

Abstract
The costs of maintaining exabytes of data produced by sequencing experiments every year has become a major issue in today’s genomics. In spite of the increasing popularity of the third generation sequencing, the existing algorithms for compressing long reads exhibit minor advantage over general purpose gzip. We present CoLoRd, an algorithm able to reduce 3rd generation sequencing data by an order of magnitude without affecting the accuracy of downstream analyzes.
In the last few years, we have seen a rapid development of the third generation sequencing. The ability to produce very long reads makes Oxford Nanopore and PacBio technologies indispensable in genome assembly [6] or identification of long structural variants [18]. Maintaining gigantic collections of sequencing reads, which often reach hundreds of gigabytes per experiment, has become a major challenge and motivated intense research on compression techniques. Due to memory constraints, which prevented from maintaining the entire read collection in main memory, the early algorithms [7, 1, 16] were only slightly better than general-purpose compressors. The more recent methods [5, 17, 11, 2] successfully tackled this issue by clustering reads originating from close genome positions. All these approaches, though, were designed for short, high-quality Illumina reads and are unsuitable for reads from 3rd generation instruments which are orders of magnitude longer and have different error profile. Among few algorithms able to compress ONT/PacBio FASTQ files, like SPRING [2], ENANO [3], or LFQC [13], none takes advantage of the data redundancy in the overlapping reads. This, together with the lossless compression of the quality stream, which is built over significantly greater alphabet than DNA (94 instead of 4 symbols) and exhibits larger noise, limits advantages over, de facto standard, gzip on 3rd generation data to tens of percent.
In the paper, we present CoLoRd, a compression algorithm for ONT and PacBio sequencing data. Its main contributions are (i) novel method for compressing the DNA component of FASTQ files and (ii) lossy processing of the quality stream. The idea of the former is based on the overlap graphs [12] often employed by long-read assemblers [9, 8]. As CoLoRd does not aim at finding true overlaps (which are critical for genome reconstruction), but those convenient for differential compression of reads, the throughput of the overlap detection can be increased at the cost of the accuracy. The algorithm determines k-mer similarities between the reads and use them for identifying closest neighbours—potential references in differential compression. The compression itself is based on anchors, i.e., exact sequence matches. The between-anchor areas are further scanned for similar fragments. As a result, the algorithm translates a read to an edit script describing differences w.r.t. similar reads (Figure 1a), which is then entropy-encoded. The presence of priority modes, i.e., memory (default), balanced, and ratio which differ mainly by the fraction of reads used as references, allows suiting the algorithm to particular needs. Since CoLoRd does not need a reference genome, it is suitable for different data types, including genomic, metagenomic, and transcriptomic. For highest flexibility, the algorithm is also equipped with a reference-based mode. By employing pseudo-reads generated from the reference genome, further improvements in a DNA compression are possible.
Figure 1: CoLoRd operation principles.

a Encoding a query read w.r.t. to the multiple reference reads. The edit script was generated during compression of a real read from Zymo data set. The colours indicate anchors (blue), matches (green), mismatches (purple), insertions (orange), deletions (red), and unmatched areas (gray). b Processing scheme of the data streams: k-mers (orange), DNA (purple), quality (green), and identifiers (red). Lightnings represent computing threads, black arrows indicate queries to data collections.
The encoding of the quality stream is done with a use of the context-based approach inspired by ENANO, complemented with the knowledge of read overlaps. In agreement with the findings on Illumina data [17], we show that the resolution of quality scores can be reduced without affecting the accuracy of downstream analyses. Therefore, quality levels are by default subject to the lossy compression. CoLoRd uses 4-level quality binning for Nanopore data with a scheme of [Q0, Q6], [Q7, Q13], [Q14, Q25] and [Q26, Q93]. For HiFi data, Q93 value is stored in a separate bin. CoLoRd calculates a representative quality in each bin such that the average read quality is preserved. The lossless mode, together with other variants, are also provided. CoLoRd was implemented in the C++ programming language and takes advantage of multiple computing threads (Figure 1b).
The advantage of CoLoRd over general-purpose (pigz v2.6, 7zip v16.02) and specialized (ENANO v1.0, SPRING v1.0) compressors was confirmed on 24 read sets representing different technologies (ONT, PacBio) with sizes ranging from 2 to 786GB (Supplementary Table 1). As presented in Figure 2a, our algorithm revealed its potential on the latest ONT Bonito-base-called and PacBio HiFi data sets. By efficiently finding overlaps in the high fidelity reads, CoLoRd reduced the size of the DNA stream by two orders of magnitude. This, accompanied by the quantization of quality levels, allowed squeezing FASTQ files to the 1/25 of their original size, which translated to 4-fold (ONT Bonito) and 10-fold (HiFi) advantage over lossless gzip compression. For instance, HiFi HG002 data set (30× coverage) was compressed from 219 to 10 gigabytes. With further accuracy improvements in 3rd generation sequencing on the horizon, even better results could be achieved. Contrarily, the high error rate of Nanopore reads produced by the older base callers, bounded the advantage of CoLoRd to lower, though, still significant levels. For experimental integrity, we additionally simulated lossy compression in gzip, 7zip, ENANO, and SPRING by binning quality levels in the input files. In this scheme, our algorithm was ahead of the rest with 2–4-fold improvement over gzip. In the reference-based mode, CoLoRd offered even better compression ratios and was superior to other algorithms using reference genomes like RENANO v1.0 [4] or CRAM v3.1 [1] (Supplementary Tables 2–3). Detailed results of investigated algorithms across selected genomic and metagenomic data sets from different technologies are presented in Figure 2c.
Figure 2: Analysis of lossless and lossy compression.
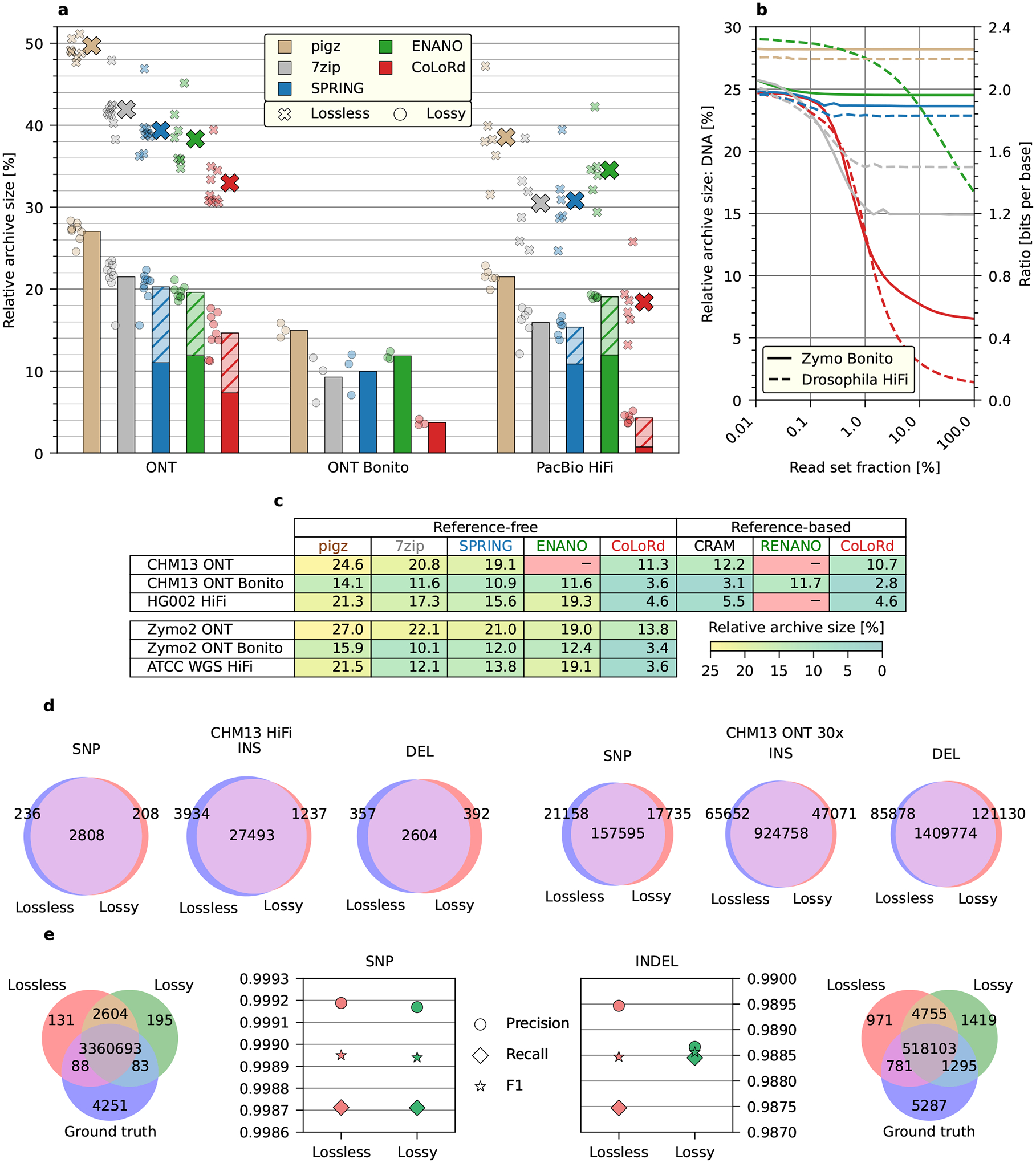
a Average performance of the algorithms with lossless (large crosses) and lossy (bars) quality compression (hatched areas represent contribution of the quality stream). The results on the individual data sets are presented as small crosses (lossless) and circles (lossy). The lossy archives for pigz, 7zip, SPRING, and ENANO were generated by binning quality scores in the input files. b Relative size of the DNA archive as a function of sequencing coverage evaluated on selected ONT and HiFi data sets. The corresponding compression ratio in bits per base is presented on the right vertical axis. c Relative sizes of lossy archives for selected genomic and metagenomic data sets in the reference-free and reference-based configurations. ENANO and RENANO were unable to process some sets. d Results of the Racon consensus generation on CHM13 ONT and CHM13 HiFi samples expressed by the number of substitutions, insertions, and deletions w.r.t. the reference genome. e Accuracy of DeepVariant SNP and INDEL calling on HG002 HiFi sample.
An essential property of CoLoRd is that with increasing coverage, the number of overlaps, thus the efficiency of the differential compression, grows as well. As the analysis of two selected data sets shows (Figure 2b), when taking 0.01% of reads, all investigated algorithms compressed the DNA stream to approximately 1/4 of its initial size. For the entire read set, however, CoLoRd offered 20-fold (Bonito Zymo with Q20 chemistry) and 100-fold (HiFi Drosophila) reduction of the DNA component, while most of the competitors exhibited minor or no improvements in the compression ratio. The only exception was 7zip which thanks to the large dictionary in LZMA algorithm was able to identify redundancies in DNA data and squeeze it to 15%–20% of the original size.
Importantly, superior compression results of the presented algorithm came at competitive running times. The lossy compression/decompression speeds at 24 threads were way above 50 MB/s, which is the throughput of PromethION—the fastest among 3rd generation instruments. Memory requirements were also reasonable with 16 GB for the aforementioned HG002 data set.
We evaluated consensus and variant calling accuracy to see how much lossy quality compression may affect downstream analyses. We mapped CHM13 HiFi reads to the CHM13 telomere-to-telomere (T2T) assembly v1.1 [14], generated the consensus with Racon v1.4.20 [19], aligned the consensus to the assembly with minimap2 v2.20 [10] and counted consensus differences (Figure 2d). The T2T assembly was derived from multiple data types including HiFi, Nanopore and Illumina reads. Our HiFi-only Racon consensus from raw reads differs from the T2T assembly at a rate of one difference per 81kb. Interestingly, lossy quality compression slightly improves the consensus accuracy to one difference per 88kb, suggesting Racon might be more compatible with our lossy scheme. We also applied the same procedure to Nanopore reads (CHM13 ONT data set downsampled to 30× coverage) and observed that lossy quality compression does not reduce the Racon consensus accuracy, either (one difference per 1.1kb for both lossless and lossy compression). To evaluate variant calling accuracy, we mapped HG002 HiFi reads to the human reference genome GRCh38 and called small variants with DeepVariant v1.1.0 [15]. When compared against the Genome-In-A-Bottle v4.2.1 truth [20], variant calls from lossy quality compression are as accurate as calls from raw reads (Figure 2e). With these experiments, also given the fact that most long-read assemblers ignore base quality at the assembly step, we conclude that lossy quality compression does not affect the accuracy of most downstream analyses. Investigation of different quality compression modes is presented in Supplementary Table 4.
Detailed results of the experiments carried out within this study (compression ratios for other CoLoRd modes, PacBio CLR data sets analysis, time and memory requirements, consensus and variant calling performance) are shown in Supplementary Data.
Here, we introduce CoLoRd, a comprehensive package for compressing Oxford Nanopore/PacBio sequencing data. Equipped with an overlap-based algorithm for compressing the DNA stream and a lossy processing of the quality information, it allows even tenfold space reduction compared to gzip, without affecting downstream analyses like variant calling or consensus generation. This opens new opportunities in the field of long read sequencing, where maintaining gigantic data volumes has become one of the major contributors to the overall costs.
Among possible CoLoRd improvements, replacing a two-pass compression scheme with online processing could be particularly helpful. This would significantly facilitate using the algorithm in streaming/pipeline configurations. A different enhancement could be redesigning the archive structure in order to take more of the multi-threading during decompression. This could potentially increase the throughput of this process several times on multi-core machines.
Methods
CoLoRd overview
CoLoRd compressor handles reads in FASTA or FASTQ format. In the former case, the input consists of two independently compressed streams: DNA data, which contribute predominantly to the file size, and sequence headers. In FASTQ files there is an additional base quality stream of the same size as DNA data.
One of the main contributions of CoLoRd is the DNA compression algorithm. DNA sequences of reads are built upon 5-element alphabet (A, C, G, T, N) and are highly redundant due to: (i) same genome fragments being covered by multiple reads and (ii) similarity between different genome regions. CoLoRd is the first method which takes advantage of the characteristics of Oxford Nanopore and PacBio data when compressing DNA stream. The algorithm processes reads one by one determining their k-mer similarity to already analyzed reads. The resulting similarity graph is then used to identify closest neighbours which are references for differential compression of the current read. The procedure is based on anchors—exact sequence matches determined with a use of m-mers (m ≤ k). The between-anchor areas are further scanned for similar fragments. As a result, the algorithm translates a read to an edit script describing differences w.r.t. similar reads. Finally, the script is entropy-encoded. This multi-step approach renders superior compression ratios of DNA data.
The quality stream consumes the same number of bytes in FASTQ file as DNA data (there is one quality symbol per base). However, base quality in 3rd generation sequencing is expressed by one of 94 values and is non-redundant, by nature. Therefore, the quality data can be compressed losslessly to much less extent than DNA. CoLoRd employs a context-based approach inspired by ENANO [3] for this purpose. However, in many analyses base quality can be discretized to fewer values or even discarded at all. Therefore, a lossy compression mode for quality was introduced in CoLoRd allowing significant reduction of this stream.
Since sequence headers contribute marginally to the sizes of FASTA/FASTQ files, they are compressed with well-established token-based method analogously as in FQSqueezer [23] or ENANO.
In the following subsections a detailed description of selected compression aspects is presented.
Similarity graph construction
The initial algorithm step is filtering k-mers of the input reads. For this purpose KMC package [28] is executed with k automatically adjusted to the data set. K-mers with less than L = 4 (possibly, sequencing errors), or more than H = 80 occurrences (non-informative) in the entire read set are discarded. Note, that the parameters differ across priority modes (memory/balanced/ratio) and technologies (ONT/HiFi/CLR). For simplicity, we give the values in a default priority mode (memory) for ONT and, optionally, HiFi, when they differ significantly. Finally, to reduce the representation, we select subset U of k-mers, such that U = {u : h(u) mod f = 0} with h being a Murmur3 hash function and f equals to 12 (ONT) or 40 (HiFi). Parameter f balances the sensitivity and the memory footprint of the procedure.
As a next step, a graph G is constructed with reads being the vertices and edges representing k-mer similarities. A weight of an edge is a number of common k-mers in reads representation. An edge is directed from a read analyzed later towards one analyzed earlier. For memory efficiency, each vertex has at most max_candidates = 8 outgoing edges to the most similar reads.
The graph is built with a use of kmers2reads association table which maps k-mers to lists of read identifiers they are contained in. The structure is initially empty. The algorithm processes reads one by one according to the following procedure:
Extract all unique k-mers from a current read r.
Create an association table M indexed by read identifiers to store number of common k-mers between r and other reads.
- For each k-mer u ∈ r ∩ U:
- Get list Lid of read identifiers containing u (if any): Lid ← kmers2reads[u].
- For each identifier id ∈ Lid update M table: M[id] ← M[id] + 1
- Indicate in kmers2reads that u was contained in r: Lid ← (Lid, id(r)).
Sort table M in a descending order w.r.t. the number of common k-mers.
Add to the graph G a new vertex representing read r.
Add directed edges from the new vertex to max_candidates reads with most k-kmers in common (use the number of k-mers as a weight).
Anchor-based DNA encoding
The compression algorithm checks if consecutive reads contain neighbours in the similarity graph G. If not, a current read r is compressed in plain mode. Otherwise, a differential compression between the read and the neighbours is performed with the use of anchors, i.e., exact matches between sequences. The anchors are determined as follows:
Establish all m-mers of r (m ≤ k, it is automatically adjusted to the data set).
If r contains less then |r|/2 unique m-mers (|r| being the read length), consider it as highly repetitive and encode in plain mode.
Otherwise, determine m-mers of all reads in the neighbourhood of r in the graph G.
Establish intersections of m-mers from r and each of its neighbours. An intersection is represented as a list of common m-mers and two lists of their positions in r and its neighbour s. The reverse complements of the neighbours are also investigated.
Using common m-mers as alphabet symbols, determine the longest common subsequence (LCS) of r and s.
Join overlapping m-mers from LCS to obtain anchors between r and s.
As a next step, r is encoded with respect to its neighbour sbest with the largest sum of anchor lengths. The algorithm alternately processes non-anchor and anchor fragments of r and sbest. Anchors are encoded as lengths and positions in sbest. For non-anchor fragments, an edit script is determined with a use of Edlib library [31]. Then, based on the entropy estimation, the decision is made whether the edit script is used for representing the analyzed read fragment. If not, two scenarios are possible. (i) If the fragment is at least 64 symbols long, it is treated as a temporary read r′ and an attempt is made to encode it with respect to an alternative candidate with the largest total anchor length w.r.t. r′. (ii) If the fragment is shorter than 64 or no alternative candidates for r′ have been found, it is encoded in plain mode. Up to 3 levels of recurrence are allowed by the procedure.
Entropy encoding of DNA data
The edit script from the previous step is a sequence of tuples of the types: start_edit_script, start_plain, start_plain_with_Ns, anchor, match, insertion, deletion, substitution, skip, alternative_ref, main_ref, plain, end_of_read. The first three of them can start the edit script and we entropy encode the starting tuple type taking as a context 4 previous read-starting-tuple types.
If start_plain or start_plain_with_Ns appears, we store also the read length. Then, we have to encode the symbols A, C, G, T, or additionally N (in the later case). We store them using an entropy coder taking as a context 8 (in case of plain read) or 4 (in case of plain read with Ns) previous symbols.
The processing of truly-edit-script-described reads is more complex. First of all we store the main reference read id and information whether the reference read is taken as it is or it is reverse complemented. Then, for each successive tuple we entropy encode its type. The context is formed of 3 previous tuple types, 2 preceding symbols in the current read and the information whether recently we have (w.r.t. reference read): medium-size insertion (from 2 to 100 symbols), long insertion, medium-size deletion, long deletion, nothing of above. The remaining of the tuple is encoded depending of the type:
alternative_ref — it can happen that the same alternative reference read id will appear more than once in the edit script for the current read. Therefore, first we encode the information whether this read id has already been seen in the current edit script. If so, we store just its short “local” id (small number). Otherwise, we store its read id and the information whether it should be reverse complemented (just to serve as a reference for the current read) or not,
anchor — the anchor length is entropy encoded,
match — nothing needs to be encoded here,
insertion — the inserted symbol is entropy encoded. The context is formed of: the symbol at the corresponding position in the reference read and 4 previous symbols,
deletion — nothing needs to be encoded here since this tuple denotes just a single-symbol deletion w.r.t. a reference read,
substitution — the symbol is entropy encoded. The context is formed of: corresponding symbol in the reference read, 3 previous symbols in the current read. Additionally, in the ratio priority, the context is extended by an information whether the 3rd and 4th previous symbols in the current read are same,
skip — this tuple type denotes that the current position in the reference read should be changed by more than 16 positions. We entropy encode the skip length,
main_ref — nothing needs to be encoded here since this tuple type denotes that switch from an alternative reference read to the main reference read,
end_of_read — nothing needs to be encoded.
Quality scores
Lossless compression
In the lossless mode, the qualities are compressed as they are. The context is formed of several parts. First, we take two previous quantized quality values. The quantization is made to reduce the alphabet from 94 possible values to 11 values. Then, we extend the context by 4 DNA symbols from the read from: the corresponding position, the two previous positions, the following position. This strategy is similar to the one used in ENANO. We, however, do not gather similar contexts. Moreover, we use information from the analysis of the DNA part of the read. In balanced and ratio modes we extend the context by an information, whether the DNA symbol was encoded as: a match, an anchor, something else.
Lossy compression
The lossy quality compression must take into consideration two major concerns. The first one is the ability to reconstruct with some resolution the information about quality of individual base calls, which may be useful in the applications like variant calling. At the same time, it is important to preserve as accurately as possible the average read quality in a case it is employed for read filtering. As a result, it was decided to map original levels into bins representing poor, moderate, good, and very good bases. The default boundaries of the bins and values representing them were established experimentally as [Q0, Q6] → Q3, [Q7,Q13] → Q10, [Q14,Q25] → Q18, [Q26,Q93] → Q35 (for HiFi data, Q93 value is stored separately due to its special meaning in some applications). The context is formed of 6 previous, quantized, quality values. It is also extended by 4 DNA symbols and information about encoding, similarly as in the lossless processing. The aforementioned lossy compression modes are referred to as 4-fixed (ONT) and 5-fixed (HiFi).
In Supplementary Figure 1 we present on selected data sets how the binning affects average read quality. In the case of Zymo2 and Zymo2 R10 samples, the average read qualities were upper-bounded by Q15, thus the error introduced by binning was small on the entire range of reported Q levels. In particular, the average read qualities were mostly underestimated by less then 0.75 (Zymo2) and 0.5 (Zymo2 R10). The different situation was in the case of lun and NA12878 data sets, where the range of Q was larger (up to Q35 and Q50, respectively). The underestimation of average quality clearly increased with rising Q, which was expected as the wide range of high quality calls [Q26,Q93] is represented by a single value Q35. Importantly, at the level Q20, which is often considered as a threshold for good calls (1% error probability), the average quality was underestimated slightly (between 0.5 and 3 in the vast majority of reads). This, however, may still be unacceptable when read filtering based on the average quality is applied. Therefore, an additional 4-avg/5-avg modes were proposed where the value representing a bin is the average of all read base calls falling into that bin. During decompression, a floor and ceiling of this value are outputted in the appropriate proportion in order to accurately reconstruct the average read quality. As this feature renders only slightly larger archives compared to the fixed bin representation, it is used by default in CoLoRd.
To understand how 4-avg mode works, let us assume that the quality part of a read is:
“##”%##&’,,+&$*.+)%((*(’*,((+*.$+(0$,.,0/(63*%’’’+−#)(/+&$$”%&$$$$$###%*0,**9;−)65−*8−(/.3+B.*>51,(2
In compression, we calculate averages of symbols in ranges [Q0, Q6], [Q7, Q13], [Q14, Q25], [Q26, Q93]. Let us denote them as a7, a14, a26, a93. They are stored with precision 1/256. During decompression we load the stored values, which for the given example are: a7 ≈ 3.3984375, a14 ≈ 9.6484375, a26 ≈ 17.8125, a93 ≈ 29.33203125. Then we decode bin numbers from the stream and output quality score [ax] or [ax] to minimize the difference between the already decoded quality scores for a bin and the stored average score. In this way we obtain the scores:
$$%$$%$%$*+*$%++*+$+*++%*++*++*$+*2$++*33+33+%$%$*+$+*2+%$%$$%$%$$%$%$$+3*++3>*+33*+2+*3+3+>*+>33+*3
The additional quality modes in CoLoRd are:
2-fixed: [Q0, Q6] → Q1, [Q7, Q93] → Q13,
2-avg: as above, but with averages as bin representatives,
none: all Q values mapped to Q0,
avg: all Q values mapped to the average read quality.
The bin boundaries and representatives in all modes can be redefined by the user according to his needs.
Reducing execution time and memory requirements
By default, all reads without N symbols are used as potential references. This requires storing them in the memory which, in spite of packing four symbols per byte, results in excessive memory requirements for large sets of reads (e.g., human genome sequencing experiments). Therefore, we introduced a mechanism which decreases the probability of using a read as a potential reference as the compression proceeds. Let R indicate the reference genome length (which is estimated from k-mers). The idea is that after processing gR,2gR,3gR, … bases of the input file, with g being some parameter, the probability of adding current read to the collection of potential compression references decreases as 1, 1/2, 1/3, …, The memory saving concerns also kmers2reads structure, which does not have to store non-reference reads.
In order to maximize the utilization of available computational resources, CoLoRd is multi-threaded. The algorithm workflow is divided into independent blocks which exchange data using synchronized queues. In order to minimize synchronization overheads, the reads are processed in larger packs. The core of the processing consists of a thread pool whose elements, depending on the current workload, perform (i) similarity graph construction and (ii) anchor-based compression. Additionally, there are four threads responsible for entropy encoding of DNA, quality, and header streams as well as loading reads from FASTA/FASTQ files.
Reference-based compression
The reference-based compression is based on the pseudo-reads generated by sliding the window over the reference genome with some overlap. The length of the window is 20 times greater than the average read length estimated during k-mers counting. The pseudo-reads are used as an initial set of reference reads in the previously described differential compression. By default, the reference genome is stored in the archive, making it self-contained. Alternatively, the genome file has to be specified explicitly during the decompression. In this case, an MD5 signature of the reference is stored in the archive to make sure that the proper reference is given.
Supplementary Material
Acknowledgements
The work was supported by National Science Centre, Poland, project DEC-2019/33/B/ST6/02040 (MK, AG, SD) and by US National Institutes of Health (grant R01HG010040, U01HG010971 and U41HG010972 to HL).
Footnotes
Code availability
The source code of CoLoRd is available at https://github.com/refresh-bio/colord. The package can be installed via Bioconda at https://anaconda.org/bioconda/colord.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Competing interests
HL is a consultant of Integrated DNA Technologies, Inc and on the Scientific Advisory Boards of Sentieon, Inc and Innozeen Inc. The remaining authors declare no competing interests.
Data availability
Oxford Nanopore data sets:
M. bovis [30]: ERR4179765; M. bovis Bonito [30]: ERR4179766; vir [24]: ERR2708427–ERR2708436; lun [22]: PRJEB30781; Sorghum [25]: SRX4104135–SRX4104138; Zymo2: https://nanopore.s3.climb.ac.uk/Zymo-GridION-EVEN-BB-SN.fq.gz; Zymo2 R10 https://s3.climb.ac.uk/nanopore/Zymo-GridION-EVEN-BB-SN-PCR-R10HC-flipflop.fq.gz; Zymo2 Bonito ERR5396170; Zymo2 PromethION https://nanopore.s3.climb.ac.uk/Zymo-PromethION-EVEN-BB-SN.fq.gz; NA12878 [6]: http://s3.amazonaws.com/nanopore-human-wgs/rel6/rel_6.fastq.gz; CHM13: https://s3-us-west-2.amazonaws.com/human-pangenomics/T2T/CHM13/nanopore/rel6/rel6.fastq.gz; CHM13 Bonito: https://s3-us-west-2.amazonaws.com/human-pangenomics/T2T/CHM13/nanopore/rel7/rel7.fastq.gz
PacBio HiFi datasets:
Mosquito: SRX8642991, SRX8642992; Drosophila [27]: SRX499318; Strawberry [26]: SRR11606867; ATCC WGS: PRJNA546278; Mouse [26]: SRR11606870; HG002 HiFi [21]: SRR10382244, SRR10382245, SRR10382248, SRR10382249; CHM13 HiFi: SRR11292120–SRR11292123
PacBio CLR datasets:
Yeast: http://gembox.cbcb.umd.edu/mhap/raw/yeast_filtered.fastq.gz;
Arabidopsis: http://gembox.cbcb.umd.edu/mhap/raw/athal_filtered.fastq.gz; Macadamia [29]: SRR11191909;
48 plex: https://downloads.pacbcloud.com/public/dataset/MicrobialMultiplexing_48plex/48-plex_sequences/;
CHM1: http://datasets.pacb.com/2014/Human54x/fastq.html; HG002 CLR [32]: SRX5590586
Other:
CHM13 T2T assembly v1.1 [14]: GCA_009914755.3;
GRCh38 assembly: GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz; Sorghum assembly: GCF_000003195.3; M. bovis assembly: GCF_000183385.1;
Genome-In-A-Bottle v4.2.1 HG002 reference variants [20]: HG002_GRCh38_1_22_v4.2.1_benchmark.vcf.gz
References
- [1].Bonfield JK & Mahoney MV Compression of FASTQ and SAM format sequencing data. PloS One 8(3), e59190 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chandak S, Tatwawadi K, Ochoa I, Hernaez M & Weissman T SPRING: a next-generation compressor for FASTQ data. Bioinformatics 35(15), 2674–2676 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dufort y Álvarez G, Seroussi G, Smircich P, Sotelo J, Ochoa I & Martín Á ENANO: Encoder for NANOpore FASTQ files. Bioinformatics 36(16), 4506–4507 (2020). [DOI] [PubMed] [Google Scholar]
- [4].Dufort y Álvarez G, Seroussi G, Smircich P, Sotelo J, Ochoa I & Martín Á RENANO: a REference-based compressor for NANOpore FASTQ files. Bioinformatics 37(24), 4862–4864 (2021). [DOI] [PubMed] [Google Scholar]
- [5].Grabowski S, Deorowicz S & Roguski Ł Disk-based compression of data from genome sequencing. Bioinformatics 31(9), 1389–1395 (2015). [DOI] [PubMed] [Google Scholar]
- [6].Jain M et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotech 36, 338–345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jones DC, Ruzzo WL, Peng X & Katze MG Compression of next-generation sequencing reads aided by highly efficient de novo assembly. Nucleic Acids Res. 40(22), e171 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Koren S, Walenz BP, Berlin K, Miller JR & Phillippy AM Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li H Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32(14), 2103–2110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li H, Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34(18), 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Liu Y, Yu Z, Dinger ME & Li J Index suffix-prefix overlaps by (w,k)-minimizer to generate long contigs for reads compression. Bioinformatics 35(12), 2066–2074 (2018). [DOI] [PubMed] [Google Scholar]
- [12].Myers E The fragment assembly string graph. Bioinformatics 21(Suppl. 2), 79–85 (2005). [DOI] [PubMed] [Google Scholar]
- [13].Nicolae M, Pathak S & Rajasekaran S LFQC: a lossless compression algorithm for FASTQ files. Bioinformatics 31(20), 3276–3281 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nurk S et al. The complete sequence of a human genome. bioRxiv preprint https://www.biorxiv.org/content/10.1101/2021.05.26.445798v1 (2021). [Google Scholar]
- [15].Poplin R et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol 36(10), 983–987 (2018). [DOI] [PubMed] [Google Scholar]
- [16].Roguski Ł & Deorowicz S DSRC 2: Industry-oriented compression of FASTQ file. Bioinformatics 30(15), 2213–2215 (2014). [DOI] [PubMed] [Google Scholar]
- [17].Roguski Ł, Ochoa I, Hernaez M & Deorowicz S FaStore: a space-saving solution for raw sequencing data. Bioinformatics 34(16), 2748–2756 (2018). [DOI] [PubMed] [Google Scholar]
- [18].Stancu MC et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun 8, 1326 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vaser R, Sovic I, Nagarajan N & Sikic M Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zook JM et al. An open resource for accurately benchmarking small variant and reference calls. Nat. Biotechnol 37(5), 561–566 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods references
- [21].Cheng H, Concepcion GT, Feng X & Li H Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Charalampous T et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat. Biotechnol 7, 783–792 (2019). [DOI] [PubMed] [Google Scholar]
- [23].Deorowicz S FQSqueezer: k-mer-based compression of sequencing data. Sci. Rep 10, 578 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Depledge DP et al. Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat. Commun 10, 754 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Deschamps S et al. A chromosome-scale assembly of the sorghum genome using nanopore sequencing and optical mapping. Nat. Commun 9, 4844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hon T et al. Highly accurate long-read HiFi sequencing data for five complex genomes. Sci. Data 7, 399 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kim K et al. Long-read, whole-genome shotgun sequence data for five model organisms. Sci. Data 1, 140045 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kokot M, Długosz M & Deorowicz S KMC 3: counting and manipulating k-mer statistics. Bioinformatics 33(17), 2759–2761 (2017). [DOI] [PubMed] [Google Scholar]
- [29].Murigneux V et al. Comparison of long-read methods for sequencing and assembly of a plant genome. GigaScience 9(12), giaa146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vereecke N et al. High quality genome assemblies of Mycoplasma bovis using a taxon-specific Bonito basecaller for MinION and Flongle long-read nanopore sequencing. BMC Bioinformatics 21, 517 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sosić M & Sikić M Edlib: a C/C++ library for fast, exact sequence alignment using edit distance. Bioinformatics 33(9), 1394–1395 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wenger AM, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol 37, 1155–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Oxford Nanopore data sets:
M. bovis [30]: ERR4179765; M. bovis Bonito [30]: ERR4179766; vir [24]: ERR2708427–ERR2708436; lun [22]: PRJEB30781; Sorghum [25]: SRX4104135–SRX4104138; Zymo2: https://nanopore.s3.climb.ac.uk/Zymo-GridION-EVEN-BB-SN.fq.gz; Zymo2 R10 https://s3.climb.ac.uk/nanopore/Zymo-GridION-EVEN-BB-SN-PCR-R10HC-flipflop.fq.gz; Zymo2 Bonito ERR5396170; Zymo2 PromethION https://nanopore.s3.climb.ac.uk/Zymo-PromethION-EVEN-BB-SN.fq.gz; NA12878 [6]: http://s3.amazonaws.com/nanopore-human-wgs/rel6/rel_6.fastq.gz; CHM13: https://s3-us-west-2.amazonaws.com/human-pangenomics/T2T/CHM13/nanopore/rel6/rel6.fastq.gz; CHM13 Bonito: https://s3-us-west-2.amazonaws.com/human-pangenomics/T2T/CHM13/nanopore/rel7/rel7.fastq.gz
PacBio HiFi datasets:
Mosquito: SRX8642991, SRX8642992; Drosophila [27]: SRX499318; Strawberry [26]: SRR11606867; ATCC WGS: PRJNA546278; Mouse [26]: SRR11606870; HG002 HiFi [21]: SRR10382244, SRR10382245, SRR10382248, SRR10382249; CHM13 HiFi: SRR11292120–SRR11292123
PacBio CLR datasets:
Yeast: http://gembox.cbcb.umd.edu/mhap/raw/yeast_filtered.fastq.gz;
Arabidopsis: http://gembox.cbcb.umd.edu/mhap/raw/athal_filtered.fastq.gz; Macadamia [29]: SRR11191909;
48 plex: https://downloads.pacbcloud.com/public/dataset/MicrobialMultiplexing_48plex/48-plex_sequences/;
CHM1: http://datasets.pacb.com/2014/Human54x/fastq.html; HG002 CLR [32]: SRX5590586
Other:
CHM13 T2T assembly v1.1 [14]: GCA_009914755.3;
GRCh38 assembly: GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz; Sorghum assembly: GCF_000003195.3; M. bovis assembly: GCF_000183385.1;
Genome-In-A-Bottle v4.2.1 HG002 reference variants [20]: HG002_GRCh38_1_22_v4.2.1_benchmark.vcf.gz