

Abstract
The essential biometal manganese (Mn) serves as a cofactor for several enzymes that are crucial for the prevention of human diseases. Whether intracellular Mn levels may be sensed and modulate intracellular signaling events has so far remained largely unexplored. The highly conserved target of rapamycin complex 1 (TORC1, mTORC1 in mammals) protein kinase requires divalent metal cofactors such as magnesium (Mg2+) to phosphorylate effectors as part of a homeostatic process that coordinates cell growth and metabolism with nutrient and/or growth factor availability. Here, our genetic approaches reveal that TORC1 activity is stimulated in vivo by elevated cytoplasmic Mn levels, which can be induced by loss of the Golgi-resident Mn2+ transporter Pmr1 and which depend on the natural resistance-associated macrophage protein (NRAMP) metal ion transporters Smf1 and Smf2. Accordingly, genetic interventions that increase cytoplasmic Mn2+ levels antagonize the effects of rapamycin in triggering autophagy, mitophagy, and Rtg1-Rtg3-dependent mitochondrion-to-nucleus retrograde signaling. Surprisingly, our in vitro protein kinase assays uncovered that Mn2+ activates TORC1 substantially better than Mg2+, which is primarily due to its ability to lower the Km for ATP, thereby allowing more efficient ATP coordination in the catalytic cleft of TORC1. These findings, therefore, provide both a mechanism to explain our genetic observations in yeast and a rationale for how fluctuations in trace amounts of Mn can become physiologically relevant. Supporting this notion, TORC1 is also wired to feedback control mechanisms that impinge on Smf1 and Smf2. Finally, we also show that Mn2+-mediated control of TORC1 is evolutionarily conserved in mammals, which may prove relevant for our understanding of the role of Mn in human diseases.
Research organism: S. cerevisiae, Human
Introduction
Manganese (Mn) is a vital trace element that is required for the normal activity of the brain and nervous system by acting, among other mechanisms, as an essential, divalent metal cofactor for enzymes such as the mitochondrial enzyme superoxide dismutase 2 (Weisiger and Fridovich, 1973), the apical activator of the DNA damage response serine/threonine kinase ATM (Chan et al., 2000) or the Mn2+-activated glutamine synthetase (Wedler and Denman, 1984). However, Mn2+ becomes toxic when enriched in the human body (Couper, 1837). While mitochondria have been proposed as a preferential organelle where Mn2+ accumulates and unfolds its toxicity by increasing oxidative stress and thus mitochondrial dysfunction (Aguirre and Culotta, 2012), the molecular mechanisms of Mn2+ toxicity in humans are also related to protein misfolding, endoplasmic reticulum (ER) stress, and apoptosis (Harischandra et al., 2019). Mn2+ homeostasis is coordinated by a complex interplay between various metal transporters for Mn2+ uptake and intracellular Mn2+ distribution and represents an essential task of eukaryotic cells, which is also of vital importance specifically for neuronal cell health (Horning et al., 2015).
Much of our knowledge on Mn2+ transport across the plasma membrane into the ER, the Golgi, endosomes, and vacuoles comes from studies in Saccharomyces cerevisiae (outlined in Figure 1A). Typically, Mn2+ is shuttled across membranes by transporters that belong to the natural resistance-associated macrophage protein (NRAMP) family, which are highly conserved metal transporters responsible for iron (Fe) and Mn2+ uptake (Supek et al., 1996). Not surprisingly, therefore, NRAMP orthologs have been found to cross-complement functions in yeast, mice, and humans (Sacher et al., 2000). One of the best-studied NRAMPs is the yeast plasma membrane protein Smf1. Interestingly, extracellular Fe or Mn2+ supplementation triggers Bsd2 adaptor protein-dependent, Rsp5-mediated ubiquitination of Smf1, which initiates its sorting through the endocytic multivesicular body pathway and subsequent lysosomal degradation (Eguez et al., 2004; Liu and Culotta, 1999). The Smf1 paralogs Smf2 and Smf3 are less well studied, but Smf2 is predominantly localized at endosomes and its levels decrease under conditions of Mn or Fe overload (Liu et al., 1997; Moreno-Cermeño et al., 2010). Within cells, the P-type ATPase Pmr1 (also known as Bsd1) represents a key transporter that shuttles Ca2+ and Mn2+ ions into the Golgi lumen. Its loss leads to increased levels of Mn2+ in the cytoplasm due to defective detoxification (Lapinskas et al., 1995). Noteworthy, several phenotypes associated with loss of Pmr1 have been shown to arise as a consequence of Mn2+ accumulation in the cytoplasm, including telomere shortening, genome instability, and bypass of the superoxide dismutase Sod1 requirement (Bolton et al., 2002; García-Rodríguez et al., 2012; Lapinskas et al., 1995; Lue et al., 2005).
Figure 1. Natural resistance-associated macrophage protein (NRAMP) transporters link Mn2+-import to rapamycin resistance.
(A) Schematical outline of yeast Mn2+ transporters and their intracellular localization. PM, plasma membrane; PVE, pre-vacuolar endosomes; ER, endoplasmic reticulum. Note that Bsd2 is a specific adaptor protein for Rsp5-mediated Smf1 and Smf2 ubiquitination in response to Mn2+ overload. The Golgi Mn2+ transporter Gdt1 is omitted for clarity. (B–D) Growth on MnCl2 and/or rapamycin-containing medium (RAP). Ten-fold dilutions of exponentially growing cells are shown. Strains and compound concentrations are indicated. Note that the medium used in (D) was supplemented with 10 mM CaCl2. Data obtained in a medium without CaCl2 are shown in Figure 1—figure supplement 3. (E) Southern blot analysis of telomere length. Genomic DNA was derived from cells grown in a medium supplemented or not with CaCl2 and cleaved by XhoI before agarose gel electrophoresis. The 1.3 kb average length of telomeres from WT cells (dashed white line, black arrow) and size marker (M) are shown. (F) Growth of pmr1∆ smf2∆ double mutants transformed with plasmids expressing yeast Smf2, Mus musculus SLC11A1, or SLC11A2 on rapamycin-containing medium. Complementary data showing that pmr1∆ rapamycin resistance is not linked to Gap1 or Tor1 localization is provided in Figure 1—figure supplement 1. Rapamycin sensitivity of bsd2∆ cells overexpressing the Vcx1-M1 transporter are shown in Figure 1—figure supplement 2.

Figure 1—figure supplement 1. pmr1∆ rapamycin resistance is not linked to Gap1 or Tor1 localization.

Figure 1—figure supplement 2. Growth of bsd2∆ mutants expressing Vcx1-M1 from plasmid pVCX1-M1 on MnCl2 and/or rapamycin (RAP) containing medium.

Figure 1—figure supplement 3. Natural resistance-associated macrophage protein (NRAMP) transporters mediate rapamycin resistance of pmr1∆ mutants.
TORC1/mTORC1 is a central, highly conserved controller of cell growth and aging in eukaryotes. It coordinates the cellular response to multiple inputs, including nutritional availability, bioenergetic status, oxygen levels, and, in multicellular organisms, the presence of growth factors (Albert and Hall, 2015; Laplante and Sabatini, 2012). In response to these diverse cues, TORC1 regulates cell growth and proliferation, metabolism, protein synthesis, autophagy, and DNA damage responses (Liu and Sabatini, 2020; Sancak et al., 2008). In S. cerevisiae, which played a pivotal role in the discovery and dissection of the TOR signaling network (Heitman et al., 1991), TORC1 is mainly localized on the surfaces of vacuoles and endosomes (Betz and Hall, 2013; Hatakeyama et al., 2019) where it integrates, among other cues, amino acid signals through the Rag GTPases and Pib2 (Nicastro et al., 2017; Tanigawa et al., 2021). In mammals, amino acids also activate the Rag GTPases, which then recruit mTORC1 to the lysosomal surface where it can be allosterically activated by the small GTPase Rheb (Ras homolog expressed in brain) that mediates the presence of growth factors and sufficient energy levels (Binda et al., 2009; Sancak et al., 2008; Sancak et al., 2010; Wedaman et al., 2003). Interestingly, mTORC1 regulates cellular Fe homeostasis (Bayeva et al., 2012), but how it may be able to sense Fe levels remains largely unknown. In addition, although TORC1/mTORC1 requires divalent metal ions to coordinate ATP at its catalytic cleft (Brunn et al., 1996; Withers et al., 1997), it is currently not known whether these or any other trace elements may play a physiological or regulatory role in controlling its activity.
In yeast, genetic evidence links high levels of cytoplasmic Mn2+ to TORC1 function, as loss of Pmr1 confers rapamycin resistance (Devasahayam et al., 2007). However, the underlying molecular mechanism(s) by which Mn2+ may mediate rapamycin resistance remains to be explored. Here, we show that Mn2+ uptake by NRAMP transporters modulates rapamycin resistance and that, in turn, TORC1 inhibition by rapamycin regulates NRAMP transporter availability. Moreover, intracellular Mn2+ excess antagonizes rapamycin-induced autophagy, mitophagy, and Rtg1-3 transcription factor complex-dependent retrograde response activation. Surprisingly, our in vitro analyses reveal that TORC1 protein kinase activity is strongly activated in the presence of MnCl2. In our attempts to understand the mechanisms underlying these observations, we discovered that Mn2+, when compared to Mg2+, significantly boosts the affinity of TORC1 for ATP. Combined, our findings also indicate that TORC1 activity is regulated by and regulates intracellular Mn2+ levels, defining Mn2+ homeostasis as a key factor in cell growth control. Importantly, our studies in human cells indicate that Mn2+-driven TORC1 activation is likely conserved throughout evolution, opening new perspectives for our understanding of Mn2+ toxicities and their role in neurodegenerative disorders and aging.
Results
NRAMP transporters regulate cytoplasmic Mn2+ levels and rapamycin resistance
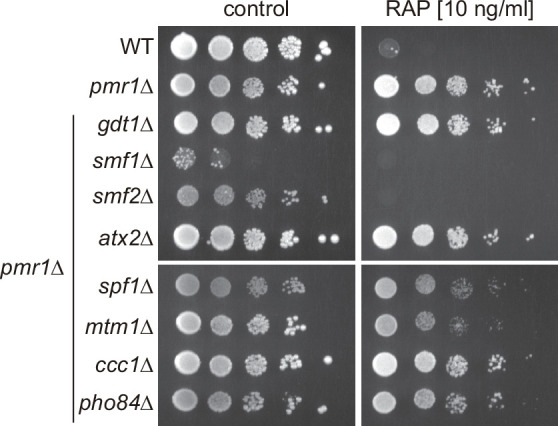
Yeast cells lacking the Golgi-localized P-type ATPase Pmr1, which transports Ca2+ and Mn2+ ions from the cytoplasm to the Golgi lumen, are resistant to the TORC1 inhibitor rapamycin (Devasahayam et al., 2006; Devasahayam et al., 2007), a phenotype that is generally associated with increased TORC1 activity. In pmr1∆ mutants, defective Mn2+ shuttling at the Golgi leads to protein sorting defects and accumulation of the general amino acid permease Gap1 at the plasma membrane (Kaufman et al., 1994). In theory, this may translate into unrestrained uptake and intracellular accumulation of amino acids, and thus hyperactivation of TORC1. However, arguing against such a model, we found cells lacking both Pmr1 and Gap1 to remain resistant to low doses of rapamycin (Figure 1—figure supplement 1A). We then asked whether loss of Pmr1 may affect the expression levels or cellular localization of TORC1. Our results indicated that GFP-Tor1 protein levels and localization to vacuolar and endosomal membranes remained unaltered in exponentially growing and rapamycin-treated WT and pmr1∆ cells (Figure 1—figure supplement 1B and C). Given the roughly fivefold increased intracellular Mn2+ levels in cells lacking Pmr1 (Lapinskas et al., 1995), we then considered the possibility that Mn2+ may have a more direct role in TORC1 activation. We, therefore, assessed the rapamycin sensitivity of cells lacking the adaptor protein Bsd2, which mediates Rsp5-dependent degradation in response to high Mn2+ levels of both the plasma membrane- and endosomal membrane-resident NRAMP Mn2+ transporters Smf1 and Smf2, respectively (Figure 1A; Liu et al., 1997). Interestingly, bsd2∆ cells were as sensitive to rapamycin as WT cells, but, unlike WT cells, could be rendered rapamycin resistant by the addition of 1 mM MnCl2 in the growth medium (Figure 1B and Figure 1—figure supplement 2). This effect was strongly reduced in the absence of Smf2 (Figure 1C), suggesting that Smf2-dependent endosomal Mn2+ export and, consequently, cytoplasmic accumulation of Mn2+ may be required for rapamycin resistance under these conditions. In line with such a model, we found that overexpression of the vacuolar membrane-resident Vcx1-M1 transporter, which imports Mn2+ into the vacuolar lumen, suppressed the Mn2+-induced rapamycin resistance of bsd2∆ mutants and increased their sensitivity to rapamycin in the absence of extracellular MnCl2 supply (Figure 1—figure supplement 2).
As schematically outlined in Figure 1A, several metal transporters have been associated with Mn2+ transport in yeast, including those localized at the plasma membrane (Smf1 and Pho84), the endosomes (Smf2 and Atx2), the Golgi (Pmr1 and Gdt1), the vacuole (Ccc1 and Vcx1), the ER (Spf1), and possibly the mitochondria (Mtm1). To identify which of these transporters contributes to the rapamycin resistance of pmr1∆ cells, we next monitored the growth of double mutants lacking Pmr1 and any of the corresponding metal transporters in the presence of rapamycin (Figure 1D). Interestingly, only loss of Smf1 or Smf2 reestablished rapamycin sensitivity in pmr1∆ cells, while loss of Gdt1 or Ccc1 even slightly enhanced the rapamycin resistance phenotype of pmr1∆ cells. Of note, growth was monitored on CaCl2-supplemented media that improved the growth of pmr1∆ cells lacking Smf1 or Smf2 without affecting their sensitivity to rapamycin, which also rules out the possibility that Ca2+ is associated with the observed rapamycin resistance phenotype (Figure 1D and Figure 1—figure supplement 3). Taken together, our data indicate that increased intracellular Mn2+ levels in pmr1∆ cells lead to rapamycin resistance and that this phenotype can be suppressed either by reducing Mn2+ import through loss of Smf1 or, possibly, by increasing Mn2+ sequestration in endosomes through loss of Smf2 as previously suggested (Luk and Culotta, 2001). To confirm our prediction that loss of Smf2 reduces cytoplasmic Mn2+ levels, we measured MnCl2-dependent telomere length shortening as an indirect proxy for cytoplasmic and nuclear Mn levels (García-Rodríguez et al., 2015). Accordingly, telomere length was significantly decreased in pmr1∆ cells (when compared to WT cells), increased in smf2∆ cells, and similar between pmr1∆ smf2∆ and WT cells (Figure 1E). These data, therefore, corroborate our assumption that loss of Smf2 suppresses the high cytoplasmic and nuclear Mn2+ levels of pmr1∆ cells.
The function of metal transporters is highly conserved across evolution as exemplified by the fact that the expression of the human Pmr1 ortholog, the secretory pathway Ca2+/Mn2+ ATPase ATP2C1/SPCA1 (ATPase secretory pathway Ca2+ transporting 1), can substitute for Pmr1 function in yeast (Muncanovic et al., 2019). A similar degree of functional conservation from lower to higher eukaryotes exists for NRAMP transporters (Sacher et al., 2000). Because Smf2 is of specific interest in the context of the present study, we asked whether the orthologous mouse proteins, that is, the divalent metal transporter SLC11A1 (NRAMP1) and SLC11A2 (DMT1) isoforms (solute carrier family 11 member 1 and 2, respectively), can restore rapamycin resistance in pmr1∆ smf2∆ double mutants. Expression of SLC11A1 did not rescue the lack of Smf2, leading to poor growth even in the absence of rapamycin. However, the SLC11A2 isoform complemented Smf2 function in these assays (Figure 1F), indicating that Pmr1 and Smf2 are evolutionarily conserved transporters that are required for Mn2+ homeostasis.
Elevated levels of intracellular Mn2+ antagonize rapamycin-induced autophagy, mitophagy, and Rtg1-3 retrograde signaling
Growth inhibition by rapamycin mimics starvation conditions and leads to the degradation and recycling of a wide spectrum of biological macromolecules via autophagy. In this context, the pmr1∆ mutant has previously been found to be defective in nutrient depletion-induced mitophagy (Kanki et al., 2010). We thus wondered if rapamycin-induced autophagic processing may also be defective in pmr1∆ mutants. We took advantage of a GFP-Atg8 fusion construct (Cheong and Klionsky, 2008) to monitor autophagy through GFP-Atg8 synthesis and processing in cells that had been subjected to rapamycin treatment for up to 6 hr. Rapamycin-induced GFP-Atg8 expression was strongly reduced in pmr1∆ cells and this phenotype was mitigated in the pmr1∆ smf2∆ double mutant (Figure 2A and B), suggesting that the initiation of autophagy is compromised by elevated cytoplasmic Mn2+ levels. Next, we assessed mitophagy by following the expression and degradation of the GFP-tagged mitochondrial membrane protein Om45 (Kanki et al., 2009). Rapamycin treatment led to a time-dependent up-regulation of Om45-GFP protein levels in all tested strains (Figure 2C and D). However, the accumulation of the cleaved GFP protein was strongly reduced in the pmr1∆ single mutant, while this effect was again suppressed in the pmr1∆ smf2∆ double mutant. Combined, our results therefore suggest that the intracellular Mn2+ flux modulates both rapamycin-induced autophagy and mitophagy.
Figure 2. Intracellular manganese (Mn) excess antagonizes rapamycin-induced autophagy, mitophagy, and Rtg1-3 retrograde signaling.

(A) Exponentially growing WT and indicated mutant strains expressing plasmid-encoded GFP-Atg8 were treated for up to 6 hr with 200 ng/ml rapamycin (RAP). GFP-Atg8 and cleaved GFP (*GFP) protein levels were analyzed by immunoblotting. Glucose-6-phosphate dehydrogenase (G-6-PDH) levels were used as a loading control. (B) Quantification of GFP-Atg8 and *GFP levels after a 6 hr rapamycin treatment. Total GFP signal (Atg8-GFP + *GFP) normalized to WT levels (left) and percentage of *GFP relative to the total GFP signal (right) are plotted. Data represent means ± SEM of independent experiments (n=4). Statistical analysis: two-tailed t-test (paired for normalized data, unpaired for GFP* percentage). *p<0.05; **p<0.01; ***p<0.001. (C) Exponentially growing WT and indicated mutant strains expressing Om45-GFP from the endogenous locus were treated and processed as in (A). (D) Quantification of Om45-GFP and *GFP levels after a 6 hr rapamycin treatment. Details as in (B) with n=3. (E) Representative fluorescence microscope images of WT, pmr1∆, smf2∆, and pmr1∆ smf2∆ mutants expressing an episomic Rtg3-GFP reporter construct. Exponentially growing cells were treated or not (control) for 30 min with 200 ng/ml rapamycin (RAP). Scale bar represents 5 µm. (F) Rtg3-GFP expressing cells were treated for up to 30 min with 200 ng/ml rapamycin. Protein levels were analyzed by immunoblotting. G-6-PDH levels were used as a loading control. (G) Quantification of Rtg3-GFP signals. Values normalized to WT levels after 15 min of rapamycin treatment (highest signal) are plotted. Data represent means ± SEM of independent experiments (n=4). Statistical analysis: paired two-tailed t-test.
To identify additional responses to elevated cytosolic Mn2+ levels, we took advantage of our previously published transcriptome analysis of pmr1∆ cells (García-Rodríguez et al., 2015). Careful analysis of these datasets revealed that the expression levels of genes activated by the heterodimeric Rtg1-Rtg3 transcription factor are reduced in pmr1∆ cells (see Supplementary file 1). Since TORC1 inhibits cytoplasmic-to-nuclear translocation of Rtg1-Rtg3 (Ruiz-Roig et al., 2012), we monitored the localization and protein levels of Rtg3-GFP in exponentially growing and rapamycin-treated WT and pmr1∆ cells. Rapamycin treatment not only induced nuclear enrichment of Rtg3-GFP as reported (Figure 2E; Ruiz-Roig et al., 2012) but also significantly increased the levels of Rtg3-GFP (Figure 2F and G). Loss of Pmr1, in contrast, significantly reduced the Rtg3-GFP levels in exponentially growing and rapamycin-treated cells, which also translated into barely visible levels of Rtg3-GFP in the nucleus (Figure 2E–G). Importantly, and in line with our finding that loss of Smf2 suppresses the high cytoplasmic and nuclear Mn2+ levels of pmr1∆ cells (see above), these latter defects in pmr1∆ cells were partially suppressed by loss of Smf2. Thus, our findings posit a model in which elevated cytoplasmic Mn2+ levels antagonize autophagy, mitophagy, and Rtg1-3-dependent retrograde signaling presumably through activation of TORC1.
MnCl2 stimulates TORC1 kinase activity in vitro
The yeast Tor1 kinase is a member of the phosphatidylinositol 3-kinase (PI3K)-related kinase (PIKK) family that can phosphorylate the human eukaryotic translation initiation factor 4E binding protein (eIF4-BP/PHAS-I) in vitro (Alarcon et al., 1999). Curiously, it does so much more efficiently when the respective in vitro kinase assays contain Mn2+ rather than Mg2+ as the sole divalent cation, a property that it appears to share with PI3-kinases (Carpenter et al., 1993; Dhand et al., 1994; Foukas et al., 2004). A similar preference for Mn2+ over Mg2+ has also been observed in mTOR kinase autophosphorylation assays (Brunn et al., 1996; Withers et al., 1997). Based on these and our observations, we decided to assess whether Mn2+ may act as a metal cofactor for TORC1 activity in vitro using TORC1 purified from yeast and a truncated form of Lst4 (Lst4Loop; Nicastro et al., 2021) as a substrate. In control experiments without divalent ions, TORC1 activity remained undetectable (Figure 3A and B). The addition of MnCl2, however, not only stimulated TORC1 in vitro in a concentration-dependent manner, but also activated TORC1 dramatically more efficiently than MgCl2 (with 25-fold lower levels of MnCl2 [38 µM] than MgCl2 [980 µM] promoting half-maximal activation of TORC1; Figure 3A and B). We next considered the possibility that Mn2+ is superior to Mg2+ in favoring the coordination of ATP in the catalytic cleft or TORC1. Supporting this idea, we found that Mn2+ significantly reduced the Km for ATP (5.3-fold) of TORC1 (Figure 3C and D). Moreover, even in the presence of saturating Mg2+ levels (i.e. 4 mM), the addition of 160 µM Mn2+ was able to enhance the VMAX almost twofold and decrease the Km for ATP of TORC1 from 50.7 to 34.4 µM, which indicates that Mn2+ can efficiently compete with Mg2+ and thereby activate TORC1. Notably, the conditions used in our in vitro kinase assays are quite comparable to the in vivo situation: accordingly, intracellular Mg2+ levels in yeast are approximately around 2 mM (van Eunen et al., 2010), while the Mn2+ levels range from 26 µM in WT cells to 170 µM in pmr1∆ cells (McNaughton et al., 2010). Our in vitro assays, therefore, provide a simple rationale for why pmr1∆ cells are resistant to rapamycin: elevated Mn2+ levels in pmr1∆ favorably boost the kinetic parameters of TORC1.
Figure 3. MnCl2 stimulates TORC1 kinase activity in vitro and in vivo.

(A) In vitro TORC1 kinase assays using [γ-32P]-ATP, recombinant Lst4Loop as substrate, and increasing concentrations (twofold dilutions) of MgCl2 or MnCl2. Substrate phosphorylation was detected by autoradiography (lower blot) and SYPRO Ruby staining is shown as loading control (upper blot). (B) Quantification of the assay shown in (A). Curve fitting and parameter calculations were performed with GraphPad Prism. Data shown are means (± SEM, n=3). (C) In vitro kinase assays (as in A) using the indicated concentrations of MgCl2 and/or MnCl2 and increasing concentrations of ATP. Substrate phosphorylation was detected by autoradiography (lower blot) and SYPRO Ruby staining is shown as loading control (upper blot). (D) Quantification of the assay shown in (C). Data shown are means (± SEM, n=3). Curve fitting and parameter calculations were performed with GraphPad Prism. KmATP are shown for each curve. VMAX [MnCl2]=1.13 ± 0.06, VMAX [MgCl2 MnCl2]=0.89 ± 0.03, VMAX [MgCl2]=0.47 ± 0.02.
TORC1 regulates NRAMP transporter protein levels
To maintain appropriate intracellular Mn2+ concentrations, cells adjust Smf1 and Smf2 protein levels through Bsd2-dependent and -independent, post-translational modifications that ultimately trigger their vacuolar degradation through the endocytic multivesicular body pathway (Liu and Culotta, 1994; Liu et al., 1997). Because NRAMP transporters are important for Mn-dependent TORC1 activation, and because TORC1 is often embedded in regulatory feedback loops to ensure cellular homeostasis (Eltschinger and Loewith, 2016), we next asked whether rapamycin-mediated TORC1 inactivation may affect the levels and/or localization of Smf1 and Smf2 using a strain that expresses N-terminally tagged Smf1 (GFP-Smf1) under its own promoter from a non-endogenous locus or a strain that expresses C-terminally tagged Smf2 (Smf2-GFP) from its endogenous locus (García-Rodríguez et al., 2015; Renz et al., 2020). Rapamycin treatment triggered a strong increase in GFP-Smf1 levels (Figure 4A and B), which is likely due to transcriptional activation of Smf1 under these conditions as published earlier (Reinke et al., 2006). In line with this interpretation, we found the respective increase to be abolished when rapamycin-treated cells were co-treated with the protein synthesis inhibitor cycloheximide (CHX). In addition, because GFP-Smf1 appeared to be degraded at a similar rate in CHX-treated cells that were treated, or not, with rapamycin, our data further indicate that TORC1 does not control Smf1 levels through post-translational control of Smf1 turnover. Interestingly, GFP-Smf1 remained predominantly at the plasma membrane in rapamycin-treated cells, even though these cells also accumulated more cleaved GFP in the vacuoles (Figure 4C). We infer from these data that TORC1 inhibition activates Smf1 expression, likely as part of a feedback control loop through which TORC1 couples its activity to Mn2+ uptake. Rapamycin-treated cells that were either co-treated or not with CHX exhibited Smf2-GFP levels that steadily decreased at a similar rate, with cleaved GFP accumulating in parallel (Figure 4D and E). Since this rate appeared to be higher than the one observed in cells treated with CHX alone, our data suggest that TORC1 antagonizes the turnover of Smf2. This was further corroborated by our fluorescence microscopy analyses revealing that the GFP signal in Smf2-GFP-expressing cells shifted from late Golgi/endosomal foci (see García-Rodríguez et al., 2015) to a predominant signal within the vacuolar lumen when cells were treated with rapamycin (Figure 4F). Whether this event is an indirect consequence of higher Mn2+ uptake under these conditions (see above), or potentially part of a local feedback control mechanism of endosomal TORC1 (Hatakeyama et al., 2019), remains to be addressed in future studies.
Figure 4. TORC1 regulates natural resistance-associated macrophage protein (NRAMP) transporter levels.

(A) GFP-Smf1 expressing cells were cultivated for 5–6 hr in a synthetic medium devoid of manganese sulfate before being treated with 200 ng/ml rapamycin (RAP), 25 µg/ml cycloheximide (CHX), or both compounds (RAP + CHX) for the indicated times. GFP-Smf1 and cleaved GFP (*GFP) protein levels were analyzed by immunoblotting using an anti-GFP antibody. Alcohol dehydrogenase (Adh1) protein levels, probed with anti-Adh1 antibodies, served as a loading control. (B) Quantification of GFP-Smf1. Percentages of GFP-Smf1 relative to the total GFP signal (GFP-Smf1 + GFP) are plotted. Data represent means ± SEM of independent experiments (n=3). Statistical analysis: unpaired two-tailed t-test. *p<0.05; **p<0.01, ***p<0.001, ****p<0.0001. (C) Microscopic analysis of GFP-Smf1 localization. Cells co-expressing GFP-Smf1 and the vacuolar marker Vph1-mCherry were grown exponentially in a manganese-free medium for 5–6 hr, then treated with 200 ng/ml rapamycin for 2 hr. Scale bar represents 5 µm. (D–E) Cells expressing Smf2-GFP from its endogenous locus were grown, treated, and processed as in (A). (F) Microscopic analysis of Smf2-GFP localization. Cells co-expressing Smf2-GFP and the vacuolar marker Vph1-mCherry were cultivated, treated, and examined as in (C).
Mn2+ activates mTORC1 signaling in human cells
To assess whether the response of TORC1 to Mn2+ is conserved among eukaryotes, we investigated if externally supplied MnCl2 activates mTORC1 signaling in mammalian cells. For this purpose, we used the human cell lines U2OS and HEK293T, which are widely used for the cellular dissection of mTORC1-mediated mechanisms in response to nutritional inputs (Bodineau et al., 2021). First, we examined the sufficiency of Mn2+ to maintain the activity of the mTORC1 pathway by incubating U2OS cells in an amino acid starvation medium supplemented with increasing amounts of MnCl2 (0–1 mM) for 2 hr. mTORC1 activity was assessed by monitoring phosphorylation of the mTORC1 downstream targets RPS6KB (ribosomal protein S6 kinase B; phospho-Threonine389) and RPS6 (ribosomal protein S6; phospho-Serine235–236). As shown in Figure 5A, and as expected, both RPS6KB and RPS6 were fully phosphorylated at those specific residues in cells incubated in the presence of amino acids and completely de-phosphorylated in cells incubated in the absence of amino acids, without MnCl2. In agreement with a positive action of Mn2+ toward mTORC1 in human cells, we observed a robust and dose-dependent increase in both RPS6KB and RPS6 phosphorylation in amino acid-starved cells incubated in the presence of MnCl2 at concentrations higher than 0.05 mM, with maximum phosphorylation observed at 0.5 mM of MnCl2. Confirming this result, we observed a similar response of RPS6KB and RPS6 phosphorylation to MnCl2 treatment in HEK293T cells, except that the maximum response was reached at 1 mM (Figure 5B). These results indicate that Mn2+ is sufficient to maintain the activity of the mTORC1 pathway in the absence of amino acid inputs, at least during short periods (2 hr). To corroborate this conclusion, we also tested the phosphorylation of additional downstream targets of mTORC1 in response to a fixed concentration of MnCl2 (0.35 mM) in U2OS cells. Amino acid starvation of U2OS cells with MnCl2 was sufficient to maintain the phosphorylation of the downstream targets of mTORC1 RPS6KB, RPS6, EIF4EBP1, and ULK1 (unc-51 like autophagy activating kinase 1) (Figure 5C). In line with our finding that MnCl2 retained AKT1 (AKT serine/threonine kinase 1) phosphorylation in amino acid-starved cells, Mn2+ has been reported to activate the mTORC1 upstream kinase AKT1, which is also a known mTORC2 downstream target (Bryan et al., 2018; Gaubitz et al., 2016). However, AKT1 phosphorylation appeared to be much less pronounced than the phosphorylation of the mTORC1 downstream targets, validating the specificity of Mn toward mTORC1. Whether Mn2+ may stimulate TORC2 activity remains to be explored.
Figure 5. MnCl2 maintains mTORC1 activity during starvation in human cells.

(A–B) Human U2OS (A) and HEK293T (B) cells were starved for all amino acids and supplemented with increasing concentrations of MnCl2 as indicated for 2 hr. Phosphorylation of the mTORC1 downstream targets RPS6KB (ribosomal protein S6 kinase B) and RPS6 (ribosomal protein S6) was assessed by immunoblot analysis. A control with cells incubated in the presence of all proteinogenic amino acids (+AA) was included as a positive control. (C) Human U2OS were starved for all amino acids (-AA) and supplemented with 0.35 mM of MnCl2 for 2 hr. Phosphorylation of RPS6KB, RPS6, EIF4EBP1, ULK1, and AKT1 was assessed by immunoblot analysis. A control with cells incubated in the presence of all proteinogenic amino acids (+AA) was included as a positive control. (D–E) Human U2OS cells were treated as in (C) for 4 hr either in the absence (D) or the presence (E) of chloroquine (CQ). Autophagic marker MAP1LC3I/II was then analyzed by immunoblot. (F–G) GFP-MAP1LC3 expressing U2OS cells were incubated as in (D). GFP-MAP1LC3 aggregation was assessed by confocal microscopy (F) and quantified using ImageJ software (G). Scale bar indicates 10 µm. Values were normalized to -AA condition. Graphs represent mean ± SD, with n=3 (*p<0.05, ANOVA followed by Tukey’s test). (H) Model of Mn2+-driven TORC1 activation. The Smf1/2 natural resistance-associated macrophage protein (NRAMP) transporter-dependent increase in cytoplasmatic Mn2+ levels favors TORC1-Mn2+ binding and ATP coordination, leading to TORC1 hyperactivation. NRAMP transporters are part of a feedback control mechanism impinged by TPRC1 (dashed lines).
Finally, to assess the physiological relevance of mTORC1 activation in response to Mn2+, we analyzed the effect of MnCl2 treatment in autophagy, a cellular process inhibited by TORC1 both in yeast and in mammalian cells (Noda and Ohsumi, 1998; Villar et al., 2017). To this end, we analyzed the autophagic marker MAP1LC3I/II (microtubule-associated protein 1 light chain 3) (Klionsky et al., 2016). During autophagy initiation, MAP1LC3I is lipidated, thereby increasing the levels of MAP1LC3II. As previously reported, the withdrawal of amino acids led to a rapid increase in MAP1LC3II levels, indicating an increase in autophagy initiation (Figure 5D). Of note, the addition of MnCl2 completely abolished the increase in MAP1LC3II levels, thus confirming that Mn2+ prevented the initiation of autophagy downstream of mTORC1. These results were also observed in cells incubated in the presence of chloroquine, an inhibitor of the fusion of autophagosomes with the lysosome, thus confirming that Mn2+ influences autophagy initiation, the process controlled by mTORC1 (Figure 5E). In addition, we analyzed the aggregation of MAP1LC3 upon autophagosome formation in U2OS cells stably expressing a GFP-MAP1LC3 construct. GFP aggregation indicates autophagosome formation in these cells. Similar to what we observed with endogenous MAP1LC3, GFP aggregation was increased during amino acid withdrawal and this increase was completely abolished by MnCl2 treatment (Figure 5F–G). This result further confirms that the Mn-dependent activation of mTORC1 in human cells is physiologically relevant for autophagy inhibition. Altogether, our results show that Mn2+ is sufficient to keep mTORC1 activated in the absence of other inputs in human cells and to inhibit autophagy downstream of mTORC1. These findings indicate that Mn2+-mediated activation of TORC1 is evolutionarily conserved from yeast to humans.
Discussion
Studies with yeast lacking the divalent Ca/Mn ion Golgi transporter Pmr1, which displays an increase in intracellular Mn2+ levels due to impaired Mn2+ detoxification (McNaughton et al., 2010), have pinpointed a functional link between Mn2+ homeostasis and TORC1 signaling (Devasahayam et al., 2006; Devasahayam et al., 2007). However, the underlying mechanism(s) of how Mn2+ impinges on TORC1 has so far remained elusive. Our study identifies Mn2+ as a divalent metal cofactor that stimulates the enzymatic activity of the TORC1 complex in vitro and suggests that Mn2+ functions similarly in vivo both in yeast and in mammalian cells. Our results support a model in which the PI3K-related protein kinase Tor1 is activated by Mg2+, but requires Mn2+ as a metal cofactor for maximal activity as this allows it to better coordinate ATP (Figure 5H). While it seemingly shares this feature with PI3Ks (Carpenter et al., 1993; Dhand et al., 1994; Foukas et al., 2004), serine/threonine protein kinases are generally known to be preferably activated by Mg2+ rather than Mn2+ (Knape et al., 2017). We deem it therefore possible that the respective Mn2+-driven activation of TORC1 is more specifically related to the architecture of its catalytic center. Resolution of this issue will therefore require future molecular dynamic modeling and/or structural analyses.
Our current study also highlights a need for coordinated control of TORC1 activity and cytoplasmic Mn2+ levels. The latter critically depend on a set of conserved NRAMP transporters, such as yeast Pmr1 and Smf1/2, and their mammalian homologs ATP2C1/SPCA1 (Muncanovic et al., 2019) and SLC11A2 (DMT1; Nevo and Nelson, 2006; and this work). In yeast, cytoplasmic Mn levels can be increased by the loss of Pmr1, which causes a defect in sequestration of Mn2+ in the Golgi. Alternatively, they can also be boosted by increased Smf1-mediated Mn2+ uptake combined with Smf2-mediated Mn2+ depletion in endosomes following the loss of Bsd2, an adaptor protein for the NEDD4 family E3 ubiquitin ligase Rsp5 (Liu et al., 1997) that normally favors vacuolar degradation of Smf1/2 (Jensen et al., 2009; Portnoy et al., 2000). In both cases, that is, loss of Pmr1 or of Bsd2 (specifically upon addition of Mn2+ in the medium), enhanced cytoplasmic Mn2+ levels mediate higher TORC1 activity and hence the resistance of cells to low doses of rapamycin. Conversely, our expression and localization studies of GFP-Smf1 and Smf2-GFP indicate that TORC1, while being controlled by Mn2+ levels, also employs feedback control mechanisms that regulate Smf1 expression and Smf2 turnover. These observations are consistent with the recent paradigm shift assigning TORC1 an essential role in cellular and organismal homeostasis (Eltschinger and Loewith, 2016) and extend this role of TORC1 to Mn metabolism. A more detailed understanding of these regulatory circuits involving TORC1 and NRAMP transporters (and vice versa) will, however, require future analyses of the specific subcellular pools of Mn2+ within different organelles using electron-nuclear double resonance spectroscopy (McNaughton et al., 2010) or inductively coupled plasma mass spectrometry (Liu et al., 2019). Of note, Mn2+ also acts as a cofactor for glutamine synthetase (Wedler and Denman, 1984) that produces the TORC1-stimulating amino acid glutamine (Jewell et al., 2015). Hence, Mn2+ homeostasis, amino acid metabolism, and TORC1 may be subjected to even more intricate, multilayered feedback regulatory circuits.
Our finding that Mn2+ activates TORC1/mTORC1 may have important implications in different biological contexts. For instance, previous work has shown that chronic exposure to high levels of Mn2+ causes autophagic dysfunction and hence accumulation of compromised mitochondria in mammalian astrocytes due to reduced nuclear localization of TFEB (transcription factor EB), a key transcription factor that coordinates the expression of genes involved in autophagy (Zhang et al., 2020). Because mTORC1 inhibits TFEB (Settembre et al., 2012) and mitochondrial quality can be improved by rapamycin-induced TFEB induction (and consequent stimulation of autophagy) (Siddiqui et al., 2015), our study now provides a simple rationale for the observed accumulation of damaged mitochondria upon Mn2+ exposure. Accordingly, excess Mn2+ antagonizes autophagy and mitophagy at least in part through mTORC1 activation and subsequent TFEB inhibition, which prevents proper disposal of hazardous and reactive oxygen species-producing mitochondria. This model also matches well with our previous observation that rapamycin restricts cell death associated with anomalous mTORC1 hyperactivation (Villar et al., 2017). Finally, our data indicate that Mn2+-driven TORC1 activation and the ensuing inhibition of auto- and mitophagy are also employed by yeast cells, which highlights the primordial nature of these processes.
Another area where our findings may be relevant relates to the use of Mn2+ to stimulate antitumor immune responses. Recent studies have shown that Mn2+ is indispensable for the host defense against cytosolic, viral double-stranded DNA as it mediates activation of the DNA sensor CGAS (cyclic GMP-AMP synthase) and its downstream adaptor protein STING1 (stimulator of interferon response cGAMP interactor 1) (Wang et al., 2018). Since Mn2+ stimulates the innate immune sensing of tumors, Mn administration has been suggested to provide an antitumoral effect and improve the treatment of cancer patients (Lv et al., 2020). Nevertheless, mTORC1 hyperactivation is known to promote tumor progression (Mossmann et al., 2018), and carcinoma and melanoma formation have previously been associated with mutations in the human PMR1 ortholog ATP2C1 that cause Hailey-Hailey disease (Mohr et al., 2011). Based on our findings, it is therefore possible that tumor formation in these patients may be causally linked to Mn2+-dependent DNA replication defects (García-Rodríguez et al., 2012), stress response (Cialfi et al., 2016), and mTORC1 activation.
In addition to the potential relevance of our findings in the context of chronic Mn2+ exposure and immune stimulation, our findings may also provide new perspectives to our understanding of specific neuropathies. For instance, exposure to Mn dust or Mn containing smoke, as a byproduct of metal welding, is well known to cause a parkinsonian-like syndrome named manganism, a toxic condition of Mn poisoning with dyskinesia (Racette et al., 2001). Interestingly, while dyskinesia has been connected to L-dopamine-mediated activation of mTORC1 (Santini et al., 2009), our findings suggest that Mn2+-driven mTORC1 hyperactivation may impair autophagy and thereby contribute to neurological diseases (Cheng et al., 2020). In line with this reasoning, compounds that inhibit mTORC1 activity, and thus stimulate autophagy, have been suggested as therapeutics for the treatment of Parkinson-like neurological symptoms (Ravikumar et al., 2004). In this context, Huntington disease (HD) is another example of patients suffering from a proteinopathy characterized by parkinsonian-like neurological syndromes (McColgan and Tabrizi, 2018). In HD patients, expansion of the polyglutamine tract in the N-terminus of the huntingtin protein leads to protein aggregation (Macdonald, 1993) and, intriguingly, HD patients exhibit reduced Mn2+ levels in the brain (Bowman et al., 2011). This raises the question of whether the respective cells aim to evade Mn-driven mTORC1 activation, for example, by reducing Mn uptake or sequestration of Mn2+ within organelles, to stimulate huntingtin protein degradation via autophagy. Finally, Mn2+ also contributes to prion formation in yeast (Chakrabortee et al., 2016), and elevated Mn2+ levels have been detected in the blood and the central nervous system of Creutzfeldt-Jakob patients (Hesketh et al., 2008). It will therefore be exciting to study the cell type-specific impact of Mn2+-driven mTORC1 activation on metabolism, genome stability, checkpoint signaling, and the immune response, all processes that play a key role in neurological diseases and aging-related processes.
Materials and methods
Antibodies
Primary antibodies against GFP (Takara Bio, 632380, AB_10013427; 1:5000), G-6-PDH (Sigma-Aldrich, A9521, AB_258454; 1:5000), Adh1 (Millipore, 126745, AB_564196; 1:200000), RPS6 (Cell Signaling Technology, 2217, AB_331355; 1:1000), phospho-RPS6 (Ser235-236) (Cell Signaling Technology, 4856, AB_2181037; 1:1000), RPS6KB (Cell Signaling Technology, 2708, AB_390722; 1:1000), phospho-RPS6KB (Thr389) (Cell Signaling Technology, 9205, AB_330944; 1:1000), EIF4EBP1 (Cell Signaling Technology, 9452, AB_331692; 1:1000), phospho-EIF4EBP1 (Thr37/46) (Cell Signaling Technology, 2855, AB_560835; 1:1000), phospho-EIF4EBP1 (Ser65) (Cell Signaling Technology, 9451, AB_330947; 1:1000), AKT1 (Cell Signaling Technology, 4691, AB_915783; 1:1000), phospho-AKT(Ser473) (Cell Signaling Technology, 4060, AB_2315049; 1:1000), phospho-AKT(Thr308) (Cell Signaling Technology, 13038, AB_2629447; 1:1000), ULK1 (Cell Signaling Technology, 8359, AB_11178668; 1:1000), phospho-ULK1(Ser757) (Cell Signaling Technology, 6888, AB_10829226; 1:1000), MAP1LC3 AB (Cell Signaling Technology, 12741, AB_2617131; 1:1000), β-actin (Cell Signaling Technology, 4967, AB_330288; 1:1000) were used.
Yeast strains, plasmids, and growth conditions
Yeast strains and plasmids used in this study are available upon request and listed in Supplementary file 2; Supplementary file 3, respectively. Gene disruption and tagging were performed with standard high-efficiency recombination methods. To generate strain MP6988, pSIVu-SMF1p-GFP-SMF1 was digested with PacI and transformed into smf1∆ strain YOL182C. Yeast cells were grown to mid-log phase in synthetic defined medium (SD; 0.17% yeast nitrogen base, 0.5% ammonium sulfate, 2% glucose, 0.2% drop-out mix) at 30°C. To induce autophagy, mitophagy, and Rtg1-Rtg3 retrograde signaling, cells were treated with 200 ng/ml rapamycin (Biosynth Carbosynth, AE27685) for the indicated time. For telomere length analyses, cells were grown for 3 days in rich medium (YPAD; 1% yeast extract, 2% peptone, 2% glucose, 0.004% adenine sulfate) with or without the addition of 10 mM CaCl2. For imaging of Rtg3-GFP, cells were grown at 26°C in SD-MSG-ura medium (0.17% yeast nitrogen base, 0.5% monosodium glutamate, 2% glucose, 0.2% drop-out mix-ura) to exponential phase and treated or not with 200 ng/ml rapamycin for 30 min. For GFP1-Smf1 and Smf2-GFP analyses, cells were grown in SD-ura medium. Overnight precultures were quickly spun and diluted into SD-ura medium devoid of manganese sulfate (0.19% yeast nitrogen base without manganese sulfate [Formedium; CYN2001], 0.5% ammonium sulfate, 2% glucose, and 0.2% drop-out mix-ura). After 5–6 hr of growth, cells were treated with either rapamycin (200 ng/ml), CHX (25 µg/ml), or both compounds for the indicated times.
Yeast cell lysate preparation and immunoblotting
Cells grown to mid-log phase were treated with 6.7% trichloroacetic acid (TCA, final concentration), pelleted, washed with 99% acetone, dried, dissolved in urea buffer (6 M urea, 50 mM Tris-HCl pH 7.5, 1% SDS, 1 mM PMSF, and 10 mM NaF) and disrupted with glass beads using a Yasui Kikai homogenizer. Cell lysates were heated at 65°C for 10 min in Laemmli SDS sample buffer, centrifuged at 15,000 g for 1 min, and the supernatants were subjected to SDS-PAGE and immunoblotted. Heat denaturation of samples was omitted to detect GFP-Smf1 and Smf2-GFP. Chemiluminescence signals were captured in an Amersham ImageQuant 800 Imager and quantified with ImageQuant TL software (Cytiva).
Drug sensitivity assays
Yeast cells grown to mid-log phase were adjusted to an initial A600 of 0.2, serially diluted 1:10, and spotted onto plates without or with rapamycin at the indicated concentrations (see figure legends). One mM MnCl2 and/or 10 mM CaCl2 were added to the medium when indicated. Plates were then incubated at 30°C for 3–4 days. Two or more biological replicates were performed for all conditions.
Analysis of telomere length
Genomic DNA was isolated from yeast strains grown in YPAD for 3 days with or without the addition of 10 mM CaCl2. DNA was digested with XhoI, separated on a 1% agarose-Tris borate EDTA gel, transferred to a Hybond XL (Amersham Biosciences) membrane, and hybridized with a 32P-labeled DNA probe specific for the terminal Y’ telomere fragment. The probe was generated by random hexanucleotide-primed DNA synthesis using a short Y’-specific DNA template, which was generated by PCR from genomic yeast DNA using the primers Y up (5’-TGCCGTGCAACAAACACTAAATCAA-3’) and Y’ low (5’-CGCTCGAGAAAGTTGGAGTTTTTCA-3’). Two biological replicates of the whole experiment were conducted.
In vitro TORC1 kinase assays
TORC1 was purified and kinase assays were performed as previously described (Nicastro et al., 2021) with minor modifications. For the kinase assays in the presence of various concentrations of magnesium or manganese, reactions were performed in a total volume of 20 µl with kinase buffer (50 mM HEPES/NaOH [pH 7.5], 150 mM NaCl), 400 ng of purified His6-Lst4Loop, 60 ng TORC1 (quantified based on the Tor1 subunit) and 640, 320, 160, 80, 40, or 20 µM MgCl2/MnCl2. The reactions were started by adding 1.4 µl of ATP mix (18 mM ATP, 3.3 mCi [γ-32P]-ATP [Hartmann Analytic, SRP-501]). For the kinase assays in the presence of various concentrations of ATP, reactions containing 4 mM MgCl2, 160 µM MnCl2, or both were started by adding 1.4 µl of stock ATP mix (5.7 mM ATP, 3.3 mCi [γ-32P]-ATP [Hartmann Analytic, SRP-501]) or twofold serial dilutions. After the addition of sample buffer, proteins were separated by SDS-PAGE, stained with SYPRO Ruby (Sigma-Aldrich, S492) (loading control), and analyzed using a phosphoimager (Typhoon FLA 9500; GE Healthcare). Band intensities from three technical replicates were quantified with ImageJ and data were analyzed with GraphPad Prism using the Michaelis-Menten non-linear fitting.
Cell culture
U2OS and HEK293T cell lines were obtained from the American Type Culture Collection (ATCC). GFP-MAP1LC3-expressing U2OS cells were kindly provided by Dr Eyal Gottlieb (Cancer Research UK, Glasgow, UK). All the cell lines were grown in high glucose DMEM base medium (Sigma-Aldrich, D6546) supplemented with 10% of fetal bovine serum, glutamine (2 mM), penicillin (Sigma-Aldrich, 100 µg/ml), and streptomycin (100 µg/ml), at 37°C, 5% CO2 in a humidified atmosphere. Amino acid starvation was performed with EBSS medium (Sigma-Aldrich, E2888) supplemented with glucose at a final concentration of 4.5 g/l. When indicated, the starvation medium was complemented with MnCl2 to a final concentration of 0.05–1 mM, chloroquine (Sigma-Aldrich; 10 µM), and amino acids, by adding MEM Amino Acids (Sigma-Aldrich, M5550), plus MEM Non-essential Amino Acid Solution (Sigma-Aldrich, M7145) and glutamine (2 mM). The identities of the cell lines bought from ATCC were authenticated by STR profiling. The identity of the GFP-MAP1LC3-expressing U2OS cells was verified by the donating research group. In addition, all cell lines were tested negative for mycoplasma contamination.
Confocal and fluorescence microscopy
Yeast cells: For imaging of GFP-Tor1, GFP-Smf1, and Smf2-GFP, images were captured with an inverted spinning disk confocal microscope (Nikon Ti-E, VisiScope CSU-W1, Puchheim, Germany) that was equipped with an Evolve 512 EM-CDD camera (Photometrics), and a 100 × 1.3 NA oil immersion Nikon CFI series objective (Egg, Switzerland). Images were then processed using the FIJI-ImageJ software. For imaging of Rtg3-GFP, images were obtained by projection of a focal plane image derived from wide-field fluorescence microscopy (DM-6000B, Leica) at 100× magnification using L5 filters and a digital charge-coupled device camera (DFC350, Leica). Pictures were processed with LAS AF (Leica).
Human cells: 8 × 105 cells were grown in coverslips for 24 hr and treated for 4 hr as indicated. Thereafter, cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) in PBS for 10 min at room temperature. For GFP-MAP1LC3 assessment, after three washes with PBS, coverslips were mounted with Prolong containing DAPI (Invitrogen). Samples were imaged with a Zeiss Apotome microscope. GFP aggregation in microscopy images was assessed using ImageJ software.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 7.0. Statistical significance was determined from at least three independent biological replicates using either Student’s t-test or ANOVA followed by Tukey’s multiple comparison test. Two-tailed Student’s t-test was used for comparison of the means of two different experimental conditions. Paired t-test was used for the pairwise comparison of normalized data. ANOVA test was used for the analyses of three variables. Differences with a p-value lower than 0.05 were considered significant. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. The number of independent experiments (n), specific statistical tests, and significance are described in the figure legends.
Acknowledgements
We thank María Díaz de la Loza and Benjamin Pillet for scientific illustration work, and V Albanèse, E de Nadal, V Goder, KD Hirschi, C Ungermann, and E Gottlieb for plasmids, yeast strains, and cell lines. Research was funded by grants from the University of Seville (2020/00001326), Junta de Andalucía/European Union Regional Funds (P20-RT-01220) and EMBO (STF-8685) to REW; the Swiss National Science Foundation (310030_166474/184671) to CDV; the Spanish Ministry of Science, Innovation and Universities (PGC2018-096244-B-I00) to RD. The author LZ was the recipient of a predoctoral grant from the Spanish Ministry of Science, Innovation and Universities (FPU19/04914).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Claudio De Virgilio, Email: Claudio.DeVirgilio@unifr.ch.
Ralf Erik Wellinger, Email: wellinger@us.es.
Maya Schuldiner, Weizmann Institute of Science, Israel.
Jonathan A Cooper, Fred Hutchinson Cancer Research Center, United States.
Funding Information
This paper was supported by the following grants:
Universidad de Sevilla 2020/00001326 to Hélène Gaillard, Ralf Erik Wellinger.
Junta de Andalucía P20-RT-01220 to Ralf Erik Wellinger.
European Molecular Biology Organization STF-8685 to Ralf Erik Wellinger.
Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung 310030_166474/184671 to Claudio De Virgilio.
Ministerio de Ciencia, Innovación y Universidades PGC2018-096244-B-I00 to Raúl V Durán.
Ministerio de Ciencia, Innovación y Universidades FPU19/04914 to Laura Zarzuela.
Additional information
Competing interests
No competing interests declared.
No competing interests declared.
Author contributions
Conceptualization, Formal analysis, Investigation, Methodology, Writing – review and editing.
Conceptualization, Formal analysis, Investigation, Visualization, Methodology, Writing – review and editing.
Investigation.
Conceptualization, Investigation, Methodology, Writing – review and editing.
Investigation.
Conceptualization, Investigation, Methodology.
Conceptualization, Investigation, Methodology, Writing – review and editing.
Conceptualization, Resources, Data curation, Formal analysis, Supervision, Funding acquisition, Methodology, Writing – review and editing.
Conceptualization, Resources, Data curation, Formal analysis, Supervision, Funding acquisition, Methodology, Writing – original draft, Writing – review and editing.
Conceptualization, Resources, Data curation, Formal analysis, Supervision, Funding acquisition, Investigation, Methodology, Writing – original draft, Project administration, Writing – review and editing.
Additional files
Data availability
All data generated or analyzed during this study are included in the manuscript and supporting files. Source data files have been provided for Figure 1, Figure 1-figure supplement 1, Figure 2, Figure 3, Figure 4 and Figure 5. The data set entitled 'Expression data of pmr1∆ mutants', originally published in García-Rodríguez et al (2012) and reused in this study is accessible at Gene Expression Omnibus under the accession code GSE29420.
The following previously published dataset was used:
Garcia-Rodriguez N, Wellinger RE. 2012. Expression data of pmr1∆ mutants. NCBI Gene Expression Omnibus. GSE29420
References
- Aguirre JD, Culotta VC. Battles with iron: manganese in oxidative stress protection. The Journal of Biological Chemistry. 2012;287:13541–13548. doi: 10.1074/jbc.R111.312181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon CM, Heitman J, Cardenas ME. Protein kinase activity and identification of a toxic effector domain of the target of rapamycin TOR proteins in yeast. Molecular Biology of the Cell. 1999;10:2531–2546. doi: 10.1091/mbc.10.8.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert V, Hall MN. mTOR signaling in cellular and organismal energetics. Current Opinion in Cell Biology. 2015;33:55–66. doi: 10.1016/j.ceb.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Bayeva M, Khechaduri A, Puig S, Chang HC, Patial S, Blackshear PJ, Ardehali H. mTOR regulates cellular iron homeostasis through tristetraprolin. Cell Metabolism. 2012;16:645–657. doi: 10.1016/j.cmet.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz C, Hall MN. Where is mTOR and what is it doing there? The Journal of Cell Biology. 2013;203:563–574. doi: 10.1083/jcb.201306041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda M, Péli-Gulli MP, Bonfils G, Panchaud N, Urban J, Sturgill TW, Loewith R, De Virgilio C. The Vam6 GEF controls TORC1 by activating the EGO complex. Molecular Cell. 2009;35:563–573. doi: 10.1016/j.molcel.2009.06.033. [DOI] [PubMed] [Google Scholar]
- Bodineau C, Tomé M, Courtois S, Costa ASH, Sciacovelli M, Rousseau B, Richard E, Vacher P, Parejo-Pérez C, Bessede E, Varon C, Soubeyran P, Frezza C, Murdoch PDS, Villar VH, Durán RV. Two parallel pathways connect glutamine metabolism and mTORC1 activity to regulate glutamoptosis. Nature Communications. 2021;12:4814. doi: 10.1038/s41467-021-25079-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton EC, Mildvan AS, Boeke JD. Inhibition of reverse transcription in vivo by elevated manganese ion concentration. Molecular Cell. 2002;9:879–889. doi: 10.1016/s1097-2765(02)00495-1. [DOI] [PubMed] [Google Scholar]
- Bowman AB, Kwakye GF, Herrero Hernández E, Aschner M. Role of manganese in neurodegenerative diseases. Journal of Trace Elements in Medicine and Biology. 2011;25:191–203. doi: 10.1016/j.jtemb.2011.08.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JC, Abraham RT. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. The EMBO Journal. 1996;15:5256–5267. doi: 10.1002/j.1460-2075.1996.tb00911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan MR, Uhouse MA, Nordham KD, Joshi P, Rose DIR, O’Brien MT, Aschner M, Bowman AB. Phosphatidylinositol 3 kinase (PI3K) modulates manganese homeostasis and manganese-induced cell signaling in a murine striatal cell line. Neurotoxicology. 2018;64:185–194. doi: 10.1016/j.neuro.2017.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter CL, Auger KR, Duckworth BC, Hou WM, Schaffhausen B, Cantley LC. A tightly associated serine/threonine protein kinase regulates phosphoinositide 3-kinase activity. Molecular and Cellular Biology. 1993;13:1657–1665. doi: 10.1128/MCB.13.3.1657-1665.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabortee S, Byers JS, Jones S, Garcia DM, Bhullar B, Chang A, She R, Lee L, Fremin B, Lindquist S, Jarosz DF. Intrinsically disordered proteins drive emergence and inheritance of biological traits. Cell. 2016;167:369–381. doi: 10.1016/j.cell.2016.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DW, Son SC, Block W, Ye R, Khanna KK, Wold MS, Douglas P, Goodarzi AA, Pelley J, Taya Y, Lavin MF, Lees-Miller SP. Purification and characterization of ATM from human placenta. Journal of Biological Chemistry. 2000;275:7803–7810. doi: 10.1074/jbc.275.11.7803. [DOI] [PubMed] [Google Scholar]
- Cheng J, Liao Y, Dong Y, Hu H, Yang N, Kong X, Li S, Li X, Guo J, Qin L, Yu J, Ma C, Li J, Li M, Tang B, Yuan Z. Microglial autophagy defect causes parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy. 2020;16:2193–2205. doi: 10.1080/15548627.2020.1719723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Klionsky DJ. Biochemical methods to monitor autophagy-related processes in yeast. Methods in Enzymology. 2008;451:1–26. doi: 10.1016/S0076-6879(08)03201-1. [DOI] [PubMed] [Google Scholar]
- Cialfi S, Le Pera L, De Blasio C, Mariano G, Palermo R, Zonfrilli A, Uccelletti D, Palleschi C, Biolcati G, Barbieri L, Screpanti I, Talora C. The loss of ATP2C1 impairs the DNA damage response and induces altered skin homeostasis: Consequences for epidermal biology in Hailey-Hailey disease. Scientific Reports. 2016;6:31567. doi: 10.1038/srep31567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couper J. On the effects of black oxide of manganese when inhaled into the lungs. British Annals of Medicine, Pharmacy, Vital Statistics, and General Science. 1837;1:41–42. [Google Scholar]
- Devasahayam G, Ritz D, Helliwell SB, Burke DJ, Sturgill TW. Pmr1, a Golgi Ca2+/Mn2+-ATPase, is a regulator of the target of rapamycin (TOR) signaling pathway in yeast. PNAS. 2006;103:17840–17845. doi: 10.1073/pnas.0604303103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devasahayam G, Burke DJ, Sturgill TW. Golgi manganese transport is required for rapamycin signaling in Saccharomyces cerevisiae. Genetics. 2007;177:231–238. doi: 10.1534/genetics.107.073577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhand R, Hiles I, Panayotou G, Roche S, Fry MJ, Gout I, Totty NF, Truong O, Vicendo P, Yonezawa K, Kasuga M, Courtneidge SA, Waterfield MD. PI 3-Kinase is a dual specificity enzyme: Autoregulation by an intrinsic protein-serine kinase activity. The EMBO Journal. 1994;13:522–533. doi: 10.1002/j.1460-2075.1994.tb06290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguez L, Chung YS, Kuchibhatla A, Paidhungat M, Garrett S. Yeast Mn2+ transporter, Smf1p, is regulated by ubiquitin-dependent vacuolar protein sorting. Genetics. 2004;167:107–117. doi: 10.1534/genetics.167.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltschinger S, Loewith R. TOR complexes and the maintenance of cellular homeostasis. Trends in Cell Biology. 2016;26:148–159. doi: 10.1016/j.tcb.2015.10.003. [DOI] [PubMed] [Google Scholar]
- Foukas LC, Beeton CA, Jensen J, Phillips WA, Shepherd PR. Regulation of phosphoinositide 3-kinase by its intrinsic serine kinase activity in vivo. Molecular and Cellular Biology. 2004;24:966–975. doi: 10.1128/MCB.24.3.966-975.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Rodríguez N, Díaz de la Loza M, Andreson B, Monje-Casas F, Rothstein R, Wellinger RE. Impaired manganese metabolism causes mitotic misregulation. The Journal of Biological Chemistry. 2012;287:18717–18729. doi: 10.1074/jbc.M112.358309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Rodríguez N, Manzano-López J, Muñoz-Bravo M, Fernández-García E, Muñiz M, Wellinger RE. Manganese redistribution by calcium-stimulated vesicle trafficking bypasses the need for P-type ATPase function. The Journal of Biological Chemistry. 2015;290:9335–9347. doi: 10.1074/jbc.M114.616334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaubitz C, Prouteau M, Kusmider B, Loewith R. TORC2 structure and function. Trends in Biochemical Sciences. 2016;41:532–545. doi: 10.1016/j.tibs.2016.04.001. [DOI] [PubMed] [Google Scholar]
- Harischandra DS, Ghaisas S, Zenitsky G, Jin H, Kanthasamy A, Anantharam V, Kanthasamy AG. Manganese-induced neurotoxicity: New insights into the triad of protein misfolding. Mitochondrial Impairment, and Neuroinflammation. Frontiers in Neuroscience. 2019;13:e00654. doi: 10.3389/fnins.2019.00654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama R, Péli-Gulli MP, Hu Z, Jaquenoud M, Garcia Osuna GM, Sardu A, Dengjel J, De Virgilio C. Spatially distinct pools of TORC1 balance protein homeostasis. Molecular Cell. 2019;73:325–338. doi: 10.1016/j.molcel.2018.10.040. [DOI] [PubMed] [Google Scholar]
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- Hesketh S, Sassoon J, Knight R, Brown DR. Elevated manganese levels in blood and CNS in human prion disease. Molecular and Cellular Neuroscience. 2008;37:590–598. doi: 10.1016/j.mcn.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Horning KJ, Caito SW, Tipps KG, Bowman AB, Aschner M. Manganese is essential for neuronal health. Annual Review of Nutrition. 2015;35:71–108. doi: 10.1146/ANNUREV-NUTR-071714-034419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LT, Carroll MC, Hall MD, Harvey CJ, Beese SE, Culotta VC. Down-Regulation of a manganese transporter in the face of metal toxicity. Molecular Biology of the Cell. 2009;20:2810–2819. doi: 10.1091/mbc.e08-10-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, Guan KL. Differential regulation of mTORC1 by leucine and glutamine. Science. 2015;347:194–198. doi: 10.1126/science.1259472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Kang D, Klionsky DJ. Monitoring mitophagy in yeast: The Om45-GFP processing assay. Autophagy. 2009;5:1186–1189. doi: 10.4161/auto.5.8.9854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Wang K, Klionsky DJ. A genomic screen for yeast mutants defective in mitophagy. Autophagy. 2010;6:278–280. doi: 10.4161/auto.6.2.10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ, Swaroop M, Murtha-Riel P. Depletion of manganese within the secretory pathway inhibits O-linked glycosylation in mammalian cells. Biochemistry. 1994;33:9813–9819. doi: 10.1021/bi00199a001. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK, Agnello M, Agostinis P, Aguilar PV, Aguirre-Ghiso J, Airoldi EM, Ait-Si-Ali S, Akematsu T, Akporiaye ET, Al-Rubeai M, Albaiceta GM, Albanese C, Albani D, Albert ML, Aldudo J, Algül H, Alirezaei M, Alloza I, Almasan A, Almonte-Beceril M, Alnemri ES, Alonso C, Altan-Bonnet N, Altieri DC, Alvarez S, Alvarez-Erviti L, Alves S, Amadoro G, Amano A, Amantini C, Ambrosio S, Amelio I, Amer AO, Amessou M, Amon A, An Z, Anania FA, Andersen SU, Andley UP, Andreadi CK, Andrieu-Abadie N, Anel A, Ann DK, Anoopkumar-Dukie S, Antonioli M, Aoki H, Apostolova N, Aquila S, Aquilano K, Araki K, Arama E, Aranda A, Araya J, Arcaro A, Arias E, Arimoto H, Ariosa AR, Armstrong JL, Arnould T, Arsov I, Asanuma K, Askanas V, Asselin E, Atarashi R, Atherton SS, Atkin JD, Attardi LD, Auberger P, Auburger G, Aurelian L, Autelli R, Avagliano L, Avantaggiati ML, Avrahami L, Awale S, Azad N, Bachetti T, Backer JM, Bae D-H, Bae J-S, Bae O-N, Bae SH, Baehrecke EH, Baek S-H, Baghdiguian S, Bagniewska-Zadworna A, Bai H, Bai J, Bai X-Y, Bailly Y, Balaji KN, Balduini W, Ballabio A, Balzan R, Banerjee R, Bánhegyi G, Bao H, Barbeau B, Barrachina MD, Barreiro E, Bartel B, Bartolomé A, Bassham DC, Bassi MT, Bast RC, Basu A, Batista MT, Batoko H, Battino M, Bauckman K, Baumgarner BL, Bayer KU, Beale R, Beaulieu J-F, Beck GR, Becker C, Beckham JD, Bédard P-A, Bednarski PJ, Begley TJ, Behl C, Behrends C, Behrens GM, Behrns KE, Bejarano E, Belaid A, Belleudi F, Bénard G, Berchem G, Bergamaschi D, Bergami M, Berkhout B, Berliocchi L, Bernard A, Bernard M, Bernassola F, Bertolotti A, Bess AS, Besteiro S, Bettuzzi S, Bhalla S, Bhattacharyya S, Bhutia SK, Biagosch C, Bianchi MW, Biard-Piechaczyk M, Billes V, Bincoletto C, Bingol B, Bird SW, Bitoun M, Bjedov I, Blackstone C, Blanc L, Blanco GA, Blomhoff HK, Boada-Romero E, Böckler S, Boes M, Boesze-Battaglia K, Boise LH, Bolino A, Boman A, Bonaldo P, Bordi M, Bosch J, Botana LM, Botti J, Bou G, Bouché M, Bouchecareilh M, Boucher M-J, Boulton ME, Bouret SG, Boya P, Boyer-Guittaut M, Bozhkov PV, Brady N, Braga VM, Brancolini C, Braus GH, Bravo-San Pedro JM, Brennan LA, Bresnick EH, Brest P, Bridges D, Bringer M-A, Brini M, Brito GC, Brodin B, Brookes PS, Brown EJ, Brown K, Broxmeyer HE, Bruhat A, Brum PC, Brumell JH, Brunetti-Pierri N, Bryson-Richardson RJ, Buch S, Buchan AM, Budak H, Bulavin DV, Bultman SJ, Bultynck G, Bumbasirevic V, Burelle Y, Burke RE, Burmeister M, Bütikofer P, Caberlotto L, Cadwell K, Cahova M, Cai D, Cai J, Cai Q, Calatayud S, Camougrand N, Campanella M, Campbell GR, Campbell M, Campello S, Candau R, Caniggia I, Cantoni L, Cao L, Caplan AB, Caraglia M, Cardinali C, Cardoso SM, Carew JS, Carleton LA, Carlin CR, Carloni S, Carlsson SR, Carmona-Gutierrez D, Carneiro LA, Carnevali O, Carra S, Carrier A, Carroll B, Casas C, Casas J, Cassinelli G, Castets P, Castro-Obregon S, Cavallini G, Ceccherini I, Cecconi F, Cederbaum AI, Ceña V, Cenci S, Cerella C, Cervia D, Cetrullo S, Chaachouay H, Chae H-J, Chagin AS, Chai C-Y, Chakrabarti G, Chamilos G, Chan EY, Chan MT, Chandra D, Chandra P, Chang C-P, Chang RC-C, Chang TY, Chatham JC, Chatterjee S, Chauhan S, Che Y, Cheetham ME, Cheluvappa R, Chen C-J, Chen G, Chen G-C, Chen G, Chen H, Chen JW, Chen J-K, Chen M, Chen M, Chen P, Chen Q, Chen Q, Chen S-D, Chen S, Chen SS-L, Chen W, Chen W-J, Chen WQ, Chen W, Chen X, Chen Y-H, Chen Y-G, Chen Y, Chen Y, Chen Y, Chen Y-J, Chen Y-Q, Chen Y, Chen Z, Chen Z, Cheng A, Cheng CH, Cheng H, Cheong H, Cherry S, Chesney J, Cheung CHA, Chevet E, Chi HC, Chi S-G, Chiacchiera F, Chiang H-L, Chiarelli R, Chiariello M, Chieppa M, Chin L-S, Chiong M, Chiu GN, Cho D-H, Cho S-G, Cho WC, Cho Y-Y, Cho Y-S, Choi AM, Choi E-J, Choi E-K, Choi J, Choi ME, Choi S-I, Chou T-F, Chouaib S, Choubey D, Choubey V, Chow K-C, Chowdhury K, Chu CT, Chuang T-H, Chun T, Chung H, Chung T, Chung Y-L, Chwae Y-J, Cianfanelli V, Ciarcia R, Ciechomska IA, Ciriolo MR, Cirone M, Claerhout S, Clague MJ, Clària J, Clarke PG, Clarke R, Clementi E, Cleyrat C, Cnop M, Coccia EM, Cocco T, Codogno P, Coers J, Cohen EE, Colecchia D, Coletto L, Coll NS, Colucci-Guyon E, Comincini S, Condello M, Cook KL, Coombs GH, Cooper CD, Cooper JM, Coppens I, Corasaniti MT, Corazzari M, Corbalan R, Corcelle-Termeau E, Cordero MD, Corral-Ramos C, Corti O, Cossarizza A, Costelli P, Costes S, Cotman SL, Coto-Montes A, Cottet S, Couve E, Covey LR, Cowart LA, Cox JS, Coxon FP, Coyne CB, Cragg MS, Craven RJ, Crepaldi T, Crespo JL, Criollo A, Crippa V, Cruz MT, Cuervo AM, Cuezva JM, Cui T, Cutillas PR, Czaja MJ, Czyzyk-Krzeska MF, Dagda RK, Dahmen U, Dai C, Dai W, Dai Y, Dalby KN, Dalla Valle L, Dalmasso G, D'Amelio M, Damme M, Darfeuille-Michaud A, Dargemont C, Darley-Usmar VM, Dasarathy S, Dasgupta B, Dash S, Dass CR, Davey HM, Davids LM, Dávila D, Davis RJ, Dawson TM, Dawson VL, Daza P, de Belleroche J, de Figueiredo P, de Figueiredo RCBQ, de la Fuente J, De Martino L, De Matteis A, De Meyer GR, De Milito A, De Santi M, de Souza W, De Tata V, De Zio D, Debnath J, Dechant R, Decuypere J-P, Deegan S, Dehay B, Del Bello B, Del Re DP, Delage-Mourroux R, Delbridge LM, Deldicque L, Delorme-Axford E, Deng Y, Dengjel J, Denizot M, Dent P, Der CJ, Deretic V, Derrien B, Deutsch E, Devarenne TP, Devenish RJ, Di Bartolomeo S, Di Daniele N, Di Domenico F, Di Nardo A, Di Paola S, Di Pietro A, Di Renzo L, DiAntonio A, Díaz-Araya G, Díaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dickey CA, Dickson RC, Diederich M, Digard P, Dikic I, Dinesh-Kumar SP, Ding C, Ding W-X, Ding Z, Dini L, Distler JH, Diwan A, Djavaheri-Mergny M, Dmytruk K, Dobson RC, Doetsch V, Dokladny K, Dokudovskaya S, Donadelli M, Dong XC, Dong X, Dong Z, Donohue TM, Doran KS, D'Orazi G, Dorn GW, Dosenko V, Dridi S, Drucker L, Du J, Du L-L, Du L, du Toit A, Dua P, Duan L, Duann P, Dubey VK, Duchen MR, Duchosal MA, Duez H, Dugail I, Dumit VI, Duncan MC, Dunlop EA, Dunn WA, Dupont N, Dupuis L, Durán RV, Durcan TM, Duvezin-Caubet S, Duvvuri U, Eapen V, Ebrahimi-Fakhari D, Echard A, Eckhart L, Edelstein CL, Edinger AL, Eichinger L, Eisenberg T, Eisenberg-Lerner A, Eissa NT, El-Deiry WS, El-Khoury V, Elazar Z, Eldar-Finkelman H, Elliott CJ, Emanuele E, Emmenegger U, Engedal N, Engelbrecht A-M, Engelender S, Enserink JM, Erdmann R, Erenpreisa J, Eri R, Eriksen JL, Erman A, Escalante R, Eskelinen E-L, Espert L, Esteban-Martínez L, Evans TJ, Fabri M, Fabrias G, Fabrizi C, Facchiano A, Færgeman NJ, Faggioni A, Fairlie WD, Fan C, Fan D, Fan J, Fang S, Fanto M, Fanzani A, Farkas T, Faure M, Favier FB, Fearnhead H, Federici M, Fei E, Felizardo TC, Feng H, Feng Y, Feng Y, Ferguson TA, Fernández AF, Fernandez-Barrena MG, Fernandez-Checa JC, Fernández-López A, Fernandez-Zapico ME, Feron O, Ferraro E, Ferreira-Halder CV, Fesus L, Feuer R, Fiesel FC, Filippi-Chiela EC, Filomeni G, Fimia GM, Fingert JH, Finkbeiner S, Finkel T, Fiorito F, Fisher PB, Flajolet M, Flamigni F, Florey O, Florio S, Floto RA, Folini M, Follo C, Fon EA, Fornai F, Fortunato F, Fraldi A, Franco R, Francois A, François A, Frankel LB, Fraser ID, Frey N, Freyssenet DG, Frezza C, Friedman SL, Frigo DE, Fu D, Fuentes JM, Fueyo J, Fujitani Y, Fujiwara Y, Fujiya M, Fukuda M, Fulda S, Fusco C, Gabryel B, Gaestel M, Gailly P, Gajewska M, Galadari S, Galili G, Galindo I, Galindo MF, Galliciotti G, Galluzzi L, Galluzzi L, Galy V, Gammoh N, Gandy S, Ganesan AK, Ganesan S, Ganley IG, Gannagé M, Gao F-B, Gao F, Gao J-X, García Nannig L, García Véscovi E, Garcia-Macía M, Garcia-Ruiz C, Garg AD, Garg PK, Gargini R, Gassen NC, Gatica D, Gatti E, Gavard J, Gavathiotis E, Ge L, Ge P, Ge S, Gean P-W, Gelmetti V, Genazzani AA, Geng J, Genschik P, Gerner L, Gestwicki JE, Gewirtz DA, Ghavami S, Ghigo E, Ghosh D, Giammarioli AM, Giampieri F, Giampietri C, Giatromanolaki A, Gibbings DJ, Gibellini L, Gibson SB, Ginet V, Giordano A, Giorgini F, Giovannetti E, Girardin SE, Gispert S, Giuliano S, Gladson CL, Glavic A, Gleave M, Godefroy N, Gogal RM, Gokulan K, Goldman GH, Goletti D, Goligorsky MS, Gomes AV, Gomes LC, Gomez H, Gomez-Manzano C, Gómez-Sánchez R, Gonçalves DA, Goncu E, Gong Q, Gongora C, Gonzalez CB, Gonzalez-Alegre P, Gonzalez-Cabo P, González-Polo RA, Goping IS, Gorbea C, Gorbunov NV, Goring DR, Gorman AM, Gorski SM, Goruppi S, Goto-Yamada S, Gotor C, Gottlieb RA, Gozes I, Gozuacik D, Graba Y, Graef M, Granato GE, Grant GD, Grant S, Gravina GL, Green DR, Greenhough A, Greenwood MT, Grimaldi B, Gros F, Grose C, Groulx J-F, Gruber F, Grumati P, Grune T, Guan J-L, Guan K-L, Guerra B, Guillen C, Gulshan K, Gunst J, Guo C, Guo L, Guo M, Guo W, Guo X-G, Gust AA, Gustafsson AB, Gutierrez E, Gutierrez MG, Gwak H-S, Haas A, Haber JE, Hadano S, Hagedorn M, Hahn DR, Halayko AJ, Hamacher-Brady A, Hamada K, Hamai A, Hamann A, Hamasaki M, Hamer I, Hamid Q, Hammond EM, Han F, Han W, Handa JT, Hanover JA, Hansen M, Harada M, Harhaji-Trajkovic L, Harper JW, Harrath AH, Harris AL, Harris J, Hasler U, Hasselblatt P, Hasui K, Hawley RG, Hawley TS, He C, He CY, He F, He G, He R-R, He X-H, He Y-W, He Y-Y, Heath JK, Hébert M-J, Heinzen RA, Helgason GV, Hensel M, Henske EP, Her C, Herman PK, Hernández A, Hernandez C, Hernández-Tiedra S, Hetz C, Hiesinger PR, Higaki K, Hilfiker S, Hill BG, Hill JA, Hill WD, Hino K, Hofius D, Hofman P, Höglinger GU, Höhfeld J, Holz MK, Hong Y, Hood DA, Hoozemans JJ, Hoppe T, Hsu C, Hsu C-Y, Hsu L-C, Hu D, Hu G, Hu H-M, Hu H, Hu MC, Hu Y-C, Hu Z-W, Hua F, Hua Y, Huang C, Huang H-L, Huang K-H, Huang K-Y, Huang S, Huang S, Huang W-P, Huang Y-R, Huang Y, Huang Y, Huber TB, Huebbe P, Huh W-K, Hulmi JJ, Hur GM, Hurley JH, Husak Z, Hussain SN, Hussain S, Hwang JJ, Hwang S, Hwang TI, Ichihara A, Imai Y, Imbriano C, Inomata M, Into T, Iovane V, Iovanna JL, Iozzo RV, Ip NY, Irazoqui JE, Iribarren P, Isaka Y, Isakovic AJ, Ischiropoulos H, Isenberg JS, Ishaq M, Ishida H, Ishii I, Ishmael JE, Isidoro C, Isobe K-I, Isono E, Issazadeh-Navikas S, Itahana K, Itakura E, Ivanov AI, Iyer AKV, Izquierdo JM, Izumi Y, Izzo V, Jäättelä M, Jaber N, Jackson DJ, Jackson WT, Jacob TG, Jacques TS, Jagannath C, Jain A, Jana NR, Jang BK, Jani A, Janji B, Jannig PR, Jansson PJ, Jean S, Jendrach M, Jeon J-H, Jessen N, Jeung E-B, Jia K, Jia L, Jiang H, Jiang H, Jiang L, Jiang T, Jiang X, Jiang X, Jiang X, Jiang Y, Jiang Y, Jiménez A, Jin C, Jin H, Jin L, Jin M, Jin S, Jinwal UK, Jo E-K, Johansen T, Johnson DE, Johnson GV, Johnson JD, Jonasch E, Jones C, Joosten LA, Jordan J, Joseph A-M, Joseph B, Joubert AM, Ju D, Ju J, Juan H-F, Juenemann K, Juhász G, Jung HS, Jung JU, Jung Y-K, Jungbluth H, Justice MJ, Jutten B, Kaakoush NO, Kaarniranta K, Kaasik A, Kabuta T, Kaeffer B, Kågedal K, Kahana A, Kajimura S, Kakhlon O, Kalia M, Kalvakolanu DV, Kamada Y, Kambas K, Kaminskyy VO, Kampinga HH, Kandouz M, Kang C, Kang R, Kang T-C, Kanki T, Kanneganti T-D, Kanno H, Kanthasamy AG, Kantorow M, Kaparakis-Liaskos M, Kapuy O, Karantza V, Karim MR, Karmakar P, Kaser A, Kaushik S, Kawula T, Kaynar AM, Ke P-Y, Ke Z-J, Kehrl JH, Keller KE, Kemper JK, Kenworthy AK, Kepp O, Kern A, Kesari S, Kessel D, Ketteler R, Kettelhut IdoC, Khambu B, Khan MM, Khandelwal VK, Khare S, Kiang JG, Kiger AA, Kihara A, Kim AL, Kim CH, Kim DR, Kim D-H, Kim EK, Kim HY, Kim H-R, Kim J-S, Kim JH, Kim JC, Kim JH, Kim KW, Kim MD, Kim M-M, Kim PK, Kim SW, Kim S-Y, Kim Y-S, Kim Y, Kimchi A, Kimmelman AC, Kimura T, King JS, Kirkegaard K, Kirkin V, Kirshenbaum LA, Kishi S, Kitajima Y, Kitamoto K, Kitaoka Y, Kitazato K, Kley RA, Klimecki WT, Klinkenberg M, Klucken J, Knævelsrud H, Knecht E, Knuppertz L, Ko J-L, Kobayashi S, Koch JC, Koechlin-Ramonatxo C, Koenig U, Koh YH, Köhler K, Kohlwein SD, Koike M, Komatsu M, Kominami E, Kong D, Kong HJ, Konstantakou EG, Kopp BT, Korcsmaros T, Korhonen L, Korolchuk VI, Koshkina NV, Kou Y, Koukourakis MI, Koumenis C, Kovács AL, Kovács T, Kovacs WJ, Koya D, Kraft C, Krainc D, Kramer H, Kravic-Stevovic T, Krek W, Kretz-Remy C, Krick R, Krishnamurthy M, Kriston-Vizi J, Kroemer G, Kruer MC, Kruger R, Ktistakis NT, Kuchitsu K, Kuhn C, Kumar AP, Kumar A, Kumar A, Kumar D, Kumar D, Kumar R, Kumar S, Kundu M, Kung H-J, Kuno A, Kuo S-H, Kuret J, Kurz T, Kwok T, Kwon TK, Kwon YT, Kyrmizi I, La Spada AR, Lafont F, Lahm T, Lakkaraju A, Lam T, Lamark T, Lancel S, Landowski TH, Lane DJR, Lane JD, Lanzi C, Lapaquette P, Lapierre LR, Laporte J, Laukkarinen J, Laurie GW, Lavandero S, Lavie L, LaVoie MJ, Law BYK, Law HK-W, Law KB, Layfield R, Lazo PA, Le Cam L, Le Roch KG, Le Stunff H, Leardkamolkarn V, Lecuit M, Lee B-H, Lee C-H, Lee EF, Lee GM, Lee H-J, Lee H, Lee JK, Lee J, Lee J-H, Lee JH, Lee M, Lee M-S, Lee PJ, Lee SW, Lee S-J, Lee S-J, Lee SY, Lee SH, Lee SS, Lee S-J, Lee S, Lee Y-R, Lee YJ, Lee YH, Leeuwenburgh C, Lefort S, Legouis R, Lei J, Lei Q-Y, Leib DA, Leibowitz G, Lekli I, Lemaire SD, Lemasters JJ, Lemberg MK, Lemoine A, Leng S, Lenz G, Lenzi P, Lerman LO, Lettieri Barbato D, Leu JI-J, Leung HY, Levine B, Lewis PA, Lezoualc'h F, Li C, Li F, Li F-J, Li J, Li K, Li L, Li M, Li M, Li Q, Li R, Li S, Li W, Li W, Li X, Li Y, Lian J, Liang C, Liang Q, Liao Y, Liberal J, Liberski PP, Lie P, Lieberman AP, Lim HJ, Lim K-L, Lim K, Lima RT, Lin C-S, Lin C-F, Lin F, Lin F, Lin F-C, Lin K, Lin K-H, Lin P-H, Lin T, Lin W-W, Lin Y-S, Lin Y, Linden R, Lindholm D, Lindqvist LM, Lingor P, Linkermann A, Liotta LA, Lipinski MM, Lira VA, Lisanti MP, Liton PB, Liu B, Liu C, Liu C-F, Liu F, Liu H-J, Liu J, Liu J-J, Liu J-L, Liu K, Liu L, Liu L, Liu Q, Liu R-Y, Liu S, Liu S, Liu W, Liu X-D, Liu X, Liu X-H, Liu X, Liu X, Liu X, Liu Y, Liu Y, Liu Z, Liu Z, Liuzzi JP, Lizard G, Ljujic M, Lodhi IJ, Logue SE, Lokeshwar BL, Long YC, Lonial S, Loos B, López-Otín C, López-Vicario C, Lorente M, Lorenzi PL, Lõrincz P, Los M, Lotze MT, Lovat PE, Lu B, Lu B, Lu J, Lu Q, Lu S-M, Lu S, Lu Y, Luciano F, Luckhart S, Lucocq JM, Ludovico P, Lugea A, Lukacs NW, Lum JJ, Lund AH, Luo H, Luo J, Luo S, Luparello C, Lyons T, Ma J, Ma Y, Ma Y, Ma Z, Machado J, Machado-Santelli GM, Macian F, MacIntosh GC, MacKeigan JP, Macleod KF, MacMicking JD, MacMillan-Crow LA, Madeo F, Madesh M, Madrigal-Matute J, Maeda A, Maeda T, Maegawa G, Maellaro E, Maes H, Magariños M, Maiese K, Maiti TK, Maiuri L, Maiuri MC, Maki CG, Malli R, Malorni W, Maloyan A, Mami-Chouaib F, Man N, Mancias JD, Mandelkow E-M, Mandell MA, Manfredi AA, Manié SN, Manzoni C, Mao K, Mao Z, Mao Z-W, Marambaud P, Marconi AM, Marelja Z, Marfe G, Margeta M, Margittai E, Mari M, Mariani FV, Marin C, Marinelli S, Mariño G, Markovic I, Marquez R, Martelli AM, Martens S, Martin KR, Martin SJ, Martin S, Martin-Acebes MA, Martín-Sanz P, Martinand-Mari C, Martinet W, Martinez J, Martinez-Lopez N, Martinez-Outschoorn U, Martínez-Velázquez M, Martinez-Vicente M, Martins WK, Mashima H, Mastrianni JA, Matarese G, Matarrese P, Mateo R, Matoba S, Matsumoto N, Matsushita T, Matsuura A, Matsuzawa T, Mattson MP, Matus S, Maugeri N, Mauvezin C, Mayer A, Maysinger D, Mazzolini GD, McBrayer MK, McCall K, McCormick C, McInerney GM, McIver SC, McKenna S, McMahon JJ, McNeish IA, Mechta-Grigoriou F, Medema JP, Medina DL, Megyeri K, Mehrpour M, Mehta JL, Mei Y, Meier U-C, Meijer AJ, Meléndez A, Melino G, Melino S, de Melo EJT, Mena MA, Meneghini MD, Menendez JA, Menezes R, Meng L, Meng L-H, Meng S, Menghini R, Menko AS, Menna-Barreto RF, Menon MB, Meraz-Ríos MA, Merla G, Merlini L, Merlot AM, Meryk A, Meschini S, Meyer JN, Mi M-T, Miao C-Y, Micale L, Michaeli S, Michiels C, Migliaccio AR, Mihailidou AS, Mijaljica D, Mikoshiba K, Milan E, Miller-Fleming L, Mills GB, Mills IG, Minakaki G, Minassian BA, Ming X-F, Minibayeva F, Minina EA, Mintern JD, Minucci S, Miranda-Vizuete A, Mitchell CH, Miyamoto S, Miyazawa K, Mizushima N, Mnich K, Mograbi B, Mohseni S, Moita LF, Molinari M, Molinari M, Møller AB, Mollereau B, Mollinedo F, Mongillo M, Monick MM, Montagnaro S, Montell C, Moore DJ, Moore MN, Mora-Rodriguez R, Moreira PI, Morel E, Morelli MB, Moreno S, Morgan MJ, Moris A, Moriyasu Y, Morrison JL, Morrison LA, Morselli E, Moscat J, Moseley PL, Mostowy S, Motori E, Mottet D, Mottram JC, Moussa CE-H, Mpakou VE, Mukhtar H, Mulcahy Levy JM, Muller S, Muñoz-Moreno R, Muñoz-Pinedo C, Münz C, Murphy ME, Murray JT, Murthy A, Mysorekar IU, Nabi IR, Nabissi M, Nader GA, Nagahara Y, Nagai Y, Nagata K, Nagelkerke A, Nagy P, Naidu SR, Nair S, Nakano H, Nakatogawa H, Nanjundan M, Napolitano G, Naqvi NI, Nardacci R, Narendra DP, Narita M, Nascimbeni AC, Natarajan R, Navegantes LC, Nawrocki ST, Nazarko TY, Nazarko VY, Neill T, Neri LM, Netea MG, Netea-Maier RT, Neves BM, Ney PA, Nezis IP, Nguyen HT, Nguyen HP, Nicot A-S, Nilsen H, Nilsson P, Nishimura M, Nishino I, Niso-Santano M, Niu H, Nixon RA, Njar VC, Noda T, Noegel AA, Nolte EM, Norberg E, Norga KK, Noureini SK, Notomi S, Notterpek L, Nowikovsky K, Nukina N, Nürnberger T, O'Donnell VB, O'Donovan T, O'Dwyer PJ, Oehme I, Oeste CL, Ogawa M, Ogretmen B, Ogura Y, Oh YJ, Ohmuraya M, Ohshima T, Ojha R, Okamoto K, Okazaki T, Oliver FJ, Ollinger K, Olsson S, Orban DP, Ordonez P, Orhon I, Orosz L, O'Rourke EJ, Orozco H, Ortega AL, Ortona E, Osellame LD, Oshima J, Oshima S, Osiewacz HD, Otomo T, Otsu K, Ou J-HJ, Outeiro TF, Ouyang D-Y, Ouyang H, Overholtzer M, Ozbun MA, Ozdinler PH, Ozpolat B, Pacelli C, Paganetti P, Page G, Pages G, Pagnini U, Pajak B, Pak SC, Pakos-Zebrucka K, Pakpour N, Palková Z, Palladino F, Pallauf K, Pallet N, Palmieri M, Paludan SR, Palumbo C, Palumbo S, Pampliega O, Pan H, Pan W, Panaretakis T, Pandey A, Pantazopoulou A, Papackova Z, Papademetrio DL, Papassideri I, Papini A, Parajuli N, Pardo J, Parekh VV, Parenti G, Park J-I, Park J, Park OK, Parker R, Parlato R, Parys JB, Parzych KR, Pasquet J-M, Pasquier B, Pasumarthi KB, Patschan D, Patterson C, Pattingre S, Pattison S, Pause A, Pavenstädt H, Pavone F, Pedrozo Z, Peña FJ, Peñalva MA, Pende M, Peng J, Penna F, Penninger JM, Pensalfini A, Pepe S, Pereira GJ, Pereira PC, Pérez-de la Cruz V, Pérez-Pérez ME, Pérez-Rodríguez D, Pérez-Sala D, Perier C, Perl A, Perlmutter DH, Perrotta I, Pervaiz S, Pesonen M, Pessin JE, Peters GJ, Petersen M, Petrache I, Petrof BJ, Petrovski G, Phang JM, Piacentini M, Pierdominici M, Pierre P, Pierrefite-Carle V, Pietrocola F, Pimentel-Muiños FX, Pinar M, Pineda B, Pinkas-Kramarski R, Pinti M, Pinton P, Piperdi B, Piret JM, Platanias LC, Platta HW, Plowey ED, Pöggeler S, Poirot M, Polčic P, Poletti A, Poon AH, Popelka H, Popova B, Poprawa I, Poulose SM, Poulton J, Powers SK, Powers T, Pozuelo-Rubio M, Prak K, Prange R, Prescott M, Priault M, Prince S, Proia RL, Proikas-Cezanne T, Prokisch H, Promponas VJ, Przyklenk K, Puertollano R, Pugazhenthi S, Puglielli L, Pujol A, Puyal J, Pyeon D, Qi X, Qian W-B, Qin Z-H, Qiu Y, Qu Z, Quadrilatero J, Quinn F, Raben N, Rabinowich H, Radogna F, Ragusa MJ, Rahmani M, Raina K, Ramanadham S, Ramesh R, Rami A, Randall-Demllo S, Randow F, Rao H, Rao VA, Rasmussen BB, Rasse TM, Ratovitski EA, Rautou P-E, Ray SK, Razani B, Reed BH, Reggiori F, Rehm M, Reichert AS, Rein T, Reiner DJ, Reits E, Ren J, Ren X, Renna M, Reusch JE, Revuelta JL, Reyes L, Rezaie AR, Richards RI, Richardson DR, Richetta C, Riehle MA, Rihn BH, Rikihisa Y, Riley BE, Rimbach G, Rippo MR, Ritis K, Rizzi F, Rizzo E, Roach PJ, Robbins J, Roberge M, Roca G, Roccheri MC, Rocha S, Rodrigues CM, Rodríguez CI, de Cordoba SR, Rodriguez-Muela N, Roelofs J, Rogov VV, Rohn TT, Rohrer B, Romanelli D, Romani L, Romano PS, Roncero MIG, Rosa JL, Rosello A, Rosen KV, Rosenstiel P, Rost-Roszkowska M, Roth KA, Roué G, Rouis M, Rouschop KM, Ruan DT, Ruano D, Rubinsztein DC, Rucker EB, Rudich A, Rudolf E, Rudolf R, Ruegg MA, Ruiz-Roldan C, Ruparelia AA, Rusmini P, Russ DW, Russo GL, Russo G, Russo R, Rusten TE, Ryabovol V, Ryan KM, Ryter SW, Sabatini DM, Sacher M, Sachse C, Sack MN, Sadoshima J, Saftig P, Sagi-Eisenberg R, Sahni S, Saikumar P, Saito T, Saitoh T, Sakakura K, Sakoh-Nakatogawa M, Sakuraba Y, Salazar-Roa M, Salomoni P, Saluja AK, Salvaterra PM, Salvioli R, Samali A, Sanchez AM, Sánchez-Alcázar JA, Sanchez-Prieto R, Sandri M, Sanjuan MA, Santaguida S, Santambrogio L, Santoni G, Dos Santos CN, Saran S, Sardiello M, Sargent G, Sarkar P, Sarkar S, Sarrias MR, Sarwal MM, Sasakawa C, Sasaki M, Sass M, Sato K, Sato M, Satriano J, Savaraj N, Saveljeva S, Schaefer L, Schaible UE, Scharl M, Schatzl HM, Schekman R, Scheper W, Schiavi A, Schipper HM, Schmeisser H, Schmidt J, Schmitz I, Schneider BE, Schneider EM, Schneider JL, Schon EA, Schönenberger MJ, Schönthal AH, Schorderet DF, Schröder B, Schuck S, Schulze RJ, Schwarten M, Schwarz TL, Sciarretta S, Scotto K, Scovassi AI, Screaton RA, Screen M, Seca H, Sedej S, Segatori L, Segev N, Seglen PO, Seguí-Simarro JM, Segura-Aguilar J, Seki E, Sell C, Seiliez I, Semenkovich CF, Semenza GL, Sen U, Serra AL, Serrano-Puebla A, Sesaki H, Setoguchi T, Settembre C, Shacka JJ, Shajahan-Haq AN, Shapiro IM, Sharma S, She H, Shen C-KJ, Shen C-C, Shen H-M, Shen S, Shen W, Sheng R, Sheng X, Sheng Z-H, Shepherd TG, Shi J, Shi Q, Shi Q, Shi Y, Shibutani S, Shibuya K, Shidoji Y, Shieh J-J, Shih C-M, Shimada Y, Shimizu S, Shin DW, Shinohara ML, Shintani M, Shintani T, Shioi T, Shirabe K, Shiri-Sverdlov R, Shirihai O, Shore GC, Shu C-W, Shukla D, Sibirny AA, Sica V, Sigurdson CJ, Sigurdsson EM, Sijwali PS, Sikorska B, Silveira WA, Silvente-Poirot S, Silverman GA, Simak J, Simmet T, Simon AK, Simon H-U, Simone C, Simons M, Simonsen A, Singh R, Singh SV, Singh SK, Sinha D, Sinha S, Sinicrope FA, Sirko A, Sirohi K, Sishi BJ, Sittler A, Siu PM, Sivridis E, Skwarska A, Slack R, Slaninová I, Slavov N, Smaili SS, Smalley KS, Smith DR, Soenen SJ, Soleimanpour SA, Solhaug A, Somasundaram K, Son JH, Sonawane A, Song C, Song F, Song HK, Song J-X, Song W, Soo KY, Sood AK, Soong TW, Soontornniyomkij V, Sorice M, Sotgia F, Soto-Pantoja DR, Sotthibundhu A, Sousa MJ, Spaink HP, Span PN, Spang A, Sparks JD, Speck PG, Spector SA, Spies CD, Springer W, Clair DS, Stacchiotti A, Staels B, Stang MT, Starczynowski DT, Starokadomskyy P, Steegborn C, Steele JW, Stefanis L, Steffan J, Stellrecht CM, Stenmark H, Stepkowski TM, Stern ST, Stevens C, Stockwell BR, Stoka V, Storchova Z, Stork B, Stratoulias V, Stravopodis DJ, Strnad P, Strohecker AM, Ström A-L, Stromhaug P, Stulik J, Su Y-X, Su Z, Subauste CS, Subramaniam S, Sue CM, Suh SW, Sui X, Sukseree S, Sulzer D, Sun F-L, Sun J, Sun J, Sun S-Y, Sun Y, Sun Y, Sun Y, Sundaramoorthy V, Sung J, Suzuki H, Suzuki K, Suzuki N, Suzuki T, Suzuki YJ, Swanson MS, Swanton C, Swärd K, Swarup G, Sweeney ST, Sylvester PW, Szatmari Z, Szegezdi E, Szlosarek PW, Taegtmeyer H, Tafani M, Taillebourg E, Tait SW, Takacs-Vellai K, Takahashi Y, Takáts S, Takemura G, Takigawa N, Talbot NJ, Tamagno E, Tamburini J, Tan C-P, Tan L, Tan ML, Tan M, Tan Y-J, Tanaka K, Tanaka M, Tang D, Tang D, Tang G, Tanida I, Tanji K, Tannous BA, Tapia JA, Tasset-Cuevas I, Tatar M, Tavassoly I, Tavernarakis N, Taylor A, Taylor GS, Taylor GA, Taylor JP, Taylor MJ, Tchetina EV, Tee AR, Teixeira-Clerc F, Telang S, Tencomnao T, Teng B-B, Teng R-J, Terro F, Tettamanti G, Theiss AL, Theron AE, Thomas KJ, Thomé MP, Thomes PG, Thorburn A, Thorner J, Thum T, Thumm M, Thurston TL, Tian L, Till A, Ting JP-Y, Titorenko VI, Toker L, Toldo S, Tooze SA, Topisirovic I, Torgersen ML, Torosantucci L, Torriglia A, Torrisi MR, Tournier C, Towns R, Trajkovic V, Travassos LH, Triola G, Tripathi DN, Trisciuoglio D, Troncoso R, Trougakos IP, Truttmann AC, Tsai K-J, Tschan MP, Tseng Y-H, Tsukuba T, Tsung A, Tsvetkov AS, Tu S, Tuan H-Y, Tucci M, Tumbarello DA, Turk B, Turk V, Turner RF, Tveita AA, Tyagi SC, Ubukata M, Uchiyama Y, Udelnow A, Ueno T, Umekawa M, Umemiya-Shirafuji R, Underwood BR, Ungermann C, Ureshino RP, Ushioda R, Uversky VN, Uzcátegui NL, Vaccari T, Vaccaro MI, Váchová L, Vakifahmetoglu-Norberg H, Valdor R, Valente EM, Vallette F, Valverde AM, Van den Berghe G, Van Den Bosch L, van den Brink GR, van der Goot FG, van der Klei IJ, van der Laan LJ, van Doorn WG, van Egmond M, van Golen KL, Van Kaer L, van Lookeren Campagne M, Vandenabeele P, Vandenberghe W, Vanhorebeek I, Varela-Nieto I, Vasconcelos MH, Vasko R, Vavvas DG, Vega-Naredo I, Velasco G, Velentzas AD, Velentzas PD, Vellai T, Vellenga E, Vendelbo MH, Venkatachalam K, Ventura N, Ventura S, Veras PS, Verdier M, Vertessy BG, Viale A, Vidal M, Vieira HLA, Vierstra RD, Vigneswaran N, Vij N, Vila M, Villar M, Villar VH, Villarroya J, Vindis C, Viola G, Viscomi MT, Vitale G, Vogl DT, Voitsekhovskaja OV, von Haefen C, von Schwarzenberg K, Voth DE, Vouret-Craviari V, Vuori K, Vyas JM, Waeber C, Walker CL, Walker MJ, Walter J, Wan L, Wan X, Wang B, Wang C, Wang C-Y, Wang C, Wang C, Wang C, Wang D, Wang F, Wang F, Wang G, Wang H-J, Wang H, Wang H-G, Wang H, Wang H-D, Wang J, Wang J, Wang M, Wang M-Q, Wang P-Y, Wang P, Wang RC, Wang S, Wang T-F, Wang X, Wang X-J, Wang X-W, Wang X, Wang X, Wang Y, Wang Y, Wang Y, Wang Y-J, Wang Y, Wang Y, Wang YT, Wang Y, Wang Z-N, Wappner P, Ward C, Ward DM, Warnes G, Watada H, Watanabe Y, Watase K, Weaver TE, Weekes CD, Wei J, Weide T, Weihl CC, Weindl G, Weis SN, Wen L, Wen X, Wen Y, Westermann B, Weyand CM, White AR, White E, Whitton JL, Whitworth AJ, Wiels J, Wild F, Wildenberg ME, Wileman T, Wilkinson DS, Wilkinson S, Willbold D, Williams C, Williams K, Williamson PR, Winklhofer KF, Witkin SS, Wohlgemuth SE, Wollert T, Wolvetang EJ, Wong E, Wong GW, Wong RW, Wong VKW, Woodcock EA, Wright KL, Wu C, Wu D, Wu GS, Wu J, Wu J, Wu M, Wu M, Wu S, Wu WK, Wu Y, Wu Z, Xavier CP, Xavier RJ, Xia G-X, Xia T, Xia W, Xia Y, Xiao H, Xiao J, Xiao S, Xiao W, Xie C-M, Xie Z, Xie Z, Xilouri M, Xiong Y, Xu C, Xu C, Xu F, Xu H, Xu H, Xu J, Xu J, Xu J, Xu L, Xu X, Xu Y, Xu Y, Xu Z-X, Xu Z, Xue Y, Yamada T, Yamamoto A, Yamanaka K, Yamashina S, Yamashiro S, Yan B, Yan B, Yan X, Yan Z, Yanagi Y, Yang D-S, Yang J-M, Yang L, Yang M, Yang P-M, Yang P, Yang Q, Yang W, Yang WY, Yang X, Yang Y, Yang Y, Yang Z, Yang Z, Yao M-C, Yao PJ, Yao X, Yao Z, Yao Z, Yasui LS, Ye M, Yedvobnick B, Yeganeh B, Yeh ES, Yeyati PL, Yi F, Yi L, Yin X-M, Yip CK, Yoo Y-M, Yoo YH, Yoon S-Y, Yoshida K-I, Yoshimori T, Young KH, Yu H, Yu JJ, Yu J-T, Yu J, Yu L, Yu WH, Yu X-F, Yu Z, Yuan J, Yuan Z-M, Yue BY, Yue J, Yue Z, Zacks DN, Zacksenhaus E, Zaffaroni N, Zaglia T, Zakeri Z, Zecchini V, Zeng J, Zeng M, Zeng Q, Zervos AS, Zhang DD, Zhang F, Zhang G, Zhang G-C, Zhang H, Zhang H, Zhang H, Zhang H, Zhang J, Zhang J, Zhang J, Zhang J, Zhang J-P, Zhang L, Zhang L, Zhang L, Zhang L, Zhang M-Y, Zhang X, Zhang XD, Zhang Y, Zhang Y, Zhang Y, Zhang Y, Zhang Y, Zhao M, Zhao W-L, Zhao X, Zhao YG, Zhao Y, Zhao Y, Zhao Y-X, Zhao Z, Zhao ZJ, Zheng D, Zheng X-L, Zheng X, Zhivotovsky B, Zhong Q, Zhou G-Z, Zhou G, Zhou H, Zhou S-F, Zhou X-J, Zhu H, Zhu H, Zhu W-G, Zhu W, Zhu X-F, Zhu Y, Zhuang S-M, Zhuang X, Ziparo E, Zois CE, Zoladek T, Zong W-X, Zorzano A, Zughaier SM. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knape MJ, Ballez M, Burghardt NC, Zimmermann B, Bertinetti D, Kornev AP, Herberg FW. Divalent metal ions control activity and inhibition of protein kinases. Metallomics. 2017;9:1576–1584. doi: 10.1039/c7mt00204a. [DOI] [PubMed] [Google Scholar]
- Lapinskas PJ, Cunningham KW, Liu XF, Fink GR, Culotta VC. Mutations in PMR1 suppress oxidative damage in yeast cells lacking superoxide dismutase. Molecular and Cellular Biology. 1995;15:1382–1388. doi: 10.1128/MCB.15.3.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X F, Culotta VC. The requirement for yeast superoxide dismutase is bypassed through mutations in BSD2, a novel metal homeostasis gene. Molecular and Cellular Biology. 1994;14:7037–7045. doi: 10.1128/mcb.14.11.7037-7045.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XF, Supek F, Nelson N, Culotta VC. Negative control of heavy metal uptake by the Saccharomyces cerevisiae BSD2 gene. The Journal of Biological Chemistry. 1997;272:11763–11769. doi: 10.1074/jbc.272.18.11763. [DOI] [PubMed] [Google Scholar]
- Liu XF, Culotta VC. Post-translation control of nramp metal transport in yeast. Journal of Biological Chemistry. 1999;274:4863–4868. doi: 10.1074/jbc.274.8.4863. [DOI] [PubMed] [Google Scholar]
- Liu Z, Xue A, Chen H, Li S. Quantitative determination of trace metals in single yeast cells by time-resolved ICP-MS using dissolved standards for calibration. Applied Microbiology and Biotechnology. 2019;103:1475–1483. doi: 10.1007/s00253-018-09587-w. [DOI] [PubMed] [Google Scholar]
- Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nature Reviews. Molecular Cell Biology. 2020;21:183–203. doi: 10.1038/s41580-019-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue NF, Bosoy D, Moriarty TJ, Autexier C, Altman B, Leng S. Telomerase can act as a template- and RNA-independent terminal transferase. PNAS. 2005;102:9778–9783. doi: 10.1073/pnas.0502252102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk EEC, Culotta VC. Manganese superoxide dismutase in Saccharomyces cerevisiae acquires its metal co-factor through a pathway involving the Nramp metal transporter, Smf2p. The Journal of Biological Chemistry. 2001;276:47556–47562. doi: 10.1074/jbc.M108923200. [DOI] [PubMed] [Google Scholar]
- Lv M, Chen M, Zhang R, Zhang W, Wang C, Zhang Y, Wei X, Guan Y, Liu J, Feng K, Jing M, Wang X, Liu YC, Mei Q, Han W, Jiang Z. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Research. 2020;30:966–979. doi: 10.1038/s41422-020-00395-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald M. A novel gene containing A trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- McColgan P, Tabrizi SJ. Huntington’s disease: a clinical review. European Journal of Neurology. 2018;25:24–34. doi: 10.1111/ene.13413. [DOI] [PubMed] [Google Scholar]
- McNaughton RL, Reddi AR, Clement MHS, Sharma A, Barnese K, Rosenfeld L, Gralla EB, Valentine JS, Culotta VC, Hoffman BM. Probing in vivo Mn2+ speciation and oxidative stress resistance in yeast cells with electron-nuclear double resonance spectroscopy. PNAS. 2010;107:15335–15339. doi: 10.1073/pnas.1009648107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr MR, Erdag G, Shada AL, Williams ME, Slingluff CL, Patterson JW. Two patients with Hailey-Hailey disease, multiple primary melanomas, and other cancers. Archives of Dermatology. 2011;147:211–215. doi: 10.1001/archdermatol.2010.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Cermeño A, Obis E, Bellí G, Cabiscol E, Ros J, Tamarit J. Frataxin depletion in yeast triggers up-regulation of iron transport systems before affecting iron-sulfur enzyme activities. The Journal of Biological Chemistry. 2010;285:41653–41664. doi: 10.1074/jbc.M110.149443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nature Reviews. Cancer. 2018;18:744–757. doi: 10.1038/s41568-018-0074-8. [DOI] [PubMed] [Google Scholar]
- Muncanovic D, Justesen MH, Preisler SS, Pedersen PA. Characterization of Hailey-Hailey Disease-mutants in presence and absence of wild type SPCA1 using Saccharomyces cerevisiae as model organism. Scientific Reports. 2019;9:12442. doi: 10.1038/s41598-019-48866-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevo Y, Nelson N. The NRAMP family of metal-ion transporters. Biochimica et Biophysica Acta. 2006;1763:609–620. doi: 10.1016/j.bbamcr.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Nicastro R, Sardu A, Panchaud N, De Virgilio C. The architecture of the rag GTPase signaling network. Biomolecules. 2017;7:E48. doi: 10.3390/biom7030048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicastro R, Raucci S, Michel AH, Stumpe M, Garcia Osuna GM, Jaquenoud M, Kornmann B, De Virgilio C. Indole-3-acetic acid is a physiological inhibitor of TORC1 in yeast. PLOS Genetics. 2021;17:e1009414. doi: 10.1371/journal.pgen.1009414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. The Journal of Biological Chemistry. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- Portnoy ME, Liu XF, Culotta VC. Saccharomyces cerevisiae expresses three functionally distinct homologues of the nramp family of metal transporters. Molecular and Cellular Biology. 2000;20:7893–7902. doi: 10.1128/MCB.20.21.7893-7902.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racette BA, McGee-Minnich L, Moerlein SM, Mink JW, Videen TO, Perlmutter JS. Welding-related parkinsonism. Neurology. 2001;56:8–13. doi: 10.1212/WNL.56.1.8. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nature Genetics. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Reinke A, Chen JCY, Aronova S, Powers T. Caffeine targets TOR complex I and provides evidence for a regulatory link between the FRB and kinase domains of Tor1p. The Journal of Biological Chemistry. 2006;281:31616–31626. doi: 10.1074/jbc.M603107200. [DOI] [PubMed] [Google Scholar]
- Renz C, Albanèse V, Tröster V, Albert TK, Santt O, Jacobs SC, Khmelinskii A, Léon S, Ulrich HD. Ubc13-Mms2 cooperates with a family of RING E3 proteins in budding yeast membrane protein sorting. Journal of Cell Science. 2020;133:jcs244566. doi: 10.1242/jcs.244566. [DOI] [PubMed] [Google Scholar]
- Ruiz-Roig C, Noriega N, Duch A, Posas F, de Nadal E. The Hog1 SAPK controls the Rtg1/Rtg3 transcriptional complex activity by multiple regulatory mechanisms. Molecular Biology of the Cell. 2012;23:4286–4296. doi: 10.1091/mbc.E12-04-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacher A, Cohen A, Nelson N. Metal ion transporters from yeast to human diseases. Comparative Biochemistry and Physiology Part A. 2000;126:e109. doi: 10.1016/S1095-6433(00)80215-9. [DOI] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Heiman M, Greengard P, Valjent E, Fisone G. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Science Signaling. 2009;2:ra36. doi: 10.1126/scisignal.2000308. [DOI] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. The EMBO Journal. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui A, Bhaumik D, Chinta SJ, Rane A, Rajagopalan S, Lieu CA, Lithgow GJ, Andersen JK. Mitochondrial quality control via the PGC1α-TFEB signaling pathway is compromised by parkin q311x mutation but independently restored by rapamycin. The Journal of Neuroscience. 2015;35:12833–12844. doi: 10.1523/JNEUROSCI.0109-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Supekova L, Nelson H, Nelson N. A yeast manganese transporter related to the macrophage protein involved in conferring resistance to mycobacteria. PNAS. 1996;93:5105–5110. doi: 10.1073/pnas.93.10.5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanigawa M, Yamamoto K, Nagatoishi S, Nagata K, Noshiro D, Noda NN, Tsumoto K, Maeda T. A glutamine sensor that directly activates TORC1. Communications Biology. 2021;4:e1093. doi: 10.1038/s42003-021-02625-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eunen K, Bouwman J, Daran-Lapujade P, Postmus J, Canelas AB, Mensonides FIC, Orij R, Tuzun I, van den Brink J, Smits GJ, van Gulik WM, Brul S, Heijnen JJ, de Winde JH, de Mattos MJT, Kettner C, Nielsen J, Westerhoff HV, Bakker BM. Measuring enzyme activities under standardized in vivo-like conditions for systems biology. The FEBS Journal. 2010;277:749–760. doi: 10.1111/j.1742-4658.2009.07524.x. [DOI] [PubMed] [Google Scholar]
- Villar VH, Nguyen TL, Delcroix V, Terés S, Bouchecareilh M, Salin B, Bodineau C, Vacher P, Priault M, Soubeyran P, Durán RV. mTORC1 inhibition in cancer cells protects from glutaminolysis-mediated apoptosis during nutrient limitation. Nature Communications. 2017;8:14124. doi: 10.1038/ncomms14124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Guan Y, Lv M, Zhang R, Guo Z, Wei X, Du X, Yang J, Li T, Wan Y, Su X, Huang X, Jiang Z. Manganese increases the sensitivity of the cGAS-STING pathway for double-stranded DNA and is required for the host defense against DNA viruses. Immunity. 2018;48:675–687. doi: 10.1016/j.immuni.2018.03.017. [DOI] [PubMed] [Google Scholar]
- Wedaman KP, Reinke A, Anderson S, Yates J, McCaffery JM, Powers T. Tor kinases are in distinct membrane-associated protein complexes in Saccharomyces cerevisiae. Molecular Biology of the Cell. 2003;14:1204–1220. doi: 10.1091/mbc.e02-09-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedler FC, Denman RB. Glutamine synthetase: the major Mn(II) enzyme in mammalian brain. Current Topics in Cellular Regulation. 1984;24:153–169. doi: 10.1016/b978-0-12-152824-9.50021-6. [DOI] [PubMed] [Google Scholar]
- Weisiger RA, Fridovich I. Mitochondrial superoxide dismutase. Journal of Biological Chemistry. 1973;248:4793–4796. doi: 10.1016/S0021-9258(19)43735-6. [DOI] [PubMed] [Google Scholar]
- Withers DJ, Ouwens DM, Nave BT, van der Zon GC, Alarcon CM, Cardenas ME, Heitman J, Maassen JA, Shepherd PR. Expression, enzyme activity, and subcellular localization of mammalian target of rapamycin in insulin-responsive cells. Biochemical and Biophysical Research Communications. 1997;241:704–709. doi: 10.1006/bbrc.1997.7878. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Yan J, Bowman AB, Bryan MR, Singh R, Aschner M. Dysregulation of TFEB contributes to manganese-induced autophagic failure and mitochondrial dysfunction in astrocytes. Autophagy. 2020;16:1506–1523. doi: 10.1080/15548627.2019.1688488. [DOI] [PMC free article] [PubMed] [Google Scholar]