whether the XBP-1-HIF-1α complex regulates gluconeogenesis genes after stroke remains to be determined.
Stimulation of Forkhead box O 1 on phosphoenolpyruvate carboxykinase expression
The transcription factor FoxO1 regulates hepatic gluconeogenesis.[41] FoxO1 binds the promotor of and increases gene expression of PCK1, the key rate-limiting gluconeogenic enzymes, and thus upregulates hepatic glucose.[7,42] Studies in hepatoma cells suggest that FoxO1 controls the transcription of reporter genes containing the PCK promoters.[9,43] A dominant-negative FoxO1 mutant prevents the dex/cAMP–induced increases in PCK expression in primary hepatocytes.[44,45] FoxO1 transcriptionally regulates PCK in the pyruvate-PEP futile cycle.[46] The resulting glucose reduction activates ER stress and it correspondingly regulates PCK2 mRNA. XBP-1s bind to FoxO1 to promote FoxO1 degradation by the 26S proteasome.[8]
Antagonism of Phosphoenolpyruvate Carboxykinase Transcriptional regulation
FoxO1 regulates gluconeogenesis in the liver by the traditional regulators of insulin and glucagon, but it is unknown whether it regulates gluconeogenesis in the brain. However, since it is a known regulator of gluconeogenesis, it is imperative to study FoxO1 regulation after stroke. XBP-1 acts as a negative regulator of gluconeogenesis in hepatic cells, whereas HIF-1α works synergistically with XBP-1 to upregulate gluconeogenesis. Our previous study showed that stroke upregulates PCK transcription, for which the net increase is expected to be an interplay between the positive HIF-1α-XBP-1 pathway, and the negative FoxO1-XBP-1.[10,30]
Neuroprotection of Therapeutic Hypothermia and Its Induction
Neuroprotection and limitations of therapeutic hypothermia
Hypothermia (TH) at 30°C–34°C core temperature is a highly effective neuroprotective treatment for ischemic stroke.[47] The mechanisms by which TH protects brain include reduced oxygen demand, preservation of ATP levels, and a general enhancement of cellular survival.[48] However, a positive outcome of TH is dependent on multiple factors such as initiation, duration, and depth of hypothermia.[16,49] In addition, temperature-induced hypothermia is fought every step of the way by the body's intrinsic physiological thermoregulatory mechanism, such as shivering and brown fat metabolism. These responses work against the clinician's efforts and may prolong the induction phase and give a less stable maintenance phase.[15,16] These and other factors reduce the clinical efficacy of temperature-induced TH and have led us to investigate alternative ways to induce TH.
Pharmacological hypothermia
PH is an alternative to physically-induced temperature reduction.[50] Dihydrocapsaicin (DHC), a capsaicinoid, is a widely used experimental PH agent.[22] DHC is an analog and congener of capsaicin in chili peppers (Capsicum), and accounts for 22% of the total capsaicinoid mixture and has nearly the same pungency as capsaicin. The mode of action of DHC involves the TRPV1 nonspecific cation channel which is expressed in warm-sensing nerve fibers in the peripheral and central nervous system.[51] TRPV1 decreases body temperature by reducing the thermoregulatory set-point at peripheral thermosensors and at the pre-optic anterior hypothalamus.[52] TRPV receptors are activated by capsaicin, DHC, and rinvanil in animal models.[23,53,54] TRPV1 activation is thought to act as a neuroprotectant by lowering body temperature to a hypothermic range.[23,24] High-dose DHC (>2.0 mg/kg/h), acting as a TRPV agonist, reduced core body temperature to 33.0 ± 0.2°C after 100 min in a rodent stroke model.[55] However, the downside was induction of hypotension and bradycardia at the high-dose (>2.0 mg/kg/h), which neutralized the benefits.[51] Low-dose DHC (0.5 mg/kg) does not show neurotoxicity,[55,56] and while it did not show cardiac complications, it was not an effective neuroprotectant.[25] We then studied the effect of low-dose DHC and other PH agents to achieve neuroprotection safely and effectively. In our preliminary study,[57,58] as compared to each agent alone, we showed a faster induction of hypothermia, within 5 min (vs. 30 min), a longer duration up to 6 h (vs. 2 h), and deeper body temperature at 34°C (vs. 35.8°C), induced by a combination of low-dose of DHC (0.5 mg/kg) and chlorpromazine and promethazine (C + P, 4 mg/kg). Importantly, there was a 40% greater reduction in infarct volume by combination therapy versus either agent alone.[58]
Antipsychotic and sedative drugs, such as the neuroleptics (C + P) have also garnered interest because of their depressant effects.[59,60] These agents served as prototypes for the phenothiazines, which we propose to use in combination with DHC. C + P were thought to induce an “artificial hibernation.”[61] We showed phenothiazines are neuroprotective after ischemic stroke,[28] likely due to their depressing effects on the nervous system where C + P alters glucose metabolism,[26] inhibits glucose uptake, and inhibits carbohydrates oxidation.[27,62,63] Impaired carbohydrate utilization occurred under normoxic conditions.[64] In diabetic models, an induced hyperglycemia was blocked by Promethazine. Other studies have indicated that the depressive effect of phenothiazines on glucose utilization was similar to anesthetics such as pentobarbitone.[65,66] It was also shown that phenothiazines cause a dose-dependent reduction in cerebral blood flow and oxygen consumption,[67] and reduced energy metabolism.[26,62] Finally, inhibitory effects of C + P on lipid peroxidation were also observed in brain tissue.[68] Taken together, the data shows a spectrum from natural hibernation, through the depressant effects of anesthetics, to the action of neuroleptics that modulate cerebral energy utilization through blood flow and glucose handling, and this line of evidence underpins our approach. It is a desirable strategy to seek to overcome the limitations of temperature-induced hypothermia and the limits of toxic PH agents by developing a combination therapy with DHC and phenothiazines.[58]
Therapeutic potential of early-initiated, intra-ischemia and prereperfusion hypothermia in stroke
Brain cells undergo irreversible damage within minutes of brain ischemia; therefore, salvage of reversible ischemic cerebral tissue remains critical and time-dependent in stroke treatment. The shorter the duration of ischemia, the more brain cells that can be saved.[69] The core tenet of treatment is to prevent the expansion of irreversible injury (i.e., the ischemic core) and salvage the reversible ischemic tissue (i.e., the ischemic penumbra)[70] as early as possible, to recanalize the cerebral artery for normal cerebral blood flow to the penumbra, and restore normal glucose metabolism, thus delaying progression to irreversible tissue injury.[71]
Induction of hypothermia can take place prior to or after reperfusion. With the premise of achieving revascularization as quickly as possible, adjuvant strategies that induce hypothermia post revascularization seem more practical. However, by using PH, it is possible to cool the body during the ischemic event, while awaiting advanced imaging, or during the administration of thrombolysis or thrombectomy. The hypothesized benefit of pre-reperfusion hypothermia would be to slow and in turn minimize cellular metabolism and core expansion while waiting for recanalization and to mitigate reperfusion injury. Previous studies on hypothermia during heart ischemia have demonstrated multiple benefits,[17,18,19,20,21,72] including reduced infarction size and improved outcomes.[73,74] Hypothermia in patients with pre-hospital cardiac arrest also improves outcomes.[75,76] A recent study has shown that pre-reperfusion ischemic hypothermia and intra-ischemia hypothermia translates to functional benefit in ischemic stroke using a pharmacological approach.[57]
Conclusions
Given the recentness of the discovery of cerebral gluconeogenesis, gluconeogenesis is either detrimental or beneficial after stroke. Therefore, gluconeogenesis may serve as an important therapeutic target after ischemic stroke. Previous studies have offered an innovative and novel neuroprotective strategy using the early initiated inter-ischemic PH to depress poststroke gluconeogenesis and its possible downstream damage mechanisms. Prior studies of cerebral gluconeogenesis suggest that impaired gluconeogenesis could be an important source of lactate after ischemic stroke. Because hypothermia preserves ATP after glucose/oxygen deprivation and also with increased neuronal metabolic demand,[27,77] early hypothermia by combination DHC/phenothiazine-induced PH may suppress the detrimental effects of gluconeogenesis, improve glucose metabolism, and reduce lactic and ROS after acute stroke. This would be a breakthrough in stroke therapeutics. Finally, because other forms of brain injury such as trauma and epilepsy (seizures) share similar damage mechanisms, depletion of ATP, acidosis, and ROS production, the results obtained in stroke, have the innovative potential to translate as therapy for other forms of acute brain injury. Past treatments that individually targeted glucose metabolism, lactic acidosis, or ROS were ineffective in clinical trials. PH is relatively easy to implement and is effective in inducing hypothermia during the early stage of stroke, which in turn decreases brain energy utilization and glucose metabolism in a holistic manner.
Financial support and sponsorship
This work was partially supported by the National Natural Science Foundation of China (82072549, 81871838, 82001277, 82101436), the Science and Technology Plan of Beijing Tongzhou District (KJ2022CX033), and the Laboratory Development Funds of Luhe Hospital (2022).
Conflicts of interest
Dr. Yuchuan Ding is an Associate Editor, Dr. Xiaokun Geng is an Editorial Board member of Brain Circulation. The article was subject to the journal's standard procedures, with peer review handled independently of them and their research groups.
References
- 1.Xue Y, Nie D, Wang LJ, Qiu HC, Ma L, Dong MX, et al. Microglial polarization: Novel therapeutic strategy against ischemic stroke. Aging Dis. 2021;12:466–79. doi: 10.14336/AD.2020.0701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li W, Yang S. Targeting oxidative stress for the treatment of ischemic stroke: Upstream and downstream therapeutic strategies. Brain Circ. 2016;2:153–63. doi: 10.4103/2394-8108.195279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li WA, Moore-Langston S, Chakraborty T, Rafols JA, Conti AC, Ding Y. Hyperglycemia in stroke and possible treatments. Neurol Res. 2013;35:479–91. doi: 10.1179/1743132813Y.0000000209. [DOI] [PubMed] [Google Scholar]
- 4.Yip J, Geng X, Shen J, Ding Y. Cerebral gluconeogenesis and diseases. Front Pharmacol. 2016;7:521. doi: 10.3389/fphar.2016.00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilkins SE, Abboud MI, Hancock RL, Schofield CJ. Targeting protein-protein interactions in the HIF system. ChemMedChem. 2016;11:773–86. doi: 10.1002/cmdc.201600012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaudhari N, Talwar P, Parimisetty A, Lefebvre d’Hellencourt C, Ravanan P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci. 2014;8:213. doi: 10.3389/fncel.2014.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang P, Tu B, Wang H, Cao Z, Tang M, Zhang C, et al. Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc Natl Acad Sci U S A. 2014;111:10684–9. doi: 10.1073/pnas.1411026111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou Y, Lee J, Reno CM, Sun C, Park SW, Chung J, et al. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat Med. 2011;17:356–65. doi: 10.1038/nm.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao J, Yu Y, Zhang Z, Chen X, Hu Z, Tong Q, et al. SCP4 promotes gluconeogenesis through FoxO1/3a dephosphorylation. Diabetes. 2018;67:46–57. doi: 10.2337/db17-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo S, Mangal R, Dandu C, Geng X, Ding Y. Role of forkhead box protein O1 (FoxO1) in stroke: A literature review. Aging Dis. 2022;13:521–33. doi: 10.14336/AD.2021.0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geng J, Zhang Y, Li S, Li S, Wang J, Wang H, et al. Metabolomic profiling reveals that reprogramming of cerebral glucose metabolism is involved in ischemic preconditioning-induced neuroprotection in a rodent model of ischemic stroke. J Proteome Res. 2019;18:57–68. doi: 10.1021/acs.jproteome.8b00339. [DOI] [PubMed] [Google Scholar]
- 12.Robbins NM, Swanson RA. Opposing effects of glucose on stroke and reperfusion injury: Acidosis, oxidative stress, and energy metabolism. Stroke. 2014;45:1881–6. doi: 10.1161/STROKEAHA.114.004889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dietrich WD, Bramlett HM. Therapeutic hypothermia and targeted temperature management for traumatic brain injury: Experimental and clinical experience. Brain Circ. 2017;3:186–98. doi: 10.4103/bc.bc_28_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J, Yenari M. Hypothermia for treatment of stroke. Brain Circ. 2015;1:14–25. [Google Scholar]
- 15.Kuczynski AM, Demchuk AM, Almekhlafi MA. Therapeutic hypothermia: Applications in adults with acute ischemic stroke. Brain Circ. 2019;5:43–54. doi: 10.4103/bc.bc_5_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han Z, Liu X, Luo Y, Ji X. Therapeutic hypothermia for stroke: Where to go? Exp Neurol. 2015;272:67–77. doi: 10.1016/j.expneurol.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 17.Götberg M, Olivecrona GK, Engblom H, Ugander M, van der Pals J, Heiberg E, et al. Rapid short-duration hypothermia with cold saline and endovascular cooling before reperfusion reduces microvascular obstruction and myocardial infarct size. BMC Cardiovasc Disord. 2008;8:7. doi: 10.1186/1471-2261-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Götberg M, Olivecrona GK, Koul S, Carlsson M, Engblom H, Ugander M, et al. A pilot study of rapid cooling by cold saline and endovascular cooling before reperfusion in patients with ST-elevation myocardial infarction. Circ Cardiovasc Interv. 2010;3:400–7. doi: 10.1161/CIRCINTERVENTIONS.110.957902. [DOI] [PubMed] [Google Scholar]
- 19.Darbera L, Chenoune M, Lidouren F, Kohlhauer M, Adam C, Bruneval P, et al. Hypothermic liquid ventilation prevents early hemodynamic dysfunction and cardiovascular mortality after coronary artery occlusion complicated by cardiac arrest in rabbits. Crit Care Med. 2013;41:e457–65. doi: 10.1097/CCM.0b013e3182a63b5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hale SL, Dave RH, Kloner RA. Regional hypothermia reduces myocardial necrosis even when instituted after the onset of ischemia. Basic Res Cardiol. 1997;92:351–7. doi: 10.1007/BF00788947. [DOI] [PubMed] [Google Scholar]
- 21.Hamamoto H, Sakamoto H, Leshnower BG, Parish LM, Kanemoto S, Hinmon R, et al. Very mild hypothermia during ischemia and reperfusion improves postinfarction ventricular remodeling. Ann Thorac Surg. 2009;87:172–7. doi: 10.1016/j.athoracsur.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu K, Khan H, Geng X, Zhang J, Ding Y. Pharmacological hypothermia: A potential for future stroke therapy? Neurol Res. 2016;38:478–90. doi: 10.1080/01616412.2016.1187826. [DOI] [PubMed] [Google Scholar]
- 23.Muzzi M, Felici R, Cavone L, Gerace E, Minassi A, Appendino G, et al. Ischemic neuroprotection by TRPV1 receptor-induced hypothermia. J Cereb Blood Flow Metab. 2012;32:978–82. doi: 10.1038/jcbfm.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu X, Wang P, Zhao Z, Cao T, He H, Luo Z, et al. Activation of transient receptor potential vanilloid 1 by dietary capsaicin delays the onset of stroke in stroke-prone spontaneously hypertensive rats. Stroke. 2011;42:3245–51. doi: 10.1161/STROKEAHA.111.618306. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Liu K, Elmadhoun O, Ji X, Duan Y, Shi J, et al. Synergistically induced hypothermia and enhanced neuroprotection by pharmacological and physical approaches in stroke. Aging Dis. 2018;9:578–89. doi: 10.14336/AD.2017.0817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zager EL, Ames A., 3rd Reduction of cellular energy requirements. Screening for agents that may protect against CNS ischemia. J Neurosurg. 1988;69:568–79. doi: 10.3171/jns.1988.69.4.0568. [DOI] [PubMed] [Google Scholar]
- 27.Dwyer DS, Liu Y, Bradley RJ. Dopamine receptor antagonists modulate glucose uptake in rat pheochromocytoma (PC12) cells. Neurosci Lett. 1999;274:151–4. doi: 10.1016/s0304-3940(99)00712-0. [DOI] [PubMed] [Google Scholar]
- 28.Geng X, Li F, Yip J, Peng C, Elmadhoun O, Shen J, et al. Neuroprotection by chlorpromazine and promethazine in severe transient and permanent ischemic stroke. Mol Neurobiol. 2017;54:8140–50. doi: 10.1007/s12035-016-0280-x. [DOI] [PubMed] [Google Scholar]
- 29.Chen J, Lee HJ, Wu X, Huo L, Kim SJ, Xu L, et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. 2015;75:554–65. doi: 10.1158/0008-5472.CAN-14-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geng X, Shen J, Li F, Yip J, Guan L, Rajah G, et al. Phosphoenolpyruvate Carboxykinase (PCK) in the brain gluconeogenic pathway contributes to oxidative and lactic injury after stroke. Mol Neurobiol. 2021;58:2309–21. doi: 10.1007/s12035-020-02251-3. [DOI] [PubMed] [Google Scholar]
- 31.Su Y, Li F. Endoplasmic reticulum stress in brain ischemia. Int J Neurosci. 2016;126:681–91. doi: 10.3109/00207454.2015.1059836. [DOI] [PubMed] [Google Scholar]
- 32.Chadwick SR, Lajoie P. Endoplasmic reticulum stress coping mechanisms and lifespan regulation in health and diseases. Front Cell Dev Biol. 2019;7:84. doi: 10.3389/fcell.2019.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niu M, Dai X, Zou W, Yu X, Teng W, Chen Q, et al. Autophagy, endoplasmic reticulum stress and the unfolded protein response in intracerebral hemorrhage. Transl Neurosci. 2017;8:37–48. doi: 10.1515/tnsci-2017-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herrmann AG, Deighton RF, Le Bihan T, McCulloch MC, Searcy JL, Kerr LE, et al. Adaptive changes in the neuronal proteome: Mitochondrial energy production, endoplasmic reticulum stress, and ribosomal dysfunction in the cellular response to metabolic stress. J Cereb Blood Flow Metab. 2013;33:673–83. doi: 10.1038/jcbfm.2012.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xin Q, Ji B, Cheng B, Wang C, Liu H, Chen X, et al. Endoplasmic reticulum stress in cerebral ischemia. Neurochem Int. 2014;68:18–27. doi: 10.1016/j.neuint.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 36.Santo EE, Paik J. FOXO in neural cells and diseases of the nervous system. Curr Top Dev Biol. 2018;127:105–18. doi: 10.1016/bs.ctdb.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown AK, Webb AE. Regulation of FOXO factors in mammalian cells. Curr Top Dev Biol. 2018;127:165–92. doi: 10.1016/bs.ctdb.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hedrick SM. The cunning little vixen: Foxo and the cycle of life and death. Nat Immunol. 2009;10:1057–63. doi: 10.1038/ni.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pereira ER, Frudd K, Awad W, Hendershot LM. Endoplasmic reticulum (ER) stress and hypoxia response pathways interact to potentiate hypoxia-inducible factor 1 (HIF-1) transcriptional activity on targets like vascular endothelial growth factor (VEGF) J Biol Chem. 2014;289:3352–64. doi: 10.1074/jbc.M113.507194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature. 2014;508:103–7. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang K, Guo X, Yan H, Wu Y, Pan Q, Shen JZ, et al. Phosphorylation of forkhead protein FoxO1 at S253 regulates glucose homeostasis in mice. Endocrinology. 2019;160:1333–47. doi: 10.1210/en.2018-00853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vander Kooi BT, Streeper RS, Svitek CA, Oeser JK, Powell DR, O’Brien RM. The three insulin response sequences in the glucose-6-phosphatase catalytic subunit gene promoter are functionally distinct. J Biol Chem. 2003;278:11782–93. doi: 10.1074/jbc.M212570200. [DOI] [PubMed] [Google Scholar]
- 43.Choi WI, Yoon JH, Song JY, Jeon BN, Park JM, Koh DI, et al. Zbtb7c is a critical gluconeogenic transcription factor that induces glucose-6-phosphatase and phosphoenylpyruvate carboxykinase 1 genes expression during mice fasting. Biochim Biophys Acta Gene Regul Mech. 2019;1862:643–56. doi: 10.1016/j.bbagrm.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 44.Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest. 2001;108:1359–67. doi: 10.1172/JCI12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou Y, Gong N, Cui Y, Wang X, Cui A, Chen Q, et al. Forkhead Box P1 (FOXP1) transcription factor regulates hepatic glucose homeostasis. J Biol Chem. 2015;290:30607–15. doi: 10.1074/jbc.M115.681627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–5. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 47.Froehler MT, Ovbiagele B. Therapeutic hypothermia for acute ischemic stroke. Expert Rev Cardiovasc Ther. 2010;8:593–603. doi: 10.1586/erc.09.129. [DOI] [PubMed] [Google Scholar]
- 48.Zhao QJ, Zhang XG, Wang LX. Mild hypothermia therapy reduces blood glucose and lactate and improves neurologic outcomes in patients with severe traumatic brain injury. J Crit Care. 2011;26:311–5. doi: 10.1016/j.jcrc.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 49.Forreider B, Pozivilko D, Kawaji Q, Geng X, Ding Y. Hibernation-like neuroprotection in stroke by attenuating brain metabolic dysfunction. Prog Neurobiol. 2017;157:174–87. doi: 10.1016/j.pneurobio.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 50.Zhong W, Yuan Y, Gu X, Kim SI, Chin R, Loye M, et al. Neuropsychological deficits chronically developed after focal ischemic stroke and beneficial effects of pharmacological hypothermia in the mouse. Aging Dis. 2020;11:1–16. doi: 10.14336/AD.2019.0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang M, Wang H, Zhao J, Chen C, Leak RK, Xu Y, et al. Drug-induced hypothermia in stroke models: Does it always protect? CNS Neurol Disord Drug Targets. 2013;12:371–80. doi: 10.2174/1871527311312030010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gavva NR, Bannon AW, Surapaneni S, Hovland DN, Jr, Lehto SG, Gore A, et al. The vanilloid receptor TRPV1 is tonically activated in vivo and involved in body temperature regulation. J Neurosci. 2007;27:3366–74. doi: 10.1523/JNEUROSCI.4833-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature. 1997;389:816–24. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 54.Fosgerau K, Weber UJ, Gotfredsen JW, Jayatissa M, Buus C, Kristensen NB, et al. Drug-induced mild therapeutic hypothermia obtained by administration of a transient receptor potential vanilloid type 1 agonist. BMC Cardiovasc Disord. 2010;10:51. doi: 10.1186/1471-2261-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cao Z, Balasubramanian A, Marrelli SP. Pharmacologically induced hypothermia via TRPV1 channel agonism provides neuroprotection following ischemic stroke when initiated 90 min after reperfusion. Am J Physiol Regul Integr Comp Physiol. 2014;306:R149–56. doi: 10.1152/ajpregu.00329.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shirakawa H, Yamaoka T, Sanpei K, Sasaoka H, Nakagawa T, Kaneko S. TRPV1 stimulation triggers apoptotic cell death of rat cortical neurons. Biochem Biophys Res Commun. 2008;377:1211–5. doi: 10.1016/j.bbrc.2008.10.152. [DOI] [PubMed] [Google Scholar]
- 57.Han Y, Geng XK, Lee H, Li F, Ding Y. Neuroprotective effects of early hypothermia induced by phenothiazines and DHC in ischemic stroke. Evid Based Complement Alternat Med. 2021;2021:1207092. doi: 10.1155/2021/1207092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guan L, Lee H, Geng X, Li F, Shen J, Ji Y, et al. Neuroprotective effects of pharmacological hypothermia on hyperglycolysis and gluconeogenesis in ischemic rats. Basel, MDPI: Biomolecules; 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burn JH. The pharmacology of chlorpromazine and promethazine. Proc R Soc Med. 1954;47:617–21. doi: 10.1177/003591575404700801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coté CJ, Karl HW, Notterman DA, Weinberg JA, McCloskey C. Adverse sedation events in pediatrics: Analysis of medications used for sedation. Pediatrics. 2000;106:633–44. doi: 10.1542/peds.106.4.633. [DOI] [PubMed] [Google Scholar]
- 61.López-Muñoz F, Alamo C, Cuenca E, Shen WW, Clervoy P, Rubio G. History of the discovery and clinical introduction of chlorpromazine. Ann Clin Psychiatry. 2005;17:113–35. doi: 10.1080/10401230591002002. [DOI] [PubMed] [Google Scholar]
- 62.MacMillan V. Effects of promethazine on the energy metabolism of normoxic and hypoxic rat brain. Stroke. 1982;13:464–9. doi: 10.1161/01.str.13.4.464. [DOI] [PubMed] [Google Scholar]
- 63.Krausz Y, Eylon L, Cerasi E. Calcium-binding proteins and insulin release. Differential effects of phenothiazines on first- and second-phase secretion and on islet cAMP response to glucose. Acta Endocrinol (Copenh) 1987;116:241–6. [PubMed] [Google Scholar]
- 64.Skinner A, Spector RG. The effect of chlorpromazine on 14-C-glucose metabolism in mouse liver and brain. Br J Pharmacol Chemother. 1968;33:129–35. doi: 10.1111/j.1476-5381.1968.tb00480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bachelard HS, Lindsay JR. Effects of neurotropic drugs on glucose metabolism in rat brain in vivo. Biochem Pharmacol. 1966;15:1053–8. doi: 10.1016/0006-2952(66)90270-x. [DOI] [PubMed] [Google Scholar]
- 66.Bachelard HS, Gaitonde MK, Vrba R. The effect of psychotropic drugs on the utilization of glucose carbon atoms in the brain, heart and liver of the rat. Biochem Pharmacol. 1966;15:1039–43. doi: 10.1016/0006-2952(66)90268-1. [DOI] [PubMed] [Google Scholar]
- 67.Berntman L, Carlsson C. Influence of “lytic cocktail” on blood flow and oxygen consumption in the rat brain. Acta Anaesthesiol Scand. 1978;22:515–8. doi: 10.1111/j.1399-6576.1978.tb01332.x. [DOI] [PubMed] [Google Scholar]
- 68.Smith DS, Rehncrona S, Siesjö BK. Inhibitory effects of different barbiturates on lipid peroxidation in brain tissue in vitro: Comparison with the effects of promethazine and chlorpromazine. Anesthesiology. 1980;53:186–94. doi: 10.1097/00000542-198009000-00002. [DOI] [PubMed] [Google Scholar]
- 69.Saver JL. Time is brain – Quantified. Stroke. 2006;37:263–6. doi: 10.1161/01.STR.0000196957.55928.ab. [DOI] [PubMed] [Google Scholar]
- 70.Paciaroni M, Caso V, Agnelli G. The concept of ischemic penumbra in acute stroke and therapeutic opportunities. Eur Neurol. 2009;61:321–30. doi: 10.1159/000210544. [DOI] [PubMed] [Google Scholar]
- 71.Chandra A, Stone CR, Du X, Li WA, Huber M, Bremer R, et al. The cerebral circulation and cerebrovascular disease III: Stroke. Brain Circ. 2017;3:66–77. doi: 10.4103/bc.bc_12_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Voorhees WD, 3rd, Abendschein DR, Tacker WA., Jr Effect of whole-body hypothermia on myocardial blood flow and infarct salvage during coronary artery occlusion in dogs. Am Heart J. 1984;107:945–9. doi: 10.1016/0002-8703(84)90833-0. [DOI] [PubMed] [Google Scholar]
- 73.Dixon SR, Whitbourn RJ, Dae MW, Grube E, Sherman W, Schaer GL, et al. Induction of mild systemic hypothermia with endovascular cooling during primary percutaneous coronary intervention for acute myocardial infarction. J Am Coll Cardiol. 2002;40:1928–34. doi: 10.1016/s0735-1097(02)02567-6. [DOI] [PubMed] [Google Scholar]
- 74.Stub D, Bernard S, Pellegrino V, Smith K, Walker T, Sheldrake J, et al. Refractory cardiac arrest treated with mechanical CPR, hypothermia, ECMO and early reperfusion (the CHEER trial) Resuscitation. 2015;86:88–94. doi: 10.1016/j.resuscitation.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 75.Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557–63. doi: 10.1056/NEJMoa003289. [DOI] [PubMed] [Google Scholar]
- 76.Han Y, Rajah GB, Hussain M, Geng X. Clinical potential of pre-reperfusion hypothermia in ischemic injury. Neurol Res. 2019;41:697–703. doi: 10.1080/01616412.2019.1609160. [DOI] [PubMed] [Google Scholar]
- 77.Heiss WD. The ischemic penumbra: Correlates in imaging and implications for treatment of ischemic stroke. The Johann Jacob Wepfer award 2011. Cerebrovasc Dis. 2011;32:307–20. doi: 10.1159/000330462. [DOI] [PubMed] [Google Scholar]