

Abstract
Caspase-2, the second mammalian caspase to be identified and the most evolutionarily conserved caspase, has eluded classification. The lack of a profound phenotype in the caspase-2–deficient mouse resulted in decreased interest in caspase-2 for many years. However, advances in the field, including the identification of a potential activation complex and the development of methods to detect active caspase-2, now illuminate our understanding of the function of this caspase. These studies suggest that caspase-2 induces death through two pathways. First, caspase-2 induces cell death independently of the mitochondrial pathway, in a manner similar to that of ced-3, a caspase in Caenorhabditis elegans. Second, caspase-2 also induces cell death upstream of the mitochondrial pathway. The choice of pathway may depend on the type of death stimulus. The placing of caspase-2 upstream and independent of mitochondrial dysfunction provides a potentially new therapeutic target for aberrant cell death.
Programmed cell death (PCD) is mediated by the caspase family of cysteine proteases. Much of our initial knowledge of PCD was derived from studies performed with the Caenorhabditis elegans model system. In C. elegans, a single caspase, ced-3, mediates death. However, in mammalian systems, there are 13 caspases. We still do not understand whether all caspases have specific functions or whether they exhibit some redundancy. Among the caspases, caspase-2, the second mammalian caspase to be identified (1, 2), is the most evolutionarily conserved (3). This suggests that caspase-2 might play a critical role in PCD in mammals; however, the function of caspase-2 remains enigmatic. There are several reasons as to why our understanding of caspase-2 function remains limited, chief among these being the lack of a profound phenotype in caspase-2–deficient mice (2, 4). After these mice were generated, studies of caspase-2 were few and far between, despite the finding that neurons from capase-2–deficient mice are resistant to β-amyloid (Aβ)-induced death (5, 6).
In the decade since the characterization of caspase-2–deficient mice, there has been a waxing and waning of interest in caspase-2. In 2002, a series of papers suggested that caspase-2 played a role upstream of the mitochondrial pathway in etoposide-mediated cell death (7–9), but other studies have convincingly shown that caspase-2 is not required (10, 11). In 2004, the identification of p53-induced protein with a death domain (PIDD) as a potential activator of caspase-2 led to a steady increase in the number of studies that addressed the function of caspase-2 (12). However, there are still many unresolved conflicts in the literature concerning caspase-2. Indeed, several studies now highlight the potential function of caspase-2 in different cell death pathways (10, 11, 13, 14). To address these conflicts and to attempt to come to a consensus concerning the function of caspase-2, we need to establish what is really known about it and how future studies of caspase-2 should be approached.
A major problem in the study of caspases is the measurement of both their activation and activity. Although these are distinct processes, they are often incorrectly viewed as being the same; however, a caspase can be activated but have its activity suppressed. The most widely used method to measure caspase activity has been the use of synthetic peptide substrates. Knowledge of the cleavage specificity of individual caspases was used to generate peptide substrates and inhibitors for each caspase, and Val-Asp-Val-Ala-Asp (VDVAD) was identified as the peptide sequence that is recognized and cleaved by caspase-2. When this motif was first identified, it was also shown to be a substrate of both caspase-3 and caspase-7 (15). A study of the peptide substrates and inhibitors of caspases has now shown convincingly that all of the various peptide reagents used are good substrates or inhibitors of caspase-3, and that they are often better indicators of caspase-3 activity than of the caspase for which they were designed (16). This means that data derived from experiments involving VDVAD do not provide any information about the specific role of caspase-2.
Another widely used method to measure the activation of caspases is the detection of cleavage fragments of the caspase. Whereas the activation of effector caspases requires that they must first be processed (or cleaved), the activation of initiator caspases does not (17). Structurally, caspase-2 is classified as an initiator caspase because it possesses a long prodomain, which contains a caspase recruitment domain (CARD). Similar to other initiator caspases, dimerization, but not cleavage, of caspase-2 is required for its activation (18). After caspase-2 has formed a dimer, auto-cleavage occurs to release a small peptide, and the resultant p37 complex is more potent than is the full-length, uncleaved p51 species. It is likely that further processing of the p37 complex to the p19 fragment is mediated by another caspase. Once this occurs, caspase-2 has minimal activity. Thus, the measurement of the cleavage of caspase-2 or other initiator caspases is not an accurate reflection of either their activity or activation state.
The best measurement of caspase activity that is currently available is the affinity ligand–capture technique, which is used to trap active caspases. With this technique, Tu et al. captured the p51 form of caspase-2, which supports a mechanism in which the first step is activation of the zymogen and shows that cleavage to the p37 form is caspase-dependent because binding of the affinity ligand to the apical (first) caspase stops the caspase cascade at that point (11). We have used this technique to capture active caspase-2 p51 in primary neurons after their treatment with Aβ or the deprivation of trophic factors from their culture media (19). The study by Tu et al. showed that caspase-2 was captured after the induction of heat shock but not after the exposure of cells to etoposide, despite previous studies that suggested a function for caspase-2 in etoposide-mediated death (11). Another study performed with caspase-2–deficient cells has confirmed the lack of a requirement for caspase-2 in etoposide-mediated cell death (10).
The activation complex of caspase-2 is thought to contain the death adaptor proteins RAIDD (RIP-associated ICH-1/CED-3–homologous protein with a death domain) and PIDD (12). In this complex, RAIDD is likely to be the central partner because it contains a CARD with which to interact with caspase-2 and a death domain with which to interact with PIDD. Although RAIDD is required for activation of caspase-2 in neurons (20), PIDD may be dispensable (19). RAIDD is also required for heat shock–mediated cell death (11). PIDD forms a complex with either RAIDD to activate caspase-2 or with nuclear factor κB (NF-κB) essential modulator (NEMO) to activate NF-κB signaling (21). The relative abundance of each of these complexes in a particular cell may be critical in determining whether death occurs. Several studies have also suggested that phosphorylation of the prodomain of caspase-2 may inhibit its activity (22, 23) because this prevents the interaction of caspase-2 with RAIDD. More studies of the caspase-2 activation complex are needed to determine the mechanism of activation.
Once caspase-2 is active, how does it induce death? Several stimuli of cell death induce a caspase-2–dependent pathway that does not require other caspases (Fig. 1). These pathways may be evolutionarily conserved from C. elegans to mammals, because ced-3 does not require other caspases to cause cell death. It is likely that Aβ-mediated death of neurons does not require other caspases because caspase-2–deficient neurons are resistant to Aβ and, although caspase-3 becomes activated in response to Aβ, it is neither necessary nor sufficient for death (5).
Fig. 1.
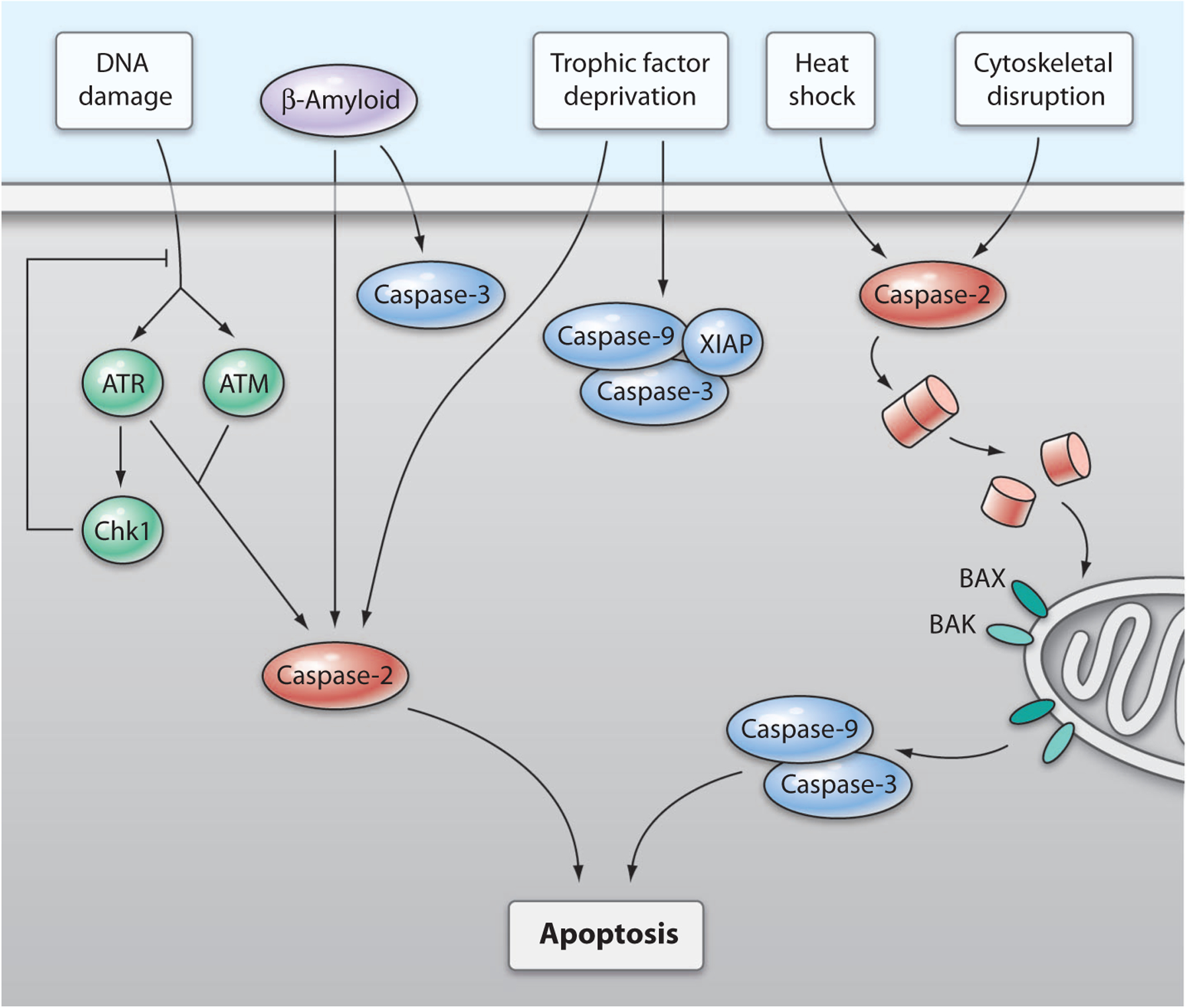
Proposed pathway of caspase-2–mediated cell death. Caspase-2 mediates cell death induced by various stimuli. Whether caspase-2 induces death directly or indirectly through the induction of mitochondrial permeabilization depends on the stimulus. DNA damage induces an ATR/ATM-dependent activation of caspase-2. Although this pathway is normally inhibited by Chk1, inhibition of Chk1 in p53-deficient cells induces a caspase-2–dependent death that is independent of mitochondrial mediators. Aβ-induced cell death in primary neurons is directly dependent on caspase-2. Although caspase-3 is also cleaved in this process, it is neither necessary nor sufficient to cause death. Deprivation of trophic factors from primary neurons induces cell death that is directly dependent on caspase-2 and also activates the apoptosome; however, the activities of caspase-9 and caspase-3 are suppressed in normal neurons by XIAP. Heat shock and cytoskeletal disruption activate caspase-2 and require induction of mitochondrial permeabilization, which is mediated by the proapoptotic Bcl-2 family members BAX and BAK, for death to occur.
Another pathway that involves caspase-2 is the DNA-damage response in p53-deficient cells (Fig. 1), which are induced to die in an ataxia telangiectasia mutated (ATM)– and ataxia telangiectasia and Rad3-related (ATR)–dependent fashion that is normally suppressed by checkpoint kinase 1 (Chk1) (13). This study was performed in zebrafish, in which morpholino oligonucleotides were used to reduce the expression of (knock down) candidate proteins, and in HeLa cells in which caspase-2 was incompletely knocked down with short hairpin RNAs. Together, these data are suggestive, but not conclusive, of there being a conserved pathway. The induction of two death pathways by a single stimulus, in which one pathway is dominant and the other is suppressed, is similar to the situation in trophic factor deprivation (TFD)–induced death in neurons. In TFD-induced death, although the caspase-2 pathway is predominant and causes cell death, apoptosome activation also occurs and is suppressed by X-linked inhibitor of apoptosis protein (XIAP), an inhibitor of caspases. In caspase-2–deficient neurons, the increased abundance of caspase-9 and the apoptosis-promoting protein second mitochondria-derived activator of caspase (Smac, also known as Diablo) overcome the inhibition of caspase activity by XIAP and restore sensitivity to TFD (24). The downstream substrates of caspase-2 in these pathways are unknown.
Caspase-2 also can act through the mitochondrial pathway in response to heat shock (10, 11) and cytoskeletal disruption (10), leading to activation of the apoptosome. Caspase-2–deficient cells are resistant to both of these stimuli. Caspase-2–mediated cleavage of the proapoptotic protein BID, which leads to mitochondrial permeabilization and apoptosome activation, occurs as a result of heat shock (25) and endoplasmic reticulum (ER) stress (14). However, other studies have failed to detect recombinant caspase-2–mediated cleavage of BID (25). It was suggested that only undetectable amounts of BID cleavage are required for heat shock–induced death; however, substantial cleavage of BID occurs in other caspase-2–dependent paradigms (10), and another interpretation might be that caspase-2 indirectly leads to cleavage of BID. In the ER stress study, the role of caspase-8 in the cleavage of BID was assessed; however, because the peptide inhibitors used were not specific, the involvement of caspase-8 in this process is uncertain (14). The link between caspase-2 and mitochondrial permeabilization still needs to be fully elucidated.
What is the best approach to study caspase-2 (and other caspases)? In the case of initiator caspases, the use of the affinity ligand–capture technique is the most appropriate measurement of their activity. This method has not been used widely for studies of apical caspases because it requires a large number of cells. Caspase-2 function can be assessed by molecular ablation, with the use of short interfering RNA knockdown, and by confirming these findings in the appropriate caspase-2–deficient cell types. As noted above, affinity ligand studies have shown that caspase-2 is the apical caspase in heat shock– and Aβ-induced cell death, and that whereas caspase-2–deficient mouse embryonic fibroblasts are resistant to heat shock and cytoskeletal disruption, caspase-2–deficient neurons are resistant to death induced by Aβ. Also critical to furthering our knowledge of caspase-2 function is the determination of the downstream substrates of caspase-2. As a therapeutic target, caspase-2 has certain advantages. In the nervous system, the expression of caspase-2 is neuron-specific; thus, inhibition or ablation of caspase-2 in the nervous system would only affect neurons. All the evidence suggests that caspase-2 is activated rapidly and is upstream or independent of mitochondria, which makes it an attractive target for ablation without disturbing mitochondrial function.
References
- 1.Kumar S, Kinoshita M, Noda M, Copeland NG, Jenkins NA, Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1 beta-converting enzyme. Genes Dev. 8, 1613–1626 (1994). [DOI] [PubMed] [Google Scholar]
- 2.Wang L, Miura M, Bergeron L, Zhu H, Yuan J, Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell 78, 739–750 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Lamkanfi M, Declercq W, Kalai M, Saelens X, Vandenabeele P, Alice in caspase land. A phylo-genetic analysis of caspases from worm to man. Cell Death Differ. 9, 358–361 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, Hara H, Moskowitz MA, Li E, Greenberg A, Tilly JL, Yuan J, Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 12, 1304–1314 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML, Caspase-2 mediates neuronal cell death induced by beta-amyloid. J. Neurosci 20, 1386–1392 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Troy CM, Shelanski ML, Caspase-2 redux. Cell Death Differ. 10, 101–107 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Lassus P, Opitz-Araya X, Lazebnik Y, Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 297, 1352–1354 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Robertson JD, Enoksson M, Suomela M, Zhivotovsky B, Orrenius S, Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J. Biol. Chem 277, 29803–29809 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Kumar S, Vaux DL, Apoptosis. A cinderella caspase takes center stage. Science 297, 1290–1291 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Ho LH, Read SH, Dorstyn L, Lambrusco L, Kumar S, Caspase-2 is required for cell death induced by cytoskeletal disruption. Oncogene 27, 3393–3404 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Tu S, McStay GP, Boucher LM, Mak T, Beere HM, Green DR, In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat. Cell Biol 8, 72–77 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Tinel A, Tschopp J, The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 304, 843–846 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R, Pascual J, Imamura S, Kishi S, Amatruda JF, Kanki JP, Green DR, D’Andrea AA, Look AT, Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 133, 864–877 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Upton JP, Austgen K, Nishino M, Coakley KM, Hagen A, Han D, Papa FR, Oakes SA, Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol. Cell. Biol 28, 3943–3951 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW, A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem 272, 17907–17911 (1997). [DOI] [PubMed] [Google Scholar]
- 16.McStay GP, Salvesen GS, Green DR, Overlapping cleavage motif selectivity of caspases: Implications for analysis of apoptotic pathways. Cell Death Differ. 15, 322–331 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Boatright KM, Salvesen GS, Mechanisms of caspase activation. Curr. Opin. Cell Biol 15, 725–731 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Baliga BC, Read SH, Kumar S, The biochemical mechanism of caspase-2 activation. Cell Death Differ. 11, 1234–1241 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Ribe E, Troy CM, Activation of caspase-2 in neuronal death: Functional implications for Alzheimer’s disease. Society for Neuroscience 37th annual meeting, San Diego, CA, 3 to 7 November 2007. [Google Scholar]
- 20.Wang Q, Maniati M, Jabado O, Pavlaki M, Troy CM, Greene LA, Stefanis L, RAIDD is required for apoptosis of PC12 cells and sympathetic neurons induced by trophic factor withdrawal. Cell Death Differ. 13, 75–83 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Tinel A, Janssens S, Lippens S, Cuenin S, Logette E, Jaccard B, Quadroni M, Tschopp J, Autoproteolysis of PIDD marks the bifurcation between pro-death caspase-2 and pro-survival NF-kappaB pathway. EMBO J. 26, 197–208 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nutt LK, Margolis SS, Jensen M, Herman CE, Dunphy WG, Rathmell JC, Kornbluth S, Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell 123, 89–103 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shin S, Lee Y, Kim W, Ko H, Choi H, Kim K, Caspase-2 primes cancer cells for TRAIL-mediated apoptosis by processing procaspase-8. EMBO J. 24, 3532–3542 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Troy CM, Rabacchi SA, Hohl JB, Angelastro JM, Greene LA, Shelanski ML, Death in the balance: Alternative participation of the caspase-2 and −9 pathways in neuronal death induced by nerve growth factor deprivation. J. Neurosci 21, 5007–5016 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonzon C, Bouchier-Hayes L, Pagliari LJ, Green DR, Newmeyer DD, Caspase-2-induced apoptosis requires bid cleavage: A physiological role for bid in heat shock-induced death. Mol. Biol. Cell 17, 2150–2157 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]