

Abstract
BACKGROUND AND AIMS:
Cholestasis is characterized by increased total bile acid (TBA) levels, which are regulated by farnesoid X receptor (FXR)/FGF15. Patients with primary sclerosing cholangitis (PSC) typically present with inflammatory bowel disease (IBD). Mast cells (MCs) (i) express FXR and (ii) infiltrate the liver during cholestasis promoting liver fibrosis. In bile-duct-ligated (BDL) MC-deficient mice (B6. Cg-KitW-sh/HNihrJaeBsmJ [KitW-sh]), ductular reaction (DR) and liver fibrosis decrease compared with BDL wild type, and MC injection exacerbates liver damage in normal mice.
APPROACH AND RESULTS:
In this study, we demonstrated that MC-FXR regulates biliary FXR/FGF15, DR, and hepatic fibrosis and alters intestinal FXR/FGF15. We found increased MC number and biliary FXR expression in patients with liver injury compared with control. Histamine and FGF19 serum levels and small heterodimer partner expression increase in patients PSC and PSC-IBD compared with healthy controls. MC injection increased liver damage, DR, inflammation, biliary senescence/senescence-associated secretory phenotype (SASP), fibrosis, and histamine in KitW-sh mice. Inhibition of MC-FXR before injection reduced these parameters. BDL and KitW-sh mice injected with MCs displayed increased TBA content, biliary FXR/FGF15, and intestinal inflammation, which decreased in BDL KitW-sh and KitW-sh mice injected with MC-FXR. MCs increased ileal FXR/FGF15 expression in KitW-sh mice that was reduced following FXR inhibition. BDL and multidrug resistance 2/ATP-binding cassette family 2 member 4 knockout (Mdr2−/−) mice, models of PSC, displayed increased intestinal MC infiltration and FXR/FGF15 expression. These were reduced following MC stabilization with cromolyn sodium in Mdr2−/− mice. In vitro, MC-FXR inhibition decreased biliary proliferation/SASP/FGF and hepatic stellate cell activation.
CONCLUSIONS:
Our studies demonstrate that MC-FXR plays a key role in liver damage and DR, including TBA regulation through alteration of intestinal and biliary FXR/FGF15 signaling.
Cholangiocytes are the target cells of cholestatic liver diseases, such as primary sclerosing cholangitis (PSC), which is characterized by increased hepatic fibrosis, ductular reaction (DR)(1,2) and impaired bile acid (BA) secretion.(3,4) A significant subset of patients with PSC present with inflammatory bowel disease (IBD), which increases their risk and incidence of colorectal and hepatobiliary cancer.(5) During PSC, cholangiocytes exhibit proliferative, inflammatory, fibrotic, and senescent phenotypes in response to liver injury, and several studies have demonstrated the critical contribution of the gut-liver axis during PSC.(6)
We have demonstrated that mast cells (MCs) infiltrate the liver during cholestatic liver injury and trigger biliary damage.(1) Following migration and activation, MCs induce DR(7) and senescence(1) through paracrine interactions with cholangiocytes. The PSC mouse models, multidrug resistance 2/ATP-binding cassette family 2 member 4 knockout (Mdr2−/−) and mice subjected to bile duct ligation (BDL), have elevated MC presence and serum histamine (HA) levels, increasing biliary expression of H1 and H2 HA receptors (HR); however, disruption of HA signaling ameliorates cholestatic liver injury.(8–10) Following BDL, MC-deficient B6.Cg-KitW-sh/HNihrJaeBsmJ (KitW-sh) mice have reduced DR, inflammation, and hepatic fibrosis compared with BDL wild type (WT), and introduction of MCs into KitW-sh mice mimics cholestatic injury,(10) demonstrating the damaging role of MCs during liver injury. Moreover, MCs induce DR through TGF-β1(11) and VEGF(8) signaling pathways.
BA synthesis and enterohepatic circulation is tightly regulated by farnesoid X receptor (FXR) and downstream targets including FGF15/FGF19 (FGF15, mouse; FGF19, human) and cytochrome p450 (Cyp) family 7 and 27 subfamily a member 1 (Cyp7a1 and Cyp27a1).(12) FXR signaling regulates the expression of many BA transporters such as Na+-dependent taurocholate cotransport peptide and apical Na+ BA transporter (ASBT).(13) Furthermore, FXR plays a protective role in intrahepatic cholestasis by antagonizing NF-κB–mediated hepatic inflammation(12,14) while increasing liver injury in extrahepatic cholestasis through dysregulation of BA transporter expression.(15) Fxr−/− mice have decreased intrahepatic bile duct mass (IBDM) and biliary proliferation following BDL, likely because of the reduced bile flow and pressure, compared with WT mice.(16) Impairment of FGF15 signaling inhibits liver regeneration and exacerbates hepatosteatosis,(17) lending to its importance in hepatic function.
Our studies found that stabilization of MCs in Mdr2−/− mice reduces total BA (TBA) levels,(7) and we, among others, have shown that MCs express FXR, ASBT and FGF receptors(18,19) and migrate into tissue in response to FGF stimulation.(20) In this study, we evaluated the role of MC-FXR in regulation of BA circulation, intestinal inflammation and liver damage in models of cholestasis.
Materials and Methods
MATERIALS
All reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless indicated otherwise. Z-guggulsterone (FXR antagonist)(21) was purchased from MilliporeSigma (Burlington, MA). Mouse pan Fibroblast growth factor (FGF) enzyme immunoassay (EIA) was purchased from R&D Systems (Minneapolis, MN). Mouse FGF15 EIA was purchased from My BioSource, Inc. (San Diego, CA) and LifeSpan Biosciences, Inc. (Seattle, WA). FGF19 EIA was purchased from Abcam (Cambrige, MA). HA EIA was purchased from Cayman Chemicals (Ann Arbor, MI). Application, dilution, and vendor information for all antibodies used in this study are detailed in Supporting Table S1. RNA was reverse transcribed and amplified using Reaction Ready First Strand cDNA Synthesis kit and RT2 quantitative PCR (qPCR) Primer Assay obtained from Qiagen (Valencia, CA). All qPCR primers are listed in Supporting Table S2. TBA colorimetric kits were purchased from Cell BioLabs, Inc. (San Diego, CA). Positive immunoreactivity was quantified with Image-Pro from Media Cybernetics, Inc. (Rockville, MD).
For in vitro studies, we used immortalized murine intrahepatic cholangiocyte cell lines.(22) Murine hepatic MCs (MC/9, American Type Culture Collection [ATCC] CRL-8306) were purchased from ATCC (Manassas, VA). Human HSC (#5300) were obtained from ScienCell (Carlsbad, CA). All cell lines were cultured and maintained by us following vendor recommendations and as published.(8) Media and reagents were purchased from Thermo Fisher (Waltham, MA).
IN VIVO MODELS
Commercially available homozygous KitW-sh (MC-deficient) 10 to 12-week-old male mice were obtained from Jackson Laboratory along with sex and age-matched WT c57BL/6J mice. WT and KitW-sh mice both display healthy liver phenotype, and WT mice have minimal MC presence.(10) To ascertain the role of MC-FXR in MC-induced damage, KitW-sh received a single injection through tail vein of MCs (MC/9, ATCC CRL-8306, 5 × 106 cells/0.1–0.2 mL sterile 1× PBS) treated with vehicle 0.1% DMSO (MC) or 10 μM Z-guggulsterone (FXR inhibitor, MC-Gugg)(21,23) for 48 hours and tagged with PKH26 Red Fluorescent Cell Linker before injection. Liver (costained with cytokeratin 19 [CK-19] to mark bile ducts), lung, and spleen were evaluated for MC presence by PKH26 detection with confocal microscopy (LEICA TCS SP5 X system, Leica Microsystems, Inc., Buffalo Grove, IL). To confirm FXR inhibition, MC-FXR, FGF15, and small heterodimer partner (SHP) expression and FGF secretion were measured by qPCR and EIA, respectively. Serum, liver, isolated cholangiocytes, small intestine, and distal ileum were collected from all animal groups (exact animal numbers are provided in the Supporting Information). Isolated cholangiocytes were collected as described.(7) Immunohistochemistry was performed on 4 to 6-μm formalin-fixed paraffin-embedded liver or small intestine sections. Immunofluorescence was performed on 4 to 6-μm optical cutting media-embedded liver, lung, or spleen.
To verify our findings in established models of cholestatic liver injury, male WT and KitW-sh mice, 10 to 12-week-old, were subjected to BDL for 7 days before euthanasia along with appropriate controls as described.(10) Additionally, Mdr2−/− mice, an established genetic model of cholestatic liver damage, treated with saline or cromolyn sodium (MC stabilizer, 24 mg/kg BW for 1 week) were used to confirm our findings.(7) Serum, liver, small intestine, and isolated cholangiocytes and hepatocytes were collected from all mice (exact animal numbers are provided in the Supporting Information).
All animal colonies were maintained following current Institutional Animal Care and Use Committee protocols approved by the Indiana University School of Medicine Laboratory Animal Resource Center, Indianapolis, IN, and Baylor Scott and White Health Animal Facility, Temple, TX. All mice were given free access to drinking water and standard chow. Animals were kept in a temperature-controlled environment with 12:12 hr light/dark cycles.
FXR/FGF19 SIGNALING AND MC ACTIVATION IN HUMAN PATIENTS
Human liver sections from healthy controls and patients with cholestatic liver diseases were used for immunohistochemistry for FXR costained with tryptase to determine the presence of activated MCs. Healthy nondiseased liver tissues were purchased from Sekisui XenoTech, LLC (Kansas City, KS). Healthy nondiseased, PSC and PSC with IBD comorbidity (PSC-IBD) serum were deidentified and provided by Dr. Burcin Ekser following transplantation. Liver sections (formalin-fixed, 4 to 5 μm thick) obtained by explant from deidentified transplant patients from patients with PSC, PSC-IBD, primary biliary cholangitis (PBC), biliary atresia, and NASH were provided by Dr. Burcin Ekser. Patient demographics are detailed in Supporting Table S3. All samples were obtained under a protocol approved by Indiana University Health; the protocol was approved by the Indiana University Institutional Review Board. Serum FGF19 (Abcam, Cambrige, MA) and HA (Cayman Chemicals, Inc., Ann Arbor, MI) levels were measured by EIA following manufacturer protocols, and levels of SHP were evaluated in total liver by qPCR from healthy nondiseased (control), PSC, and PSC-IBD samples. For all patient sample analysis, written informed consent was obtained from each patient and the study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki as described and approved by the Institutional Review Board, Indiana University. No donor organs were obtained from executed prisoners or other institutionalized persons.
Further detailed information for experiments performed in vivo and in vitro are detailed in the Supporting Methods.
STATISTICAL ANALYSIS
Data are presented as mean ± SEM. Groups were analyzed by the Student’s unpaired t test when two groups were analyzed. Welch one-way ANOVA was used when more than two groups are analyzed, followed by appropriate post hoc test with GraphPad Prism 9 (San Diego, CA). P < 0.05 was considered significant.
Results
PATIENTS WITH CHOLESTATIC LIVER DISEASE HAVE INCREASED MC PRESENCE AND FXR EXPRESSION
FXR agonists are currently being studied as therapeutic options for patients with cholestatic liver disease(4,12); however, the effect of FXR on hepatic MC presence has not been defined. In patients, FXR expression (brown; found in hepatocytes and cholangiocytes) increased in cholangiocytes from late-stage PSC, PSC-IBD, late-stage PBC, biliary atresia, and NASH compared with control tissue, and the up-regulation of FXR is accompanied by elevated expression of MC tryptase (red staining) found near bile ducts (Fig. 1A). Further, serum levels of FGF19 (Fig. 1B) and mRNA expression of SHP (Fig. 1C) increased in patients with PSC-IBD compared with controls. These results support the clinical implications of MC infiltration during human cholestatic liver disease and the up-regulation of FXR expression and activation of downstream signaling during the progression of chronic liver injuries, including PSC-IBD.
FIG. 1.
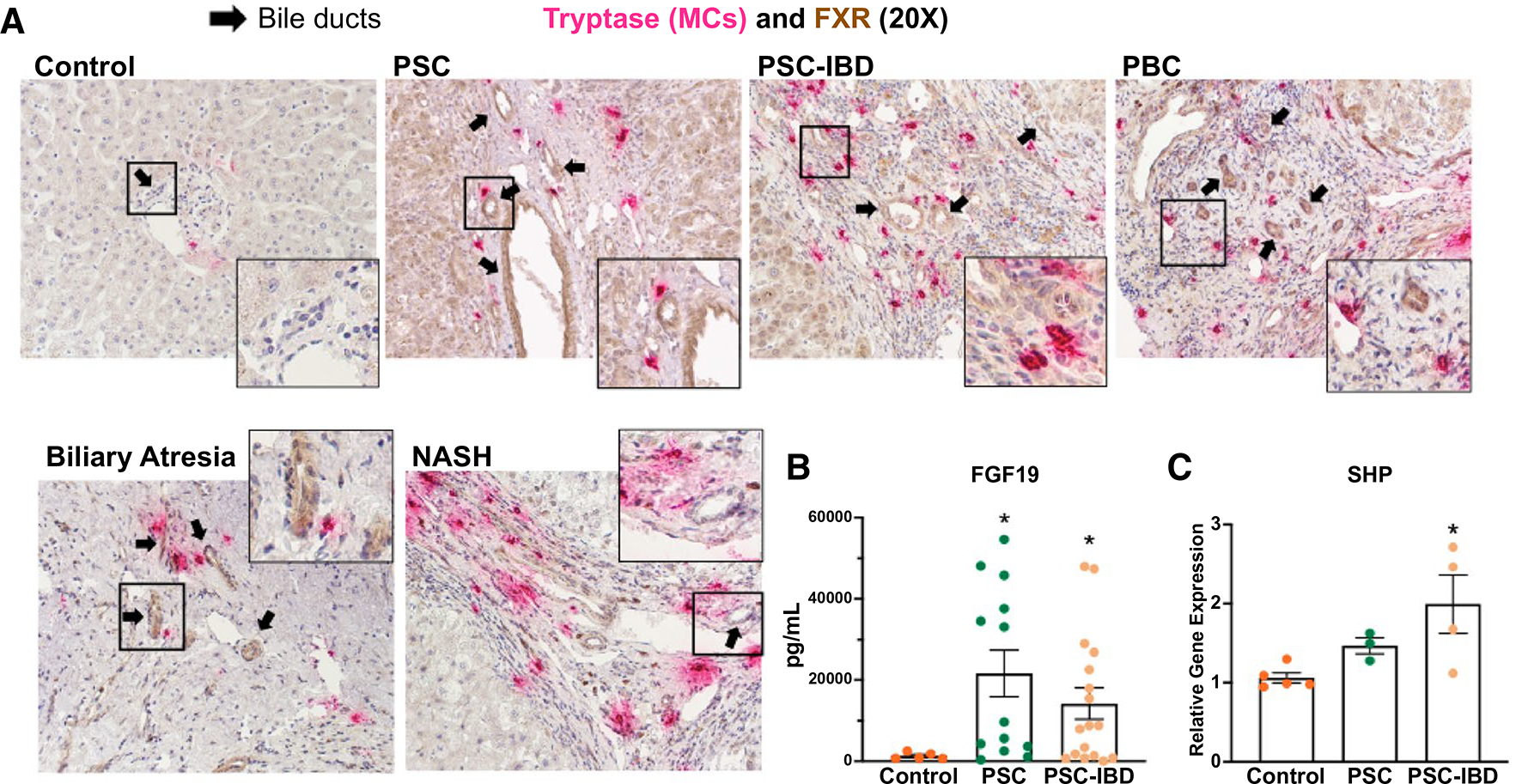
(A) Hepatic FXR expression and MC presence increase in human liver disease explant patients. Livers from nondiseased control, PSC, PSC-IBD, PBC, biliary atresia, and NASH were stained using immunohistochemistry for FXR (brown), costained with tryptase (red) to detect activated MC presence. Enhanced biliary FXR immunoreactivity (marked by black arrows) is present in all diseased tissue explants compared with control, with a subset (PSC, PSC-IBD, and PBC) demonstrating increased hepatocyte FXR expression, indicating activated hepatic FXR function. Tryptase expression also increased in all models of disease compared with control. (B) Patients with PSC (n = 14) and PSC-IBD (n = 17) have increased serum FGF19 compared with healthy controls (n = 5). (C) Patients with PSC (n = 3) and PSC-IBD (n = 4) also have increased hepatic SHP expression compared with controls (n = 5). Data are mean ± SEM of n = 2 (serum) or n = 3 (qPCR) experiments per patient sample. *P < 0.05 vs. control. Representative images are presented as ×20 and zoom boxes are ×80.
TBA CONTENT AND HEPATIC FXR/FGF SIGNALING ARE DECREASED IN CHOLESTATIC MODELS LACKING MCS OR HA SIGNALING
To establish a rationale for our study, we examined changes in TBA content and FXR/FGF signaling in established cholestatic models, including BDL and Mdr2−/− mice. With regard to TBA content, we found that BDL WT mice had elevated serum TBA content compared with WT mice; however, BDL KitW-sh mice had reduced serum TBA content compared with BDL WT mice (Fig. 2A). BDL WT mice have significantly increased hepatic FXR mRNA expression compared with WT mice (Fig. 2B), whereas BDL KitW-sh mice have lower hepatic FXR expression compared with both WT and BDL WT mice, indicating that MCs are involved in the regulation of the hepatic FXR expression. Furthermore, we found that panFGF serum secretion was minimal in WT and KitW-sh mice (Fig. 2C); however, panFGF serum levels increased in BDL WT mice compared with WT mice, whereas BDL KitW-sh mice displayed decreased panFGF serum secretion compared with BDL WT (Fig. 2C). Finally, we verified increased FGF15 expression and immunoreactivity by immunofluorescence in bile ducts of BDL WT mice compared with WT mice, which was reduced in BDL KitW-sh mice (Fig. 2D). In Mdr2−/− mice, we found increased FXR expression (Supporting Fig. S1A), biliary FGF15 immunoreactivity (Supporting Fig. S1B), and panFGF serum content (Supporting Fig. S1C) that were reduced in Mdr2−/− mice treated with cromolyn sodium.
FIG. 2.
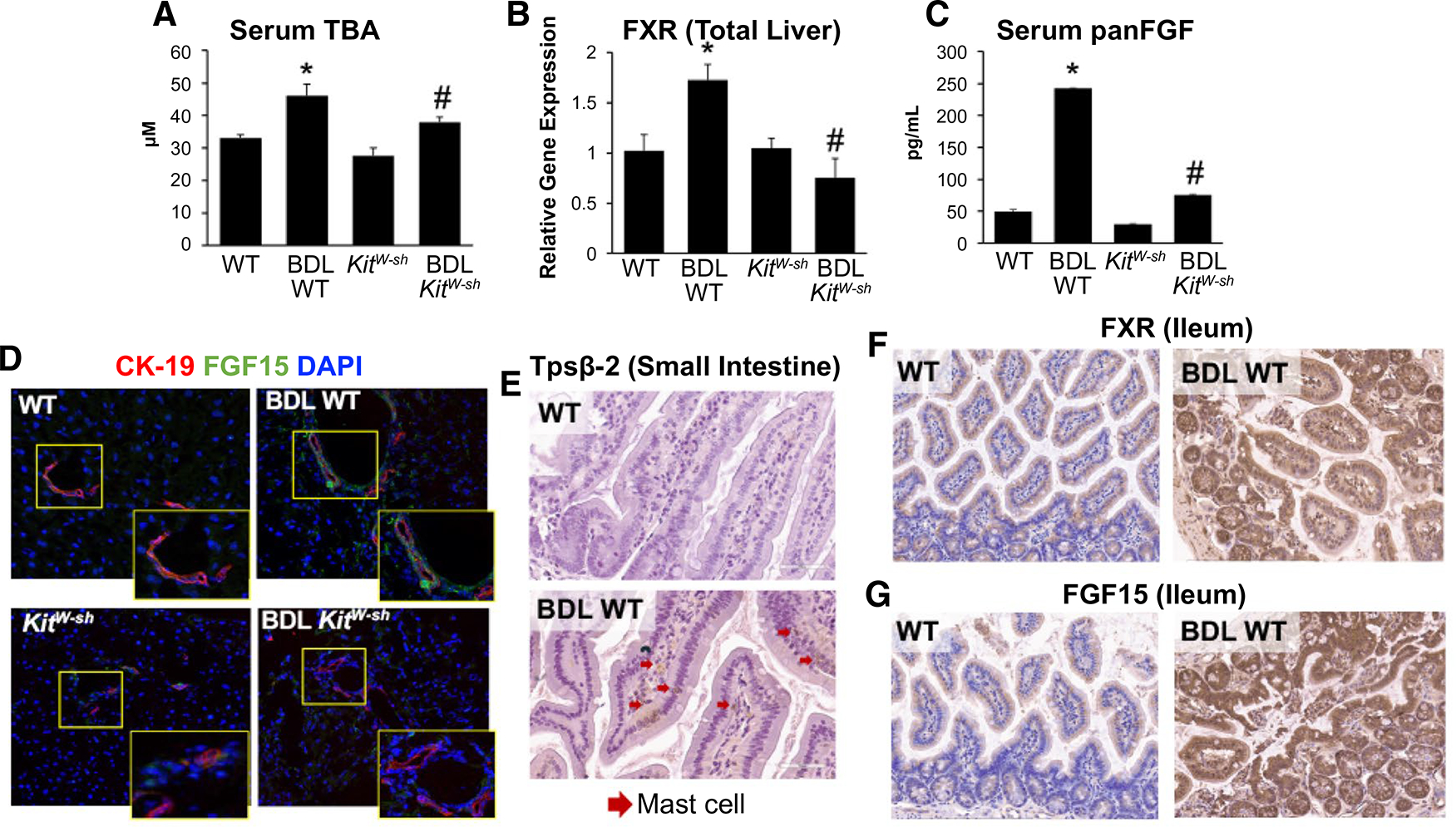
(A) MCs alter TBA and FXR/FGF signaling in an established model of cholestatic MC-deficient mice. Serum TBA content is increased in BDL WT compared with WT mice, but in BDL KitW-sh mice, TBA content is reduced. Serum TBA was unchanged between control groups. Hepatic FXR is unchanged in WT and KitW-sh mice; however, expression is elevated in BDL WT mice compared with WT and KitW-sh. (B) BDL KitW-sh mice exhibited reduced hepatic FXR expression compared with BDL WT mice. (C) BDL WT mice have elevated serum panFGF secretion compared with WT and KitW-sh, which is reduced in BDL KitW-sh mice; no significant changes were found between control groups. (D) Biliary FGF15 (green) expression was elevated in BDL WT mice shown by immunofluorescence, costained with CK-19 to mark bile ducts (red) but reduced in BDL KitW-sh mice with no changes between control groups. (E) In BDL WT small intestine, there is increased MC presence detected by tryptase β−2 (Tpsβ−2) immunoreactivity and marked by red arrows. In distal ileum from BDL WT there was increased (F) FXR and (G) FGF15 immunoreactivity compared with WT mice. Data are mean ± SEM of n = 4 experiments for qPCR, panFGF EIA, and TBA from 4–6 mice per group. *P < 0.05 vs. WT; #P < 0.05 vs. BDL WT. Representative images are presented as ×20 for FXR and ×40 for FGF15 and Tpsβ−2.
MC INFILTRATION ALTERS INTESTINAL FXR/FGF15 IN CHOLESTATIC MICE
We found that BDL WT and Mdr2−/− mice have increased intestinal MC infiltration compared with their WT controls shown by tryptase β−2 (Fig. 2E and Supporting Fig. S1E). Along with increased MC presence, we found elevated expression of FXR and FGF15 in distal ileum from BDL WT mice compared with their WT controls (Fig. 2F,G). When Mdr2−/− mice were treated with cromolyn sodium to reduce MC activation, intestinal MC infiltration, FXR, and FGF15 expression were all reduced (Supporting Fig. S1D–F). In distal ileum from BDL WT, we also found increased mRNA expression of FGF15 and SHP, whereas ASBT decreased compared with WT mice (Supporting Fig. S2A), and, in small intestine, stem cell factor (SCF), IL-1β, TGF-β1, and chemokine (C-C motif) ligand (CCL) 3 mRNA expression are increased following BDL compared with WT mice. These parameters were decreased in BDL KitW-sh mice lacking MCs (Supporting Fig. S2B)
VALIDATION OF MC-FXR/FGF EXPRESSION AND MC INJECTION/MIGRATION AND DAMAGE OF MODEL
Cultured murine MCs were treated with vehicle (MC) or Z-guggulsterone (MC-Gugg), tagged with PKH26, and injected into KitW-sh mice through tail vein. MC-Gugg have decreased FXR FGF15 and SHP (P < 0.07) mRNA expression (Fig. 3A–C) and panFGF secretion (Fig. 3D) compared with vehicle-treated MCs. Similar to our previous findings,(10) we found that MCs (marked with PKH26, red) migrate to the liver and reside in close proximity to bile ducts (stained with CK-19, green) (Fig. 3E,F). Concurrent with our previous work, we found minimal MC presence in lung and spleen from our MC-Gugg–injected mice (data not shown).(10) By hematoxylin and eosin staining, we verified that MC injection induces periportal inflammation and lobular damage that is ameliorated in KitW-sh mice treated with MC-Gugg (Fig. 3G).
FIG. 3.
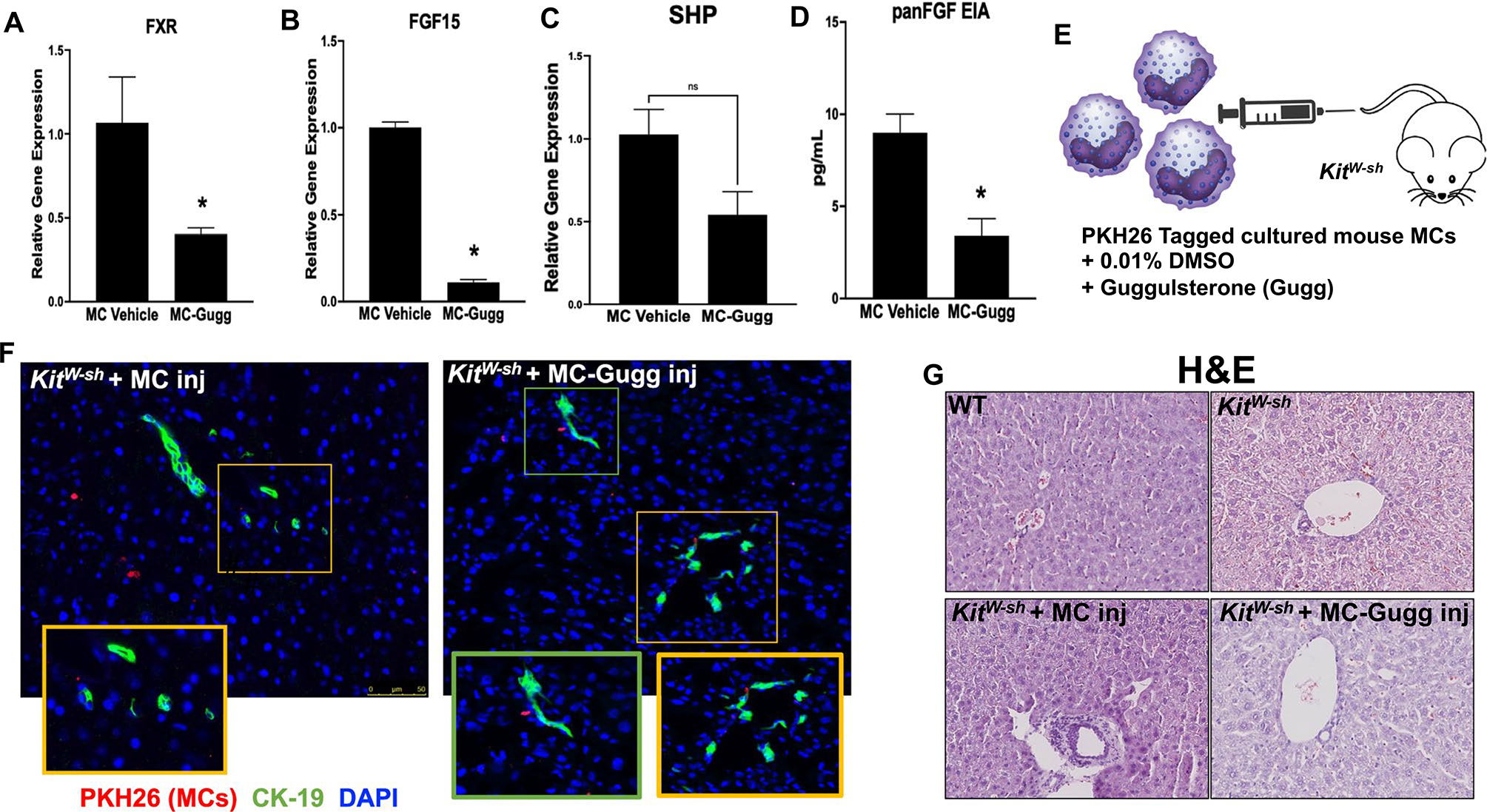
Validation of model and hepatic damage in KitW-sh mice. Cultured MCs (MC/9, ATCC CRL-8306) were stimulated with 0.1% DMSO (vehicle) or 10 μM Z-guggulsterone for 48 hours and (A) FXR, (B) FGF15, (C) SHP mRNA expression, and (D) panFGF secretion were measured. Following treatment, MC-Gugg have reduced (A-C) FXR, FGF15, and SHP mRNA levels, measured by qPCR, and (D) panFGF secretion, measured by EIA, compared with vehicle-treated MCs. (E) Cultured MCs were treated with 0.1% DMSO or guggulsterone (Gugg) and tagged with PKH26 before injection into KitW-sh mice through tail vein. MC migration to the liver was confirmed in KitW-sh mice injected with both (F) vehicle-treated MCs and Z-guggulsterone–treated MCs (MC-Gugg) by immunofluorescence imaging of PKH26 (red) and CK-19 to mark bile ducts (green). (G) Hematoxylin and eosin (H&E) staining demonstrates increased hepatic damage and ductular inflammation in KitW-sh + MC that is absent in WT and KitW-sh mice. Hepatic damage is reduced in KitW-sh + MC-Gugg mice. Data are mean ± SEM of n = 4 experiments from n = 2 biological replicates for qPCR and panFGF EIA. *P < 0.05 vs. MC-vehicle. Representative images are presented as ×20 for immunofluorescence, with ×40 zoom boxes, and ×10 for H&E. Abbreviation: Inj, injury.
MCS ALTER ENTEROHEPATIC FXR/FGF AXIS AND INTESTINAL INFLAMMATION
In KitW-sh mice injected with MCs, compared with WT, there was increased (1) liver and serum TBA content (Fig. 4A,B) and (2) hepatic and biliary FXR expression (Fig. 4C,D) that was reduced in KitW-sh mice injected with MC-Gugg. Downstream of FXR, hepatocyte Cyp7a1 expression also increased in KitW-sh mice injected with MCs; however, hepatocyte Cyp27a1 was reduced (Fig. 4E) compared with WT, and when KitW-sh mice were treated with MC-FXR, Cyp7a1 was reduced, whereas Cyp27a1 remained unchanged (Fig. 4E). Immunoreactivity of FGF15 (Fig. 4G) and liver and serum FGF15 secretion (Fig. 4H,I) all increased in KitW-sh mice injected with MCs compared with WT and KitW-sh mice. All of these parameters were decreased when KitW-sh mice were injected with MC-Gugg (Fig. 4G–I). There were minimal changes seen between WT and KitW-sh mice for TBA levels and FXR and FGF15 expression.
FIG. 4.
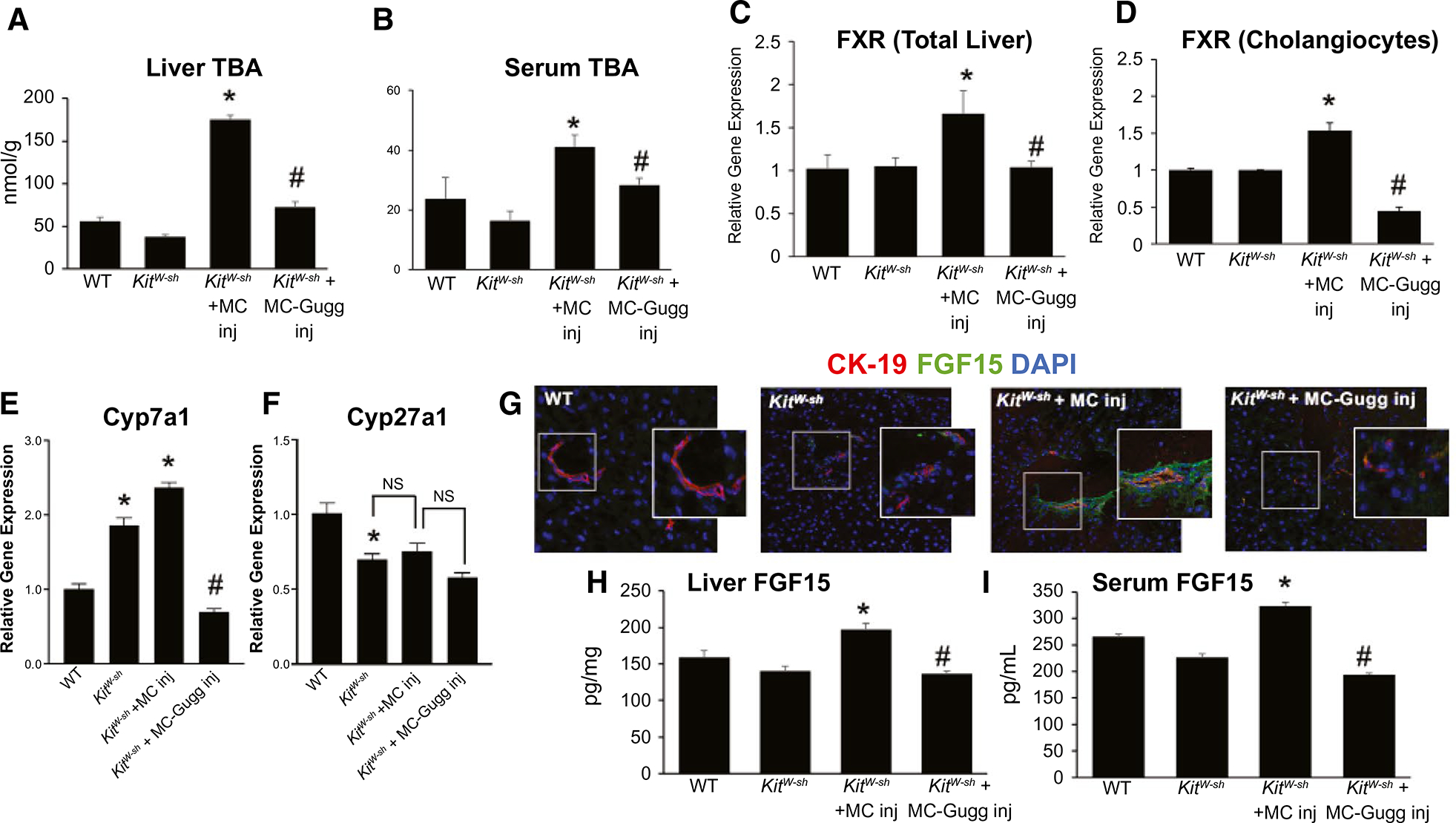
MC-FXR regulates hepatic and biliary FXR/FGF signaling. Introduction of MCs in KitW-sh mice increased (A) hepatic and (B) circulating TBA content; however, both TBA levels were reduced with MC-FXR inhibition. (A,B) No significant changes were noted between WT and KitW-sh groups. (C) Hepatic and (D) biliary FXR expression increased in KitW-sh mice injected with MCs compared with WT and KitW-sh mice, and inhibition of MC-FXR reduces both hepatic and biliary FXR expression in KitW-sh + MC-Gugg mice. The expression of (E) hepatocyte Cyp7a1 increased in both control KitW-sh mice and KitW-sh mice injected with MCs compared with WT, whereas (F) hepatocyte Cyp27a1 decreased in all groups compared with WT. (E,F) In KitW-sh mice injected with MC-Gugg, Cyp7a1 expression decreased and Cyp27a1 levels remained unchanged compared with control. (G) Biliary FGF15 (green) expression, costained with CK-19 to mark bile ducts (red), and (H) hepatic and (I) serum FGF15 secretion increased in KitW-sh mice injected with MCs, that was subsequently reduced in mice injected with MC-Gugg (G-I). Minimal changes were noted between control groups (G-I). Data are mean ± SEM of n = 8 experiments for qPCR and n = 4 experiments for FGF15 and panFGF EIA from 6–8 mice and n = 4 experiments from 6–8 mice per group for serum and liver TBA. *P < 0.05 vs. WT, #P < 0.05 vs. KitW-sh + MC. Representative images are presented as ×20 with ×40 zoom boxes. Abbreviation: Inj, injury.
Interestingly, in accordance with our hepatic findings, distal ileum FXR and FGF15 expressions are minimal in WT and elevated in KitW-sh mice (Fig. 5A,B). MC injection into KitW-sh mice increased intestinal FXR and FGF15 expression (Fig. 5A,B), which was reduced when KitW-sh mice were injected with MCs lacking FXR (Fig. 5A,B), demonstrating an impact of MC-FXR on hepatic and intestinal FXR/FGF15 signaling in MC-deficient mice. We found that small intestine mRNA levels of SCF, IL-1β, TGF-β1, and CCL3 were elevated following MC injection in KitW-sh compared with WT and KitW-sh mice (Fig. 5C). These inflammatory markers decreased when FXR was inhibited in MCs before injection (Fig. 5C). These findings indicate an important role for MCs in both FXR/FGF15 enterohepatic circulation and intestinal inflammation.
FIG. 5.
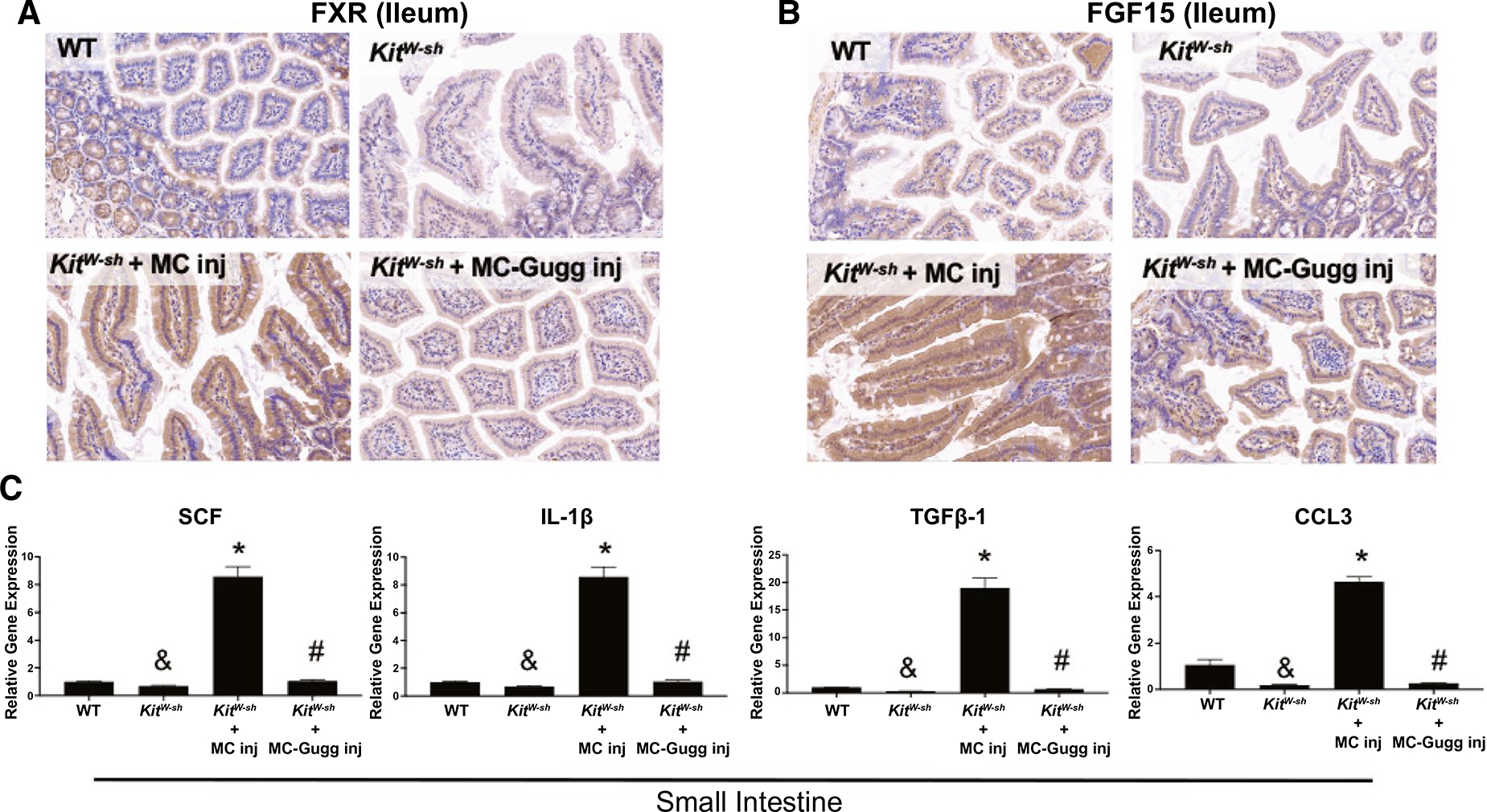
Intestinal FXR/FGF15 and inflammation are regulated by MC-FXR. Introduction of MCs in KitW-sh mice increases ileal (A) FXR and (B) FGF15 expression compared with WT and KitW-sh mice and inhibition of MC-FXR decreases (A) FXR and (B) FGF15 expression, as shown in KitW-sh + MC-Gugg mice. (C) MC injection increased small intestine expression of SCF, IL-1β, TGF-β1, and CCL3 in KitW-sh compared with WT and KitW-sh mice; however, intestinal inflammation was reduced when MC-FXR was inhibited. Representative images are presented as ×20. Data are mean ± SEM of n = 4 experiments for qPCR from 6–8 mice (MC injection group). *P < 0.05 vs. WT and KitW-sh, #P < 0.05 vs. KitW-sh + MC, &P < 0.05 vs. WT.
INHIBITION OF MC-FXR PREVENTS DR, INFLAMMATION, BILIARY SENESCENCE, SENESCENCE-ASSOCIATED SECRETORY PHENOTYPE, AND HEPATIC FIBROSIS
Introduction of MCs into KitW-sh mice increased IBDM (Fig. 6A) and F4/80-positive Kupffer cells (Fig. 6B), which also confirmed by semiquantification (Fig. 6C,D), compared with WT and KitW-sh mice. MC injection also enhanced hepatic inflammatory marker expression of IL-1β, CCL5, chemokine (C-X-C motif) ligand (CXCL) 2, and CXCL5 (Fig. 6E). Inhibition of MC-FXR reduces IBDM and hepatic inflammation compared with KitW-sh + MC mice (Fig. 6A–E). KitW-sh mice injected with MCs had increased biliary proliferation shown by Ki-67 (Supporting Fig. S3A) and biliary senescence demonstrated by immunofluorescence for p16 in liver sections and mRNA expression of p18 and p21 in isolated cholangiocytes (Supporting Fig. S3B,C) compared with WT and KitW-sh mice. Inhibition of MC-FXR before injection into KitW-sh mice reduced these parameters (Supporting Fig. S3A–C).
FIG. 6.
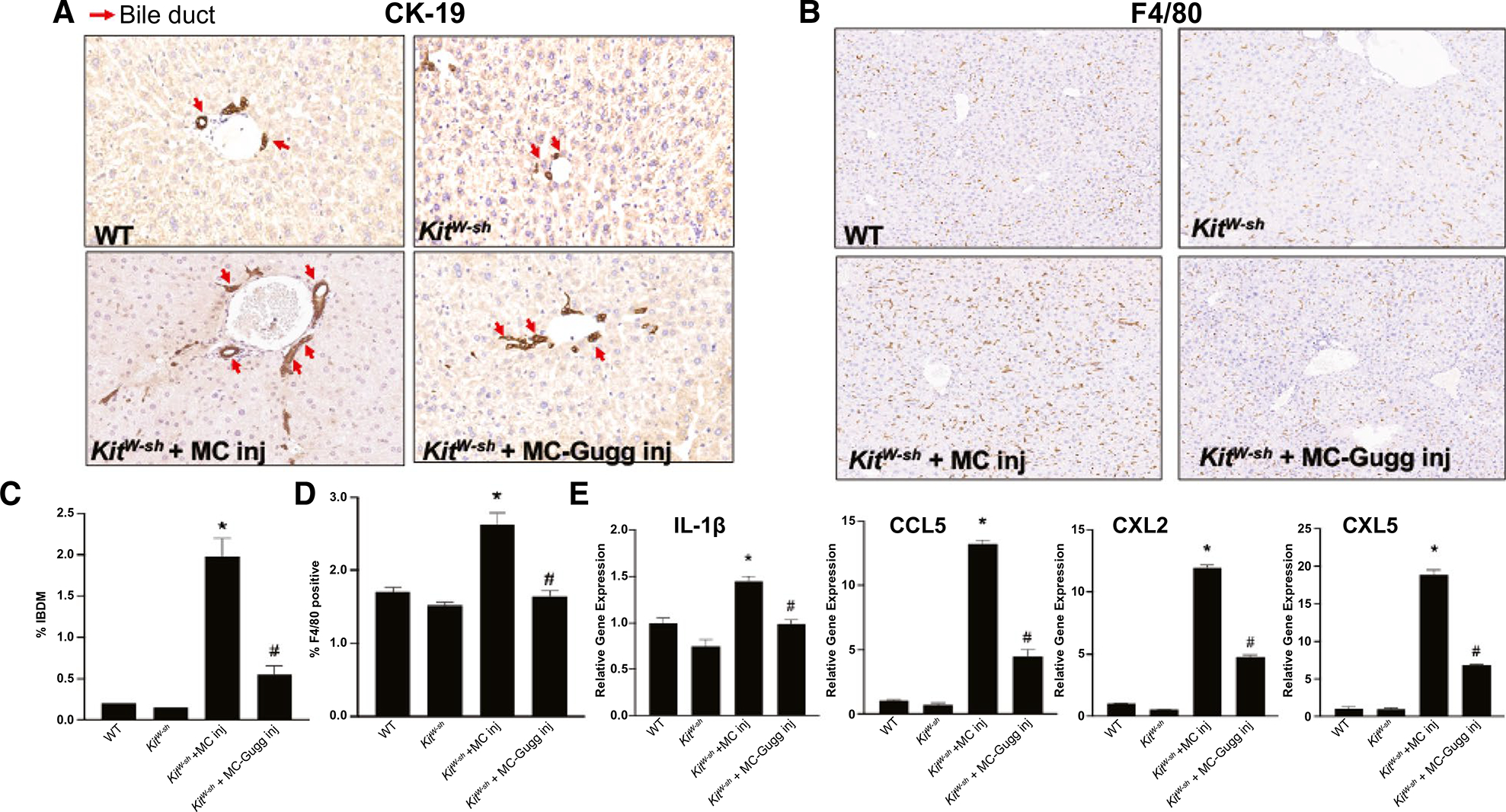
Inhibition of MC-FXR reduces IBDM and inflammation. The effect of MC-FXR inhibition on IBDM and liver inflammation was measured by immunohistochemistry for (A) CK-19 and (B) EGF-like module-containing mucin-like hormone receptor-like 1 mouse homolog (F4/80), respectively, with (C,D) semiquantification. WT and KitW-sh mice display minimal IBDM and F4/80 positive Kupffer cells that increased following MC injection in KitW-sh + MC mice. Inhibition of MC-FXR reduced both IBDM and inflammation. (A) Red arrows indicate CK-19 positive bile ducts. (E) WT and KitW-sh mice have minimal expression of hepatic inflammatory markers IL-1β, CCL5, CXCL2, and CXCL5, whereas KitW-sh + MC mice have increased hepatic inflammatory marker expression that is reduced in KitW-sh + MC-FXR mice. Data are mean ± SEM of n = 10–15 representative images for immunoreactivity semiquantification and of n = 4 experiments for qPCR from 6–8 mice per group. *P < 0.05 vs. WT, #P < 0.05 vs. KitW-sh + MC. All representative images are presented as ×10. Abbreviation: Inj, injury.
KitW-sh + MC mice display increased collagen deposition as shown by increased fast green-sirius red stain and semiquantification compared with KitW-sh and WT mice 3 days after injection; however, inhibition of MC-FXR resulted in reduced collagen deposition and hepatic fibrosis compared with KitW-sh + MC mice (Supporting Fig. S4A). Further, WT and KitW-sh mice have minimal HSC presence shown by desmin that is increased in KitW-sh + MC and found in the surrounding bile ducts and portal area (Supporting Fig. S4B). KitW-sh + MC mice display elevated expression of alpha smooth muscle actin (α-SMA) compared with WT and KitW-sh, whereas in KitW-sh + MC-Gugg, HSC presence and α-SMA expression is reduced (Supporting Fig. S4B,C).
INHIBITION OF MC-FXR REDUCES BILIARY SENESCENCE-ASSOCIATED SECRETORY PHENOTYPES AND HA/H1 SIGNALING
Ingenuity Pathway Analysis (IPA) analysis demonstrated FXR and HA signaling crosstalk through inflammatory cytokines/senescence-associated secretory phenotypes (SASP), TGF-β1 and IL-1β, and H1HR (Fig. 7A). Biliary mRNA TGF-β1 and IL-1β expression increased in KitW-sh + MC compared with WT and KitW-sh mice, whereas inhibition of MC-FXR reduced these parameters (Fig. 7B). In human patients with PSC and PSC-IBD, we found increased HA secretion compared with control patients (Fig. 7C). Furthermore, WT and KitW-sh mice have minimal serum HA levels that were elevated in KitW-sh + MC; however, when MC-FXR was inhibited, HA levels are reduced (Fig. 7D). We have shown that HA activates cholangiocytes through paracrine interactions through H1HR signaling.(24) WT and KitW-sh have minimal biliary mRNA expression of H1HR (Fig. 7E). Following MC injection, KitW-sh have increased biliary H1HR mRNA expression compared with WT and KitW-sh mice, indicating activation of biliary HA signaling; however, KitW-sh + MC-Gugg mice have reduced biliary H1HR expression compared with KitW-sh + MC mice (Fig. 7E). Combined, these results demonstrate that MC-FXR signaling has a synergistic relationship with systemic and autocrine HA signaling through regulation of biliary HA and SASP through H1HR.
FIG. 7.
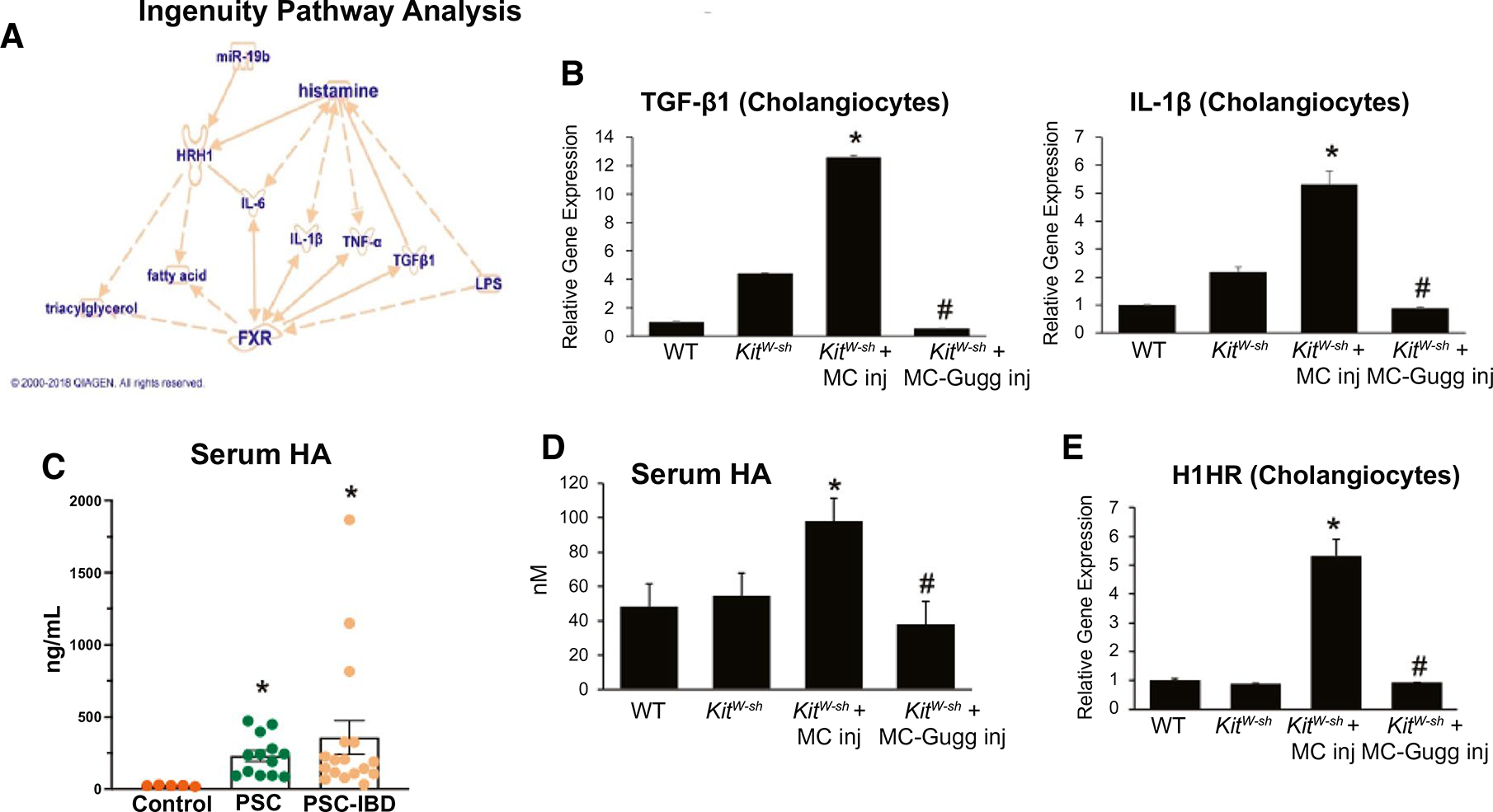
FXR and HA signaling crosstalk through IL-1β, TGF-β1, and H1HR. (A) IPA demonstrated a link between HA and FXR signaling crosstalk through IL-1β, TGF-β1, and H1HR. (B) TGF-β1 and IL-1β expression increased in isolated cholangiocytes from KitW-sh + MC compared with WT and KitW-sh mice by qPCR and inhibition of MC-FXR reduced biliary TGF-β1 and IL-1β expression. (C) Serum HA increases in patients with PSC (n = 14) and PSC-IBD (n = 17) compared with controls. (D) WT and KitW-sh have minimal HA serum secretion, which is elevated in KitW-sh + MC mice. (D) Serum HA is reduced in KitW-sh + MC-Gugg mice. (E) MC injection increases biliary H1HR expression in KitW-sh + MC compared with WT and KitW-sh mice and inhibition of MC-FXR reduces biliary H1HR expression. Data are mean ± SEM of n = 8 experiments for qPCR, n = 4 experiments for HA EIA from 6–8 mice per group, and n = 2 experiments per patient sample for HA EIA. *P < 0.05 vs. control group, #P < 0.05 vs. KitW-sh + MC. Abbreviation: Inj, injury.
INHIBITION OF MC-FXR BLOCKS BILIARY HA SIGNALING, IN VITRO
Similar to our in vivo findings, we found that in vitro murine cholangiocytes express minimal mRNA expression of IL-1β, TGF-β1, and H1HR under basal conditions; however, after stimulation with murine MC-vehicle supernatant, biliary IL-1β, TGF-β1 and H1HR increased (Supporting Fig. S5A,B). Inhibition of MC-FXR reduced biliary expression of IL-1β, TGF-β1, and H1HR (Supporting Fig. S5A,B). Correspondingly, HA secretion increased in murine cholangiocytes treated with murine MC-vehicle supernatants, but was reduced (not significant) when murine MCs were pretreated with the FXR inhibitor (Supporting Fig. S5C). Next, we found that following murine MC-vehicle supernatant stimulation, cholangiocytes have increased mRNA expression of FGF15 compared with basal cholangiocytes (Supporting Fig. S5D). Inhibition of MC-FXR reduced biliary expression of FGF15 compared with murine cholangiocytes treated with MC-vehicle supernatants (Supporting Fig. S5D). Taken together, these results confirm our IPA findings and in vivo studies, wherein MC-FXR regulates systemic and hepatic MC activation through activation of IL-1β, TGF-β1, and H1HR signaling. We conclude that MCs, through activation of FXR, regulate biliary FXR/FGF15 signaling and IL-1β, TGF-β1, and H1HR expression in a synergistic manner.
Cholangiocytes stimulated with MC-vehicle supernatant had increased proliferation compared with basal cholangiocytes as measured by bromodeoxyuridine-positive semiquantification and 3-(4,5-dimethylthiaz ol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophe nyl)-2H-tetrazolium assay (Supporting Fig. S6A,B). Treatment with MC-Gugg supernatant resulted in decreased biliary proliferation (Supporting Fig. S6A,B). Taken together, we have found that MC-FXR is capable of activating an inflammatory and proliferative cholangiocyte phenotype in vitro, mimicking hallmarks of in vivo DR seen in cholestatic liver diseases.(25)
In the final sets of experiments, human HSCs stimulated with basal-treated MC supernatants had increased α-SMA and fibronectin-1 mRNA expression compared with basal treatment and inhibition of MC-FXR decreased these parameters (Supporting Fig. S6C), demonstrating a direct interaction of MC-FXR and HSCs and further supporting our in vivo studies.
Discussion
We identified a signaling pathway wherein MC-specific FXR/FGF signaling regulates DR and hepatic fibrosis, recapitulating cholestatic liver injury. We confirm previous work establishing the role for MCs in the induction and development of cholestatic liver damage characterized by DR, biliary senescence/SASP, and fibrosis in MC-deficient KitW-sh mice, which are phenotypically normal. Our study reports the significant impact of MCs on TBA signaling and biliary and intestinal FXR/FGF axis during liver injury.
MCs are innate immune cells that, upon receptor activation, induce an inflammatory response through various mediators, including HA and FGF.(26) Our group demonstrated that MCs infiltrate the liver following injury and perpetuate cholestatic damage(8,9,24) and that stabilization of MCs in cholestatic models results in amelioration of DR, inflammation, and hepatic fibrosis.(27) Aside from traditional receptors, MCs express a number of other receptors and binding sites, including FXRβ and ASBT.(19) Our current work demonstrates that MC-derived FXR/FGF signaling regulates biliary damage and hepatic fibrosis. In support of this, during organ transplantation, MCs can produce a variety of both proinflammatory and anti-inflammatory mediators as well as release factors like FGF-2, which increases cell-to-cell interactions during regeneration.(28) In addition, FGF may induce MC recruitment, as demonstrated in a study of prostate cancer in which the authors found that stimulation of FGF-2 induced MC recruitment and promoted angiogenesis in mice and inhibition of FGF-2 reduced tumor growth.(20)
We found that inhibition of MC-FXR signaling ameliorates biliary senescence and SASP through IL-1β/TGF-β1 and HA/H1HR signaling. Interestingly, most mediators secreted by MCs are also SASP components, and several studies have implicated TGF-β1 in liver disease progression.(9,11,29) We found that MCs induce IL-1β expression that was reduced when FXR was inhibited; however, a study by Xiong et al. found that obeticholic acid (OCA; FXR agonist), in combination with lipopolysaccharide, ameliorated liver damage and inflammation by decreasing hepatic IL-1β.(30)
During canonical enterohepatic circulation, intestinal FXR/FGF15 signaling becomes activated following an elevation of serum TBAs, which results in down-regulation of ileal ASBT and inhibition of hepatic BA synthesis enzymes.(4) Fxr−/− mice have reduced intrahepatic BA pressure following BDL surgery, resulting in lower IBDM compared with BDL WT.(31) We found that MC injection increased TBA content and hepatic FXR/FGF15 signaling in MC-deficient mice, and these features were blunted after inhibition of MC-FXR, pinpointing MCs as critical regulators of TBA and hepatic FXR/FGF15 signaling. Interestingly, we found that intestinal FXR/FGF15 expression was increased following MC injection in MC-deficient mice and reduced in MC-FXR injected and untreated MC-deficient mice. This could be due to KitW-sh phenotype, which lack melanocytes and interstitial cells, but warrants further investigation. In contrast to our findings, Verbeke et al. found that vehicle-treated BDL rats had low expression of SHP, a downstream target of FXR activation, in ileum and jejunum that was rescued in ileum only following oral gavage of OCA.(32) Nonetheless, in our current study, BDL WT and Mdr2−/− mice displayed elevated intestinal FXR and FGF15 expression compared with WT controls, due to the complexity of the intestinal FXR/FGF15 axis this also requires further investigation.
Previous studies found that BAs regulate the release of specific mediators from MCs, including the release of HA(33,34); however, our studies show that MCs influence hepatic and intestinal BA signaling. Our findings are not surprising considering that MCs express BA transporters and ligands,(19) which may interact with surrounding cells to induce proliferation, senescence, or inflammation or alter BA synthesis. MC HA and intestinal H1HR activation contribute to chenodeoxycholic acid (CDCA)–induced colonic chloride secretion in rats, whereas MC-deficient rats had decreased CDCA-induced chloride secretion,(33) supporting the role for MCs in intestinal BA signaling. Interestingly, BA activation of FXR in rat colon induced mucosal MC release of nerve growth factor, which was reduced following FXR inhibition and silencing,(35) supporting our proposed crosstalk between BA signaling, FXR, and MC mediators. Furthermore, human colonic transcriptomic analysis showed up-regulation of BA signaling pathways in patients with PSC-IBD compared with those with IBD and healthy controls(36); however, MC presence and activation must be assessed in intestinal tissue from patients with PSC-IBD in order to confirm MC and FXR intestinal crosstalk. It is unlikely that MCs synthesize BAs, and thus, regulation of MC mediators like FGF15 or HA may act as a secondary mechanism to control BA circulation and signaling.
We demonstrate that inhibition of MC-FXR reduces biliary FXR and FGF15 in vivo and in vitro, which may result in reduced cholehepatic shunting of BAs; however, further analysis is needed to confirm this. Our data show that inhibition of MC-FXR results in a reduction of TBA levels that are otherwise elevated following cholestatic liver injury.(37) Quist et al. found that CDCA induces HA release from MCs in vitro, but cholic acid impeded HA release, thus supporting the concept that BAs and MCs may interact to influence HA signaling.(34)
It has been shown that there is dysregulation of FXR and FGF signaling in various liver diseases including NAFLD, PBC and PSC.(16,38,39) OCA is currently being used in clinical trials and shows potential for improved liver histology and lipid absorption in patients with PSC and NASH, respectively, and provides clinical benefits to patients with BA diarrhea(40–42); however, the overall benefits of FXR agonism are controversial because of adverse events seen in patients with decompensated cirrhotic PSC and PBC.(3) This may be due to the increased expression of biliary FXR in patients with liver injury, as shown in our study. If FXR is already enhanced in cholangiocytes, administering OCA may be counterproductive by inducing biliary senescence or increasing damage during cholestatic injury; however, further studies should be performed to investigate this possibility. Furthermore, gut microbiota influences BA signaling and intestinal inflammation and the subsequent positive or negative effects of FXR during diseases like NAFLD or NASH.(43) In support of our study, in intestinal Fxr−/− knockout mice fed high-fat diet (HFD) and treated with antibiotics, HFD-associated pathologies were reduced,(44) further implicating the complex role of FXR signaling during liver disease. Interestingly, OCA but not fexaramine (FXR agonist) ameliorated altered gut-barrier vascularization in experimental cirrhotic mice (carbon tetrachloride or BDL), but both, OCA and fexaramine, were able to reduce bacterial translocation from small intestine to the liver.(45) It has been recently shown that MC presence and BA receptor increase in the duodenum of patients with irritable bowel syndrome, which, along with our study, indicates a role for MC regulation of BA receptor expression throughout the intestinal tract.(46) The role of MCs in the regulation of epithelial barrier, inflammation, and gut-liver axis during cholestatic liver injury is a complicated future direction for this study.
In conclusion, we have generated a model of DR and hepatic fibrosis by injecting MCs into phenotypically normal MC-deficient mice and confirmed our findings using the established BDL and PSC mouse model. Our data demonstrate that MCs regulate biliary injury and DR through alteration of TBA levels and FXR/FGF signaling through biliary SASP and HA/H1HR pathways (Fig. 8). In human samples, we found that both FXR in bile ducts and MC presence are up-regulated in cholestatic liver damage, demonstrating the close interaction between MCs and biliary FXR during liver disease. Although controversial, our results demonstrate that specific inhibition of MC-FXR ameliorates cholestatic liver injury by reducing TBA levels, biliary FXR/FGF15, and subsequent HA/H1HR signaling. Our study also confirms a role for MC-FXR in the regulation of intestinal MC infiltration, inflammation, and FXR/FGF15 in cholestatic mice (Fig. 8). Further investigation into the contribution of MC mediators is needed to fully identify clinical therapies; however, our data suggest that MCs may be a targetable link for patients with PSC alone or PSC coupled with IBD.
FIG. 8.
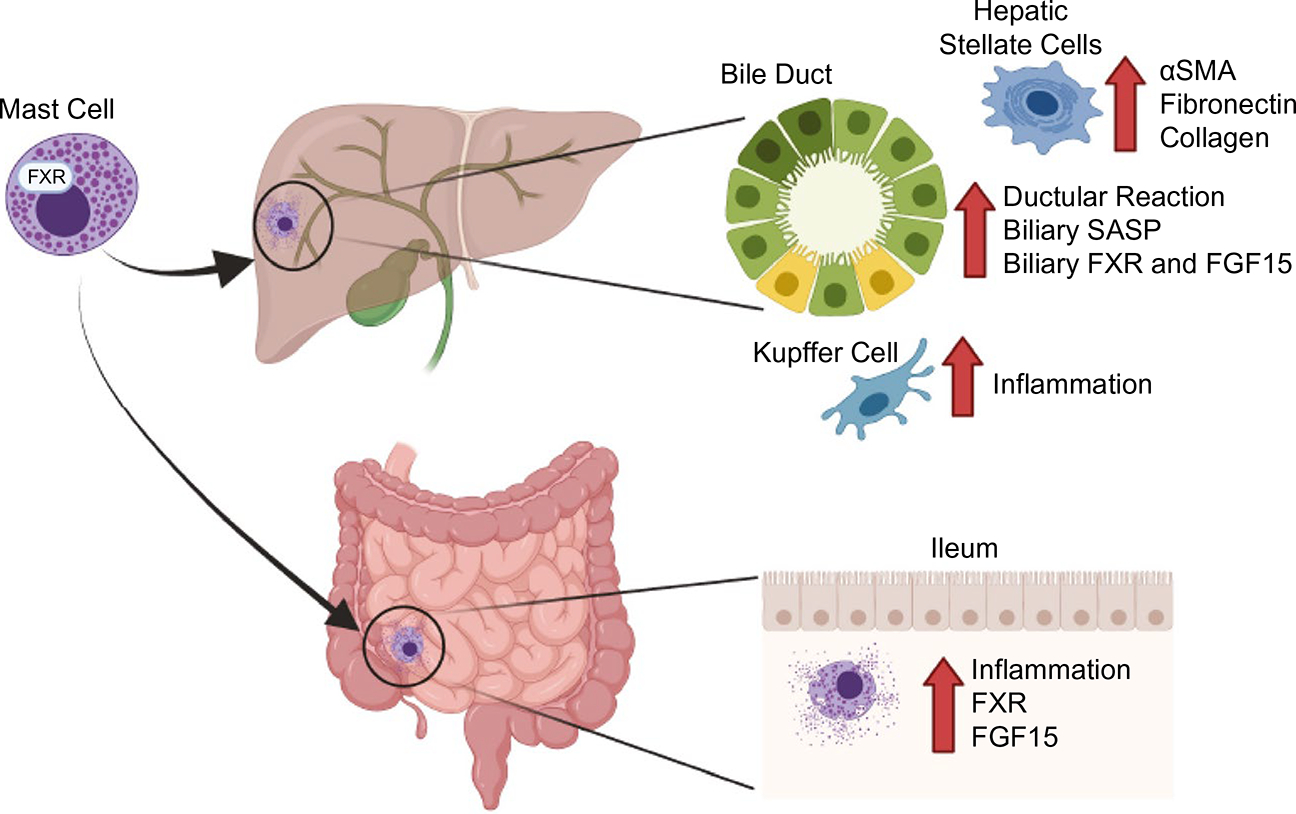
Graphical abstract demonstrating the role of MC-FXR during cholestatic liver damage. Patients with cholestatic liver diseases, such as PSC, often present with comorbidities like IBD. We found that MCs, which express FXR and secrete FGFs and HA, infiltrate the liver and contribute to increased DR, biliary SASP, and FXR/FGF15 during cholestatic liver injury. Furthermore, MCs can activate HSCs to increase α-SMA, fibronectin-1, and collagen, exacerbating liver damage. In this study, we found that MCs infiltrate the intestine of cholestatic liver injury mouse models, increasing intestinal inflammation and altering the FXR/FGF15 axis, leading to dysregulated BA signaling. MCs may serve as an ideal target for therapeutic interventions for patients presenting with cholestatic liver injury and intestinal inflammation. Created with BioRender.com.
Supplementary Material
Acknowledgments
Supported by the Hickam Endowed Chair, Gastroenterology, Medicine, Indiana University and the Indiana University Health – Indiana University School of Medicine Strategic Research Initiative, the SRCS and RCS Award and VA Merit awards to G.A. (5I01BX000574) and H.F. (1I01BX003031) from the United States Department of Veteran’s Affairs, Biomedical Laboratory Research and Development Service, and NIH grants DK108959 and DK119421 to H.F., DK11003 to S.G., and DK054811 to G.A. and S.G. This material is the result of work supported by resources at the Central Texas Veterans Health Care System, Temple, TX, and Richard L. Roudebush VA Medical Center, Indianapolis, IN. The content is the responsibility of the author(s) alone and does not necessarily reflect the views or policies of the Department of Veterans Affairs or the United States Government.
Abbreviations:
- α-SMA
alpha smooth muscle actin
- ASBT
apical Na+bile acid transporter
- ATCC
American Type Culture Collection
- BA
bile acid
- BDL
bile duct ligation
- CCL
chemokine (C-C motif) ligand
- CDCA
chenodeoxycholic acid
- CK-19
cytokeratin-19
- CXCL
chemokine (C-X-C motif) ligand
- Cyp
cytochrome p450
- DR
ductular reaction
- EIA
enzyme immunoassay
- FGF
fibroblast growth factor
- FXR
farnesoid X receptor
- Gugg
guggulsterone
- HA
histamine
- HR
histamine receptor
- IBD
inflammatory bowel disease
- IBDM
intrahepatic bile duct mass
- IPA
Ingenuity Pathway Analysis
- KitW-sh
B6.Cg-KitW-sh/HNihrJaeBsmJ
- MC
mast cell
- Mdr2−/−
multidrug resistance 2/ATP-binding cassette family 2 member 4 knockout
- OCA
obeticholic acid
- PBC
primary biliary cholangitis
- PSC
primary sclerosing cholangitis
- qPCR
quantitative PCR
- SASP
senescence-associated secretory phenotype
- SCF
stem cell factor
- SHP
small heterodimer partner
- TBA
total bile acid
- WT
wild type
Footnotes
Potential conflict of interest: Nothing to report.
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.32028/suppinfo.
REFERENCES
- 1).Meadows V, Kennedy L, Hargrove L, Demieville J, Meng F, Virani S, et al. Downregulation of hepatic stem cell factor by Vivo-Morpholino treatment inhibits mast cell migration and decreases biliary damage/senescence and liver fibrosis in Mdr2(−/−) mice. Biochim Biophys Acta Mol Basis Dis 2019;1865:165557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4)−/− mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J Hepatol 2005;43:1045–1054. [DOI] [PubMed] [Google Scholar]
- 3).Eaton JE, Vuppalanchi R, Reddy R, Sathapathy SK, Ali B, Kamath PS. Liver injury in patients with cholestatic liver disease treated with obeticholic acid. Hepatology 2020;71:1511–1514. [DOI] [PubMed] [Google Scholar]
- 4).Meadows V, Kennedy L, Kundu D, Alpini G, Francis H. Bile acid receptor therapeutics effects on chronic liver diseases. Front Med (Lausanne) 2020;7:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Mertz A, Nguyen NA, Katsanos KH, Kwok RM. Primary sclerosing cholangitis and inflammatory bowel disease comorbidity: an update of the evidence. Ann Gastroenterol 2019;32:124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Kummen M, Hov JR. The gut microbial influence on cholestatic liver disease. Liver Int 2019;39:1186–1196. [DOI] [PubMed] [Google Scholar]
- 7).Jones H, Hargrove L, Kennedy L, Meng F, Graf-Eaton A, Owens J, et al. Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis Mdr2(−/−) mice. Hepatology 2016;64:1202–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Kennedy L, Hargrove L, Demieville J, Karstens W, Jones H, DeMorrow S, et al. Blocking H1/H2 histamine receptors inhibits damage/fibrosis in Mdr2(−/−) mice and human cholangiocarcinoma tumorigenesis. Hepatology 2018;68:1042–1056. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9).Kennedy L, Meadows V, Demieville J, Hargrove L, Virani S, Glaser S, et al. Biliary damage and liver fibrosis are ameliorated in a novel mouse model lacking l-histidine decarboxylase/histamine signaling. Lab Invest 2020;100:837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Hargrove L, Kennedy L, Demieville J, Jones H, Meng F, DeMorrow S, et al. Bile duct ligation-induced biliary hyperplasia, hepatic injury, and fibrosis are reduced in mast cell-deficient Kit(W-sh) mice. Hepatology 2017;65:1991–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Kyritsi K, Kennedy L, Meadows V, Hargrove L, Demieville J, Pham L, et al. Mast cells induce ductular reaction mimicking liver injury in mice through mast cell–derived transforming growth factor beta1 signaling. Hepatology 2021;73:2397–2410. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12).Yuan ZQ, Li KW. Role of farnesoid X receptor in cholestasis. J Dig Dis 2016;17:501–509. [DOI] [PubMed] [Google Scholar]
- 13).Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res 2009;50:2340–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008;48:1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Renga B, Mencarelli A, D’Amore C, Cipriani S, D’Auria MV, Sepe V, et al. Discovery that theonellasterol a marine sponge sterol is a highly selective FXR antagonist that protects against liver injury in cholestasis. PLoS One 2012;7:e30443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Wagner M, Fickert P, Zollner G, Fuchsbichler A, Silbert D, Tsybrovskyy O, et al. Role of farnesoid X receptor in determining hepatic ABC transporter expression and liver injury in bile duct-ligated mice. Gastroenterology 2003;125:825–838. [DOI] [PubMed] [Google Scholar]
- 17).Alvarez-Sola G, Uriarte I, Latasa MU, Fernandez-Barrena MG, Urtasun R, Elizalde M, et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut 2017;66:1818–1828. [DOI] [PubMed] [Google Scholar]
- 18).Akimoto S, Ishikawa O, Iijima C, Miyachi Y. Expression of basic fibroblast growth factor and its receptor by fibroblast, macrophages and mast cells in hypertrophic scar. Eur J Dermatol 1999;9:357–362. [PubMed] [Google Scholar]
- 19).Meng F, Kennedy L, Hargrove L, Demieville J, Jones H, Madeka T, et al. Ursodeoxycholate inhibits mast cell activation and reverses biliary injury and fibrosis in Mdr2(−/−) mice and human primary sclerosing cholangitis. Lab Invest 2018;98:1465–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Ronca R, Tamma R, Coltrini D, Ruggieri S, Presta M, Ribatti D. Fibroblast growth factor modulates mast cell recruitment in a murine model of prostate cancer. Oncotarget 2017;8:82583–82592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Urizar NL, Liverman AB, Dodds DT, Silva FV, Ordentlich P, Yan Y, et al. A natural product that lowers cholesterol as an antagonist ligand for FXR. Science 2002;296:1703–1706. [DOI] [PubMed] [Google Scholar]
- 22).Ueno Y, Alpini G, Yahagi K, Kanno N, Moritoki Y, Fukushima K, et al. Evaluation of differential gene expression by microarray analysis in small and large cholangiocytes isolated from normal mice. Liver Int 2003;23:449–459. [DOI] [PubMed] [Google Scholar]
- 23).Cui J, Huang LI, Zhao A, Lew JL, Yu J, Sahoo S, et al. Guggulsterone is a farnesoid X receptor antagonist in coactivator association assays but acts to enhance transcription of bile salt export pump. J Biol Chem 2003;278:10214–10220. [DOI] [PubMed] [Google Scholar]
- 24).Kennedy L, Meadows V, Kyritsi K, Pham L, Kundu D, Kulkarni R, et al. H2 histamine receptor Vivo-Morpholino treatment ameliorates large bile duct damage in mice deficient in ATP binding cassette subfamily B member 4 (Abcb4−/−) via down-regulation of cAMP/ERK signaling. Am J Pathol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Sato K, Marzioni M, Meng F, Francis H, Glaser S, Alpini G. Ductular reaction in liver diseases: pathological mechanisms and translational significances. Hepatology 2019;69:420–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Weiskirchen R, Meurer SK, Liedtke C, Huber M. Mast cells in liver fibrogenesis. Cells 2019;8:1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Aller MA, Martinez V, Arias A, Nava MP, Cuervas-Mons V, Vergara P, et al. Mast cell-mediated splanchnic cholestatic inflammation. Clin Res Hepatol Gastroenterol 2019;43:561–574. [DOI] [PubMed] [Google Scholar]
- 28).Elieh Ali Komi D, Ribatti D. Mast cell-mediated mechanistic pathways in organ transplantation. Eur J Pharmacol 2019;857:172458. [DOI] [PubMed] [Google Scholar]
- 29).Shevchenko OP, Kurabekova RM, Tsirulnikova OM. The role of transforming growth factor beta 1 under diseases of liver. [in Russian] Klin Lab Diagn 2017;62:161–164. [PubMed] [Google Scholar]
- 30).Xiong X, Ren Y, Cui Y, Li R, Wang C, Zhang Y. Obeticholic acid protects mice against lipopolysaccharide-induced liver injury and inflammation. Biomed Pharmacother 2017;96:1292–1298. [DOI] [PubMed] [Google Scholar]
- 31).Marschall HU, Wagner M, Bodin K, Zollner G, Fickert P, Gumhold J, et al. Fxr−/− mice adapt to biliary obstruction by enhanced phase I detoxification and renal elimination of bile acids. J Lipid Res 2006;47:582–592. [DOI] [PubMed] [Google Scholar]
- 32).Verbeke L, Farre R, Verbinnen B, Covens K, Vanuytsel T, Verhaegen J, et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am J Pathol 2015;185:409–419. [DOI] [PubMed] [Google Scholar]
- 33).Gelbmann CM, Schteingart CD, Thompson SM, Hofmann AF, Barrett KE. Mast cells and histamine contribute to bile acid-stimulated secretion in the mouse colon. J Clin Invest 1995;95:2831–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Quist RG, Ton-Nu HT, Lillienau J, Hofmann AF, Barrett KE. Activation of mast cells by bile acids. Gastroenterology 1991;101:446–456. [DOI] [PubMed] [Google Scholar]
- 35).Li WT, Luo QQ, Wang B, Chen X, Yan XJ, Qiu HY, et al. Bile acids induce visceral hypersensitivity via mucosal mast cell-to-nociceptor signaling that involves the farnesoid X receptor/nerve growth factor/transient receptor potential vanilloid 1 axis. FASEB J 2019;33:2435–2450. [DOI] [PubMed] [Google Scholar]
- 36).Quraishi MN, Acharjee A, Beggs AD, Horniblow R, Tselepis C, Gkoutos G, et al. A pilot integrative analysis of colonic gene expression, gut microbiota, and immune infiltration in primary sclerosing cholangitis-inflammatory bowel disease: association of disease with bile acid pathways. J Crohns Colitis 2020;14:935–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Wang R, Sheps JA, Liu L, Han J, Chen PSK, Lamontagne J, et al. Hydrophilic bile acids prevent liver damage caused by lack of biliary phospholipid in Mdr2(−/−) mice. J Lipid Res 2019;60:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Wunsch E, Milkiewicz M, Wasik U, Trottier J, Kempińska-Podhorodecka A, Elias E, et al. Expression of hepatic fibroblast growth factor 19 is enhanced in primary biliary cirrhosis and correlates with severity of the disease. Sci Rep 2015;5:13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Armstrong LE, Guo GL. Role of FXR in liver inflammation during nonalcoholic steatohepatitis. Curr Pharmacol Rep 2017;3:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Bowlus CL, Pockros PJ, Kremer AE, Pares A, Forman LM, Drenth JPH, et al. Long-term obeticholic acid therapy improves histological endpoints in patients with primary biliary cholangitis. Clin Gastroenterol Hepatol 2019;18:1170–1178.e6. [DOI] [PubMed] [Google Scholar]
- 41).Makri E, Cholongitas E, Tziomalos K. Emerging role of obeticholic acid in the management of nonalcoholic fatty liver disease. World J Gastroenterol 2016;22:9039–9043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).Walters JR, Johnston IM, Nolan JD, Vassie C, Pruzanski ME, Shapiro DA. The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid. Aliment Pharmacol Ther 2015;41:54–64. [DOI] [PubMed] [Google Scholar]
- 43).Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: pathophysiological basis for therapy. J Hepatol 2020;72:558–577. [DOI] [PubMed] [Google Scholar]
- 44).Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest 2015;125:386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Sorribas M, Jakob MO, Yilmaz B, Li H, Stutz D, Noser Y, et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J Hepatol 2019;71:1126–1140. [DOI] [PubMed] [Google Scholar]
- 46).Miura K, Oshima T, Ito C, Horikawa T, Yamada M, Tomita T, et al. Vitamin D receptor is overexpressed in the duodenum of patients with irritable bowel syndrome. J Gastroenterol Hepatol 2021;36:951–958. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.