

Abstract
Synaptotagmin-1 (Syt1) is a vesicular calcium sensor required for synchronous neurotransmitter release, composed of a single-pass transmembrane domain linked to two C2 domains (C2A and C2B) that bind calcium, acidic lipids, and SNARE proteins that drive fusion of the synaptic vesicle with the plasma membrane. Despite its essential role, how Syt1 couples calcium entry to synchronous release is poorly understood. Calcium binding to C2B is critical for synchronous release, and C2B additionally binds the SNARE complex. The C2A domain is also required for Syt1 function, but it is not clear why. Here, we asked what critical feature of C2A may be responsible for its functional role and compared this to the analogous feature in C2B. We focused on highly conserved poly-lysine patches located on the sides of C2A (K189-192) and C2B (K324-327). We tested effects of charge-neutralization mutations in either region (Syt1K189-192A and Syt1K326-327A) side by side to determine their relative contributions to Syt1 function in cultured cortical neurons from mice of either sex and in single-molecule experiments. Combining electrophysiological recordings and optical tweezers measurements to probe dynamic single C2 domain–membrane interactions, we show that both C2A and C2B polybasic patches contribute to membrane binding, and both are required for evoked release. The size of the readily releasable vesicle pool and the rate of spontaneous release were unaffected, so both patches are likely required specifically for synchronization of release. We suggest these patches contribute to cooperative membrane binding, increasing the overall affinity of Syt1 for negatively charged membranes and facilitating evoked release.
SIGNIFICANCE STATEMENT Synaptotagmin-1 is a vesicular calcium sensor required for synchronous neurotransmitter release. Its tandem cytosolic C2 domains (C2A and C2B) bind calcium, acidic lipids, and SNARE proteins that drive fusion of the synaptic vesicle with the plasma membrane. How calcium binding to Synaptotagmin-1 leads to release and the relative contributions of the C2 domains are unclear. Combining electrophysiological recordings from cultured neurons and optical tweezers measurements of single C2 domain–membrane interactions, we show that conserved polybasic regions in both domains contribute to membrane binding cooperatively, and both are required for evoked release, likely by increasing the overall affinity of Synaptotagmin-1 for acidic membranes.
Keywords: calcium-triggered exocytosis, exocytosis, membrane–protein interactions, neurotransmitter release, synaptotagmin
Introduction
Synaptotagmin-1 (Syt1) is a major neuronal calcium sensor for synchronous neurotransmitter release (Geppert et al., 1994; Fernández-Chacón et al., 2001; Xu et al., 2007; Chapman, 2008; Südhof, 2013b). In mice, knock-out (KO) of syt1 is lethal at birth, with nearly complete elimination of synchronous release from cultured hippocampal neurons collected from syt1−/− newborns (Geppert et al., 1994). In humans, mutations in Syt1 result in severe neurodevelopmental disorders (Bradberry et al., 2020). Syt1 is a synaptic vesicle protein comprising a short N-terminal luminal sequence, a single-pass transmembrane domain, and a cytoplasmic linker region followed by two tandem C2 domains (C2A and C2B) at the C terminus that bind calcium, acidic lipids, and SNARE proteins (Chapman, 2008). Despite its critical role in neurotransmitter release, how Syt1 couples calcium entry to synchronous membrane fusion is not well understood (Chapman, 2008; Südhof, 2013a; Park and Ryu, 2018; Rizo, 2018; Brunger et al., 2018b). In particular, the relative contributions of the two tandem C2 domains of Syt1 to the overall function of the protein are not clear. Here, we show that conserved polybasic patches in both C2 domains are required for efficient membrane binding and evoked release.
The C2 domains of Syt1 are eight-stranded β-sandwich structures with two protruding loops that form the Ca2+ binding pockets, with three and two Ca2+ ions thought to bind to C2A and C2B, respectively, via a series of highly conserved aspartates (Chapman, 2008). Calcium binding leads hydrophobic residues at the tips of the calcium-binding loops to bury into the membrane for both C2A and C2B, with preference for bilayers containing phosphatidylserine (PS) or phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) for the former and latter, respectively (Chapman and Davis, 1998; Bai et al., 2000, 2004a; Bradberry et al., 2019; Nyenhuis et al., 2019). Calcium binding mutations in C2B result in stronger phenotypes than in C2A, suggesting calcium binding to C2B is essential for triggering synchronous release, whereas calcium binding to C2A may have a more faciliatory role (Fernández-Chacón et al., 2001, 2002; Mackler et al., 2002; Robinson et al., 2002; Stevens and Sullivan, 2003; Nishiki and Augustine, 2004; Bowers and Reist, 2020b). C2 domains also interact with SNARE proteins in both calcium-dependent and independent manners, but there is still some debate on which interactions are most relevant physiologically (Chapman et al., 1995; Rickman et al., 2004; Brewer et al., 2015; Park et al., 2015; Zhou et al., 2015). Early biochemical (Chapman et al., 1996) and recent structural studies suggest only the C2B domain binds the neuronal SNARE complex and that binding is maintained in the presence of membranes (Davis et al., 1999; Wang et al., 2016), although there is some debate about whether the binding is calcium dependent (Chapman et al., 1996; Tucker et al., 2003, 2004) or independent (Zhou et al., 2015, 2017). Both the C2A and C2B domains have a highly conserved poly-lysine patch located on the side, K189-192 for the former and K324-237 for the latter (Fig. 1A,B). The role of the polybasic patch in C2B has been studied in the past, with conflicting results. Reist and colleagues (Mackler and Reist, 2001; Loewen et al., 2006) showed that replacing three of the C2B domain polybasic region lysines with glutamines (syt1K379,380,384Q) in Drosophila third instar larvae resulted in an ∼40% decrease in evoked release and a doubling of spontaneous release at the neuromuscular junction (NMJ), effects attributed to vesicle priming defects in the mutant. Using autaptic cultures of syt1 knock-out neonatal mice, hippocampal neurons expressing wild-type or mutant Syt1, Li et al. (2006) found that partial neutralization of the C2B domain polybasic region of Syt1 (K326A, K327A, the KAKA substitution), similarly resulted in a 50% decrease in evoked release but with little effect on spontaneous release or the readily releasable pool (RRP) size. Using a similar preparation, Borden et al. (2005) also reported mild effects for the K326A, K327A substitution, an ∼50% reduction in the fraction of vesicles released per stimulus and an unchanged RRP size. Both studies also found a reduction in the normalized EPSC amplitude as a function of extracellular calcium concentration. By contrast, rescue of syt−/− neonatal autaptic hippocampal neuronal cultures with a Syt1 K325A,K327A mutant resulted in a much more severe, fivefold to sixfold reduction in synchronous release and an ∼60% reduction in the RRP size (Chang et al., 2018). Chang et al. (2018) also found this mutation disrupts tight attachment of synaptic vesicles to the active zone plasma membrane. It is not clear if the discrepancies among the reported results are because of differences in the species used, the preparation of the cultures, and/or the stimulation methods used.
Figure 1.
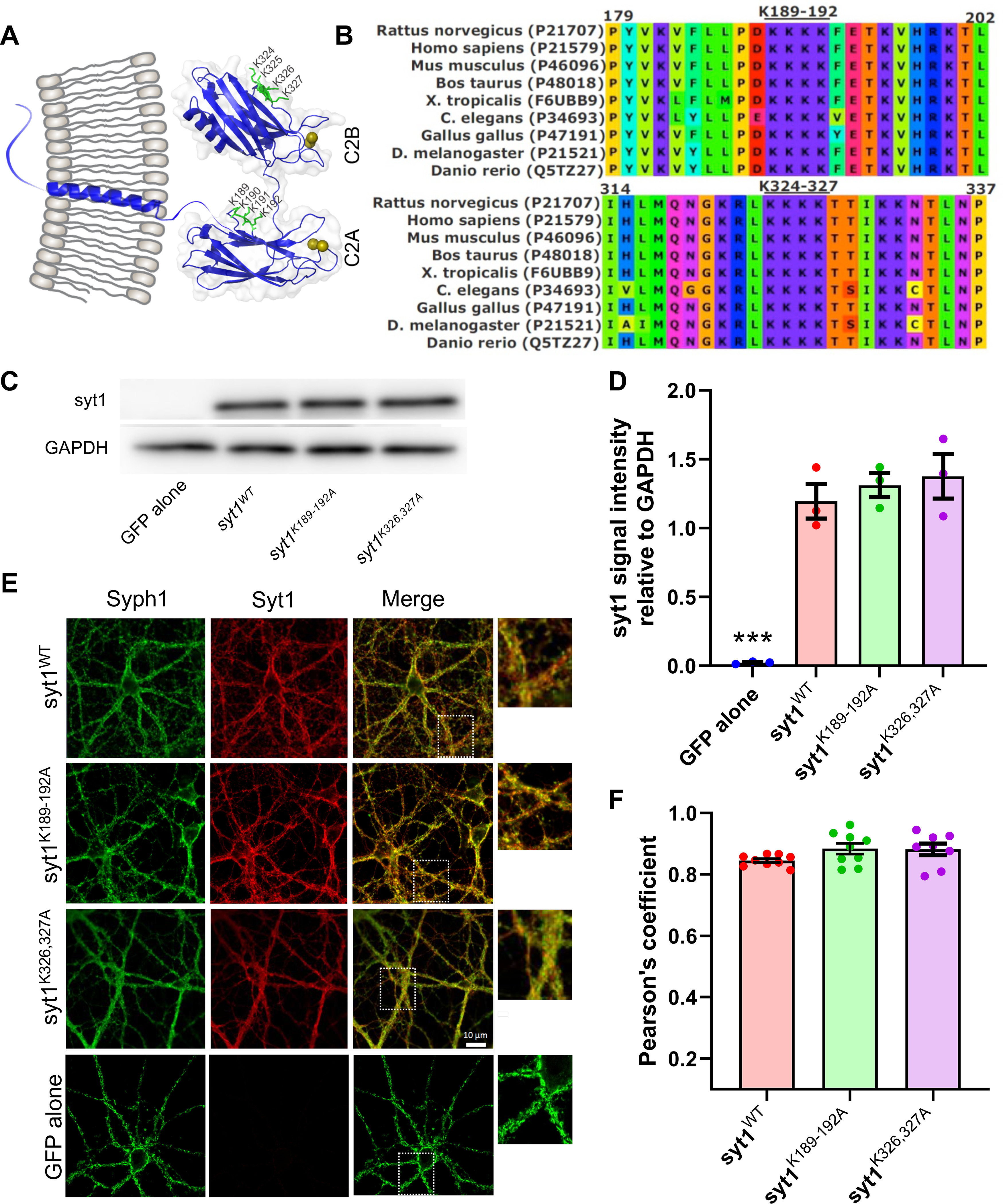
Expression and targeting of Syt1 polybasic patch mutants are similar to those of wild-type Syt1. A, Schematic of the structure of Syt1, with the polybasic patches marked with ball-and-stick representation in green. The numbering refers to the mouse sequence. Calcium ions are depicted as dark yellow spheres. The C2A and C2B domains are rendered from Protein Data Bank entry 5CCG using PyMOL (PyMOL Molecular Graphics System, Schrödinger), whereas the rest of the molecule is schematically drawn using CorelDRAW. B, Multiple alignment of synaptotagmin-1 protein sequences from various species as indicated, using Clustal (Sievers et al., 2011). The uniprot access codes are shown in parentheses (https://www.uniprot.org/). C, Western blot analysis of the expression of wild-type or mutant Syt1 transgenes in syt1−/− mouse neonatal cortical cultures. A representative result from three separate experiments is shown. D, Quantification of the Western blots, showing that all syt1 constructs were expressed at similar levels. For each condition, the integrated pixel intensity for the Syt1 band was normalized to that of GAPDH, and the mean ± SEM from three independent experiments is shown; ***p < 0.01, n.s. at 5% level (1-way ANOVA, followed by Dunnett's test to compare mutants against WT Syt1; WT vs Syt1K189-192A, p = 0.81; WT vs Syt1K326327A, p = 0.56). E, Exogenously expressed wild-type or mutant Syt1 are correctly targeted. Immunofluorescence signals of Syt1 variants were compared with those of Syph1, a synaptic vesicle marker. The boxed regions in the third column are shown expanded on the right. F, Quantification of colocalization of Syph1 and Syt1 immunofluorescence signals using Pearson's correlation coefficient (a value of 1 indicates perfect colocalization). There were no significant differences among the groups (1-way ANOVA, followed by Dunnett's test to compare mutants against WT Syt1; WT vs Syt1K189-192A, p = 0.14; WT vs Syt1K326327A, p = 0.18).
The C2A domain has received less attention since the finding that disrupting Ca2+ binding to C2A results in less severe phenotypes (Fernández-Chacón et al., 2002; Robinson et al., 2002; Stevens and Sullivan, 2003) than a similar disruption in the C2B domain (Mackler et al., 2002; Nishiki and Augustine, 2004). In addition, biochemical studies reported that the C2B domain, but not the C2A domain, binds phosphoinositides in a calcium-independent manner through its poly-lysine patch (Bai et al., 2004a; Bradberry et al., 2019; Nyenhuis et al., 2019). However, recent work suggests Ca2+ binding to C2A (Striegel et al., 2012; Shields et al., 2020) and the ensuing insertion of the hydrophobic residues at the tips of the C2A loops (Bowers and Reist, 2020a) are required for evoked release, at least at the Drosophila larvae NMJ, consistent with biochemical experiments showing robust Ca2+-dependent membrane binding and penetration of the C2A domain to negatively charged membranes (Chapman and Davis, 1998; Davis et al., 1999; Chapman, 2008). In addition, domain deletion and swapping experiments suggested that the C2A domain is essential for Syt1 function in Drosophila (Lee et al., 2013) and more recently in mice (Courtney et al., 2019).
Despite these recent results showing the importance of the C2A domain, the function of the poly-lysine patch in C2A remains unclear. Neutralization of the C2A polybasic patch in Drosophila larvae NMJ resulted in a 2.4-fold increase in spontaneous release frequency but left evoked release intact (Mace et al., 2009). By contrast, injection of a small peptide including the polybasic sequence into the squid giant nerve terminals inhibited transmitter release when the nerve was stimulated, suggesting this region to be important for evoked release (Bommert et al., 1993). Similarly, injection of the entire C2A domain into PC12 cells resulted in inhibition of Ca2+-triggered release, an effect attributed to the poly-lysine motif through alanine-scanning mutagenesis (Thomas and Elferink, 1998). However, in other studies, recombinant C2A did not inhibit release from PC12 cells (Shin et al., 2002; Tucker et al., 2003).
In summary, the reported effects of mutations of the conserved polybasic patches in the two C2 domains of Syt1 are conflicting. We decided to test effects of charge-neutralization mutations in the two poly-basic domains side by side to determine their relative contributions to Syt1 function in cultured cortical mouse neurons and in biophysical single-molecule experiments. Combining electrophysiological recordings and single-molecule optical tweezer (OT) measurements to probe dynamic C2 domain–membrane interactions, we show that both C2A and C2B polybasic patches contribute to membrane binding, and both are required for evoked release. There was no measurable effect on the size of the RRP of vesicles or spontaneous release. We suggest these patches contribute to cooperative binding to membranes, increasing the overall affinity of C2AB for negatively charged membranes and facilitating evoked release.
Materials and Methods
Neuron preparation, Syt1 lentivirus production and transduction
Primary cortical neurons from pups of either sex, born to heterozygous syt1 KO parents, were isolated at postnatal day 0–1. All procedures are in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocols were reviewed and approved by the Animal Care and Use Committee at the University of Wisconsin–Madison. In brief, cortices were dissected from mouse brain and digested for 25 min at 37°C in 0.25% trypsin-EDTA (Corning). After mechanical dissociation, cortical neurons were plated on 12 mm glass coverslips whose surfaces were pretreated with poly-D-lysine (Thermo Fisher Scicentific) for at least 1 h at room temperature. Cultures were maintained in Neurobasal A-based (Invitrogen) medium with B27 (2%, Thermo Fisher Scientific) and GlutaMAX (2 mm, Invitrogen) at 37°C incubator.
For virus infection experiments, WT and mutant forms of syt1 DNA were subcloned into a FUGW transfer plasmid (catalog #14883, Addgene) modified with a synapsin promoter and an IRES-expressed soluble GFP marker. Lentiviral particles were generated as previously described (Courtney et al., 2019). In brief, transfer plasmids that carry WT or mutated syt1 were cotransfected with packaging and helper (pCD/NL-BH*ΔΔΔ and VSV-G encoding pLTR-G) plasmids into HEK 293/T cells. Lentivirus was collected from the media 48–72 h after transfection and concentrated by ultracentrifugation. These viruses were first titrated and then infected neurons on day in vitro (DIV) 3–5.
Electrophysiology
Cortical neurons (DIV 14–19) with GFP fluorescence were patched at room temperature using a MultiClamp 700B amplifier (Molecular Devices). Recording pipettes were pulled from borosilicate glass (Sutter Instruments) and filled with the pipette internal solution composed of the following (in mm): 130 KCl, 1 EGTA, 10 HEPES, 2 ATP, 0.3 GTP, and 5 sodium phosphocreatine, pH 7.35, and 290 mOsm, which generates a 3–5 ΜΩ resistance in a bath solution containing the following (in mm): 128 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 30 d-glucose, and 25 HEPES, pH 7.3, and 305 mOsm. Patched neurons for data collection were held at −70 mV with access resistance () <15 ΜΩ at any time point. GABAA-receptor mediated currents were pharmacologically isolated by including d-AP5 (50 µm; Abcam) and CNQX (20 µm; Abcam) in the bath solution. Data were obtained using a Digidata 1440A Digitizer (Molecular Devices) and Clampex 10 software (Molecular Devices) at 5 kHz. IPSCs were captured with 5 mm QX-314 in pipette solution and evoked by a single stimulus via a concentric bipolar electrode (125/25 µm extended tip, 50 mm length; model no. 30201, FHC) placed ∼300 µm away from the neural soma. For every recording, we first applied current pulses at low frequency (0.03 Hz) using a constant-current A385 stimulus isolator (World Precision Instruments). We adjusted the stimulator tip position and current amplitude until a stable postsynaptic current was obtained for every pulse. Stimulator currents used were ∼0.5–1.0 mA for all conditions, among which no particular differences were noted. The input resistance (the sum of the series and membrane resistances) was comparable for all conditions, 130.8 ± 8.1, 132.4 ± 8.2, 132.1 ± 5.9, 128.6 ± 10 MΩ (mean ± SEM) for syt1−/− neurons expressing GFP alone, wild-type syt1, syt1K189-192A, or syt1K326,327A, respectively. For miniature IPSC (mIPSC) measurements, tetrodotoxin (TTX, 1 µm) was included in the bath solution to inhibit all action potentials. For every cell, 300 s of data were recorded, and miniature events were identified and analyzed by the template-matching algorithm in Clampfit software.
The RRP size was quantified by osmotic shock (Rosenmund and Stevens, 1996). In brief, 0.5 m sucrose in bath solution was puffed by a Picospritzer III (Parker Hannifin) through a fused silica needle (28 gauge, World Precision Instruments), which was placed ∼500 µm away from the soma of the aimed neuron. Recorded neurons were fully covered by sucrose and treated for 15 s, yielding a distinct fast and slow phase of release. The phasic response was integrated to determine the RRP size. All RRP recordings were performed in the presence of 20 µm CNQX, 50 µm d-AP5, and 1 µm tetrodotoxin in the bath. Both the experiments and the analyses were performed blindly.
Western blotting
Cortical neurons (14–19 DIV) expressing WT or mutant forms of syt1 were treated with lysis buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 2% SDS, 0.1% Triton X-100, 10 mm EDTA), supplemented with protease inhibitors (complete mini EDTA-free, one tablet/50 ml lysis buffer, Roche). The lysed neurons were mixed with 4× Laemmli sample buffer and heated at 70°C for 10 min (stored at −20°C until use). The total proteins were separated by SDS-PAGE gel, and then transferred to PVDF membrane. The membranes were blocked with 5% milk and incubated with an anti-syt1 primary antibody (3 µg/ml; catalog #mAb48, Developmental Studies Hybridoma Bank) and a GAPDH primary antibody (1:1000; catalog #2118, Cell Signaling Technology) in TBS-T with 1% milk at 4°C overnight. After washing in TBS-T, blots were incubated with Goat anti-Mouse IgG2b Cross-Adsorbed Secondary Antibody HRP (1:2000; catalog #M32407, Invitrogen) or Goat anti-Rabbit IgG (H L)-HRP conjugate (1:1000; catalog #172-1019, Bio-Rad) in TBS-T for 1 h at room temperature, washed in TBS-T, and then imaged with a CCD gel imaging device (GE).
Immunocytochemistry and confocal microscopy
Cortical neurons (DIV 14–18) on coverslips were fixed by incubation with prewarmed 4% paraformaldehyde for 10 min at 37°C. Cell membranes were permeabilized by 0.2% saponin for 10 min, rinsed in PBS, and blocked for 1 h at room temperature in PBS buffer supplemented with 5% normal goat serum, 5% BSA, and 0.02% saponin. Cells were then incubated with Syt1 monoclonal antibody (3 µg/ml; catalog #mAb48, Developmental Studies Hybridoma Bank) and synaptophysin 1 primary antibody (guinea pig pAb, 1:500; catalog #101004, Synaptic Systems) in PBS containing 1% BSA and 0.02% saponin at 4°C overnight. Cells were washed in PBS containing 0.02% saponin and incubated with Goat Anti-Mouse IgG2b Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 (5 µg/ml; catalog #A-21145, Invitrogen) and Goat anti-Guinea Pig IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 (5 µg/ml; catalog #A-21450, Invitrogen) for 1 h at room temperature. Stained neurons were imaged on a Zeiss LSM 880 confocal microscope equipped with a 60× oil objective. Syt1 and synaptophysin colocalizations were quantified by Pearson's correlation coefficient using the Coloc 2 plug-in in FIJI software (Schindelin et al., 2012). For every condition, eight to nine viewfields from three to four slides, prepared from at least two different breeders, were imaged. On a given slide, areas with reasonably dense yet flat distributions of dendrites were randomly selected and imaged. There were no noticeable differences in cell density or morphology for any of the conditions. Entire images were subjected to colocalization analysis.
Protein expression and purification
The wild-type rat Syt1 construct used for the single-molecule assay and its preparation were described in detail previously (Ma et al., 2017). The Syt1 mutants were created from the wild-type StyI construct using PCR mutagenesis and prepared similarly to wild-type Syt1. Briefly, all the Syt1 constructs contain an N-terminal Avi-Tag, a flexible polypeptide linker, the C2AB domain, and a unique cysteine residue added to the C terminus. The DNA coding sequences of Syt1 proteins were cloned into pET SUMO vector, which contains a His tag and a sumo protein at the N terminus. All Syt1 proteins were expressed in BL21 E. coli cells and purified using the His tag. Protein biotinylation was carried in vitro using BirA ligase, which catalyzes biotinylation of a lysine residue in the Avi-Tag. The SUMO tag was then removed by the SUMO protease. The purified Syt1 protein was cross-linked to the DNA handle by mixing the two at a 50:1 molar ratio under an oxidization condition.
Circular dichroism spectroscopy
Circular dichroism (CD) measurements were conducted on a Chirascan Circular Dichroism Spectrometer (Applied Photophysics) equipped with a temperature controller. Data were collected from 10 µm samples of WT and mutant Syt1 C2AB domains in 10 mm Tris-HCl. pH 8.0, 100 mm NaCl buffer (with 1 mm EGTA or 1 mm CaCl2) over a wavelength range of 200–280 nm, with 1 nm increments, in a 1 mm path length cell at 25°C. All samples were allowed to equilibrate for 10 min before CD analysis. Each spectrum represents the average of three separate spectral recordings. The contribution of buffer with or without liposomes to the measured ellipticity was subtracted as a blank. Molar ellipticity values were calculated using the expression , where is the ellipticity (millidegrees), is the protein concentration (mol/L), is the cuvette path length (cm), and is the number of amino acid residues in the protein. Temperature denaturation experiments were performed at a wavelength of 217 nm by increasing the temperature from 20 to 100°C in 5°C increments, a 2 min temperature equilibration time, and a 3 s averaging time. The fraction of unfolded protein at each temperature was calculated by using the formula , where is the observed mean residue ellipticity, and and are the mean residue ellipticities of the unfolded and-folded states, respectively. and were estimated by extrapolation of the linear regions of the extremes of the denaturation curves. The midpoint of the melting transition () was estimated from the LogIC50 after fitting the data to a Boltzmann's sigmoidal equation.
Single-molecule assay for Syt1-membrane interactions
The dual-trap high-resolution optical tweezers are home built and calibrated as previously described (Sirinakis et al., 2012). Briefly, a 1064 nm laser (CW Nd:YVO4 laser with a maximum power of 5 W) beam is expanded by ∼10-fold in diameter by two telescopes. Between the telescopes, the laser beam is split into two orthogonally polarized beams. One beam is steered by a mirror attached to a nano-positioning stage, which is used to move the optical trap in the sample plane. The two beams are then focused by a water immersion 60× objective (NA = 1.2) to form two optical traps inside the central channel of a microfluidic flow cell. The outgoing laser beams are collected and collimated by a second identical objective, split again by polarization, and projected onto two position-sensitive detectors to detect bead positions using back-focal-plane interferometry (Sirinakis et al., 2012).
Statistical analysis
One-way ANOVA or the nonparametric Kruskal–Wallis test (adjusted for multiple comparisons using Dunnett's test for the former and the Dunn–Sidak procedure for the latter) were used for null-hypothesis significance testing, as indicated in figure legends. For data in Figure 2, C and F (see Fig. 4B), we additionally quantified the effect size using an estimation graphic (Ho et al., 2019; Extended Data Figs. 2-1 and 4-1).
Figure 2.
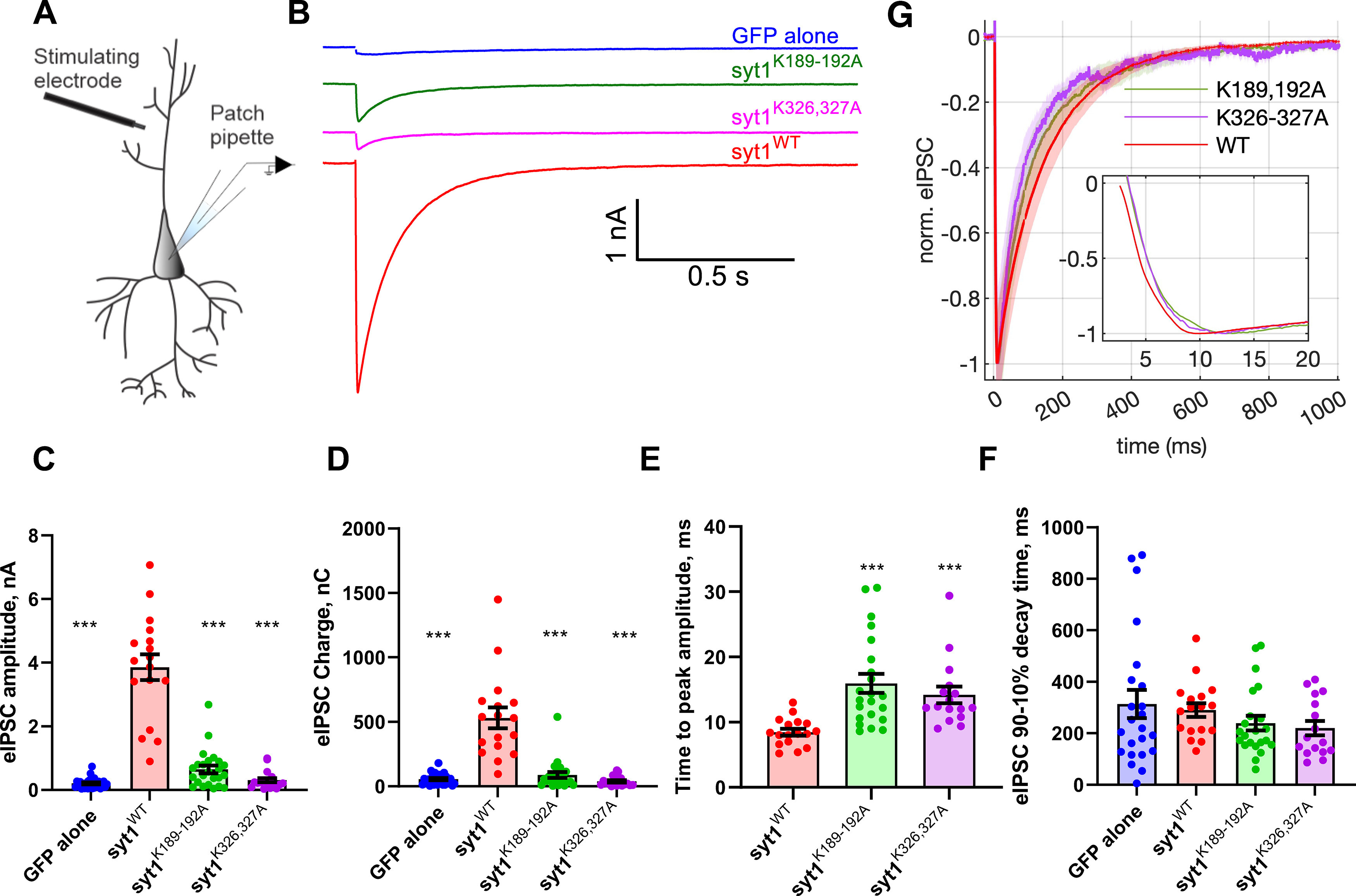
Syt1 C2A and C2B polybasic patch mutations dramatically reduce evoked release. A, Schematic of the recording configuration. B, Representative examples of IPSCs recorded from cultured syt1−/− cortical neurons expressing the indicated transgenes. Neurons expressing the Syt1 C2A (Syt1K189-192A) or C2B (Syt1K326-327A) polybasic patch mutations had greatly diminished responses compared with neurons expressing wild-type Syt1. Neurons lacking Syt1 expression had nearly all evoked release abolished. C–F, Quantification of evoked release parameters. eIPSC amplitudes were (mean ± SEM; in nA) GFP alone, 0.21 ± 0.03; WT, 3.9 ± 0.40; syt1K189-192A, 0.65 ± 0.12; syt1K326,327A, 0.31 ± 0.07 (C). eIPSC charges (time integrals of the IPSC traces, mean ± SEM; in nC) were GFP alone, 55.48 ± 8.12; WT, 531.5 ± 80.84; syt1K189-192A, 88.98 ± 22.38; syt1K326,327A, 38.97 ± 9.30 (D). Time to reach the negative eIPSC peak from the time stimulation is applied (mean ± SEM; in ms) GFP alone, 82.91 ± 8.12 (data not shown); WT, 8.471 ± 0.51; syt1K189-192A, 15.95 ± 1.47; syt1K326,327A, 14.19 ± 1.29 (E). Five traces (GFP alone), two traces (syt1K189-192A), and two traces (syt1K326,327A) with very little response were excluded from analysis as the time to peak could not be determined accurately. F, eIPSC decay times were (mean ± SEM; ms) GFP alone, 263.8 ± 50.34; WT, 290.3 ± 26.80; syt1K189-192A, 228.1 ± 27.53; syt1K326,327A, 178.6 ± 29.19 (n = 28, 17, 24, and 18 cells tested for GFP alone, WT syt1, syt1K189-192A, and syt1K326,327A, respectively). The same traces excluded from analysis in E were also excluded here. G, Averaged eIPSCs, normalized to the negative peak value for the conditions as indicated. The shaded patches represent SEM. Inset, Short time scales. Cells were prepared from 5 syt1−/− pups. For C–F we used Kruskal–Wallis test, followed by Dunn's multiple comparisons test to compare mutants against WT Syt1; *p < 0.05, **p < 0.01, ***p < 0.001, respectively. See Extended Data Figure 2-1 for effect size estimation of evoked release parameters.
Figure 4.

Polybasic patch mutations do not affect the readily releasable pool. A, Representative current traces elicited by application of a 0.5 m sucrose solution for 15 s, indicated by the gray bars above each trace, for syt1 KO neurons expressing the indicated transgenes. B, Integral of the hypertonic sucrose-induced currents to estimate the size of the RRP. The RRP size is indistinguishable for Syt1WT, Syt1K189-192A, and Syt1K326-327A, but is lower by ∼2.4-fold for neurons lacking Syt1. Sucrose-induced total charges (RRP sizes) were (mean ± SEM; nC) GFP alone, 2.82 ± 0.28; WT, 6.59 ± 0.375; syt1K189-192A, 6.16 ± 0.43; syt1K326,327A, 6.13 ± 0.54 (n = 24, 23, 23, and 21 cells tested for GFP alone, syt1 WT, syt1K189-192A, and syt1K326,327A, respectively). We used the Kruskal–Wallis test, followed by Dunn's multiple comparisons test to compare mutants against WT Syt1. ***p < 0.001. Extended Data Figure 4-1 shows estimation of effect sizes.
Results
Synaptotagmin-1 polybasic patch mutants are expressed at wild-type levels and trafficked correctly
To assess the roles of the polybasic patches in its two C2 domains, we expressed Syt1 with mutations designed to neutralize the poly-lysine patch in the C2A (K189-192A) domain or the C2B (K326A,K327A) domain, designated as syt1K189-192A or syt1K326,327A, respectively, in cultured cortical neurons from syt1−/− mice (Geppert et al., 1994). We expressed wild-type syt1 as a positive control or GFP alone as a negative control. Transgenes expressing wild-type or mutant Syt1 resulted in similar protein expression levels as assessed by Western blot analysis, whereas Syt1 was undetectable for the negative control (Fig. 1C,D). As shown in Figure 1, E and F, all three Syt1 variants were correctly trafficked to synaptic vesicles as their immunofluorescence signals had a high correlation with those of Synaptophysin-1 (Syph1), a synaptic vesicle marker (Courtney et al., 2019).
Both synaptotagmin-1 C2A and C2B domain polybasic patches are required for evoked release
Next, we measured evoked IPSCs (eIPSCs) from cultured cortical neurons from Syt-1 KO mice expressing the transgenes using field stimulation (Fig. 2A). Evoked release was nearly abolished in KO neurons expressing GFP alone (Fig. 2B), consistent with previous reports (Geppert et al., 1994). Expression of wild-type syt1 restored evoked release. By contrast, evoked release was greatly diminished in neurons expressing syt1K326,327A. Interestingly, release was also largely reduced in neurons expressing syt1K189-192A.
Effect size estimation (Ho et al., 2019) for evoked release parameters. A, The mean differences in eIPSC amplitude for three comparisons against the shared control (cells expressing WT Syt1) are shown as Cumming estimation plots. The raw data are plotted on the top axes. Bottom, Mean differences are plotted as bootstrap sampling distributions. Each mean difference is depicted as a dot. Each 95% confidence interval is indicated by the ends of the vertical error bars. The unpaired mean difference (in nA) between WT and GFP is −3.65 [95.0% CI, −4.39, −2.87], between WT and K189-192A is −3.21 [95.0% CI, −3.98, −2.38], and between WT and K326-327A is −3.55 [95.0% CI, −4.31, −2.77]. B, Effect size estimation as in A, for eIPSC charge. The unpaired mean difference (in nC) between WT and GFP is −476 [95.0% CI, −664, −351], between WT and K189-192A is −443 [95.0% CI, −642, −310], and between WT and K326-327A is −493 [95.0% CI, −690, −369]. C, Effect size estimation as in A, for time-to-peak values. The unpaired mean difference (in ms) between WT and K189-192A is 7.48 [95.0% CI, 4.8, 10.8], between WT and K326-327A is 5.72 [95.0% CI, 3.64, 9.25]. The unpaired mean difference between WT and GFP alone is 74.4 [95.0% CI, 43.0, 117]. The values for GFP alone are not displayed, as they are much larger and make it difficult to visualize the differences between WT and mutants. D, Effect size estimations for eIPSC decay times. The unpaired mean difference (in ms) between WT and GFP alone is 23.4 [95.0% CI, −82.3, 153], between WT and K189-192A is −51.0 [95.0% CI, −125, 24.6], and between WT and K326-327A is −92.0 [95.0% CI, −169, −15.8]. In all cases, 5000 bootstrap samples were taken; the confidence interval is bias corrected and accelerated. Traces with a very weak response were excluded from analysis for C and D because the time to peak or the decay time could not be determined reliably (Fig. 2). Download Figure 2-1, EPS file (1.5MB, eps) .
Quantification of evoked release parameters indicated the peak amplitude of the IPSCs and the charge transfer (integral of the IPSC traces) were significantly lower in neurons expressing GFP alone or the mutants than those expressing WT Syt1 (Fig. 2C,D, Extended Data Fig. 2-1). The time to reach the (negative) eIPSC peak following stimulation was slower for cells expressing either polybasic patch mutant compared with those expressing WT Syt1 (Fig. 2E, Extended Data Fig. 2-1), but much faster than those expressing GFP (mean ± SEM = 82.9 ± 19.1 ms, n = 23). Compared with neurons expressing WT Syt1, the decay of the eIPSC traces was slightly faster for cells expressing the polybasic patch mutants (Fig. 2F,G, Extended Data Fig. 2-1). However, the slow eIPSC decays for GABAergic neurons in culture may be dominated by receptor dynamics (Jones and Westbrook, 1996) obscuring true release kinetics.
We normalized the amplitudes of averaged IPSCs to compare the kinetics of the evoked responses (Fig. 2G). Compared with neurons expressing WT Syt1, there was a slight delay for the onset of release and the (negative) peak was reached slightly later for the mutants (Fig. 2G, inset), consistent with the analysis of individual traces above. Overall, these results indicate that the poly-lysine patch in either C2 domain is critical for evoked release.
Spontaneous release and the readily releasable pool of vesicles are not affected by Syt1 polybasic patch mutations
We next tested whether the polybasic patch mutations affected spontaneous release. Miniature IPSCs were recorded from resting sty1−/− neurons expressing GFP alone, wild-type Syt1, or the polybasic patch mutants Syt1K189-192A or Syt1K326,327A (Fig. 3). Although the miniature IPSC frequency was elevated twofold to threefold in neurons lacking Syt1 as reported previously (Littleton et al., 1993; Liu et al., 2014; Bai et al., 2016; Vevea and Chapman, 2020), the frequency was similar in neurons expressing wild-type or mutant Syt1. Furthermore, there were no significant differences for mIPSC amplitudes, rise times, or decay times among the experimental groups (Fig. 3C–E). Thus, the polybasic patch mutations do not affect spontaneous release significantly in this preparation.
Figure 3.
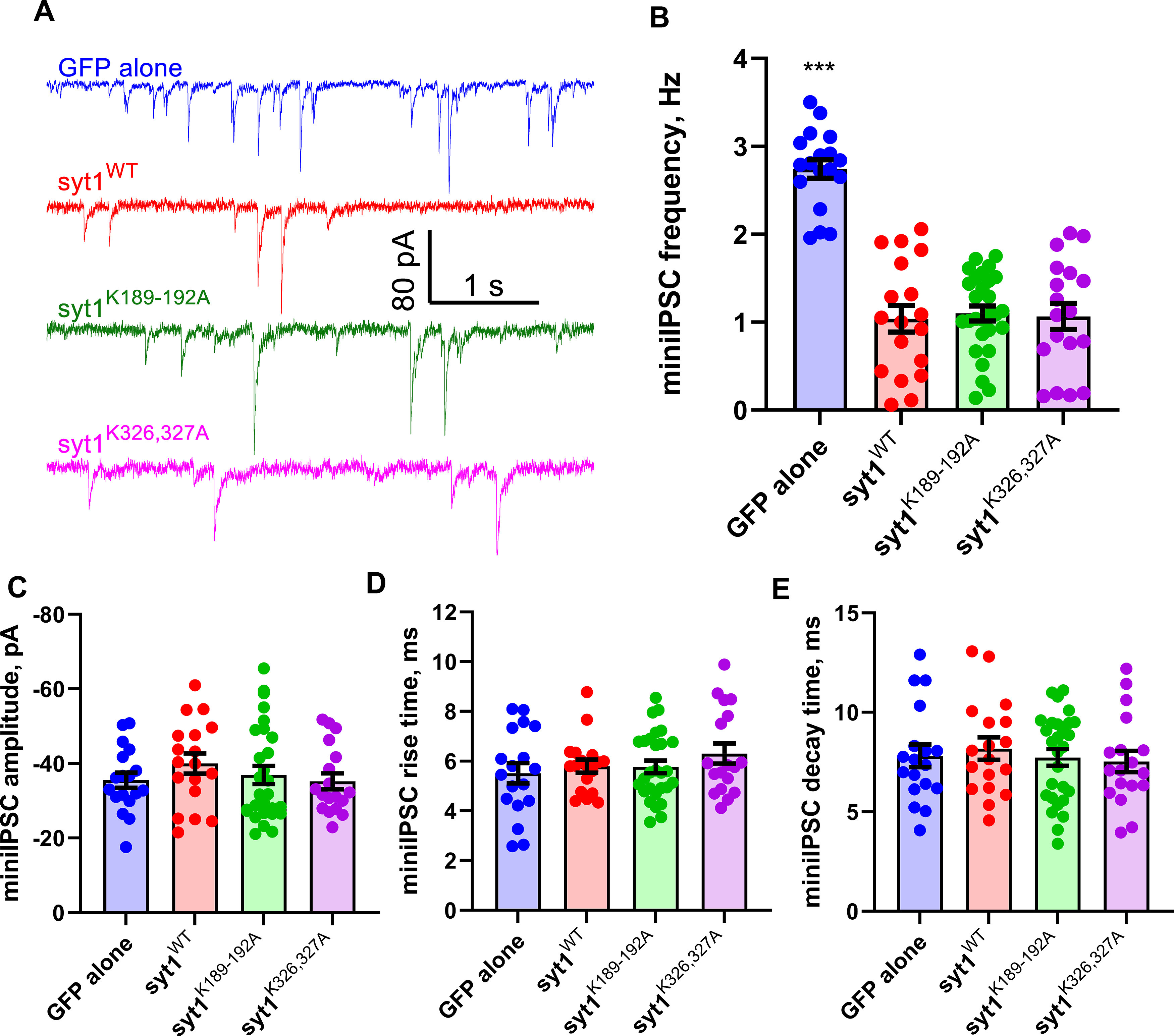
Syt1 C2A and C2B polybasic patch mutations do not affect spontaneous release. A, Representative current traces from voltage-clamped, resting cortical mouse syt−/− neurons expressing the indicated transgenes. B–E, Quantification of mIPSC parameters from traces such as the ones shown in A. Apart from an increase in the mIPSC frequency for neurons lacking Syt1 (B), there are no significant differences among the experimental groups for mIPSC amplitude (C), rise time (D), or decay time (E). mIPSC frequencies were (mean ± SEM; Hz) GFP alone, 2.74 ± 0.10; WT, 1.04 ± 0.15; syt1K189-192A, 1.1 ± 0.09; syt1K326,327A, 1.07 ± 0.15. mIPSC amplitudes were (mean ± SEM; pA) GFP alone, −35.54 ± 2.04; WT, −40.00 ± 2.69; syt1K189-192A, −36.95 ± 2.42; syt1K326,327A, −35.22 ± 2.13. mIPSC rise times were (mean ± SEM; ms) GFP alone 5.52 ± 0.41; WT, 5.80 ± 0.27; syt1K189-192A. 5.77 ± 0.26; syt1K326,327A, 6.31 ± 0.40. mIPSC decay times were (mean ± SEM; ms) GFP alone, 7.81 ± 0.57; WT, 8.183 ± 0.56; syt1K189-192A, 7.74 ± 0.42; syt1K326,327A, 7.536 ± 0.53 (n = 18, 18, 28, and 18 cells tested for GPF alone, WT syt1, syt1K189-192A, and syt1K326,327A, respectively. Cells were prepared from three syt1−/− pups. For B–E, we used Kruskal–Wallis test, followed by Dunn's multiple comparisons test to compare mutants against WT Syt1; ***p < 0.001.
The reduction in evoked release we observed in neurons expressing the polybasic patch mutants Syt1K189-192A or Syt1K326-327A could be because of a defect in the size of the RRP of synaptic vesicles. To test this possibility, we applied hypertonic sucrose to the four groups of neurons (Fig. 4) and integrated the phasic currents to estimate the total RRP size (Rosenmund and Stevens, 1996). In neurons lacking Syt1, there was a twofold to threefold reduction in the RRP size, whereas the RRP sizes were similar for syt1−/− neurons expressing wild-type Syt1 or the polybasic patch mutants (Fig. 4, Extended Data Fig. 4-1). Thus, the polybasic patch mutations do not substantially affect RRP size measured by hypertonic sucrose application.
Effect size estimation for readily releasable pool sizes. The mean differences in hypertonic sucrose-induced RRP size estimates for three comparisons against the shared control (cells expressing WT Syt1) are shown as a Cumming estimation plot. The raw data are plotted on the top axes. Bottom, mean differences are plotted as bootstrap sampling distributions. Each mean difference is depicted as a dot. Each 95% confidence interval is indicated by the ends of the vertical error bars. The unpaired mean difference (in nC) between WT and GFP is −3.77 [95.0% CI, −4.66, −2.84], between WT and K189-192A is −0.433 [95.0% CI, −1.5, 0.669], and between WT and K326-327A is −0.466 [95.0% CI, −1.59, 0.987]. Download Figure 4-1, EPS file (518.8KB, eps) .
Polybasic mutations in either C2 domain impairs membrane binding
To uncover the molecular mechanism by which the polybasic mutations impair evoked neurotransmitter release, we measured the membrane binding affinities and kinetics of both mutant C2AB domains, using a recently developed single-molecule approach (Ma et al., 2017) based on OTs. A single Syt1 C2AB domain was tethered between a silica bead and a polystyrene bead, forming a dumbbell in solution suspended by optical traps (Fig. 5A). The silica bead was coated with a lipid bilayer (Ma et al., 2017) that contained 85 mol% 1-palmitoyl-2-oleoylphosphatidylcholine (POPC), 10 mol% 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), 5 mol% PI(4,5)P2 and 0.03% mol% biotin-PEG-DSPE to mimic the plasma membrane. The Syt1 C2AB domain was attached to the lipids through biotin-streptavidin interaction via an N-terminal 73 amino acid flexible polypeptide linker and pulled from the C terminus via a 2260 bp DNA handle. Dynamic C2AB-membrane binding and unbinding events were detected by the corresponding changes in the extension of the protein-DNA tether. We previously determined the membrane binding affinities of the wild-type tandem C2AB domain (Fig. 5B) and the individual C2B domain of Syt1 (Ma et al., 2017). Here, we applied the single-molecule approach to characterize membrane binding of Syt1 with the polybasic patch mutations in either C2 domain or a calcium-binding mutation in the C2B domain. All our experiments were conducted in the presence of 100 µm Ca2+ in the solution.
Figure 5.
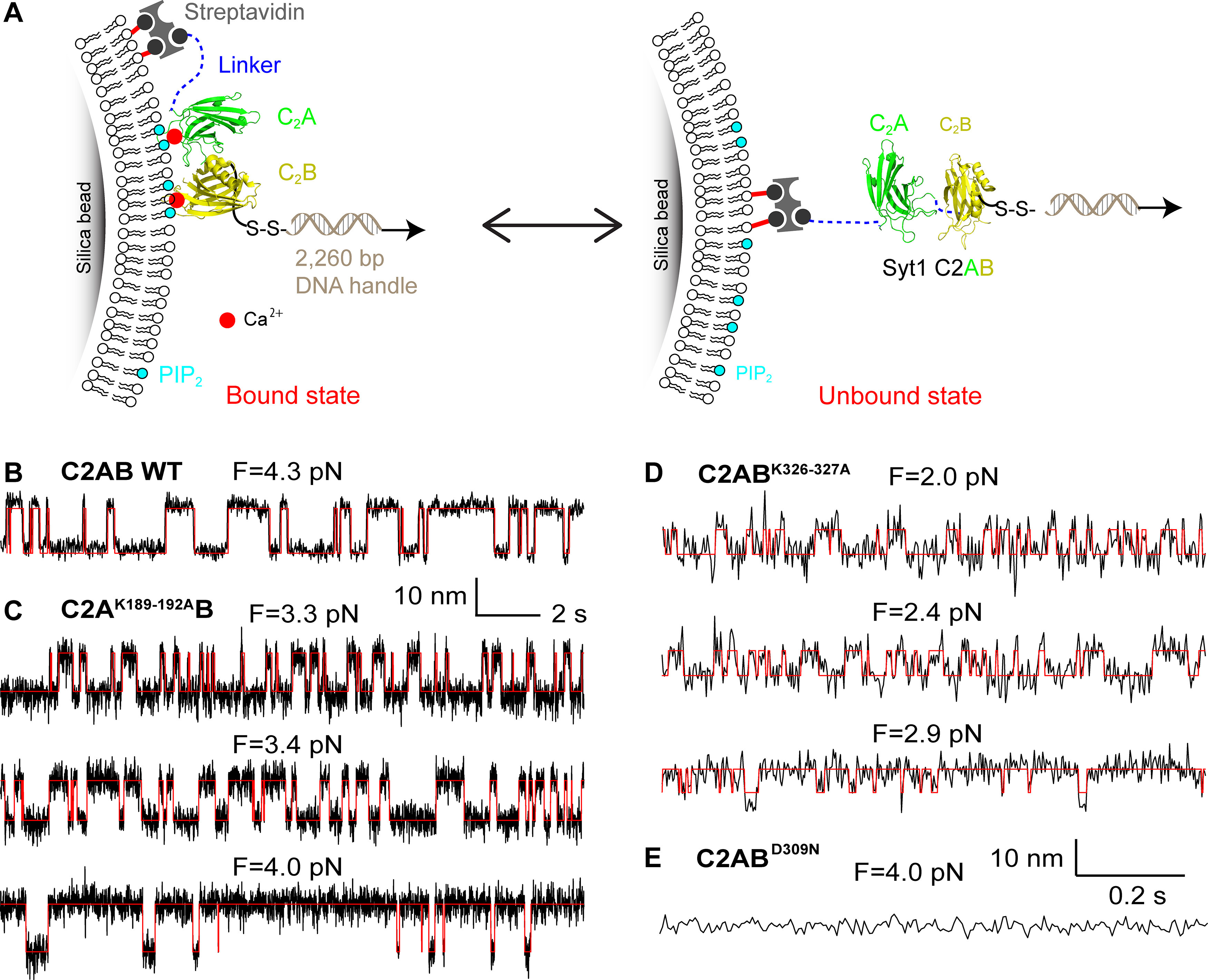
The neutralization mutations in Syt1 C2AB domain impair its membrane binding as revealed by optical tweezers. A, Schematic of the experimental setup. The Syt1 construct was directly attached to the supported bilayer via biotin-streptavidin interactions at the N terminus and cross-linked to a DNA handle via a disulfide bond. The other end of the DNA was attached to a polystyrene bead (data not shown). Membrane binding and unbinding of the C2AB domain was detected by the corresponding extension change of the protein-DNA tether. The bilayer is composed of 85 mol% POPC, 10 mol% DOPS, 5 mol% PI(4,5)P2, and 0.03% mol% biotin-PEG-DSPE. B–E, Extension-time trajectories at the indicated constant mean forces showing dynamic C2AB binding of wild-type StyI C2AB (B), C2AK189-192AB (C), or C2ABK326-327A (D), or no membrane binding of C2ABD309N (E). Note that the extension-time trajectories in B and C and D and E share the same scale bars.
We bacterially expressed and purified the soluble C2AB domains with a polybasic patch mutation in the C2A (C2AK189-192AB) or the C2B domain (C2ABK326-327A). All recombinant proteins were well folded, and the mutations did not affect the thermal stability of the proteins (Fig. 6). We also tested the mechanical stability of these proteins by monitoring the force-induced unfolding dynamics using OT. The unfolding forces and the associated extension changes were indistinguishable for the wild-type or mutant C2 domains, either for the individual domains or when they were in tandem, as shown in Figure 7.
Figure 6.
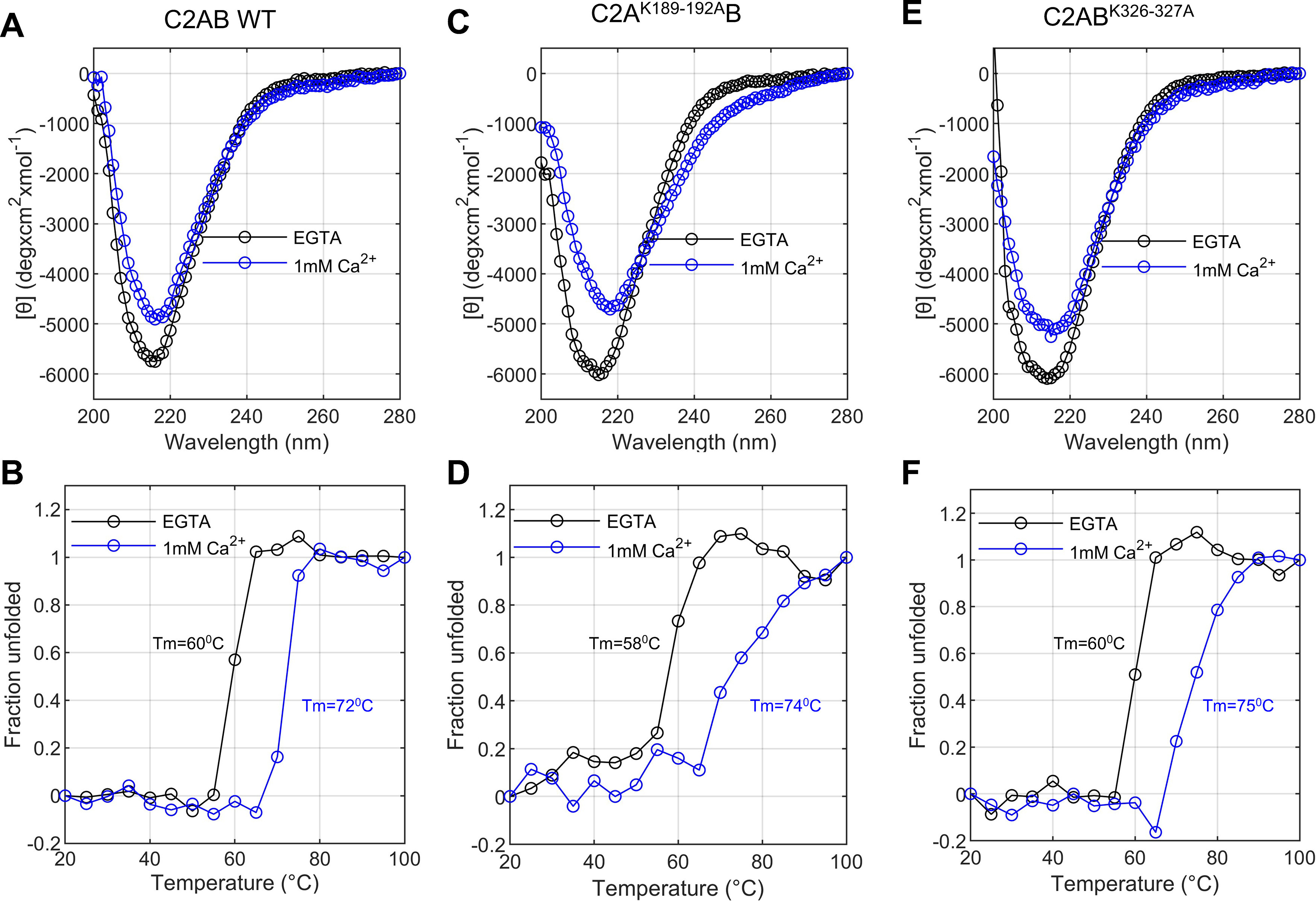
Circular dichroism analysis shows the purified recombinant proteins are well folded, and the polybasic patch mutations do not affect protein stability. A, C, E, Molar ellipticity as a function of wavelength for purified recombinant wild-type Syt1 C2AB domains (A) and Syt1 C2AB domains bearing mutations in the C2A (C2AK189-192AB; C) or the C2B (C2ABK326-327A; E) polybasic patch as indicated. Data were collected in the absence (1 mm EGTA, black) or presence of 1 mm Ca2+ (blue) at 25°C. Each spectrum represents the average of three separate recordings. B, D, F, Thermal denaturation curves for WT (B), C2AK189-192AB (D), or C2ABK326-327A (F), measured at 217 nm.
Figure 7.

The charge neutralization mutations do not significantly alter folding of the C2 domains. A, Diagram showing force-induced unfolding of C2 domains to access their structure and stability based on the unfolding force and extension change associated with the C2 domain unfolding. B, Force-extension curves obtained by pulling a single Syt1 C2AB, C2A, or C2B domain with a trap separation speed of 10 nm/s. The C2 domain was attached to the lipid bilayer coated on the silica bead in the presence of 100 µm Ca2+. Different C2 transitions are marked with red dashed ovals for reversible membrane binding and unbinding, magenta arrows for C2A domain unfolding, and red arrows for C2B domain unfolding. The C2 domain unfolding force (Fu) and its associated extension change (Δx) were determined for each C2 unfolding event, as indicated. C, Unfolding force histograms of C2A and C2B domains in the wild-type or mutant Syt1 C2AB constructs. The total number of unfolding events (N) and the average unfolding force (F) and its SD (in parenthesis) are shown. The close average unfolding force for the mutant or wild-type C2A or C2B domain indicates that the mechanical stability of the C2 domain is not altered by charge neutralization mutations. D, Extension change histograms of C2A and C2B domains. Comparisons of the extension changes between wild-type and mutant C2 domains suggest that the charge neutralization mutations barely affect the structures of the C2 domains.
We detected reversible membrane binding and unbinding transitions of C2AK189-192AB in the force range of 2.6–4.2 pN as is indicated by the two-state extension flickering at constant mean forces (Fig. 5C). Here, the states with higher and lower average extensions represent the unbound and the membrane-bound states, respectively. The state probabilities and binding and unbinding rates were determined by hidden Markov modeling (Zhang et al., 2016). As expected, the probability of the mutant C2 domain being in the unbound state increases as force increases, whereas the unbinding rate and binding rate approximately exponentially increases and decreases, respectively (Figs. 5C, 8). The analysis also revealed an equilibrium force with equal probabilities being in both states that represents the membrane binding affinity of the protein. C2AK189–192AB has a smaller equilibrium force (3.5 pN) than that of wild-type Syt1 (4.7 pN; Table 1), which indicates that neutralization of the basic patch in C2A reduces the membrane binding affinity of Syt1. Detailed data analysis (Zhang et al., 2016; Ma et al., 2017) revealed that C2AK189-192AB binds the membrane with an energy of 9.1 (±0.9) kBT (mean ± SEM), compared with the 12.8 (±0.8) kBT binding energy for the wild-type C2AB. The reduction in the binding energy is mainly caused by a decrease in the membrane association rate constant, with insignificant change in the dissociation rate constant. These observations suggest that the four lysine residues in C2A help Syt1 C2AB domain bind to the membrane through their long-range electrostatic interactions with the membrane, and neutralization of the basic residues reduces the binding rate constant. We similarly measured the membrane binding affinity and kinetics of C2ABK326-327A. Neutralization of the two lysine residues in C2B significantly reduces the equilibrium force to 2.1 (±0.4) pN and membrane binding energy to 5.9 (±0.4) kBT (Figs. 5D, 8). Interestingly, the mutations decrease the binding energy mainly by increasing the dissociation rate constant, with only a small decrease in the association rate constant (Table 1). Finally, we tested membrane binding by C2ABD309N as a control that impairs Ca2+-dependent StyI C2AB membrane binding (Fernandez et al., 2001) and neurotransmitter release (Nishiki and Augustine, 2004). Our single-molecule assay did not detect any membrane binding by C2ABD309N under these conditions (Fig. 5E), confirming that calcium promotes membrane binding of the C2AB domain. In conclusion, both the C2A domain K189-192A and the C2B domain K326-327A mutations likely impair neurotransmitter release by reducing membrane binding of the Syt1 C2AB domain required for release.
Figure 8.

Force-dependent C2AB unbinding probabilities and binding and unbinding rates. All the experimental measurements (symbols) are simultaneously nonlinearly fit by a theoretical model to account for the effect of force on protein binding and unbinding (curves) to derive the membrane binding energy and the rate constants (see above, Materials and Methods) for Syt1 C2AK189-192AB (top two rows) and C2ABK326-327A (bottom two rows).
Table 1.
Comparison of membrane binding energy and kinetics of wild-type and mutant Sty1 C2AB domains
| Syt1 | Equilibrium force (pN) | Binding energy with tether (Eb) (kBT) | Binding energy without tether (Eon) (kBT) | Log10[kb (s−1)] | Log10[kon (M−1s−1)] | Log10[kub (s−1)] |
|---|---|---|---|---|---|---|
| WT | 4.7 (0.2) | 10.8 (0.8) | 12.8 (0.8) | 4.6 (0.4) | 5.4 (0.4) | 0 (0.2) |
| K189-192A | 3.5 (0.6) | 7.1 (0.9) | 9.1 (0.9) | 3.5 (0.2) | 4.3 (0.2) | 0.4 (0.3) |
| K326-327A | 2.1 (0.4) | 3.9 (0.4) | 5.9 (0.4) | 3.9 (0.1) | 4.7 (0.1) | 2.2 (0.1) |
The flexible tether linking the C2 domain(s) to the bilayer on the silica bead increases the likelihood of rebinding, affecting the apparent binding energy. We used a previously developed model to account for this effect, as in Lu et al. (Ma et al., 2017).
Discussion
Two lines of evidence led to the idea that the C2B domain is the functionally more important C2 subunit of Syt1, with the C2A domain mostly playing a faciliatory role. First, impairing calcium binding to C2A has a milder effect than impairing calcium-binding to the C2B domain (Fernández-Chacón et al., 2001, 2002; Mackler et al., 2002; Robinson et al., 2002; Stevens and Sullivan, 2003; Nishiki and Augustine, 2004). Second, the C2B domain of Syt1, but not the C2A domain, interacts with the SNARE proteins (Rickman et al., 2004; Brewer et al., 2015; Zhou et al., 2015; Wang et al., 2016; Zhou et al., 2017; but see Shao et al., 1997; Fernandez et al., 1998; Park et al., 2015) that drive membrane fusion (Südhof and Rothman, 2009). However, recent work has challenged the merely facilitatory role attributed to C2A, as it was found that the C2A domain is crucial for robust evoked release in flies (Striegel et al., 2012; Lee et al., 2013; Bowers and Reist, 2020a) and in mice (Courtney et al., 2019). Here, we asked what feature of the C2A domain may be responsible for its critical role.
Because previous work suggested that calcium binding to the C2A domain may not be an essential functional feature of this C2 domain, we explored the role of another potentially important region, a highly conserved stretch with four tandem lysines (residues 189–192). The C2B domain similarly possesses a conserved polybasic patch on one side (residues 324–327). Separate functional studies of these two polybasic regions led to inconsistent, or even conflicting results. Neutralization of the C2A polybasic region through mutagenesis only increased spontaneous release in the Drosophila larvae NMJ (Mace et al., 2009), whereas injection of peptides into the squid giant nerve terminals (Bommert et al., 1993) or PC12 cells (Thomas and Elferink, 1998) suggested the C2A polybasic patch is essential for evoked release. Charge inversion or neutralization mutations of the C2B polybasic region resulted in a 40–50% reduction in evoked release but with divergent effects on spontaneous release at the Drosophila NMJ (Mackler and Reist, 2001; Loewen et al., 2006) and in mouse hippocampal neuronal cultures (Borden et al., 2005; Li et al., 2006; Chang et al., 2018) or a fivefold to sixfold reduction in evoked release accompanied with an ∼60% reduction in the RRP size, also in mouse hippocampal neuronal cultures (Chang et al., 2018). These discrepancies can be because of differences in the species or preparations used, but as there is no consensus among these results, we decided to test the roles of both C2 domain polybasic patches side by side to reveal their individual contributions in mammalian synapses.
We found that evoked release was reduced ∼6 and >10-fold in syt1−/− mouse cortical neuronal cultures expressing charge-neutralization mutations in the C2A and C2B polybasic patches, respectively, compared with those expressing WT Syt1. The mutations also slightly slowed the initial release kinetics. The severe reductions in the amount of evoked release were not because of differences in expression levels or the sizes of the RRPs. Consistent with our results, some previous studies also reported no effect on the RRP size of the K326A,K327A mutation both for cultured hippocampal (Borden et al., 2005; Li et al., 2006) and cortical neurons (Courtney et al., 2019). However, another study (Chang et al., 2018), using a different mutation in the C2B polybasic patch (K325A,K327A), found an ∼60% reduction in the RRP size. It is possible that the disagreement is because of the different mutations introduced, but other causes cannot be excluded because differences in RRP estimates can arise depending on culture conditions (Liu et al., 2009, 2013) and methods used for the RRP estimates (Neher, 2015; Kaeser and Regehr, 2017; Silva et al., 2021). The polybasic patch mutations did not affect spontaneous release here, yet we had previously found that Syt1 is a contributor to spontaneous release in GABAergic hippocampal neurons, albeit a minor one (Courtney et al., 2018). This suggests that the polybasic patch mutations may not be important for spontaneous release, the assay may not be sensitive enough to detect minor differences, spontaneous GABA release mechanisms may differ between hippocampal and cortical preparations, or any combination of these. In summary, both polybasic patches in C2A and C2B are important for evoked release in mammalian synapses. Because the RRP sizes were not different, the most likely explanation for the reduction in evoked release in the mutants was a reduction in release probability.
We reasoned that the effects of the C2A and C2B domain polybasic patch mutations we observed on evoked release could be because of the disruption of putative direct interactions of these patches with acidic membranes (Bai et al., 2004a; Vennekate et al., 2012; Bradberry et al., 2019; Nyenhuis et al., 2019). Other potential interactions are likely to be less relevant. For example, potential interactions between voltage-gated calcium channels (Cavs) and Syt1 were reported (Zhong et al., 1999; Cohen et al., 2003). However, coexpressing syt1 or syt1K189-192A with Cav in oocytes produced the same minor effects on channel properties (Cohen et al., 2003). Later work showed that Cavs are recruited to liquid-like condensates at the active zone via their C termini (X. Wu et al., 2019, 2021) through multiple low-affinity and redundant interactions, most prominently with RIM and RIM-BP (Hibino et al., 2002; Kaeser et al., 2011; Liu et al., 2011; Rizalar et al., 2021); direct Syt1-Cav interactions are not thought to play a significant role (Nanou and Catterall, 2018; Rizalar et al., 2021). The C2 domain mutations tested in our study are also unlikely to affect Syt1–SNARE interactions, for the following reasons. First, crystal structures reveal a C2B-ternary SNARE complex binding interface that is away from the polybasic patch (Zhou et al., 2015, 2017), allowing simultaneous binding of C2B to membranes and the SNARE complex (Wang et al., 2016). Second, the neutralization mutations minimally affect binding of Syt1 to the binary t-SNARE complex (Rickman et al., 2004; Bai et al., 2004b; Chapman, 2008). For example, the mutations K326-327A reduce binding of the soluble recombinant C2AB domains of Syt1 to the t-SNARE acceptor complex formed by Syntaxin1 and SNAP25 (Rickman et al., 2004), but the stoichiometry of binding is unaffected and the mutations only shift the from 3 µM to 10 µM in solution (Bai et al., 2004b). This would translate to an ∼1.2 kT decrease in the free energy of binding to the t-SNAREs, much smaller than the ∼6.9 kT reduction we observed for binding to PI(4,5)P2 containing membranes (12.8 kT vs 5.9 kT for wild-type vs K326-327A mutant; Table 1). Third, extensive experiments have demonstrated a key role for Syt1-membrane interactions in the presence of SNARE proteins both in biochemical experiments (Honigmann et al., 2013; Schupp et al., 2016; Wang et al., 2016) and in neurons (Chang et al., 2018). Overall, these studies suggested that the defects we observed in neurons expressing syt1K189-192A or syt1K326,327A could be mainly because of the disruption of interactions between C2 domain polybasic patches and acidic membranes.
To probe C2 domain–membrane interactions, we used a single-molecule assay developed recently (Ma et al., 2017). Bulk assays that probe protein–membrane interactions are convenient and very valuable but suffer from a number of challenges. First, they often cannot resolve intermediates, energetics, and kinetics of protein–membrane binding because of difficulties in synchronizing the reactions and in applying force to proteins or membranes (Zhang et al., 2013). Second, Syt1 C2 domains can bridge membranes (Hui et al., 2011; Kuo et al., 2011; Seven et al., 2013) and/or oligomerize (Bai et al., 2000; Fukuda and Mikoshiba, 2000; Tagliatti et al., 2020), likely affecting the bulk measurements. Such interactions may well be physiologically important, but their effects are difficult to disentangle from direct binding-unbinding events in bulk experiments. Our single-molecule measurements avoid possible complications from membrane bridging as there is only one membrane to bind to. Furthermore, multimerization of Syt1 C2B domains are absent in our assay. Thus, single-molecule measurements directly probe C2 domain–membrane interactions in the absence of potential challenges faced in bulk measurements.
The C2B domain polybasic patch is known to bind acidic lipids, preferably PI(4,5)P2, and this interaction can be detected in bulk assays in the absence of calcium (Bai et al., 2004a; Bradberry et al., 2019; Nyenhuis et al., 2019). By analogy, the C2A domain polybasic patch could contribute to membrane binding. Indeed, using single-molecule atomic force microscopy, Takahashi et al. (2010) reported reduced binding of the C2AB domain to supported bilayers when the C2A polybasic patch was neutralized, although no binding energies could be extracted from the measurements carried under far from equilibrium conditions. In addition to the two polybasic patches, Syt1 C2A and C2B domains can also bind membranes through their calcium-binding loops. When bound, calcium ions coordinate between highly conserved aspartates in the C2A and C2B calcium-binding loops and head groups of negatively charged lipids in the membrane (Chapman, 2008). Hydrophobic residues at the tips of the calcium-binding loops in both C2 domains penetrate into the membrane in the presence of calcium, fortifying membrane binding (Chapman and Davis, 1998; Bai et al., 2000; Chapman, 2008). Thus, there are at least four membrane binding sites on Syt1, two on each C2 domain—a calcium-dependent site and a polybasic patch. Our results show that the contributions of these sites to overall binding is not additive, suggesting highly cooperative interactions.
We note that some membrane binding activities that the single-molecule OT assay fails to detect have been reported using ensemble measurements. Notably, membrane binding of a C2AB construct carrying a mutation that prevents calcium binding to the C2B domain can be detected in bulk assays in the presence of calcium (Hui et al., 2006; but see Fernandez et al., 2001), presumably via calcium-dependent C2A–membrane interactions (Chapman and Davis, 1998; Davis et al., 1999; Chapman, 2008). Similarly, membrane binding for the C2AB domain can be detected using bulk liposome binding assays in the absence of calcium (Bai et al., 2004a; Bradberry et al., 2019; Nyenhuis et al., 2019), but not in our OT assay (Ma et al., 2017). Finally, in the presence of calcium, the C2A domain avidly binds membranes containing PS in bulk experiments (Chapman and Davis, 1998; Davis et al., 1999; Chapman, 2008), but the OT assay fails to detect the interaction (Ma et al., 2017). These apparent discrepancies are actually expected for several reasons. First and most likely, some of these apparent discrepancies may be because of the relatively low amount of PS (10 mole %) we included in the bilayers. Second, bulk assays are often more sensitive to detect weak interactions (per molecule), because the ensemble average of signals from a large number of molecules are detected (Leckband and Israelachvili, 2001). Third, the OT assay measures dynamic binding and unbinding events under a load, and these must occur within a certain range of force and time scales to be detectable. By contrast, many bulk measurements probe equilibrium properties. That is, even if a bulk equilibrium assay reports a low dissociation coefficient , the dynamics may not be readily detectable in the OT assay. Subtler is the effect of applied load on dissociation kinetics; if unbinding is accelerated under sufficiently low applied forces, binding-unbinding events may not be detectable in the OT assay, even if such events can be detected in single-molecule fluorescence assays where no load is applied. Finally, Syt1–membrane interactions may be promoted by high membrane curvature (Martens et al., 2007; Hui et al., 2009) so that binding to small liposomes in bulk experiments may be stronger than binding to a relatively flat supported bilayer in our OT assay.
The results above are important for our interpretation of the contribution of the C2A and C2B domain polybasic patches to Syt1–membrane interactions. In particular, although the C2A domain binds acidic membranes (detected using bulk assays), binding has not been reported in the absence of calcium, at least in the presence of moderate amounts of PS (Chapman and Davis, 1998; Davis et al., 1999; Chapman, 2008), leading to the idea that the C2A polybasic patch does not contribute significantly to the overall Syt1–membrane interactions. Our results clearly demonstrate that both the C2A and the C2B domain polybasic patches significantly enhance calcium-dependent lipid binding of the C2AB domain. Given the preference of the C2A domain for binding PS over PI(4,5)P2, and the fact that the inner leaflet of the plasma membrane contains ∼20 mole % PS (van Meer et al., 2008), about twice the amount we used in our OT assay, it is likely that the C2A domain polybasic patch–plasma membrane interactions are even stronger in vivo and physiologically relevant.
How can we explain the cooperativity between the four binding sites? The simplest idea is that multiple attractive interactions increase the binding rate and dramatically slow the overall kinetics of unbinding, even if individual binding sites each contribute a relatively small interaction energy. This is a well-known phenomenon for polymer adsorption to surfaces (O'Shaughnessy and Vavylonis, 2005). Even if individual segments on a random polymer adsorb onto a surface with a small binding energy of order and come off rapidly because of thermal energy, as one segment unbinds, another can bind, maintaining multiple binding sites occupied at any given moment. For long polymers the probability that all segments unbind at the same time is very small, and adsorption is essentially irreversible. In the case of Syt1 C2AB domains, the cooperativity of the binding sites is certainly more complicated, as the domains are well folded and the surface they bind is soft, allowing multiple bound-state configurations. This complexity is likely reflected in our finding that neutralizations of the C2A or the C2B polybasic sites both lead to a reduction of the overall binding energy but through different pathways. In the former case, the binding rate was >10-fold slower compared with wild-type C2AB, with a modest effect on the unbinding rate. For the latter, the unbinding rate was accelerated two orders of magnitude, with a smaller effect on the binding rate.
Our results are broadly consistent with the observation that Syt1 mutations that inhibit calcium-binding to the C2A domain while partially mimicking the charge neutralization by calcium binding (D232N) do not reduce synchronous release, and even enhance it (Yoshihara and Littleton, 2002; Stevens and Sullivan, 2003; Pang et al., 2006). In these mutants, the calcium-dependent binding site on C2A would be replaced by a partial calcium mimic, turning it to a calcium-independent binding site [Note, however, that these mutants do not display increased calcium-independent binding to anionic lipids (Bai et al., 2000), so the actual picture is likely to be more complex.]. By contrast, inhibiting calcium binding by an aspartate-to-glutamate (D→E) substitution removes the calcium-dependent binding site on C2A altogether and results in a dramatic decrease in evoked release (Striegel et al., 2012). The fact that similar D→N substitutions in the C2B domain calcium-binding site result in severe inhibition of evoked release (Mackler et al., 2002) likely reflect the fact that the C2B domain binds the SNARE complex and that calcium binding to the C2B domain likely leads to a reorientation of the C2B-SNARE complex (Bai et al., 2016; Brunger et al., 2018a; Z. Wu et al., 2021), its dissociation (Rothman et al., 2017; Voleti et al., 2020), or some other specific rearrangement required for triggering membrane fusion.
Cooperativity between the C2 domains of Syt1 was noted long ago, but the nature of this cooperativity has proven difficult to pin down (Chapman, 2008; Evans et al., 2016). Our results provide new insights into this question and show that both inter- and intra-C2 domain binding sites of Syt1 contribute to membrane binding in a highly cooperative manner.
Footnotes
This work was supported by the National Institutes of Health–National Institute of Neurological Disorders and Stroke (Grant R01 NS113236 to E.K. and R35 NS097362 to E.R.C.), National Institute of General Medical Sciences (Grant R35 GM131714 to Y.Z.), National Eye Institute (Grant R01EY010542 to E.K.), and National Institute of Mental Health (Grant R01 MH61876 to E.R.C.). E.R.C. is an investigator at the Howard Hughes Medical Institute. We thank the members of the Karatekin, Chapman, and Zhang labs for discussions and David Zenisek (Yale University) for discussions and feedback on the manuscript.
The authors declare no competing financial interests.
References
- Bai H, Xue R, Bao H, Zhang L, Yethiraj A, Cui Q, Chapman ER (2016) Different states of synaptotagmin regulate evoked versus spontaneous release. Nat Commun 7:10971. 10.1038/ncomms10971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Earles CA, Lewis JL, Chapman ER (2000) Membrane-embedded synaptotagmin penetrates cis or trans target membranes and clusters via a novel mechanism. J Biol Chem 275:25427–25435. 10.1074/jbc.M906729199 [DOI] [PubMed] [Google Scholar]
- Bai J, Tucker WC, Chapman ER (2004a) PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol 11:36–44. 10.1038/nsmb709 [DOI] [PubMed] [Google Scholar]
- Bai J, Wang CT, Richards DA, Jackson MB, Chapman ER (2004b) Fusion pore dynamics are regulated by synaptotagmin*t-SNARE interactions. Neuron 41:929–942. 10.1016/S0896-6273(04)00117-5 [DOI] [PubMed] [Google Scholar]
- Bommert K, Charlton MP, DeBello WM, Chin GJ, Betz H, Augustine GJ (1993) Inhibition of neurotransmitter release by C2-domain peptides implicates synaptotagmin in exocytosis. Nature 363:163–165. 10.1038/363163a0 [DOI] [PubMed] [Google Scholar]
- Borden CR, Stevens CF, Sullivan JM, Zhu Y (2005) Synaptotagmin mutants Y311N and K326/327A alter the calcium dependence of neurotransmission. Mol Cell Neurosci 29:462–470. 10.1016/j.mcn.2005.03.015 [DOI] [PubMed] [Google Scholar]
- Bowers MR, Reist NE (2020a) The C2A domain of synaptotagmin is an essential component of the calcium sensor for synaptic transmission. PLoS One 15:e0228348. 10.1371/journal.pone.0228348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers MR, Reist NE (2020b) Synaptotagmin: mechanisms of an electrostatic switch. Neurosci Lett 722:134834. 10.1016/j.neulet.2020.134834 [DOI] [PubMed] [Google Scholar]
- Bradberry MM, Bao H, Lou X, Chapman ER (2019) Phosphatidylinositol 4,5-bisphosphate drives Ca(2+)-independent membrane penetration by the tandem C2 domain proteins synaptotagmin-1 and Doc2β. J Biol Chem 294:10942–10953. 10.1074/jbc.RA119.007929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradberry MM, Courtney NA, Dominguez MJ, Lofquist SM, Knox AT, Sutton RB, Chapman ER (2020) Molecular basis for synaptotagmin-1-associated neurodevelopmental disorder. Neuron 107:52–64.e7. 10.1016/j.neuron.2020.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer KD, Bacaj T, Cavalli A, Camilloni C, Swarbrick JD, Liu J, Zhou A, Zhou P, Barlow N, Xu J, Seven AB, Prinslow EA, Voleti R, Häussinger D, Bonvin AMJJ, Tomchick DR, Vendruscolo M, Graham B, Südhof TC, Rizo J (2015) Dynamic binding mode of a Synaptotagmin-1-SNARE complex in solution. Nat Struct Mol Biol 22:555–564. 10.1038/nsmb.3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Choi UB, Lai Y, Leitz J, Zhou Q (2018a) Molecular mechanisms of fast neurotransmitter release. Annu Rev Biophys 47:469–497. 10.1146/annurev-biophys-070816-034117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Leitz J, Zhou Q, Choi UB, Lai Y (2018b) Ca(2+)-triggered synaptic vesicle fusion initiated by release of inhibition. Trends Cell Biol 28:631–645. 10.1016/j.tcb.2018.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Trimbuch T, Rosenmund C (2018) Synaptotagmin-1 drives synchronous Ca(2+)-triggered fusion by C2B-domain-mediated synaptic-vesicle-membrane attachment. Nat Neurosci 21:33–40. 10.1038/s41593-017-0037-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER (2008) How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem 77:615–641. 10.1146/annurev.biochem.77.062005.101135 [DOI] [PubMed] [Google Scholar]
- Chapman ER, Davis AF (1998) Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J Biol Chem 273:13995–14001. 10.1074/jbc.273.22.13995 [DOI] [PubMed] [Google Scholar]
- Chapman ER, Hanson PI, An S, Jahn R (1995) Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. J Biol Chem 270:23667–23671. 10.1074/jbc.270.40.23667 [DOI] [PubMed] [Google Scholar]
- Chapman ER, An S, Edwardson JM, Jahn R (1996) A novel function for the second C2 domain of synaptotagmin - Ca2+-triggered dimerization. J Biol Chem 271:5844–5849. 10.1074/jbc.271.10.5844 [DOI] [PubMed] [Google Scholar]
- Cohen R, Elferink LA, Atlas D (2003) The C2A domain of synaptotagmin alters the kinetics of voltage-gated Ca2+ channels Ca(v)1.2 (Lc-type) and Ca(v)2.3 (R-type). J Biol Chem 278:9258–9266. 10.1074/jbc.m210270200 [DOI] [PubMed] [Google Scholar]
- Courtney NA, Briguglio JS, Bradberry MM, Greer C, Chapman ER (2018) Excitatory and inhibitory neurons utilize different Ca(2+) sensors and sources to regulate spontaneous release. Neuron 98:977–991.e5. 10.1016/j.neuron.2018.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney NA, Bao H, Briguglio JS, Chapman ER (2019) Synaptotagmin 1 clamps synaptic vesicle fusion in mammalian neurons independent of complexin. Nat Commun 10:4076. 10.1038/s41467-019-12015-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AF, Bai J, Fasshauer D, Wolowick MJ, Lewis JL, Chapman ER (1999) Kinetics of synaptotagmin responses to Ca2+ and assembly with the core SNARE complex onto membranes. Neuron 24:363–376. 10.1016/s0896-6273(00)80850-8 [DOI] [PubMed] [Google Scholar]
- Evans CS, He Z, Bai H, Lou X, Jeggle P, Sutton RB, Edwardson JM, Chapman ER (2016) Functional analysis of the interface between the tandem C2 domains of synaptotagmin-1. Mol Biol Cell 27:979–989. 10.1091/mbc.E15-07-0503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez I, Ubach J, Dulubova I, Zhang X, Südhof TC, Rizo J (1998) Three-dimensional structure of an evolutionarily conserved N-terminal domain of syntaxin 1A. Cell 94:841–849. 10.1016/s0092-8674(00)81742-0 [DOI] [PubMed] [Google Scholar]
- Fernandez I, Araç D, Ubach J, Gerber SH, Shin O, Gao Y, Anderson RG, Südhof TC, Rizo J (2001) Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid binding machine. Neuron 32:1057–1069. 10.1016/s0896-6273(01)00548-7 [DOI] [PubMed] [Google Scholar]
- Fernández-Chacón R, Königstorfer A, Gerber SH, García J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Südhof TC (2001) Synaptotagmin I functions as a calcium regulator of release probability. Nature 410:41–49. 10.1038/35065004 [DOI] [PubMed] [Google Scholar]
- Fernández-Chacón R, Shin OH, Königstorfer A, Matos MF, Meyer AC, Garcia J, Gerber SH, Rizo J, Südhof TC, Rosenmund C (2002) Structure/function analysis of Ca2+ binding to the C2A domain of synaptotagmin 1. J Neurosci 22:8438–8446. 10.1523/JNEUROSCI.22-19-08438.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Mikoshiba K (2000) Calcium-dependent and -independent hetero-oligomerization in the synaptotagmin family. J Biochem 128:637–645. 10.1093/oxfordjournals.jbchem.a022796 [DOI] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC (1994) Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79:717–727. 10.1016/0092-8674(94)90556-8 [DOI] [PubMed] [Google Scholar]
- Hibino H, Pironkova R, Onwumere O, Vologodskaia M, Hudspeth AJ, Lesage F (2002) RIM binding proteins (RBPs) couple Rab3-interacting molecules (RIMs) to voltage-gated Ca(2+) channels. Neuron 34:411–423. 10.1016/s0896-6273(02)00667-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho J, Tumkaya T, Aryal S, Choi H, Claridge-Chang A (2019) Moving beyond P values: data analysis with estimation graphics. Nat Methods 16:565–566. 10.1038/s41592-019-0470-3 [DOI] [PubMed] [Google Scholar]
- Honigmann A, van den Bogaart G, Iraheta E, Risselada HJ, Milovanovic D, Mueller V, Müllar S, Diederichsen U, Fasshauer D, Grubmüller H, Hell SW, Eggeling C, Kühnel K, Jahn R (2013) Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nat Struct Mol Biol 20:679–686. 10.1038/nsmb.2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Bai J, Chapman ER (2006) Ca2+-triggered simultaneous membrane penetration of the tandem C2-domains of synaptotagmin I. Biophys J 91:1767–1777. 10.1529/biophysj.105.080325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Gaffaney JD, Wang Z, Johnson CP, Evans CS, Chapman ER (2011) Mechanism and function of synaptotagmin-mediated membrane apposition. Nat Struct Mol Biol 18:813–821. 10.1038/nsmb.2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui EF, Johnson CP, Yao J, Dunning FM, Chapman ER (2009) Synaptotagmin-mediated bending of the target membrane is a critical step in Ca(2)+-regulated fusion. Cell 138:709–721. 10.1016/j.cell.2009.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL (1996) The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci 19:96–101. 10.1016/S0166-2236(96)80037-3 [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Regehr WG (2017) The readily releasable pool of synaptic vesicles. Curr Opin Neurobiol 43:63–70. 10.1016/j.conb.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Südhof TC (2011) RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell 144:282–295. 10.1016/j.cell.2010.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo W, Herrick DZ, Cafiso DS (2011) Phosphatidylinositol 4,5-bisphosphate alters synaptotagmin 1 membrane docking and drives opposing bilayers closer together. Biochemistry 50:2633–2641. 10.1021/bi200049c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leckband D, Israelachvili J (2001) Intermolecular forces in biology. Q Rev Biophys 34:105–267. 10.1017/s0033583501003687 [DOI] [PubMed] [Google Scholar]
- Lee J, Guan Z, Akbergenova Y, Littleton JT (2013) Genetic analysis of synaptotagmin C2 domain specificity in regulating spontaneous and evoked neurotransmitter release. J Neurosci 33:187–200. 10.1523/JNEUROSCI.3214-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shin OH, Rhee JS, Araç D, Rah JC, Rizo J, Südhof T, Rosenmund C (2006) Phosphatidylinositol phosphates as co-activators of Ca2+ binding to C2 domains of synaptotagmin 1. J Biol Chem 281:15845–15852. 10.1074/jbc.M600888200 [DOI] [PubMed] [Google Scholar]
- Littleton JT, Stern M, Schulze K, Perin M, Bellen HJ (1993) Mutational analysis of Drosophila synaptotagmin demonstrates its essential role in Ca(2+)-activated neurotransmitter release. Cell 74:1125–1134. 10.1016/0092-8674(93)90733-7 [DOI] [PubMed] [Google Scholar]
- Liu H, Dean C, Arthur CP, Dong M, Chapman ER (2009) Autapses and networks of hippocampal neurons exhibit distinct synaptic transmission phenotypes in the absence of synaptotagmin I. J Neurosci 29:7395–7403. 10.1523/JNEUROSCI.1341-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Chapman ER, Dean C (2013) “Self” versus “non-self” connectivity dictates properties of synaptic transmission and plasticity. PLoS One 8:e62414. 10.1371/journal.pone.0062414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HS, Bai H, Xue RH, Takahashi H, Edwardson JM, Chapman ER (2014) Linker mutations reveal the complexity of synaptotagmin 1 action during synaptic transmission. Nat Neurosci 17:670–677. 10.1038/nn.3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KS, Siebert M, Mertel S, Knoche E, Wegener S, Wichmann C, Matkovic T, Muhammad K, Depner H, Mettke C, Bückers J, Hell SW, Müller M, Davis GW, Schmitz D, Sigrist SJ (2011) RIM-binding protein, a central part of the active zone, is essential for neurotransmitter release. Science 334:1565–1569. 10.1126/science.1212991 [DOI] [PubMed] [Google Scholar]
- Loewen CA, Lee SM, Shin YK, Reist NE (2006) C2B polylysine motif of synaptotagmin facilitates a Ca2+-independent stage of synaptic vesicle priming in vivo. Mol Biol Cell 17:5211–5226. 10.1091/mbc.e06-07-0622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Cai Y, Li Y, Jiao J, Wu Z, O'Shaughnessy B, De Camilli P, Karatekin E, Zhang Y (2017) Single-molecule force spectroscopy of protein-membrane interactions. Elife 6:e30493. 10.7554/eLife.30493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace KE, Biela LM, Sares AG, Reist NE (2009) Synaptotagmin I stabilizes synaptic vesicles via its C(2)A polylysine motif. Genesis 47:337–345. 10.1002/dvg.20502 [DOI] [PubMed] [Google Scholar]
- Mackler JM, Reist NE (2001) Mutations in the second C2 domain of synaptotagmin disrupt synaptic transmission at Drosophila neuromuscular junctions. J Comp Neurol 436:4–16. [PubMed] [Google Scholar]
- Mackler JM, Drummond JA, Loewen CA, Robinson IM, Reist NE (2002) The C(2)B Ca(2+)-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature 418:340–344. 10.1038/nature00846 [DOI] [PubMed] [Google Scholar]
- Martens S, Kozlov MM, McMahon HT (2007) How synaptotagmin promotes membrane fusion. Science 316:1205–1208. 10.1126/science.1142614 [DOI] [PubMed] [Google Scholar]
- Nanou E, Catterall WA (2018) Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron 98:466–481. 10.1016/j.neuron.2018.03.017 [DOI] [PubMed] [Google Scholar]
- Neher E (2015) Merits and limitations of vesicle pool models in view of heterogeneous populations of synaptic vesicles. Neuron 87:1131–1142. 10.1016/j.neuron.2015.08.038 [DOI] [PubMed] [Google Scholar]
- Nishiki T, Augustine GJ (2004) Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J Neurosci 24:8542–8550. 10.1523/JNEUROSCI.2545-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyenhuis SB, Thapa A, Cafiso DS (2019) Phosphatidylinositol 4,5 bisphosphate controls the cis and trans interactions of synaptotagmin 1. Biophys J 117:247–257. 10.1016/j.bpj.2019.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shaughnessy B, Vavylonis D (2005) Non-equilibrium in adsorbed polymer layers. J Phys: Condens Matter 17:R63–R99. 10.1088/0953-8984/17/2/R01 [DOI] [Google Scholar]
- Pang ZPP, Shin OH, Meyer AC, Rosenmund C, Südhof TC (2006) A gain-of-function mutation in synaptotagmin-1 reveals a critical role of Ca2+-dependent soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex binding in synaptic exocytosis. J Neurosci 26:12556–12565. 10.1523/JNEUROSCI.3804-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Ryu JK (2018) Models of synaptotagmin-1 to trigger Ca(2+)-dependent vesicle fusion. FEBS Lett 592:3480–3492. 10.1002/1873-3468.13193 [DOI] [PubMed] [Google Scholar]
- Park Y, Seo JB, Fraind A, Pérez-Lara A, Yavuz H, Han K, Jung SR, Kattan I, Walla PJ, Choi M, Cafiso DS, Koh DS, Jahn R (2015) Synaptotagmin-1 binds to PIP(2)-containing membrane but not to SNAREs at physiological ionic strength. Nat Struct Mol Biol 22:815–823. 10.1038/nsmb.3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman C, Archer DA, Meunier FA, Craxton M, Fukuda M, Burgoyne RD, Davletov B (2004) Synaptotagmin interaction with the syntaxin/SNAP-25 dimer is mediated by an evolutionarily conserved motif and is sensitive to inositol hexakisphosphate. J Biol Chem 279:12574–12579. 10.1074/jbc.M310710200 [DOI] [PubMed] [Google Scholar]
- Rizalar FS, Roosen DA, Haucke V (2021) A presynaptic perspective on transport and assembly mechanisms for synapse formation. Neuron 109:27–41. 10.1016/j.neuron.2020.09.038 [DOI] [PubMed] [Google Scholar]
- Rizo J (2018) Mechanism of neurotransmitter release coming into focus. Protein Sci 27:1364–1391. 10.1002/pro.3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson IM, Ranjan R, Schwarz TL (2002) Synaptotagmins I and IV promote transmitter release independently of Ca(2+) binding in the C(2)A domain. Nature 418:336–340. 10.1038/nature00915 [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996) Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16:1197–1207. 10.1016/s0896-6273(00)80146-4 [DOI] [PubMed] [Google Scholar]
- Rothman JE, Krishnakumar SS, Grushin K, Pincet F (2017) Hypothesis—buttressed rings assemble, clamp, and release SNAREpins for synaptic transmission. FEBS Lett 591:3459–3480. 10.1002/1873-3468.12874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schupp M, Malsam J, Ruiter M, Scheutzow A, Wierda KD, Söllner TH, Sørensen JB (2016) Interactions between SNAP-25 and synaptotagmin-1 are involved in vesicle priming, clamping spontaneous and stimulating evoked neurotransmission. J Neurosci 36:11865–11880. 10.1523/JNEUROSCI.1011-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seven AB, Brewer KD, Shi L, Jiang QX, Rizo J (2013) Prevalent mechanism of membrane bridging by synaptotagmin-1. Proc Natl Acad Sci U S A 110:E3243–3252. 10.1073/pnas.1310327110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X, Li C, Fernandez I, Zhang X, Südhof TC, Rizo J (1997) Synaptotagmin-syntaxin interaction: the C2 domain as a Ca2+-dependent electrostatic switch. Neuron 18:133–142. 10.1016/s0896-6273(01)80052-0 [DOI] [PubMed] [Google Scholar]
- Shields MC, Bowers MR, Kramer HL, Fulcer MM, Perinet LC, Metz MJ, Reist NE (2020) The role of the C2A domain of synaptotagmin 1 in asynchronous neurotransmitter release. PLoS One 15:e0232991. 10.1371/journal.pone.0232991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin OH, Rizo J, Südhof TC (2002) Synaptotagmin function in dense core vesicle exocytosis studied in cracked PC12 cells. Nat Neurosci 5:649–656. 10.1038/nn869 [DOI] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li WZ, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva M, Tran V, Marty A (2021) Calcium-dependent docking of synaptic vesicles. Trends Neurosci 44:579–592. 10.1016/j.tins.2021.04.003 [DOI] [PubMed] [Google Scholar]
- Sirinakis G, Ren YX, Gao Y, Xi ZQ, Zhang YL (2012) Combined versatile high-resolution optical tweezers and single-molecule fluorescence microscopy. Rev Sci Instrum 83:093708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM (2003) The synaptotagmin C2A domain is part of the calcium sensor controlling fast synaptic transmission. Neuron 39:299–308. 10.1016/s0896-6273(03)00432-x [DOI] [PubMed] [Google Scholar]
- Striegel AR, Biela LM, Evans CS, Wang Z, Delehoy JB, Sutton RB, Chapman ER, Reist NE (2012) Calcium binding by synaptotagmin's C2A domain is an essential element of the electrostatic switch that triggers synchronous synaptic transmission. J Neurosci 32:1253–1260. 10.1523/JNEUROSCI.4652-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC (2013a) A molecular machine for neurotransmitter release: synaptotagmin and beyond. Nat Med 19:1227–1231. [DOI] [PubMed] [Google Scholar]
- Südhof TC (2013b) Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80:675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323:474–477. 10.1126/science.1161748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliatti E, Bello OD, Mendonça PRF, Kotzadimitriou D, Nicholson E, Coleman J, Timofeeva Y, Rothman JE, Krishnakumar SS, Volynski KE (2020) Synaptotagmin 1 oligomers clamp and regulate different modes of neurotransmitter release. Proc Natl Acad Sci U S A 117:3819–3827. 10.1073/pnas.1920403117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Shahin V, Henderson RM, Takeyasu K, Edwardson JM (2010) Interaction of synaptotagmin with lipid bilayers, analyzed by single-molecule force spectroscopy. Biophys J 99:2550–2558. 10.1016/j.bpj.2010.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Elferink LA (1998) Functional analysis of the C2A domain of synaptotagmin 1: implications for calcium-regulated secretion. J Neurosci 18:3511–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker WC, Edwardson JM, Bai J, Kim HJ, Martin TF, Chapman ER (2003) Identification of synaptotagmin effectors via acute inhibition of secretion from cracked PC12 cells. J Cell Biol 162:199–209. 10.1083/jcb.200302060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker WC, Weber T, Chapman ER (2004) Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304:435–438. 10.1126/science.1097196 [DOI] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9:112–124. 10.1038/nrm2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennekate W, Schröder S, Lin CC, van den Bogaart G, Grunwald M, Jahn R, Walla PJ (2012) Cis- and trans-membrane interactions of synaptotagmin-1. Proc Natl Acad Sci U S A 109:11037–11042. 10.1073/pnas.1116326109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vevea JD, Chapman ER (2020) Acute disruption of the synaptic vesicle membrane protein synaptotagmin 1 using knockoff in mouse hippocampal neurons. Elife 9:e56469. 10.7554/eLife.56469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voleti R, Jaczynska K, Rizo J (2020) Ca2+-dependent release of synaptotagmin-1 from the SNARE complex on phosphatidylinositol 4,5-bisphosphate-containing membranes. Elife 9:e57154. 10.7554/eLife.57154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Li Y, Ma C (2016) Synaptotagmin-1 C2B domain interacts simultaneously with SNAREs and membranes to promote membrane fusion. Elife 5:e14211. 10.7554/eLife.14211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Cai Q, Shen Z, Chen X, Zeng M, Du S, Zhang M (2019) RIM and RIM-BP form presynaptic active-zone-like condensates via phase separation. Mol Cell 73:971–984.e5. 10.1016/j.molcel.2018.12.007 [DOI] [PubMed] [Google Scholar]
- Wu X, Ganzella M, Zhou J, Zhu S, Jahn R, Zhang M (2021) Vesicle tethering on the surface of phase-separated active zone condensates. Mol Cell 81:13–24.e7. 10.1016/j.molcel.2020.10.029 [DOI] [PubMed] [Google Scholar]
- Wu Z, Dharan N, McDargh ZA, Thiyagarajan S, O'Shaughnessy B, Karatekin E (2021) The neuronal calcium sensor Synaptotagmin-1 and SNARE proteins cooperate to dilate fusion pores. Elife 10:e68215. 10.7554/eLife.68215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Mashimo T, Südhof TC (2007) Synaptotagmin-1, -2, and -9: ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54:567–581. 10.1016/j.neuron.2007.05.004 [DOI] [PubMed] [Google Scholar]
- Yoshihara M, Littleton JT (2002) Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron 36:897–908. 10.1016/s0896-6273(02)01065-6 [DOI] [PubMed] [Google Scholar]
- Zhang XM, Ma L, Zhang YL (2013) High-resolution optical tweezers for single-molecule manipulation. Yale J Biol Med 86:367–383. [PMC free article] [PubMed] [Google Scholar]
- Zhang YL, Jiao J, Rebane AA (2016) Hidden Markov modeling with detailed balance and its application to single protein folding. Biophys J 111:2110–2124. 10.1016/j.bpj.2016.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Yokoyama CT, Scheuer T, Catterall WA (1999) Reciprocal regulation of P/Q-type Ca2+ channels by SNAP-25, syntaxin and synaptotagmin. Nat Neurosci 2:939–941. 10.1038/14721 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, Uervirojnangkoorn M, Zeldin OB, Brewster AS, Sauter NK, Cohen AE, Soltis SM, Alonso-Mori R, Chollet M, Lemke HT, Pfuetzner RA, Choi UB, Weis WI, Diao J, Südhof TC, Brunger AT (2015) Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature 525:62–67. 10.1038/nature14975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Zhou P, Wang AL, Wu D, Zhao M, Südhof TC, Brunger AT (2017) The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 548:420–425. 10.1038/nature23484 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect size estimation (Ho et al., 2019) for evoked release parameters. A, The mean differences in eIPSC amplitude for three comparisons against the shared control (cells expressing WT Syt1) are shown as Cumming estimation plots. The raw data are plotted on the top axes. Bottom, Mean differences are plotted as bootstrap sampling distributions. Each mean difference is depicted as a dot. Each 95% confidence interval is indicated by the ends of the vertical error bars. The unpaired mean difference (in nA) between WT and GFP is −3.65 [95.0% CI, −4.39, −2.87], between WT and K189-192A is −3.21 [95.0% CI, −3.98, −2.38], and between WT and K326-327A is −3.55 [95.0% CI, −4.31, −2.77]. B, Effect size estimation as in A, for eIPSC charge. The unpaired mean difference (in nC) between WT and GFP is −476 [95.0% CI, −664, −351], between WT and K189-192A is −443 [95.0% CI, −642, −310], and between WT and K326-327A is −493 [95.0% CI, −690, −369]. C, Effect size estimation as in A, for time-to-peak values. The unpaired mean difference (in ms) between WT and K189-192A is 7.48 [95.0% CI, 4.8, 10.8], between WT and K326-327A is 5.72 [95.0% CI, 3.64, 9.25]. The unpaired mean difference between WT and GFP alone is 74.4 [95.0% CI, 43.0, 117]. The values for GFP alone are not displayed, as they are much larger and make it difficult to visualize the differences between WT and mutants. D, Effect size estimations for eIPSC decay times. The unpaired mean difference (in ms) between WT and GFP alone is 23.4 [95.0% CI, −82.3, 153], between WT and K189-192A is −51.0 [95.0% CI, −125, 24.6], and between WT and K326-327A is −92.0 [95.0% CI, −169, −15.8]. In all cases, 5000 bootstrap samples were taken; the confidence interval is bias corrected and accelerated. Traces with a very weak response were excluded from analysis for C and D because the time to peak or the decay time could not be determined reliably (Fig. 2). Download Figure 2-1, EPS file (1.5MB, eps) .
Effect size estimation for readily releasable pool sizes. The mean differences in hypertonic sucrose-induced RRP size estimates for three comparisons against the shared control (cells expressing WT Syt1) are shown as a Cumming estimation plot. The raw data are plotted on the top axes. Bottom, mean differences are plotted as bootstrap sampling distributions. Each mean difference is depicted as a dot. Each 95% confidence interval is indicated by the ends of the vertical error bars. The unpaired mean difference (in nC) between WT and GFP is −3.77 [95.0% CI, −4.66, −2.84], between WT and K189-192A is −0.433 [95.0% CI, −1.5, 0.669], and between WT and K326-327A is −0.466 [95.0% CI, −1.59, 0.987]. Download Figure 4-1, EPS file (518.8KB, eps) .